

Summary
Isolation of leukemia stem cells presents a challenge due to the heterogeneity of the immunophenotypic markers commonly used to identify blood stem cells. Several studies have reported that relative levels of reactive oxygen species (ROS) can be used to enrich for stem cell populations, suggesting a potential alternative to surface antigen-based methods. Here, we describe a protocol to enrich for stem cells from human acute myeloid leukemia specimens using relative levels of ROS. This protocol provides consistent enrichment of leukemia stem cells.
For complete details on the use and execution of this protocol, please refer to Lagadinou et al. (2013) and Pei et al. (2018).
Subject areas: Flow cytometry/mass cytometry, Cancer, Metabolism, Stem cells
Graphical abstract
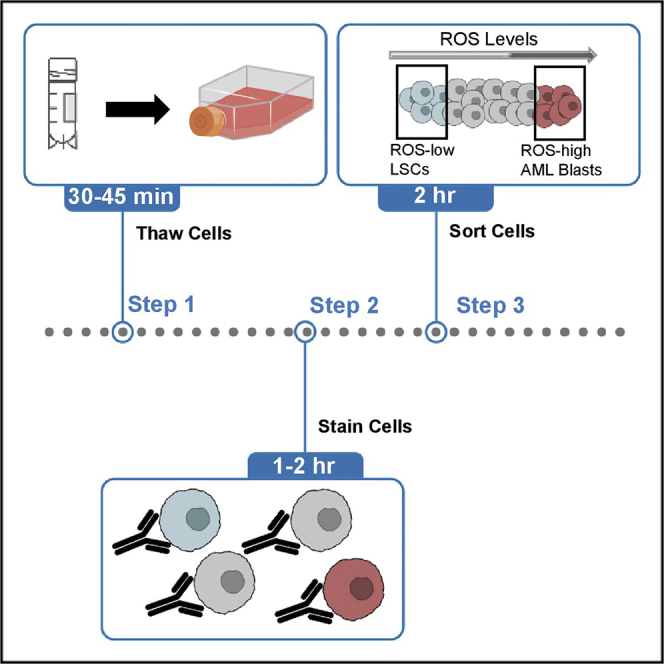
Highlights
-
•
Low levels of reactive oxygen species are a hallmark of stem cells
-
•
Relative levels of reactive oxygen species allow for enrichment of leukemia stem cells
-
•
This protocol is an alternative to surface antigen-based methods
Isolation of leukemia stem cells presents a challenge due to the heterogeneity of the immunophenotypic markers commonly used to identify blood stem cells. Several studies have reported that relative levels of reactive oxygen species (ROS) can be used to enrich for stem cell populations, suggesting a potential alternative to surface antigen-based methods. Here, we describe a protocol to enrich for stem cells from human acute myeloid leukemia specimens using relative levels of ROS. This protocol provides consistent enrichment of leukemia stem cells.
Before you begin
This protocol combines the use of cell surface markers with live cell dyes that detect reactive oxygen species (ROS) to enrich of primary human LSCs. It is important to note that ROS dyes have been previously used to enrich for other stem cell populations (Diehn et al., 2009; Jang and Sharkis, 2007; Smith et al., 2000). While cell surface markers and ROS dyes are commonly used to understand the biology of AML this protocol was developed to allow the combination of these approaches to enrich for human LSCs from primary AML specimens.
Preparation of media and reagents
Serum-free custom media
Prepare 500 mL of metabolomics media (Invitria) in 0.45 μm vacuum filter jar. Supplement with 5 mL of Pen/Strep (Thermo Fisher) and 5 mL of 2-Mercaptoethanol (Sigma-Aldrich). Dilute low-density lipoprotein (LDL) (Sigma-Aldrich) by adding 1,455 μL of cold PBS to 10 mg stock. Add LDL to a final concentration of 10 μg/mL to the media.
