

Abstract
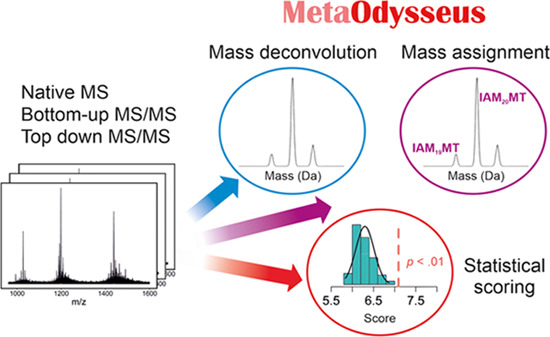
Identification of metal-binding sites in proteins and understanding metal-coupled protein folding mechanisms are aspects of high importance for the structure-to-function relationship. Mass spectrometry (MS) has brought a powerful adjunct perspective to structural biology, obtaining from metal-to-protein stoichiometry to quaternary structure information. Currently, the different experimental and/or instrumental setups usually require the use of multiple data analysis software, and in some cases, they lack some of the main data analysis steps (MS processing, scoring, identification). Here, we present a comprehensive data analysis pipeline that addresses charge-state deconvolution, statistical scoring, and mass assignment for native MS, bottom-up, and native top-down with emphasis on metal–protein complexes. We have evaluated all of the approaches using assemblies of increasing complexity, including free and chemically labeled proteins, from low- to high-resolution MS. In all cases, the results have been compared with common software and proved how MetaOdysseus outperformed them.
Keywords: metalloprotein, zinc, Cys-rich, mass spectrometry, R package
Introduction
Mass spectrometry (MS) has become a powerful approach for structural and physicochemical characterization of metal–protein complexes.1−11 Among all amino acid residues responsible for d-block metal-ion binding, cysteine (Cys) is the most frequent residue in structural sites building and stabilizing the protein structure.12 It participates in many catalytic and chemical/posttranslational reactions explained by its high nucleophilic character. The identification of redox Cys state and metal coordination position has been a subject of many research focusing on the structural characterization of metalloproteins and mechanisms of their folding.6,12,13 Of all techniques used for that purpose, electrospray ionization mass spectrometry (ESI-MS) is currently one of the most informative. Particularly, native ESI-MS has become an indispensable tool for the speciation of metal–protein complexes, especially when studying spectroscopic silent metal ions (e.g., Zn2+).1−3,5,7,10,11 In around 10% of the proteome, Zn2+ ions are playing catalytic, structural, and regulatory roles.2,12−16 Cysteine chemical labeling, improved ESI sources together with the recent advances in the transmission technology of Orbitrap and time-of-flight (TOF) mass analyzers have permitted the elucidation of the Cys oxidation state in the native-like structure.2−6,14,17 The different reactivity between a free Cys residue and metal-binding Cys enables its identification with the use of an appropriate electrophile.14
A great example of a protein class, which is intensively studied in terms of their structure and transient species is the metallothionein (MT) family, whose representatives were used in this study.1−4,11,14 In particular, mammalian MTs are a low molecular mass (∼6 kDa) involved in the homeostatic control of Zn2+ and Cu+ ions, controlling metal fluctuations in the cytosol, nucleus, and mitochondria.18−20 They contain at least a dozen of MT proteins, and they represent one of the major cellular zinc buffering system.21
The process of data analysis involves three steps. (i) Preprocessing raw mass spectra thorough baseline subtraction, smoothing, and peak picking. The spectra should be noise-filtered while preserving all of the information. (ii) Charge deconvolution. All of the peak series have to be identified, and the correct charge state should be assigned. (iii) Identification of the deconvoluted mass with a peptide/protein search engine. The identifications need to be accompanied by a scoring system. Certainly, a great number of deconvolution algorithms have been developed, mainly classified into peak assignment or fitting algorithms. Peak assignment algorithms are based on performing a peak detection and assign a charge state to each peak using multiple charge states (e.g., MaxEnt,22,23 AutoMass,24Z-score25) or isotope peak spacing (e.g., THRASH,26Z-score25). Simulation algorithms simulate multiple hypothetical mass and charge distributions, and that mass-charge distribution that fits the data best is taken to be the most correct (e.g., CHAMP,27 Massign,28 UniDec,29 PeakSeeker30).
While peak assignment constitutes fast and simple algorithms, often are not suitable for complex spectra because of the difficulty in peak picking, and do not produce quantitative results. On the other hand, simulation algorithms are quantitative but computationally intensive. Regardless of the algorithm used for ESI deconvolution, the quality of the results is judged with a scoring system that is usually algorithm-dependent. Recently, a universal score (UniScore) based on the evaluation of critical steps during deconvolution has been released, promoting an unbiased scoring system to compare between multiple deconvolution algorithms.31
Protein/peptide database search and statistical modeling tools are well established in peptide-based bottom-up proteomics. Looking at finding new peptide-spectrum matches (PSMs) or integrating searching engine with scoring models has resulted in a variety of software, in addition to the commercial offerings Mascot32 or SEQUEST.33 This includes freely available programs such as MetaMorpheus,34 MS-GF+,35 MaxQuant,36 OMSSA,37 or X!Tandem.38 Commonly, bottom-up MS/MS spectra are scored without charge deconvolution.
Nevertheless, protein-based top-down MS generates a high degree of complexity that presents a challenge for data analysis, more pronounced for native top-down MS.3,39 The sequential pipeline analysis requires a peak picking, charge deconvolution, proteoform identification, and quantification steps. However, most of the software’s available focus is exclusively on each step of the pipeline, and scarce are those that include all steps.40 For instance, algorithms for charge deconvolution are based on isotope spacing, including MS-Deconv41 or TopFD42 in addition to the first high-resolution top-down deconvolution algorithm THRASH,26 and commercially included SNAP43 algorithm by Bruker Daltonics and the Xtract44 algorithm by Thermo Scientific. Following mass spectra deconvolution, common data analysis tools for identification are ProSight PTM,45 MS-TopDown,46 MS-Align+,47 or TopPIC.42 A tool that incorporates all of the steps is MASH Suite Pro,48 which accommodates enhance-THRASH, MS-Deconv,41 and TopFD42 algorithms for spectral deconvolution and MS-Align+47 or TopPic42 for database searching.
