

Abstract
Stromal interaction molecules (STIMs), including STIM1 and STIM2, are single-pass transmembrane proteins that are located predominantly in the endoplasmic reticulum (ER). They serve as calcium ion (Ca2+) sensors within the ER. In the central nervous system (CNS), they are involved mainly in Orai-mediated store-operated Ca2+ entry (SOCE). The key molecular components of the SOCE pathway are well-characterized, but the molecular mechanisms that underlie the regulation of this pathway need further investigation. Numerous intracellular target proteins that are located in the plasma membrane, ER, cytoskeleton, and cytoplasm have been reported to play essential roles in concert with STIMs, such as conformational changes in STIMs, their translocation, the stabilization of their interactions with Orai, and the activation of other channels. The present review focuses on numerous regulators, such as Homer, SOCE-associated regulatory factor (SARAF), septin, synaptopodin, golli proteins, partner of STIM1 (POST), and transcription factors and proteasome inhibitors that regulate STIM-Orai interactions in the CNS. Further we describe novel roles of STIMs in mediating Ca2+ influx via other than Orai pathways, including TRPC channels, VGCCs, AMPA and NMDA receptors, and group I metabotropic glutamate receptors. This review also summarizes recent findings on additional molecular targets of STIM proteins including SERCA, IP3Rs, end-binding proteins (EB), presenilin, and CaMKII. Dysregulation of the SOCE-associated toolkit, including STIMs, contributes to the development of neurodegenerative disorders (e.g., Alzheimer's disease, Parkinson's disease, and Huntington's disease), traumatic brain injury, epilepsy, and stroke. Emerging evidence points to the role of STIM proteins and several of their molecular effectors and regulators in neuronal and glial physiology and pathology, suggesting their potential application for future therapeutic strategies.
Keywords: STIM, SOCE components, glutamate receptors, Ca2+ channels, calcium signaling, STIM regulators and effectors, store-operated calcium entry (SOCE), central nervous system
Introduction
Calcium ion (Ca2+) is a second messenger of crucial importance to neurons as it participates in the transmission of the depolarizing signals and contributes to synaptic activity and apoptosis. Cytoplasmic Ca2+ level in neurons is regulated in a comprehensive way via the components localized in the plasma membrane (PM) such as ion channels, exchangers, and pumps, as well as the components localized in the mitochondria, endoplasmic reticulum (ER), Golgi apparatus, and nucleus (Brini et al., 2014).
Plasma membrane Ca2+ channels in neurons are divided into three major groups according to their mechanism of action: voltage-gated Ca2+ channels (VGCC), receptor-operated Ca2+ channels (ROC: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors [AMPARs] and N-methyl-D-aspartate receptors [NMDARs]), and store-operated channels (SOC; Orai, transient receptor potential cation [TRPC] channels, arachidonate-regulated Ca2+ [ARC] channels) (Brini et al., 2014). In neurons, Ca2+ entry from the extracellular space is mediated via VGCCs and glutamate receptors (ionotropic: AMPAR, NMDAR, and metabotropic: mGluR) and is complemented by store-operated Ca2+ entry (SOCE) (Brini et al., 2014). Noteworthy, SOCE is the main Ca2+ source in resting neurons, while after depolarization Ca2+ influx is mediated mainly via VGCC, NMDAR and AMPAR (Brini et al., 2014). mGluR mediates both rapid transient depolarization and prolonged depolarization (Brini et al., 2014). Two systems responsible for Ca2+ extrusion from the cytoplasm to the extracellular milieu are PM Ca2+ adenosine triphosphatase (PMCA) and PM Na+/Ca2+ exchanger (NCX). While NCX affinity to Ca2+ is low, its capacity is high. Contrary, PMCA is characterized by opposite properties (Blaustein et al., 2002; Brini and Carafoli, 2011; Brini et al., 2014).
Mitochondria are also essential components of neuronal Ca2+ toolkit. They modulate intensity and duration of Ca2+ signals following extracellular stimuli (Duszyński et al., 2006). Since they have the ability to accumulate Ca2+, they function as Ca2+ buffers. Mitochondria localized in close proximity to Ca2+ channels are exposed to high Ca2+ level and can accumulate Ca2+ efficiently. This decreases local Ca2+ level and results in depletion of ER Ca2+ stores and activation of SOCE (Duszyński et al., 2006; Spät and Szanda, 2017). Special communication between mitochondria and the ER also enables Ca2+ release from the ER to mitochondria and its accumulation in the mitochondrial matrix. Increased Ca2+ concentration in the mitochondrial matrix stimulates the energy metabolism and boosts the activity of the tricarboxylic acid cycle enzymes, providing reducing equivalents to the respiratory chain and thus influencing the production of ATP (Brini et al., 2014). Calcium influx during SOCE results in mitochondrial Ca2+ uptake, which in turn boosts mitochondrial energy metabolism. If Ca2+ overload appears, it may cause cell apoptosis (Spät and Szanda, 2017). Thus, mitochondria link cell metabolism with Ca2+ signaling and homeostasis (Duszyński et al., 2006).
Neuronal Ca2+ signaling also appears to be pivotal in the nucleus. Cell depolarization propagates Ca2+ to the nucleus where they target the CREB transcription factor and DREAM transcriptional repressor, thereby affecting the transcription of many genes (Dick and Bading, 2010).
In neurons, the ER constitutes a vital Ca2+ storage organelle. Release of Ca2+ from the ER occurs via ryanodine receptor (RyR) and inositol-1,4,5-trisphosphate 3 (IP3) receptor (IP3R). Ca2+ release through IP3R occurs in response to mGluR activation in the PM. In turn, an elevated level of cytoplasmic Ca2+ is the major trigger for Ca2+ release via RyR in the mechanism known as Ca2+-induced Ca2+ release (CICR) (Brini et al., 2014). The decreased Ca2+ level in the ER is refilled by SOCE.
SOCE is based on the influx of Ca2+ from the extracellular environment through channels of the PM and the replenishment of these ions in the ER when their levels decrease because of release into the cytoplasm (Blaustein and Golovina, 2001; Putney, 2003). The depletion of ER Ca2+ stores is detected by stromal interaction molecules (STIMs), including STIM1 and STIM2 proteins, that are sensors of Ca2+ levels in the ER (Liou et al., 2005; Roos et al., 2005; Zhang et al., 2005). After the activation of IP3Rs, the drop in Ca2+ concentration in the ER (Berridge et al., 2000) causes the oligomerization of STIM proteins and their movement toward ER-PM junctions (Liou et al., 2005; Zhang et al., 2005; Wu et al., 2006; Serwach and Gruszczynska-Biegala, 2019). At these junctions, STIM proteins form complexes with proteins of Ca2+ release-activated channels (CRACs) that are formed by Orais or SOCs that consist of Orais and TRPC channels, leading to the activation of these channels (Liou et al., 2005; Mercer et al., 2006; Soboloff et al., 2006c; Liao et al., 2008; Salido et al., 2009; Saul et al., 2014; Albarran et al., 2016). Two types of Ca2+ currents are caused by Ca2+ store depletion: ICRAC (mediated by the activation of Orai1 and STIM1) and ISOC (involving Orai1, TRPC1, and STIM1; Desai et al., 2015). Channel activation results in Ca2+ influx from the extracellular milieu to the cytoplasm (Prakriya et al., 2006), and then Ca2+ is taken to the ER by sarco/endoplasmic reticulum Ca2+-adenosine triphosphatase (SERCA) pump.
The interaction between STIM proteins and Orai1 is widely known to be essential for the proper function of SOCE in non-excitable cells. SOCE is a ubiquitous cell signaling pathway that is also present in many other tissues, including the rodent and human brain (Moccia et al., 2015) where it is involved in the regulation of intracellular ionic equilibrium and determines the excitability of neurons (Emptage et al., 2001; Gemes et al., 2011; Sun et al., 2014; Majewski and Kuznicki, 2015).
STIM proteins were originally described in non-excitable cells. They are now known to be present in most cells, including excitable cells, such as neurons, where STIM2 protein is predominantly expressed (Berna-Erro et al., 2009; Skibinska-Kijek et al., 2009; Gruszczynska-Biegala et al., 2011; Steinbeck et al., 2011). The primary function of STIM2 in neurons was suggested to be the regulation of resting levels of Ca2+ in the ER and Ca2+ leakage (Gruszczynska-Biegala et al., 2011; Gruszczynska-Biegala and Kuznicki, 2013). The main function of STIM1 in neurons appears to involve the activation of SOCE (Gruszczynska-Biegala et al., 2011). Various studies have also identified STIM proteins in neuroglial cells, such as astroglia, tumor cells of astroglial origin, oligodendrocyte progenitor cells (OPCs), and microglia (Kettenmann and Bruce, 2013; Verkhratsky and Parpura, 2014; Kraft, 2015; Molnár et al., 2016). Although both STIM1 and STIM2 are expressed in astroglia, STIM1 is thought to be the more abundant isoform in these cells (Gruszczynska-Biegala et al., 2011; Kraft, 2015). Gruszczynska-Biegala et al. showed that Stim1 mRNA levels in both astroglial and neuronal cortical cultures were similar (Gruszczynska-Biegala et al., 2011).
In addition to interactions with and gating Orai, STIMs were found to recognize numerous interaction partners other than Orai. Thus, the present review focuses on the most important highly divergent target molecules of STIM proteins including positive and negative effectors and regulators in the central nervous system (CNS), mainly in neurons and glia. Recent data revealed a key role for STIM in several physiological and pathological conditions, including hypoxic/ischemic neuronal injury, traumatic brain injury (TBI), epilepsy, Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD; Berna-Erro et al., 2009; Gemes et al., 2011; Steinbeck et al., 2011; Sun et al., 2014; Zhang et al., 2014, 2015; Popugaeva et al., 2015; Rao et al., 2015; Vigont et al., 2015; Tong et al., 2016; Czeredys et al., 2018; Serwach and Gruszczynska-Biegala, 2019). Therefore, studies of SOCE and STIM proteins may elucidate pathogenic mechanisms that are involved in the development of these diseases. Consequently, positive and negative modulators of STIM protein function or translocation may have many potential therapeutic applications. Thus, we also briefly discuss the pathophysiological significance of STIM protein interactions with their target proteins.
STIM Protein Structure
STIM1 and STIM2 proteins are encoded by the STIM1 and STIM2 genes, respectively, in humans (Williams et al., 2001). They are type 1 transmembrane proteins that are localized in the ER, although STIM1 was also found in the PM (Williams et al., 2001; Liou et al., 2005; Roos et al., 2005; Keil et al., 2010). Both isoforms contain luminal and cytosolic domains (Figure 1; Soboloff et al., 2012; Moccia et al., 2015). The ER luminal N-terminal domain consists of a conserved cysteine pair, a Ca2+-binding canonical EF-hand (cEF) domain, a non-Ca2+-binding hidden EF-hand (hEF) domain, a sterile α-motif (SAM) with one (for STIM2) or two (for STIM1) N-glycosylation sites, and a transmembrane domain (TMD). The cytosolic C-terminus includes three coiled-coil regions (CC1, CC2, and CC3) with a STIM–Orai-activating region (SOAR). The SOAR contains four α-helices (Sα1, Sα2, Sα3, and Sα4) and a KIKKKR sequence, which is required for the activation of Orai1 (Yuan et al., 2009; Yang et al., 2012). The CRAC activation domain (CAD) and Orai1-activating small fragment (OASF) are both larger than the SOAR, contain a CC1 region, and activate Orai1 (Muik et al., 2009; Park et al., 2009). SOAR function is inhibited by an inhibitory helix that is localized in Cα3 (Yang et al., 2012). Downstream of SOAR is an acidic inhibitory domain (ID) that also mediates the fast Ca2+-dependent inactivation of Orai1 (Lee et al., 2009). The C-terminus tail of STIM proteins also contains a proline/serine-rich (PS) domain, a microtubule-interacting domain, and a polybasic lysine-rich domain that is responsible for phospholipid interaction in the PM (Soboloff et al., 2012).
Figure 1.
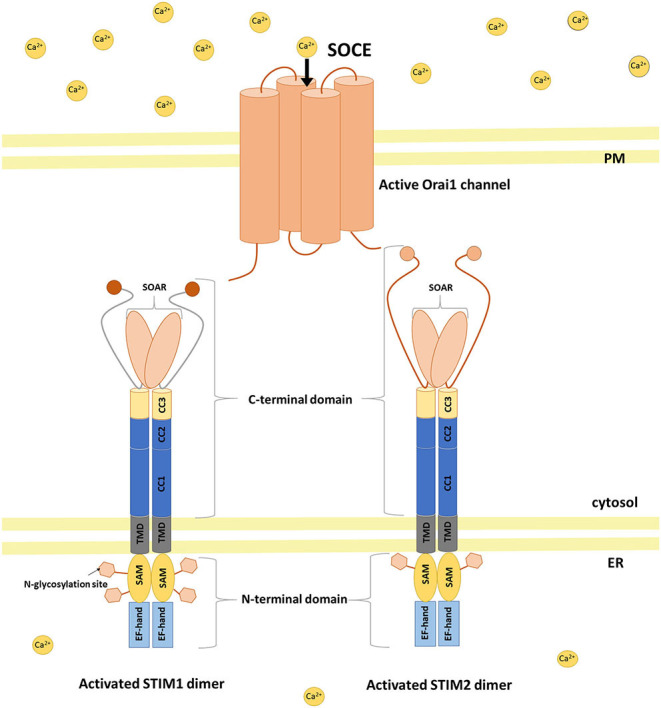
STIM activation and coupling with Orai1 in a mechanism of SOCE. Activation of STIM dimers is initiated by low Ca2+ concentration in the ER. STIMs oligomerize and migrate to ER–PM junctions where they activate SOC channels (e.g., Orai1) causing Ca2+ influx from the extracellular milieu to the cytoplasm and then refilling ER Ca2+ stores. SAM sterile alpha motif, TMD transmembrane domain, CC coiled-coil, SOAR STIM–Orai-activating region.
STIM1 and STIM2 proteins diverge significantly within the C-terminus (Figure 1). Strong evidence indicates that STIM proteins associate in vivo, and these interactions may be mediated by an association between CC regions of C-terminal ends of these proteins (Soboloff et al., 2006a). Notably, STIM2 has lower affinity for Ca2+ sensing compared with STIM1 because of three amino acid substitutions in cEF that allow STIM2 to detect smaller decreases in Ca2+ inside the ER lumen (Brandman et al., 2007; Hoth and Niemeyer, 2013).
STIM Proteins in the Physiology of Neurons, Autophagy, and Neurodegenerative Diseases
Physiology
By activating neuronal SOCE, STIM proteins play a pivotal role in the physiology of neurons (Serwach and Gruszczynska-Biegala, 2019). In neurons deprived of STIM2, the amount of mushroom dendritic spines, which are vital for memory storage, was decreased (Sun et al., 2014; Garcia-Alvarez et al., 2015; Yap et al., 2017). STIM2 also co-localizes with Ca2+/calmodulin-dependent protein kinase II (CaMKII) in dendritic spines and regulates its phosphorylation (Yap et al., 2017). STIM2-mediated SOCE sustained CaMKII activation and thus is important in the maintenance of dendritic spines. STIM2-Orai2-TRPC6 complexes regulate SOCE in mice hippocampal synapses and thus influence the number of dendritic spines. Orai-STIM2 complexes play an essential role in the formation of new synapses (Korkotian et al., 2017). All these studies demonstrate a vital role of STIM-mediated SOCE in both formation and maintenance of mushroom spines, and suggest its role in synaptic plasticity. Neuronal SOCE indeed takes part in long-term potentiation (LTP) and long-term depression (LTD), processes responsible for memory and learning (Serwach and Gruszczynska-Biegala, 2019). In Purkinje neurons (PNs), slow excitatory post-synaptic currents (EPSCs) were the result of TRPC3 activity. Lack of STIM1 resulted in no ER Ca2+ release and slow EPSCs (Hartmann et al., 2014). Thus, STIM1 is considered to refill the dendritic ER Ca2+ stores only under resting conditions. In resting cells, STIM1-mediated SOCE also caused an ubiquitination and degradation of Sp4 transcription factor (Lalonde et al., 2014). These results underlie an essential homeostatic function of STIM1-mediated SOCE in resting neurons.
Autophagy
Autophagy is stimulated in response to various types of cellular stress, including ER stress, oxidative stress, starvation of nutrients and growth factors, hypoxia and mitochondrial damage (Kroemer et al., 2010). Moderate ER stress can improve the ability of the ER to process unfolded or misfolded proteins and maintain cell survival. However, if the stress is prolonged or extensive, homeostasis within the cell is disrupted leading to apoptosis, which is primarily mediated by Ca2+ overload, or autophagy (Berridge et al., 2000; Bernales et al., 2006; Ding et al., 2007; Høyer-Hansen and Jäättelä, 2007). Autophagy, e.g., caused by oxygen and glucose deprivation and reoxygenation, may maintain cellular homeostasis, but its excessive level may lead to autophagic neuronal necrosis and apoptosis (Ahsan et al., 2019; Zhou et al., 2019). Differences in cytosolic Ca2+ levels associated with autophagy and apoptosis have been demonstrated in several cell lines. Calcium is mostly considered as an activator of autophagy, but there are some reports that Ca2+ suppresses autophagy (Høyer-Hansen et al., 2007; Cárdenas et al., 2010; Law et al., 2010; Parys et al., 2012; Wong et al., 2013).