Thawing media
Prepare thawing media by supplementing IMDM (Thermo Fisher) with 5% fetal bovine serum (FBS) (Corning). Thawing media should be stored a 4°C for 1 month.
DNase I stock
For 2,000 U/mL stock of DNase (Sigma-Aldrich), dissolve 20,000 units of DNase in 10 mL of PBS. Store in aliquots of 250 μL and store at −20°C.
FACS buffer
Prepare calcium and magnesium free PBS with 2% FBS.
FACS-DAPI solution
Suspend 5 mg of DAPI (Sigma-Aldrich) in 4.6 mL of sterile water for a final concentration of 5 mM. Store in −20°C. Make the FACS-DAPI solutions by adding 1 μL of DAPI stock to 50 mL of FACS buffer. Keep FACS-DAPI for up to 1 month at +4°C
Red cell lysis buffer
Prepare 10× lysis buffer by combining 82 g NH4Cl, 10 g KCHO3, 2 mL 0.5 M EDTA, 998 mL of sterile water. Filter solution with 0.22 μm filter and store final stock at 4°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human CD45 BB515 (HI30) | BD | Cat#564585; RRID: AB_2732068 |
| Mouse anti-human CD3 PE-Cy7 (SP34-2) | BD | Cat#557749; AB_396855 |
| Mouse anti-human CD19 PE (HIB19) | BD | Cat#555413; RRID: AB_395813 |
| OKT3 | Sigma-Aldrich | 86022706-DNA-5UG |
| Biological samples | ||
| Primary AML specimens | University of Colorado Hematologic Malignancies Tissue bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| IMDM | Thermo Fisher | 12440-053 |
| FBS | Corning | 35077CV |
| Pen/strep | thermosphere | 15140122 |
| Serum-free metabolomics media | Invitra | Custom https://invitria.com/custom-media-formulation/ |
| 2-Mercaptoethanol | Sigma-Aldrich | M3148-100ML |
| Low-density lipoprotein | Sigma-Aldrich | 437644-10MG |
| PBS | Corning | 21040CV |
| SCF | PeptroTech | 300-07-100ug |
| FLT3 | PeptroTech | 300-19-100ug |
| IL3 | PeptroTech | 200-03-100ug |
| DNase I | Sigma-Aldrich | 4536282001 |
| DAPI | Sigma-Aldrich | D9542-5MG |
| CellRox deep red | thermosphere | C10422 |
| Human methylcellulose | R&D Systems | HSC005 |
| Busulfan | Sigma-Aldrich | B2635-25G |
| NaCl | Sigma-Aldrich | S7653-250G |
| Experimental models: organisms/strains | ||
| NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ (NSGS) mice | The Jackson Laboratory | Cat# JAX:013062, RRID: IMSR_JAX:013062 |
| Software and algorithms | ||
| FlowJo v10 | FlowJo LLC | https://www.flowjo.com |
Step-by-step method details
Thawing primary cells
Timing: 30–45 min
This step describes a method to thaw a primary acute myeloid leukemia (AML) cell sample that was in cryogenic storage. This will ensure that cell viability is maintained while preventing clumping of cells. If using a fresh sample, this step is not necessary.
-
1.
Warm primary cells that were stored in liquid nitrogen until completely thawed in a 37°C water bath. This thawing protocol has been successfully used on cell numbers ranging from 5–200 million total cells.
-
2.
Transfer cells to 15 mL tube.
-
3.
Introduce 100 μL of DNase I (−20°C) dropwise to the cells that are still in freezing media. Gently agitate the tube to mix.
-
4.
Incubate the cells in 37°C for 90 s with the DNase.
-
5.
While gently rocking the 15 mL tube, slowly add 10 mL thawing media. Rocking the tube while adding media will help mix the AML cells with the media and decrease clumping of cells. Invert the tube to further mix.
-
6.
Centrifuge the cells at 580 × g for 7 min at 20°C.
-
7.
