

Abstract
Pulmonary mycoses are difficult to treat and detrimental to patients. Fungal infections modulate the lung immune response, induce goblet cell hyperplasia and metaplasia, and mucus hypersecretion in the airways. Excessive mucus clogs small airways and reduces pulmonary function by decreasing oxygen exchange, leading to respiratory distress. The forkhead box protein A2 (FOXA2) is a transcription factor that regulates mucus homeostasis in the airways. However, little is known whether pulmonary mycosis modulates FOXA2 function. Herein, we investigated whether Blastomyces dermatitidis and Histoplasma capsulatum–infected canine and feline lungs and airway epithelial cells could serve as higher animal models to examine the relationships between fungal pneumonia and FOXA2-regulated airway mucus homeostasis. The results indicate that fungal infection down-regulated FOXA2 expression in airway epithelial cells, with concomitant overexpression of mucin 5AC (MUC5AC) and mucin 5B (MUC5B) mucins. Mechanistic studies reveal that B. dermatitidis infection, as well as β-glucan exposure, activated the Dectin-1–SYK–epidermal growth factor receptor–AKT/extracellular signal-regulated kinase 1/2 signaling pathway that inhibits the expression of FOXA2, resulting in overexpression of MUC5AC and MUC5B in canine airway cells. Further understanding of the role of FOXA2 in mucus hypersecretion may lead to novel therapeutics against excessive mucus in both human and veterinary patients with pulmonary mycosis.
Dimorphic fungi, including Blastomyces dermatitidis, Histoplasma capsulatum, Coccidiodes immitis, and Coccidiodes posadasii, are important etiologic pathogens of pulmonary mycoses. B. dermatitidis is present in the soil and wet decaying wood of eastern North America and central United States.1, 2, 3 H. capsulatum is prevalent in Ohio and Mississippi river valleys, and commonly found in soil, and in bird and bat droppings.4,5 C. immitis and C. posadasii are commonly found in the soil of desert southwestern United States and in both Central and South America.6 These fungal pathogens primarily infect immunocompromised patients, but are also observed in healthy individuals.3,4,6 Because fungal infections are rare and symptoms are not specific, definitive diagnosis is challenging and typically delayed by 1 month in 50% of human patients, preventing proper treatment in a timely manner.7,8 In patients with B. dermatitidis who are immunosuppressed because of organ transplant or AIDS, mortality rates range between 30% and 40%8, 9, 10 and increase dramatically to 60% in individuals who develop acute respiratory distress syndrome.9 Even in patients who eventually recover, antifungal regimens can last 6 to 12 months, with potential severe adverse effects such as nephrotoxicity.2,11 Pulmonary mycosis occurs through inhalation of fungal particles (eg, spores, yeasts, or mycelial fragments), or through breach of barrier.3,4,6 Not surprisingly, pneumonia is the most common complication in patients with blastomycosis.2 From the initial site of infection, fungi disseminate to other body organs, including the central nervous, skeletal, and reproductive systems, resulting in higher morbidity and mortality.1,5
Although fungal infection causes goblet cell metaplasia and hyperplasia, and mucus hypersecretion and accumulation,12, 13, 14 it is mechanistically understudied. Compared with those in healthy lungs, airways in diseased lungs frequently exhibit mucin hypersecretion, surface mucus dehydration, and ciliary dysfunction that hinder the clearing of microbial pathogens, setting the stage for chronic microbial infections.15,16 Despite the grim outcome of muco-obstructive diseases, there is still a dearth of effective muco-active therapies. Therefore, unraveling the regulatory mechanism of mucus production could be the key to improving treatment of muco-obstructive diseases. Mucin glycoproteins are major constituents of airway mucus, and they support hydration of mucus gels by increasing their viscosity and elasticity.15,17 Among all mucins, the gel-forming mucin 5AC (MUC5AC) and mucin 5B (MUC5B) predominate in both healthy and diseased airways. MUC5AC is secreted by goblet cells in both tracheal and bronchial epithelium, whereas MUC5B is expressed in submucosal glands of trachea and bronchi, and in surface secretory cells throughout the airway down to the level of preterminal bronchioles.17 As part of the mucus layer, MUC5AC and MUC5B protect the airway epithelium by trapping and removing inhaled microbes and pollutants, as well as regulating the innate immune response. MUC5B, but not MUC5AC, has been shown to be required for mucociliary clearance and controlling infections in the airways and middle ear, and for maintaining immune homeostasis in mouse lungs.18 Interestingly, contrary to Roy et al,18 overexpression of MUC5AC confers increased resistance to infection by the PR8/H1N1 influenza virus.19 Furthermore, regardless of ciliary beating, IL-13–stimulated airway epithelium fails to transport MUC5AC-containing gels, which are tethered to mucus-producing cells,20 suggesting that mucin composition is a major functional determinant in mucociliary clearance.
Mucus hypersecretion and failure in clearance are prominent pathologic features of muco-obstructive diseases, including cystic fibrosis, chronic obstructive pulmonary disease (COPD), chronic bronchitis (CB), primary ciliary dyskinesis, and asthma.15,21,22 Studies in mouse models and cultured human airway epithelial cells have shown that both type 2 T helper (Th2) cell–derived cytokines (eg, IL-4 and IL-13)-STAT6-SAM pointed domain–containing Ets transcription factor (SPDEF)23,24 and epidermal growth factor receptor (EGFR) signaling pathways25 positively regulate goblet cell metaplasia and hyperplasia, and mucus production. Activated STAT6-SPDEF and EGFR pathways converge to down-regulate the forkhead box protein A2 (FOXA2), a key transcription factor that regulates lung morphogenesis, surfactant protein production, goblet cell differentiation, and mucin expression.26, 27, 28 More importantly, FOXA2 expression is depleted in the airways of human patients with bronchopulmonary dysplasia and bronchiectasis,27 asthma,17 and COPD.27 FOXA2 expression is also attenuated by cigarette smoke, the most important cause of COPD.29 Infections by respiratory bacterial pathogens Pseudomonas aeruginosa, Mycoplasma pneumoniae, and Bordetella bronchiseptica, as well as exposure to P. aeruginosa exotoxin pyocyanin consistently reduce the expression of FOXA2 in human, mouse, and canine airways.28,30, 31, 32, 33, 34 FOXA2 is also depleted in human COPD lungs34,35 and in canine patients with naturally developed COPD and CB.35
Imbalance among cytokine responses often plays an important role in driving and worsening airway disease pathogenesis. Type 1 T helper (Th1) cytokines, including interferon-γ (IFN-γ) and tumor necrosis factor-α/β (TNF-α/β), elevate airway hyperresponsiveness and activate antigen-presenting cells, including macrophages.36 Th2 cytokines, including IL-4 and IL-13, promote goblet cell metaplasia and mucin biosynthesis.25,37,38 IL-17 produced by type 17 T helper (Th17) cells can trigger MUC5AC and MUC5B expression in tracheobronchial epithelium,39 as well as augment neutrophilic infiltration and Th2-mediated eosinophilic airway inflammation.40,41 Also, proinflammatory milieu, including pyrogenic cytokine IL-1β and chemokine IL-8, increases mucin hyperproduction by activating airway epithelial cells.42,43 Although B. dermatitidis and H. capsulatum have been reported to modulate cytokine productions,44, 45, 46, 47 how these changes affect mucin production in the airways is not well-characterized.
Similar to humans, the lungs of companion animals are continuously bombarded by environmental pollutants and infectious agents. Many clinical features of respiratory diseases in companion animals resemble human patients, including CB, COPD, goblet cell metaplasia and hyperplasia, mucus hypersecretion, and chronic lung infections.35 The prevalence of blastomycosis and histoplasmosis in dogs and cats is comparable to humans.48 Notably, geographic prevalence of canine pulmonary mycoses overlaps with that of humans, allowing it to be a potential epidemiologic marker to predict outbreaks in human.11,49 However, it is unclear whether fungal infections modulate FOXA2 function. In this study, we explored whether B. dermatitidis and H. capsulatum–infected canine and feline lungs and airway cells can serve as higher animal models to examine FOXA2 function during fungal pneumonia.
Materials and Methods
Chemicals, Reagents, and Antibodies
Unless stated otherwise, chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO). The Saccharomyces cerevisiae β-glucan (CAS 9012 to 72-0; EMD Millipore, Burlington, MA) is a major structural element of fungal cell wall composed of linear β1,3-glucan with a small number of β1,6-glucan branches. Antibodies were used at dilutions recommended by manufacturers and optimized as needed. Antibodies were diluted in 5% bovine serum albumin. Antibodies against FOXA2 [sc-10160; immunohistochemistry (IHC) 1:250; Western blot 1:1000], MUC5AC (sc-20118; IHC 1:250; Western blot 1:1000), MUC5B (sc-20119; IHC 1:500; Western blot 1:1000), STAT6 (sc-1689; Western blot 1:1000), phosphorylated STAT6 (sc-71793; Western blot 1:1000), EGFR (sc-03; Western blot 1:1000), phosphorylated EGFR (pEGFR; sc-101668; Western blot 1:1000), AKT (Western blot 1:1000), phosphorylated AKT (pAKT; sc-16646; Western blot 1:1000), and glyceraldehyde-3-phosphate dehydrogenase (sc-166545; Western blot 1:1000) were purchased from Santa Cruz Biotechnology (Dallas, TX). Antibodies against SYK (13,198; Western blot 1:1000), phosphorylated SYK (2710; Western blot 1:1000), extracellular signal-regulated kinase (ERK) 1/2 (4695; Western blot 1:1000), phosphorylated ERK1/2 (pERK1/2; 4370; Western blot 1:1000), and histone H3 (9717; Western blot 1:1000) were purchased from Cell Signaling Technology (Danvers, MA). Specific inhibitors AG1478 (658,552), LY294002 (440,202), and PD98059 (513,000) were purchased from Sigma Aldrich. Piceatannol (SC-200610) was purchased from Santa Cruz Biotechnology. Specific Ambion siRNA silencers for CLEC7A (encoding Dectin-1) (AM16708-249228) and SYK (AM16708-138814) genes were purchased from Thermo Fisher Scientific (Waltham, MA).
Canine Airway Epithelial Cell Cultures, Fungal Cultures, and Infection
The immortalized canine airway carcinoma cell line (BACA) was provided by Dr. Timothy Fan (Department of Veterinary Clinical Medicine, College of Veterinary Medicine (CVM), University of Illinois at Urbana-Champaign (UIUC). BACA cells were cultured in the Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution in 5% CO2 at 37°C. After reaching 95% confluency, cells were exposed to either live or heat-killed B. dermatitidis strain SCB-2 (Gilchrist et Stokes; ATCC, Manassas, VA; ATCC 26199) for 24 hours at a multiplicity of infection of 1:1 or 5:1, or to β-glucan at indicated concentrations. B. dermatitidis ATCC 26199 (SCB-2) was maintained as yeast on BD Middlebrook 7H10 agar supplemented with oleic acid–albumin complex (Sigma-Aldrich) slants at 39°C. Histoplasma capsulatum ATCC 26032 (G217B) was grown at 37°C in Ham's F12 medium supplemented with glucose (18.2 g/L), glutamic acid (1 g/L), HEPES (6g/L), and cysteine (8.4 mg/L). The yeasts were heat killed by incubating at 65°C for 30 minutes.
