

Abstract
Oral squamous cell carcinoma (OSCC) is one of the most painful cancers, which interferes with orofacial function including talking and eating. We report that legumain (Lgmn) cleaves protease-activated receptor-2 (PAR2) in the acidic OSCC microenvironment to cause pain. Lgmn is a cysteine protease of late endosomes and lysosomes that can be secreted; it exhibits maximal activity in acidic environments. The role of Lgmn in PAR2-dependent cancer pain is unknown. We studied Lgmn activation in human oral cancers and oral cancer mouse models. Lgmn was activated in OSCC patient tumors, compared with matched normal oral tissue. After intraplantar, facial or lingual injection, Lgmn evoked nociception in wild-type (WT) female mice but not in female mice lacking PAR2 in NaV1.8-positive neurons (Par2Nav1.8), nor in female mice treated with a Lgmn inhibitor, LI-1. Inoculation of an OSCC cell line caused mechanical and thermal hyperalgesia that was reversed by LI-1. Par2Nav1.8 and Lgmn deletion attenuated mechanical allodynia in female mice with carcinogen-induced OSCC. Lgmn caused PAR2-dependent hyperexcitability of trigeminal neurons from WT female mice. Par2 deletion, LI-1, and inhibitors of adenylyl cyclase or protein kinase A (PKA) prevented the effects of Lgmn. Under acidified conditions, Lgmn cleaved within the extracellular N terminus of PAR2 at Asn30↓Arg31, proximal to the canonical trypsin activation site. Lgmn activated PAR2 by biased mechanisms in HEK293 cells to induce Ca2+ mobilization, cAMP formation, and PKA/protein kinase D (PKD) activation, but not β-arrestin recruitment or PAR2 endocytosis. Thus, in the acidified OSCC microenvironment, Lgmn activates PAR2 by biased mechanisms that evoke cancer pain.
SIGNIFICANCE STATEMENT Oral squamous cell carcinoma (OSCC) is one of the most painful cancers. We report that legumain (Lgmn), which exhibits maximal activity in acidic environments, cleaves protease-activated receptor-2 (PAR2) on neurons to produce OSCC pain. Active Lgmn was elevated in OSCC patient tumors, compared with matched normal oral tissue. Lgmn evokes pain-like behavior through PAR2. Exposure of pain-sensing neurons to Lgmn decreased the current required to generate an action potential through PAR2. Inhibitors of adenylyl cyclase and protein kinase A (PKA) prevented the effects of Lgmn. Lgmn activated PAR2 to induce calcium mobilization, cAMP formation, and activation of protein kinase D (PKD) and PKA, but not β-arrestin recruitment or PAR2 endocytosis. Thus, Lgmn is a biased agonist of PAR2 that evokes cancer pain.
Keywords: asparaginyl endopeptidase, cancer pain, legumain, oral cancer, protease, protease-activated receptor-2
Introduction
Up to 90% of cancer patients endure pain; oral cancer is one of the most painful (van den Beuken-van Everdingen et al., 2007). Pain often overwhelms oral cancer patients in the final months of life; these patients suffer most while speaking, drinking, or eating, and their quality of life plummets (Connelly and Schmidt, 2004; Kolokythas et al., 2007). Oral squamous cell carcinoma (OSCC) pain worsens with disease progression and responds poorly to opioids. Development of an alternative to opioids is stymied by our poor understanding of the mechanism driving cancer pain. While the etiology of oral cancer pain is not well understood, it is known that OSCC secretes mediators that sensitize and activate nociceptors within the cancer microenvironment and generate pain. These mediators include endothelin, ATP, nerve growth factor, and proteases; proteases produce pain by cleaving protease-activated receptor-2 (PAR2) on nociceptors (Pickering et al., 2008; Schmidt et al., 2007; Lam and Schmidt, 2010; Ye et al., 2011, 2014b; Lam et al., 2012). The proteases that activate PAR2 in the OSCC microenvironment are unknown. Anaerobic metabolism in tumors and inflamed tissues acidifies extracellular fluid. Legumain (Lgmn; asparaginyl endopeptidase) is a cysteine protease of late endosomes and lysosomes with an acidic pH optimum. Although Lgmn has been implicated in tumor metastasis (Kembhavi et al., 1993), and patients with metastatic oral cancer report greater pain (Connelly and Schmidt, 2004), it is unknown whether Lgmn causes PAR2-dependent cancer pain.
PAR2 is a G-protein-coupled receptor (GPCR) expressed by nociceptors that mediates neurogenic inflammation and pain (Steinhoff et al., 2000; Vergnolle et al., 2001). Proteases activate PAR2 through distinct mechanisms. The canonical agonists trypsin, tryptase, and kallikrein cleave within the extracellular N terminus of PAR2, which exposes a tethered ligand that binds to and activates the cleaved receptor (Nystedt et al., 1995; Böhm et al., 1996a; Corvera et al., 1997; Angelo et al., 2006). The biased agonists cathepsin S and elastase cleave at different sites within the PAR2 N terminus, leading to distinct pathways of PAR2 signaling and trafficking (Ramachandran et al., 2009; Zhao et al., 2014). PAR2 couples to signaling pathways that sensitize and activate pain-related ion channels, including the transient receptor potential vanilloid (TRPV1, TRPV4) and ankyrin (TRPA1) channels, yielding sustained sensitization of nociceptors and chronic pain (Amadesi et al., 2006; Dai et al., 2007; Zhao et al., 2015). Since PAR2 is upstream of these pronociceptive channels, blockade of proteases and of PAR2 is advantageous for the management of cancer pain. Therefore, there is a need to identify proteases that remain active in the acidified extracellular millieu of tumors and to determine whether they activate PAR2 to produce pain. Some proteases known to activate PAR2 show diminished activity under acidic conditions (Hachem et al., 2003, 2005). Moreover, proteases are often profiled in diseased tissues by measurement of protein or mRNA, not activity.
Here, we report that Lgmn is a unique activator of PAR2. We found that Lgmn is reproducibly and robustly activated in OSCC patients and mice with OSCC. Under acidified conditions, Lgmn cleaved PAR2 at a distinct site and activated PAR2 by biased mechanisms, leading to hyperexcitability of nociceptors and nociceptive behavior in mice. A Lgmn inhibitor prevented OSCC pain in mice. Thus, we have identified Lgmn as a novel mediator and therapeutic target for OSCC pain.
Materials and Methods
Reagents.
We used recombinant human Lgmn (440,000 ng/ml, catalog #2199-CY-010, R & D System), Lgmn substrate Z-Ala-Ala-Asn-AMC (I-1865; Bachem), DMEM and HBSS (ThermoFisher), PAR2 agonist 2-Furoyl-LIGRLO-NH2 and PAR1 agonist TFLLR-NH2 (Tocris), Lgmn-generated PAR2 activating peptide (RSSKGR; GL Biochem), and other reagents (Sigma) unless otherwise specified.
Lgmn activation and inhibition.
Lgmn was activated per manufacturer protocol. Lgmn activation was confirmed by incubating Lgmn (1 ng/µl) with Z-Ala-Ala-Asn-AMC (200 μm) in dilution buffer [250 mm NaCl, 50 mm 2-(N-morpholino) ethanesulfonic acid (MES); pH 5.0] or MES-HBSS (HBSS, 50 mm MES pH 5.0, 5.5 or 6.0). Fluorescence (excitation 340 nm, emission 460 nm) was measured every 30 s for 10 min in a Flexstation three plate reader (Molecular Devices). Lgmn (1 ng/µl) was assayed for activity in the presence of the Lgmn inhibitors QDD100531 or QDD123427 (1 pm –10 μm) and PAR2 antagonists I-343 (10 μm) or GB88 (10 μm; Farmer, 2013; Lieu et al., 2016; Jimenez-Vargas et al., 2018). Rich Williams provided the Lgmn inhibitors. The specificity of QDD100531 was demonstrated in Ness et al. 2015 (compound 9h in Supplementary Table 1); QDD100531 showed no reactivity to other proteases including cathepsin S, cathepsin B, caspase-3, caspase-8, or USP17 (Ness et al., 2015). QDD123427 showed similar specificity (R. Williams, personal communication). GB88 exhibited specific antagonist activity against four PAR2 agonists differing in structure and mechanism; selectivity of GB88 for PAR2 over PAR1 and PAR4 was also demonstrated (Suen et al., 2012). I-343, a member of the I-191 family of full PAR2 antagonists, inhibited inositol phosphate-1 (IP1) generation induced by the PAR2 agonists trypsin and 2-Furoyl-LIGRLO-NH2, but not IP1 accumulation induced by ATP (Farmer, 2013; Jiang et al., 2018; Jimenez-Vargas et al., 2018). For studies in mice, activated Lgmn was diluted in 50 mm MES and 250 mm NaCl, at pH 5.0 (dilution buffer) to a concentration of 300 ng/20 µl. For the in vivo experiments with LI-1 (10 mm, 100 µl, diluted in DMSO; Lee and Bogyo, 2010) the inhibitor was injected into the tail vein 2 h before injection of Lgmn. LI-1 was a gift from Matthew Bogyo. It is a covalent Lgmn inhibitor that exhibits >20,000-fold selectivity for Lgmn over cathepsin B, cathepsin L, and caspase-3. It has previously been shown to inhibit all Lgmn activity in vivo within 1 h of administration (Edgington-Mitchell et al., 2016).
OSCC patients.
Patients were screened and enrolled through New York University (NYU) Oral Cancer Center after consent. Detailed demographic information (age, sex, ethnicity, cancer location, primary tumor stage, and evidence of metastasis) was collected. During surgical resection, tumor and matched normal oral mucosa specimens were collected (normal was harvested at anatomically matched contralateral site). Specimens were frozen in liquid nitrogen and maintained at −80°C. The Committee on Human Research at NYU Langone Medical Center approved human studies.
Mice.
Female C57BL/6J (#000664) and NU/J Foxn1nu athymic mice (#002019), four to eight weeks, were from The Jackson Laboratory. Female C57BL/6J and F2rl1−/− (B6. Cg-F2rl1tm1Mslb/J) mice (#004993), four to eight weeks, from The Jackson Laboratory, were used for trigeminal ganglia (TG) dissociation. F2rl1 conditional knock-out (KO) C57BL/6 mice were generated by genOway as described (Jimenez-Vargas et al., 2018). Lgmn−/− C57BL/6N mice were a gift from Thomas Reinheckel (Matthews et al., 2010). The NYU Institutional Animal Care and Use Committee approved mouse studies.
Analysis of total and active Lgmn in tissues.
Snap frozen human and murine tissues were sonicated in 50 mm citrate pH 5.5, 0.5% CHAPS, 0.1% Triton X-100, and 4 mm DTT (10 µl/mg tissue). Solids were cleared by centrifugation and protein concentration was measured by BCA assay (Pierce). Protein was diluted in citrate buffer (50 µg/20 µl buffer), and LE28 was added from a 100× DMSO stock (1 μm final; Edgington et al., 2013). Samples were incubated at 37°C for 15 min, and the reaction was quenched with 5× sample buffer [200 mm Tris-Cl (pH 6.8), 8% SDS, 0.04% bromophenol blue, 5% β-mercaptoethanol, and 40% glycerol]. Protein was resolved on a 15% polyacrylamide gel under reducing conditions. LE28 binding was detected by scanning the gel for Cy5 fluorescence using a Typhoon 5 (GE Healthcare). Proteins were transferred to nitrocellulose membranes for immunoblotting with a goat anti-human Lgmn antibody (R & D AF2199, 1:1000 diluted in 50% Li-Cor blocking buffer and 50% PBS-T containing 0.05% Tween 20). Donkey-anti goat-HRP (1:10,000; A15999; Invitrogen) was used for detection with Clarity Western ECL Substrate (Bio-Rad). Actin (Sigma A5060) and ponceau stain were controls.
Tongue xenograft cancer model.
An orthotopic xenograft tongue cancer model was created by injecting HSC-3 into the tongue (Lam et al., 2012). NU/J Foxn1nu athymic mice were injected in the left lateral tongue under anesthesia [1 × 105 HSC-3 human tongue OSCC cells suspended in 20 µl vehicle (1:1 mixture of DMEM and Matrigel; Corning, reference #354234), or vehicle alone]. After two weeks, the resulting xenografted tumors and vehicle-injected tongues were excised and snap frozen for protein analysis as above. Goat anti-mouse Lgmn (R & D AF2058) and donkey anti-goat IR-800 (Li-Cor) were used in the immunoblot.
Paw xenograft cancer model.
The plantar surface of the right hind paw of NU/J Foxn1nu athymic mice were inoculated with 1 × 105 HSC-3 in 20 μl of DMEM and Matrigel (Ye et al., 2011, 2014a). The paw xenograft model permits measurement of mechanical and heat hypersensitivity of the paw. By 14 d after inoculation, a visible tumor developed in the paw. After measuring baseline mechanical and thermal withdrawal thresholds, HSC-3 were inoculated into the hind paw. Mechanical and thermal withdrawal were measured at post inoculation days 3, 6, 10, and 13. On post inoculation day 14, LI-1 (10 mm, 100 µl) was injected into the tail vein. Mechanical and thermal withdrawal were measured at 1, 3, 6, 12, 24, and 48 h after injection of LI-1 into the paw cancer mouse model.