However, the role of STIM1/Orai1 in autophagy and apoptosis in the CNS is still unclear, and there is not much work on the subject, thus underscoring the need for further research in this field. Proteasome inhibitors MG-132 and LA promoted the autophagy-mediated degradation of STIM1 and STIM2 and thus reduced SOCE in neurons (Kuang et al., 2016). The opposite is true in HEK293 cells where the stability of STIM1 was not affected by proteasome inhibitors, although thapsigargin-induced surface levels of STIM1 and SOCE were increased in cells pretreated with MG-132 (Keil et al., 2010). These differences are likely to be due to the different conditions for treating cells with protease inhibitors and may also exist depending on the type of cells used. Further, Kondratskyi et al. demonstrated that, in prostate cancer cells, SOCE inhibitor (ML-9) stimulates autophagosome formation and inhibits autophagosome degradation independent of SOCE and STIM1 (Kondratskyi et al., 2014). On the other hand, in prostate cancer cells (DU145 and PC3), resveratrol has been proposed to induce autophagy by regulating the function of STIM1 and then SOCE. Indeed, STIM1 overexpression restores resveratrol-induced reduction of SOCE as well as autophagic cell death induced by ER stress (Selvaraj et al., 2016). In line with this, STIM1 and SOCE have been shown to positively regulate oxidized low-density lipoprotein-induced autophagy in endothelial progenitor cells (Yang et al., 2017). Recently, hypoxia-induced Ca2+ release from the ER in neuron-like PC12 cells was modulated by STIM1/Orai1 (Hu et al., 2020). In addition, STIM1/Orai1 signaling induced by α2-adrenergic receptor agonist, dexmedetomidine, following hypoxia was mediated by a decrease of [Ca2+]i, leading to a reduction of autophagy. The results suggest that dexmedetomidine may have neuroprotective effects against oxidative stress, autophagy, and neuronal apoptosis after oxygen-glucose deprivation and reoxygenation injury through modulation of Ca2+-STIM1/Orai1 signaling (Hu et al., 2020). In turn, STIM1 has been shown to be not essential in hypoxia-mediated autophagy in both SHSY-5Y and HSG cells (Sukumaran et al., 2015). The available data suggest that STIMs influence autophagy differently depending on cell type and triggers of autophagy.
Neurodegenerative Diseases
Dysregulation of neuronal SOCE and changes in STIM expression levels are associated with various pathological conditions of the CNS such as hypoxic/ischemic neuronal injury, TBI, epilepsy, AD, PD and HD (Figure 2; Serwach and Gruszczynska-Biegala, 2019). Many studies have demonstrated the role of STIM2 in hypoxic/ischemic neuronal injury (Soboloff et al., 2006b; Vig et al., 2006; Berna-Erro et al., 2009). Hippocampal neurons, both in slices and in culture, showed reduced ER Ca2+ level during hypoxia, and STIM2 reduced Ca2+ overload during ischemic challenge (Berna-Erro et al., 2009). Stim2 knockout (KO) mice were better protected against cerebral ischemia (Berna-Erro et al., 2009). Thus, it seems that the absence of STIM2 may potentially constitute a protective strategy against stroke. STIM1 and STIM2 have also been implicated in epilepsy as they are up-regulated both in CA1 and CA3 regions of chronic epileptic mice (Steinbeck et al., 2011). Non-selective SOCE inhibitors rhythmized epileptic burst activity in epileptic hippocampal slices, suggesting that SOCE blockage may potentially bring positive effect in patients with epilepsy. STIM2 has also been shown to be overexpressed after TBI (Rao et al., 2015). The downregulation of STIM2 improved neuronal survival in models of TBI, decreasing neuronal apoptosis and preserving neurological function by alleviating mitochondrial disfunction and Ca2+ overload. STIM2 downregulation not only decreased Ca2+ release from the ER, but also reduced SOCE and dropped mitochondrial Ca2+ level, restoring its morphology and function. Downregulation of STIM2 has a neuroprotective effect and may be a target in TBI treatment (Rao et al., 2015). Dysregulation of SOCE also contributes to PD. A neurotoxin, which mimics PD in vitro, decreased level of TRPC1 and its interaction with STIM1, thus increasing neuronal death. Pharmacological inhibition of SOCE appears to be neuroprotective representing a potential target for PD drug discovery (Pchitskaya et al., 2018). In HD transgenic mice, over-activation of synaptic SOCE and enhancement of STIM2 expression resulted in the disruption of dendritic spines. STIM2 knockdown has been shown to normalize SOCE and prevent loss of dendritic spines. It seems that pharmacological modulation of SOCE and its components have neuroprotective effects in HD patients. On the other hand, in mice models of AD, impairment of SOCE and reduction of synaptic STIM2 proteins contributed to the destabilization of dendritic spines (Sun et al., 2014; Zhang et al., 2016). Since stabilization of dendritic spines is considered to prevent memory loss in AD patients, the modification of STIM proteins and SOCE may be a potential therapeutic target in the treatment of memory loss in these patients.
Figure 2.

Impact of Ca2+ overload on STIM expression level and SOCE in the development of CNS disorders. In hypoxic/ischemic neuronal injury, TBI, epilepsy, PD, and HD Ca2+ overload is associated with increased STIM expression level and SOCE and thus decreased ER Ca2+ level. Contrary, in AD Ca2+ overload results in decreased STIM expression level and SOCE and thus increased ER Ca2+ level. SOCE blockage and STIM downregulation seem to be neuroprotective in hypoxic/ischemic neuronal brain injury, epilepsy, TBI, HD, and PD, while in AD increased expression level of STIMs and SOCE enhancement appear to be neuroprotective.
Changes in neuronal SOCE may vary among pathological states of the CNS. SOCE blockage and STIM downregulation seem to be neuroprotective in hypoxic/ischemic neuronal brain injury, epilepsy, TBI, HD and PD, while in AD STIMs and SOCE appear to be neuroprotective (Figure 2).
Misfolded proteins and the associated ER stress are common features of some neurodegenerative diseases such as PD, AD and HD. These properties can further induce autophagy or apoptosis in neurons (Ghavami et al., 2014; Remondelli and Renna, 2017). Given the high sensitivity of neurons to ER Ca2+ store disturbances, STIM and SOCE have been proposed as potential targets for neuroprotection by reversing ER and mitochondrial stress-induced damage. Interestingly, blockade of SOCE reduced apoptosis mediated by oxidative stress in hippocampal neuronal HT-22 cells (Rao et al., 2013). Hawkins et al. demonstrated that in lymphocyte cells oxidative stress favors STIM1 trafficking and puncta formation, which confirms that STIM1 is regulated by the redox state (Hawkins et al., 2010). In turn, the formation of the STIM1 puncta, their translocation to the PM and the subsequent SOCE in HEK cells were disrupted by mitochondrial depolarization in mitofusin 2 dependent manner. These effects have been shown to be overcome by overexpression of STIM1 (Singaravelu et al., 2011). Consequently, STIM1 in 401L neuroblastoma cells provided protection against ER stress and mitochondrial oxidative stress causing cell death (Zhang and Thomas, 2016). Experiments performed on embryonic fibroblasts also reported that STIM1 rescue protected from oxidative stress and enabled cell survival by impairing the translocation of the apoptosis-inducing factor into the nucleus (Henke et al., 2012). All these findings suggest that STIM may indeed provide protection against cell death mediated by the ER and oxidative stress, which often precede neurodegeneration.
STIM-Binding Ca2+ Channels
According to the current state of knowledge, neuronal STIM proteins regulate both CRAC and SOC. Other channel proteins, such as L-type voltage-gated Ca2+ channels (VGCCs; Cav1.2, Cav1.3) and receptor/ligand-activated Ca2+ channels (AMPARs and NMDARs), couple or engage in an interplay with STIMs in the CNS in a modulatory way as part of SOCE signaling (Figure 3).
Figure 3.

Schematic overview of key regulators and effectors of STIM proteins in the CNS. Negative regulators (—|): SARAF prevents STIM1 activation and inhibits the STIM1-Orai association. SARAF silencing increases TRPC1-mediated Ca2+ entry. The PS1–γ-secretase complex cleaves the STIM1 transmembrane domain. Homer1a dissociates the STIM1-Orai1 complex. Lower dSEPT7 expression increases the amount of dSTIM-dOrai clusters. Positive regulators (→): The glutamate-mediated activation of mGluRs results in Ca2+ release from ER stores via IP3Rs and activates STIM-Orai coupling. EB3 forms complexes with STIM2, which promotes the formation of mushroom spines in hippocampal neurons. SEPT1/4 regulates the number of ER-PM junctions and enhances STIM1-Orai1 interactions. The STIM1-POST complex binds to SERCA and promotes ER Ca2+ refilling. Golli proteins interact with STIM1 and TRPC1 and thus enhance SOCE. SP interacts with STIM and Orai and determines synaptic plasticity. Positive effectors (+): STIM proteins increase Ca2+ influx via Orai, TRPC, and AMPARs. STIM2-mediated SOCE activates CaMKII and thus stabilizes mushroom spines. Negative effectors (–): STIM proteins decrease Ca2+ influx via L-type VGCCs and NMDARs.
Orai
As mentioned above, emptying ER Ca2+ stores causes STIM protein oligomerization and translocation of the oligomers toward the PM where they form complexes, known as puncta, with an Ca2+ selective ion channel protein, Orai1. We previously showed that the depletion of Ca2+ from the ER by thapsigargin, a selective SERCA inhibitor, increased the number of puncta-like structures with Yellow Fluorescence Protein (YFP)-STIM1 and Orai1 but not those with YFP-STIM2 and Orai1 (Klejman et al., 2009; Gruszczynska-Biegala et al., 2011). In contrast, a reduction of extracellular Ca2+ levels with ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) triggered the puncta formation of both YFP-STIM1/Orai1 and YFP-STIM2/Orai1. Other results showed that endogenous STIM1 and STIM2 can interact with Orai1, which was observed in a co-immunoprecipitation assay and in situ proximity ligation assay (PLA; Gruszczynska-Biegala and Kuznicki, 2013). The higher association between endogenous STIM2 and Orai1 in cortical neurons occurred in the presence of BAPTA-AM, membrane permeable Ca2+ chelator, and in a low-Ca2+ medium but not in the presence of thapsigargin. When SOCE was induced, the greatest number of PLA signals that corresponded to integrated STIM1 and Orai1 puncta was visible. The interaction between them was quantified and correlated well with the number of exogenous complexes that formed under the same conditions (Klejman et al., 2009; Gruszczynska-Biegala et al., 2011; Gruszczynska-Biegala and Kuznicki, 2013).
We can conclude that STIM1 and STIM2 can activate Orai1 channels and play different roles in neuronal SOCE (Berna-Erro et al., 2009; Klejman et al., 2009; Keil et al., 2010; Gruszczynska-Biegala et al., 2011; Gruszczynska-Biegala and Kuznicki, 2013; Sun et al., 2014; Majewski and Kuznicki, 2015). In rat cortical neurons, STIM1 mainly forms complexes with Orai1 and activates SOCE only after Ca2+ is completely emptied from the ER. This demonstrates its role in maintaining the level of Ca2+ in the ER. In contrast, STIM2 forms a hetero-complex with Orai1 to allow the regulation of resting intracellular Ca2+ levels and activation of constitutive Ca2+ influx after a slight decrease in Ca2+ levels in the ER.
In the rat cortex, SOCE is mainly mediated by Orai1-STIM1 complexes. In the mouse brain, in contrast, it is triggered either by Orai2 and STIM2 (cortex and hippocampus; Berna-Erro et al., 2009; Sun et al., 2014) or by Orai2 and STIM1 (cerebellum; Hartmann et al., 2014). Likewise, STIM2-gated SOC channels in dendritic mushroom spines are formed by a complex of Orai2 and TRPC6 (Zhang et al., 2016; Popugaeva et al., 2020). In turn, both STIM1 and STIM2 are involved in SOCE in sensory neurons in dorsal root ganglia. Moreover, both Orai1 and Orai3 contribute to SOCE in these neurons, where they may form homomultimers to mediate SOCE (Wei et al., 2017).
In addition to a canonical function as an ER Ca2+ level refilling toolkit, STIM1–Orai1-mediated SOCE was also shown to regulate gene expression and proliferation in mouse and human neural progenitor cells (NPCs) and thus is thought to be a key regulator of neurogenesis in mammalian cells (Somasundaram et al., 2014; Gopurappilly et al., 2018). The knockdown of Orai1 or STIM1 diminishes SOCE in NPCs. SOCE is not observed in NPCs from transgenic mice that lack Orai1 or STIM1 or in knock-in (KI) mice that express a Orai1 mutant. The deletion or suppression of STIM1 and Orai1 diminishes the proliferation of embryonic and adult NPCs both in vitro and in vivo in the subventricular zone (SVZ) in the adult mouse brain (Somasundaram et al., 2014). Domenichini et al. showed that SOCE in SVZ cells is mediated not only by STIM1 and Orai1 but also by TRPC1 (Domenichini et al., 2018). The pharmacological blockade of this process in mouse SVZ cells decreases proliferation and impairs self-renewal by shifting the type of cell division from symmetric to asymmetric, thereby reducing stem cell population (Domenichini et al., 2018). In human NPCs, SOCE has been shown to be significantly attenuated by the short-hairpin RNA/micro RNA targeting of STIM1 (Gopurappilly et al., 2018). Gopurapilly et al. investigated global gene expression in human NPCs with STIM1 knockdown and showed that signaling pathways that are associated with DNA replication and cell proliferation were downregulated, whereas post-synaptic cell signaling was upregulated in these cells (Gopurappilly et al., 2018). To understand the functional relevance of these gene expression alterations, these authors also measured the self-renewal capacity of NPCs with STIM1 knockdown and found a substantially smaller neurosphere size and number and a decrease in differentiation toward cells with a neuronal lineage. These findings demonstrate that STIM1-mediated SOCE in human NPCs regulates gene expression alterations, which is likely to modulate the differentiation and self-renewal of NPCs (Gopurappilly et al., 2018).
STIM1 and ORAI1 have been shown to be involved in Ca2+ signaling in both astroglia and glioblastoma cells. The knockdown of both STIM1 and Orai1 or Orai1 alone resulted in a reduction of SOCE in rat astrocytes (Moreno et al., 2012). The silencing of STIM1 or Orai1 was shown to reduce SOCE and CRAC currents in human glioblastoma cells (Motiani et al., 2013). Surprisingly, Ronco et al. found that Orai3 but not Orai1 is a dominant Orai homolog in astroglia (Ronco et al., 2014). Recent studies ascribed a role to STIM1, Orai1, and Orai3 in astroglial SOCE (Gao et al., 2016; Kwon et al., 2017). Additionally, the activation of SOCE in spinal astroglia promotes the production of proinflammatory cytokines, such as tumor necrosis factor α (TNF-α) and interleukin-6 (IL-6). The production of TNF-α and IL-6 was decreased by the knockdown of Orai1 or STIM1 (Gao et al., 2016). Interestingly, Orai1 and STIM2 knockdown minimized lipopolysaccharide (LPS)-induced TNF-α and IL-6 production (Gao et al., 2016). This research may provide a basis for assessing SOCE and its components for the treatment of chronic pain and other neurological diseases that are associated with astroglial overactivation.
Studies that focused on the optic nerve in mice identified Orai1 and STIM1 but not STIM2 in astrocytes, whereas Orai1, STIM1, and STIM2 in oligodendrocytes suggested that STIM1 may be localized in the cell soma, and STIM2 may be localized in myelin (Papanikolaou et al., 2017).
In cultured rat microglia, SOCE is mediated by Orai channels rather than TRPC channels (Ohana et al., 2009). Siddiqui et al. identified high levels of Orai1 and STIM1 in microglial podosomes, structures that are responsible for cell motility (Siddiqui et al., 2012). Another research group confirmed the expression of STIM1, Orai1, Orai2, and Orai3 in cultured mouse microglia and showed that the downregulation of STIM1 and Orai1 reduced SOCE in these cells (Heo et al., 2015). Michaelis et al. studied the role of Orai1 and STIM proteins in microglia using cells that were obtained from knockout mice (Michaelis et al., 2015). The results showed that SOCE was reduced in the absence of Orai1 or STIM proteins. SOCE was nearly absent in Stim1−/− microglia and substantially reduced in Orai1−/− microglia, whereas a less pronounced effect was observed in Stim2−/− microglia (Michaelis et al., 2015). Orai1 and STIM1 appear to be major components of microglial SOCE, and STIM2 is also a constituent of this signaling pathway in these cells (Kraft, 2015). Interestingly, recent studies showed that STIM1- and Orai1-mediated SOCE regulate phagocytic activity and cytokine release in primary murine microglia (Heo et al., 2015). Phagocytic activity, as well as LPS stimulation-mediated proinflammatory cytokine release (e.g., TNF-α and IL-6), was inhibited by SOCE inhibitors and STIM1 and Orai1 knockdown (Heo et al., 2015). This research suggests that STIM1 may be a new regulatory target for the prevention of an excessive proinflammatory response of microglia in neurodegenerative disorders.
TRPC Channels
In addition to Orai activation, STIM proteins may cause Ca2+ influx via TRPC channels, which are found in cells from all regions of the brain and spinal cord, with high TRPV (TRP channel subfamily V), TRPC, and TRPM (TRP channel subfamily M) expression (Verkhratsky et al., 2014). Nevertheless, TRPC1 and Orai1 activation is mediated by different STIM1 domains. TRPC1 is involved in SOCE, but like the other TRPC channels, it is unable to generate a current that resembles Ca2+-selective ICRAC (Albarrán et al., 2016). TRPC1 function depends on Orai1-mediated Ca2+ influx, which triggers the recruitment of TRPC1 into the PM, where it is activated by STIM1. TRPC1 is thought to modify the initial Ca2+ signal that is caused by Orai1 activation (Ambudkar et al., 2017).
TRPC1-mediated SOCE is essential for neuronal survival (Wang and Poo, 2005; Bollimuntha et al., 2006; Selvaraj et al., 2012). The STIM1-TRPC1 interaction is thought to be neuroprotective in both in vitro and in vivo models of PD (Selvaraj et al., 2012; Sun et al., 2017, 2018). In the human SH-SY5Y neuroblastoma cell line, SOCE mainly depends on the activation of TRPC1. A neurotoxin that caused the selective loss of dopaminergic neurons (DNs) in the substantia nigra pars compacta (SNpc) decreases TRPC1 expression, the TRPC1-STIM1 interaction, and SOCE but not Orai expression (Selvaraj et al., 2012). TRPC1 overexpression prevents the neurotoxin-mediated loss of SOCE and decreases ER Ca2+ levels and the unfolded protein response (UPR). Additionally, TRPC1-mediated Ca2+ entry activates the neuroprotective AKT pathway. STIM1 or TRPC1 but not TRPC3 silencing increases the UPR. Consistent with these results, Trpc1−/− mice have a higher UPR and lower number of DNs, similar to PD patients. The overexpression of TRPC1 in mice increased DN survival after neurotoxin treatment.