Canine and Feline Lung Specimens
The fungal pneumonia cases (Table 1) and lung tissue sections from feline and canine animals were provided by the UIUC CVM Veterinary Diagnostic Laboratory. Healthy control lungs were found to be free of infection on necropsy, and the animals were euthanized of unrelated causes. Animals with fungal pneumonia either died or were euthanized because of infection with B. dermatitidis, H. capsulatum, or C. immitis. Lung tissues were paraffin embedded, divided into sections and stained.
Table 1.
Clinical Cases of Dogs and Cats with Fungal Pneumonia Analyzed in This Study
| Case no. | Species, breed | Etiologic agent | FOXA2 depletion | PAS | MUC5AC | MUC5B |
|---|---|---|---|---|---|---|
| 19-30783 | Canine, papillon | None (control lung) | No | − | Normal | Normal |
| 18-H49300 | Canine, poodle mix | Blastomyces dermatitidis | Yes | + | + | + |
| 18-H40147 | Canine, German shepard | B. dermatitidis | Yes | + | + | + |
| 19-30783 | Feline, Persian | None (control lung) | No | − | Normal | Normal |
| 17-H54030 | Feline, domestic longhair | Histoplasma capsulatum | Yes | + | + | + |
| 18-34071 | Feline, domestic shorthair | H. capsulatum | Yes | + | + | + |
+, positive staining for PAS and MUC5AC/MUC5B; −, lack of PAS and MUC5AC and MUC5B staining; FOXA2, forkhead box protein A2; PAS, periodic acid-Schiff.
GMS, Periodic Acid-Schiff, and IHC Staining
Grocott methenamine silver (GMS) staining was performed according to the instruction provided by the manufacturer (HT100A-1 KT; Sigma-Aldrich). Periodic acid-Schiff (395B; Sigma-Aldrich) and IHC staining were performed as we have previously published.28,31, 32, 33, 34, 35,50 For IHC, lung sections were probed with primary antibodies and detected by using an ABC kit (PK-6101 or PK-105; Vector Laboratories, Burlingame, CA). Images were captured using an Olympus DP70 light microscope (Central Valley, PA).
Western Blot Analysis
Protein extracts were prepared from BACA cells using the M-PER Mammalian Protein Extraction Reagent (78,501; Thermo Fisher Scientific). Protein concentration was determined using the BCA Protein Assay Kit (23,227; Thermo Fisher Scientific). Proteins were separated using 10% SDS-PAGE, transferred to nitrocellulose membranes (88,018; Thermo Fisher Scientific), and probed with normal or phospho-specific antibodies against FOXA2, STAT6, SYK, phosphorylated SYK, EGFR, pEGFR, AKT, pAKT, ERK1/2, and pERK1/2. Histone H3 and glyceraldehyde-3-phosphate dehydrogenase were used as loading controls. Mucins were resolved on 1% agarose and 3% to 6% acrylamide hybrid gels in the tris–borate-ethylenediaminetetraacetic acid (EDTA; TBE) buffer,34,35,51 and transferred onto polyvinylidene difluoride membranes (88518; Thermo Fisher Scientific). Western blots were probed with the horseradish peroxidase–conjugated secondary antibodies and visualized using the ECL Western Blotting Detection System (RPN2132; Amersham Biosciences, Pistacaway, NJ).
Measurement of Intracellular ROS
Reactive oxygen species (ROS) levels in BACA cells exposed to B. dermatitidis or β-glucan were measured using the Cellular Reactive Oxygen Species Assay kit (ab186027; Abcam, Cambridge MA), according to procedures provided by the manufacturer.
HBEGF Neutralization Assays
BACA cells (0.5 × 106) were seeded on 6-well plates overnight. After three washes with sterile phosphate-buffered saline (PBS), the cells were incubated in fetal bovine serum–free DMEM. Subsequently, the cells were treated for 2 hours with 1 μg of anti–heparin-binding epidermal growth factor (EGF)-like growth factor (HBEGF) primary antibody (SC-74441; Santa Cruz Biotechnology) or control mouse IgG (SC-2025; Santa Cruz Biotechnology) dissolved in 1% bovine serum albumin–PBS solution. Then, the cells were exposed to heat-killed B. dermatitidis for 24 hours in DMEM with 10% fetal bovine serum, and total cell lysates were prepared for Western blot analysis.
Immunofluorescence Analysis
Dual-immunofluorescence staining of FOXA2 with MUC5AC or MUC5B was conducted using BACA cells cultured on coverslips. The cells were incubated with or without 10 μmol/L SYK inhibitor piceatannol for 1 hour before 24 hours of stimulation by heat-killed B. dermatitidis (multiplicity of infection 5:1). BACA cells were fixed in 4% paraformaldehyde for 30 minutes. Then, the cells were blocked with 1% goat serum in PBS for 1 hour and incubated for 16 hours with primary antibody against FOXA2, MUC5AC, or MUC5B (1:500 dilution). The cells were then incubated with the Alexa Fluor 488– or Alexa Fluor 594–conjugated secondary antibody (1:500 dilution) for 1 hour. Subsequently, the cells were mounted with the ProLong Gold Antifade Mountant with DAPI (P36942; Invitrogen, Carlsbad, CA) and imaged using an A1R confocal microscope (Nikon, Melville, NY). Images were quantified by the ImageJ software version 1.52a (Java 1.8.0_112; NIH, Bethesda, MD; http://imagej.nih.gov/ij).
Gene Knockdown by siRNA
BACA cells were seeded at densities of 0.5 × 106 and 0.5 × 105 on 6- and 96-well plates, respectively, and cultured overnight. They were subsequently washed with sterile PBS and incubated with siRNA transfection reagent composed of Lipofectamine RNAiMAX (Invitrogen; number 13778030), specific siRNAs against CLEC7A and SYK, and Opti-MEM (Thermo Fisher Scientific; number 31785070). After 4 hours of transfection, transfected cells were washed three times with PBS and cultured in DMEM containing fetal bovine serum and penicillin-streptomycin for 24 hours. On day 3, the cells were exposed to B. dermatitidis or β-glucan for 24 hours in penicillin-streptomycin–free DMEM before analysis for ROS, FOXA2, and mucins.
Quantitative Real-Time PCR Analysis of the Expression of Cytokine Genes in BACA Cells
Ninety percent confluent BACA cells were incubated with either live or heat-killed B. dermatitidis, or with β-glucan. After 24 hours of incubation, BACA cells were lysed by the Trizol reagent (Ambion, Naugatuck, CT; 15596018). Lysates were mixed with equal volume of 100% ethanol and centrifuged. The supernatants were collected for total RNA extraction by using the Direct-zol RNA miniprep plus (Zymo Research, Tustin, CA; R2070). cDNA synthesis was performed by using the SuperScript III First-Strand Synthesis System (Invitrogen; number 18080051). Synthesized cDNAs were used for quantitative real-time PCR to examine relative mRNA levels of IL-1β, TNF-α, IL-33, IL-4, IL-13, IL-8, and IL-17 genes after normalizing to glyceraldehyde-3-phosphate dehydrogenase. Quantitative real-time PCR was performed using Power SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA; number 4367659) with specific primers (Table 2).
Table 2.
Quantitative Real-Time PCR Primer Sequences
| Cytokines | Sense | Antisense |
|---|---|---|
| IL-1β | 5′-CAAGTCTCCCACCAGCTCTGTA-3′ | 5′-GGGCTTCTTCAGCTTCTCCAA-3′ |
| IL-4 | 5′-TCACCAGCACCTTTGTCCAC-3′ | 5′-CGCTTGTGTTCTTTGGAGCA-3′ |
| IL-8 | 5′-CTCTCTGTGAAGCTGCAGTTCTG-3′ | 5′-GGAAAGGTGTGGAGTGTGTTTTT-3′ |
| IL-13 | 5′-TGTGGAGCGTCAACCTGA-3′ | 5′-TGCGTTCACTGGAAATCTGC-3′ |
| IL-17 | 5′-GGAATCTGCACCGCAATGAGGAC-3′ | 5′-CGCAGAACCAGGATCTCTTGCTGG-3′ |
| IL-33 | 5′-GATGTTGATGGCCAGACGTT-3′ | 5′-CCTGGTCTGGCAATTGGTTT-3′ |
| TNF-α | 5′-TCTCGAACCCCAAGTGACAAG-3′ | 5′-CAACCCATCTGACGGCACTA-3′ |
| GAPDH | 5′-TGGGCTACACTGAGCACCAG-3′ | 5′-GGGTGTCGCTGTTGAAGTCA-3′ |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TNF-α, tumor necrosis factor-α.
Statistical Analysis
Quantitative data were expressed as the means ± SEM. Statistical significance comparison for samples with equal variances was determined using the parametric t-test for two unpaired samples. Statistical significance was expressed as not significant, P ≤ 0.05, P ≤ 0.01, or P ≤ 0.001.
Results
Retrospective Analysis of Fungal Pneumonia Cases at the UIUC CVM Veterinary Diagnostic Laboratory
We analyzed fungal infections in canines and felines that were identified by 18s rRNA PCR, mycology culture, or lung biopsy presented to the UIUC CVM Veterinary Diagnostic Laboratory between January 2014 and June 2019. The number of fungal infection cases ranged from 0 to 14, with the highest caseload occurring in 2015 (Supplemental Figure S1A). Most cases for culture and 18s rRNA were sampled from unknown areas, followed by respiratory tract and skin. Of the 63 total fungal cases, 15 were pulmonary mycosis (Supplemental Figure S1B). The 15 cases of fungal pneumonia were further classified by year (Supplemental Figure S1C). The year with the highest number of pulmonary mycoses was 2018, with 6 cases, followed by 2015, with 5 cases. Eight of the respiratory mycoses were caused by B. dermatitidis, followed by H. capsulatum (3 cases) and C. immitis (1 case), and 3 cases were unidentified (Supplemental Figure S1D). On the basis of lung tissue availability (Table 1), we examined the role of FOXA2 in mucus hypersecretion during fungal pneumonia in canines and felines.
FOXA2 Expression Is Down-Regulated with Concomitant Up-Regulation of Mucins in Airways of Dogs and Cats with Fungal Pneumonia
Recently, we demonstrated that diseased canine lungs (COPD and CB) with bacterial infection showed FOXA2 depletion, accompanied by increased expression of MUC5AC and MUC5B.35 We hypothesized that pulmonary fungal infection would deplete FOXA2 and cause mucus hypersecretion. We compared healthy canine and feline lungs with those infected by B. dermatitidis and H. capsulatum, respectively. Cases used are displayed in Table 1. Healthy canine and feline lung tissues showed no evidence of fungal organisms with GMS staining (Table 1, Figures 1 and 2, and Supplemental Figures S2 and S3). In contrast, fungal-infected canine and feline lung tissues were positive for intralesional round typical yeast bodies under GMS staining, indicating the presence of B. dermatitidis or H. capsulatum, respectively (Figures 1 and 2 and Supplemental Figures S2 and S3). Airways from healthy lungs had basal levels of mucins, as evidenced by the weak staining with periodic acid-Schiff, and low immunoreactivity with IHC for MUC5AC and MUC5B. In contrast, B. dermatitidis and H. capsulatum–infected airway epithelia exhibited increased expression of mucins (Figures 1 and 2 and Supplemental Figures S2 and S3). FOXA2 expression was inversely correlated with MUC5AC and MUC5B in healthy and infected lungs. IHC analysis indicated that FOXA2 was strongly expressed in healthy canine and feline lungs, but was severely depleted in the bronchial surface epithelia of the infected lungs, which overlapped the area of MUC5AC and MUC5B overexpression (Figures 1 and 2 and Supplemental Figures S2 and S3). Some residual FOXA2 expression could be seen in the basal epithelial cells of infected airways, serving as internal positive controls (Figures 1 and 2 and Supplemental Figures S2 and S3). These results indicate that fungal infections deplete the expression of airway FOXA2 that is inversely associated with increased MUC5AC and MUC5B.
Figure 1.