4-Nitroquinoline 1-oxide (4NQO)-induced OSCC model.
An OSCC mouse model was generated by exposing mice to 4NQO (100 μg/ml) in drinking water for 16 weeks (Lam et al., 2012). Functional allodynia (gnaw-time) was measured with dolognawmeters (Dolan et al., 2010). Before administration of 4NQO, mice were examined to confirm the absence of oral abnormalities. 4NQO administration and dolognawmeter training over 15 sessions overlapped; baseline gnaw-time was calculated from the final 5 sessions. Functional allodynia was measured after 28 weeks. The tongue was harvested and a 1- to 2-mm coronal section was dissected from the most clinically suspicious region, fixed in 10% neutral buffer formalin, and processed for paraffin embedding and slide preparation. Four 5-μm hematoxylin and eosin (H & E)-stained tongue sections were evaluated for OSCC. Two pathologists blinded to group identity performed histopathologic analysis. Only mice with histologically confirmed OSCC were included in the analysis of nociception.
Mechanical and thermal nociception in the hind paw.
To assess mechanical nociception, mice were placed on a platform with a metal mesh floor and acclimated for 1 h. Paw withdrawal threshold was measured with von Frey filaments (Stoelting; Pickering et al., 2008). Withdrawal threshold was defined as the gram-force sufficient to elicit left hindpaw withdrawal. Withdrawal threshold for each animal was determined as the mean of three trials for each animal. Thermal hyperalgesia was measured with a paw thermal stimulator (IITC Life Sciences; Yamano et al., 2017). Mice were placed in a plastic chamber on a 25°C glass surface. A radiant heat source was focused on the left hind paw and withdrawal latency was measured as the mean of three trials taken at least 5 min apart in each mouse. The cutoff latency was established at 20 s. Lgmn (300 ng in 20 µl) or vehicle (control in 20 µl) was administered by intraplantar injection into the left hind paw under 1% isoflurane. Injections were made 1 h before the withdrawal tests (days 0, 1, and 4).
Facial mechanical nociception.
Mice were placed individually in a transparent, mesh-floor, box and acclimated for 1 h every other day for two weeks. We measured withdrawal responses to mechanical stimulation of the left cheek with von Frey filaments ranging from 0.008 to 4 g-force (11 filaments in total) in ascending order (Deseure et al., 2003). We applied the von Frey filament to the cheek, defined by the area below the eye, between the nose and the ear. Each fiber was applied once; however, if the response to a von Frey filament was equivocal or the mouse was moving, the same von Frey filament was reapplied to the same area of the cheek 10 s after the first stimulus, or when the mouse stopped moving. The interval between applications of von Frey filaments of different intensities was 5 min. The facial nociception score was reported as a numerical average of the 11 responses in the following response categories: 0: no response; 1: detection, the mouse is aware of the filament that stimulates the face; the mouse turns its head slightly to the object; 2: reaction, the mouse turn its head away quickly, pulls it backward or reacts with a single face wipe; 3: escape/attack, the mouse quickly escapes from the object, attacks the object with its paw or mouth, or reacts with two facial swipes; 4: multiple facial grooming, the mouse responds to the filament simulation with more than three facial wipes continuously. Hair on the left cheek was removed before subcutaneous injections of Lgmn. The whiskers were not trimmed. Lgmn (300 ng in 20 µl) was injected subcutaneously to the left cheek under 1% isoflurane. Injections were made 1 h before the facial mechanical withdrawal test at days 0, 1, and 4.
Orofacial behavior.
The dolognawmeter quantifies a behavioral index of orofacial nociception (Dolan et al., 2010). The device measures the time taken to gnaw through a dowel and is a validated index of orofacial nociception in mice with OSCC. Mice were trained for 15 training sessions in the dolognawmeter or until the coefficient of variance of the time required to gnaw was below 0.2. A baseline gnaw-time (mean of the final five training sessions) was established for each mouse. After baseline gnaw-times were determined, treatment or drug injections were initiated and the mice underwent behavioral testing. Each response was analyzed relative to the mouse's baseline. Activated Lgmn, 300 ng in 20 µl, was injected into the tongue under isoflurane. The injection was performed at days 0, 1, and 4. One hour after injection, the mice were tested with a dolognawmeter.
Quantification of Lgmn in OSCC cells.
Lgmn was measured in HSC-3 and DOK cells by ELISA. HSC-3 or DOK cells (∼5000/well of a 12-well plate) were cultured for 72 h (∼70% confluency; Lam et al., 2012). Medium was removed, cells were washed with 5 ml PBS without Ca2+ and Mg2+, and DMEM (500 µl) was added to each well. After 48 h, medium was collected and centrifuged (1500 rpm, 4 min, 4°C). Cells were homogenized with 100 µl of RIPA buffer/well (Thermo Scientific, product #89901). Cell lysate was collected and centrifuged (1500 rpm, 10 min, 4°C). The pellet was discarded. A RayBio Human Lgmn ELISA kit (RayBiotech, code ELH-Lgmn-1) was used for Lgmn quantification. The standard curve was generated using the following concentrations: 7000, 2800, 1120, 448, 179, 72, 29, and 0 pg/ml of Lgmn provided with the kit. The optical densities of the standards and samples were read at 450 nm wavelength using a Promega GloMax luminometer (Promega BioSystems, Model E9032). Alternatively, cells were live-labeled with LE28 (1 μm, 0.1% DMSO) for 4 h, lysed on ice in PBS containing 0.1% Triton X-100, and cleared by centrifugation. Supernatants collected overnight in serum-free media were concentrated using an Amicon Filter with a 3-kDa cutoff. Total protein from whole-cell lysates or supernatants (∼60 µg) were resolved by SDS-PAGE. Gels were scanned for Cy5 fluorescence and subject to Lgmn immunoblotting.
Lgmn immunofluorescence in cancer cells.
HSC-3 and DOK cells were grown on cover slips at 37°C, 5% CO2 for 48 h. Cells were washed with PBS and fixed in 4% paraformaldehyde at room temperature for 15 min. Cells were incubated with 3% bovine serum albumin (BSA) in PBS to block non-specific binding, then incubated with mouse monoclonal anti-Lgmn antibody (Santa Cruz Biotechnology, B-8: sc-133234, lot #A0610), 1:50, 4°C, overnight. Cells were washed in PBS and then incubated with goat anti-mouse secondary antibody conjugated to Alexa Fluor 488 (Life Technology, A11029), 1:300, room temperature for 3.5 h. Nuclei were stained with Hoechst (Thermo Scientific, product #62249, lot #RG2244203). The cover slips were mounted on slides in Fluoromount G (Electron Microscope Sciences). A laser scanning confocal microscope (LSM 700, Carl Zeiss) was used to obtain fluorescent images. The images were captured with a Zeiss Plan-Apochromat 63×/1.40 Oil DIC M27 objective lens for Lgmn signal quantification or 20× DIC objective lens for capturing the representative images. The fluorescent signal intensity of each cell was measured by a blinded researcher using NIH ImageJ. Controls included the following: (1) staining of the spleen from wild-type (WT) and Lgmn−/− mice; (2) preabsorption of the primary anti-Lgmn antibody with Lgmn; and (3) omission of the primary antibody. For the preabsorption negative control, the primary anti-Lgmn antibody (0.07 nm, equivalent to 1:50 dilution) was incubated in 10× higher concentration of activated Lgmn (0.7 nm) at 37°C for 48 h versus cells stained with the primary anti-Lgmn antibody, which was incubated in activated Lgmn vehicle. WT and Lgmn−/− mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine (intraperitoneal) and transcardially perfused with 25 ml cold PBS, followed by 25 ml of 10% neutral formalin solution. The spleen was postfixed in 10% neutral formalin solution for 24 h, cryoprotected in 30% (v/v) sucrose in PBS for 2 d at 4°C, and embedded in Tissue-TekR optimum cutting temperature (OCT) compound (Sakura Finetek). Sections (10 µm) were processed for immunofluorescence staining.
Dissociation of TG neurons.
Mice were anesthetized with isoflurane, bilateral TG were removed and neurons were enzymatically dispersed as described (Ono et al., 2015). Ganglia were cut into 8–10 segments and incubated for 30 min in collagenase and dispase (12 mg collagenase, 14 mg dispase in 3 ml HBSS). Neurons were triturated and plated onto laminin/poly-L-ornithine-coated coverslips. Neurons were cultured in Leibovitz medium containing 10% fetal calf serum, with penicillin and streptomycin for 24 h (37°C, 5% CO2).
Electrophysiological recording.
Hyperexcitability of small TG neurons (≤20 µm, <30 pF) was quantified by measuring rheobase (Scheff et al., 2018). Whole-cell patch-clamp recordings were made using Axon patch 200B amplifier (Molecular Devices). Digidata 1440A (Molecular Devices) was used for data acquisition and pulse generation. Rheobase was measured using 250 ms square pulses starting from −10 pA with steps of 10 pA until the action potential threshold was reached. The resting membrane potentials were recorded at stable conditions without current injection. Neurons with resting membrane potentials more positive than −40 mV were excluded from analysis. Input resistance was estimated from current-clamp recordings of the voltage response to 250 ms of 10 pA hyperpolarizing current. Pipette resistance was 4–5 mΩ in the following external solution: 140 mm NaCl, 5 mm KCl, 10 mm HEPES, 10 mm glucose, 1 mm MgCl2, and 2 mm CaCl2; pH was adjusted to 7.3–7.4 with NaOH. Pipette solution consisted of the following: 110 mm K-gluconate, 30 mm KCl, 10 mm HEPES, 1 mm MgCl2, and 2 mm CaCl2; pH was adjusted to 7.25 with KOH. Rheobase was measured after 10 min incubation with Lgmn (20 ng/ml in external solution, pH 5.5), Lgmn (20 ng/ml) plus LI-1 (10 μm), or Lgmn vehicle (Lgmn activation buffer pH 5.5). To study the signaling pathways of Lgmn induced hyperexcitability, perforated patch-clamp was performed to avoid dialysis of cytoplasmic constituents. Amphotericin B (240 µg/ml) was used in the pipette solution. Neurons were preincubated with GF109203X (GFX, Tocris) or PKI-tide (both 1 μm, 30 min, 37°C) before challenge with Lgmn (20 ng/ml) or Lgmn vehicle. Rheobase was measured 10 min after incubation with Lgmn or vehicle.
Cell lines.
Human embryonic kidney (HEK293) cells stably expressing the human (h)PAR2 with extracellular N-terminal FLAG and intracellular C-terminal HA11 epitopes (HEK-FLAG-PAR2-HA cells) have been described (Böhm et al., 1996b). PAR1 and PAR2 were deleted from HEK293 cells using CRISPR/Cas9 (Ungefroren et al., 2017). Cells were maintained in DMEM with 10% fetal bovine serum and hygromycin (100 µg/ml, 5% CO2, 37°C). Dysplastic oral keratinocyte, DOK, cell number 94122104 from Sigma-Aldrich was cultured in DMEM/F12 (Invitrogen), 10% fetal bovine serum, 50 IU/ml penicillin/streptomycin and 5 µg/ml hydrocortisone. Human OSCC, HSC-3, cell number JCRB0623, was from Japanese Collection of Research Bioresources Cell Bank. HSC-3 was cultured in DMEM, 10% fetal bovine serum, and 50 IU/ml penicillin/streptomycin. DOK and HSC-3 cell lines were maintained at 37°C with 5% CO2.
PAR2 cleavage.
A peptide corresponding to hPAR2 amino acids 21–50 was synthesized by American Peptide Company and dissolved in water at 300 μm. The peptide was diluted to 200 μm with Lgmn activation buffer (50 mm sodium acetate, 100 mm NaCl, pH 4.5) in the presence and absence of Lgmn (200 nm; final volume 25 µl). After overnight incubation at 37°C, the reactions were quenched with 25 µl of 50% acetonitrile containing 0.1% trifluoroacetic acid (TFA). Samples (2 µl) were subject to LCMS analysis with a Shimadzu LCMS 2020 fitted with a Phenomenex Luna 3 µm C8(2) column (100 Å, 100 × 2 mm). A gradient of 0% – 60% acetonitrile over 10 min with 0.05% TFA was used for separation.
Immunofluorescence in HEK cells.
HEK-FLAG-PAR2-HA cells (45,000) were plated in eight-well ibiTreat µ-slides and incubated overnight. Cells were washed with HBSS and incubated with Lgmn (100 nm final in HBSS pH 5.5) or trypsin (10 nm final in HBSS pH 7.4; 1 h 37°C). Cells were fixed in 4% paraformaldehyde on ice for 10 min. Cells were incubated with blocking buffer [3% normal horse serum (NHS) and 0.1% saponin] for 30 min at RT. Primary antibodies in blocking buffer [rabbit anti-FLAG (1:250, Rockland) and mouse anti-HA (1:250; Ray Biotech)] were incubated overnight at 4°C. Cells were washed with PBS and secondary antibodies in blocking buffer (donkey anti-rabbit-Alexa Fluor 488 and donkey anti-mouse-Alexa Fluor 647; 1:500; ThermoFisher) were added for 1 h at RT. After washing with PBS, DAPI was added for 5 min followed by additional washing. Cells were imaged immediately on a Leica SP8 confocal microscope.