Here, to fulfill the deficits encountered for the available MS-based software, we developed MetaOdysseus, a comprehensive data analysis pipeline in R language that integrates native MS, bottom-up and native top-down protein analysis with special focus on metal–protein complexes. The characterization of metal–protein complexes, involving determination of the metal–protein stoichiometry, identification of metal-binding sites, or characterization of metal-coupled folding events, requires combining multiple MS-based approaches.2,3,7,14
Although much effort has been put in to develop new proteomic software, less attention has been paid to integrate native MS and proteomics into a single software. Moreover, the available common software presents deficits for the data analysis of metal–protein complexes at the peptide and intact protein levels. For native MS analysis of intact protein complexes, although many deconvolution algorithms have been released, still a global pipeline for the data analysis is missing and thus, we incorporated: several preprocessing schemes; two deconvolution algorithms, one based on peak assignment and other on mass spectra simulation; the UniScore metrics promoting the global evaluation of the deconvolution; and a targeted mass assignment engine. We demonstrated how deconvolution results (measured as the UniDec metric) obtained with MetaOdysseus agreed closely with the UniDec algorithm. For peptide-level MS analysis, MetaOdysseus surpassed Mascot and MS-GF+, providing a higher number of PSMs, and equal or higher sensitivity at the same false discovery rate (FDR) and at every precision value. For native top-down analysis, we compared deconvolution results obtained with MetaOdysseus and with the eTHRASH algorithm included in the MASH Suite pro and demonstrated that MetaOdysseus yielded a higher true positive rate and a lower false-negative rate. To benchmark the identification issue, we compared our software with two common free tools for matching a single-candidate sequence against MS spectra: ProSight Lite45 and MS-Align+. While ProSight Lite requires the definition of the exact position of PTMs, MS-Align+ identifies unexpected PTMs and thus appears capable of top-down analysis of metalloproteins. However, there are two major limitations that hamper its use for it: (i) the Cys protection parameters are fixed, with or without modification and only allows two types of modification, carbamidomethylation or carboxymethylation. Many research studies focused on metal-binding proteins used other reagents for Cys modification and, in the case of metal-binding Cys proteins, incomplete Cys modification is commonly observable.3,4,6 (ii) The maximum number of unexpected or blind PTMs is two, which limits a proteoform to bind a maximum of two metal ions and excludes its analysis for a large part of metalloproteins. Although both ProSight Lite and MetaOdysseus provided similar identification, MetaOdysseus is a time-saving solution that runs automatically in comparison to the required manual pinpointing with ProSight Lite. Altogether, this research shows that MetaOdysseus provides a global solution for the analysis of metal–protein complexes.
Materials and Methods
Overview of Software
The software presented herein was implemented in R programming language and incorporates a set of algorithms for processing, charge-state deconvolution, target mass assignment, and statistical evaluation for different MS experiments (native MS, bottom-up, and native top-down) and ionization sources (ESI and MALDI). File format. The MS raw data or preprocessed data (centroided and/or smoothed and background removed) can be imported as xy, mzXML, or mzML with or without preprocessing (smoothing, centroiding, background removal).
Preparation of Mass Spectra (Optional)
The mass spectra are smoothed and the background is removed with asymmetric least squares. For smoothing, we incorporated three algorithms: (1) Savitzky–Golay filters;49 (2) finite-difference approach;50 and (3) discrete wavelet transform.51
Peak Detection (Optionally)
Here, we performed a peak detection with a continuous wavelet transform-based peak detection algorithm.52 It convolves the mass spectra with a Mexican Hat wavelet and the true peaks are registered as local maxima.
Charge-State Deconvolution
For ESI-MS spectrum where multiple charge states for each specie is expected, we focused on addressing the problem of assigning mass series through two algorithms, one based on peak assignment adapted from the Z-score25 algorithm, and the other based on simulation spectra. The peak assignment algorithm can be used for high-resolution mass spectra, and thus, a mass with only one charge state can be also deconvoluted. The simulation algorithm was incorporated to account for peaks with lower resolution that are widening due to the incomplete desolvation or adduct formation, common observations of native mass spectra.30
Mass Assignment
A theoretical in silico mass pattern is generated from the protein sequence, and with a simple cross-correlation mass-to-mass matching algorithm, we solve the mass assignment problem. For the generation of theoretical masses, we added features addressing metalloprotein studies, that is, the incorporation of several Cys-labeling reagents and metal ions with partial labeling modification.
Statistical Scoring
Here, a scoring scheme was incorporated for each MS experiment to evaluate the deconvolution results and/or the mass assignment. For native MS, we integrated the UniScore for the simulation algorithm that evaluates the quality of the deconvolution with a universal score that is software-independent.31 After the mass assignment, each feature is reported with their scored mass error, DScore, UScore, FScore, CCScore, and MScore along with the global UniScore. For MALDI-MS analysis of enzymatically digested proteins (peptide-mass fingerprinting, PMF), a PMF score is computed based on similarity descriptors,53 and its confidence interval is calculated with bootstrapping. An empirical p-value is computed producing a list of decoy protein sequences via permutation tests and calculating a histogram of decoy PMF after interrogation against the experimental spectrum. For bottom-up MS/MS, the quality of the single peptide-spectrum matches (PSMs) is evaluated with a peak match probability scored based on computing probabilities from a binomial distribution and the probability is log-transformed. The problem of multiple PSMs is addressed with the estimation of the false discovery rate using the target-decoy approach (TDA).54 We used a conservative TDA approach using a separate target-decoy database search and building the decoy database with a randomized target protein sequence. For native top-down MS/MS, the quality of the charge-state deconvolution algorithm based on peak assignment was defined as the total score S defined as ∑sm/z, where sm/zis the (log(SNR))m/z for each charge state zi. A single-spectrum confidence score of the mass assignment is computed as the p-value obtained via permutation tests for the probability log-transformed from a binomial distribution. In addition to the summary given below, a detailed description of each step is provided in the Supporting Information.