Airway expression of forkhead box protein A2 (FOXA2) is inversely correlated with mucin abundance in canine pulmonary mycosis. Paraffin-embedded lung sections from healthy and fungal-infected animals were stained with Grocott methenamine silver (GMS), periodic acid-Schiff (PAS), and immunohistochemistry (IHC) for the presence of fungal pathogens and the expression of FOXA2 and mucins. Healthy control lung sections stained negative for fungal organisms and mucin secretion with GMS and PAS, with strong expression of FOXA2 in bronchial surface epithelia (black arrows). Fungal-infected canine lungs were positive for yeast bodies under GMS staining (black spots; black arrows) in the submucosa and bronchial lumens, accompanied by positive PAS staining (pink spots; black arrows) in surface epithelia, which colocalized to the regions expressing MUC5AC and MUC5B in IHC (black arrows). FOXA2 expression was down-regulated in the bronchial surface epithelia of the infected lung tissues (black arrows), with successful positive controls shown in basal epithelial cells (red arrows). Additional images can be found in the Supplemental Figure S2. Scale bars = 50 μm.
Figure 2.

Airway expression of forkhead box protein A2 (FOXA2) is inversely correlated with mucin abundance in feline pulmonary mycosis. Paraffin-embedded lung sections from healthy and fungal-infected animals were stained with Grocott methenamine silver (GMS), periodic acid-Schiff (PAS), and immunohistochemistry (IHC) for the presence of fungal pathogens and the expression of FOXA2 and mucins. Healthy control lung sections stained negative for fungal organisms and showed basal levels of mucin secretion with GMS and PAS, with strong expression of FOXA2 in bronchial surface epithelia (black arrows) and basal cells (red arrows). Fungal-infected feline lungs were positive for yeast bodies under GMS staining (black spots; black arrows) in the submucosa and bronchial lumens, accompanied by positive PAS staining (pink spots; black arrows) in surface epithelia, which colocalized to the regions expressing MUC5AC and MUC5B in IHC (black arrows). FOXA2 expression was depleted in the bronchial surface epithelia of the infected lung tissues (black arrows), with successful positive controls shown in basal epithelial cells (red arrows). Additional images can be found in the Supplemental Figure S3. Scale bars: 50 μm (all other panels); 200 μm (MUC5AC left panel).
B. dermatitidis Infection and β-Glucan Exposure Deplete FOXA2 Expression in Cultured Canine Bronchial Epithelial Cells
To gain insight into the mechanisms by which fungal pneumonia causes mucin overexpression, we used BACA cells to examine the expression dynamics of FOXA2 and mucins. BACA cells were exposed either to live or heat-killed B. dermatitidis strain SCB-2 at both multiplicity of infection of 1:1 and 5:1, and the expression of FOXA2 and mucins was assessed at 24 hours after treatment. Control BACA cells treated with PBS showed strong expression of FOXA2, with basal levels of MUC5AC and MUC5B (Figure 3A). Although the heat-killed B. dermatitidis reduced the expression of FOXA2 while increasing the expression of both mucins, the effect was more modest when compared with the cells infected by live B. dermatitidis, which showed severely reduced FOXA2 and significantly higher levels of both mucins (Figure 3A). Additional replicates of Western blots are provided in the Supplemental Figure S4A.
Figure 3.

Blastomyces dermatitidis and β-glucan induce mucin expression in the immortalized canine airway carcinoma (BACA) cells. BACA cells at 90% confluency were challenged with live (LV) or heat-killed (HK) B. dermatitidis strain SCB-2 at the indicated multiplicity of infection (MOI), or indicated concentrations of β-glucan for 24 hours. Control (Ctrl) cells were exposed to the same volume of sterile phosphate-buffered saline. Cytoplasmic and nuclear protein extracts were separated on acrylamide-agarose (mucins) and acrylamide [FOXA2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and H3] gels, respectively, and probed with specific antibodies against each protein. GAPDH and H3 were used as loading controls. The experiments were independently performed three times with similar results. Western blots from one typical experiment are shown. Additional replicates of Western blots can be found in Supplemental Figure S4. Expression levels of MUC5AC, MUC5B, and forkhead box protein A2 (FOXA2) were measured by ImageJ software version 1.52a. Densitometry data of FOXA2, MUC5AC, and MUC5B expression are expressed as means ± SEM from all three experiments (A and B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (t-test).
β-Glucan is a highly immunogenic component of fungal cell wall capable of inducing host innate immune responses.52 However, the glucan component of the infectious yeast form of B. dermatitidis (eg, cultured at 37°C to 39°C) consists of about 95% α-glucan and 5% β-glucan, whereas in the mycelial form, it consists of about 60% α-glucan and 40% β-glucan.53 Interestingly, a recent study with antibody-directed targeting of radioisotope against (1→ 3)-β-glucan successfully inhibited blastomycosis in mice,54,55 clearly indicating the importance of β-glucan in B. dermatitidis survival. To assess whether β-glucan modulates FOXA2 and mucin expression, BACA cells were treated with 0.1, 1, or 10 μg/mL β-glucan and assessed after 48 hours. β-Glucan decreased the expression of FOXA2, with concomitant increases in the expression of both MUC5AC and MUC5B (Figure 3B). Additional replicates of Western blots are provided in Supplemental Figure S4B. Collectively, these findings support the hypothesis that lung infection by pathogenic fungi depletes FOXA2 expression, resulting in mucus hypersecretion in canines and felines (Figures 1 and 2 and Supplemental Figures S2 and S3).
H. capsulatum Infection Depletes FOXA2 Expression in Cultured Canine Bronchial Epithelial Cells
To determine the broader applicability of canine fungal pneumonia as a model to study FOXA2 function, we examined FOXA2 inactivation and mucin production in BACA cells exposed to either live or heat-killed H. capsulatum strain G217B. Similar to B. dermatitidis, infection by live H. capsulatum, as well as exposure to heat-killed yeast, severely depleted FOXA2 expression and increased expression of both MUC5B and MUC5AC (Figure 4). These results indicate that FOXA2 is a conserved target of inhibition by pathogenic fungi.
Figure 4.

Histoplasma capsulatum induces mucin expression in the immortalized canine airway carcinoma (BACA) cells. BACA cells at 90% confluency were challenged with live (LV) or heat-killed (HK) H. capsulatum strain G217B at the indicated multiplicity of infection (MOI). Control (Ctrl) cells were exposed to the same volume of sterile phosphate-buffered saline. Cytoplasmic and nuclear protein extracts were separated on acrylamide-agarose (mucins) and acrylamide [forkhead box protein A2 (FOXA2), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and H3] gels, respectively, and probed with specific antibodies against each protein. GAPDH and H3 were used as loading controls. The experiments were independently performed three times with similar results. Western blots from one typical experiment are shown.
B. dermatitidis Infection and β-Glucan Exposure Activate the EGFR–Phosphatidylinositol 3-Kinase/AKT but Not STAT6 Signaling to Antagonize FOXA2
Previously, we have shown that infection by respiratory bacterial pathogens, including P. aeruginosa,28,30,31,34,35 B. bronchiseptica,35 and M. pneumoniae,30 inhibits the expression of FOXA2, resulting in excessive production of MUC5AC and MUC5B. Specifically, P. aeruginosa and its oxidation-reduction active exotoxin pyocyanin activate both Th2 cytokines–STAT6-SPDEF and EGFR-AKT/ERK1/2 to inhibit the expression of FOXA2. Similarly, M. pneumoniae inhibits FOXA2 by activating both the STAT3-STAT6 and ROS-EGFR signaling cascades. Therefore, we examined whether the loss of FOXA2 expression was caused by B. dermatitidis and β-glucan activating the aforementioned antagonistic signaling pathways. Various membrane, cytoplasmic, and nuclear protein extracts (Figure 3) were probed with antibodies against normal and phosphorylated proteins. The results indicate that both live and heat-killed B. dermatitidis (Figure 5A), as well as β-glucan (Figure 5C) activated EGFR-AKT and EGFR-ERK1/2, but not STAT6 (Figure 5, B and D). The increase in the levels of pEGFR, pAKT, and pERK1/2 inversely correlated with the expression pattern of FOXA2 and positively correlated with the expression of both MUC5AC and MUC5B (Figure 3 and Supplemental Figure S4).
Figure 5.

Blastomyces dermatitidis and β-glucan activate the epidermal growth factor receptor (EGFR)–phosphatidylinositol 3-kinase/AKT signaling that antagonizes forkhead box protein A2. Membrane, cytoplasmic, and nuclear protein extracts of the immortalized canine airway carcinoma (BACA) cells from Figure 4 were probed with regular or phospho-specific antibody against each protein. Total EGFR, AKT, extracellular signal-regulated kinase (ERK) 1/2, and STAT6 were used as loading controls for their respective phosphorylated counterparts. H3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as additional loading controls. A and B: Live (LV) and heat-killed (HK) B. dermatitidis increased the levels of phosphorylated EGFR (pEGFR), phosphorylated AKT (pAKT), and phosphorylated ERK1/2 (pERK1/2) but not phosphorylated STAT6 (pSTAT6). C and D: β-Glucan increased the levels of pEGFR, pAKT, and pERK1/2, but not pSTAT6. The experiments were independently performed three times with similar results. Western blots from one typical experiment are shown. Ctrl, control; MOI, multiplicity of infection.
Inhibition of EGFR-AKT/ERK1/2 Signaling Restores FOXA2 Expression and Attenuates Mucin Production Induced by B. dermatitidis and β-Glucan
To confirm that B. dermatitidis and β-glucan enhance EGFR-AKT– and EGFR-ERK1/2–mediated signaling, the expression of FOXA2 and both mucins was assessed in BACA cells pretreated for 1 hour with specific inhibitors for EGFR (AG1478), AKT (LY294002), and ERK1/2 (PD98059), before challenge by B. dermatitidis and β-glucan. LY294002 is a highly selective inhibitor of phosphatidylinositol 3-kinase, a kinase that activates AKT. More importantly, all three inhibitors, especially PD98059, restored the expression of FOXA2, which was accompanied by reduced expression of MUC5AC and MUC5B in BACA cells treated with either live or heat-killed B. dermatitidis, or with β-glucan (Figure 6). Additional replicates of mucin and FOXA2 Western blots are provided in Supplemental Figure S5. These results indicate that pulmonary blastomycosis activates both EGFR-ERK1/2 and EGFR–phosphatidylinositol 3-kinase–AKT signaling cascades to suppress the expression of FOXA2, resulting in excessive mucus.
Figure 6.

Inhibition of epidermal growth factor receptor (EGFR)–AKT/extracellular signal-regulated kinase (ERK) 1/2 signaling cascades restores forkhead box protein A2 (FOXA2) to attenuate the ability of Blastomyces dermatitidis and β-glucan to induce mucin hypersecretion. Immortalized canine airway carcinoma (BACA) cells were pre-exposed to 20 μmol/L of EGFR inhibitor AG1478 (AG), AKT inhibitor LY294002 (LY), and ERK1/2 inhibitor PD98059 (PD) for 1 hour before the addition of sterile phosphate-buffered saline (negative control) or live (LV) and heat-killed (HK) B. dermatitidis and β-glucan, respectively. The expression of FOXA2, MUC5AC, and MUC5B was examined by specific antibodies and quantified by densitometry. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control for MUC5AC and MUC5B. Histone H3 was used as a loading control for FOXA2. AG, LY, and PD restored the expression of FOXA2 with concomitant down-regulation of MUC5AC and MUC5B in B. dermatitidis–infected or β-glucan exposed BACA cells. The ERK inhibitor showed the highest restorative effect on FOXA2 expression, resulting in most significant reduction in mucin expression. The experiments were repeated three times with similar results. Additional replicates of Western blots can be found in Supplemental Figure S5. Protein expressions were compared by the t-test. Densitometry data of MUC5AC and MUC5B expression are expressed as means ± SEM from all three experiments (A–C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (t-test).
Inhibition of Dectin-1 and SYK Ameliorates FOXA2 Inhibition by B. dermatitidis and β-Glucan
Dectin-1 is a C-type lectin-like transmembrane pattern-recognition receptor on mammalian cells that binds β-glucan and stimulates the innate immune cells in response to fungal pathogens.52 On binding to β-glucan, Dectin-1 signaling activates the tyrosine protein kinase SYK to potentiate various antifungal responses, including cytokine and chemokine production, respiratory burst, and phagocytosis.52 Because B. dermatitidis and β-glucan induce mucin expression in BACA cells, we hypothesized that Dectin-1 and SYK may modulate the expression of FOXA2. SYK phosphorylation was increased by both B. dermatitidis and by β-glucan (Figure 7A). Inhibition of CLEC7A (which encodes Dectin-1) and SYK, mediated by siRNA, restored FOXA2 expression in BACA cells treated with either live or heat-killed B. dermatitidis, or with β-glucan. FOXA2 restoration was accompanied by significant attenuation of MUC5AC. The expression of MUC5B was also reduced, although at lower levels (Figure 7, B and C).
Figure 7.

Blastomyces dermatitidis and β-glucan activates Dectin-1–SYK signaling to inhibit forkhead box protein A2 (FOXA2) expressions in BACA cells. The expression of CLEC7A and SYK genes in the immortalized canine airway carcinoma (BACA) cells (90% confluency) was inhibited by specific siRNA before exposure to live (LV) or heat-killed (HK) B. dermatitidis strain SCB-2 at the indicated multiplicity of infection (MOI), or to indicated concentrations of β-glucan for 24 hours. Control (Ctrl) cells were exposed to the same volume of sterile phosphate-buffered saline. Cytoplasmic and nuclear protein extracts were separated on acrylamide-agarose gels (for mucins) and normal 10% acrylamide gels [for SYK, phosphorylated SYK (pSYK), FOXA2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and H3], and probed with normal or phospho-specific antibodies against each protein. Total SYK, GAPDH, and H3 were used as loading controls. A: Both live and heat-killed B. dermatitidis and β-glucan increased the levels of pSYK. B and C: Inhibition of CLEC7A (B) and SYK (C) by siRNA partially restored FOXA2 expression, which was accompanied by reduction in MUC5AC and MUC5B. The experiments were independently performed three times with similar results. Western blots from a typical experiment are shown. β-glu, β-glucan.
Immunofluorescence was used to validate the above results. Because both live and heat-killed B. dermatitidis inhibited FOXA2 (Figures 3, 5, 6, and 7), we used heat-killed fungus for the immunofluorescence studies for its experimental convenience. Heat-killed B. dermatitidis reduced FOXA2 expression in the nuclei of mucin-secreting BACA cells and increased expression of MUC5AC and MUC5B (Figure 8). More importantly, siRNA-mediated knockdown of SYK prevented both depletion of FOXA2 and overexpression of MUC5AC and MUC5B by heat-killed B. dermatitidis (Figure 8). Collectively, these results indicate that induction of Dectin-1–SYK signaling inhibits FOXA2, resulting in overexpression of mucins.
Figure 8.