STIM1 was also shown to inactivate Ca2+ entry via VGCCs, which is detrimental to DNs. Thus, the STIM1-TRPC1 interaction was thought to inhibit Ca2+ influx via VGCC channels, thereby protecting DNs (Selvaraj et al., 2012). Subsequent research showed that TRPC1 regulates L-type VGCCs in SNpc neurons (Sun et al., 2017). The STIM1-TRPC1 interaction after store depletion reduced DN activity in wildtype (WT) but not Trpc1−/− mice. In Trpc1−/− SNpc neurons, L-type VGCC Ca2+ currents increased, STIM1-Cav1.3 interactions were attenuated, and the number of DNs decreased. After TRPC1 activation, L-type Ca2+ currents and Cav1.3 opening probability decreased, whereas they increased after STIM1/TRPC1 silencing. Additionally, store depletion increased the Cav1.3-TRPC1-STIM1 association. TRPC1 appears to suppress Cav1.3 activation proving that STIM1 is essential for DN survival (Sun et al., 2017).
Sun et al. showed that mesenchymal stem cell (MSC)-derived DNs, similar to native neurons, utilize TRPC1-mediated SOCE (Sun et al., 2018). Similar to SH-SY5Y cells, neurotoxin treatment in MSC-derived DNs decreased TRPC1 expression and SOCE. TRPC inhibition alleviated dopamine release and MSC-derived DN viability. These results indicate that ER Ca2+ levels that are maintained by TRPC1-mediated SOCE are neuroprotective. Neurotoxin exposure may cause alterations of SOCE and TRPC1-mediated Ca2+ homeostasis that may further induce ER stress and the UPR, leading to neurodegeneration. These results demonstrate that MSC-derived DNs are similar to native DNs, which potentially broadens the prospect of their usage for regenerative therapy in PD patients (Sun et al., 2018).
STIM1 and TRPC have also been shown to be involved in SOCE in astroglia. Antisense oligonucleotides that targeted the Trpc1 gene reduced SOCE in murine astrocytes (Golovina, 2005). An anti-TRPC1 antibody lessened SOCE in rat astrocytes (Malarkey et al., 2008). In spinal astrocytes, SOCE was predominantly subserved by TRPC3 (Miyano et al., 2010). Some studies ascribed these differences between Orai- and TRPC-mediated SOCE to the stage of astroglial development, suggesting that SOCE in immature and maturating astroglia is predominantly mediated by Orai, whereas SOCE in mature cells is predominantly mediated by TRPC1 (Kettenmann and Bruce, 2013; Verkhratsky and Parpura, 2014). Another study speculated that Orai1 and Orai3 are expressed in astroglial cells with abundant SOCE, whereas TRPC1 is restricted to astroglia where this process is attenuated (Kwon et al., 2017). STIM proteins, Orai1, and TRPM3 were identified as constituents of SOCE in astrocytes and oligodendrocytes of the mouse optic nerve (Papanikolaou et al., 2017). The developmental downregulation of Orai1 is consistent with TRPC channels as major components of mature astrocytes and oligodendrocytes, suggesting a potential role for Orai/STIM SOCE in immature glia and TRPM3 in mature glia (Verkhratsky et al., 2014).
Müller glia, a type of retinal glial cell, expresses STIM1 and requires the synergistic activation of both TRPC1 and Orai channels. The precise mechanism by which Orai and TRPC1 are activated by STIM1 has not been ascertained in these cells (Molnár et al., 2016).
VGCCs
VGCCs are Ca2+ channels that are present primarily in electrically excitable cells (as neurons), known as transducers of electrical activity that enable Ca2+ influx in response to subthreshold depolarizing stimuli or action potentials (Harraz and Altier, 2014). They are assembled from the pore-forming α1 subunit and accessory β and α2δ-like subunits (Heine et al., 2020). Various isoforms of the α1 subunit differ in voltage and Ca2+ sensitivity, which defines the specific kinetic properties of the channel. Thus, VGCCs are classified into low and high voltage-activated. The accessory β and α2δ-like subunits play a role in membrane trafficking and the modulation of kinetic properties of high voltage-activated Ca2+ channels (Campiglio and Flucher, 2015; Brockhaus et al., 2018). α2δ-like subunit and chemotaxis receptor domain containing 1 (Cachd1) alter the kinetic properties and surface expression of VGCCs (Cottrell et al., 2018; Dahimene et al., 2018). Cachd1 also impacts the gating and trafficking of low voltage-activated Ca2+ channels (Cottrell et al., 2018).
In excitable cells, VGCCs are the main route of Ca2+ entry in response to depolarizing stimuli. The main VGCC subtype that is present in neuronal, cardiac, and smooth muscle cells is Cav1.2, whereas Cav1.3 is the predominant subtype in DNs (Sun et al., 2017). Both VGCC subtypes were shown to be suppressed by STIM1 (Park et al., 2010; Wang et al., 2010; Harraz and Altier, 2014; Sun et al., 2017, 2018). Two independent studies of excitable cells showed that ER Ca2+ store depletion alleviates depolarization-mediated Cav1.2 activity, whereas the Cav1.2 response increases after functional impairments in STIM1 (Park et al., 2010; Wang et al., 2010). STIM1-Cav1.2 interactions are directly mediated by the SOAR domain (i.e., the same domain that activates SOCs) of STIM1 and C-terminus of the Cav1.2 α1 subunit (Harraz and Altier, 2014; Pascual-Caro et al., 2018). The influence of STIM1 on VGCCs is also associated with an increase in channel internalization from the PM. Despite reporting similar results, two studies suggested different inhibitory mechanisms. Park et al. proposed a mechanism that involves the attenuation of VGCC expression, whereas Wang et al. suggested a potential role for Orai1 in the inhibitory STIM1-Cav1.2 interaction because the simultaneous inhibition of both Orai1 and STIM1 was necessary to suppress Cav1.2 activity (Park et al., 2010; Wang et al., 2010).
The ability of STIM1 to regulate Orai1 and Cav1.2 is tissue specific. STIM1 appears to stimulate SOCE in non-excitable cells and inhibit VGCCs in excitable cells (Harraz and Altier, 2014). STIM1 was also shown to control the plasticity of L-type VGCC-dependent dendritic spines (Dittmer et al., 2017; Sather and Dittmer, 2019). The activation of neuronal STIM1 induces changes in the ER structure, which depends on L-type VGCCs. The NMDAR activation of L-type VGCCs triggers Ca2+ release from the ER, which in turn causes STIM1 aggregation and its coupling with L-type VGCCs and then inhibits the activation of this channel, thus increasing ER spine content and stabilizing mushroom spines (Dittmer et al., 2017). STIM1 deficiency is associated with AD and triggers SH-SY5Y cell death by upregulating Cav1.2 (Pascual-Caro et al., 2018). Thus, STIM1 KO cells may constitute an in vitro model to study the pathogenesis of AD and may be useful for understanding the role of STIM1 in neurodegeneration. In turn, as mentioned in section TRPC Channels above, TRPC1 in DNs facilitates STIM1-Cav1.3 interactions to suppress Cav1.3 activity, thereby reducing apoptosis and protecting DNs against neurotoxin-induced insults that lead to PD (Sun et al., 2017).
AMPA Receptors
AMPARs belong to the family of ionotropic glutamate receptors. They are thought to be the most significant mediators of excitatory neurotransmission in the CNS (Rogawski, 2011). They are assembled from four subunits (GluA1-4) and are mainly permeable to Na+ and K+ and to Ca2+ to a lesser extent. The subunit composition of AMPARs varies depending on the stage of development, region, and cell type (Song and Huganir, 2002). Phosphorylation of the GluA1 C-terminal tail regulates activity-dependent synaptic transport and channel features of the receptor (Esteban et al., 2003). Ser-831 and Ser-845 are two phosphorylation sites of GluA1 that have been well-characterized. Phosphorylation at Ser-831 by CaMKII and protein kinase C (PKC) regulates channel conductance, whereas cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA)-dependent phosphorylation at Ser-845 enhances channel open probability and promotes AMPAR internalization (Derkach et al., 1999). The PKA-dependent phosphorylation of GluA1 depends on the PKA scaffold AKAP150, which places PKA in proximity to its synaptic targets (Garcia-Alvarez et al., 2015) and cAMP-mediated dissociation of the regulatory subunit (rPKA) from the catalytic subunit (cPKA; Garcia-Alvarez et al., 2015). Both Ser-845 and Ser-831 phosphorylation sites play a role in LTP and LTD, forms of synaptic plasticity that are responsible for learning and memory (Esteban et al., 2003; Makino and Malinow, 2011).
Interestingly, STIM2 was shown to interact with AMPARs through a mechanism that is not associated with SOCE (Garcia-Alvarez et al., 2015). Recent research showed that STIM2 induces the cAMP/PKA-dependent delivery of GluA1 to the PM (Garcia-Alvarez et al., 2015). These authors suggested that STIM2 couples PKA to AMPARs and promotes the phosphorylation of GluA1 at Ser-845. They revealed a strong interaction between STIM2 and cPKA and weak STIM2 binding to rPKA and AKAP150, which may clarify the mechanism of interaction. Surprisingly, STIM2 and the phosphorylation of GluA1 at Ser-831 are negatively correlated. In STIM2-silenced neurons, phosphorylation at Ser-831 is enhanced. These findings indicate that STIM2 regulates the phosphorylation of GluA1 at both Ser-845 and Ser-831 but in a different manner. In turn, STIM1 overexpression was shown to increase GluA1 phosphorylation at Ser-845 in hippocampal synaptoneurosomes (Majewski et al., 2017).
Our previous study found that STIM proteins in primary rat cortical neurons may also interact with AMPARs in a SOCE-dependent manner, meaning that when ER Ca2+ stores are depleted, Ca2+ may enter through Orais, TRPC channels, and AMPARs (Gruszczynska-Biegala et al., 2016). The SOCE inhibitors ML-9 and SKF96365 decreased AMPA-induced Ca2+ influx, and the competitive AMPAR antagonists CNQX and NBQX inhibited SOCE. The induction of SOCE by thapsigargin resulted in AMPAR activation either directly through the recruitment of AMPARs to the PM or indirectly through unknown mechanisms. We also confirmed that both STIM1 and STIM2 proteins directly interacted with the GluA1 and GluA2 subunits of AMPARs. Moreover, STIM-AMPAR complexes appear to be located in ER-PM junctions (Gruszczynska-Biegala et al., 2016).
NMDA Receptors
NMDARs are ligand-gated ion channels that mediate Ca2+ influx when activated by glutamate, the main excitatory neurotransmitter in the mammalian CNS. NMDARs are tetramers that are composed of two glycine-binding NR1 (GluN1) and two glutamate-binding NR2 (GluN2) subunits (Cull-Candy and Leszkiewicz, 2004). Synaptic NMDARs consist mainly of NR1-NR2A or NR1-NR2A-NR2B receptors, and somatic NMDARs consist mainly of NR1-NR2B (Cull-Candy and Leszkiewicz, 2004). Ca2+ influx through NMDARs plays an important role in neuronal development, the formation of basal excitatory synaptic transmission, cell survival, and different forms of synaptic plasticity, such as LTP and LTD (Malenka and Bear, 2004; Nakazawa et al., 2004; Kerchner and Nicoll, 2008).
Pathologically high levels of glutamate and NMDA cause excitotoxicity by allowing high levels of Ca2+ to enter the cell (Sattler and Tymianski, 2000). Excessive NMDAR stimulation and prolonged increases in intracellular Ca2+ concentrations cause Ca2+ overload, which is considered a main cause of neuronal death in various neurodegenerative diseases that are associated with excitotoxicity, such as HD and AD (Marambaud et al., 2009; Pchitskaya et al., 2018; Serwach and Gruszczynska-Biegala, 2019). Attenuating intracellular Ca2+ overload is thus essential for limiting neuronal cell death under neuropathological conditions.
In hippocampal pyramidal neurons, SOCE can be activated by synaptic NMDAR stimulation, thus demonstrating its involvement in synaptic plasticity, such as LTP (Baba et al., 2003; Dittmer et al., 2017). Interestingly, glutamate release from neuronal terminals and NMDAR activation also induce SOCE in the PM of adjacent astrocytes possibly involving astrocytic mGluR5 (Lim et al., 2018). We recently reported that NMDARs contribute to Ca2+ influx in SOCE in rat cortical neurons (Gruszczynska-Biegala et al., 2020). The glutamate depolarization of neurons activates Ca2+ influx through NMDARs and L-type VGCCs, releases Ca2+ from the ER (Simpson et al., 1995; Emptage et al., 1999, 2001; Dittmer et al., 2017), and aggregates and activates STIM1, which then triggers SOCE (Rae et al., 2000; Emptage et al., 2001; Dittmer et al., 2017). In recent work, we found that endogenous STIM1 and STIM2 interact in situ and in vitro and co-localize with endogenous NMDAR subunits in rat cortical neurons. Emptying Ca2+ from ER stores induces a decrease in the physical association between endogenous STIM1 and NR2B and between STIM2 and NR2A (Gruszczynska-Biegala et al., 2020). Additionally, STIMs were shown to modulate NMDA-evoked intracellular Ca2+ levels by interacting with them. These data suggest that STIM1 and STIM2 are negative regulators of NMDA-induced intracellular Ca2+ elevations in cortical neurons (Gruszczynska-Biegala et al., 2020), in which they have been shown to inhibit the activity of L-type VGCCs (Park et al., 2010; Wang et al., 2010; Dittmer et al., 2017).
STIM Interacting Proteins
Most of the aforementioned studies on STIM function in the CNS focused on its canonical function in ER Ca2+ store refilling that results from the activation of SOCE. Recently, an increasing number of proteins have been reported to play a vital role in the regulation of Orai1-, TRPC-, and STIM1-associated Ca2+ signaling in both a SOCE-dependent and SOCE-independent manner in the CNS. This section focuses on several of the most important partners of STIM proteins that may also contribute to essential molecular processes in the CNS (Figure 3).
mGluRs
Group I metabotropic glutamate receptors (mGluRs) are widely distributed in the CNS and play a key role in synaptic transmission and plasticity. Excessive mGluR activation was shown in acute and chronic neurodegenerative disorders such as PD, AD, and HD (Crupi et al., 2019). The stimulation of group I mGluRs activates two signaling pathways (Hartmann et al., 2014). The first signaling pathway activates phospholipase C (PLC), thus inducing the formation of IP3, which interacts with IP3Rs to release Ca2+ from ER stores (Hartmann et al., 2014). The second signaling pathway involves the formation of slow excitatory post-synaptic potentials (EPSPs; Batchelor et al., 1994) and is mediated by TRPC3 (Hartmann et al., 2008).
STIM proteins also interact with TRPC3, and the STIM-mGluR association has been widely investigated. Hartman et al. showed that following mGluR1 activation, STIM1 proteins oligomerize and evoke SOCE through TRPC3 channels (Hartmann et al., 2008, 2014). Consistent with this, Ng et al. found that the activation of group I mGluRs with 3,5-dihydroxyphenylglycine (DHPG) in hippocampal neurons stimulated STIM1 oligomerization and its transport to the PM (Ng et al., 2011). Another research group showed that STIM1 in cerebellar PNs participates in mGluR1-dependent synaptic transmission and thus regulates cerebellar motor behavior (Hartmann et al., 2014). In mice with the PN-specific deletion of STIM1, mGluR1-dependent signaling was eliminated, and both IP3-dependent Ca2+ release from the ER and TRPC3-mediated EPSCs were attenuated. The disruption of these two pathways abolished cerebellar motor behavior (Hartmann et al., 2014). The role of STIM1 in synaptic plasticity was also investigated in hippocampal slices from adult transgenic Tg(STIM1)Ibd mice. STIM1 overexpression appears to disrupt mGluR LTD that is induced by DHPG (Majewski et al., 2017).
A recent analysis by Tellios et al. showed that in the absence of neuronal nitric oxide synthase (nNOS)-derived NO signaling along with the higher expression of mGluR1, STIM1 expression and cluster density are elevated in PNs (Tellios et al., 2020). These authors suggested that the overactivation of mGluR1 results in ER Ca2+ depletion and chronic STIM1 oligomerization. Because of the unlimited opening of TRPC3 channels, Ca2+ entry through STIM1-gated TRPC3 channels may be elevated, further leading to a reduction of dendritic spine integrity in PNs (Tellios et al., 2020). In contrast, under physiological conditions, nNOS/NO signaling maintains Ca2+ homeostasis in neurons by reducing its influx through mGluR1 and inducing the S-nitrosylation of STIM1. As STIMs and Orais as well as NMDARs and mGluRs are expressed in the microvascular endothelial cells of the brain (LeMaistre et al., 2012; Negri et al., 2020), we can suspect that these endothelial receptors may also be regulated by STIMs. Especially since the Ca2+ response to glutamate by activating mGluR1 and mGluR5 is initiated by endogenous Ca2+ release from the ER through IP3R3 and sustained by SOCE, resulting in a rapid NO release (Negri et al., 2020).
Homer Family Proteins
Homer family proteins are post-synaptic scaffolding proteins that regulate glutamatergic signaling and intracellular Ca2+ concentrations in neurons (Chen et al., 2012). Homer was reported in neurological disorders, including AD, TBI, and schizophrenia (Luo et al., 2012). These proteins are divided into two major groups: short-form Homer1a proteins and long-form Homer1b/c, Homer2, and Homer3 proteins. Both groups have N-terminal EVH1 domain that is involved in protein interactions, and long-form proteins also have a C-terminal coiled-coil domain that is involved in self-association (Chen et al., 2012). Homer1b/c has been shown to alter SOCE through an association with STIM1 and Orai1 in human platelets. This interaction between STIM1, Homer1b/c, and Orai1 is enhanced by thapsigargin (Jardin et al., 2012). Thapsigargin also induces Homer1, STIM1, and L-type VGCC associations in HEK-293 cells (Dionisio et al., 2015).
In the CNS, SOCE antagonists and STIM1-targeted small-interfering RNA (siRNA) increase the expression of Homer1a mRNA and the amount of Homer1a protein (Li et al., 2013). The knockdown of Homer1a expression partially reverses this effect. Moreover, SOCE inhibition appears to protect neurons against oxidative stress through the upregulation of Homer1a expression (Li et al., 2013). SOCE inhibitors also prevented mitochondrial dysfunction and activation of mitochondrial apoptotic factors after MPP+ injury. Since mitochondrial dysfunction is thought to play a crucial role in PD, it seems that SOCE may be a potential target in the treatment of PD patients (Li et al., 2013).