On-cell Westerns.
HEK-FLAG-PAR2-HA cells were plated on poly-d-lysine (PDL)-coated 96-well plates (30,000 cells/well) and incubated overnight. Cells were washed two times in HBSS (pH 7.4) and placed in MES-HBSS (pH 5.0) for Lgmn assays or HBSS (pH 7.4) for trypsin assays. Cells were incubated with Lgmn (1 or 10 ng/µl final concentration) in MES-HBSS (pH 5.0), trypsin (10 nm final concentration) in HBSS (pH 7.4), or vehicle (buffer control) for 30 min at 37°C. Cells were washed with HBSS and fixed with 4% paraformaldehyde in PBS for 20 min on ice. Cells were washed three times in PBS and incubated with blocking buffer (PBS + 3% NHS) for 1 h at room temperature. Cells were incubated with mouse anti-FLAG antibody (1:500, Cell Signaling) in PBS + 1% NHS overnight at 4°C. Cells were washed three times in PBS, incubated with donkey anti-mouse Alexa Fluor 790 (1:1000, A11371, ThermoFisher) in PBS + 1% NHS for 1 h at room temperature. Cells were washed 1 time in PBS and incubated with the nuclear stain SYTO 82 Orange (1 μm, ThermoFisher) in saline for 30 min. Cells were washed three times with saline and then imaged on an GE HealthcareTyphoon imaging system (GE). FLAG immunofluorescence intensity was quantified using NIH ImageJ and was normalized to nuclear fluorescent intensity to correct possible cell loss.
Measurement of intracellular Ca2+.
HEK-FLAG-PAR2-HA cells were plated on PDL-coated 96-well plates (25,000 cells/well) and incubated overnight. Cells were loaded with fura-2 AM (1 μm, Cayman Chemicals) in loading buffer (150 mm NaCl, 2.6 mm KCl, 0.1 mm CaCl2, 1.18 mm MgCl2, 10 mm D-glucose, 10 mm HEPES, 4 mm probenecid, 0.5% BSA, pH 7.4) for 1 h at 37°C. Cells were washed 2 times in HBSS and then placed in MES-HBSS (pH 5.0, 5.5, 6.0, or 7.4) for the Lgmn assays or HBSS (pH 7.4) for trypsin assays. Fluorescence was measured with 340 or 380 nm excitation and 530 nm emission with a Flexstation three plate reader. Baseline fluorescence was measured for 45 s (Zhao et al., 2015). Cells were challenged with Lgmn (1 or 10 ng/µl final concentration) in MES-HBSS (pH 5.0, 5.5, 6.0, or 7.4), trypsin (10 nm final concentration) in HBSS (pH 7.4), or vehicle (buffer control), and fluorescence was measured for an additional 180 s. To confirm that Lgmn-induced Ca2+ responses were because of activation of PAR2, cells were incubated with the PAR2 antagonists I-343 (10 μm) or GB88 (10 μm; Farmer, 2013; Lieu et al., 2016; Jimenez-Vargas et al., 2018), the PAR1 antagonist SCH79797 (200 nm; Ahn et al., 2000), or vehicle in HBSS + 1% DMSO for 1 h at 37°C before the Ca2+ assay. To confirm the requirement for protease activity, Lgmn was incubated with the Lgmn inhibitors QDD100531 (1 μm) or QDD123427 (100 nm; Ness et al., 2015) in HBSS +1% DMSO for 1 h before the Ca2+ assay. Cells were maintained with the inhibitors. To assess the requirement for hydrolyzis of the Asn30↓Arg31 site, a mutant PAR2 in which Asn30 was replaced with Ala (PAR2-ΔN30A) was generated (Twist Biosciences, San Francisco, CA). cDNA (5 µg) encoding PAR2-ΔN30A was expressed in HEK-PAR2-KO cells using polyethylenimine (ratio 1:6, DNA:PEI), and cells were studied after 48 h. To assess the capacity of Lgmn or trypsin to desensitize PAR2-mediated Ca2+ signaling, HEK-FLAG-PAR2-HA cells were preincubated with Lgmn (1 or 10 ng/µl final concentration) in MES-HBSS (pH 5.0), trypsin (10 nm final concentration) in HBSS (pH 7.4), or vehicle (buffer control) for 10 min at 37°C. Cells were washed and recovered in HBSS (pH 7.4) for 20 min at 37°C. Cells were then challenged with trypsin (10 nm). Desensitization of Ca2+ signals to the second challenge with trypsin was calculated as a percentage of the responses in cells preincubated with the vehicle control.
FRET assays of cAMP, protein kinase D (PKD), and ERK.
Genetically encoded FRET biosensors targeted to the cytosol were used to assess cAMP, PKD and ERK activation in living cells in real time (Zhao et al., 2015). HEK-FLAG-PAR2-HA cells were transfected with cDNA (5 µg) encoding the cAMP biosensor Cyto-EPAC (Jimenez-Vargas et al., 2018), the PKD biosensor Cyto-DKAR (Zhao et al., 2019), or the ERK biosensor Cyto-EKAR (Yarwood et al., 2017) using polyethylenimine (ratio 1:6, DNA:PEI). Cells were plated on PDL-coated 96-well plates (25,000 cells/well) and incubated overnight. Cells were washed 2× in HBSS and placed in MES-HBSS (pH 5.0) for the Lgmn assays. The cyan (470 nm) and yellow fluorescent protein (535 nm) emission ratios were measured with a CLARIOstarPlus plate reader (BMG). Baseline fluorescent ratios were recorded for 5 min. Cells were challenged with Lgmn (1 or 10 ng/µl) in MES-HBSS (pH 5.0) or vehicle (buffer control), and FRET was measured for an additional 30 min. FRET ratios were normalized to the MES-HBSS vehicle control.
BRET assays of β-arrestin recruitment.
HEK293 cells were transfected with cDNA encoding the PAR2-RLuc8 (1 µg) and β-arrestin-1-YFP (4 µg) with polyethylenimine (ratio 1:6, DNA:PEI; Jensen et al., 2013). Cells were plated on PDL-coated 96-well white walled plates (30,000 cells/well) and incubated overnight. Cells were washed 2× in HBSS and placed in MES-HBSS (pH 5.0) for the Lgmn assays or HBSS (pH 7.4) for trypsin assays. Coelenterazine-h (5 μm, Nanolight, Pinetop AZ) was added to the cells and the cells were challenged with Lgmn (1 or 10 ng/µl) or trypsin (10 nm). RLuc8 luminescence (480 nm) and YFP fluorescence (530 nm) emission were measured using a CLARIOstarPlus plate reader. Baseline fluorescence ratios were recorded for 2.5 min. The BRET ratio was normalized to vehicle control and baseline.
Experimental design and statistical analysis.
We used GraphPad Prism 7 and 8 (GraphPad Prism, GraphPad Software) for the statistical analysis. Results are expressed as mean ± SEM. For cell-based assays, triplicate measurements were made from four to five experiments; differences were evaluated by one-way or two-way ANOVA and Dunnett's or Tukey's multiple comparisons test. One-way ANOVA and Tukey's or Sidak's multiple comparisons and Student's t test were used for in vivo behavioral experiments and comparison of rheobase. Unpaired Student's t test was used to compare values between two groups.
Results
Lgmn is activated in human and murine OSCC
To determine whether Lgmn is activated in OSCCs, we collected OSCC specimens and matched normal oral mucosa from seven patients (Table 1). Specimens were incubated with a fluorescently quenched activity-based probe (LE28) selective for Lgmn (Edgington et al., 2013). Two LE28-labeled species of 36 kDa and 25 kDa were activated in OSCC versus normal tissue (Fig. 1A–D). Immunoprecipitation with a Lgmn-specific antibody confirmed the identity of mature forms of Lgmn (Fig. 1D). Immunoblotting revealed total levels of mature Lgmn 36 kDa increased in all OSCC versus normal tissue (Fig. 1A). The 56 kDa inactive Lgmn zymogen (pro-Lgmn) was detected in all specimens. Total Lgmn and Lgmn activity of 36 kDa were upregulated in a murine OSCC xenograft model (human OSCC cells (HSC-3) inoculated in nude mouse tongues) versus normal (Fig. 2A–C).
Table 1.
Patient profiles
| Patient # | Sex | Age | Ethnicity | Tumor location | Primary tumor stage | Nodal status |
|---|---|---|---|---|---|---|
| 1 | F | 71 | Hispanic | Mandibular gingiva | pT4a | pN0 |
| 2 | M | 57 | Hispanic | Mandibular gingiva | pT2 | pN2a |
| 3 | M | 66 | Hispanic | Floor of mouth, mandibular gingiva | pT4a | pN0 |
| 4 | F | 77 | White/Non-Hispanic | Mandibular gingiva | pT4a | pN0 |
| 5 | F | 50 | Asian | Tongue | pT1 | pN0 |
| 6 | M | 93 | Asian | Mandibular gingiva | pT2 | pN0 |
| 7 | F | 81 | White/Non-Hispanic | Maxillary gingiva | pT2 | pN0 |
Figure 1.

Lgmn activation in human OSCC. A, Active Lgmn labeled by LE28 (upper panel) as shown by in-gel fluorescence and total Lgmn immunoreactivity (IR; lower panel) as shown by Western blotting in OSCC biopsies and patient-matched normal oral mucosa. The gel (upper panel) was transferred to nitrocellulose and immunoblotted for total Lgmn levels (lower panel). B, Densitometry of the 36-kDa species labeled by LE28, displayed as averages for all normal and OSCC samples (t(12) = 3.124, **p = 0.0088, when 36-kDa Lgmn activity in matched normal oral mucosa and SCC is compared, unpaired Student's t test). C, Densitometry of the 25-kDa species labeled by LE28, displayed as averages for all normal and OSCC samples (t(12) = 2.367, *p = 0.0356, when 25-kDa Lgmn activity in matched normal oral mucosa and SCC is compared, unpaired Student's t test). D, Immunoprecipitation of LE28-labeled tumor sample with a Lgmn-specific antibody to confirm the 36- and 25-kDa species (n = 7).
Figure 2.

Lgmn in xenograft model of OSCC. A, B, Active Lgmn labeled by LE28 (A), shown by in-gel fluorescence, and total Lgmn immunoreactivity (IR; B), shown by Western blotting of lysates from HSC-3 xenografts or control tongues. The gel from (A) was transferred to nitrocellulose and immunoblotted for total Lgmn levels (B). C, Densitometry of active and total Lgmn from A, B, respectively (n = 3–5, t(6) = 2.592, *p = 0.0411, when active Lgmn in normal mouse tongue and xenograft cancer is compared, and t(6) = 2.818, *p = 0.0304, when total Lgmn in normal mouse tongue xenograft cancer is compared, unpaired Student's t tests).
We examined Lgmn expression in HSC-3 and dysplastic oral keratinocytes (DOK, non-cancer cell line) by immunofluorescence and ELISA. Immunoreactive Lgmn was detected in cytosolic granules of HSC-3 and DOK cells, and expression was confirmed by ELISA of cell lysate and conditioned medium (supernatant; Fig. 3A,F,G). The intensity of Lgmn staining was higher in HSC-3 (Fig. 3B). Controls for the selectivity of the Lgmn antibody included absence of staining of spleen from Lgmn−/− mice (Fig. 3C), abolition of Lgmn staining by Lgmn antibody preabsorption with Lgmn (Fig. 3D), and lack of staining when the Lgmn antibody was omitted (Fig. 3E). The Lgmn protein concentration in lysate and supernatant from HSC-3 was higher than the concentration from DOK (Fig. 3F,G). The levels of Lgmn in lysate (Fig. 3H,I) and supernatant (Fig. 3J,K) were higher in HSC-3 compared with DOK. Immunoblotting revealed upregulation of Lgmn 56-kDa zymogen and Lgmn 36-kDa mature form in HSC-3 versus DOK (Fig. 3I). HSC-3 cells also secreted more pro-Lgmn than DOKs (Fig. 3J,K). These results suggest that OSCC cells express and release more Lgmn than dysplastic keratinocytes.
Figure 3.

Expression of Lgmn in OSCC cells. A, Localization of immunoreactive Lgmn (red) in DOK and HSC-3. B, Lgmn signal intensity in DOK and HSC-3 was quantified in individual cells by NIH ImageJ (t(52) = 8.11, **p = 8.53E-11, when Lgmn signal of DOK is compared with HSC-3, n = 20 and 34 cells in DOK and HSC-3, respectively, unpaired Student's t test). C, Localization of immunoreactive Lgmn in the spleen of WT and Lgmn−/− mice. D, Preabsorption of Lgmn antibody with Lgmn eliminated HSC-3 staining. E, Omission of the anti-Lgmn antibody resulted in lack of HSC-3 staining. F, G, Quantification of Lgmn protein by ELISA in DOK and HSC-3 cell lysate and supernatant. The concentration of Lgmn in HSC-3 cell lysate and supernatant was three times higher than that of DOK. N = 6 experiments in each group (in F, t(10) = 5.70, **p = 0.0002, when the Lgmn concentration in supernatant from HSC-3 is compared with the Lgmn protein concentration in supernatant from DOK, unpaired Student's t test. In G, t(10) = 6.49, **p = 0.000069, when the Lgmn concentration in lysate from HSC-3 is compared with the Lgmn concentration in lysate from DOK, unpaired Student's t test). Scale bar in A, C, D, E: 50 µm. H, Labeling of active Lgmn with LE28 in DOK and HSC-3 cell lysate, as shown by in-gel fluorescence. I, Lgmn immunoblot in DOK and HSC-3 cell lysate in H, with ponceau stain and actin immunoblot to verify equal loading. J, Labeling of Lgmn with LE28 in DOK and HSC-3 cell supernatant, as shown by in-gel fluorescence. K, Immunoblot of Lgmn from DOK and HSC-3 cell supernatant.