Inhibition of SYK restores forkhead box protein A2 (FOXA2) ability to attenuate excessive mucin expression. The expression of SYK gene in the immortalized canine airway carcinoma (BACA) cells (90% confluency) was inhibited by specific siRNA before exposure to heat-killed (HK) Blastomyces dermatitidis strain SCB-2 at the indicated multiplicity of infection (MOI) for 24 hours. Control cells were exposed to the same volume of sterile phosphate-buffered saline. BACA cells were double stained with primary antibodies against FOXA2 and MUC5AC and MUC5B. Nuclei in airway cells were stained with DAPI (blue). Labeled cells were visualized by secondary antibodies conjugated to Alexa Fluor 594 (red; FOXA2) and to Alexa Fluor 488 (cyan-green; MUC5AC and MUC5B). A: Colocalization of FOXA2 expression with MUC5AC in BACA cells. B: Colocalization of FOXA2 expression with MUC5B in BACA cells. FOXA2 and mucin expression were visualized using a Nikon A1 confocal microscope. Experiments were independently performed three times. Fluorescence intensity of FOXA2, MUC5AC, and MUC5B was analyzed with the ImageJ software version 1.52a. At least 500 cells from each experiment were counted. Data are expressed as means ± SEM from all three experiments (A and B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (t-test). Original magnification, ×100 (A and B).
Induction of EGFR-AKT/ERK1/2 Signaling by SYK Is Mediated through ROS and HBEGF Following Fungal Stimulation
Next, we used the heat-killed B. dermatitidis to examine the signaling hierarchy between SYK and EGFR in the context of FOXA2 inhibition. As expected, heat-killed B. dermatitidis activated EGFR-AKT/ERK1/2 signaling (Figure 9A). More importantly, SYK knockdown by siRNA reduced the levels of pEGFR, pAKT, and pERK1/2, suggesting that SYK acted upstream of the EGFR signaling during inhibition of FOXA2 by B. dermatitidis.
Figure 9.

Dectin-1 (Dec) and SYK act upstream of epidermal growth factor receptor (EGFR) signaling through up-regulation of reactive oxygen species (ROS). A: SYK acts upstream of EGFR-AKT/extracellular signal-regulated kinase (ERK) 1/2. Immortalized canine airway carcinoma (BACA) cells were pre-exposed to the siRNA against SYK before challenged with heat-killed (HK) Blastomyces dermatitidis strain SCB-2 (multiplicity of infection 5:1) for 24 hours. Control cells were exposed to the same volume of sterile phosphate-buffered saline. Membrane, cytoplasmic, and nuclear protein extracts were probed with regular or phospho-specific antibody against each protein. Total EGFR, AKT, and ERK1/2 were used as loading controls for their respective phosphorylated counterparts. H3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as additional loading controls. B: Live (LV) and HK B. dermatitidis and β-glucan induced intracellular levels of ROS that were attenuated by siRNA against CLEC7A (Dec) and SYK genes, or specific inhibitors against EGFR [AG1478 (AG)], AKT [LY294002 (LY)], and ERK1/2 [PD98059 (PD)] proteins. ROS levels were measured using the Oxiselect in vitro ROS/RNS assay kit. The experiments were performed independently three times in triplicate. C: Glutathione (GSH) reduces the expression of mucins by heat-killed B. dermatitidis. Additional replicates of Western blots can be found in Supplemental Figure S6. Expression levels of MUC5AC, MUC5B, and forkhead box protein A2 (FOXA2) were measured by using the ImageJ software version 1.52a. Densitometry data are shown. D: GSH attenuates SYK and EGFR signaling. The expression of each protein was determined by normal or phospho-specific antibodies. Total SYK, EGFR, Akt, ERK1/2, GAPDH, and H3 were used as loading controls. Data are expressed as means ± SEM from three experiments (B and C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (t-test). pAKT, phosphorylated AKT; pEGFR, phosphorylated EGFR; pERK1/2, phosphorylated ERK1/2; pSYK, phosphorylated SYK.
Previously, it has been reported that activation of the Dectin-1–SYK–phospholipase C–γ2-ERK axis increases intracellular ROS.52,56 The intracellular ROS augments EGFR signaling following binding of its ligands57,58 and is involved in the activation of mucin expression.33 We hypothesized that activation of Dectin-1–SYK signaling by B. dermatitidis leads to increased intracellular ROS. In turn, ROS acts together with EGFR ligand(s) to activate EGFR-AKT/ERK1/2 signaling, which inhibits FOXA2. First, the levels of intracellular ROS were assessed in BACA cells exposed to either live or heat-killed B. dermatitidis, or to β-glucan with or without siRNA knockdown of CLEC7A and SYK, as well as specific inhibitors for EGFR (AG1478), AKT (LY294002), and ERK1/2 (PD98059). ROS production was increased by both B. dermatitidis and β-glucan but was significantly reduced by SYK gene expression knockdown and pretreatment of each of the inhibitors (Figure 9B). Interestingly, although ROS was decreased in both live and heat-killed B. dermatitidis–exposed BACA cells after inhibition of Dectin-1 and AKT, the levels of reduction were not statistically significant. However, in BACA cells exposed to β-glucan, the inhibition of these effectors led to significant reduction of ROS production after inhibition of Dectin-1 but not AKT (Figure 9B). Collectively, these results indicate that Dectin-1–SYK plays an important role in generation of intracellular ROS in response to B. dermatitidis and β-glucan.
To confirm the role of ROS, we examined whether the antioxidant glutathione (GSH) could reduce the ability of heat-killed B. dermatitidis to suppress FOXA2 expression. GSH restored FOXA2 expression and down-regulated both mucins (Figure 9C); replicate Western blots can be found in Supplemental Figure S6. In addition, GSH reduced the levels of phosphorylated SYK, pEGFR, pAKT, and pERK1/2 induced by heat-killed B. dermatitidis (Figure 9D). Collectively, these results indicate that in response to fungal infection, SYK acts upstream and activates EGFR-AKT/ERK1/2 signaling through intracellular ROS, resulting in FOXA2 inhibition and mucin hypersecretion.
As previously mentioned, on engaging with the ligands, EGFR signaling is potentiated by intracellular ROS.57,58 Seven EGFR ligands are known: the high-affinity ligands EGF, transforming growth factor-α (TGF-α), HBEGF, and betacellulin; and the low-affinity ligands amphiregulin, epiregulin, and epigen.59 Of the four high-affinity ligands examined only HBEGF was induced by heat-killed B. dermatitidis and participated in mucin overexpression in BACA cells (Figure 10), consistent with a previous report.60 Western blot analysis with specific antibodies did not detect expression of TGF-α, EGF, and amphiregulin (data not shown). Induction of HBEGF by heat-killed B. dermatitidis was attenuated by siRNA-mediated knockdown of CLEC7A and SYK, indicating that HBEGF-EGFR acts downstream of Dectin-1 and SYK (Figure 10A). Because ROS promotes EGFR activation when the cells are stimulated by EGFR ligands,57 we examined if GSH could attenuate HBEGF induction by heat-killed B. dermatitidis. GSH was able to partially attenuate the expression of HBEGF (Figure 10A). To further authenticate these findings, we measured the mucin expression stimulated by heat-killed B. dermatitidis with or without HBEGF neutralizing antibody. Neutralization of HBEGF with antibody restored the expression of FOXA2 while down-regulating the expression of MUC5AC and MUC5B (Figure 10B); replicate Western blots can be found in Supplemental Figure S7. In contrast, control mouse IgG was unable to neutralize the inhibitory effects of heat-killed B. dermatitidis on FOXA2, resulting in excessive mucins (Figure 10, B and C). Collectively, these results indicate that HBEGF acts together with SYK-induced ROS to potentiate EGFR-AKT/ERK1/2 signaling that results in FOXA2 inhibition and increased mucin expression during B. dermatitidis infection.
Figure 10.
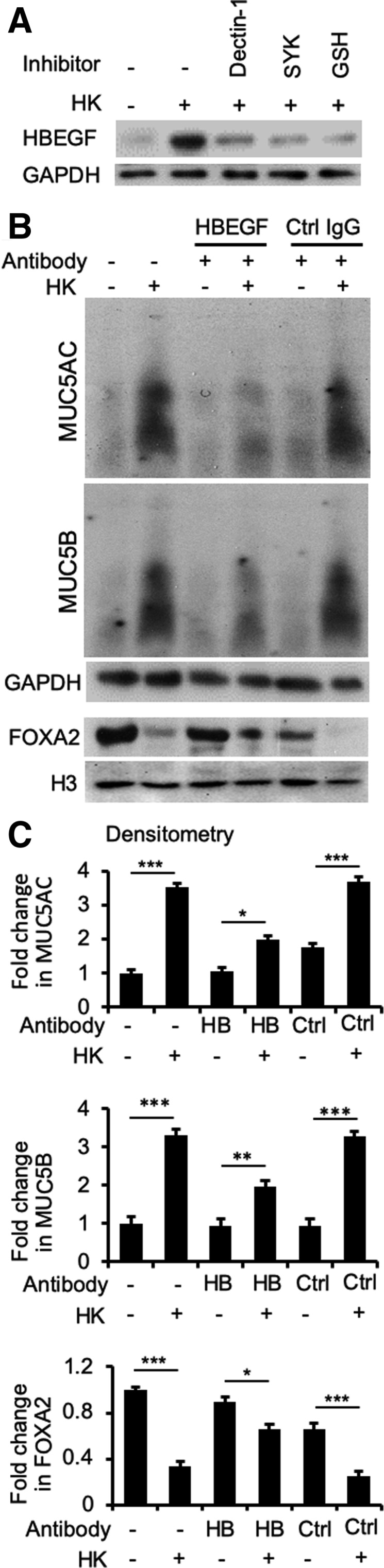
Heparin-binding EGF-like growth factor (HBEGF) bridges SYK and epidermal growth factor receptor signaling in airway epithelial cells exposed to Blastomyces dermatitidis. A: Immortalized canine airway carcinoma (BACA) cells were pre-exposed to the SYK inhibitor piceatannol (20 μmol/L) or glutathione (5 mmol/L) for 1 hour before challenged with heat-killed (HK) B. dermatitidis strain SCB-2 [multiplicity of infection (MOI) 5:1] for 24 hours. Control cells were exposed to the same volume of sterile phosphate-buffered saline. Membrane, cytoplasmic, and nuclear protein extracts were probed with antibody against each protein. H3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as additional loading controls. B and C: Antibody neutralization of HBEGF restores forkhead box protein A2 (FOXA2) to attenuate mucin expression. BACA cells were pretreated with anti-HBEGF antibody (HB; 1 μg) or an irrelevant control mouse IgG (Ctrl; 1 μg) for 2 hours before 24 hours of exposure to heat-killed B. dermatitidis (MOI 5:1). GAPDH and H3 were used as loading controls. The experiments were independently performed three times with similar results. Western blots from one typical experiment are shown. Additional replicates of Western blots can be found in Supplemental Figure S7. Expression levels of MUC5AC, MUC5B, and FOXA2 were measured by using the ImageJ software version 1.52a. Densitometry data are expressed as means ± SEM from all three experiments (C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (t-test).
B. dermatitidis and β-Glucan Modulate the Expression of Cytokine Genes that Mediate Mucin Production
B. dermatitidis infection has been long known to modulate cytokine production in host cells that enable chronic pulmonary infection and persistent inflammatory responses.2,61 Herein, we examined the expression of specific cytokine/chemokine genes known to regulate mucin biosynthesis in BACA cells exposed to either live or heat-killed B. dermatitidis, or to β-glucan. These include the Th1 cytokine gene TNF-α,45 Th17 cytokine gene IL-17,62 Th2 cytokine genes IL-4,63 IL-13,47 and IL-33,64 and the pyrogenic proinflammatory cytokine genes IL-1β62 and IL-8.65 IL-1β has been shown to be important for the induction of Th17 cells during fungal infection.66 We found that TNF-α was only moderately induced in BACA cells infected with live B. dermatitidis but not with heat-killed fungus nor β-glucan (Figure 11). Expression of IL-17 was up-regulated by heat-killed B. dermatitidis and β-glucan, but not by live B. dermatitidis. In contrast, Th2 cytokine gene IL-4 was not significantly altered (Figure 11) and IL-13 was not detected (data not shown). Both live and heat-killed B. dermatitidis consistently and significantly inhibited the expression of IL-33, involved in enhancing the induction of Th2 cytokines (Figure 11). Expression of IL-1β was up-regulated by both live and heat-killed B. dermatitidis, and by β-glucan, whereas IL-8 was up-regulated by heat-killed B. dermatitidis and β-glucan only (Figure 11). Collectively, these results suggest that IL-1β, IL-17, and IL-8 participate in the induction of mucin hypersecretion during B. dermatitidis–mediated lung disease.
Figure 11.