Homer1a affects STIM1-Orai1 associations, inhibits SOCE, and alleviates glutamate-induced cell death after oxidative stress injury (Rao et al., 2016). Both thapsigargin-induced and glutamate-mediated STIM1-Orai1 associations are attenuated by Homer1a overexpression. After thapsigargin-induced ER Ca2+ store depletion, the association between STIM1 and Homer1a decreases, whereas no change occurs in the Homer1a-Orai1 association. These interactions between STIM1, Homer1a, and Orai1 are similar to interactions between STIM1 and Homer1b/c or STIM1, Homer1b/c, and Orai1 (Jardin et al., 2012; Dionisio et al., 2015; Rao et al., 2016). Therefore, Homer1a might dissociate STIM1-Orai1 complexes and downregulate SOCE through negative competition with Homer1b/c (Rao et al., 2016). The regulation of Homer1a and SOCE inhibition may be a potential therapeutic target for the treatment of neurological disorders, the etiology of which is associated with oxidative stress.
SARAF
SOCE-associated regulatory factor (SARAF) is a 339-amino-acid type I transmembrane protein that has exceptionally high transcript levels in neuronal tissues (Palty et al., 2012). It is assembled from a single N-terminal transmembrane domain and a C-terminal domain that is located in the ER or PM (Palty et al., 2012; Albarran et al., 2016). The activation of SARAF involves the intraluminal region, whereas the interaction with SARAF engages the cytosolic region (Jha et al., 2013). Gene product of TMEM66, SARAF was identified as a biomarker linked to AD (Twine et al., 2011).
SARAF modulates STIM1-regulated Ca2+ entry, which includes both SOCE and Ca2+ influx through ARC channels (Palty et al., 2012; Albarran et al., 2016). SARAF prevents the spontaneous activation of STIM1, regulates STIM1–Orai1-mediated SOCE, facilitates the slow Ca2+-dependent inactivation of SOCE, and promotes STIM1 deoligomerization after Ca2+ store refilling (Palty et al., 2012; Jha et al., 2013). SARAF was also reported to modulate cytosolic and ER Ca2+ levels (Palty et al., 2012). The molecular mechanism of action of SARAF was described by Jha et al., who showed that SARAF interacts with the C-terminal inhibitory domain (CTID) of STIM1 to mediate the slow Ca2+-dependent inactivation (SCDI) of Orai1-forming CRAC (Jha et al., 2013). Under resting conditions, when intracellular Ca2+ stores are filled with Ca2+, CTID facilitates the access of SARAF to the SOAR to keep STIM1 in an inactive state, resulting in the inhibition of STIM1-Orai communication. Upon Ca2+ store depletion, SARAF dissociates from STIM1 to allow the activation of STIM1-Orai1 complexes at ER-PM junctions, thereby leading to SOCE (Jha et al., 2013).
SARAF is constitutively expressed in the PM. It was also shown to modulate Ca2+ entry through ARC channels in the SH-SY5Y neuroblastoma cell line (Albarran et al., 2016). ARC channels are heteropentameric complexes that consist of three Orai1 and two Orai3 subunits and PM-resident STIM1. The overexpression of SARAF in neuroblastoma cells attenuated the arachidonic acid (AA)-induced Ca2+ response, and the transfection of SARAF siRNA enhanced AA-stimulated Ca2+ influx via ARC channels. The results suggest that SARAF is a negative regulator of AA-induced Ca2+ signaling (Albarran et al., 2016).
SARAF was also shown to interact with TRPC1 in a STIM1-independent manner in both STIM1-defficient NG115-401L and endogenous STIM1-expressing SH-SY5Y neuroblastoma cells (Albarrán et al., 2016). The silencing of SARAF expression in STIM1-deficient cells increased TRPC1-mediated Ca2+ entry. In cells that endogenously expressed STIM1, the interaction between SARAF and TRPC1 was not associated with STIM1. This regulation of Ca2+ entry is thought to protect the cell from Ca2+ overload and adjust the influx of Ca2+, which was previously reported for the regulation of SOCE and ARC channels (Albarran et al., 2016). The silencing of SARAF expression did not influence TRPC6-mediated Ca2+ entry, in contrast to TRPC1, meaning that SARAF is unlikely to regulate the TRPC6 function. SARAF appears to play a negative regulatory role in both STIM1–Orai1- and STIM1–TRPC1-mediated Ca2+ entry by destabilizing STIM1/Orai1 complexes (Palty et al., 2012). Notably, a recent study reported a reduction of the expression of STIM1 and SARAF in the ischemic cortex, indicating that SARAF may be a new neuroprotective target for the treatment of stroke (La Russa et al., 2020).
Septins
Septins are a class of evolutionary conserved GTPases that are assembled into hexameric or octameric complexes organized into linear filaments or other higher-order structures. They function as diffusion barriers and intracellular scaffolds in cells during various cellular processes (Mostowy and Cossart, 2012). The altered septin function may contribute to synaptic dysfunction in neurodegenerative diseases (Marttinen et al., 2015).
Septins were shown to regulate SOCE in both non-excitable mammalian cells (Sharma et al., 2013) and Drosophila neurons (Deb and Hasan, 2016, 2019; Deb et al., 2016, 2020). In Drosophila, septins are classified into several groups: dSEPT1, dSEPT2, dSEPT4, dSEPT5 and dSEPT7. The dSEPT7, dSEPT1, and dSEPT4 groups form linear hexamers. dSEPT1 and dSEPT4 occupy the central position, and dSEPT7 is localized in terminal positions of the hexamer. These hexamers are then linked and form linear septin filaments (Mostowy and Cossart, 2012; Mavrakis et al., 2014). The molecular mechanism of SOCE regulation and contribution of different subgroups of septins to the regulation of SOCE is complex.
The simultaneous knockdown of dSEPT1 and dSEPT4 reduced SOCE in Drosophila flight circuit neurons (Deb et al., 2016). The knockdown of these subgroups results in the loss of septin filaments and loss of the diffusion barrier, which has a negative influence on Orai activation by STIM (Deb and Hasan, 2016). dSEPT1 and dSEPT4 appear to function as positive regulators of SOCE in Drosophila neurons (Deb et al., 2016; Deb and Hasan, 2019). On the other hand, reduction of dSEPT7 had no significant influence on SOCE in Drosophila neurons. Nevertheless, the reduction of dSEPT7 in primary neurons that had low levels of Drosophila STIM1 (dSTIM1) improved SOCE (Deb et al., 2016). Additionally, STIM1 is necessary for SOCE through Orai channels also in SEPT7 knockdown human neural progenitor cells (hNPCs; Deb et al., 2020). In resting neurons with low dSEPT7 expression, the intensity of dSTIM and resulting dSTIM-dOrai clusters that are observed near the ER–PM region increased (Deb et al., 2016, 2020). Similar STIM1 and Orai1 reorganization was shown at the cell surface in SEPT7 knockdown neurons that were differentiated from hNPCs (Deb et al., 2020). Deb et al. suggested that the partial reduction of dSEPT7 leaves hexameric complexes intact but results in the formation of smaller septin filaments because filament elongation requires dSEPT7 (Deb et al., 2016). Shorter SEPT7 filaments support dSTIM migration to the peripheral ER in resting neurons, promoting Orai channel opening independently from either ER-Ca2+ store depletion or Ca2+ release through IP3Rs. Similarly, SEPT4 regulates the number of the ER-PM junctions and enhances STIM1-Orai1 interactions within junctions in human cells (Katz et al., 2019). Store-independent dOrai Ca2+ influx results in higher cytosolic Ca2+ concentrations in resting neurons (Deb and Hasan, 2016). The loss of dSEPT7 influences the constitutive activation of Orai channels in resting neurons by uncoupling septin heteromers from ER-PM junctions, thus allowing the STIM interaction with Orai (Deb et al., 2016, 2020).
Septins (e.g., dSEPT1, dSEPT4, and SEPT7) appear to perform antagonistic rather than synergistic functions in Orai channel activation by STIM. This discrepancy may be caused by a different assembly of septins during complex formation and by differences in subsequent filament structure (Deb and Hasan, 2019). Altogether, these results could link alterations of septin expression to impairments in STIM1-dependent SOCE in human neurodegenerative diseases.
Golli Proteins
Golli proteins are isoforms of myelin basic protein (MBP) that are abundantly expressed in immune cells and the CNS (Paez et al., 2011). It is upregulated in adult OPCs and microglia in multiple sclerosis lesions (Filipovic et al., 2002). Myelin abnormalities have been implicated in neurodegenerative and neuropsychiatric diseases and Golli-MBP expression was increased in the aging brains (Siu et al., 2015). Golli proteins were shown to be components of remyelination that is caused by treatment with taxol in demyelinating transgenic mice, thus demonstrating their role in early stages of OPC proliferation and migration (Moscarello et al., 2002). Although no evidence suggests the occurrence of SOCE in oligodendroglia cells, this process was detected in OPCs and brain slices and shown to be mediated by STIM1 and TRPC1. Additionally, SOCE in these cells is positively modified by golli proteins that interact with STIM1 and TRPC1 (Paez et al., 2011). Changes in golli protein expression alter VGCCs and SOCs to mediate the migration and proliferation in OPCs that influence their maturation and survival (Paez et al., 2009a,b; Paez et al., 2007). Another study showed that golli protein overexpression increases the mitogen-stimulated proliferation of OPCs through the activation of SOCE, which is essential for cell division (Paez et al., 2009a). In OPCs, the proliferation of golli protein-KO cells was less robust, and the duration of the cell cycle increased. Golli proteins were also reported to increase apoptotic cell death, which was associated with an increase in Ca2+ influx through VGCCs (Paez et al., 2009a). Notably, the C-terminus of STIM1 was also shown to bind to MBP in a brain extract in a pull-down assay, which is likely an epitope that is shared with golli protein (Walsh et al., 2010).
POST and SERCA
Several molecular mechanisms are responsible for the increase in cytosolic Ca2+ concentrations after cell depolarization, including SERCA, PMCA, NCX, and mitochondrial Ca2+ uptake. Among these mechanisms, SERCA and PMCA are regulated by STIM1, combined with an adaptor protein called partner of STIM1 (POST; Krapivinsky et al., 2011). After ER Ca2+ store depletion, the STIM1-POST complex binds to SERCA and keeps it close to Ca2+ entry sites on the PM to promote the refilling of ER Ca2+ stores (Krapivinsky et al., 2011). The STIM1-POST complex also inhibits PMCA activity, which is associated with Ca2+ outflow from the cytoplasm to the extracellular space (Ritchie et al., 2012). STIM1 appears to play opposing roles at the same time. However, SERCA contributes more to cytosolic Ca2+ clearance than PMCA, especially in PNs (Fierro et al., 1998). Thus, after PM depolarization, STIM1 reduces cytoplasmic Ca2+ concentrations. Additionally, Ryu et al. showed that STIM1 contributes to SERCA-dependent cytosolic Ca2+ clearance in the soma of firing PNs (Ryu et al., 2017). These authors proposed that the STIM1-POST complex may pull SERCA into the vicinity of VGCCs. If so, then SERCA could buffer Ca2+ influx more rapidly after depolarization, which can optimize the Ca2+-clearing and -buffering function of SERCA and prevent excessive cytosolic Ca2+ concentrations during repetitive firing (Ryu et al., 2017).
STIM1 mediates the sequestration of cytosolic Ca2+ ions by SERCA. It may also regulate neuronal excitability. Nevertheless, still unknown is whether this occurs only with high-firing neurons, such as PNs, or also with slow-firing neurons, such as pyramidal and cortical neurons (Ryu et al., 2017).
In this context it is worth noting that the function of SERCA can be also regulated by ER lumen residents calreticulin and ERp57 oxidoreductase (Li and Camacho, 2004). Thus, we suspect that these proteins may interact with STIM in neurons, especially since they are expressed therein (Coe and Michalak, 2010). ERp57 and STIM1 formed complexes in vivo and in vitro to inhibit STIM oligomerization and SOCE in mouse embryonic fibroblasts (Prins et al., 2011). Interestingly, in megakaryocytes from healthy individuals, calreticulin regulated SOCE activation through interaction with ERp57 and STIM1. In megakaryocytes from patients with mutated calreticulin, destabilization of the complex between calreticulin, ERp57 and STIM1 was observed, leading to enhanced SOCE and thus to abnormal cell proliferation (Di Buduo et al., 2020). ERp57 is associated not only with myeloproliferative neoplasms, but also with many disease states of CNS, such as prion disorders and AD, where ERp57 and calreticulin have been shown to prevent amyloid aggregation (Coe and Michalak, 2010). Similarly, SERCA-mediated Ca2+ dysregulation is associated with neuropathological conditions, such as affective disorders and neurogenerative diseases (Britzolaki et al., 2020).
IP3Rs
Receptors that activate PLC cause the formation of IP3, which triggers both Ca2+ release from ER stores through IP3Rs and Ca2+ influx from the extracellular milieu, which is mediated by SOCE. IP3Rs are thought to regulate SOCE by mediating ER Ca2+ release. Under physiological conditions, store depletion causes STIM and IP3R accumulation near the PM, an association between STIM and Orai, and the activation of SOCE. In Drosophila neurons with mutant IP3Rs, SOCE was attenuated (Chakraborty et al., 2016; Deb et al., 2016) and this attenuation was reversed by STIM and Orai overexpression. The authors speculated that after ER Ca2+ store depletion in Drosophila neurons, IP3R translocation to the ER-PM junction triggers the coupling of STIM to Orai, leading to the activation of SOCE (Chakraborty et al., 2016).
The same research group also reported the enhancement of spontaneous Ca2+ influx from the extracellular milieu and loss of SOCE in Drosophila pupal neurons with mutant IP3Rs (Chakraborty and Hasan, 2017). Both spontaneous Ca2+ influx and the attenuation of SOCE were reversed by dOrai and dSTIM overexpression. Additionally, the expression of VGCCs decreased, and the expression of trp mRNAs and TRPC protein increased in mutant neurons, suggesting that these channels might be associated with the increase in spontaneous Ca2+ influx. Spontaneous Ca2+ influx likely compensates for the loss of SOCE in Drosophila IP3R mutant neurons and maintains intercellular Ca2+ homeostasis (Chakraborty and Hasan, 2017). The overexpression of dSTIM in insulin-producing neurons and aminergic neurons also improves SOCE and restores flight in a flightless Drosophila IP3R mutant (Agrawal et al., 2010). These authors suggested that IP3R-mediated Ca2+ release couples to SOCE via dSTIM/dOrai in Drosophila flight circuit neurons, thereby allowing dSTIM to compensate for impairments in IP3R function (Agrawal et al., 2010).
No evidence has been reported that the contribution of IP3R to SOCE in Drosophila occurs in mammalian neurons. However, in some mammalian cells, IP3Rs have been shown to co-localize with Orai1 (Lur et al., 2011) and interact with STIM1, Orai1, and TRPCs (Hong et al., 2011). The expression of IP3R isoform (IP3R3) was shown to be significantly lower in STIM1-deficient SH-SY5Y cells, meaning that STIM1 is a positive regulator of ITPR3 gene expression in these cells (Pascual-Caro et al., 2020). IP3R3 is a Ca2+ channel that is localized mainly at the ER-mitochondrion junction, which transfers Ca2+ from the ER to mitochondria (Ivanova et al., 2014). Thus, STIM1 deficiency leads to a decrease in mitochondrial Ca2+ concentrations, leading to cell death. The overexpression of IP3R3 restores mitochondrial Ca2+ homeostasis and bioenergetics, ATP production, and cell survival in STIM1-KO neuronal-like cells (Pascual-Caro et al., 2020). These results provide evidence of a novel STIM1-IP3R3-mediated pathway of mitochondrial Ca2+ levels, the dysregulation of which contributes to neurodegeneration. Mitochondria from AD patients have lower Ca2+ uptake (Kumar et al., 1994), which is attributed to lower IP3R3 and STIM1 levels (Pascual-Caro et al., 2020).
EB1 and EB3
The dynamic structure of dendritic spines is preserved mainly by actin filaments, and microtubules (MTs) are cytoskeleton-organizing components localized in dendrites and axons (Majewski and Kuznicki, 2015; Wu et al., 2017). Microtubules have been shown to enter dendritic spines and trigger spine head enlargement (Gu et al., 2008; Hu et al., 2008). This transport of MTs into dendritic spines appears to be involved in mechanisms of synaptic plasticity. Microtubule plus-ends contain end-binding (EB) proteins, which are divided into three types: EB1, EB2, and EB3 and have been shown to interact with STIM1 (Akhmanova and Steinmetz, 2010).
EB1/EB3-STIM1 complexes mediate ER movement in non-excitable cells (Grigoriev et al., 2008; Honnappa et al., 2009; Asanov et al., 2013). The STIM1-EB association sequesters STIM1 in MTs and prevents the excessive activation of SOCE (Chang et al., 2018). STIM1 regulates the dynamics of EB1/EB3, coupling the ER to MTs within filopodia and thus controlling growth cones in the nascent nervous system (Pavez et al., 2019). Additionally, recent research demonstrated that EB3 forms complexes with STIM2 that promote the formation of mushroom spines in hippocampal neurons, and the disintegration of these complexes results in the loss of mushroom spines (Pchitskaya et al., 2017). The overexpression of EB3 increases the proportion of mushroom spines and rescues their deficiency in hippocampal neurons in an AD mouse model. EB3 overexpression also rescues the loss of mushroom spines after STIM2 knockdown, whereas STIM2 overexpression does not restore mushroom spines after EB3 depletion. Neither STIM2 overexpression nor the activation of hippocampal TRPC6 increases spine neuronal SOCs or the proportion of mushroom spines in WT neurons. EB3 recruits various proteins to dendritic spines during synaptic plasticity, and STIM2 may be one of these cargo proteins (Pchitskaya et al., 2017). EB3 is involved in the regulation of dendritic spine morphology partly through its association with STIM2. Therefore, targeting EB3-STIM2 complexes may stabilize dendritic spines in AD patients (Pchitskaya et al., 2017).