PAR2 expression on NaV1.8-positive nociceptors is necessary for Lgmn-induced mechanical allodynia
To investigate whether Lgmn causes allodynia by activating PAR2 on nociceptors, we administered Lgmn (300 ng intraplantar injection) for 3 d (0, 1, 4 d) to WT (C57BL/6J) and Par2Nav1.8 mice, which lack PAR2 in NaV1.8-positive neurons. We measured paw withdrawal responses to stimulation of the plantar surface with von Frey filaments at baseline and 1 h after each Lgmn injection (Fig. 4A). Lgmn induced mechanical allodynia on all 3 d in WT mice; however, mechanical allodynia was attenuated in Par2Nav1.8 mice by 51% on day 0 (Fig. 4C). To confirm that the nociceptive action of Lgmn required enzymatic activity and to test the analgesic potential of a Lgmn inhibitor, we administered the Lgmn-selective inhibitor, LI-1 (10 mm, 100 µl, i.v.; Edgington-Mitchell, 2016) to WT mice 120 min before intraplantar injection of Lgmn and measured paw withdrawal at 1, 3, 6, and 24 h after Lgmn injection (Fig. 4B). LI-1 attenuated Lgmn-induced mechanical allodynia by 44% 1 h after Lgmn injection (Fig. 4D). Thus, Lgmn-induced mechanical allodynia in mice requires PAR2 expression on Nav1.8-expressing nociceptors and Lgmn enzymatic activity.
Figure 4.

Lgmn-evoked nociception. A, Experiment timeline to test the effect of Lgmn (red arrow) on paw, facial mechanical nociception, and oral function on WT and Par2Nav1.8 mice. B, Experiment timeline to test the effect of Lgmn (red arrow) and LI-1 (blue arrow) on paw mechanical nociception on WT mice. C, Effects of Lgmn or vehicle (Veh) on paw withdrawal in WT and Par2Nav1.8 mice. Arrows indicate time of Lgmn or vehicle injection. Relative to WT Veh. mice, withdrawal thresholds were significantly lower in WT Lgmn mice, but not Par2Nav1.8 mice, at days 0, 1, and 4 (interaction F(9,64) = 2.47, p = 0.02; **p = 0.0002 at d0, **p = 0.0033 at d1, **p = 0.002 at d4, respectively, when WT mice treated with 300 ng Lgmn are compared with WT mice treated with Lgmn vehicle, n = 5 in each group, two-way ANOVA, Tukey's multiple comparisons). D, Effects of LI-1 on Lgmn-evoked mechanical allodynia in WT mice. Blue arrow indicates time of LI-1 or vehicle administration. Red arrow indicates time of Lgmn or Lgmn vehicle injection. Withdrawal thresholds were measured at 1, 3, 6, and 24 h after Lgmn injection. The effect of Lgmn varied with time (interaction F(16,100) = 2.98, p = 0.005, two-way ANOVA, n = 5 in each group, Tukey's multiple comparisons). One hour after injection of LI-1, mean withdrawal threshold was lower in Lgmn versus Lgmn vehicle (**p = 0.0002). LI-1 prevented the nociceptive effect of Lgmn at 3 and 6 h after Lgmn injection (#p = 0.04 and ##p = 0.009, respectively, when Lgmn plus LI-1 is compared with Lgmn plus LI-1 vehicle), but not 1 h after LI-1 injection (p = 0.30). Lgmn Veh. at pH 5 reduced the withdrawal threshold more than Lgmn Veh. at pH 7.5 at 6 h after paw injection (F(4,100) = 24.5, $p = 0.04, when Lgmn Veh. at pH 5.0 is compared with Lgmn Veh. at pH 7.5, two-way ANOVA, Tukey's multiple comparisons). E, Lgmn (300 ng) was injected into the cheek at days 0, 1, and 4 following baseline facial withdrawal measurements. Arrows indicate Lgmn injection into the cheek. Lgmn induced facial mechanical allodynia in WT but not Par2Nav1.8 mice. The means of the facial nociception score were significantly increased in WT mice versus Par2Nav1.8 mice at days 0, 1, and 4 (F(3,36) = 71.69, **p = 2.98E-15 at d0, d1, and d4, when WT 300 ng Lgmn is compared with Par2Nav1.8 300 ng Lgmn, n = 5 in each group, two-way ANOVA, Sidak's multiple comparisons). F, Lgmn (300 ng) significantly increased gnaw-time at 1 d after injection in WT mice versus baseline, but not in Par2Nav1.8 mice (F(3,33) = 3.26, ##p = 0.0045 at d1, when WT 300 ng Lgmn is compared with baseline, n = 8 and 6 in WT and Par2Nav1.8 mice, respectively, two-way ANOVA, Tukey's multiple comparisons).
PAR2 expression on NaV1.8-positive nociceptors is necessary for Lgmn-induced orofacial nociception
We used reflexive and operant assays to test whether Lgmn induces nociception in the orofacial region. We injected Lgmn into the cheek and measured facial withdrawal to stimulation with von Frey filaments. Lgmn (300 ng) was injected subcutaneously for 3 d (0, 1, 4 d) to WT and Par2Nav1.8 mice. Withdrawal was measured at baseline and 1 h after each injection (Fig. 4A). Lgmn induced facial mechanical allodynia on all 3 d in WT mice, but the nociceptive effect was attenuated in Par2Nav1.8 mice by 81% on day 0 (Fig. 4E). For operant behavioral testing, Lgmn (300 ng) was injected into the tongue. Dolognawmeters quantified a behavioral index of nociception 1 h after injection (Fig. 4A). Lgmn induced orofacial dysfunction in WT but not Par2Nav1.8 mice (Fig. 4F). Thus, expression of PAR2 in Nav1.8-expressing nociceptors is necessary for Lgmn-induced pain.
Lgmn mediates OSCC nociception
We used LI-1 to study whether Lgmn secreted from HSC-3 contributes to nociception in the xenograft paw cancer model. After measuring baseline mechanical withdrawal and thermal latency in the paws of NU/J Foxn1nu athymic mice, we inoculated HSC-3 cells, which highly express Lgmn (Fig. 5A). Postinoculation withdrawal measurements verified cancer-generated mechanical allodynia or thermal hyperalgesia. Mice were then treated with LI-1, and mechanical and thermal nociception were assessed (Fig. 5A). LI-1 reversed OSCC-induced mechanical withdrawal at 3, 6, 12, and 24 h after injection (Fig. 5B), and reversed thermal hyperalgesia at 3 and 24 h postinjection (Fig. 5C). LI-1 had no effect 48 h postinjection. An OSCC mouse model was generated with 4NQO administered over 16 weeks in Lgmn−/−, Par2Nav1.8 and WT mice. Oral mechanical allodynia was measured with dolognawmeters at week 28. Tongues were removed, sectioned, stained with H & E, and reviewed independently by two pathologists (Fig. 5D). OSCC was confirmed in all groups (Fig. 5E). OSCC incidence was >80% at 28 weeks after the administration of 4NQO in all groups (Fig. 5F). Lgmn−/− and Par2Nav1.8 showed significantly less mechanical allodynia than the WT mice (Fig. 5G).
Figure 5.

Contributions of Lgmn and PAR2 to OSCC pain. A, The experimental protocol included baseline measurements of withdrawal threshold to mechanical stimulation and withdrawal latency to thermal stimulation, HSC-3 inoculation, injection of LI-1, and measurements of nociception. B, Mechanical allodynia. HSC-3 inoculation (black arrow) produced mechanical allodynia after 10 d. LI-1 (blue arrow) reversed cancer-induced mechanical nociception versus vehicle control after 3, 6, 12, and 24 h following injection but not after 48 h (F(1,77) = 32.42, **p = 0.0003 at 3 h, **p = 0.0015 at 6 h, **p = 0.0068 at 12 h, **p = 2.15E-07 at 24 h, when Veh. control is compared with LI-1, n = 8 in each group, two-way ANOVA, Sidak's multiple comparisons). C, Thermal hyperalgesia. HSC-3 inoculation (black arrow) produced thermal hyperalgesia beginning at 3 d. LI-1 (blue arrow), versus vehicle control, reduced thermal hyperalgesia after 3 and 24 h but not after 48 h (F(10,140) = 24.45, **p = 0.0029 at 3 h, *p = 0.0278 at 24 h, when Veh. control is compared with LI-1, n = 8 in each group, two-way ANOVA, Sidak's multiple comparisons). D, The experimental protocol included administration of 4NQO to the mice for 16 weeks, measurement of OSCC nociception (gnaw-time) with the dolognawmeter, and histologic analysis of the tongue to confirm cancer. E, Representative histologic images of the tongue from C57BL/6J mice that did not receive 4NQO, and of the tongues from C57BL/6J, Par2Nav1.8, and Lgmn−/− mice at 28 weeks following 4NQO administration. Arrows indicate tongue carcinoma. Scale bar: 100 µm. F, The percentage of C57BL/6J, Par2Nav1.8, and Lgmn−/− mice that developed tongue cancer at 28 weeks after 4NQO administration. G, Change of gnaw-time versus baseline (percentage change of gnaw-time at baseline was set as 0%; data not shown) of C57BL/6J (n = 10), Par2Nav1.8 (n = 13), and Lgmn−/− (n = 10) mice with 4NQO-induced tongue cancer (F(1,30) = 16.28, *p = 0.029, when C57BL/6J is compared with Par2Nav1.8, *p = 0.024, when C57BL/6J is compared with Lgmn−/−, two-way ANOVA with Tukey's multiple comparisons).
Lgmn induces PAR2-dependent hyperexcitability of trigeminal neurons
To determine whether Lgmn causes PAR2-dependent hyperexcitability of TG neurons, we measured rheobase in WT mice using whole-cell patch-clamp. Neurons were studied in acidic buffer (external solution, pH 5.5). Lgmn (20 ng/ml, 10 min) decreased rheobase versus vehicle. There was no significant difference between the resting membrane potentials of TG neurons from WT mice pretreated with Lgmn versus Lgmn vehicle (Lgmn: −55.7 ± 2.2 mV, n = 13; Lgmn vehicle: −56.6 ± 3.9 mV; n = 11, t(22) = 0.207, p = 0.8378, unpaired Student's t test). The mean input resistance was increased in TG neurons pretreated with Lgmn compared with Lgmn vehicle, but there was no statistically significant difference (Lgmn: 659.3 ± 93.9 MΩ, n = 13; Lgmn vehicle: 526.9 ± 79.9 MΩ; n = 11, t(22) = 1.052, p = 0.3443, unpaired Student's t test). To investigate requirement for activity, we preincubated Lgmn with LI-1 (10 μm, 10 min) or vehicle. LI-1 prevented the effect of Lgmn on rheobase (Fig. 6A,B). To investigate the contribution of PAR2, we analyzed Lgmn-induced hyperexcitability in TG neurons from Par2−/− mice. Lgmn (20 ng/ml, 10 min) did not affect rheobase of Par2−/− neurons (Fig. 6C,D).
Figure 6.

PAR2 mediated Lgmn-induced hyperexcitability in TG neurons. A, Representative raw traces of whole-cell patch-clamp recordings showing membrane potential response at rheobase of TG neurons from WT mice treated with Lgmn vehicle, Lgmn, and Lgmn + LI-1. B, Rheobase of TG neurons in different treatment groups. Lgmn vehicle: 110.0 ± 31.4 pA, n = 11; Lgmn: 26.9 ± 22.1 pA, n = 13; Lgmn + LI-1: 94.0 ± 42.4 pA, n = 10 (F(2,31) = 23.14, ****p = 1.23E-6, when Lgmn and Lgmn vehicle are compared, ****p = 6.93E-5, when Lgmn and Lgmn + LI-1 are compared, one-way ANOVA with Tukey's multiple comparisons). C, Representative raw traces of whole-cell patch-clamp recordings showing membrane potential response at rheobase of TG neurons from Par2−/− mice treated with Lgmn and Lgmn vehicle. D, Rheobase of TG neurons from Par2−/− mice. Lgmn vehicle: 65.3 ± 26.0 pA, n = 13; Lgmn: 62.0 ± 30.1 pA, n = 10. E, Lgmn induced hyperexcitability and PKC-dependent or PKA-dependent pathways. Perforated patch-clamp recordings were used to measure rheobase of TG neurons. Neurons were preincubated with GFX 1 μm and PKI-tide 1 µm before Lgmn or Lgmn vehicle treatments. Rheobase was measured after neurons were challenged with Lgmn and Lgmn vehicle. Lgmn vehicle, 87.7 ± 12.5 pA, n = 9; Lgmn, 35.6 ± 8.5 pA, n = 12; GFX + Lgmn vehicle, 84.1 ± 8.5 pA, n = 12; GFX + Lgmn, 80.0 ± 9.7 pA, n = 10; PKI-tide + Lgmn vehicle, 89.0 ± 8.7 pA, n = 10; PKI-tide + Lgmn, 86.0 ± 11.3 pA, n = 10 (F(5,57) = 4.93, **p = 0.0052, when Lgmn and Lgmn vehicle are compared, *p = 0.0198 when the Lgmn and Lgmn + GFX are compared, **p = 0.0198, when Lgmn and Lgmn + PKI-tide are compared, one-way ANOVA with Tukey's multiple comparisons).