Mass Spectrometry Measurements
ESI-MS analysis was performed on a Bruker Maxis Impact (Bruker Daltonik GmbH, Bremen, Germany) calibrated with a commercial ESI-TOF Tuning mix (Sigma-Aldrich). Proteins were buffer-exchanged to 20 mM ammonium acetate using PD-10 desalting columns (Sigma-Aldrich). Protein concentrations were adjusted to 5–15 μM and directly injected by a syringe pump with a 2 μL/min flow rate. The following MS parameters were used for direct injection electrospray ionization-mass spectrometry: collision energy, 10 eV; collision RF 600 Vpp endplate offset potential, 500 V; capillary potential, 2500 V; nebulizer gas, (N2); pressure, 1.5 bar; drying gas (N2) flow rate, 4 L/min; drying temperature, 180 °C. The transfer parameters were: funnel 1 RF, 400 Vpp; isCID energy, 0 eV; multiple RF, 400 Vpp. The quadrupole ion energy was 5 eV, and the low mass for transmission was set at m/z 300. The mass range was set from 50 to 3000 m/z. Native top-down experiments were performed with a data-dependent MS/MS acquisition (DDA) with auto MS/MS with a collision energy of 200 eV, or with a data-independent acquisition (DIA) using broad-band CID (bbCID) with increasing collision energy of 50–200 eV. A MALDI-TOF/TOF MS Bruker UltrafleXtreme instrument (Bruker Daltonik GmbH, Bremen, Germany) was used for MALDI-MS analysis. MALDI instrument was controlled by flexControl v 3.4 and flexAnalysis v 3.4 software. 2,5-Dihydroxybenzoic acid (DHB) was used as MALDI-TOF matrix for protein analysis. The saturated matrix solution was prepared in 30% acetonitrile and 0.1% trifluoroacetic acid. MALDI-MS analysis of proteins was performed in a linear positive mode in the 2–20 kDa range. The mass spectra were typically acquired by averaging 2000 subspectra from a total of 2000 laser shots per spot. Laser power was set at 5–10% above the threshold. The calibration was done using standard peptide and protein calibration mixture obtained from Bruker (Bruker Daltonik GmbH, Bremen, Germany).
Mass Spectrometry Data
Several MS datasets were obtained from different mass spectrometers to demonstrated the MetaOdysseus’s capabilities: (1) native MS, native top-down MS/MS and bottom-up MS/MS data acquired in a regular quadrupole time-of-flight (Bruker Maxis Impact); (2) external native MS data acquired in an Orbitrap Eclipse tribrid by Robinson’s group;55 (3) peptide-mass fingerprinting with a MALDI-TOF/TOF MS Bruker UltrafleXtreme instrument.
Cysteine Residues Labeling, Reconstitution and Trypsin Digestion of Metallothionein
Freshly SEC-purified apo-MT proteins (25 μM) were incubated with freshly prepared 10 mM alkylator (IAM, IAA, NEM, ethyl iodoacetate) for 15 min in the dark in 100 mM (NH4)2CO3. The alkylation reaction was stopped by dry ice. All solutions and plastic tubes were previously degassed by purging with nitrogen. To eliminate the excess of alkylation reagents, samples were desalted with ZipTip μ-C18 (Merck Millipore) and eluted with 10 μL of Milli-Q water/ACN solution (50/50, v/v). To obtain Zn7MT, an appropriate molar equivalent of 500 μM ZnSO4 over particular apo-MT was added. In solution trypsin digestion was carried out with proteomics-grade trypsin (Sigma-Aldrich) freshly prepared in 1 mM HCl at a 1:20 trypsin/protein (w:w) ratio at 37 °C for 15 min. The reaction was stopped by freezing the sample on dry ice. Aliquots were immediately analyzed or stored at −20 °C.
Expression and Purification of Metallothionein Proteins
MT1e, MT2, and MT3 were expressed and purified as previously described and are detailed in the Supporting Information.(2,56) Designed pTYB21 plasmids encoding MT1e, MT2, and MT3 were deposited in Addgene plasmid repository (https://www.addgene.org) as plasmid #105702, plasmid #105693, and plasmid #105710, respectively.
Data Availability
All data generated or analyzed during this study and an R script that follows the results section are deposited in https://github.com/ManuelPerisDiaz/Data_MetaOdysseus. The MetaOdysseus R package is deposited in GitHub and can be freely downloaded from the following link: https://github.com/ManuelPerisDiaz/MetaOdysseus.
Results and Discussion
Case 1: Deconvolution and Assignment of ESI-MS Spectra
We benchmarked MetaOdysseus here with the application to systems of increasing complexity, from isotopically resolved single protein to an unresolved peak distribution mix of proteins: (1) high- and low-resolution native ESI-MS spectra for ∼6 kDa metal-binding proteins apo-MT2 and Zn7MT2; (2) high- and low-resolution native ESI-MS spectra for chemically labeled Cys-iodoacetamide (IAM) or Cys-labeled with N-ethylmaleimide (NEM) Zn7MT2; (3) external dataset of native MS spectra of 17–175 kDa soluble proteins (myoglobin, BSA, ADH, and Herceptin) analyzed with an Orbitrap Eclipse tribrid.55 The R-based software may be used sequentially for processing, charge-state deconvolution, and mass assignment of mass spectra, or it may independently use any of its modules. For example, deconvolution can be performed externally, and the mass assignment problem solved within MetaOdysseus. A qualitative comparison of the preprocessing algorithms incorporated in MetaOdysseus is exemplified for low-resolution apo-MT2, which showed that the finite-difference penalty followed by asymmetric least squares provided the mass spectra with the most reduced background noise (Figure S2). Subsequently, deconvolution was successfully obtained in all of the cases, fitting the simulated component spectra to the experimental one with R2 ranging from 0.85 to 0.99 (Tables S1 and S2). However, as it has been demonstrated, R2 is not a proper statistic to evaluate the quality of the fit because of the potential for overfitting the raw data. Thus, another scoring metric is required to confidently assess the quality of the deconvolution. Here, we incorporated the recently presented universal score (UniScore),31 defined as the weighted average of the deconvolution score (DScore) for each mass. DScore is calculated with four components that can capture the fit of each charge state (UScore), peak shape (MScore), gaussian charge-state distribution (CSScore), and the symmetry and resolution of the peaks (Fscore). As it can be seen, the UniScore obtained varied widely from 38 to 97, in comparison to the narrower distribution of R2 values. Not only the broadening of the UniScore distribution but also the low correlation (Pearson’s correlation coefficient 0.22) between R2 and the UniScore indicates that both metrics provide complementary information and that R2 tends to overfit. Deconvolution results obtained with the simulation approach incorporated in MetaOdysseus were compared with the UniDec algorithm.