Synaptopodin
In cultured neurons, STIM1 interacts with anchoring proteins in the dendritic spine apparatus that consists of laminar smooth ER stacks. Synaptopodin (SP) is localized between ER stacks of the spine apparatus. This cytosolic actin and α-actinin-binding protein has been shown to be essential for the formation of this organelle (Deller et al., 2003). Synaptopodin is more common in spines with large-volume spine heads, where it regulates synaptic plasticity by controlling spine head enlargement during LTP in the CA1 region of the hippocampus and enhances glutamate-induced Ca2+ release in dendritic spines of cultured hippocampal neurons (Deller et al., 2003; Vlachos et al., 2009; Korkotian et al., 2014). Synaptopodin deficiency alleviated the AD symptoms in the 3xTg mice and restores normal synaptic plasticity (Aloni et al., 2019).
Synaptopodin was recently shown to regulate activity-dependent Ca2+ signaling by recruiting STIM1 to the post-synaptic density (PSD; Korkotian et al., 2014; Segal and Korkotian, 2014). In primary hippocampal neurons, SP co-localizes with STIM1 (Korkotian et al., 2014). The localization of STIM1 in spines depended on SP, in which this protein preferentially located STIM1 to mushroom spines, where this association was especially evident (Korkotian et al., 2014). These results indicate that SP interacts with STIM and Orai and thus may regulate the functionality of Ca2+ stores and determine synaptic plasticity.
As SP belongs to actin-binding proteins, it would be interesting to investigate whether STIM could also directly interact with actin in dendritic spines. It is worth mentioning the regulatory role of actin in spine morphogenesis and stabilization that is necessary for memory formation, the role in mechanisms related to synaptic plasticity and the contribution to AD pathology (Basu and Lamprecht, 2018; Pelucchi et al., 2020). Previous research has demonstrated that STIM1 may interact with actin, and actin remodeling was required to move STIM to the PM after store depletion in human platelets. Furthermore, the polymerization of actin filaments was necessary for association of STIM1 with TRPC1 (López et al., 2006). Similarly, actin fibers were shown to be involved in an alternatively spliced long variant of STIM1 oligomerization that precedes activation of Orai1 in myoblasts (Darbellay et al., 2011). Notably, Trebak's group reported that STIM1 controls formation of actin stress fibers, independently of Orai1 and Ca2+, thus thrombin-mediated disruption of endothelial barrier function (Shinde et al., 2013).
Presenilins and CaMKII
Familial Alzheimer's disease (FAD) is caused by a dominant inherited mutation of presenilins (PSs; PS1 and PS2) and amyloid-β (Aβ) precursor protein (APP; Chakroborty and Stutzmann, 2014). Presenilins constitute catalytic components of the γ-secretase complex, which cleaves transmembrane APP to produce Aβ. PS1 mutations have been shown to change APP cleavage in favor of producing Aβ. This peptide accumulates, causing neuronal death in the cerebral cortex and hippocampal neurons, contributing to cognitive impairment and other pathological hallmarks of AD (Chakroborty and Stutzmann, 2014).
Endogenous PS1 and STIM1 have been shown to interact in human SH-SY5Y neuroblastoma cells and mouse primary cortical neurons (Tong et al., 2016). Tong et al. defined STIM1 as a new substrate of γ-secretase in a PS model of AD. The PS1-accociated γ-secretase complex cleaves the STIM1 transmembrane domain, reducing ORAI activation and diminishing SOCE (Tong et al., 2016). Dendritic spines in hippocampal neurons with mutant PS1 are destabilized, which is reversed by both a γ-secretase inhibitor and STIM1 overexpression. Although, the cleavage of STIM2 has not yet been established, its structural similarity to STIM1 suggests that it may also be a target of γ-secretase. Ryazantseva et al. reported the enhancement of SOCE in hippocampal neurons with a PS1 ΔE9 mutation (Ryazantseva et al., 2018). This PS1 mutation excludes 28 amino acids from the proteolytic cleavage site, resulting in the accumulation of uncleaved proteins. STIM1 accumulation results in its enhanced relocation to the PM, increasing the Orai1-TRPC association and enhancing SOCE (Ryazantseva et al., 2018). In turn, SOCE was attenuated directly in neurons from transgenic mice expressing human mutant PS1 A246E (Herms et al., 2003). The effect of PS1 on STIM1 appears to differ depending on the type of mutation. Greotti et al. reported that both PS1 and PS2 in SH-SY5Y cells reduce SOCE by reducing STIM1 expression levels (Greotti et al., 2019). However, this reduction does not depend on γ-secretase activity. Lower amounts of STIM1 protein were also found in FAD PS-expressing cells that were treated with a γ-secretase inhibitor. Moreover, chronic ER Ca2+ depletion or alterations of STIM1 expression levels did not affect SOCE under resting conditions (Greotti et al., 2019). These results may be considered an adaptive consequence of a prolonged reduction of ER Ca2+ levels.
Interestingly, Aβ itself decreases both STIM1 and STIM2 expression. Several studies have reported a link between Aβ-mediated STIM2 downregulation and the loss of synapses in animal models of AD (Bojarski et al., 2009; Fonseca et al., 2015; Popugaeva et al., 2015; Sanati et al., 2019). STIM2 protects mushroom spines from toxic effects of amyloid oligomers in vitro and in vivo in models of amyloid synaptotoxicity (Popugaeva et al., 2015). Sanati et al. reported that gold nanoparticles (AuNPs) reversed deteriorations of memory and spatial learning in Aβ-treated rat hippocampal neurons (Sanati et al., 2019). AuNPs delay the elongation of Aβ and dissociate existing Aβ to less toxic form, enhanced the expression of STIM protein, and potentiated the cAMP/PKA signaling cascade that modulates synaptic plasticity and influences learning and memory (Waltereit and Weller, 2003; Sanati et al., 2019). Additionally, AuNPs may directly increase cAMP levels and consequently recruit STIM2 and PKA to potentiate GluR1-dependent synaptic plasticity (Sanati et al., 2019).
In PS1 KI neurons, STIM2-mediated SOCE activates CaMKII and thus stabilizes mushroom spines (Sun et al., 2014). STIM2 is abundantly expressed in dendritic spines of hippocampal neurons, where it co-localizes with CaMKII. STIM2 overexpression rescues SOCE, restores CaMKII activity, and prevents dendritic spine loss. The conditional deletion of STIM2 reduced synaptic SOCE, thereby causing the loss of mushroom spines and eventually leading to the death of hippocampal neurons in mice that expressed FAD-associated PS1 variants (Sun et al., 2014). On the other hand, STIM2 was reported to inhibit ICRAC and SOCE amplitude and enhance intracellular Ca2+ stores through PS1 M146V mutant expression in a cellular model of AD (Ryazantseva et al., 2013). Mushroom spine loss also occurred in an APP-KI mouse model of AD, which was reported to be attributable to the accumulation of Aβ in the medium of APP-KI neurons (Zhang et al., 2015). Aβ overactivates mGluR5, leading to higher ER Ca2+ levels, the downregulation of STIM2 expression, impairments in synaptic SOCE, and lower CaMKII activity (Sun et al., 2014; Popugaeva et al., 2015; Zhang et al., 2015). The pharmacological inhibition of mGluR5 or overexpression of STIM2 restores synaptic SOCE and prevents mushroom spine loss. Downregulation of the synaptic STIM2–SOCE–CaMKII pathway causes the loss of mushroom spines in both PS1-KI and APP-KI models of AD. Moreover, TRPC6 and Orai2 have been shown to form complexes with STIM2 in hippocampal dendritic spines (Zhang et al., 2016). Thus, TRPC6 activation, STIM2 overexpression, and SOCE positive modulators can rescue mushroom spine loss in hippocampal neurons from both PS-KI and APP-KI mouse models of AD (Sun et al., 2014; Zhang et al., 2016).
Other STIM-Interacting Molecules
Recent studies have shown other regulators of STIM proteins, such as transcription factors and proteasome inhibitors that may negatively modulate their function. Here, we describe three such regulators.
NEUROD2
Neurogenic differentiation factor 2 (NEUROD2) is one of the most important neurogenic transcription factors in the CNS (Guner et al., 2017), which mutation is associated with schizophrenia (Dennis et al., 2019). Contrary to previous research that showed that NEUROD2 is a transcriptional activator (Fong et al., 2012; Bayam et al., 2015), a recent study suggested that it may also limit Stim1 expression in cortical neurons and consequently regulate Ca2+ influx in SOCE (Guner et al., 2017). Using a chromatin immunoprecipitation and sequencing approach in mouse postnatal cerebral cortical tissue, NEUROD2 was found to bind to an intronic element within the Stim1 gene. The knockdown of Neurod2 expression in cortical neurons increased STIM1 protein expression and resulted in the upregulation of SOCE, whereas its overexpression decreased SOCE. NEUROD2 activity is induced by Ca2+ influx via VGCCs, and depolarization-mediated Ca2+ influx via VGCCs appears to activate NEUROD2, which in turn fine-tunes the expression of STIM1 and SOCE and results in the STIM1-dependent inhibition of L-type VGCCs (Guner et al., 2017).
Sp4
Sp4 is a transcription factor that regulates neuronal morphogenesis and function. Its stability depends on membrane potential. Sp4 level was increased in the brains of AD patients and reduced in the brains of bipolar disorder patients (Boutillier et al., 2007; Pinacho et al., 2011). A recent study reported that the maximal activation of SOCE under resting conditions promotes the degradation of Sp4 in cerebellar granule neurons in vitro (Lalonde et al., 2014). The lowering of extracellular K+ levels reduces neuronal excitability and stimulates the depletion of ER Ca2+ stores, resulting in STIM1 migration to the ER-PM junction and SOCE activation. SOCE inhibitors prevent the ubiquitination and degradation of Sp4 during low extracellular K+ levels. STIM1 knockdown also inhibits the degradation of Sp4, whereas a constitutively active STIM1 mutant (STIM1D76A) decreased Sp4 protein levels after depolarization (Lalonde et al., 2014). Neurons that were transfected with STIM1D76A were less likely to show immunopositive nuclei with Sp4 than WT STIM1. These findings suggest that STIM1 regulates Sp4 protein, meaning that Sp4 is a downstream effector of STIM1 and STIM1-mediated SOCE. We can assume that dysregulation of STIM1-mediated SOCE may induce abnormal Sp4 expression and promote the development of disorders such as bipolar disorder or AD.
Proteasome Inhibitors
A recent study showed that sub-lethal doses of proteasome inhibitors, such as MG-132 and clasto-lactacystin-β-lactone (LA), decreased STIM1 and STIM2 levels in primary rat cortical neurons but did not affect either Orai1 or TRPC1 (Kuang et al., 2016). The loss of STIM1 and STIM2 proteins was also observed in SH-SY5Y neuroblastoma cells that had low levels of the proteasome subunit β type 5. Additionally, MG-132 and LA promoted autophagy and STIM1/STIM2 mobilization to lysosomes. Thus, inhibition of the ubiquitin-proteasome system, common in neurodegenerative disorders, may disrupt Ca2+ homeostasis by suppressing SOCE.
Concluding Remarks
Calcium homeostasis in the CNS is vital for cell maintenance. As a second messenger, Ca2+ participates in plenty physiological processes and its level is regulated in a comprehensive way via the components localized in the PM (ion channels, exchangers, and pumps), as well as the components localized in the mitochondria, ER, Golgi apparatus, and nucleus. Under pathological conditions, Ca2+ homeostasis is dysregulated, with increased cytoplasm, mitochondrial, and changed ER Ca2+ concentration leading to apoptosis (Ureshino et al., 2019). Since the increasing evidence of the relevance of Ca2+ homeostasis in neuroprotection, we focused on the expression and function of Ca2+ signaling-related proteins, STIM partners and effectors, in terms of the effects on Ca2+ regulation and its potential use in the alleviation of the symptoms of neurodegenerative diseases.
Since the discovery of STIM and Orai proteins 15 years ago, they have been found to be the main components of SOCE but not the only components. Experimental evidence that was reviewed herein clearly demonstrates that STIM-Orai-mediated SOCE in the CNS is influenced by several regulators and STIMs have several effectors. We summarized the existing knowledge of target molecules of STIM proteins (Table 1 and Figure 3).
Table 1.
STIM target molecules.
| Interacting protein | Identity of binding protein | Subcellular location | Function | STIM isoform |
|---|---|---|---|---|
| Orai | Ca2+ channel | PM | Positive effector | STIM1 STIM2 |
| TRP | Ca2+ channel | PM | Positive effector | STIM1 |
| VGCC | Ca2+ channel | PM | Negative effector | STIM1 |
| AMPAR | Ionotropic receptor | PM | Positive effector | STIM1 STIM2 |
| NMDAR | Ionotropic receptor | PM | Negative effector | STIM1 STIM2 |
| mGluR1 | Metabotropic receptor | PM | Positive regulator | STIM1 |
| septin | GTPase | PM lipids (cytoskeleton) | Positive/negative regulator | dSTIM |
| Homer | Scaffolding protein | PSD (cytoskeleton) | Negative regulator | STIM1 |
| SP | Actin-binding protein | PSD (cytoskeleton) | Positive regulator | STIM1 |
| CaMKII | Kinase | PSD (cytoskeleton) | Positive effector | STIM2 |
| EB1, EB3 | Microtubule-binding protein | Cytoplasm (cytoskeleton) | Positive effector/regulator | STIM1 STIM2 |
| Golli proteins | Myelin basic protein | Cytoplasm | Positive regulator | STIM1 |
| SARAF | Transmembrane regulatory factor | ER (predominant), PM | Negative regulator | STIM1 |
| POST | Transmembrane protein | ER (predominant), PM | Positive regulator | STIM1 |
| SERCA | Ca2+-ATPase | ER | Positive effector | STIM1 |
| IP3R | Ca2+ channel | ER | Positive regulator/effector |
dSTIM STIM1 |
| PS1 | Ca2+-leak channel, endoprotease |
ER | Negative regulator | STIM1 STIM2 |
| NEUROD2 | Transcription factor | Nucleus | Negative regulator | STIM1 |
| Sp4 | Transcription factor | Nucleus | Negative effector | STIM1 |
| MG-132/LA | Proteasome inhibitors | Cytoplasm/ nucleus |
Negative regulator | STIM1 STIM2 |
PM, plasma membrane; ER, endoplasmic reticulum; PSD, post-synaptic density.
We can distinguish both positive (SEPT1, SEPT4, golli proteins, SP, POST, and EB) and negative (Homer, SARAF, SEPT7, PS1, NEUROD2, and proteasome inhibitors) regulators of STIM that affect STIM expression and structure and its movement to the PM or activation of Orai channels (Table 1). The majority of these regulators have an impact on STIM-Orai interactions, but both golli proteins and SARAF also influence STIM-TRPC associations. Interestingly, some of the regulators appear to function in two ways. SEPT1 and SEPT4 increase STIM–Orai-dependent SOCE, whereas SEPT7 inhibits STIM migration to ER-PM junctions and Orai activation. In turn, EB3 can restore the loss of mushroom spines after knocking down STIM2 (i.e., a regulator of STIM). Conversely, the dynamics of EB1/EB3 are regulated by STIM1 (i.e., an effector of STIM). Notably, in contrast to golli proteins in immune cells where it negatively regulates STIM-dependent SOCE, golli proteins in the brain appear to have a positive regulatory action on SOCE activity that is mediated by STIM1. We assume that the action of golli proteins differs depending on the type of cell tested. However, the exact molecular mechanisms that underlie the STIM1-golli protein interaction have not yet been defined.
The main effectors of STIM proteins are Orai and TRPC, which together constitute molecular components of SOCE. The evidence gathered in this review suggests that disturbances of the STIM-Orai- and STIM-TRPC-mediated SOCE pathway contribute to the pathogenesis of diverse neurodegenerative diseases. The STIM-Orai association is also vital for the regulation of neurogenesis in mammalian cells and production of proinflammatory cytokines in astroglia and murine microglia. In turn, the STIM-TRPC interaction is essential for the survival of DNs in animal models of PD. In addition to Orai and TRPC, STIM proteins can also control Ca2+ influx via other molecular channels, including L-type VGCCs, and receptors, such as AMPARs, NMDARs, mGluR1, and mGluR5. The interaction between STIM proteins and their molecular targets can both increase and decrease Ca2+ influx from the extracellular milieu to the cytoplasm. Associations between STIMs and Orai, TRPC, AMPARs, mGluR1, and mGluR5 elevate Ca2+ influx, whereas Ca2+ influx via L-type VGCCs and NMDARs is inhibited by STIMs. Interestingly, maximal SOCE activation occurs under resting conditions, whereas VGCC and NMDAR activation requires cell membrane depolarization. Depolarization activates Ca2+ influx via both NMDARs and L-type VGCCs and decreases Ca2+ content in the ER, thereby enhancing STIM1-mediated SOCE and decreasing Ca2+ influx via VGCCs and NMDARs. Thus, STIM proteins in neurons may regulate Ca2+ influx under both resting and action potential.
The relationship between STIM proteins and glutamate receptors is essential for different forms of synaptic plasticity. In primary rat hippocampal neurons, STIM2 promotes the phosphorylation and surface delivery of AMPARs, contributing to LTP. SOCE can be activated by synaptic NMDARs, thus also influencing LTP. Additionally, STIM1 was shown to control the plasticity of L-type VGCC-dependent dendritic spines in PNs and strengthen mGluR1-dependent synaptic transmission, thereby regulating cerebellar motor behavior. Other STIM protein effectors and regulators that reside outside the PM also contribute to synaptic plasticity. Synaptopodin recruits STIM1 to the PSD and regulates the function of Ca2+ stores and plasticity of spine heads during LTP. After cell depolarization, the STIM1-POST complex binds to SERCA and keeps it in close proximity to L-type VGCCs to promote ER Ca2+ replenishment during repetitive firing, regulating neuronal excitability and plasticity. The STIM1-POST complex appears to prevent excessive Ca2+ concentrations in the cytoplasm. In turn, the STIM1-IP3R3 interaction regulates basal Ca2+ levels in mitochondria. Interestingly, the dysregulation of both cytosolic and mitochondrial Ca2+ levels contributes to neurodegeneration in AD. In turn, the STIM2-EB3 and STIM2-CaMKII complexes promote the formation of mushroom spines and stabilize them. Targeting these complexes could be a novel way of stabilizing dendritic spines and thus improve memory in AD patients.