To evaluate the signaling pathway that mediates effects of Lgmn on excitability, perforated patch-clamp recordings were made from TG neurons pretreated with inhibitors of protein kinase C (PKC; GFX, 1 μm; Coultrap et al., 1999) or protein kinase A (PKA; PKI-tide, 1 μm; Ohlstein et al., 1990) for 30 min at 37°C before treatment with Lgmn (20 ng/ml) or vehicle. GFX and PKI-tide prevented Lgmn-induced hyperexcitability (Fig. 6E). Thus, Lgmn causes hyperexcitability of TG nociceptors through Lgmn enzymatic activity, expression of PAR2, PKC activity, and PKA activity.
Lgmn cleaves PAR2
To determine whether Lgmn can cleave PAR2 and identify the cleavage site, Lgmn (200 nm in acetate buffer, pH 4.5) was incubated with a peptide corresponding to residues 21–50 (S21CSGTIQGTNRSSKGRSLIGKVDGTSHVTG50) of the extracellular N terminus of hPAR2 (200 μm). Digest was analyzed by HPLC and mass spectrometry. Cleavage products were detected corresponding to PAR231–50 and PAR221–30 fused to PAR221–50 (Fig. 7A–C; Table 2). Thus, Lgmn cleaves the N terminus of hPAR2 at Asn30↓Arg31 consistent with its preference for asparagine residues and ability to ligate peptides with C-terminal asparagine residues to free N termini (Mikula et al., 2017).
Figure 7.

Lgmn cleavage of PAR2. A, Lgmn PAR2 cleavage sites and PAR2 N terminus indicating known cleavage sites. B, C, HPLC chromatograms (B) and product identification by mass spectrometry (C) of degradation of a fragment of PAR2 (hPAR221–50) by Lgmn, showing that Lgmn cleaves PAR2 at Asn30↓Arg31. D, Localization of PAR2 using antibodies to extracellular N-terminal FLAG and intracellular C-terminal HA epitopes (inset) in HEK-FLAG-PAR2-HA cells incubated with vehicle (control), Lgmn, or trypsin. Arrows denote plasma membrane localization; arrowheads denote endosomal localization. Scale bar: 20 µm. E, F, On-cell Western showing that trypsin and Lgmn remove the extracellular N-terminal FLAG epitope from HEK-FLAG-PAR2-HA cells. E, Quantification of triplicate observations from n = 5 individual experiments (F(5,22) = 16.03, **p = 0.0034, Lgmn 1 ng/µl compared with vehicle, ***p = 0.0002, Lgmn 100 ng/µg compared with vehicle, ****p = 2.5E-7 trypsin compared with vehicle, one-way ANOVA with Dunnett's test, n = 5). F, Representative images of FLAG immunoreactivity and Syto Orange stain.
Table 2.
Masses of hPAR2 N-terminal peptide cleavage products identified by mass spectrometry
| Peptide | Expected | Found |
|---|---|---|
| Substrate: SCSGTIQGTNRSSKGRSLIGKVdGTSHVTG | M: 2991.268 | |
| (M + 2)/2: 1496.64 | 1496.45 | |
| (M + 3)/2: 998.10 | 998.10 | |
| (M + 4)/4: 748.83 | 748.80 | |
| Product 1: RSSKGRSLIGKVdGTSHVTG | M: 2042.263 | |
| (M + 2)/2: 1022.14 | 1022.45 | |
| (M + 3)/3: 681.76 | 681.95 | |
| (M + 4)/4: 511.57 | ||
| Product 2: SCSGTIQGTNSCSGTIQGTNRSSKGRSLIGKVdGTSHVTG•H2O | M: 3958.274 | |
| (M + 2)/2: 1980.15 | ||
| (M + 3)/3: 1320.43 | 1320.10 | |
| (M + 4)/4: 990.58 | 990.40 |
To determine whether Lgmn cleaves intact PAR2 at the plasma membrane, hPAR2 with an extracellular FLAG epitope and intracellular HA epitope was expressed in HEK293 cells (Fig. 7D). HEK-FLAG-PAR2-HA cells were incubated with Lgmn (100 nm, HBSS pH 5.0), trypsin (10 nm, HBSS pH 7.4), or vehicle (buffer control). FLAG and HA were localized by immunofluorescence and confocal microscopy. In vehicle-treated cells, FLAG and HA colocalized at the plasma membrane (Fig. 7D). After incubation with Lgmn, FLAG was depleted from the plasma membrane whereas HA was retained at the plasma membrane, consistent with PAR2 cleavage and removal of the extracellular FLAG epitope. After incubation with trypsin, FLAG was depleted from the plasma membrane, and HA was detected within endosomes, consistent with PAR2 cleavage and endocytosis (Böhm et al., 1996a).
On-cell Western was used to quantify removal of the FLAG epitope. In Lgmn incubated cells (1 or 10 ng/µl, MES-HBSS pH 5.0, 30 min, 37°C), FLAG immunoreactivity was reduced 26 ± 7% (1 ng/µl) or 34 ± 2% (10 ng/µl) versus vehicle-treated cells (Fig. 7E,F). After trypsin incubation (10 nm, HBSS pH 7.4, 30 min, 37°C), FLAG immunoreactivity was reduced 53 ± 7% versus vehicle-treated cells. Nuclear stain (Syto Orange) confirmed that proteases did not remove cells from the plate (Fig. 7F).
Thus, Lgmn can cleave intact PAR2 at the surface of HEK cells and remove the extracellular FLAG epitope. Lgmn cleaves PAR2 at Asn30↓Arg31, proximal to the trypsin cleave site (Arg36↓Ser37). Lgmn does not evoke endocytosis of PAR2.
Lgmn activity and Lgmn-induced Ca2+ signaling are pH dependent
Trypsin, tryptase, and kallikreins cleave PAR2 at Arg36↓Ser37 and induce coupling to Gαq and mobilization of intracellular Ca2+ (Böhm et al., 1996a; Corvera et al., 1999; Oikonomopoulou et al., 2006). To examine whether Lgmn can mobilize Ca2+, HEK-FLAG-PAR2-HA cells were challenged with Lgmn (1 or 10 ng/µl, MES-HBSS pH 5.0, 5.5, 6.0 or HBSS pH 7.4), trypsin (10 nm, HBSS pH 7.4), or vehicle (buffer control). Change in intracellular Ca2+ was measured using fura-2 AM. Trypsin increased Ca2+, reflected by increased F340/380 nm emission, which rapidly declined toward basal (Fig. 8A). At pH 7.4, 10 ng/µl Lgmn induced a small increase in Ca2+, whereas 1 ng/µl Lgmn did not elicit a Ca2+ response (Fig. 8A). At a lower pH of 5.0, 1 and 10 ng/µl Lgmn caused sustained and concentration-dependent increases in Ca2+ (Fig. 8B,E). At pH 5.0 and 5.5, 10 ng/µl Lgmn increased Ca2+, compared with pH 6.0 and 7.4 (Fig. 8C,D); however, at pH 5, 1 ng/µl Lgmn increased Ca2+ compared with pH 5.5 and 6.0 (Fig. 8E,F). Lgmn activity assays confirmed the acidic pH optimum of Lgmn, which was active at pH 5.0 and 5.5 but not pH >6.0 (Fig. 8G). Lgmn increased Ca2+, in the absence of extracellular Ca2+, indicating intracellular mobilization (Fig. 8H). The Lgmn inhibitors, QDD100531 (1 μm) and QD123427 (100 nm), prevented Lgmn-evoked (1 ng/µl) Ca2+ signals (Fig. 8I,J), and caused concentration-dependent inhibition of activity (Fig. 8K; Ness et al., 2015).
Figure 8.
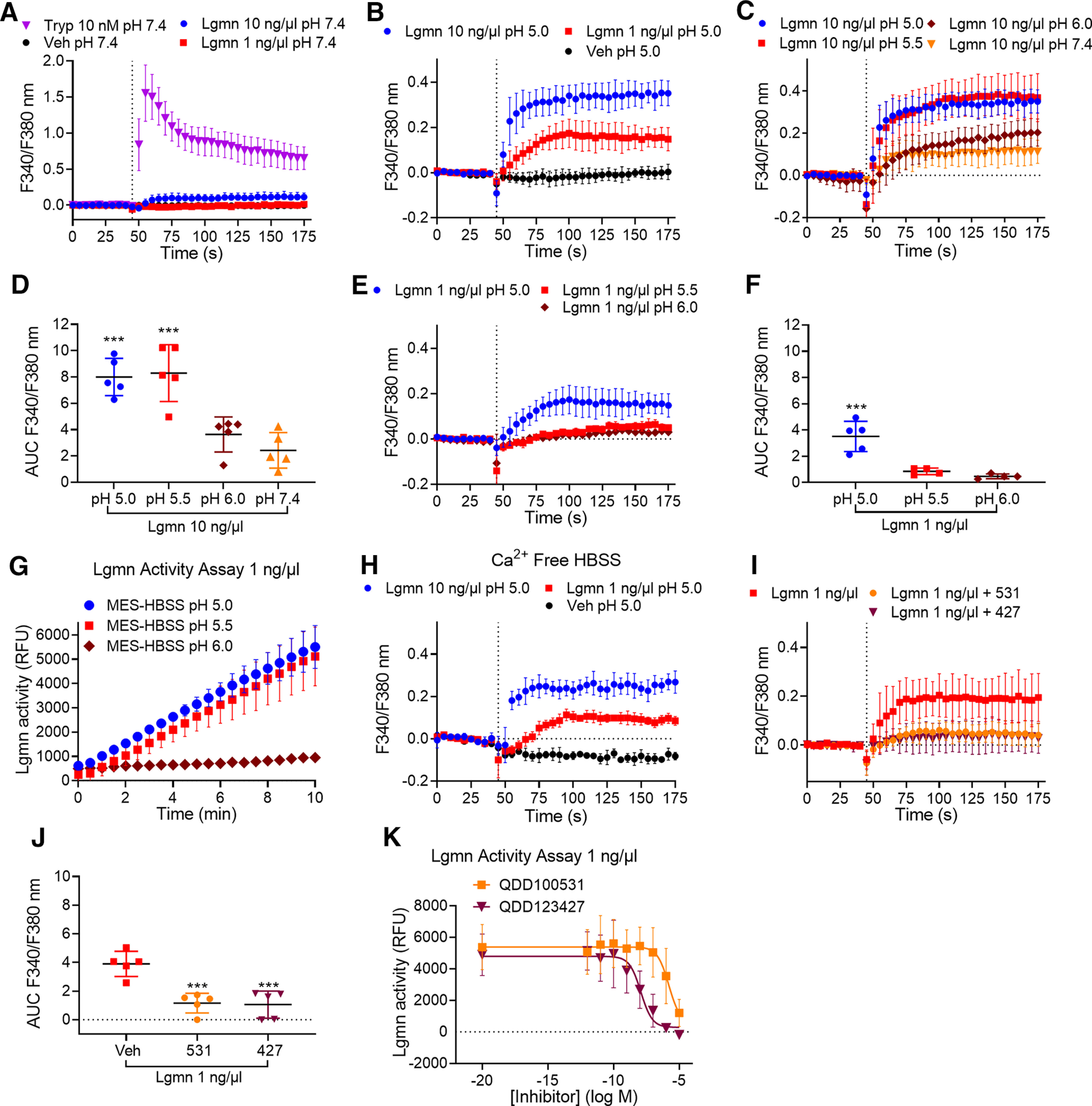
pH dependence of Lgmn activity and stimulation of Ca2+ signaling. Time course (A–C) and area under curve (AUC; D) of trypsin-evoked and Lgmn-evoked Ca2+ signaling in HEK-FLAG-PAR2-HA cells at pH 7.4, 6.0, 5.5, and 5.0 (F(3,16) = 17.5 ***p = 0.003, pH 5.0 compared with pH 7.4, ***p = 0.0001, pH 5.5 compared with pH 7.4, one-way ANOVA with Tukey's test, n = 5). E, F, Time course (E) and AUC (F) of Lgmn (1 ng/µl)-evoked Ca2+ signaling in HEK-FLAG-PAR2-HA cells at pH 6.0, 5.5, and 5.0 (F(2,10) = 22.68, ***p = 0.0009 for pH 5.0 compared with pH 5.5 and ***p = 0.0003 for pH 5.0 compared with pH 6.0, one-way ANOVA with Tukey's test, n = 5). G, Lgmn activity assays at pH 5.0, 5.5, and 6.0. H, Effects of depletion of extracellular Ca2+ on Lgmn responses. I, J, Time course (I) and AUC (J) of the effects of the Lgmn inhibitors QDD100531 (531) and QD123427 (427) on Lgmn Ca2+ signals (F(2,12) = 22.32, ***p = 0.0002 for 531 and ***p = 0.0002 for 427 compared with vehicle, one-way ANOVA with Tukey's test, n = 5). K, Lgmn activity assay in the presence of graded concentrations of Lgmn inhibitors QDD100531 and QDD123427 (pH 5.0).