Aspirate the media and add 70 μL of DNase to the pellet, resuspend by flicking the tube.
-
8.
While gently rocking the 15 mL tube, slowly add 10 mL thawing media. Invert the tube to mix.
-
9.
Centrifuge the cells at 580 × g for 7 min at 20°C.
-
10.
Aspirate the media and add 50 μL of DNase to the pellet, resuspend by flicking the tube.
-
11.
While gently rocking the 15 mL tube, slowly add 10 mL thawing media. Invert the tube to mix.
-
12.
Centrifuge the cells at 580 × g for 7 min at 20°C.
Staining
This step describes the methods to stain the cells in preparation for sorting using antibodies and dyes.
Here we provide optimized antibody dilutions for approximately 50–200 million AML cells and the instrumentation we have worked with; however, it is important to titrate and optimize antibody concentrations for each instrument (see Troubleshooting Problem 1). Data shown in this protocol was generated using a BD FACSAria. These dilutions likely provide a good starting point.
-
13.
Aspirate remaining thawing media from the cell pellet.
-
14.
Resuspend cells in 80 μL of FACS buffer. Distribute 5 μL for unstained and single antibody controls into six, 5 mL round bottom polypropylene tubes, transfer the remaining 50 μL of cell suspension into a 7th tube. If the AML specimen is limited the user may choose to use less cells for staining controls. In this case, the number of cells used for staining controls can be adjusted as needed.
-
15.
Count cells (the remaining protocol was optimized for 50–200 million cells). 1–10 μL of cells is often sufficient for cell counting. It should be noted that manual counting is recommended as AML specimens often contain heterogenous cell types which are not as accurately counted using some automated counting systems.
-
16.
Add 45 μL of FACS buffer to each control tube so each tube contains 50 μL of total volume. Adjust volume of 7th tube with sample of interest to achieve cell concentration of 0.1–0.5 million cells/μL (ex. 50 million cells in no less than 100 μL of FACS buffer).
-
17.
Add antibodies to the appropriate tubes shown in Table 1. Recommended starting dilutions for antibodies are CD45-FITC (1:20), CD3-PeCy7 (1:40), CD19-Pe (1:20) (see Troubleshooting Problem 1).
-
18.
Incubate the tubes for 20 min at 4°C.
-
19.
Wash cells in 500 μL of FACS buffer and centrifuge at 580 × g for 5 min at 20°C.
-
20.
Aspirate the FACS buffer and unbound antibodies from the cell pellet.
-
21.
Prepare a 5 μM working stock of CellROX in serum-free metabolomics media. Add 500 μL to 1 mL per tube. For control tubes use serum-free metabolomics media without CellROX. CellROX is a cell permeable fluorogenic probe that exhibits photostable fluorescence upon oxidation by ROS. Other ROS dyes can and have been be used including 2′,7′-dichlorodihydrofluorescein diacetate (DCF) (Lagadinou et al., 2013); however, we normally observe better resolution of the low and high populations using CellROX.
-
22.
Incubate the tubes for 30 min at 37°C.
-
23.
Centrifuge the cells at 580 × g for 5 min at 20°C and aspirate the media.
-
24.
Wash the sample and control tubes with 1 mL of FACS buffer and spin at 580 × g for 5 min.
-
25.
Aspirate the remaining FACS buffer from the pellet and resuspend each tube in either FACS buffer or FACS-DAPI preparation according to Table 1.
-
26.
Pass the cells through a cell strainer to ensure a single cell suspension. Count the cells and adjust the total volume to give a final cell concentration of 20 million cells/mL.
-
27.
Transfer all samples on ice to cell sorter or flow cytometry facility. This protocol has been successfully executed using a BD FACSAria and a Sony SH800S cell sorter; however, it should be applicable to other sorters with ability to excite around 644 nm and detect around 665 nm emission.
Table 1.