In the second step, once the mass spectra were deconvolved, the masses and charges of the components were assigned. To do so, masses deconvoluted from the zero-charge mass distribution were interrogated against a constructed theoretical ion library based on the protein sequence. For every mass deconvolved from the mass spectra, DScore, UScore, MScore, CSScore, and Fscore are reported along with the identifications with its mass error (Table S3). To give an example, the most intensive deconvoluted mass for the apo-MT2 sample was assigned as apo-MT2, and the mass deconvoluted has a DScore of 0.75 and a global UniScore of 69 (Tables S1, S3, and Figure S3). The charge-state series of this component showed [apo-MT2 + zH]z+ with z = 4–6 (Figures S4 and S3). The high UScore of 0.90 indicated that the signals corresponding to these charge states are mostly captured by the deconvoluted mass and do not overlap with adjacent charge states. However, the points lost from the MScore indicate fluctuations in the peak shape between the different charge states. The CSScore and FScore of 100 indicate that the charge-state distributions are well separated and are symmetric. Another example is the high-resolution nMS of Zn7MT2 after reaction with the Cys-labeling reagent IAM (Figure 1A). The deconvolved neutral mass spectrum was scored with a UniScore of 62, and two masses were assigned as Zn0IAM19MT2 and Zn0IAM20MT2 with DScore values of 68 and 63, respectively (Figure 1B). In general, the distribution of R2 and UniScore for both software agrees closely, as we did not find differences in the mean (p-value (Wilcoxon test) 0.4 and 0.98, respectively) (Figure 1C,D and Table S4). These results demonstrate that MetaOdysseus provides a quantitative deconvolution and an accurate mass assignment algorithm for low- and high-resolution native MS.
Figure 1.

High-resolution MS for a Zn7MT2 IAM Cys-labeled (A) and the deconvolved neutral mass spectrum (B) with assigned DScores above each peak and the global UniScore. Comparison between MetaOdysseus and UniDec for the R2 (C) and UniScore (D) values obtained.
Case 2: Deconvolution and Mass Assignment of MALDI-MS Spectra
The second case approached is the peak picking of linear and reflector mode of MALDI-TOF spectra and the mass assignment problem. Two experiments were used to evaluate the software: (1) analysis of intact and chemically Cys-labeled apo- and Zn7MT2 proteins with a set of common alkylators (iodoacetic acid (IAA), NEM, IAM, and ethyl iodoacetate (ET)) and (2) peptide-mass fingerprinting (PMF) of 10 proteins (apo-MT2, Zn4-7MT2, apo-MT3, and Zn4-7MT3) after enzymatic digestion. The preprocessing algorithms included clearly removed the background and baseline fluctuations common for MALDI ion source and detected the true signals57 (Figure S5).
For the first experiment, the mass assignment algorithm identified a variable range of Cys-labeled residues for apo-MT2. The extent of the modification obtained under experimental conditions showed the following order: IAM (19–20 M, where M denotes modifications), NEM (16–20 M), ethyl iodoacetate (2–9 M), and IAA (0–4 M) (Table S5 and Figure S6a–d). As expected, there is a different extent and statistical distribution for the alkylation reaction, with different populations of modifications.2,4 For example, the hydrophilic character and small size of IAM result in possible access to those Cys residues that are buried in the compact native structure. In the case of Zn7MT2, most of the Cys residues (up to 13 out of 20) were protected by the Zn(Cys)4 coordination environment toward alkylation by the nucleophiles ethyl iodoacetate, IAA and IAM (Table S5 and Figure S6e–h). However, the use of NEM in high excess resulted in a large number of modifications (16–20 M) with an alkylation profile identical to the one obtained for apo-MT2 (Figure S6b,f).
In the second experiment, after mass deconvolution, the mass assignment algorithm produced a list of peptide masses and their mathematically possible Cys modifications. Tryptic peptides were identified with multiple IAM modifications (Table S6). For Cys-rich metalloproteins, likely the peptide generated harbors multiple Cys residues. For example, three peptides from the region [32–43] for apo-MT2 were identified, 5M [32–44], 4M [32–44], and 2M [32–44] (nM, where n indicates the number of modifications) (Table S6). The software positively identified peptide masses with isotopically well-resolved patterns as well as could identify low abundant signals from the background (Figure S7).
The quality of the deconvoluted and mass assigned mass spectra for all 10 datasets was assessed with a global PMF score,53 and its confidence interval was estimated with bootstrapping. The correlogram showed a low correlation between the mass coverage (MC) and the hit ratio (p < 0.005), and thus, in this case, the PMF score is mostly correlated by the hit ratio (Figure S8 and Table S7). For instance, a higher PMF score was obtained for Zn5MT3 than Zn4MT3, even though the latter one achieved higher mass coverage. To validate a particular PMF score, we constructed a list of decoy sequences via the permutation of the protein sequence and computing a p-value (Figure S9). In any case, the PMF score was statistically significant (p < 0.005).
To compare the performance obtained with MetaOdysseus, we processed the datasets within BioTools software (Bruker Daltonics), performing a peak picking using averaging-based peak finding SNAP algorithm and a targeted database searching against the protein sequence with Sequence Editor software (Bruker Daltonics). Then, we calculated a confusion matrix for each dataset. Using all 10 datasets, we observed how MetaOdysseus obtained higher accuracy and precision with lower FDR (p < 0.05) (Figure 2). Moreover, we did not find differences in the F1 score, suggesting MetaOdysseus performance equal to or better than BioTools.
Figure 2.

Comparison between MetaOdysseus and Bruker Daltonics for the analysis of enzymatically digested for Zn0-7MT2 and Zn0-7MT3 proteins analyzed with MALDI-MS; * stands for a level of significance 0.05, n.s stands for no significance level for the mean comparison between the groups, FDR and F1 stand for false discovery rates and Fscore, respectively.