Lgmn induces Ca2+ signaling through PAR2
To determine whether Lgmn induces Ca2+ signaling through PAR2, we used specific antagonists for PAR2 and cells genetically deleted for PAR2. PAR2 antagonists, I-343 and GB88 (10 μm; Farmer, 2013; Lieu et al., 2016; Jimenez-Vargas et al., 2018) abolished Lgmn-stimulated (1 and 10 ng/µl) Ca2+ responses (Fig. 9A–C). I-343 and GB88 did not inhibit the enzymatic activity of Lgmn (1 ng/µl), which was slightly increased in the presence of these antagonists (Fig. 9D). To determine whether Lgmn induced a Ca2+ increase through PAR1, we used the specific PAR1 antagonist SCH79797 (Ahn et al., 2000). SCH79797 (200 nm) did not alter Lgmn-induced Ca2+ responses (Fig. 9E,F). To confirm the pharmacologic evidence that PAR2, and not PAR1, mediated Lgmn-evoked Ca2+ signals, PAR2 or PAR1 was deleted from HEK293 cells using CRISPR/Cas9 (Ungefroren et al., 2017). In PAR2-KO cells, the PAR2 selective agonist 2-Furoyl-LIGRLO-NH2 did not increase Ca2+ (Fig. 9G); however, the PAR1 selective agonist TFLLR-NH2 increased Ca2+ in PAR2-KO cells (Fig. 9H). In PAR1-KO cells, TFLLR-NH2 did not increase Ca2+ except at a high concentration (10 μm; Fig. 9I), whereas 2-Furoyl-LIGRLO-NH2 increased Ca2+ in PAR1-KO cells (Fig. 9J). Lgmn (10 ng/µl) mobilized Ca2+ in PAR1-KO but not PAR2-KO cells (Fig. 9K).
Figure 9.

Lgmn signals through PAR2. A–C, Time course (A, B) and area under curve (C) of the effects of the PAR2 antagonists I-343 and GB88 on Lgmn Ca2+ signals (F(2,12) = 9.89 for Lgmn 10 ng/µl with **p = 0.0036 for I-343 and **p = 0.0052 for GB88 compared with vehicle, F(2,12) = 6.84 for Lgmn 1 ng/µl with **p = 0.0073 for I-343 and *p = 0.0384 for GB88 compared with vehicle, one-way ANOVA with Tukey's test, n = 5). D, Lgmn activity assays in the presence of the PAR2 antagonists I-343 and GB88 (pH 5.0). Triplicate observations from n = 5 individual experiments. E, F, Time course (E) and area under curve (F) of the effects of the PAR1 antagonist SCH79797 on Lgmn Ca2+ signals. G–J, Effects of the PAR2 agonist 2-Furoyl-LIGRLO-NH2 and the PAR1 agonist TFLLR-NH2 on Ca2+ signals in PAR2-KO (G, H) and PAR1-KO HEK293 (I, J) cells. K, Time course of Lgmn Ca2+ signals in HEK-PAR1-KO and HEK-PAR2-KO cells. L, Time course of Lgmn Ca2+ signals in HEK-PAR2ΔR30A cells. M, Effects of potential PAR2 activating peptide R31SSKGR36 Ca2+ signaling in HEK-FLAG-PAR2-HA cells.
To confirm that Lgmn activates PAR2 by cleavage at the Asn30↓Arg31 site, we generated a mutant receptor in which the Asn30 residue was mutated to Ala, which would not be recognized by Lgmn. PAR2-ΔN30A was transfected into PAR2-KO HEK cells. 2-Furoyl-LIGRLO-NH2 (10 μm) robustly increased Ca2+ in HEK-PAR2ΔN30A cells, whereas Lgmn (10 ng/µl) had no effect (Fig. 9L). To determine whether Lgmn activates PAR2 by exposure of a tethered ligand, we synthesized a hexapeptide, R31SSKGR36, corresponding to a potential tethered ligand revealed by Lgmn cleavage of PAR2 at the Asn30↓Arg31 site. However, R31SSKGR36 (10 μm to 0.01 nm) did not alter Ca2+ in HEK-PAR2 cells (Fig. 9M). These data confirm that the Lgmn-driven Ca2+ response is PAR2 dependent but does not involve exposure of a tethered ligand domain.
Lgmn desensitizes PAR2, but does not induce an association between PAR2 and β-arrestin-1
Processes that terminate PAR2 signaling at the plasma membrane include β-arrestin-mediated desensitization of PAR2, PAR2 cleavage and removal of activation sites and tethered ligand domains, and PAR2 endocytosis (Böhm et al., 1996a; Déry et al., 1999; DeWire et al., 2007). Since Lgmn cleaves PAR2 proximal to the trypsin site, subsequent inhibition of trypsin signaling would likely reflect PAR2 desensitization. To examine desensitization, HEK-FLAG-PAR2-HA cells were incubated with Lgmn (1 or 10 ng/µl, MES-HBSS pH 5.0), trypsin (10 nm, HBSS pH 7.4), or vehicle (buffer control; Fig. 10A). Cells were washed and recovered in HBSS pH 7.4 for 20 min and then challenged with trypsin (10 nm). In cells preincubated with vehicle, trypsin challenge at 30 min robustly increased Ca2+ (Fig. 10B,C). Initial challenge with trypsin also increased Ca2+, but response to a second challenge at 30 min was reduced by 53.7 ± 6.3% versus the response in vehicle-treated cells, consistent with desensitization and endocytosis of PAR2. Initial challenge with Lgmn (10 ng/µl) slightly increased Ca2+, but the response to a second challenge at 30 min was reduced by 49.9 ± 6.9% versus response in vehicle-treated cells, consistent with desensitization of PAR2.
Figure 10.

Lgmn desensitization of PAR2 Ca2+ signaling and recruitment of β-arrestin-1. A, HEK-FLAG-PAR2-HA cells were exposed to trypsin or Lgmn for 10 min, washed, and then challenged with trypsin 20 min after washing. B, Time course of Ca2+ signaling. C, Recovery of trypsin responses [area under curve (AUC) from B] in cells pretreated with vehicle, trypsin, or Lgmn (F(4,20) = 1.61, *p = 0.0116 for trypsin, and *p = 0.0372 for Lgmn 10 ng/µl compared with vehicle, one-way ANOVA with Tukey's test n = 5). D, E, BRET assays of recruitment of β-arrestin-1 to PAR2. D, Time courses. E, AUC (F(3,8) = 96.95, **** p = 1.25E-6 for trypsin versus vehicle, one-way ANOVA with Tukey's test, n = 5). All assays were done in triplicate.
Given that Lgmn desensitizes PAR2, we sought to determine whether Lgmn recruits β-arrestin-1 to PAR2. After trypsin cleavage, PAR2 becomes phosphorylated by GPCR kinases and interacts with β-arrestins, which mediate desensitization and endocytosis (Corvera et al., 1999). However, after cathepsin S or elastase cleavage, PAR2 neither recruits β-arrestins nor induces endocytosis (Zhao et al., 2014, 2015). We showed that Lgmn does not induce PAR2 endocytosis (Fig. 7D). It is unknown whether PAR2 associates with β-arrestin-1 following Lgmn cleavage. Thus, we examined BRET between PAR2-Rluc8 and β-arrestin-1-YFP following treatment with Lgmn. Trypsin (10 nm), but not Lgmn (1 or 10 ng/µl), stimulated PAR2-Rluc8/β-arrestin-1-YFP BRET (Fig. 10D,E). These results accord with the inability of Lgmn to evoke PAR2 endocytosis.
Lgmn activates PAR2-mediated cAMP formation and activation of PKD and ERK in HEK293 cells
After activation by trypsin, PAR2 couples to Gαq, leading to mobilization of intracellular Ca2+, generation of cAMP, and activation of ERK and PKD (DeFea et al., 2000; Amadesi et al., 2009). ERK contributes to sensitization of nociceptors (Ji et al., 1999), and PKD promotes mobilization of PAR2 from Golgi and recovery of responses to extracellular proteases (Amadesi et al., 2009; Zhao et al., 2019). To examine whether Lgmn-activated PAR2 couples to a similar range of downstream effectors, we expressed FRET biosensors of cytosolic cAMP (Cyto-Epac), cytosolic PKD (Cyto-DKAR), and cytosolic ERK (Cyto-EKAR) in HEK-FLAG-PAR2-HA cells. Lgmn caused concentration-dependent activation of cAMP (Fig. 11A,B), PKD (Fig. 11C,D), and ERK (Fig. 11E,F) within the cytosol. cAMP and PKD responses were robust; however, the ERK response was small and detected only after treatment with a higher Lgmn concentration.
Figure 11.

Lgmn activation of cAMP, PKD and ERK signaling and recruitment of β-arrestin-1. A–F, FRET assays of cytosolic cAMP (A, B), cytosolic PKD (C, D), and cytosolic ERK (E, F) in HEK-FLAG-PAR2-HA cells. A, C, E, Time courses. B, Area under curve (AUC) for cytosolic cAMP (F(2,12) = 26.43, ****p = 0.00,004 for Lgmn 10 ng/µl compared with vehicle, one-way ANOVA with Tukey's test, n = 5). D, AUC for cytosolic DKAR (F(2,12) = 69.77, ****p = 2.5E-7 for Lgmn 10 ng/µl and *p = 0.0117 for Lgmn 1 ng/µl compared with vehicle, one-way ANOVA with Tukey's test, n = 5). F, AUC for cytosolic ERK (F(2,12) = 10.99, **p = 0.0015 for Lgmn 10 ng/µl compared with vehicle, one-way ANOVA with Tukey's test, n = 5).
Discussion
We report that Lgmn is secreted from OSCC cells and is robustly and reproducibly activated in human and mouse OSCCs compared with normal mucosa. Under acidic conditions, Lgmn cleaves and activates PAR2 by biased mechanisms to evoke sustained hyperexcitability of nociceptors. We confirmed that PAR2 and Lgmn contribute to OSCC pain in OSCC mouse models that recapitulate the progression of OSCC observed in humans; genes for Lgmn and PAR2 on nociceptors were deleted in these mice. Lgmn contributes to cancer hallmarks including proliferation, invasion, and metastasis (Murthy et al., 2005; Vasiljeva et al., 2006; Li et al., 2013; Ohno et al., 2013; Edgington-Mitchell et al., 2015). While Lgmn has been reported to produce bone cancer pain through neurotrophin receptors (Yao et al., 2017), the role of the Lgmn/PAR2 axis has not been described and could be therapeutically exploited.
The mechanism responsible for Lgmn activation in oral cancer is unresolved. Lgmn is synthesized as pro-Lgmn and traffics through the endoplasmic reticulum and Golgi (Dall and Brandstetter, 2016). Lgmn is packaged and activated in the acidic environment of lysosomes (Dall and Brandstetter, 2016). At pH >6.0, acidic residues unfold and lose proteolytic activity. pH in cancers varies (5.4–6.7; Meyer et al., 1948; Vaupel et al., 1981; Newell et al., 1993; Gillies et al., 1994); the Lgmn activation mechanism remains obscure (Dall and Brandstetter, 2012). Exosomes released from OSCC might exhibit a pH low enough to activate Lgmn. A single report reveals acidic exosomes in cancer patients (Logozzi et al., 2019). Cells from human OSCCs, including the cell line used in this study (HSC-3), secrete exosomes (Dayan et al., 2012; Li et al., 2019). Stabilization between a RGD motif in the catalytic domain and the integrin αvβ3 might also activate Lgmn at a higher pH (Liu et al., 2012).
We demonstrated that Lgmn causes cancer-associated nociception through PAR2 activation on Nav1.8-expressing neurons. While all nociceptors express Nav1.8, some non-nociceptors, including low-threshold mechanoreceptors that mediate touch sensation, also express Nav1.8 (Shields et al., 2012). Since Lgmn activates cathepsins (Edgington-Mitchell et al., 2016), which can also activate PAR2 (Zhao et al., 2014), it is possible that Lgmn activates PAR2 directly or indirectly (Edgington-Mitchell et al., 2016). However, we found that Lgmn directly cleaves a fragment of hPAR2 at a unique Asn30↓Arg31 site, consistent with known Lgmn selectivity. Site mutation prevented Lgmn-evoked signaling, confirming this mechanism of proteolytic activation. Lgmn evoked hypersensitivity of TG neurons from WT mice (determined by patch clamp). These effects of Lgmn were prevented by a Lgmn inhibitor and absent in neurons from mice lacking PAR2, confirming necessity of PAR2 activation. The nociceptive behavior we measured accords with Lgmn-induced neuronal hypersensitivity. Lgmn induced nociceptive responses in anatomic regions innervated by DRG (paw) and TG (craniofacial) neurons; a Lgmn inhibitor and selective deletion of PAR2 in Nav1.8 neurons attenuated nociceptive responses. A Lgmn inhibitor eliminated chronic mechanical and thermal nociception in mice inoculated with OSCC cells.