Required samples for flow cytometry
| Tube Name | CD45-FITC | CD3-PeCy7 | CD19-Pe | DAPI | CellROX |
|---|---|---|---|---|---|
| Unstained | |||||
| CD45-FITC only | X | ||||
| CD3-PeCy7 only | X | ||||
| CD19-Pe only | X | ||||
| DAPI only | X | ||||
| CellROX only | X | ||||
| Stained cells | X | X | X | X | X |
Gating and sorting
This step describes the critical methods for sorting primary cells into ROS-low and ROS-high populations. These sorting methods will distinguish between functional AML LSCs (ROS low) and AML blasts (ROS high). These methods will also exclude lymphocytes which can sometimes appear in ROS-low populations. Gating strategy shown in Figure 1. Representative ROS profiles are shown in Figure 2A.
-
28.
Start up and set up cell sorter using best practices. Protocol as described is run on FACSAria with 70 μm nozzle at 70 PSI sheath pressure and sort rates of 500–12,000 events/s and done under purity masking.
CRITICAL: Sort time should be kept as short as possible with a recommended maximum time of 2 h per AML specimens. Further, it is highly recommended that sorting is performed at 4°C.
-
29.Run each control, starting with unstained sample to optimize the voltage, gating, and compensation matrix.
-
a.Use the unstained sample to identify sample and single cell populations
-
b.Use DAPI to gate on live cells.
-
c.Use CD3 and CD19 to exclude lymphocytes (see Troubleshooting Problem 2).
-
d.Use CD45 vs. SSC to gate for AML blasts (see Troubleshooting Problem 3).
-
e.Use CellROX to gate for target ROS-low and ROS-high populations. These gates should cover the lowest 20% of cells fluorescing with CellROX and the highest 20% of cells fluorescing with CellROX, respectively.
-
f.Cells are collected in a 5–15 mL conical tubes that have 1–2 mL of metabolomics media.
-
a.
Figure 1.

Scheme for ROS-low LSCs and ROS-high AML blasts cell sorting and expected results
(A) Flow scheme demonstrating ROS sorting to enrich ROS-low LSCs and AML blasts. Step 1 is FSC versus SSC which is used to isolate cells. Step 2 is FSC-A versus FSC-H used to isolate single cells. Step 3 is FSC versus DAPI which is used to isolated DAPI- live cells. Step 4 is CD19 versus CD3 used to exclude lymphocytes (CD3-CD19-). Step 5 is CD45 versus SSC used to isolate AML blasts. Step 6 is CellROX staining, CellROX deep red shown here.
(B) Example of the number of colonies formed from ROS-low and ROS-high cells. Data adapted from Jones et al. (2018).
(C) Example of engraftment into NSGS mice from ROS-low and ROS-high cells. Data from Pei et al. (2018).
Figure 2.

Scheme for ROS-low LSCs and ROS-high AML blasts cell sorting and expected results
(A) Example of ROS profiles from 12 primary AML specimens. These data were collected using CellROX deep red.
(B) Example of blast gating from 6 AML specimens using CD45 versus SSC.
Population validation (optional)
After sorting, to validate the stemness of the ROS-low and ROS-high populations, colony forming assays and engraftment assays can be performed (Lagadinou et al., 2013; Pei et al., 2018).
-
30.Colony forming assay – The colony forming assay is a readout of stem and progenitor function. This assay has several caveats including that some AML specimens will not form colonies and colonies can be formed by progenitors’ cells as well as stem cells. The strength of this assay is that it requires low cell numbers.
-
a.Suspend approximately 100,000 cells from ROS-low and ROS-high populations in 4 mL of human methylcellulose and mix.
-
b.Place 1 mL of each into three 35 mm cell culture dish.
-
c.Incubate all the dishes in a secondary large bioassay dish (245 mm × 245 mm) with a small volume of water in an extra 35 mm dish to maintain humidity in the bioassay dish.
-
d.Incubate at 37°C for 10–14 days until colonies are visible.