Case 3: Deconvolution and Assignment of MS/MS Spectra
Bottom-Up Approach
A dataset of MS/MS spectra collected for enzymatically digested Zn0-7MT3 proteins was employed to demonstrate how MetaOdysseus solves the spectrum matching problem for peptide identification from the bottom-up approach. Given a set MS/MS spectrum (40) and a sequence database, we were able to obtain a 90% (36 out 40) of peptide-spectrum match (PSM) (Table S8). To evaluate the quality of the PSMs, a simple peak match probability score based on computing probabilities from a binomial distribution was generated for each mass spectrum. Accounting for false-positives filtering by FDR threshold (1%) estimated with a target-decoy method resulted in 33% of PSMs (Table S9 and Figure S10). We investigated how MetaOdysseus compares to Mascot and MS-GF+, in terms of their percentage of PSMs, sensitivity, specificity, and precision. PSMs (80 and 70%) were achieved by MS-GF+ and Mascot, which after filtering by 1% FDR remained with 15 and 10%, respectively (Figure 3 and Table S10). We found that at every precision value for correct peptide identification, MetaOdysseus provided an increase in sensitivity over MS-GF+ and Mascot (Figure 4A). The receiver-operating characteristic (ROC) analysis at different FDR cutoffs revealed that MetaOdysseus provided at least equal sensitivity to MS-GF+ for different FDRs and better sensitivity in all cases in comparison to Mascot (Figure 4B). MetaOdysseus achieved the highest accuracy calculated as the area under the curve (AUC) with values of 0.80, 0.68, and 0.50 for MetaOdysseus, MS-GF+, and Mascot, respectively. The highest sensitivity obtained with MetaOdysseus is remarkable at low FDR thresholds (Figure 4C). MetaOdysseus can be also used only for identification, importing already preprocessed mass spectra. Since MS-GF+ only works for identification without peak picking step, we evaluated the identification ability of MetaOdysseus and MS-GF+ with an already preprocessed set of mass spectra. We have observed how MetaOdysseus achieved equal or higher sensitivity at the same FDR (Figure S11A). The precision–recall curve showed that the sensitivity achieved with MetaOdysseus was similar or higher at every precision value (Figure S11B). The ROC curves showed that for a particular range of specificity, MS-GF+ is more sensitive; however, a similar accuracy measured as AUC (0.68) was obtained for both software (Figure S11C). In terms of the number of PSMs, 24% against 15% obtained with MS-GF+ was achieved by MetaOdysseus.
Figure 3.

Relationship between the peptide-spectrum matches (PSMs) and the false discovery rate (FDR) obtained for the different software compared (MetaOdysseus, MS-GF+, and Bruker Daltonics) for the bottom-up MS analysis.
Figure 4.
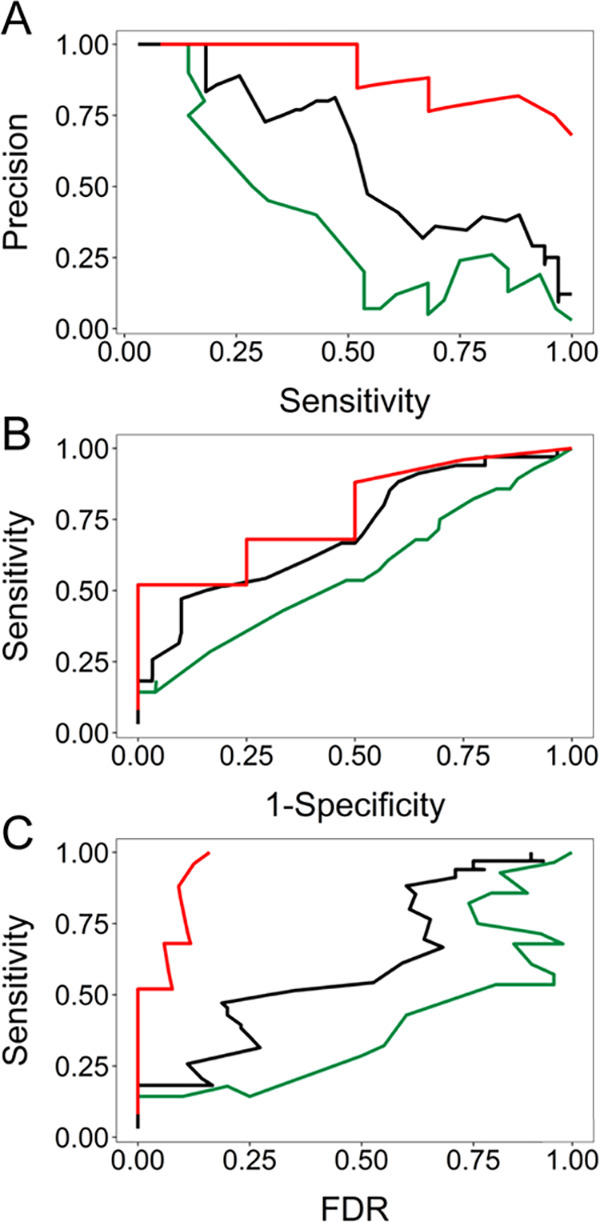
Comparison between MetaOdysseus (red line), MS-GF+ (black line), and Mascot (green line) for the peptide-spectrum matches (PSMs) results obtained in terms of their sensitivity, specificity, and precision. (A) Precision–recall curves for the different FDR cutoffs. (B) Receiver-operating characteristic (ROC) analysis at different FDR cutoffs. (C) Analysis of the sensitivity achieved at different FDR cutoffs obtained.
Native Top-Down Protein Characterization
Here, we report how MetaOdysseus solves the assignment process of a given set of MS/MS spectra of Zn7MT1e obtained by collision-induced dissociation experiments using different collisional activation energies. In general, the presence of the seven Zn2+ coordinating all of the 20 Cys residues provides a folded and stable structure. Mass deconvolution returned the charge states and masses for the metal–protein complexes present in the mass spectra (Table S11). Results were compared to the eTHRASH algorithm incorporated in the MASH suite pro, considering as a reference method the manual inspection of the data (Table S12). Deconvolution with MetaOdysseus provided a higher number of true positives (58 vs 17) and a lower number of false positives (11 vs 19) (Figure 5A). Calculated confusion matrix showed that MetaOdysseus provided higher TPR (0.98 vs 0.30) and lower FNR (0.38 vs 0.66). Following spectral deconvolution, we focused on protein identification. Isolation and fragmentation of the m/z 1614.11 that corresponds to the [Zn7MT1e]4+ ion yielded CID fragment ion corresponding to b17 and y31 containing one and three metal ions where charge is retained by the N- and C-terminus, respectively (Table S13). Interestingly, the y31 fragment derived from backbone dissociation between α- and β-domain contained three Zn2+, probing the higher stability of Zn–S bonds and folding of the α-domain.14,56 On the other hand, fragment b17 with four Cys residues in the sequence contained one Zn2+. Analysis of the CID fragments from the data-independent acquisition (DIA) experiment showed that except the common b17 fragment, most of them derived from the α-domain (C-terminus). This can be attributed to the higher thermodynamic stability and lower kinetic lability of the Zn–S bonds for α-domain (Table S13).14,56 We detected the fragment y36 corresponding to the Zn4Cys11 cluster within α-domain, which suggests four Zn2+ coordinates in the α-domain as featured for mammalian MTs (Figure 5B). To these identifications, we assigned a statistical significance by computing first a score based on the binomial distribution probability and then by the estimation of an empirical p-value via permutation tests (Figure 5C). We compared the database searching with ProSight Lite, which required to pinpoint manually the position of the custom modification, that is, Zn2+ in here.58 So, we added to the N- and C-terminus from zero to seven Zn2+, which leads to a total of 49 possible solutions. This manual inspection validated the results found automatically with MetaOdysseus and did not detect any new feature.