Blastomyces dermatitidis and β-glucan alter the transcript levels of cytokine genes that regulate mucin expression. Immortalized canine airway carcinoma (BACA) cells were infected with live (LV) or heat-killed (HK) B. dermatitidis strain SCB-2 at the multiplicity of infection of 5:1, or to β-glucan (10 μg/mL) for 24 hours. The expression of IL-1β, TNF-α, IL-33, IL-4, IL-13, IL-8, and IL-17 genes was analyzed by quantitative real-time PCR and normalized to the GAPDH, and compared against BACA cells treated with phosphate-buffered saline (-). The experiments were independently performed three times in triplicate. Typical results from one of the experiments are shown. The data represent means ± SEM of triplicated samples in each group. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 (t-test).
Discussion
In this study, we examined the feasibility of using fungal-infected canine and feline lungs as model to study the interactions between FOXA2 inactivation and mucus hypersecretion. Both canine and feline lungs infected with B. dermatitidis and H. capsulatum show dramatic reduction in FOXA2 expression, accompanied by mucin hypersecretion. Based collectively on histopathologic analysis of lung tissues and in vitro molecular analysis in BACA cells, we propose that B. dermatitidis infection activates the antagonistic SYK-ROS-HBEGF-EGFR-AKT/ERK1/2 signaling pathway to down-regulate FOXA2 expression, resulting in mucin overexpression (Figure 12). As far as we know, this is the first study to mechanistically link SYK-ROS-HBEGF-EGFR-AKT/ERK1/2 to FOXA2 inactivation and dysregulation of mucus homeostasis in pulmonary mycosis, especially in canine and feline species.
Figure 12.

Proposed model of forkhead box protein A2 (FOXA2) inactivation and disruption of mucus homeostasis by Blastomyces dermatitidis. EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; HBEGF, heparin-binding EGF-like growth factor; ROS, reactive oxygen species.
Albeit less common than viral and bacterial respiratory infections, pulmonary mycosis can be life-threatening in patients with compromised immune system and muco-obstructive diseases.2 Pulmonary mycosis caused by inhaled fungal particles disseminates to other organs, including skin, bone, and central nervous systems.3,4,6 In limited cases seen at the UIUC CVM Veterinary Diagnostic Laboratory between 2014 and 2019, B. dermatitidis was the most common etiologic agent. This was expected as B. dermatitidis is endemic to Illinois, sometimes nicknamed Chicago's disease.1, 2, 3 H. capsulatum was the second most common fungal agent seen in our study, which is also expected as it is endemic to the midwestern United States.4,5 Pulmonary manifestations of fungal infections are most common. For example, >80% and 90% of blastomycosis cases in humans and dogs, respectively, are of respiratory diseases.49,67 The US Centers for Disease Control and Prevention estimates one to two cases of blastomycosis per 100,000 people per year in reportable states.68 Because of limitation in diagnostic methods, the US Centers for Disease Control and Prevention number is likely an underestimation. Another confounding factor is that pulmonary symptoms of blastomycosis are similar to those caused by viral or bacterial pneumonia, resulting in frequent misdiagnosis. Canine blastomycosis appears at a higher rate compared with human cases,48 with significant geographic overlap in regional prevalence.68 Therefore, apart from our experimental evidence showing that both canine and feline fungal infections could serve as natural disease models to study the mechanisms of FOXA2 inactivation and mucus hypersecretion in human pulmonary mycosis, monitoring of disease outbreak in these companion animals may serve as a sentinel to predict potential changes in the ecologic environment and circulating endemic strains in humans, which may improve diagnosis, earlier treatment, and clinical outcome.
Most recently, we have demonstrated the feasibility of canine models to study mucin hypersecretion caused by P. aeruginosa and B. bronchiseptica.35 One of the advantages of the canine and feline models is that they allow the study of pathogenesis of asthma, COPD, CB, and canine respiratory disease complex in natural hosts that better mimic chronic lung diseases in humans. There has been much debate as to whether artificially induced respiratory diseases in rodents could recapitulate the full spectrum of human lung diseases.35,69,70 Mouse lung differs from human lung in three major ways: (i) the relatively large size, (ii) the dearth of submucosal glands, and (iii) the presence of high numbers of Clara (club) cells in airways. Therefore, when compared against human lung, which has smaller airways and higher number of mucus glands, the disease pathogenesis (eg, goblet cell hyperplasia and metaplasia, excessive mucus, and FOXA2 depletion) and treatment response could be subdued in mice. In contrast, chronic lung diseases naturally develop in canine and feline, and may represent higher animal models of human respiratory diseases,71 including the study of FOXA2-mediated mucus homeostasis and mucociliary clearance. Our current study demonstrates that blastomycosis and histoplasmosis down-regulate the expression of FOXA2, resulting in mucin overexpression in canine and feline airways. Although excessive mucus is found in Aspergillus-mediated allergic bronchopulmonary aspergillosis72 and in a rat model of Pneumocystis infection,14 mucus hypersecretion has not been thoroughly examined in pulmonary blastomycosis and histoplasmosis. Chest radiography has shown that most human patients with blastomycosis and other pulmonary mycoses have airway infiltration,67 which is suggestive of mucus hypersecretion. Collectively, these findings indicate that FOXA2 is a conserved target of inactivation in the airways of humans, canines, and felines by fungal and bacterial pathogens.
In a previous study, a mixture of β-glucan and supernatant of Aspergillus fumigatus culture has been shown to increase MUC5AC gene transcripts through the TGF-α–EGFR signaling in the immortalized human bronchial epithelial NCI-H292 cells.73 In addition, an experimental model of A. fumigatus–induced asthma was shown to elevate IL-4 and IL-13 levels,74 suggesting that fungal sensitization enhances downstream IL13RA1/IL4R-STAT6-SPDEF signaling. Similarly, Pneumocystis activates the transcription of MUC5B gene, partially through inhibition of FOXA2 by STAT6 in rat lungs.14 There are some similarities and differences between our results when compared with these aforementioned studies. Similar to A. fumigatus study, we show that EGFR signaling is involved in response to B. dermatitidis and β-glucan. However, instead of TGF-α, BACA cells respond to B. dermatitidis and elevate mucin expression driven by the SYK-ROS-HBEGF-EGFR-AKT/ERk1/2 pathway. No signals for the expression of TGF-α, EGF, or amphiregulin, were detected by Western blotting (data not shown). However, we cannot rule out the lack of cross-reactivity of the antibodies for canine proteins. In addition, B. dermatitidis and β-glucan failed to activate STAT6 in BACA cells. One possible explanation is that Th2 cytokines (eg, IL-13) secreted by innate lymphoid cell-2 and Th2 cells, which are needed to activate STAT6, are absent in BACA cells infected with B. dermatitidis. However, this is less likely as we have previously shown that infection by P. aeruginosa or exposure to its oxidation-reduction active toxin pyocyanin could activate STAT6 in BACA cells, even in the absence of innate lymphoid cell-2 and Th2 cells.35 Further analysis is needed to determine whether the lack of STAT6 response is specific to the interactions between B. dermatitidis and BACA cells.
β-Glucan is an immunogenic fungal cell wall component that stimulates the pulmonary defense system, including the production of mucins.12 Although the infectious yeast phase of B. dermatitidis expresses minimal amount of β-glucan,54,55 it is important for survival and virulence.54,55 Binding of β-glucan to toll-like receptor 4 on human airway epithelial cells increases MUC5B expression through the activation of p38 mitogen-activated protein kinase and NF-κB.12 Toll-like receptor 2 expression is not induced by β-glucan,12 but cooperates with Dectin-1 to enhance the recognition of β-glucan.75,76 Aspergillus fumigatus up-regulates the production of proinflammatory cytokines, antimicrobial peptides, and ROS in a toll-like receptor 2–Dectin-1-dependent manner.77 However, there are no reports of β-glucan inducing mucin expression through Dectin-1–mediated signaling. SYK, recruiting to Dectin-1, activating ERK and phosphatidylinositol 3-kinase/AKT,56 and producing intracellular ROS.56 Intracellular ROS, including hydrogen peroxide and superoxide anion, act as secondary messengers and microbicidal agents,56 as well as maintain the activated state of pEGFR by inactivating phosphatases and transmitting signals to downstream targets.57 We show that in response to B. dermatitidis and β-glucan, SYK down-regulates FOXA2 expression and elevates mucin production through ROS-HBEGF-EGFR-AKT/ERK1/2 signaling. Attenuation of EGFR-mediated signaling and mucin expression by GSH indicates that ROS likely functions as the secondary messenger of SYK that activates EGFR and downstream kinase cascades. In addition, our results show that HBEGF acts as an EGFR ligand downstream of SYK-mediated signaling. Therefore, ROS and HBEGF collectively relay the signaling from SYK to EGFR in response to B. dermatitidis and β-glucan.
The intensity of FOXA2 inhibition was in the order of live yeast > heat-killed yeast > β-glucan. These observations suggest that an active infection process, mediated by additional factors expressed by live B. dermatitidis, modulates FOXA2 expression. One such candidate is blastomyces adhesin-1 (BAD1), a secreted yeast-phase–specific host cell adhesion factor2,45 that down-regulates TNF-α production from macrophages and polymorphonuclear leukocytes. In addition, the serine protease dipeptidyl peptidase-IV cleaves the human chemokine CXCL-2, which modulates neutrophil activity and aggravates various symptoms of blastomycosis.78 Similarly, serine proteases of A. fumigatus activate TNF-α converting enzyme that enhances TGF-α secretion and airway mucin expression.73
Th1 cytokines, including IFN-γ and TNF-α, are important in host resistance against B. dermatitidis by recruitment of immune cells, development of host adaptive immunity in the lung, and enhancement of fungal clearance by macrophages and neutrophils.45,79 With respect to mucin biosynthesis, IFN-γ negatively regulates mucin expression by inhibiting STAT-6,80 a key transcription factor mediating Th2 cytokine (eg, IL-4 and IL-13) response. Previous studies have shown that FOXA2 regulates the level of pulmonary Th2 cytokines, including IL-4 and IL-13.81 FOXA2-deficient airway epithelial cells up-regulate the expression and secretion of IL-33 in an ATP-dependent manner, which acts on pulmonary CD4+ Th2 cells, innate lymphoid cells, and semi-invariant NKT cells with interacting monocytes and macrophages that result in IL-13 production, airway mucus cell metaplasia, and mucus formation.81 The importance of macrophages in IL-33 signaling is also observed in H. capsulatum infection.64 We did not observe significant alteration in the expression of either Th1 (TNF-α) or Th2 cytokine genes (IL-4 and IL-13), although the expression of FOXA2 was inhibited during fungal infection. In fact, both live and heat-killed B. dermatitidis suppress the expression of IL-33 (Figure 11). These results suggest that airway epithelial cells may not play a role in the induction of both Th1 and Th2 axes in mediating mucin biosynthesis during B. dermatitidis infection.
Our results suggest that the induction of mucin production in canine airways by B. dermatitidis infection is caused by Th17 (IL-17), pyrogenic cytokine IL-1β, and chemokine IL-8. Previous studies have shown that both IL-1β and IL-17 stimulate mucin biosynthesis in airway epithelial cells independent of Th1 and Th2 cytokine response.82 In both cystic fibrosis and non–cystic fibrosis bronchiectasis disease, such as COPD, IL-1β promotes the production of proinflammatory mediators, including IL-8, which exacerbate neutrophilic inflammation, and eventually, persistent mucus hypersecretion in the diseased airways,83 through the production of neutrophil elastase, a potent stimulator of mucin biosynthesis.84 In addition, previous studies have shown that IL-1β and IL-17 are required to boost immunity against B. dermatitidis.62 Furthermore, Th17-mediated antifungal immunity is augmented by IL-1β, probably through recruitment and activation of CD4+ T cells.62 Collectively, our results together with the aforementioned studies indicate the importance of FOXA2-mediated homeostasis in mucin biosynthesis, where healthy levels of mucus protect against microbial infection by mucociliary escalator function,18,19 whereas excessive mucus leads to detrimental chronic colonization and infection by microbes.
There are some parallels between pulmonary mycosis caused by B. dermatitidis and tuberculosis caused by Mycobacterium tuberculosis. Because B. dermatitidis and M. tuberculosis have similar route of transmission and pulmonary pathophysiological characteristics (eg, pyogranulomatous response), blastomycosis is often misdiagnosed as tuberculosis.67 Majority of B. dermatitidis and M. tuberculosis is eliminated by macrophages and neutrophils. However, pathogens that survived initial immune assaults can persist and replicate in macrophages.85,86 These intracellular infections cause pulmonary infiltrate with granulomas, accompanying necrosis and fibrosis in adjacent tissues.87,88 Although B. dermatitidis and M. tuberculosis are sequestered in granulomas, these microbes can be reactivated by dysregulation of host immune system.89 To eliminate these pathogens, cell-mediated immunity is critically important.46,67,90, 91, 92 Individuals with incapacitated cellular immunity (eg, patients with AIDS and those who are undergoing anticancer chemotherapy and anti-inflammatory treatment) can readily develop blastomycosis, infection by other emerging dimorphic fungi,9,10 or tuberculosis, with increased morbidity and mortality. Macrophage and dendritic cells with ingested B. dermatitidis and M. tuberculosis are pivotal in alerting and priming antigen-specific T cells, which produce both Th1 (TNF-α and IFN-γ) and Th17 (eg, IL-17) cytokines93,94 that augment activation and higher recruitment of the phagocytes. In addition, the pyrogenic proinflammatory cytokine, IL-1β, enhances Th17 cells and resistance against B. dermatitidis infection,62 whereas IFN-γ, TNF-α, and IL-2 producing CD4+ and CD8+ T cells are important in defense against M. tuberculosis.95 Collectively, these cytokines orchestrate the immune system to control disease progression. In contrast, Th2 cytokines, such as IL-4 and IL-13, attenuate host immunity against B. dermatitidis and M. tuberculosis. It has been shown that IL-4 facilitates reactivation of latent M. tuberculosis by IFN-γ production96 and macrophage-mediated phagocytosis.97 Similarly, pulmonary blastomycosis elevates IL-4 and lowers IFN-γ levels in plasma.63 Inverse regulation of IL-4 versus IFN-γ and IL-17 is further supported by the study demonstrating that CD4+ T-cell–mediated immunity against B. dermatitidis was increased by IFN-γ and IL-17 but not by IL-4 in mice.46,94
In the context of mucin biosynthesis, persistent production of proinflammatory cytokines IL-1β and TNF-α exhibits mucogenic effects via FOXA2 depletion in airway epithelial cells,98 implicating that the early stages of blastomycosis and tuberculosis may be accompanied by higher mucin production. Also, IL-1β facilitates ectodomain shedding of pro-HBEGF, which helps HBEGF recognition by EGFR.99 On the basis of our results, IL-1β is predicted to promote binding of HBEGF to EGFR that causes mucin hyperproduction in airway epithelial cells. During progressive stage or reoccurrence of blastomycosis and tuberculosis, elevated levels of Th2 cytokines63,96 could antagonize FOXA2 through IL-4/IL-13R–mediated signaling, leading to increased mucus production. Moreover, FOXA2-depleted airway epithelial cells can amplify Th2 responses81 that may aggravate progression of blastomycosis and tuberculosis by inhibiting Th1 response. Collectively, antagonistic effects of Th1 and Th2 cytokines, further modulated by Th17, on FOXA2 may aggravate mucus accumulation in airways infected by B. dermatitidis and M. tuberculosis.
Finally, as shown through GMS staining (Figures 2 and 3 and Supplemental Figures S1 and S2), B. dermatitidis invades into submucosal lung tissues rather than colonizing the surface epithelia. It is likely that FOXA2 agonists, such as the recently reported incretin mimetic exenatide34 and muco-active agents,98 could attenuate excessive mucus secretion and improve mucociliary clearance in airway colonized by B. dermatitidis and H. capsulatum, thus reducing the risk of superinfection by other microbial pathogens. However, improvement of mucociliary clearance may not be effective in eliminating fungal pathogens that have penetrated into lung tissues. Therefore, FOXA2 agonists can be considered for adjuvant therapeutics in combination with existing antifungals to combat infection while improving physiological functions of the lungs. Future studies will focus on this combinatorial therapeutic approach.
Acknowledgments
We thank Prof. George Deepe (University of Cincinnati College of Medicine) for kindly providing Histoplasma Capsulatum, Prof. Timothy Fan (College of Veterinary Medicine, University of Illinois at Urbana-Champaign) for kindly providing the BACA cell line, and Karen Doty for assistance with the Grocott methenamine silver staining.
Footnotes
Supported by the University of Illinois College of Veterinary Medicine Companion Animal Research Grant Program (James R. Harkness Fund) and NIH grants HL090699 (G.W.L.) and HL142626A1 (G.W.L.).
W.C., A.X.Y., and A.S contributed equally to this work.
Disclosures: None declared.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2020.09.013.
Supplemental Data
Supplemental Figure S1.