-
e.Colonies are then counted using a light microscope.
-
a.
-
31.Engraftment assay – Engraftment into immune deficient mice is the gold-standard assay for assessing stem cell function (Bonnet and Dick, 1997; Lapidot et al., 1994).
-
a.Use NSGS mice conditioned with 25 mg/kg busulfan via intraperitoneal injection utilizing 28G syringe 24 h prior to transplant.
-
b.Treat 100,000–2,000,000 cells primary ROS-low or ROS-high populations with OKT3 to prevent graft versus host disease (Wunderlich et al., 2014).
-
c.Warm the mice under a heat lamp and then restrain the mouse in a restrainer so that the tail is out. Clean the tail with alcohol wipe.
-
d.Inject primary cells in a 0.1 mL volume of FACS Buffer intravenously through the tail vein using a 28G syringe.
-
e.After 6–12 weeks, euthanize mice and collect the bone marrow.
-
f.To collect bone marrow, femurs and tibias are dissected from mice and stored on ice until all samples have been collected. The marrow is flushed out of the bone using 1 mL of FACS buffer.
-
g.Spin samples at 580 × g for 5 min and aspartate FACS buffer.
-
h.Red blood cells are then lysed by incubating samples at 20°C for 1 min in RBC lysis buffer.
-
i.Spin samples at 580 × g for 5 min and aspartate RBC lysis buffer.
-
j.Resuspend samples in FACS buffer and filter through cell strainer to remove any clumping material.
-
k.Spin samples at 580 × g for 5 min and aspartate FACS buffer.
-
l.Resuspend cell in FACS buffer containing antibodies including CD45, CD34, CD38, and CD123 to measure human leukemic cells and stem cells via flow cytometry.
-
a.
Expected outcomes
Upon successful completion of this protocol the user should have acquired populations enriched for human LSCs and AML blasts. These cell populations can be used for various downstream applications. To validate enrichment of LSC and AML blast population, two assays can be employed. Colony forming assays requires less cells but has several caveats. Colony forming assays is a readout of stem and progenitor function. Also, some AML specimens do not form colonies. If this assay is successful, ROS-low LSCs are expected to form more colonies than ROS-high AML blasts (Figure 1B) (Jones et al., 2019; Jones et al., 2018; Pei et al., 2020). The gold-standard assay is engraftment into immune deficient mice (Bonnet and Dick, 1997; Lapidot et al., 1994). It is expected that ROS-low LSCs preferentially engraft into immune deficient mice when compared to ROS-high AML blasts (Figure 1C) (Lagadinou et al., 2013; Pei et al., 2018).
Limitations
The limitations of this protocol are consistent with many protocols that rely on primary AML specimens. For successful completion of this protocol it is important to start with a reliable source of viable primary AML specimens. Further, flow sorting primary human AML specimens can be inherently challenging as it can be difficult to determine blast gates and distinguishing between lymphocytes and aberrant lymphocyte marker expression on AML cells (see Troubleshooting section for more details).
The major limitation specific to this protocol is that it relies on the use of a metabolic dye to detect ROS levels which can change over time. When using this protocol, it is important to begin sorting for ROS-low LSCs and ROS-high AML blasts shortly after the staining protocol is complete. Waiting a prolonged period of time can result is the dye being metabolized or excreted from the cells and ultimately in inaccurate population enrichment. It is important to note that the time it takes to metabolize the ROS dye varies between patient specimens therefore careful monitoring of the cells while sorting is required. A decrease in overall signal will be observed if the dye is being metabolized or excreted from the cells.
Troubleshooting
Problem 1
Due to variability in AML specimen cell number and available cytometers the antibody concentrations outlined in this protocol may not be optimal for an individual’s application.
Potential solution
Before using this protocol, it is essential to titrate and optimize all antibody and dye concentrations using best practices for flow cytometry.