Figure 5.

Native top-down experiments. (A) Venn diagram for the TP and FP for the deconvoluted masses obtained with MetaOdysseus and with eTHRASH. (B) Assignment of the CID fragments for the native data-independent top-down experiments at different collision energies. (C) Statistical significance assessment for the protein identification from the spectra presented in (B). The score was computed based on a binomial distribution probability and then its significance estimated with an empirical p-value via permutation tests.
Integration of the Native MS and Proteomics for a Comprehensive Data Analysis
The characterization of metal–protein complexes, including the metal–protein stoichiometry, mapping the metal-binding sites, or studying their metal-coupled folding, requires combining several experimental protocols.2,3,7,14 The interplay between metal ions and proteins and its inherently dynamic nature challenges their investigation. In our recent study, we attempted to develop an experimental approach that combined native MS and proteomics approaches to elucidate the metal–protein stoichiometry and topology of different Zn7-xMT2 species.2,14 Having prepared a metal–protein complex (e.g., Zn4MT2), the first step is to record mass spectra under nondenaturing conditions. The native-like structure is likely retained because of the kinetic trapping effect, which allowed us to determine the stoichiometry. Afterward, the protein was labeled with IAM, a Cys-alkylating reagent. In principle, one may determine the number of Cys residues coordinating metal ions by the simple premise that a metal-bound thiolate exhibits lower reactivity toward nucleophiles than a free Cys residue. Thus, the mass spectra for Zn4MT2 IAM-labeled confirmed the [Zn4IAM6-10MT2]4+ ions, in agreement with MALDI-MS analysis where the modification profile centered at 9–10 modifications.2,14 Because MT2 contains 20 Cys residues, we may hypothesize a minimum of the remaining 10 Cys coordinate four Zn2+. To identify the ligands binding Zn2+, a peptide-mass fingerprint was first run. It localized IAM moieties in the N-terminal and central fragment of the protein. Combined with a bottom-up approach, the results suggested that the first four Zn2+ are partially redistributed between both α- and β-domains.14 Rusell’s group has also employed a combination of native MS, bottom-up, and top-down approaches to identify Cd2+-binding sites in MT2.3 These examples summarize how the tools presented in this software can be joined into an integrated pipeline for the comprehensive characterization of a dynamic protein–ligand system.
Conclusions
Although many software applications are available for mass spectrometry analysis, technical limitations encountered when analyzing metal–protein complexes pursued us to develop MetaOdysseus. We sought to design an automatic and free tool to assist in the data processing analysis for multiple MS-based platforms and experimental approaches focused on metal–protein complexes. Overall, four main steps are in the pipeline, mass spectra preprocessing (baseline correction and background reduction), charge-state deconvolution, statistical scoring, and mass assignment. We have demonstrated its capabilities for situations of different complexities, including native MS (TOF and Orbitrap mass analyzers) analysis of metal-free and chemically labeled Zn2+-loaded proteins; MALDI-TOF analysis of these intact proteins and enzymatically digested and, bottom-up MS and native top-down analysis with collision-induced dissociation experiments. MetaOdysseus robustly handles the aforementioned situations. The performance obtained was compared to different existent software applications for each possible scenario. In most cases, MetaOdysseus outperformed its counterpart software, demonstrating its usefulness. Notwithstanding, the R software’s easy adaptability will lead to future updates incorporating new applications related to structural studies of metal-binding proteins.
Acknowledgments
This research was supported by the National Science Centre of Poland (NCN) under the Opus grant no. 2016/21/B/NZ1/02847 (to A.K.) and Preludium grant no. 2018/31/N/ST4/01909 (to M.D.P.-D.). The research was partially supported by the European Research Council (ERC) under the European Union′s Horizon 2020 research and innovation program (grant no. 759585), the Czech Science Agency (grant no. 19-13766J), and CEITEC 2020 (LQ1601). M.D.P.-D. thanks the Erasmus+ program and the Polish National Agency for Academic Exchange under the PROM program (grant no. PPI/PRO/2018/1/00007/U/00).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00651.