Animal fungal mycosis cases at the University of Illinois College of Veterinary Medicine Veterinary Diagnostic Laboratory. A: Number of fungal cases verified by 18S rRNA, mycology culture, or lung biopsy by year. B: Organ systems sampled for 18S rRNA PCR, mycology culture, or lung biopsy from 2014 to 2019. C: Number of respiratory fungal infections confirmed by 18S rRNA PCR, mycology culture, or pathology between 2014 and 2019. D: Cases of fungal pneumonia analyzed in this study by species from 2017 to 2019. B. dermatitidis, Blastomyces dermatitidis; C. immitis, Coccidiodes immitis; H. capsulatum, Histoplasma capsulatum.
Supplemental Figure S2.

Airway expression of forkhead box protein A2 (FOXA2) is inversely correlated with mucin abundance in canine pulmonary mycosis. Paraffin-embedded lung sections from healthy and fungal-infected animals were stained with Grocott methenamine silver (GMS), periodic acid-Schiff (PAS), and immunohistochemistry (IHC) for the presence of fungal pathogens and the expression of FOXA2 and mucins. Healthy control lung sections stained negative for fungal organisms and mucin secretion with GMS and PAS, with strong expression of FOXA2 in bronchial surface epithelia (black arrows). Fungal-infected canine lungs were positive for yeast bodies under GMS staining (black spots; black arrows) in the submucosa and bronchial lumens, accompanied by positive PAS staining (pink spots; black arrows) in surface epithelia, which colocalized to the regions expressing MUC5AC and MUC5B in IHC staining (black arrows). FOXA2 expression was down-regulated in the bronchial surface epithelia of the infected lungs (black arrows), with successful internal positive controls shown in basal epithelial cells (red arrows). Scale bars = 50 μm.
Supplemental Figure S3.

Airway expression of forkhead box protein A2 (FOXA2) is inversely correlated with mucin abundance in feline pulmonary mycosis. Paraffin-embedded lung sections from healthy and fungal-infected animals were stained with Grocott methenamine silver (GMS), periodic acid-Schiff (PAS), and immunohistochemistry (IHC) for the presence of Histoplasma capsulatum and the expression of FOXA2 and mucins. Healthy control lung sections stained negative for fungal organisms under GMS staining (with yet some visible non-specific background staining), along with basal expression of mucins confirmed by PAS and IHC methods, and strong expression of FOXA2 in bronchial surface epithelia (black arrows). In contrast, fungal-infected feline lungs were positive for yeast bodies under GMS staining (black spots; black arrows) in the bronchial lumens, accompanied by positive PAS staining (pink spots; black arrows) in surface epithelia, which colocalized to the regions expressing MUC5AC and MUC5B in IHC (black arrows). FOXA2 expression was depleted (black arrows). Scale bars = 50 μm.
Supplemental Figure S4.

Blastomyces dermatitidis and β-glucan induce mucin expression in the immortalized canine airway carcinoma (BACA) cells. These are replicates of Western blots shown in Figure 3. BACA cells at 90% confluency were challenged with live (LV) or heat-killed (HK) B. dermatitidis strain SCB-2 at the indicated multiplicity of infection (MOI), or indicated concentrations of β-glucan for 24 hours. Control (Ctrl) cells were exposed to the same volume of sterile phosphate-buffered saline. Cytoplasmic and nuclear protein extracts were separated on acrylamide-agarose (mucins) and acrylamide [forkhead box protein A2 (FOXA2), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and H3] gels, respectively, and probed with specific antibodies against each protein. GAPDH and H3 were used as loading controls.
Supplemental Figure S5.

Inhibition of epidermal growth factor receptor (EGFR)–AKT/extracellular signal-regulated kinase (ERK) 1/2 signaling cascades restores forkhead box protein A2 (FOXA2) and attenuates mucin hypersecretion induced by Blastomyces dermatitidis and β-glucan. These are replicates of Western blots shown in Figure 6. Immortalized canine airway carcinoma (BACA) cells were pre-exposed to 20 μmol/L of EGFR inhibitor AG1478 (AG), AKT inhibitor LY294002 (LY), and ERK1/2 inhibitor PD98059 (PD) or sterile phosphate-buffered saline (-) for 1 hour before the addition of live (LV) and heat-killed (HK) B. dermatitidis and β-glucan, respectively. The expression of FOXA2, MUC5AC, and MUC5B was examined by specific antibodies. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and H3 were used as loading controls.
Supplemental Figure S6.

The antioxidant glutathione (GSH) restores forkhead box protein A2 (FOXA2) expression to attenuate MUC5AC and MUC5B production induced by heat-killed (HK) Blastomyces dermatitidis. These are replicates of Western blots shown in Figure 9C. BACA cells (90% confluency) were exposed to heat-killed B. dermatitidis SCB-2 at the multiplicity of infection 5:1 for 24 hours, in the presence or absence of GSH (5 μmol/L). Control (-) cells were exposed to the same volume of sterile phosphate-buffered saline. Cytoplasmic and nuclear protein extracts were separated on acrylamide-agarose gels (mucins) and normal 10% acrylamide gels [FOXA2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and H3], and probed with specific antibodies against each protein. GAPDH and H3 were used as loading controls.
Supplemental Figure S7.