Studying STIM proteins and their partners in different subcellular compartments enables us to understand a wide range of processes that are regulated by these proteins. Future studies should examine the ways in which these regulators act in concert to modulate STIM activity during Ca2+ influx into the cell. The precise molecular mechanisms of action of STIMs together with these all regulators (e.g., in the activation/inactivation of Orai channels) also remain to be explored. Such studies will help to elucidate the pathological mechanisms that are involved in the development of various neurodegenerative diseases (e.g., AD, PD, and HD), affective disorders (schizophrenia, bipolar disorder), chronic pain, oxidative trauma, brain trauma, stroke, and epilepsy. Therefore, it appears that STIM proteins and their modulators/effectors may have potential therapeutic applications for the treatment of these diseases.
Author Contributions
KS wrote and commented on the manuscript and prepared the figures. JG-B designed the manuscript, contributed to writing it, and prepared the final version. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Barbara Zabłocka and Dr. Magdalena Czeredys for critically reading the manuscript.
Glossary
Abbreviations
- AD
Alzheimer's disease
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- APP
amyloid-β precursor protein
- Ca2+
calcium ion
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CNS
central nervous system
- CRAC
Ca2+ release-activated channel
- DNs
dopaminergic neurons
- dSTIM1
Drosophila STIM1
- EB
end-binding protein
- ER
endoplasmic reticulum
- FAD
Familial Alzheimer's disease
- HD
Huntington's disease
- IP3
inositol-1,4,5-trisphosphate 3
- IP3R
IP3 receptor
- KI
knockin
- KO
knockout
- LTD
long-term depression
- LTP
long-term potentiation
- MBP
myelin basic protein
- mGluR
metabotropic glutamate receptor
- MT
microtubule
- NEUROD2
Neurogenic differentiation factor 2
- NMDAR
N-methyl-D-aspartate receptor
- nNOS
neuronal nitric oxide synthase
- NPC
neural progenitor cell
- OASF
Orai1-activating small fragment
- OPC
oligodendrocyte progenitor cell
- PD
Parkinson's disease
- PLA
proximity ligation assay
- PLC
phospholipase C
- PM
plasma membrane
- PMCA
plasma membrane Ca2+ adenosine triphosphatase
- PN
Purkinje neuron
- POST
partner of STIM1
- PS
presenilin
- PSD
post-synaptic density
- SARAF
SOCE-associated regulatory factor
- SERCA
sarco/endoplasmic reticulum Ca2+-adenosine triphosphatase
- siRNA
small-interfering RNA
- SOAR
STIM–Orai-activating region
- SOC
store-operated channel
- SOCE
store-operated Ca2+ entry
- SP
synaptopodin
- STIM
stromal interaction molecule
- TRPC
transient receptor potential cation channel subfamily C
- TRPM
transient receptor potential cation channel subfamily M
- VGCC
voltage-gated Ca2+ channel
- WT
wildtype.
Footnotes
Funding. This study was supported by funds from the National Science Centre (research project no. 2017/26/E/NZ3/01144 to JG-B).
References
- Agrawal N., Venkiteswaran G., Sadaf S., Padmanabhan N., Banerjee S., Hasan G. (2010). Inositol 1,4,5-trisphosphate receptor and dSTIM function in Drosophila insulin-producing neurons regulates systemic intracellular calcium homeostasis and flight. J. Neurosci. 30, 1301–1313. 10.1523/JNEUROSCI.3668-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahsan A., Zheng Y. R., Wu X. L., Tang W. D., Liu M. R., Ma S. J., et al. (2019). Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci. Ther. 25, 976–986. 10.1111/cns.13136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A., Steinmetz M. O. (2010). Microtubule +TIPs at a glance. J. Cell Sci. 123, 3415–3419. 10.1242/jcs.062414 [DOI] [PubMed] [Google Scholar]
- Albarrán L., López J. J., Gómez L. J., Salido G. M., Rosado J. A. (2016). SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem. J. 473, 3581–3595. 10.1042/BCJ20160348 [DOI] [PubMed] [Google Scholar]
- Albarran L., Lopez J. J., Woodard G. E., Salido G. M., Rosado J. A. (2016). Store-operated Ca2+ entry-associated regulatory factor (SARAF) plays an important role in the regulation of arachidonate-regulated Ca2+ (ARC) channels. J Biol Chem. 291, 6982–6988. 10.1074/jbc.M115.704940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloni E., Oni-Biton E., Tsoory M., Moallem D. H., Segal M. (2019). Synaptopodin deficiency ameliorates symptoms in the 3xTg mouse model of Alzheimer's disease. J. Neurosci 39, 3983–3992. 10.1523/JNEUROSCI.2920-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar I. S., de Souza L. B., Ong H. L. (2017). TRPC1, Orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium 63, 33–39. 10.1016/j.ceca.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanov A., Sherry R., Sampieri A., Vaca L. (2013). A relay mechanism between EB1 and APC facilitate STIM1 puncta assembly at endoplasmic reticulum-plasma membrane junctions. Cell Calcium 54, 246–256. 10.1016/j.ceca.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Baba A., Yasui T., Fujisawa S., Yamada R. X., Yamada M. K., Nishiyama N., et al. (2003). Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J. Neurosci. 23, 7737–7741. 10.1523/JNEUROSCI.23-21-07737.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Lamprecht R. (2018). The role of actin cytoskeleton in dendritic spines in the maintenance of long-term memory. Front. Mol. Neurosci. 11, 143. 10.3389/fnmol.2018.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor A. M., Madge D. J., Garthwaite J. (1994). Synaptic activation of metabotropic glutamate receptors in the parallel fibre-Purkinje cell pathway in rat cerebellar slices. Neuroscience 63, 911–915. 10.1016/0306-4522(94)90558-4 [DOI] [PubMed] [Google Scholar]
- Bayam E., Sahin G. S., Guzelsoy G., Guner G., Kabakcioglu A., Ince-Dunn G. (2015). Genome-wide target analysis of NEUROD2 provides new insights into regulation of cortical projection neuron migration and differentiation. BMC Genomics 16:681. 10.1186/s12864-015-1882-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna-Erro A., Braun A., Kraft R., Kleinschnitz C., Schuhmann M. K., Stegner D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal. 2:ra67. 10.1126/scisignal.2000522 [DOI] [PubMed] [Google Scholar]
- Bernales S., McDonald K. L., Walter P. (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423. 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. J., Lipp P., Bootman M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. 10.1038/35036035 [DOI] [PubMed] [Google Scholar]
- Blaustein M. P., Golovina V. A. (2001). Structural complexity and functional diversity of endoplasmic reticulum Ca(2+) stores. Trends Neurosci. 24, 602–608. 10.1016/S0166-2236(00)01891-9 [DOI] [PubMed] [Google Scholar]
- Blaustein M. P., Juhaszova M., Golovina V. A., Church P. J., Stanley E. F. (2002). Na/Ca exchanger and PMCA localization in neurons and astrocytes: functional implications. Ann. N. Y. Acad. Sci. 976, 356–366. 10.1111/j.1749-6632.2002.tb04762.x [DOI] [PubMed] [Google Scholar]
- Bojarski L., Pomorski P., Szybinska A., Drab M., Skibinska-Kijek A., Gruszczynska-Biegala J., et al. (2009). Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer's disease. Biochim. Biophys. Acta 1793, 1050–1057. 10.1016/j.bbamcr.2008.11.008 [DOI] [PubMed] [Google Scholar]
- Bollimuntha S., Ebadi M., Singh B. B. (2006). TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 1099, 141–149. 10.1016/j.brainres.2006.04.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutillier S., Lannes B., Buée L., Delacourte A., Rouaux C., Mohr M., et al. (2007). Sp3 and sp4 transcription factor levels are increased in brains of patients with Alzheimer's disease. Neurodegener. Dis. 4, 413–423. 10.1159/000107701 [DOI] [PubMed] [Google Scholar]
- Brandman O., Liou J., Park W. S., Meyer T. (2007). STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339. 10.1016/j.cell.2007.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M., Cal,ì T., Ottolini D., Carafoli E. (2014). Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. 10.1007/s00018-013-1550-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M., Carafoli E. (2011). The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold. Spring Harb. Perspect. Biol. 3:a004168. 10.1101/cshperspect.a004168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britzolaki A., Saurine J., Klocke B., Pitychoutis P. M. (2020). A role for SERCA pumps in the neurobiology of neuropsychiatric and neurodegenerative disorders. Adv. Exp. Med. Biol. 1131, 131–161. 10.1007/978-3-030-12457-1_6 [DOI] [PubMed] [Google Scholar]
- Brockhaus J., Schreitmüller M., Repetto D., Klatt O., Reissner C., Elmslie K., et al. (2018). α-neurexins together with α2δ-1 auxiliary subunits regulate Ca2+ influx through Ca v 2.1 channels. J. Neurosci. 38, 8277–8294. 10.1523/JNEUROSCI.0511-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiglio M., Flucher B. E. (2015). The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J. Cell. Physiol. 230, 2019–2031. 10.1002/jcp.24998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas C., Miller R. A., Smith I., Bui T., Molgó J., Müller M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. 10.1016/j.cell.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Deb B. K., Chorna T., Konieczny V., Taylor C. W., Hasan G. (2016). Mutant IP3 receptors attenuate store-operated Ca2+ entry by destabilizing STIM-Orai interactions in Drosophila neurons. J. Cell Sci. 129, 3903–3910. 10.1242/jcs.191585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Hasan G. (2017). Spontaneous Ca2+ influx in drosophila pupal neurons is modulated by IP3-receptor function and influences maturation of the flight circuit. Front. Mol. Neurosci. 10:111. 10.3389/fnmol.2017.00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S., Stutzmann G. E. (2014). Calcium channelopathies and Alzheimer's disease: insight into therapeutic success and failures. Eur. J. Pharmacol. 739, 83–95. 10.1016/j.ejphar.2013.11.012 [DOI] [PubMed] [Google Scholar]
- Chang C. L., Chen Y. J., Quintanilla C. G., Hsieh T. S., Liou J. (2018). EB1 binding restricts STIM1 translocation to ER-PM junctions and regulates store-operated Ca entry. J. Cell Biol. 217, 2047–2058. 10.1083/jcb.201711151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Fei F., Jiang X. F., Zhang L., Qu Y., Huo K., et al. (2012). Down-regulation of Homer1b/c attenuates glutamate-mediated excitotoxicity through endoplasmic reticulum and mitochondria pathways in rat cortical neurons. Free Radic. Biol. Med. 52, 208–217. 10.1016/j.freeradbiomed.2011.10.451 [DOI] [PubMed] [Google Scholar]
- Coe H., Michalak M. (2010). ERp57, a multifunctional endoplasmic reticulum resident oxidoreductase. Int. J. Biochem. Cell Biol. 42, 796–799. 10.1016/j.biocel.2010.01.009 [DOI] [PubMed] [Google Scholar]
- Cottrell G. S., Soubrane C. H., Hounshell J. A., Lin H., Owenson V., Rigby M., et al. (2018). CACHD1 is an α2δ-like protein that modulates Ca V 3 voltage-gated calcium channel activity. J. Neurosci. 38, 9186–9201. 10.1523/JNEUROSCI.3572-15.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crupi R., Impellizzeri D., Cuzzocrea S. (2019). Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 12:20. 10.3389/fnmol.2019.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S. G., Leszkiewicz D. N. (2004). Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004:re16. 10.1126/stke.2552004re16 [DOI] [PubMed] [Google Scholar]
- Czeredys M., Vigont V. A., Boeva V. A., Mikoshiba K., Kaznacheyeva E. V., Kuznicki J. (2018). Huntingtin-associated protein 1A regulates store-operated calcium entry in medium spiny neurons from transgenic YAC128 mice, a model of Huntington's disease. Front. Cell. Neurosci. 12:381. 10.3389/fncel.2018.00381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahimene S., Page K. M., Kadurin I., Ferron L., Ho D. Y., Powell G. T., et al. (2018). The α 2 δ-like protein cachd1 increases N-type calcium currents and cell surface expression and competes with α 2 δ-1. Cell Rep. 25, 1610.e5–1621.e5. 10.1016/j.celrep.2018.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbellay B., Arnaudeau S., Bader C. R., Konig S., Bernheim L. (2011). STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 194, 335–346. 10.1083/jcb.201012157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb B. K., Chakraborty P., Gopurappilly R., Hasan G. (2020). SEPT7 regulates Ca2+ entry through Orai channels in human neural progenitor cells and neurons. Cell Calcium 90:102252. 10.1016/j.ceca.2020.102252 [DOI] [PubMed] [Google Scholar]
- Deb B. K., Hasan G. (2016). Regulation of store-operated Ca2+ entry by septins. Front. Cell Dev. Biol. 4:142. 10.3389/fcell.2016.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb B. K., Hasan G. (2019). SEPT7-mediated regulation of Ca2+ entry through Orai channels requires other septin subunits. Cytoskeleton 76, 104–114. 10.1002/cm.21476 [DOI] [PubMed] [Google Scholar]
- Deb B. K., Pathak T., Hasan G. (2016). Store-independent modulation of Ca(2+) entry through Orai by Septin 7. Nat. Commun. 7:11751. 10.1038/ncomms11751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller T., Korte M., Chabanis S., Drakew A., Schwegler H., Stefani G. G., et al. (2003). Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 100, 10494–10499. 10.1073/pnas.1832384100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis D. J., Han S., Schuurmans C. (2019). bHLH transcription factors in neural development, disease, and reprogramming. Brain Res. 1705, 48–65. 10.1016/j.brainres.2018.03.013 [DOI] [PubMed] [Google Scholar]
- Derkach V., Barria A., Soderling T. R. (1999). Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 96, 3269–3274. 10.1073/pnas.96.6.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P. N., Zhang X., Wu S., Janoshazi A., Bolimuntha S., Putney J. W., et al. (2015). Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 8:ra74. 10.1126/scisignal.aaa8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Buduo C. A., Abbonante V., Marty C., Moccia F., Rumi E., Pietra D., et al. (2020). Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 135, 133–144. 10.1182/blood.2019001103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick O., Bading H. (2010). Synaptic activity and nuclear calcium signaling protect hippocampal neurons from death signal-associated nuclear translocation of FoxO3a induced by extrasynaptic N-methyl-D-aspartate receptors. J. Biol. Chem. 285, 19354–19361. 10.1074/jbc.M110.127654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W. X., Ni H. M., Gao W., Hou Y. F., Melan M. A., Chen X., et al. (2007). Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 282, 4702–4710. 10.1074/jbc.M609267200 [DOI] [PubMed] [Google Scholar]
- Dionisio N., Smani T., Woodard G. E., Castellano A., Salido G. M., Rosado J. A. (2015). Homer proteins mediate the interaction between STIM1 and Cav1.2 channels. Biochim. Biophys. Acta 1853, 1145–1153. 10.1016/j.bbamcr.2015.02.014 [DOI] [PubMed] [Google Scholar]
- Dittmer P. J., Wild A. R., Dell'Acqua M. L., Sather W. A. (2017). STIM1 Ca2+ sensor control of L-type Ca2+-channel-dependent dendritic spine structural plasticity and nuclear signaling. Cell Rep. 19, 321–334. 10.1016/j.celrep.2017.03.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenichini F., Terrié E., Arnault P., Harnois T., Magaud C., Bois P., et al. (2018). Store-operated calcium entries control neural stem cell self-renewal in the adult brain subventricular zone. Stem Cells 36, 761–774. 10.1002/stem.2786 [DOI] [PubMed] [Google Scholar]
- Duszyński J., Kozie,ł R., Brutkowski W., Szczepanowska J., Zabłocki K. (2006). The regulatory role of mitochondria in capacitative calcium entry. Biochim. Biophys. Acta 1757, 380–387. 10.1016/j.bbabio.2006.04.017 [DOI] [PubMed] [Google Scholar]
- Emptage N., Bliss T. V., Fine A. (1999). Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22, 115–124. 10.1016/S0896-6273(00)80683-2 [DOI] [PubMed] [Google Scholar]
- Emptage N. J., Reid C. A., Fine A. (2001). Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29, 197–208. 10.1016/S0896-6273(01)00190-8 [DOI] [PubMed] [Google Scholar]
- Esteban J. A., Shi S. H., Wilson C., Nuriya M., Huganir R. L., Malinow R. (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143. 10.1038/nn997 [DOI] [PubMed] [Google Scholar]
- Fierro L., DiPolo R., Llano I. (1998). Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J. Physiol. 510 (Pt 2), 499–512. 10.1111/j.1469-7793.1998.499bk.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic R., Rakic S., Zecevic N. (2002). Expression of Golli proteins in adult human brain and multiple sclerosis lesions. J. Neuroimmunol. 127, 1–12. 10.1016/S0165-5728(02)00070-X [DOI] [PubMed] [Google Scholar]
- Fong A. P., Yao Z., Zhong J. W., Cao Y., Ruzzo W. L., Gentleman R. C., et al. (2012). Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev. Cell 22, 721–735. 10.1016/j.devcel.2012.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca A. C., Moreira P. I., Oliveira C. R., Cardoso S. M., Pinton P., Pereira C. F. (2015). Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol. Neurobiol. 51, 610–622. 10.1007/s12035-014-8740-7 [DOI] [PubMed] [Google Scholar]
- Gao X., Xia J., Munoz F. M., Manners M. T., Pan R., Meucci O., et al. (2016). STIMs and Orai1 regulate cytokine production in spinal astrocytes. J. Neuroinflammation 13:126. 10.1186/s12974-016-0594-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alvarez G., Lu B., Yap K. A., Wong L. C., Thevathasan J. V., Lim L., et al. (2015). STIM2 regulates PKA-dependent phosphorylation and trafficking of AMPARs. Mol. Biol. Cell 26, 1141–1159. 10.1091/mbc.E14-07-1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G., Bangaru M. L., Wu H. E., Tang Q., Weihrauch D., Koopmeiners A. S., et al. (2011). Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J. Neurosci. 31, 3536–3549. 10.1523/JNEUROSCI.5053-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S., Shojaei S., Yeganeh B., Ande S. R., Jangamreddy J. R., Mehrpour M., et al. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49. 10.1016/j.pneurobio.2013.10.004 [DOI] [PubMed] [Google Scholar]
- Golovina V. A. (2005). Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J. Physiol. 564, 737–749. 10.1113/jphysiol.2005.085035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopurappilly R., Deb B. K., Chakraborty P., Hasan G. (2018). Stable STIM1 knockdown in self-renewing human neural precursors promotes premature neural differentiation. Front. Mol. Neurosci. 11:178. 10.3389/fnmol.2018.00178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greotti E., Capitanio P., Wong A., Pozzan T., Pizzo P., Pendin D. (2019). Familial Alzheimer's disease-linked presenilin mutants and intracellular Ca2+ handling: a single-organelle, FRET-based analysis. Cell Calcium 79, 44–56. 10.1016/j.ceca.2019.02.005 [DOI] [PubMed] [Google Scholar]