Detailed materials and protocol for expression and purification of MTs; detailed list of R packages embedded in MetaOdysseus; detailed explanation for the charge deconvolution algorithm with the simulation method; detailed explanation for the charge deconvolution algorithm with the peak assignment method; implementation of the universal deconvolution score UniScore; calculation of the PMF score and validation by permutation tests; protocol for the comparison between MetaOdysseus and Bruker Daltonics for MALDI-MS; scoring PSMs and filtering by FDR strategy in a bottom-up MS; details for the comparison between MetaOdysseus, Mascot, and MS-GF+ for a bottom-up MS; details for the comparison between MetaOdysseus and MASH suite Pro for mass deconvolution of top-down MS; deconvolution results obtained with MetaOdysseus for a set of ESI-MS spectra (Table S1); software parameters used in the MetaOdysseus for deconvolution results in Table S1 (Table S2); mass assignment of the deconvolved spectrum obtained with MetaOdysseus in Tables S1 and S2 (Table S3); deconvolution results obtained with UniDec for the samples assayed (Table S4); charge deconvolution overview (Figure S1); comparison of combined algorithms for ESI-MS spectrum processing performed over apo-MT2 (Figure S2); deconvolution scoring scheme based on UniDec (Figure S3); analysis of ESI-MS spectra for apo-MT2 and Zn7MT2 (Figure S4); MALDI-TOF-MS analysis of NEM-labeled apo-MT2 (Figure S5); annotated MALDI-TOF-MS spectra of chemically labeled apo- and Zn7MT2 using of the R package (Table S5); processed MALDI-TOF-MS spectrum for apo-MT2 and Zn7MT2 chemically labeled by a set of alkylation reagents (Figure S6); annotated peptide-mass fingerprint for IAM-labeled apo-MT2 (Table S6); peptide-mass fingerprint (PMF) of chemically labeled apo-MT2 and apo-MT3 obtained by MALDI-TOF-MS (Figure S7); scores obtained for annotated peptide-mass fingerprint for apo-MT2 and apo-MT3 (Table S7); correlogram for the PMF scores hit ratio, sequence coverage, and mass coverage for the 10 datasets analyzed (Zn0MT2···Zn7MT2, Zn0MT3··· Zn7MT2) with MALDI-MS (Figure S8); permutation tests to calculate the p-value from the null distributions for the PMF scores obtained for the decoy list scored against the experimental MALDI-MS spectrum for Zn0MT2···Zn7MT2 and Zn0MT3···Zn7MT3 proteins (Figure S9); peptide-spectrum matches (PSMs) for the dataset of MS/MS spectra collected for enzymatically digested Zn0-7MT3 proteins (Table S8); deconvolution and assignment of the bottom-up MS/MS spectra with MetaOdysseus (Table S9); evaluation of the relationship between the false positives (FP) and the score achieved for the peptide-spectrum matches computes as a peak match probability scored based on the probabilities obtained from a binomial distribution (Figure S10); peptide-spectrum matches (PSM) obtained with MS-GF+ for the set of enzymatically digested Zn0-7MT3 proteins (Table S10); comparison between MetaOdysseus (red line) and MS-GF+ (black line) for the peptide-spectrum matches (PSMs) results obtained in terms of their sensitivity, specificity, and precision (Figure S11); mass deconvolution for the native top-down MS/MS obtained with MetaOdysseus using the peak assignment algorithm (Table S11); mass deconvolution for the native top-down MS/MS obtained with MASH Suite Pro using the eTRASH algorithm (Table S12); and mass assignment for the deconvolved masses from the native top-down MS/MS obtained with MetaOdysseus (Table S13) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yu X.; Wojciechowski M.; Fenselau C. Assessment of metals in reconstituted metallothioneins by electrospray mass spectrometry. Anal. Chem. 1993, 65, 1355–1359. 10.1021/ac00058a010. [DOI] [PubMed] [Google Scholar]
- Drozd A.; Wojewska D.; Peris–Díaz M. D.; Jakimowicz P.; Krężel A. Crosstalk of the structural and zinc buffering properties of mammalian metallothionein-2. Metallomics 2018, 10, 595–613. 10.1039/C7MT00332C. [DOI] [PubMed] [Google Scholar]
- Chen S. H.; Russell W. K.; Russell D. H. Combining chemical labeling, bottom-up and top-down ion-mobility mass spectrometry to identify metal-binding sites of partially metalated metallothionein. Anal. Chem. 2013, 85, 3229–3237. 10.1021/ac303522h. [DOI] [PubMed] [Google Scholar]
- Irvine G. W.; Santolini M.; Stillman M. J. Selective cysteine modification of metal-free human metallothionein 1a and its isolated domain fragments: Solution structural properties revealed via ESI-MS. Protein Sci. 2017, 26, 960–971. 10.1002/pro.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace N. J.; Weerapana E. A competitive chemical-proteomic platform to identify zinc-binding cysteines. ACS Chem. Biol. 2014, 9, 258–265. 10.1021/cb400622q. [DOI] [PubMed] [Google Scholar]
- Scotcher J.; Clarke D. J.; Weidt S. K.; Mackay C. L.; Hupp T. R.; Sadler P. J.; Langridge-Smith P. R. Identification of two reactive cysteine residues in the tumor suppressor protein p53 using top-down FTICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 888–897. 10.1007/s13361-011-0088-x. [DOI] [PubMed] [Google Scholar]
- Martin E. M.; Kondrat F. D. L.; Stewart A. J.; Scrivens J. H.; Sadler P.; Blindauer C. A. Native electrospray mass spectrometry approaches to probe the interaction between zinc and an anti-angiogenic peptide from histidine-rich glycoprotein. Sci. Rep. 2018, 8, 8646 10.1038/s41598-018-26924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel K.; Natan E.; Hall Z.; Fersht A. R.; Robinson C. V. Intrinsically disordered p53 and its complexes populate compact conformations in the gas phase. Angew. Chem., Int. Ed. 2013, 52, 361–365. 10.1002/anie.201203047. [DOI] [PubMed] [Google Scholar]
- Jurneczko E.; Cruickshank F.; Porrini M.; Clarke D. J.; Campuzano I. D. G.; Morris M.; Nikolova P. V.; Barran P. E. Probing the conformational diversity of cancer-associated mutations in p53 with ion-mobility mass spectrometry. Angew. Chem., Int. Ed. 2013, 52, 4370–4374. 10.1002/anie.201210015. [DOI] [PubMed] [Google Scholar]
- Arlt C.; Flegler V.; Ihling C. H.; Schäfer M.; Thondorf I.; Sinz A. An integrated mass spectrometry based approach to probe the structure of the full-length wild-type tetrameric p53 tumor suppressor. Angew. Chem., Int. Ed. 2016, 128, 1–6. 10.1002/ange.201510990. [DOI] [PubMed] [Google Scholar]
- Pérez–Zúñiga C.; Leiva–Presa À.; Austin R. N.; Capdevila M.; Palacios Ò. Pb(II) binding to the brain specific mammalian metallothionein isoform MT3 and its isolated αMT3 and βMT3 domains. Metallomics 2019, 11, 349–361. 10.1039/C8MT00294K. [DOI] [PubMed] [Google Scholar]
- Padjasek M.; Kocyła A.; Kluska K.; Kerber O.; Ba-Tran J.; Krężel A. Structural zinc binding sites shaped for greater works: structure-function relations in classical zinc finger, hook and clasp domains. J. Inorg. Biochem. 2020, 204, 110955 10.1016/j.jinorgbio.2019.110955. [DOI] [PubMed] [Google Scholar]