OSCC patients complain of mechanical-induced and function-induced pain and not spontaneous pain (Connelly and Schmidt, 2004; Kolokythas et al., 2007). Our operant orofacial pain assay and automated device to perform the assay (dolognawmeter) quantifies a behavioral index of mechanical allodynia during gnawing (comparable to chewing in humans). Lgmn-induced mechanical allodynia and heat hyperalgesia in OSCC patients might involve PAR2 sensitization of TRPV4 and TRPV1 ion channels, respectively (Grant et al., 2007; Sipe et al., 2008). TRPV4 mediates mechanosensation, while TRPV1 responds to heat and acids (Caterina et al., 1999; Liedtke and Friedman, 2003; Liedtke et al., 2003; Suzuki et al., 2003a,b). TRPV4 and TRPV1 are sensitized by adenylyl cyclase-dependent, PKA-dependent, and PKCε-dependent mechanisms, which yield ion channel phosphorylation (Numazaki et al., 2002; Amadesi et al., 2006; Zhao et al., 2019). We showed that Lgmn cleavage of PAR2 activates adenylyl cyclase and cAMP formation; cAMP unleashes catalytic subunits of PKA, which subsequently phosphorylate TRPV channels. We also showed that Lgmn alters rheobase through PKC. Lgmn robustly activates PKD, which likely contributes to PAR2 trafficking from Golgi to plasma membrane (Zhao et al., 2019).
HEK293 cell experiments revealed that Lgmn mobilizes intracellular calcium, stimulates formation of cAMP, and activates PKD and ERK. Selective inhibitors of Lgmn abolished the calcium responses; we therefore infer that protease activity is necessary. Lgmn-evoked signals were detected only under mildly acidic conditions, consistent with the acidic pH optimum of Lgmn. We infer that Lgmn-evoked calcium signaling required cleavage/activation of PAR2 because PAR2 antagonism or deletion and mutation of the cleavage site abolished signals. PAR1 antagonism or deletion had no effect. Further studies are needed to reveal mechanisms by which Lgmn activated PAR2 signals to regulate channel activity and nociception. Trypsin activation of PAR2 involves exposure of a tethered ligand domain; peptides mimicking the tethered ligand activate the receptor (Hollenberg et al., 1996). Lgmn activation does not involve a tethered ligand; a synthetic peptide corresponding to the revealed N terminus was inactive. Trypsin-activated PAR2 recruits β-arrestins and then internalizes; Lgmn did not promote β-arrestin recruitment or receptor endocytosis. Thus, like cathepsin S and elastase (Zhao et al., 2014, 2015), Lgmn activates PAR2 by biased mechanisms to evoke pain.
Our findings are relevant for OSCC patients with pain. While the role of PAR2 in OSCC pain is clear, antagonism of PAR2 as a pain therapy approach is challenging. Access to the PAR2 binding pocket frustrates development of a clinically viable PAR2 antagonist (Goh et al., 2009; Suen et al., 2014; Boitano et al., 2015). Moreover, PAR2 continues to signal following cleavage and endocytosis (Jimenez-Vargas et al., 2018); however, we showed that Lgmn-cleaved PAR2 was not endocytosed. Furthermore, blockade of the Lgmn/PAR2 axis with a Lgmn inhibitor abrogates OSCC pain in mice. Accordingly, a pain therapy strategy that utilizes blockade of Lgmn is physiologically expedient and holds great clinical potential.
Footnotes
This work was supported by National Institutes of Health Grants NS102722, DE026806, DK118971, and DE029951 (to N.W.B., B.L.S.) and the Department of Defense Grant W81XWH1810431 (to N.W.B., B.L.S.). L.E.E.-M. was supported by the Priority-Driven Collaborative Cancer Research Grant GNT1157171 (co-funded by Cancer Australia and Cure Cancer), a Grimwade Fellowship from the Russell and Mab Grimwade Miegunyah Fund at The University of Melbourne, and the Australian Research Council Discovery Early Career Researcher Award Fellowship DE180100418.
N.W.B. is a founding scientist of Endosome Therapeutics Inc, and research in N.W.B.'s laboratory is partly supported by Takeda Pharmaceuticals International. J.C.D. fabricates dolognawmeter assay devices through Gnatheon Scientific LLC. All other authors declare no competing financial interests.
References
- Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M (2000) Inhibition of cellular action of thrombin by N3-cyclopropyl-7-[[4-(1-methylethyl)phenyl]methyl]-7H-pyrrolo[3, 2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochem Pharmacol 60:1425–1434. 10.1016/s0006-2952(00)00460-3 [DOI] [PubMed] [Google Scholar]
- Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, Bunnett NW (2006) Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 575:555–571. 10.1113/jphysiol.2006.111534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadesi S, Grant AD, Cottrell GS, Vaksman N, Poole DP, Rozengurt E, Bunnett NW (2009) Protein kinase D isoforms are expressed in rat and mouse primary sensory neurons and are activated by agonists of protease-activated receptor 2. J Comp Neurol 516:141–156. 10.1002/cne.22104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo PF, Lima AR, Alves FM, Blaber SI, Scarisbrick IA, Blaber M, Juliano L, Juliano MA (2006) Substrate specificity of human kallikrein 6: salt and glycosaminoglycan activation effects. J Biol Chem 281:3116–3126. 10.1074/jbc.M510096200 [DOI] [PubMed] [Google Scholar]
- Böhm SK, Khitin LM, Grady EF, Aponte G, Payan DG, Bunnett NW (1996a) Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem 271:22003–22016. 10.1074/jbc.271.36.22003 [DOI] [PubMed] [Google Scholar]
- Böhm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M, Nelken NA, Coughlin SR, Payan DG, Bunnett NW (1996b) Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J 314:1009–1016. 10.1042/bj3141009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitano S, Hoffman J, Flynn AN, Asiedu MN, Tillu DV, Zhang Z, Sherwood CL, Rivas CM, DeFea KA, Vagner J, Price TJ (2015) The novel PAR2 ligand C391 blocks multiple PAR2 signalling pathways in vitro and in vivo. Br J Pharmacol 172:4535–4545. 10.1111/bph.13238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D (1999) A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398:436–441. 10.1038/18906 [DOI] [PubMed] [Google Scholar]
- Connelly ST, Schmidt BL (2004) Evaluation of pain in patients with oral squamous cell carcinoma. J Pain 5:505–510. 10.1016/j.jpain.2004.09.002 [DOI] [PubMed] [Google Scholar]
- Corvera CU, Déry O, McConalogue K, Böhm SK, Khitin LM, Caughey GH, Payan DG, Bunnett NW (1997) Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J Clin Invest 100:1383–1393. 10.1172/JCI119658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvera CU, Déry O, McConalogue K, Gamp P, Thoma M, Al-Ani B, Caughey GH, Hollenberg MD, Bunnett NW (1999) Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J Physiol 517:741–756. 10.1111/j.1469-7793.1999.0741s.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Sun H, Tenner TE Jr, Machu TK (1999) Competitive antagonism of the mouse 5-hydroxytryptamine3 receptor by bisindolylmaleimide I, a “selective” protein kinase C inhibitor. J Pharmacol Exp Ther 290:76–82. [PubMed] [Google Scholar]
- Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K (2007) Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117:1979–1987. 10.1172/JCI30951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E, Brandstetter H (2012) Activation of legumain involves proteolytic and conformational events, resulting in a context- and substrate-dependent activity profile. Acta Crystallogr Sect F Struct Biol Cryst Commun 68:24–31. 10.1107/S1744309111048020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E, Brandstetter H (2016) Structure and function of legumain in health and disease. Biochimie 122:126–150. 10.1016/j.biochi.2015.09.022 [DOI] [PubMed] [Google Scholar]
- Dayan D, Salo T, Salo S, Nyberg P, Nurmenniemi S, Costea DE, Vered M (2012) Molecular crosstalk between cancer cells and tumor microenvironment components suggests potential targets for new therapeutic approaches in mobile tongue cancer. Cancer Med 1:128–140. 10.1002/cam4.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW (2000) Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 148:1267–1281. 10.1083/jcb.148.6.1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déry O, Thoma MS, Wong H, Grady EF, Bunnett NW (1999) Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-Arrestin-dependent endocytosis of a proteinase receptor. J Biol Chem 274:18524–18535. 10.1074/jbc.274.26.18524 [DOI] [PubMed] [Google Scholar]
- Deseure K, Koek W, Adriaensen H, Colpaert FC (2003) Continuous administration of the 5-hydroxytryptamine1A agonist (3-chloro-4-fluoro-phenyl)-[4-fluoro-4-[[(5-methyl-pyridin-2-ylmethyl)-amino]-methyl]piperidin-1-yl]-methadone (F 13640) attenuates allodynia-like behavior in a rat model of trigeminal neuropathic pain. J Pharmacol Exp Ther 306:505–514. 10.1124/jpet.103.050286 [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK (2007) Beta-arrestins and cell signaling. Annu Rev Physiol 69:483–510. 10.1146/annurev.physiol.69.022405.154749 [DOI] [PubMed] [Google Scholar]
- Dolan JC, Lam DK, Achdjian SH, Schmidt BL (2010) The dolognawmeter: a novel instrument and assay to quantify nociception in rodent models of orofacial pain. J Neurosci Methods 187:207–215. 10.1016/j.jneumeth.2010.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington LE, Verdoes M, Ortega A, Withana NP, Lee J, Syed S, Bachmann MH, Blum G, Bogyo M (2013) Functional imaging of legumain in cancer using a new quenched activity-based probe. J Am Chem Soc 135:174–182. 10.1021/ja307083b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington-Mitchell LE. (2016) Pathophysiological roles of proteases in gastrointestinal disease. Am J Physiol Gastrointest Liver Physiol 310:G234–G239. 10.1152/ajpgi.00393.2015 [DOI] [PubMed] [Google Scholar]
- Edgington-Mitchell LE, Rautela J, Duivenvoorden HM, Jayatilleke KM, van der Linden WA, Verdoes M, Bogyo M, Parker BS (2015) Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid-derived suppressor cells in breast cancer. Oncotarget 6:27008–27022. 10.18632/oncotarget.4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington-Mitchell LE, Wartmann T, Fleming AK, Gocheva V, van der Linden WA, Withana NP, Verdoes M, Aurelio L, Edgington-Mitchell D, Lieu T, Parker BS, Graham B, Reinheckel T, Furness JB, Joyce JA, Storz P, Halangk W, Bogyo M, Bunnett NW (2016) Legumain is activated in macrophages during pancreatitis. Am J Physiol Gastrointest Liver Physiol 311:G548–G560. 10.1152/ajpgi.00047.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer LJ. (2013) Imidazopyridazines useful as inhibitors of the PAR-2 signaling pathway. U.S. Patent Application WO 2015/048245.