- Grigoriev I., Gouveia S. M., van der Vaart B., Demmers J., Smyth J. T., Honnappa S., et al. (2008). STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 18, 177–182. 10.1016/j.cub.2007.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J., Kuznicki J. (2013). Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J. Neurochem. 126, 727–738. 10.1111/jnc.12320 [DOI] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J., Pomorski P., Wisniewska M. B., Kuznicki J. (2011). Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 6:e19285. 10.1371/journal.pone.0019285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J., Sladowska M., Kuznicki J. (2016). AMPA receptors are involved in store-operated calcium entry and interact with STIM proteins in rat primary cortical neurons. Front. Cell. Neurosci. 10:251. 10.3389/fncel.2016.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J., Strucinska K., Maciag F., Majewski L., Sladowska M., Kuznicki J. (2020). STIM Protein-NMDA2 receptor interaction decreases NMDA-dependent calcium levels in cortical neurons. Cells 9:160. 10.3390/cells9010160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Firestein B. L., Zheng J. Q. (2008). Microtubules in dendritic spine development. J. Neurosci. 28, 12120–12124. 10.1523/JNEUROSCI.2509-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guner G., Guzelsoy G., Isleyen F. S., Sahin G. S., Akkaya C., Bayam E., et al. (2017). NEUROD2 regulates stim1 expression and store-operated calcium entry in cortical neurons. eNeuro 4, 1–17. 10.1523/ENEURO.0255-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz O. F., Altier C. (2014). STIM1-mediated bidirectional regulation of Ca(2+) entry through voltage-gated calcium channels (VGCC) and calcium-release activated channels (CRAC). Front. Cell Neurosci. 8:43. 10.3389/fncel.2014.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J., Dragicevic E., Adelsberger H., Henning H. A., Sumser M., Abramowitz J., et al. (2008). TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59, 392–398. 10.1016/j.neuron.2008.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J., Karl R. M., Alexander R. P., Adelsberger H., Brill M. S., Rühlmann C., et al. (2014). STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82, 635–644. 10.1016/j.neuron.2014.03.027 [DOI] [PubMed] [Google Scholar]
- Hawkins B. J., Irrinki K. M., Mallilankaraman K., Lien Y. C., Wang Y., Bhanumathy C. D., et al. (2010). S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 190, 391–405. 10.1083/jcb.201004152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M., Heck J., Ciuraszkiewicz A., Bikbaev A. (2020). Dynamic compartmentalization of calcium channel signalling in neurons. Neuropharmacology 169:107556. 10.1016/j.neuropharm.2019.02.038 [DOI] [PubMed] [Google Scholar]
- Henke N., Albrecht P., Pfeiffer A., Toutzaris D., Zanger K., Methner A. (2012). Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem. 287, 42042–42052. 10.1074/jbc.M112.417212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo D. K., Lim H. M., Nam J. H., Lee M. G., Kim J. Y. (2015). Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell. Signal. 27, 177–186. 10.1016/j.cellsig.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Herms J., Schneider I., Dewachter I., Caluwaerts N., Kretzschmar H., Van Leuven F. (2003). Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J. Biol. Chem. 278, 2484–2489. 10.1074/jbc.M206769200 [DOI] [PubMed] [Google Scholar]
- Hong J. H., Li Q., Kim M. S., Shin D. M., Feske S., Birnbaumer L., et al. (2011). Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12, 232–245. 10.1111/j.1600-0854.2010.01138.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honnappa S., Gouveia S. M., Weisbrich A., Damberger F. F., Bhavesh N. S., Jawhari H., et al. (2009). An EB1-binding motif acts as a microtubule tip localization signal. Cell 138, 366–376. 10.1016/j.cell.2009.04.065 [DOI] [PubMed] [Google Scholar]
- Hoth M., Niemeyer B. A. (2013). The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 71, 237–271. 10.1016/B978-0-12-407870-3.00010-X [DOI] [PubMed] [Google Scholar]
- Høyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., et al. (2007). Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 25, 193–205. 10.1016/j.molcel.2006.12.009 [DOI] [PubMed] [Google Scholar]
- Høyer-Hansen M., Jäättelä M. (2007). Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 14, 1576–1582. 10.1038/sj.cdd.4402200 [DOI] [PubMed] [Google Scholar]
- Hu X., Viesselmann C., Nam S., Merriam E., Dent E. W. (2008). Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 28, 13094–13105. 10.1523/JNEUROSCI.3074-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. D., Tang C. L., Jiang J. Z., Lv H. Y., Wu Y. B., Qin X. D., et al. (2020). Neuroprotective effects of dexmedetomidine preconditioning on oxygen-glucose deprivation-reoxygenation injury in PC12 cells via regulation of Ca2+-STIM1/Orai1 signaling. Curr. Med. Sci. 40, 699–707. 10.1007/s11596-020-2201-5 [DOI] [PubMed] [Google Scholar]
- Ivanova H., Vervliet T., Missiaen L., Parys J. B., De Smedt H., Bultynck G. (2014). Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 1843, 2164–2183. 10.1016/j.bbamcr.2014.03.007 [DOI] [PubMed] [Google Scholar]
- Jardin I., Albarrán L., Bermejo N., Salido G. M., Rosado J. A. (2012). Homers regulate calcium entry and aggregation in human platelets: a role for Homers in the association between STIM1 and Orai1. Biochem. J. 445, 29–38. 10.1042/BJ20120471 [DOI] [PubMed] [Google Scholar]
- Jha A., Ahuja M., Maléth J., Moreno C. M., Yuan J. P., Kim M. S., et al. (2013). The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 202, 71–79. 10.1083/jcb.201301148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Z. B., Zhang C., Quintana A., Lillemeier B. F., Hogan P. G. (2019). Septins organize endoplasmic reticulum-plasma membrane junctions for STIM1-ORAI1 calcium signalling. Sci. Rep. 9:10839. 10.1038/s41598-019-46862-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil J. M., Shen Z., Briggs S. P., Patrick G. N. (2010). Regulation of STIM1 and SOCE by the ubiquitin-proteasome system (UPS). PLoS ONE 5:e13465. 10.1371/journal.pone.0013465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner G. A., Nicoll R. A. (2008). Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 9, 813–825. 10.1038/nrn2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H. R., Bruce R. (2013). Neuroglia. Oxford: Oxford University Press; 10.1093/med/9780199794591.001.0001 [DOI] [Google Scholar]
- Klejman M. E., Gruszczynska-Biegala J., Skibinska-Kijek A., Wisniewska M. B., Misztal K., Blazejczyk M., et al. (2009). Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem. Int. 54, 49–55. 10.1016/j.neuint.2008.10.005 [DOI] [PubMed] [Google Scholar]
- Kondratskyi A., Yassine M., Slomianny C., Kondratska K., Gordienko D., Dewailly E., et al. (2014). Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis. 5:e1193. 10.1038/cddis.2014.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkotian E., Frotscher M., Segal M. (2014). Synaptopodin regulates spine plasticity: mediation by calcium stores. J. Neurosci. 34, 11641–11651. 10.1523/JNEUROSCI.0381-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkotian E., Oni-Biton E., Segal M. (2017). The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 595, 125–140. 10.1113/JP272645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R. (2015). STIM and ORAI proteins in the nervous system. Channels 9, 245–252. 10.1080/19336950.2015.1071747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G., Krapivinsky L., Stotz S. C., Manasian Y., Clapham D. E. (2011). POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. U.S.A. 108, 19234–19239. 10.1073/pnas.1117231108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Mariño G., Levine B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang X. L., Liu Y., Chang Y., Zhou J., Zhang H., Li Y., et al. (2016). Inhibition of store-operated calcium entry by sub-lethal levels of proteasome inhibition is associated with STIM1/STIM2 degradation. Cell Calcium 59, 172–180. 10.1016/j.ceca.2016.01.007 [DOI] [PubMed] [Google Scholar]
- Kumar U., Dunlop D. M., Richardson J. S. (1994). Mitochondria from Alzheimer's fibroblasts show decreased uptake of calcium and increased sensitivity to free radicals. Life Sci. 54, 1855–1860. 10.1016/0024-3205(94)90142-2 [DOI] [PubMed] [Google Scholar]
- Kwon J., An H., Sa M., Won J., Shin J. I., Lee C. J. (2017). Orai1 and Orai3 in combination with stim1 mediate the majority of store-operated calcium entry in astrocytes. Exp. Neurobiol. 26, 42–54. 10.5607/en.2017.26.1.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Russa D., Frisina M., Secondo A., Bagetta G., Amantea D. (2020). Modulation of cerebral store-operated calcium entry-regulatory factor (SARAF) and peripheral Orai1 following focal cerebral ischemia and preconditioning in mice. Neuroscience 441, 8–21. 10.1016/j.neuroscience.2020.06.014 [DOI] [PubMed] [Google Scholar]
- Lalonde J., Saia G., Gill G. (2014). Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci. Signal. 7:ra51. 10.1126/scisignal.2005242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law B. Y., Wang M., Ma D. L., Al-Mousa F., Michelangeli F., Cheng S. H., et al. (2010). Alisol, B., a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 9, 718–730. 10.1158/1535-7163.MCT-09-0700 [DOI] [PubMed] [Google Scholar]
- Lee K. P., Yuan J. P., Zeng W., So I., Worley P. F., Muallem S. (2009). Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc. Natl. Acad. Sci. U.S.A. 106, 14687–14692. 10.1073/pnas.0904664106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMaistre J. L., Sanders S. A., Stobart M. J., Lu L., Knox J. D., Anderson H. D., et al. (2012). Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J. Cereb. Blood Flow Metab. 32, 537–547. 10.1038/jcbfm.2011.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Chen W., Zhang L., Liu W. B., Fei Z. (2013). Inhibition of store-operated calcium entry attenuates MPP(+)-induced oxidative stress via preservation of mitochondrial function in PC12 cells: involvement of Homer1a. PLoS ONE 8:e83638. 10.1371/journal.pone.0083638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Camacho P. (2004). Ca2+-dependent redox modulation of SERCA 2b by ERp57. J. Cell Biol. 164, 35–46. 10.1083/jcb.200307010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Erxleben C., Abramowitz J., Flockerzi V., Zhu M. X., Armstrong D. L., et al. (2008). Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. U.S.A. 105, 2895–2900. 10.1073/pnas.0712288105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D., Mapelli L., Canonico P. L., Moccia F., Genazzani A. A. (2018). Neuronal activity-dependent activation of astroglial calcineurin in mouse primary hippocampal cultures. Int. J. Mol. Sci. 19:2997. 10.3390/ijms19102997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. 10.1016/j.cub.2005.05.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López J. J., Salido G. M., Pariente J. A., Rosado J. A. (2006). Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 281, 28254–28264. 10.1074/jbc.M604272200 [DOI] [PubMed] [Google Scholar]
- Luo P., Li X., Fei Z., Poon W. (2012). Scaffold protein Homer 1: implications for neurological diseases. Neurochem. Int. 61, 731–738. 10.1016/j.neuint.2012.06.014 [DOI] [PubMed] [Google Scholar]
- Lur G., Sherwood M. W., Ebisui E., Haynes L., Feske S., Sutton R., et al. (2011). InsP3receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences. Biochem. J. 436, 231–239. 10.1042/BJ20110083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski L., Kuznicki J. (2015). SOCE in neurons: Signaling or just refilling? Biochim. Biophys. Acta 1853, 1940–1952. 10.1016/j.bbamcr.2015.01.019 [DOI] [PubMed] [Google Scholar]
- Majewski Ł., Maciag F., Boguszewski P. M., Wasilewska I., Wiera G., Wójtowicz T., et al. (2017). Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1071–1087. 10.1016/j.bbamcr.2016.11.025 [DOI] [PubMed] [Google Scholar]
- Makino H., Malinow R. (2011). Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron 72, 1001–1011. 10.1016/j.neuron.2011.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey E. B., Ni Y., Parpura V. (2008). Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia 56, 821–835. 10.1002/glia.20656 [DOI] [PubMed] [Google Scholar]
- Malenka R. C., Bear M. F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. 10.1016/j.neuron.2004.09.012 [DOI] [PubMed] [Google Scholar]
- Marambaud P., Dreses-Werringloer U., Vingtdeux V. (2009). Calcium signaling in neurodegeneration. Mol. Neurodegener. 4:20. 10.1186/1750-1326-4-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marttinen M., Kurkinen K. M., Soininen H., Haapasalo A., Hiltunen M. (2015). Synaptic dysfunction and septin protein family members in neurodegenerative diseases. Mol. Neurodegener. 10:16. 10.1186/s13024-015-0013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M., Azou-Gros Y., Tsai F. C., Alvarado J., Bertin A., Iv F., et al. (2014). Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat. Cell Biol. 16, 322–334. 10.1038/ncb2921 [DOI] [PubMed] [Google Scholar]
- Mercer J. C., Dehaven W. I., Smyth J. T., Wedel B., Boyles R. R., Bird G. S., et al. (2006). Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 281, 24979–24990. 10.1074/jbc.M604589200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis M., Nieswandt B., Stegner D., Eilers J., Kraft R. (2015). STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 63, 652–663. 10.1002/glia.22775 [DOI] [PubMed] [Google Scholar]
- Miyano K., Morioka N., Sugimoto T., Shiraishi S., Uezono Y., Nakata Y. (2010). Activation of the neurokinin-1 receptor in rat spinal astrocytes induces Ca2+ release from IP3-sensitive Ca2+ stores and extracellular Ca2+ influx through TRPC3. Neurochem. Int. 57, 923–934. 10.1016/j.neuint.2010.09.012 [DOI] [PubMed] [Google Scholar]
- Moccia F., Zuccolo E., Soda T., Tanzi F., Guerra G., Mapelli L., et al. (2015). Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front. Cell Neurosci. 9:153. 10.3389/fncel.2015.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár T., Yarishkin O., Iuso A., Barabas P., Jones B., Marc R. E., et al. (2016). Store-operated calcium entry in Müller Glia is controlled by synergistic activation of TRPC and orai channels. J. Neurosci. 36, 3184–3198. 10.1523/JNEUROSCI.4069-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno C., Sampieri A., Vivas O., Peña-Segura C., Vaca L. (2012). STIM1 and Orai1 mediate thrombin-induced Ca(2+) influx in rat cortical astrocytes. Cell Calcium 52, 457–467. 10.1016/j.ceca.2012.08.004 [DOI] [PubMed] [Google Scholar]
- Moscarello M. A., Mak B., Nguyen T. A., Wood D. D., Mastronardi F., Ludwin S. K. (2002). Paclitaxel (Taxol) attenuates clinical disease in a spontaneously demyelinating transgenic mouse and induces remyelination. Mult. Scler 8, 130–138. 10.1191/1352458502ms776oa [DOI] [PubMed] [Google Scholar]
- Mostowy S., Cossart P. (2012). Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 13, 183–194. 10.1038/nrm3284 [DOI] [PubMed] [Google Scholar]
- Motiani R. K., Hyzinski-García M. C., Zhang X., Henkel M. M., Abdullaev I. F., Kuo Y. H., et al. (2013). STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 465, 1249–1260. 10.1007/s00424-013-1254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M., Fahrner M., Derler I., Schindl R., Bergsmann J., Frischauf I., et al. (2009). A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J. Biol. Chem. 284, 8421–8426. 10.1074/jbc.C800229200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K., McHugh T. J., Wilson M. A., Tonegawa S. (2004). NMDA receptors, place cells and hippocampal spatial memory. Nat. Rev. Neurosci. 5, 361–372. 10.1038/nrn1385 [DOI] [PubMed] [Google Scholar]
- Negri S., Faris P., Pellavio G., Botta L., Orgiu M., Forcaia G., et al. (2020). Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 77, 2235–2253. 10.1007/s00018-019-03284-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A. N., Krogh M., Toresson H. (2011). Dendritic EGFP-STIM1 activation after type I metabotropic glutamate and muscarinic acetylcholine receptor stimulation in hippocampal neuron. J. Neurosci. Res. 89, 1235–1244. 10.1002/jnr.22648 [DOI] [PubMed] [Google Scholar]
- Ohana L., Newell E. W., Stanley E. F., Schlichter L. C. (2009). The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels 3, 129–139. 10.4161/chan.3.2.8609 [DOI] [PubMed] [Google Scholar]
- Paez P. M., Fulton D., Colwell C. S., Campagnoni A. T. (2009b). Voltage-operated Ca(2+) and Na(+) channels in the oligodendrocyte lineage. J. Neurosci. Res. 87, 3259–3266. 10.1002/jnr.21938 [DOI] [PubMed] [Google Scholar]
- Paez P. M., Fulton D., Spreuer V., Handley V., Campagnoni A. T. (2011). Modulation of canonical transient receptor potential channel 1 in the proliferation of oligodendrocyte precursor cells by the golli products of the myelin basic protein gene. J. Neurosci. 31, 3625–3637. 10.1523/JNEUROSCI.4424-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez P. M., Fulton D. J., Spreuer V., Handley V., Campagnoni C. W., Campagnoni A. T. (2009a). Regulation of store-operated and voltage-operated Ca2+ channels in the proliferation and death of oligodendrocyte precursor cells by golli proteins. ASN Neuro 1.e00003. 10.1042/AN20090003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez P. M., Spreuer V., Handley V., Feng J. M., Campagnoni C., Campagnoni A. T. (2007). Increased expression of golli myelin basic proteins enhances calcium influx into oligodendroglial cells. J. Neurosci. 27, 12690–12699. 10.1523/JNEUROSCI.2381-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R., Raveh A., Kaminsky I., Meller R., Reuveny E. (2012). SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 149, 425–438. 10.1016/j.cell.2012.01.055 [DOI] [PubMed] [Google Scholar]
- Papanikolaou M., Lewis A., Butt A. M. (2017). Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Struct. Funct. 222, 2993–3005. 10.1007/s00429-017-1380-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., et al. (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. 10.1016/j.cell.2009.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. Y., Shcheglovitov A., Dolmetsch R. (2010). The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105. 10.1126/science.1191027 [DOI] [PubMed] [Google Scholar]