Antibody neutralization of heparin-binding EGF-like growth factor (HBEGF) restores forkhead box protein A2 (FOXA2) to attenuate mucin expression during exposure to heat-killed (HK) Blastomyces dermatitidis. These are replicates of Western blots shown in Figure 10B. BACA cells were pretreated with anti-HBEGF antibody (1 μg) or an irrelevant control mouse IgG (Ctrl IgG; 1 μg) for 2 hours before 24 hours of exposure to heat-killed B. dermatitidis (multiplicity of infection 5:1). Control (-) cells were exposed to the same volume of sterile phosphate-buffered saline. Cytoplasmic and nuclear protein extracts were separated on acrylamide-agarose gels (mucins) and normal 10% acrylamide gels [FOXA2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and H3], and probed with specific antibodies against each protein. GAPDH and H3 were used as loading controls.
References
- 1.Bariola J., Vyas K. Pulmonary blastomycosis. Semin Respir Crit Care Med. 2011;32:745–753. doi: 10.1055/s-0031-1295722. [DOI] [PubMed] [Google Scholar]
- 2.McBride J.A., Gauthier G.M., Klein B.S. Clinical manifestations and treatment of blastomycosis. Clin Chest Med. 2017;38:435–449. doi: 10.1016/j.ccm.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith J., Gauthier G. New developments in blastomycosis. Semin Respir Crit Care Med. 2015;36:715–728. doi: 10.1055/s-0035-1562898. [DOI] [PubMed] [Google Scholar]
- 4.Azar M.M., Hage C.A. Clinical perspectives in the diagnosis and management of histoplasmosis. Clin Chest Med. 2017;38:403–415. doi: 10.1016/j.ccm.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Wheat L.J., Azar M.M., Bahr N.C., Spec A., Relich R.F., Hage C. Histoplasmosis. Infect Dis Clin North Am. 2016;30:207–227. doi: 10.1016/j.idc.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Kollath D.R., Miller K.J., Barker B.M. The mysterious desert dwellers: coccidioides immitis and Coccidioides posadasii, causative fungal agents of coccidioidomycosis. Virulence. 2019;10:222–233. doi: 10.1080/21505594.2019.1589363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman S.W., Lin A.C., Hendricks K.A., Nolan R.L., Currier M.M., Morris K.R., Turner H.R. Endemic blastomycosis in Mississippi: epidemiological and clinical studies. Semin Respir Infect. 1997;12:219–228. [PubMed] [Google Scholar]
- 8.Lemos L.B., Baliga M., Guo M. Blastomycosis: the great pretender can also be an opportunist: initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194–203. doi: 10.1053/adpa.2002.34575. [DOI] [PubMed] [Google Scholar]
- 9.Gauthier G., Safdar N., Klein B., Andes D. Blastomycosis in solid organ transplant recipients. Transpl Infect Dis. 2007;9:310–317. doi: 10.1111/j.1399-3062.2007.00227.x. [DOI] [PubMed] [Google Scholar]
- 10.Pappas P.G., Pottage J.C., Powderly W.G., Fraser V.J., Stratton C.W., McKenzie S., Chmel H., Bonebrake F.C., Blum R., Shafer R.W. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847–853. doi: 10.7326/0003-4819-116-10-847. [DOI] [PubMed] [Google Scholar]
- 11.Limper A.H., Knox K.S., Sarosi G.A., Ampel N.M., Bennett J.E., Catanzaro A., Davies S.F., Dismukes W.E., Hage C.A., Marr K.A. An official American Thoracic Society statement: treatment of fungal infections in adult pulmonary and critical care patients. Am J Respir Crit Care Med. 2011;183:96–128. doi: 10.1164/rccm.2008-740ST. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y.D., Bae C.H., Song S.Y., Choi Y.S. Effect of β-Glucan on MUC4 and MUC5B Expression in Human Airway Epithelial Cells: International Forum of Allergy & Rhinology. Int Forum Allergy Rhinol. 2015;5:708–715. doi: 10.1002/alr.21549. [DOI] [PubMed] [Google Scholar]
- 13.Li E., Landers C.T., Tung H.-Y., Knight J.M., Marshall Z., Luong A.U., Rodriguez A., Kheradmand F., Corry D.B. Fungi in mucoobstructive airway diseases. Ann Am Thorac Soc. 2018;15:S198–S204. doi: 10.1513/AnnalsATS.201803-154AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rojas D.A., Iturra P.A., Méndez A., Ponce C.A., Bustamante R., Gallo M., Bórquez P., Vargas S.L. Increase in secreted airway mucins and partial Muc5b STAT6/FoxA2 regulation during Pneumocystis primary infection. Sci Rep. 2019;9:2078. doi: 10.1038/s41598-019-39079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.John F., Burton D. Airway mucus function and dysfuction. N Engl J Med. 2010;363:2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucher R.C. Muco-obstructive lung diseases. N Engl J Med. 2019;380:1941–1953. doi: 10.1056/NEJMra1813799. [DOI] [PubMed] [Google Scholar]
- 17.Bonser L.R., Erle D.J. Airway mucus and asthma: the role of MUC5AC and MUC5B. J Clin Med. 2017;6:112. doi: 10.3390/jcm6120112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy M.G., Livraghi-Butrico A., Fletcher A.A., McElwee M.M., Evans S.E., Boerner R.M., Alexander S.N., Bellinghausen L.K., Song A.S., Petrova Y.M. Muc5b is required for airway defence. Nature. 2014;505:412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehre C., Worthington E.N., Liesman R.M., Grubb B.R., Barbier D., O'Neal W.K., Sallenave J.-M., Pickles R.J., Boucher R.C. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci U S A. 2012;109:16528–16533. doi: 10.1073/pnas.1206552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonser L.R., Zlock L., Finkbeiner W., Erle D.J. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J Clin Invest. 2016;126:2367–2371. doi: 10.1172/JCI84910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curran D.R., Cohn L. Advances in mucous cell metaplasia: a plug for mucus as a therapeutic focus in chronic airway disease. Am J Respir Cell Mol Biol. 2010;42:268–275. doi: 10.1165/rcmb.2009-0151TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesan S., Comstock A.T., Sajjan U.S. Barrier function of airway tract epithelium. Tissue Barriers. 2013;1:e24997. doi: 10.4161/tisb.24997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G., Korfhagen T.R., Xu Y., Kitzmiller J., Wert S.E., Maeda Y., Gregorieff A., Clevers H., Whitsett J.A. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest. 2009;119:2914–2924. doi: 10.1172/JCI39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park K.-S., Korfhagen T.R., Bruno M.D., Kitzmiller J.A., Wan H., Wert S.E., Hershey G.K.K., Chen G., Whitsett J.A. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest. 2007;117:978–988. doi: 10.1172/JCI29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhen G., Park S.W., Nguyenvu L.T., Rodriguez M.W., Barbeau R., Paquet A.C., Erle D.J. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol. 2007;36:244–253. doi: 10.1165/rcmb.2006-0180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi W., Choe S., Lau G.W. Inactivation of FOXA2 by respiratory bacterial pathogens and dysregulation of pulmonary mucus homeostasis. Front Immunol. 2020;11:515. doi: 10.3389/fimmu.2020.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan H., Kaestner K.H., Ang S.-L., Ikegami M., Finkelman F.D., Stahlman M.T., Fulkerson P.C., Rothenberg M.E., Whitsett J.A. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development. 2004;131:953–964. doi: 10.1242/dev.00966. [DOI] [PubMed] [Google Scholar]
- 28.Hao Y., Kuang Z., Xu Y., Walling B.E., Lau G.W. Pyocyanin-induced mucin production is associated with redox modification of FOXA2. Respir Res. 2013;14:82. doi: 10.1186/1465-9921-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du C., Lu J., Zhou L., Wu B., Zhou F., Gu L., Xu D., Sun Y. MAPK/FoxA2-mediated cigarette smoke-induced squamous metaplasia of bronchial epithelial cells. Int J Chron Obstruct Pulmon Dis. 2017;12:3341. doi: 10.2147/COPD.S143279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao Y., Kuang Z., Jing J., Miao J., Mei L.Y., Lee R.J., Kim S., Choe S., Krause D.C., Lau G.W. Mycoplasma pneumoniae modulates STAT3-STAT6/EGFR-FOXA2 signaling to induce overexpression of airway mucins. Infect Immun. 2014;82:5246–5255. doi: 10.1128/IAI.01989-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao Y., Kuang Z., Walling B.E., Bhatia S., Sivaguru M., Chen Y., Gaskins H.R., Lau G.W. Pseudomonas aeruginosa pyocyanin causes airway goblet cell hyperplasia and metaplasia and mucus hypersecretion by inactivating the transcriptional factor FoxA2. Cell Microbiol. 2012;14:401–415. doi: 10.1111/j.1462-5822.2011.01727.x. [DOI] [PubMed] [Google Scholar]
- 32.Jeffries J.L., Jia J., Choi W., Choe S., Miao J., Xu Y., Powell R., Lin J., Kuang Z., Gaskins H.R., Lau G.W. Pseudomonas aeruginosa pyocyanin modulates mucin glycosylation with sialyl-Lewisx to increase binding to airway epithelial cells. Mucosal Immunol. 2015;9:1039–1050. doi: 10.1038/mi.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y., Duan C., Kuang Z., Hao Y., Jeffries J.L., Lau G.W. Pseudomonas aeruginosa pyocyanin activates NRF2-ARE-mediated transcriptional response via the ROS-EGFR-PI3K-AKT/MEK-ERK MAP kinase signaling in pulmonary epithelial cells. PLoS One. 2013;8:e72528. doi: 10.1371/journal.pone.0072528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi W., Choe S., Lin J., Borchers M.T., Kosmider B., Vassallo R., Limper A.H., Lau G.W. Exendin-4 restores airway mucus homeostasis through the GLP1R-PKA-PPARγ-FOXA2-phosphatase signaling. Mucosal Immunol. 2020;13:637–651. doi: 10.1038/s41385-020-0262-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi W., Yang A.X., Waltenburg M.A., Choe S., Steiner M., Radwan A., Lin J., Maddox C.W., Stern A.W., Fredrickson R.L., Lau G.W. FOXA2 depletion leads to mucus hypersecretion in canine airways with respiratory diseases. Cell Microbiol. 2018;21:e12957. doi: 10.1111/cmi.12957. [DOI] [PubMed] [Google Scholar]
- 36.Cui J., Pazdziorko S., Miyashiro J.S., Thakker P., Pelker J.W., DeClercq C., Jiao A., Gunn J., Mason L., Leonard J.P. TH1-mediated airway hyperresponsiveness independent of neutrophilic inflammation. J Allergy Clin Immunol. 2005;115:309–315. doi: 10.1016/j.jaci.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 37.Cohn L., Homer R.J., MacLeod H., Mohrs M., Brombacher F., Bottomly K. Th2-induced airway mucus production is dependent on IL-4Rα, but not on eosinophils. J Immunol. 1999;162:6178–6183. [PubMed] [Google Scholar]
- 38.Dabbagh K., Takeyama K., Lee H.-M., Ueki I.F., Lausier J.A., Nadel J.A. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol. 1999;162:6233–6237. [PubMed] [Google Scholar]
- 39.Chen Y., Thai P., Zhao Y.-H., Ho Y.-S., DeSouza M.M., Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 40.Wakashin H., Hirose K., Maezawa Y., Kagami S-i, Suto A., Watanabe N., Saito Y., Hatano M., Tokuhisa T., Iwakura Y. IL-23 and Th17 cells enhance Th2-cell–mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178:1023–1032. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]
- 41.Wilson R.H., Whitehead G.S., Nakano H., Free M.E., Kolls J.K., Cook D.N. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180:720–730. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bautista M.V., Chen Y., Ivanova V.S., Rahimi M.K., Watson A.M., Rose M.C. IL-8 regulates mucin gene expression at the posttranscriptional level in lung epithelial cells. J Immunol. 2009;183:2159–2166. doi: 10.4049/jimmunol.0803022. [DOI] [PubMed] [Google Scholar]
- 43.Gray T., Coakley R., Hirsh A., Thornton D., Kirkham S., Koo J.-S., Burch L., Boucher R., Nettesheim P. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1β in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2004;286:L320–L330. doi: 10.1152/ajplung.00440.2002. [DOI] [PubMed] [Google Scholar]
- 44.Kroetz D.N., Deepe G.S. The role of cytokines and chemokines in Histoplasma capsulatum infection. Cytokine. 2012;58:112–117. doi: 10.1016/j.cyto.2011.07.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finkel-Jimenez B., Wüthrich M., Klein B.S. BAD1, an essential virulence factor of Blastomyces dermatitidis, suppresses host TNF-α production through TGF-β-dependent and-independent mechanisms. J Immunol. 2002;168:5746–5755. doi: 10.4049/jimmunol.168.11.5746. [DOI] [PubMed] [Google Scholar]
- 46.Wüthrich M., Filutowicz H.I., Klein B.S. Mutation of the WI-1 gene yields an attenuated Blastomyces dermatitidis strain that induces host resistance. J Clin Invest. 2000;106:1381–1389. doi: 10.1172/JCI11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wüthrich M., Warner T., Klein B.S. IL-12 is required for induction but not maintenance of protective, memory responses to Blastomyces dermatitidis: implications for vaccine development in immune-deficient hosts. J Immunol. 2005;175:5288–5297. doi: 10.4049/jimmunol.175.8.5288. [DOI] [PubMed] [Google Scholar]
- 48.Furcolow M.L., Busey J.F., Menges R.W., Chick E.W. Prevalence and incidence studies of human and canine blastomycosis II: yearly incidence studies in three selected states, 1960–1967. Am J Epidemiol. 1970;92:121–131. doi: 10.1093/oxfordjournals.aje.a121184. [DOI] [PubMed] [Google Scholar]