Problem 2
Some AML specimens exhibit aberrant expression of the lymphocyte marker CD3. It can be challenging to determine which cells is lymphocytes and which cells are AML cells in these cases.
Potential solution
The use of back-gating to distinguish true lymphocytes based on cell granularity and size is a possible way to determine if a population of cells are lymphocytes or AML cells. Mutational analysis can also be used to distinguish the AML cells from normal cells.
Problem 3
Determining the blast population and distinguishing between normal and leukemic cells can be challenging and sometimes not possible with flow cytometry alone.
Potential solution
Below are examples of blast gating strategies for various AML specimens (Figure 2B). However, sometimes it is not possible to distinguish cell populations by flow cytometry alone. This is especially true when comparing blasts to normal monocytes. In this case it is best to measure the mutational landscape of each population to definitively determine if each population is leukemic or normal.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Courtney L. Jones, Courtney.Jones@uhnresearch.ca
Materials availability
This study to not generate new unique reagents.
Data and code availability
This study did not generate data or code.
Acknowledgments
The authors thank the University of Colorado Hematology Clinical Trials Unit (HCTU) for help in acquisition of patient samples. This work was supported by an award from The Leukemia and Lymphoma Society, and The Princess Margaret Cancer Centre, The Princess Margaret Cancer Foundation, and the Ontario Ministry of Health (C.L.J.); the Evans MDS Foundation young investigator award (B.M.S.); National Institutes of Health (NIH) National Cancer Institute (NCI) grant R01 5 R01 CA200707 (C.T.J); and a Leukemia and Lymphoma Society Specialized Center of Research (SCOR) grant (principal investigator, C.T.J.). C.T.J. is supported by the Nancy Carroll Allen Endowed Chair.
Author contributions
B.M.S. and C.L.J. performed experiments and analysis used in the generation of figures. B.M.S., C.O.B., C.T.J., and C.L.J. wrote and reviewed this manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Brett M. Stevens, Email: brett.stevens@cuanschutz.edu.
Courtney L. Jones, Email: courtney.jones@uhnresearch.ca.
References
- Bonnet D., Dick J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N., Qian D., Lam J.S., Ailles L.E., Wong M. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y.Y., Sharkis S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C.L., Stevens B.M., D'Alessandro A., Culp-Hill R., Reisz J.A., Pei S., Gustafson A., Khan N., DeGregori J., Pollyea D.A. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood. 2019;134:389–394. doi: 10.1182/blood.2019898114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C.L., Stevens B.M., D'Alessandro A., Reisz J.A., Culp-Hill R., Nemkov T., Pei S., Khan N., Adane B., Ye H. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724–740.e4. doi: 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadinou E.D., Sach A., Callahan K., Rossi R.M., Neering S.J., Minhajuddin M., Ashton J.M., Pei S., Grose V., O'Dwyer K.M. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T., Sirard C., Vormoor J., Murdoch B., Hoang T., Caceres-Cortes J., Minden M., Paterson B., Caligiuri M.A., Dick J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- Pei S., Minhajuddin M., Adane B., Khan N., Stevens B.M., Mack S.C., Lai S., Rich J.N., Inguva A., Shannon K.M. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018;23:86–100.e6. doi: 10.1016/j.stem.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei S., Pollyea D.A., Gustafson A., Stevens B.M., Minhajuddin M., Fu R., Riemondy K.A., Gillen A.E., Sheridan R.M., Kim J. Monocytic subclones confer resistance to venetoclax-based therapy in acute myeloid leukemia patients. Cancer Discov. 2020;10:536–551. doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Ladi E., Mayer-Proschel M., Noble M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc. Natl. Acad. Sci. U S A. 2000;97:10032–10037. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M., Brooks R.A., Panchal R., Rhyasen G.W., Danet-Desnoyers G., Mulloy J.C. OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood. 2014;123:e134–e144. doi: 10.1182/blood-2014-02-556340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate data or code.