- Parys J. B., Decuypere J. P., Bultynck G. (2012). Role of the inositol 1,4,5-trisphosphate receptor/ Ca2+-release channel in autophagy. Cell Commun. Signal. 10:17. 10.1186/1478-811X-10-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Caro C., Berrocal M., Lopez-Guerrero A. M., Alvarez-Barrientos A., Pozo-Guisado E., Gutierrez-Merino C., et al. (2018). STIM1 deficiency is linked to Alzheimer's disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca. J. Mol. Med. 96, 1061–1079. 10.1007/s00109-018-1677-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Caro C., Orantos-Aguilera Y., Sanchez-Lopez I., de Juan-Sanz J., Parys J. B., Area-Gomez E., et al. (2020). STIM1 deficiency leads to specific down-regulation of ITPR3 in SH-SY5Y cells. Int. J. Mol. Sci. 21:6598. 10.3390/ijms21186598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavez M., Thompson A. C., Arnott H. J., Mitchell C. B., D'Atri I., Don E. K., et al. (2019). STIM1 is required for remodeling of the endoplasmic reticulum and microtubule cytoskeleton in steering growth cones. J. Neurosci. 39, 5095–5114. 10.1523/JNEUROSCI.2496-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchitskaya E., Kraskovskaya N., Chernyuk D., Popugaeva E., Zhang H., Vlasova O., et al. (2017). Stim2-Eb3 association and morphology of dendritic spines in hippocampal neurons. Sci. Rep. 7:17625. 10.1038/s41598-017-17762-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchitskaya E., Popugaeva E., Bezprozvanny I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. 10.1016/j.ceca.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelucchi S., Stringhi R., Marcello E. (2020). Dendritic spines in alzheimer's disease: how the actin cytoskeleton contributes to synaptic failure. Int. J. Mol. Sci. 21:908. 10.3390/ijms21030908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinacho R., Villalmanzo N., Lalonde J., Haro J. M., Meana J. J., Gill G., et al. (2011). The transcription factor SP4 is reduced in postmortem cerebellum of bipolar disorder subjects: control by depolarization and lithium. Bipolar Disord. 13, 474–485. 10.1111/j.1399-5618.2011.00941.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E., Chernyuk D., Bezprozvanny I. (2020). Reversal of calcium dysregulation as potential approach for treating Alzheimer's disease. Curr. Alzheimer Res. 17, 344–354. 10.2174/1567205017666200528162046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E., Pchitskaya E., Speshilova A., Alexandrov S., Zhang H., Vlasova O., et al. (2015). STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 10:37. 10.1186/s13024-015-0034-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. 10.1038/nature05122 [DOI] [PubMed] [Google Scholar]
- Prins D., Groenendyk J., Touret N., Michalak M. (2011). Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 12, 1182–1188. 10.1038/embor.2011.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney J. W. (2003). Capacitative calcium entry in the nervous system. Cell Calcium 34, 339–344. 10.1016/S0143-4160(03)00143-X [DOI] [PubMed] [Google Scholar]
- Rae M. G., Martin D. J., Collingridge G. L., Irving A. J. (2000). Role of Ca2+ stores in metabotropic L-glutamate receptor-mediated supralinear Ca2+ signaling in rat hippocampal neurons. J. Neurosci. 20, 8628–8636. 10.1523/JNEUROSCI.20-23-08628.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao W., Peng C., Zhang L., Su N., Wang K., Hui H., et al. (2016). Homer1a attenuates glutamate-induced oxidative injury in HT-22 cells through regulation of store-operated calcium entry. Sci. Rep. 6:33975. 10.1038/srep33975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao W., Zhang L., Peng C., Hui H., Wang K., Su N., et al. (2015). Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim. Biophys. Acta 1852, 2402–2413. 10.1016/j.bbadis.2015.08.014 [DOI] [PubMed] [Google Scholar]
- Rao W., Zhang L., Su N., Wang K., Hui H., Wang L., et al. (2013). Blockade of SOCE protects HT22 cells from hydrogen peroxide-induced apoptosis. Biochem. Biophys. Res. Commun. 441, 351–356. 10.1016/j.bbrc.2013.10.054 [DOI] [PubMed] [Google Scholar]
- Remondelli P., Renna M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10:187. 10.3389/fnmol.2017.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. F., Samakai E., Soboloff J. (2012). STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. EMBO J. 31, 1123–1133. 10.1038/emboj.2011.495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski M. A. (2011). Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr. 11, 56–63. 10.5698/1535-7511-11.2.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronco V., Grolla A. A., Glasnov T. N., Canonico P. L., Verkhratsky A., Genazzani A. A., et al. (2014). Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 55, 219–229. 10.1016/j.ceca.2014.02.016 [DOI] [PubMed] [Google Scholar]
- Roos J., DiGregorio P. J., Yeromin A. V., Ohlsen K., Lioudyno M., Zhang S., et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445. 10.1083/jcb.200502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazantseva M., Goncharova A., Skobeleva K., Erokhin M., Methner A., Georgiev P., et al. (2018). Presenilin-1 delta E9 mutant induces STIM1-driven store-operated calcium channel hyperactivation in hippocampal neurons. Mol. Neurobiol. 55, 4667–4680. 10.1007/s12035-017-0674-4 [DOI] [PubMed] [Google Scholar]
- Ryazantseva M., Skobeleva K., Kaznacheyeva E. (2013). Familial Alzheimer's disease-linked presenilin-1 mutation M146V affects store-operated calcium entry: does gain look like loss? Biochimie 95, 1506–1509. 10.1016/j.biochi.2013.04.009 [DOI] [PubMed] [Google Scholar]
- Ryu C., Jang D. C., Jung D., Kim Y. G., Shim H. G., Ryu H. H., et al. (2017). STIM1 regulates somatic Ca2+ signals and intrinsic firing properties of cerebellar purkinje neurons. J. Neurosci. 37, 8876–8894. 10.1523/JNEUROSCI.3973-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salido G. M., Sage S. O., Rosado J. A. (2009). TRPC channels and store-operated Ca(2+) entry. Biochim. Biophys. Acta 1793, 223–230. 10.1016/j.bbamcr.2008.11.001 [DOI] [PubMed] [Google Scholar]
- Sanati M., Khodagholi F., Aminyavari S., Ghasemi F., Gholami M., Kebriaeezadeh A., et al. (2019). Impact of Gold Nanoparticles on amyloid β-induced Alzheimer's disease in a rat animal model: involvement of STIM proteins. ACS Chem. Neurosci. 10, 2299–2309. 10.1021/acschemneuro.8b00622 [DOI] [PubMed] [Google Scholar]
- Sather W. A., Dittmer P. J. (2019). Regulation of voltage-gated calcium channels by the ER calcium sensor STIM1. Curr. Opin. Neurobiol. 57, 186–191. 10.1016/j.conb.2019.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R., Tymianski M. (2000). Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 78, 3–13. 10.1007/s001090000077 [DOI] [PubMed] [Google Scholar]
- Saul S., Stanisz H., Backes C. S., Schwarz E. C., Hoth M. (2014). How ORAI and TRP channels interfere with each other: interaction models and examples from the immune system and the skin. Eur. J. Pharmacol. 739, 49–59. 10.1016/j.ejphar.2013.10.071 [DOI] [PubMed] [Google Scholar]
- Segal M., Korkotian E. (2014). Endoplasmic reticulum calcium stores in dendritic spines. Front. Neuroanat. 8:64. 10.3389/fnana.2014.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S., Sun Y., Sukumaran P., Singh B. B. (2016). Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 55, 818–831. 10.1002/mc.22324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S., Sun Y., Watt J. A., Wang S., Lei S., Birnbaumer L., et al. (2012). Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 122, 1354–1367. 10.1172/JCI61332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwach K., Gruszczynska-Biegala J. (2019). STIM proteins and glutamate receptors in neurons: role in neuronal physiology and neurodegenerative diseases. Int. J. Mol. Sci. 20:2289. 10.3390/ijms20092289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Quintana A., Findlay G. M., Mettlen M., Baust B., Jain M., et al. (2013). An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 499, 238–242. 10.1038/nature12229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde A. V., Motiani R. K., Zhang X., Abdullaev I. F., Adam A. P., González-Cobos J. C., et al. (2013). STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry. Sci. Signal. 6:ra18. 10.1126/scisignal.2003425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui T. A., Lively S., Vincent C., Schlichter L. C. (2012). Regulation of podosome formation, microglial migration and invasion by Ca(2+)-signaling molecules expressed in podosomes. J. Neuroinflammation 9:250. 10.1186/1742-2094-9-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson P. B., Challiss R. A., Nahorski S. R. (1995). Neuronal Ca2+ stores: activation and function. Trends Neurosci. 18, 299–306. 10.1016/0166-2236(95)93919-O [DOI] [PubMed] [Google Scholar]
- Singaravelu K., Nelson C., Bakowski D., de Brito O. M., Ng S. W., Di Capite J., et al. (2011). Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J. Biol. Chem. 286, 12189–12201. 10.1074/jbc.M110.174029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu C. R., Balsor J. L., Jones D. G., Murphy K. M. (2015). Classic and Golli Myelin Basic Protein have distinct developmental trajectories in human visual cortex. Front. Neurosci. 9:138. 10.3389/fnins.2015.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinska-Kijek A., Wisniewska M. B., Gruszczynska-Biegala J., Methner A., Kuznicki J. (2009). Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol. Exp. 69, 413–428. [DOI] [PubMed] [Google Scholar]
- Soboloff J., Rothberg B. S., Madesh M., Gill D. L. (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565. 10.1038/nrm3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J., Spassova M. A., Dziadek M. A., Gill D. L. (2006a). Calcium signals mediated by STIM and Orai proteins–a new paradigm in inter-organelle communication. Biochim. Biophys. Acta 1763, 1161–1168. 10.1016/j.bbamcr.2006.09.023 [DOI] [PubMed] [Google Scholar]
- Soboloff J., Spassova M. A., Hewavitharana T., He L. P., Xu W., Johnstone L. S., et al. (2006b). STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr. Biol. 16, 1465–1470. 10.1016/j.cub.2006.05.051 [DOI] [PubMed] [Google Scholar]
- Soboloff J., Spassova M. A., Tang X. D., Hewavitharana T., Xu W., Gill D. L. (2006c). Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665. 10.1074/jbc.C600126200 [DOI] [PubMed] [Google Scholar]
- Somasundaram A., Shum A. K., McBride H. J., Kessler J. A., Feske S., Miller R. J., et al. (2014). Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 34, 9107–9123. 10.1523/JNEUROSCI.0263-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I., Huganir R. L. (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588. 10.1016/S0166-2236(02)02270-1 [DOI] [PubMed] [Google Scholar]
- Spät A., Szanda G. (2017). The role of mitochondria in the activation/maintenance of SOCE: store-operated Ca2+ entry and mitochondria. Adv. Exp. Med. Biol. 993, 257–275. 10.1007/978-3-319-57732-6_14 [DOI] [PubMed] [Google Scholar]
- Steinbeck J., Nadine H., Opatz J., Gruszczynska-Biegala J., Schneider L., Theiss S., et al. (2011). Store-operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp. Neurol. 232, 185–194. 10.1016/j.expneurol.2011.08.022 [DOI] [PubMed] [Google Scholar]
- Sukumaran P., Sun Y., Vyas M., Singh B. B. (2015). TRPC1-mediated Ca2+ entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 6:e1674. 10.1038/cddis.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Zhang H., Liu J., Popugaeva E., Xu N. J., Feske S., et al. (2014). Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. 10.1016/j.neuron.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Selvaraj S., Pandey S., Humphrey K. M., Foster J. D., Wu M., et al. (2018). MPP+ decreases store-operated calcium entry and TRPC1 expression in Mesenchymal Stem Cell derived dopaminergic neurons. Sci. Rep. 8:11715. 10.1038/s41598-018-29528-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Zhang H., Selvaraj S., Sukumaran P., Lei S., Birnbaumer L., et al. (2017). Inhibition of L-type Ca2+ channels by TRPC1-STIM1 complex is essential for the protection of dopaminergic neurons. J. Neurosci. 37, 3364–3377. 10.1523/JNEUROSCI.3010-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellios V., Maksoud M. J. E., Xiang Y. Y., Lu W. Y. (2020). Nitric oxide critically regulates purkinje neuron dendritic development through a metabotropic glutamate receptor type 1-mediated mechanism. Cerebellum 19, 510–526. 10.1007/s12311-020-01125-7 [DOI] [PubMed] [Google Scholar]
- Tong B. C., Lee C. S., Cheng W. H., Lai K. O., Foskett J. K., Cheung K. H. (2016). Familial Alzheimer's disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 9:ra89. 10.1126/scisignal.aaf1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twine N. A., Janitz K., Wilkins M. R., Janitz M. (2011). Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer's disease. PLoS ONE 6:e16266. 10.1371/journal.pone.0016266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ureshino R. P., Erustes A. G., Bassani T. B., Wachilewski P., Guarache G. C., Nascimento A. C., et al. (2019). The Interplay between Ca2+ signaling pathways and neurodegeneration. Int. J. Mol. Sci. 20:6004 10.3390/ijms20236004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A., Parpura V. (2014). Store-operated calcium entry in neuroglia. Neurosci. Bull 30, 125–133. 10.1007/s12264-013-1343-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A., Reyes R. C., Parpura V. (2014). TRP channels coordinate ion signalling in astroglia. Rev. Physiol. Biochem. Pharmacol. 166, 1–22. 10.1007/112_2013_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., et al. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223. 10.1126/science.1127883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigont V., Kolobkova Y., Skopin A., Zimina O., Zenin V., Glushankova L., et al. (2015). Both Orai1 and TRPC1 are involved in excessive store-operated calcium entry in striatal neurons expressing mutant huntingtin exon 1. Front. Physiol. 6:337. 10.3389/fphys.2015.00337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos A., Korkotian E., Schonfeld E., Copanaki E., Deller T., Segal M. (2009). Synaptopodin regulates plasticity of dendritic spines in hippocampal neurons. J. Neurosci. 29, 1017–1033. 10.1523/JNEUROSCI.5528-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. M., Doherty M. K., Tepikin A. V., Burgoyne R. D. (2010). Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 430, 453–460. 10.1042/BJ20100650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R., Weller M. (2003). Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol. Neurobiol. 27, 99–106. 10.1385/MN:27:1:99 [DOI] [PubMed] [Google Scholar]
- Wang G. X., Poo M. M. (2005). Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature 434, 898–904. 10.1038/nature03478 [DOI] [PubMed] [Google Scholar]
- Wang Y., Deng X., Mancarella S., Hendron E., Eguchi S., Soboloff J., et al. (2010). The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109. 10.1126/science.1191086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D., Mei Y., Xia J., Hu H. (2017). Orai1 and Orai3 mediate store-operated calcium entry contributing to neuronal excitability in dorsal root ganglion neurons. Front. Cell. Neurosci. 11:400. 10.3389/fncel.2017.00400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. T., Manji S. S., Parker N. J., Hancock M. S., Van Stekelenburg L., Eid J. P., et al. (2001). Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem. J. 357, 673–685. 10.1042/0264-6021:3570673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A., Grubb D. R., Cooley N., Luo J., Woodcock E. A. (2013). Regulation of autophagy in cardiomyocytes by Ins(1,4,5)P(3) and IP(3)-receptors. J. Mol. Cell Cardiol. 54, 19–24. 10.1016/j.yjmcc.2012.10.014 [DOI] [PubMed] [Google Scholar]
- Wu M. M., Buchanan J., Luik R. M., Lewis R. S. (2006). Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 174, 803–813. 10.1083/jcb.200604014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Whiteus C., Xu C. S., Hayworth K. J., Weinberg R. J., Hess H. F., et al. (2017). Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. U.S.A. 114, E4859–E4867. 10.1073/pnas.1701078114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Yu J., Li D., Yu S., Ke J., Wang L., et al. (2017). Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy 13, 82–98. 10.1080/15548627.2016.1245261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Jin H., Cai X., Li S., Shen Y. (2012). Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. U.S.A. 109, 5657–5662. 10.1073/pnas.1118947109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K. A., Shetty M. S., Garcia-Alvarez G., Lu B., Alagappan D., Oh-Hora M., et al. (2017). STIM2 regulates AMPA receptor trafficking and plasticity at hippocampal synapses. Neurobiol. Learn. Mem. 138, 54–61. 10.1016/j.nlm.2016.08.007 [DOI] [PubMed] [Google Scholar]
- Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., Muallem S. (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343. 10.1038/ncb1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Thomas D. W. (2016). Stromal Interaction Molecule 1 rescues store-operated calcium entry and protects NG115-401L cells against cell death induced by endoplasmic reticulum and mitochondrial oxidative stress. Neurochem. Int. 97, 137–145. 10.1016/j.neuint.2016.04.002 [DOI] [PubMed] [Google Scholar]
- Zhang H., Sun S., Wu L., Pchitskaya E., Zakharova O., Fon Tacer K., et al. (2016). Store-operated calcium channel complex in postsynaptic spines: a new therapeutic target for Alzheimer's disease treatment. J. Neurosci. 36, 11837–11850. 10.1523/JNEUROSCI.1188-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wu L., Pchitskaya E., Zakharova O., Saito T., Saido T., et al. (2015). Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer's disease. J. Neurosci. 35, 13275–13286. 10.1523/JNEUROSCI.1034-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Song J. N., Wu Y., Zhao Y. L., Pang H. G., Fu Z. F., et al. (2014). Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats. Neuroreport 25, 507–513. 10.1097/WNR.0000000000000127 [DOI] [PubMed] [Google Scholar]
- Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., et al. (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. 10.1038/nature04147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Xu H., Liu B., Dun L., Lu C., Cai Y., et al. (2019). Suppression of lncRNA RMRP ameliorates oxygen-glucose deprivation/re-oxygenation-induced neural cells injury by inhibiting autophagy and PI3K/Akt/mTOR-mediated apoptosis. Biosci. Rep. 39:BSR20181367. 10.1042/BSR20181367 [DOI] [PMC free article] [PubMed] [Google Scholar]