- 49.Arceneaux K., Taboada J., Hosgood G. Blastomycosis in dogs: 115 cases (1980-1995) J Am Vet Med Assoc. 1998;213:658–664. [PubMed] [Google Scholar]
- 50.Caldwell C.C., Chen Y., Goetzmann H.S., Hao Y., Borchers M.T., Hassett D.J., Young L.R., Mavrodi D., Thomashow L., Lau G.W. Pseudomonas aeruginosa exotoxin pyocyanin causes cystic fibrosis airway pathogenesis. Am J Pathol. 2009;175:2473–2488. doi: 10.2353/ajpath.2009.090166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz B.L., Packer N.H., Karlsson N.G. Small-scale analysis of O-linked oligosaccharides from glycoproteins and mucins separated by gel electrophoresis. Anal Chem. 2002;74:6088–6097. doi: 10.1021/ac025890a. [DOI] [PubMed] [Google Scholar]
- 52.Goodridge H.S., Wolf A.J., Underhill D.M. β-Glucan recognition by the innate immune system. Immunol Rev. 2009;230:38–50. doi: 10.1111/j.1600-065X.2009.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanetsuna F., Carbonell L.M. Cell wall composition of the yeastlike and mycelial forms of Blastomyces dermatitidis. J Bacteriol. 1971;106:946–948. doi: 10.1128/jb.106.3.946-948.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Theel E.S., Doern C.D. Point-counterpoint: β-d-glucan testing is important for diagnosis of invasive fungal infections. J Clin Microbiol. 2013;51:3478–3483. doi: 10.1128/JCM.01737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helal M., Allen K.J., Van Dijk B., Nosanchuk J.D., Snead E., Dadachova E. Radioimmunotherapy of blastomycosis in a mouse model with a (1→ 3)-β-glucans targeting antibody. Front Microbiol. 2020;11:147. doi: 10.3389/fmicb.2020.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plato A., Willment J.A., Brown G.D. C-type lectin-like receptors of the dectin-1 cluster: ligands and signaling pathways. Int Rev Immunol. 2013;32:134–156. doi: 10.3109/08830185.2013.777065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bae Y.S., Kang S.W., Seo M.S., Baines I.C., Tekle E., Chock P.B., Rhee S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 58.Weng M.-S., Chang J.-H., Hung W.-Y., Yang Y.-C., Chien M.-H. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. J Exp Clin Cancer Res. 2018;37:61. doi: 10.1186/s13046-018-0728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh B., Carpenter G., Coffey R.J. EGF receptor ligands: recent advances. F1000Res. 2016;5 doi: 10.12688/f1000research.9025.1. F1000 Faculty Rev-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Q., Li H., Yao Y., Xia D., Zhou J. The overexpression of heparin-binding epidermal growth factor is responsible for Th17-induced airway remodeling in an experimental asthma model. J Immunol. 2010;185:834–841. doi: 10.4049/jimmunol.0901490. [DOI] [PubMed] [Google Scholar]
- 61.Sterkel A.K., Lorenzini J.L., Fites J.S., Vignesh K.S., Sullivan T.D., Wuthrich M., Brandhorst T., Hernandez-Santos N., Deepe G.S., Jr., Klein B.S. Fungal mimicry of a mammalian aminopeptidase disables innate immunity and promotes pathogenicity. Cell Host Microbe. 2016;19:361–374. doi: 10.1016/j.chom.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wüthrich M., LeBert V., Galles K., Hu-Li J., Ben-Sasson S.Z., Paul W.E., Klein B.S. Interleukin 1 enhances vaccine-induced antifungal T-helper 17 cells and resistance against Blastomyces dermatitidis infection. J Infect Dis. 2013;208:1175–1182. doi: 10.1093/infdis/jit283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brummer E., Hanson L.H., Stevens D.A. IL-4, IgE, and interferon-γ production in pulmonary blastomycosis: comparison in mice untreated, immunized, or treated with an antifungal (SCH 39304) Cell Immunol. 1993;149:258–267. doi: 10.1006/cimm.1993.1153. [DOI] [PubMed] [Google Scholar]
- 64.Verma A., Kroetz D.N., Tweedle J.L., Deepe G. Type II cytokines impair host defense against an intracellular fungal pathogen by amplifying macrophage generation of IL-33. Mucosal Immunol. 2015;8:380–389. doi: 10.1038/mi.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merkhofer R.M., O'Neill M.B., Xiong D., Hernandez-Santos N., Dobson H., Fites J.S., Shockey A.C., Wuethrich M., Pepperell C.S., Klein B.S. Investigation of genetic susceptibility to blastomycosis reveals interleukin-6 as a potential susceptibility locus. MBio. 2019;10 doi: 10.1128/mBio.01224-19. e01224-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zielinski C.E., Mele F., Aschenbrenner D., Jarrossay D., Ronchi F., Gattorno M., Monticelli S., Lanzavecchia A., Sallusto F. Pathogen-induced human TH 17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 67.Saccente M., Woods G.L. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367–381. doi: 10.1128/CMR.00056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herrmann J.A., Kostiuk S.L., Dworkin M.S., Johnson Y.J. Temporal and spatial distribution of blastomycosis cases among humans and dogs in Illinois (2001–2007) J Am Vet Med Assoc. 2011;239:335–343. doi: 10.2460/javma.239.3.335. [DOI] [PubMed] [Google Scholar]
- 69.Gelfand E.W. Mice are a good model of human airway disease. Am J Respir Crit Care Med. 2002;166:5–6. doi: 10.1164/rccm.2204023. [DOI] [PubMed] [Google Scholar]
- 70.Persson C.G. Mice are not a good model of human airway disease. Am J Respir Crit Care Med. 2002;166:6–7. doi: 10.1164/rccm.2204001. [DOI] [PubMed] [Google Scholar]
- 71.Luna C., Sibila O., Agusti C., Torres A. Animal models of ventilator-associated pneumonia. Eur Respir J. 2009;33:182–188. doi: 10.1183/09031936.00046308. [DOI] [PubMed] [Google Scholar]
- 72.Shah A., Panjabi C. Allergic bronchopulmonary aspergillosis: a perplexing clinical entity. Allergy Asthma Immunol Res. 2016;8:282–297. doi: 10.4168/aair.2016.8.4.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oguma T., Asano K., Tomomatsu K., Kodama M., Fukunaga K., Shiomi T., Ohmori N., Ueda S., Takihara T., Shiraishi Y. Induction of mucin and MUC5AC expression by the protease activity of Aspergillus fumigatus in airway epithelial cells. J Immunol. 2011;187:999–1005. doi: 10.4049/jimmunol.1002257. [DOI] [PubMed] [Google Scholar]
- 74.Blease K., Jakubzick C., Westwick J., Lukacs N., Kunkel S.L., Hogaboam C.M. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J Immunol. 2001;166:5219–5224. doi: 10.4049/jimmunol.166.8.5219. [DOI] [PubMed] [Google Scholar]
- 75.Yadav M., Schorey J.S. The β-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood. 2006;108:3168–3175. doi: 10.1182/blood-2006-05-024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gantner B.N., Simmons R.M., Canavera S.J., Akira S., Underhill D.M. Collaborative induction of inflammatory responses by dectin-1 and toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun W.-K., Lu X., Li X., Sun Q.-Y., Su X., Song Y., Sun H.-M., Shi Y. Dectin-1 is inducible and plays a crucial role in Aspergillus-induced innate immune responses in human bronchial epithelial cells. Eur J Clin Microbiol Infect Dis. 2012;31:2755–2764. doi: 10.1007/s10096-012-1624-8. [DOI] [PubMed] [Google Scholar]
- 78.Lorenzini J., Scott Fites J., Nett J., Klein B.S. Blastomyces dermatitidis serine protease dipeptidyl peptidase IVA (DppIVA) cleaves ELR+ CXC chemokines altering their effects on neutrophils. Cell Microbiol. 2017;19:e12741. doi: 10.1111/cmi.12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morrison C.J., Stevens D.A. Enhanced killing of Blastomyces dermatitidis by gamma interferon-activated murine peripheral blood polymorphonuclear neutrophils. Int J Immunopharmacol. 1989;11:855–862. doi: 10.1016/0192-0561(89)90106-9. [DOI] [PubMed] [Google Scholar]
- 80.Heller N.M., Matsukura S., Georas S.N., Boothby M.R., Rothman P.B., Stellato C., Schleimer R.P. Interferon-γ inhibits STAT6 signal transduction and gene expression in human airway epithelial cells. Am J Respir Cell Mol Biol. 2004;31:573–582. doi: 10.1165/rcmb.2004-0195OC. [DOI] [PubMed] [Google Scholar]
- 81.Chen G., Wan H., Luo F., Zhang L., Xu Y., Lewkowich I., Wills-Karp M., Whitsett J.A. Foxa2 programs Th2 cell-mediated innate immunity in the developing lung. J Immunol. 2010;184:6133–6141. doi: 10.4049/jimmunol.1000223. [DOI] [PubMed] [Google Scholar]
- 82.Fujisawa T., Chang M.M.-J., Velichko S., Thai P., Hung L.-Y., Huang F., Phuong N., Chen Y., Wu R. NF-κB mediates IL-1β–and IL-17A–induced MUC5B expression in airway epithelial cells. Am J Respir Cell Mol Biol. 2011;45:246–252. doi: 10.1165/rcmb.2009-0313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muselet-Charlier C., Roque T., Boncoeur E., Chadelat K., Clement A., Jacquot J., Tabary O. Enhanced IL-1β-induced IL-8 production in cystic fibrosis lung epithelial cells is dependent of both mitogen-activated protein kinases and NF-κB signaling. Biochem Biophys Res Commun. 2007;357:402–407. doi: 10.1016/j.bbrc.2007.03.141. [DOI] [PubMed] [Google Scholar]
- 84.Nadel J.A. Role of neutrophil elastase in hypersecretion during COPD exacerbations, and proposed therapies. Chest. 2000;117:386S–389S. doi: 10.1378/chest.117.5_suppl_2.386s. [DOI] [PubMed] [Google Scholar]
- 85.Bradsher R.W., Jr. The endemic mimic: blastomycosis an illness often misdiagnosed. Trans Am Clin Climatol Assoc. 2014;125:188. [PMC free article] [PubMed] [Google Scholar]
- 86.Kumar A., Kunoor A., Eapen M., Singh P.K., Chowdhary A. Blastomycosis misdiagnosed as tuberculosis, India. Emerg Infect Dis. 2019;25:1776. doi: 10.3201/eid2509.190587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wallis R., Broder M., Wong J., Hanson M., Beenhouwer D. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis. 2004;38:1261–1265. doi: 10.1086/383317. [DOI] [PubMed] [Google Scholar]
- 88.Randhawa H.S., Chowdhary A., Kathuria S., Roy P., Misra D.S., Jain S., Chugh T.D. Blastomycosis in India: report of an imported case and current status. Med Mycol. 2013;51:185–192. doi: 10.3109/13693786.2012.685960. [DOI] [PubMed] [Google Scholar]
- 89.Mohan V.P., Scanga C.A., Yu K., Scott H.M., Tanaka K.E., Tsang E., Tsai M.C., Flynn J.L., Chan J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–1855. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cozad G.C., Chang C.-T. Cell-mediated immunoprotection in blastomycosis. Infect Immun. 1980;28:398–403. doi: 10.1128/iai.28.2.398-403.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morozumi P.A., Brummer E., Stevens D.A. Protection against pulmonary blastomycosis: correlation with cellular and humoral immunity in mice after subcutaneous nonlethal infection. Infect Immun. 1982;37:670–678. doi: 10.1128/iai.37.2.670-678.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Martino M., Lodi L., Galli L., Chiappini E. Immune response to Mycobacterium tuberculosis: a narrative review. Front Pediatr. 2019;7:350. doi: 10.3389/fped.2019.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brummer E., Hanson L., Restrepo A., Stevens D. In vivo and in vitro activation of pulmonary macrophages by IFN-gamma for enhanced killing of Paracoccidioides brasiliensis or Blastomyces dermatitidis. J Immunol. 1988;140:2786–2789. [PubMed] [Google Scholar]
- 94.Monin L., Griffiths K., Slight S., Lin Y-y, Rangel-Moreno J., Khader S.A. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal Immunol. 2015;8:1099–1109. doi: 10.1038/mi.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang X., Zhang J., Liang J., Zhang Y., Teng X., Yuan X., Fan X. Protection against Mycobacterium tuberculosis infection offered by a new multistage subunit vaccine correlates with increased number of IFN-γ+ IL-2+ CD4+ and IFN-γ+ CD8+ T cells. PLoS One. 2015;10:e0122560. doi: 10.1371/journal.pone.0122560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heitmann L., Abad Dar M., Schreiber T., Erdmann H., Behrends J., Mckenzie A.N., Brombacher F., Ehlers S., Hölscher C. The IL-13/IL-4R α axis is involved in tuberculosis-associated pathology. J Pathol. 2014;234:338–350. doi: 10.1002/path.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Varin A., Mukhopadhyay S., Herbein G., Gordon S. Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood. 2010;115:353–362. doi: 10.1182/blood-2009-08-236711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song K.S., Lee W.-J., Chung K.C., Koo J.S., Yang E.J., Choi J.Y., Yoon J.-H. Interleukin-1β and tumor necrosis factor-α induce MUC5AC overexpression through a mechanism involving ERK/p38 mitogen-activated protein kinases-MSK1-CREB activation in human airway epithelial cells. J Biol Chem. 2003;278:23243–23250. doi: 10.1074/jbc.M300096200. [DOI] [PubMed] [Google Scholar]
- 99.Takenobu H., Yamazaki A., Hirata M., Umata T., Mekada E. The stress- and inflammatory cytokine-induced ectodomain shedding of heparin-binding epidermal growth factor-like growth factor is mediated by p38 MAPK, distinct from the 12-O-tetradecanoylphorbol-13-acetate-and lysophosphatidic acid-induced signaling cascades. J Biol Chem. 2003;278:17255–17262. doi: 10.1074/jbc.M211835200. [DOI] [PubMed] [Google Scholar]