- Gillies RJ, Liu Z, Bhujwalla Z (1994) 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. Am J Physiol 267:C195–C203. 10.1152/ajpcell.1994.267.1.C195 [DOI] [PubMed] [Google Scholar]
- Goh FG, Ng PY, Nilsson M, Kanke T, Plevin R (2009) Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br J Pharmacol 158:1695–1704. 10.1111/j.1476-5381.2009.00415.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, Lopez CB, Joseph EK, Levine JD, Liedtke W, Vanner S, Vergnolle N, Geppetti P, Bunnett NW (2007) Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 578:715–733. 10.1113/jphysiol.2006.121111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachem JP, Crumrine D, Fluhr J, Brown BE, Feingold KR, Elias PM (2003) pH directly regulates epidermal permeability barrier homeostasis, and stratum corneum integrity/cohesion. J Invest Dermatol 121:345–353. 10.1046/j.1523-1747.2003.12365.x [DOI] [PubMed] [Google Scholar]
- Hachem JP, Behne M, Aronchik I, Demerjian M, Feingold KR, Elias PM, Mauro TM (2005) Extracellular pH Controls NHE1 expression in epidermis and keratinocytes: implications for barrier repair. J Invest Dermatol 125:790–797. 10.1111/j.0022-202X.2005.23836.x [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M, al-Ani B (1996) Proteinase-activated receptor-2 in rat aorta: structural requirements for agonist activity of receptor-activating peptides. Mol Pharmacol 49:229–233. [PubMed] [Google Scholar]
- Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, Murphy JE, Alemi F, Cottrell GS, Korbmacher C, Lambert NA, Bunnett NW, Corvera CU (2013) The bile acid receptor TGR5 does not interact with β-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem 288:22942–22960. 10.1074/jbc.M113.455774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ (1999) Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci 2:1114–1119. 10.1038/16040 [DOI] [PubMed] [Google Scholar]
- Jiang Y, Yau MK, Lim J, Wu KC, Xu W, Suen JY, Fairlie DP (2018) A potent antagonist of protease-activated receptor 2 that inhibits multiple signaling functions in human cancer cells. J Pharmacol Exp Ther 364:246–257. 10.1124/jpet.117.245027 [DOI] [PubMed] [Google Scholar]
- Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, Herenbrink CK, Yeatman HR, Edgington-Mitchell L, Porter CJH, Halls ML, Canals M, Veldhuis NA, Poole DP, McLean P, Hicks GA, et al. (2018) Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci USA 115:E7438–E7447. 10.1073/pnas.1721891115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kembhavi AA, Buttle DJ, Knight CG, Barrett AJ (1993) The two cysteine endopeptidases of legume seeds: purification and characterization by use of specific fluorometric assays. Arch Biochem Biophys 303:208–213. 10.1006/abbi.1993.1274 [DOI] [PubMed] [Google Scholar]
- Kolokythas A, Connelly ST, Schmidt BL (2007) Validation of the University of California San Francisco oral cancer pain questionnaire. J Pain 8:950–953. 10.1016/j.jpain.2007.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DK, Schmidt BL (2010) Serine proteases and protease-activated receptor 2-dependent allodynia: a novel cancer pain pathway. Pain 149:263–272. 10.1016/j.pain.2010.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DK, Dang D, Zhang J, Dolan JC, Schmidt BL (2012) Novel animal models of acute and chronic cancer pain: a pivotal role for PAR2. J Neurosci 32:14178–14183. 10.1523/JNEUROSCI.2399-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Bogyo M (2010) Development of near-infrared fluorophore (NIRF)-labeled activity-based probes for in vivo imaging of legumain. ACS Chem Biol 5:233–243. 10.1021/cb900232a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhou Y, Liu J, Su X, Qin H, Huang S, Huang X, Zhou N (2019) Potential markers from serum-purified exosomes for detecting oral squamous cell carcinoma metastasis. Cancer Epidemiol Biomarkers Prev 28:1668–1681. 10.1158/1055-9965.EPI-18-1122 [DOI] [PubMed] [Google Scholar]
- Li N, Liu Q, Su Q, Wei C, Lan B, Wang J, Bao G, Yan F, Yu Y, Peng B, Qiu J, Yan X, Zhang S, Guo F (2013) Effects of legumain as a potential prognostic factor on gastric cancers. Med Oncol 30:621. 10.1007/s12032-013-0621-9 [DOI] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM (2003) Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA 100:13698–13703. 10.1073/pnas.1735416100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Tobin DM, Bargmann CI, Friedman JM (2003) Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci USA 100 [Suppl 2]:14531–14536. 10.1073/pnas.2235619100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu T, Savage E, Zhao P, Edgington-Mitchell L, Barlow N, Bron R, Poole DP, McLean P, Lohman RJ, Fairlie DP, Bunnett NW (2016) Antagonism of the proinflammatory and pronociceptive actions of canonical and biased agonists of protease-activated receptor-2. Br J Pharmacol 173:2752–2765. 10.1111/bph.13554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Bajjuri KM, Liu C, Sinha SC (2012) Targeting cell surface alpha(v)beta(3) integrin increases therapeutic efficacies of a legumain protease-activated auristatin prodrug. Mol Pharm 9:168–175. 10.1021/mp200434n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logozzi M, Capasso C, Di Raimo R, Del Prete S, Mizzoni D, Falchi M, Supuran CT, Fais S (2019) Prostate cancer cells and exosomes in acidic condition show increased carbonic anhydrase IX expression and activity. J Enzyme Inhib Med Chem 34:272–278. 10.1080/14756366.2018.1538980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews SP, Werber I, Deussing J, Peters C, Reinheckel T, Watts C (2010) Distinct protease requirements for antigen presentation in vitro and in vivo. J Immunol 184:2423–2431. 10.4049/jimmunol.0901486 [DOI] [PubMed] [Google Scholar]
- Meyer KA, Kammerling EM, Amtman L, Koller M, Hoffman SJ (1948) pH studies of malignant tissues in human beings. Cancer Res 8:513–518. [PubMed] [Google Scholar]
- Mikula KM, Tascón I, Tommila JJ, Iwaï H (2017) Segmental isotopic labeling of a single-domain globular protein without any refolding step by an asparaginyl endopeptidase. FEBS Lett 591:1285–1294. 10.1002/1873-3468.12640 [DOI] [PubMed] [Google Scholar]
- Murthy R, Xiong H, Nunez R, Cohen AC, Barron B, Szklaruk J, Madoff DC, Gupta S, Wallace MJ, Ahrar K, Hicks ME (2005) Yttrium 90 resin microspheres for the treatment of unresectable colorectal hepatic metastases after failure of multiple chemotherapy regimens: preliminary results. J Vasc Interv Radiol 16:937–945. 10.1097/01.RVI.0000161142.12822.66 [DOI] [PubMed] [Google Scholar]
- Ness KA, Eddie SL, Higgins CA, Templeman A, D'Costa Z, Gaddale KK, Bouzzaoui S, Jordan L, Janssen D, Harrison T, Burkamp F, Young A, Burden R, Scott CJ, Mullan PB, Williams R (2015) Development of a potent and selective cell penetrant Legumain inhibitor. Bioorg Med Chem Lett 25:5642–5645. 10.1016/j.bmcl.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Newell K, Franchi A, Pouyssegur J, Tannock I (1993) Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc Natl Acad Sci USA 90:1127–1131. 10.1073/pnas.90.3.1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M (2002) Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem 277:13375–13378. 10.1074/jbc.C200104200 [DOI] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Larsson AK, Strömbeck B, Sundelin J (1995) Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem 232:84–89. 10.1111/j.1432-1033.1995.tb20784.x [DOI] [PubMed] [Google Scholar]
- Ohlstein EH, Vickery L, Sauermelch C, Willette RN (1990) Vasodilation induced by endothelin: role of EDRF and prostanoids in rat hindquarters. Am J Physiol 259:H1835–H1841. 10.1152/ajpheart.1990.259.6.H1835 [DOI] [PubMed] [Google Scholar]
- Ohno Y, Nakashima J, Izumi M, Ohori M, Hashimoto T, Tachibana M (2013) Association of legumain expression pattern with prostate cancer invasiveness and aggressiveness. World J Urol 31:359–364. 10.1007/s00345-012-0977-z [DOI] [PubMed] [Google Scholar]
- Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I, Blaber M, Blaber SI, Scarisbrick I, Andrade-Gordon P, Cottrell GS, Bunnett NW, Diamandis EP, Hollenberg MD (2006) Proteinase-activated receptors, targets for kallikrein signaling. J Biol Chem 281:32095–32112. 10.1074/jbc.M513138200 [DOI] [PubMed] [Google Scholar]
- Ono K, Ye Y, Viet CT, Dang D, Schmidt BL (2015) TRPV1 expression level in isolectin B4-positive neurons contributes to mouse strain difference in cutaneous thermal nociceptive sensitivity. J Neurophysiol 113:3345–3355. 10.1152/jn.00973.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering V, Gupta JR, Quang P, Jordan RC, Schmidt BL (2008) Effect of peripheral endothelin-1 concentration on carcinoma-induced pain in mice. Eur J Pain 12:293–300. 10.1016/j.ejpain.2007.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, Hollenberg MD (2009) Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol 76:791–801. 10.1124/mol.109.055509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff NN, Bhattacharya A, Dowse E, Dang RX, Dolan JC, Wang S, Kim H, Albertson DG, Schmidt BL (2018) Neutrophil-mediated endogenous analgesia contributes to sex differences in oral cancer pain. Front Integr Neurosci 12:52. 10.3389/fnint.2018.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BL, Pickering V, Liu S, Quang P, Dolan J, Connelly ST, Jordan RC (2007) Peripheral endothelin A receptor antagonism attenuates carcinoma-induced pain. Eur J Pain 11:406–414. 10.1016/j.ejpain.2006.05.007 [DOI] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD (2012) Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 153:2017–2030. [DOI] [PubMed] [Google Scholar]
- Sipe WE, Brierley SM, Martin CM, Phillis BD, Cruz FB, Grady EF, Liedtke W, Cohen DM, Vanner S, Blackshaw LA, Bunnett NW (2008) Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol 294:G1288–G1298. 10.1152/ajpgi.00002.2008 [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW (2000) Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 6:151–158. 10.1038/72247 [DOI] [PubMed] [Google Scholar]
- Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, Fairlie DP (2012) Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110). Br J Pharmacol 165:1413–1423. 10.1111/j.1476-5381.2011.01610.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen JY, Cotterell A, Lohman RJ, Lim J, Han A, Yau MK, Liu L, Cooper MA, Vesey DA, Fairlie DP (2014) Pathway-selective antagonism of proteinase activated receptor 2. Br J Pharmacol 171:4112–4124. 10.1111/bph.12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M (2003a) Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278:22664–22668. 10.1074/jbc.M302561200 [DOI] [PubMed] [Google Scholar]
- Suzuki M, Watanabe Y, Oyama Y, Mizuno A, Kusano E, Hirao A, Ookawara S (2003b) Localization of mechanosensitive channel TRPV4 in mouse skin. Neurosci Lett 353:189–192. 10.1016/j.neulet.2003.09.041 [DOI] [PubMed] [Google Scholar]
- Ungefroren H, Witte D, Mihara K, Rauch BH, Henklein P, Jöhren O, Bonni S, Settmacher U, Lehnert H, Hollenberg MD, Kaufmann R, Gieseler F (2017) Transforming growth factor-β1/activin receptor-like kinase 5-mediated cell migration is dependent on the protein proteinase-activated receptor 2 but not on proteinase-activated receptor 2-stimulated Gq-calcium signaling. Mol Pharmacol 92:519–532. 10.1124/mol.117.109017 [DOI] [PubMed] [Google Scholar]
- van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, Schouten HC, van Kleef M, Patijn J (2007) Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol 18:1437–1449. 10.1093/annonc/mdm056 [DOI] [PubMed] [Google Scholar]
- Vasiljeva O, Papazoglou A, Krüger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, Peters C, Reinheckel T (2006) Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res 66:5242–5250. 10.1158/0008-5472.CAN-05-4463 [DOI] [PubMed] [Google Scholar]
- Vaupel PW, Frinak S, Bicher HI (1981) Heterogeneous oxygen partial pressure and pH distribution in C3H mouse mammary adenocarcinoma. Cancer Res 41:2008–2013. [PubMed] [Google Scholar]
- Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, Cirino G, Gerard N, Basbaum AI, Andrade-Gordon P, Hollenberg MD, Wallace JL (2001) Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat Med 7:821–826. 10.1038/89945 [DOI] [PubMed] [Google Scholar]
- Yamano S, Viet CT, Dang D, Dai J, Hanatani S, Takayama T, Kasai H, Imamura K, Campbell R, Ye Y, Dolan JC, Kwon WM, Schneider SD, Schmidt BL (2017) Ex vivo nonviral gene delivery of μ-opioid receptor to attenuate cancer-induced pain. Pain 158:240–251. 10.1097/j.pain.0000000000000750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao P, Ding Y, Han Z, Mu Y, Hong T, Zhu Y, Li H (2017) Suppression of asparaginyl endopeptidase attenuates breast cancer-induced bone pain through inhibition of neurotrophin receptors. Mol Pain 13:1744806917708127. 10.1177/1744806917708127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarwood RE, Imlach WL, Lieu T, Veldhuis NA, Jensen DD, Klein Herenbrink C, Aurelio L, Cai Z, Christie MJ, Poole DP, Porter CJH, McLean P, Hicks GA, Geppetti P, Halls ML, Canals M, Bunnett NW (2017) Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc Natl Acad Sci USA 114:12309–12314. 10.1073/pnas.1706656114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Dang D, Zhang J, Viet CT, Lam DK, Dolan JC, Gibbs JL, Schmidt BL (2011) Nerve growth factor links oral cancer progression, pain, and cachexia. Mol Cancer Ther 10:1667–1676. 10.1158/1535-7163.MCT-11-0123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Bae SS, Viet CT, Troob S, Bernabé D, Schmidt BL (2014a) IB4(+) and TRPV1(+) sensory neurons mediate pain but not proliferation in a mouse model of squamous cell carcinoma. Behav Brain Funct 10:5. 10.1186/1744-9081-10-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Ono K, Bernabé DG, Viet CT, Pickering V, Dolan JC, Hardt M, Ford AP, Schmidt BL (2014b) Adenosine triphosphate drives head and neck cancer pain through P2X2/3 heterotrimers. Acta Neuropathol Commun 2:62. 10.1186/2051-5960-2-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, Kocan M, Sostegni S, Haerteis S, Baraznenok V, Henderson I, Lindström E, Guerrero-Alba R, Valdez-Morales EE, Liedtke W, McIntyre P, Vanner SJ, Korbmacher C, Bunnett NW (2014) Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem 289:27215–27234. 10.1074/jbc.M114.599712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, Bunnett NW (2015) Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem 290:13875–13887. 10.1074/jbc.M115.642736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Pattison LA, Jensen DD, Jimenez-Vargas NN, Latorre R, Lieu T, Jaramillo JO, Lopez-Lopez C, Poole DP, Vanner SJ, Schmidt BL, Bunnett NW (2019) Protein kinase D and Gβγ mediate sustained nociceptive signaling by biased agonists of protease-activated receptor-2. J Biol Chem 294:10649–10662. 10.1074/jbc.RA118.006935 [DOI] [PMC free article] [PubMed] [Google Scholar]