- Kochańczyk T.; Nowakowski M.; Wojewska D.; Kocyła A.; Ejchart A.; Koźmiński W.; Krężel A. Metal-coupled folding as the driving force for the extreme stability of Rad50 zinc hook dimer assembly. Sci. Rep. 2016, 6, 36346 10.1038/srep36346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris-Díaz M. D.; Guran R.; Zitka O.; Adam V.; Krężel A. Metal and affinity-specific dual labeling of cysteine-rich proteins for identification of metal-binding sites. Anal. Chem. 2020, 10.1021/acs.analchem.0c01604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreini C.; Banci L.; Bertini I.; Rosato A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. 10.1021/pr050361j. [DOI] [PubMed] [Google Scholar]
- Andreini C.; Banci L.; Bertini I.; Rosato A. Zinc through the three domains of life. J. Proteome Res. 2006, 5, 3173–3178. 10.1021/pr0603699. [DOI] [PubMed] [Google Scholar]
- Rose R. J.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods 2012, 9, 1084–1086. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- Maret W. Metals on the move: zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. BioMetals 2011, 24, 411–418. 10.1007/s10534-010-9406-1. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Maret W.; Vallee B. L. Differential fluorescence labeling of cysteinyl clusters uncovers high tissue levels of thionein. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 5556–5559. 10.1073/pnas.101123298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova M. D.; Ivanova I. A.; Cherian M. G. Metallothionein and apoptosis during differentiation of myoblasts to myotubes: protection against free radical toxicity. Toxicol. Appl. Pharmacol. 1999, 159, 175–184. 10.1006/taap.1999.8755. [DOI] [PubMed] [Google Scholar]
- Krężel A.; Maret W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237. 10.3390/ijms18061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrige A. G.; Seddon M. J.; Green B. N.; Jarvis S. A.; Skilling J.; Staunton J. Disentangling electrospray spectra with maximum entropy. Rapid Commun. Mass Spectrom. 1992, 6, 707–711. 10.1002/rcm.1290061115. [DOI] [Google Scholar]
- Ferrige A. G.; Seddon M. J.; Jarvis S.; Skilling J.; Aplin R. Maximum entropy deconvolution in electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 1991, 5, 374–377. 10.1002/rcm.1290050810. [DOI] [Google Scholar]
- Tseng Y.-H.; Uetrecht C.; Yang S.-C.; Barendregt A.; Heck A. J. R.; Peng W.-P. Game-theory-based search engine to automate the mass assignment in complex native electrospray mass spectra. Anal. Chem. 2013, 85, 11275–11283. 10.1021/ac401940e. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Marshall A. G. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 1998, 9, 225–233. 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- Horn D. M.; Zubarev R. A.; McLafferty F. W. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J. Am. Soc. Mass Spectrom. 2000, 11, 320–332. 10.1016/S1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- Stengel F.; Baldwin A. J.; Bush M. F.; Hilton G. R.; Lioe H.; Basha E.; Jaya N.; Vierling E.; Benesch J. L. P. Dissecting heterogeneous molecular chaperone complexes using a mass spectrum deconvolution approach. Chem. Biol. 2012, 19, 599–607. 10.1016/j.chembiol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgner N.; Robinson C. V. Massign: An assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Anal. Chem. 2012, 84, 2939–2948. 10.1021/ac300056a. [DOI] [PubMed] [Google Scholar]
- Marty M. T.; Baldwin A. J.; Marklund E. G.; Hochberg G. K. A.; Benesch J. L. P.; Robinson C. V. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 2015, 87, 4370–4376. 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Trnja M. J.; Roh S.-H.; Robinson P. J. J.; Shiau C.; Fujimori D. G.; Chiu W.; Burlingame A. L.; Guan S. Improved peak detection and deconvolution of native electrospray mass spectra from large protein complexes. J. Am. Soc. Mass Spectrom. 2015, 26, 2141–2151. 10.1007/s13361-015-1235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty M. T. Universal score for deconvolution of intact protein and native electrospray mass spectra. Anal. Chem. 2020, 92, 4395–4401. 10.1021/acs.analchem.9b05272. [DOI] [PubMed] [Google Scholar]
- Perkins D.; Pappin D.; Creasy D.; Cottrell J. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Eng J.; McCormack A.; Yates J. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Solntsev S. K.; Shortreed M. R.; Frey B. L.; Smith L. M. Enhanced global post-translational modification discovery with MetaMorpheus. J. Proteome Res. 2018, 17, 1844–1851. 10.1021/acs.jproteome.7b00873. [DOI] [PubMed] [Google Scholar]
- Kim S.; Pevzner P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Geer L. Y.; Markey S. P.; Kowalak J. A.; Wagner L.; Xu M.; Maynard D. M.; Yang X.; Shi W.; Bryant S. H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 3, 958–964. 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Li H.; Nguyen H. H.; Loo R. R. O.; Campuzano I. D. G.; Loo J. A. An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem. 2018, 10, 139–148. 10.1038/nchem.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcllwain S. J.; Wu Z.; Wetzel M.; Belongia D.; Jin Y.; Wenger K.; Ong I. M.; Ge Y. Enhancing top-down proteomics data analysis by combining deconvolution results through a machine learning strategy. J. Am. Soc. Mass Spectrom. 2020, 31, 1104–1113. 10.1021/jasms.0c00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Inbar Y.; Dorrestein P. C.; Wynne C.; Edwards N.; Souda P.; Whitelegge J. P.; Bafna V.; Pevzner P. A. Deconvolution and database search of complex tandem mass spectra of intact proteins: A combinatorial approach. Mol. Cell. Proteomics 2010, 9, 2772–2782. 10.1074/mcp.M110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou Q.; Xun L. K.; Liu X. W. TopPIC: a software tool for top-down mass spectrometry based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. 10.1093/bioinformatics/btw398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster C.Mass spectrometry method for accurate mass determination of unknown ions. US Patent US6188064B12001.
- Liu X.; Sirotkin Y.; Shen Y.; Anderson G.; Tsai Y. S.; Ting Y. S.; Goodlet D. R.; Smith R. D.; Bafna V.; Pevzner P. A. Protein identification using top-down spectra. Mol. Cell. Proteomics 2012, 11, M111.008524 10.1074/mcp.M111.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDuc R. D.; Taylor G. K.; Kim Y. B.; Januszyk T. E.; Bynum L. H.; Sola J. V.; Garavelli J. S.; Kelleher N. L. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004, 32, W340–W345. 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. M.; Pesavento J. J.; Mizzen C. A.; Kelleher N. L.; Pevzner P. A. Interpreting top-down mass spectra using spectral alignment. Anal. Chem. 2008, 80, 2499–2505. 10.1021/ac702324u. [DOI] [PubMed] [Google Scholar]
- Liu X. W.; Hengel S.; Wu S.; Tolic N.; Pasa-Tolic L.; Pevzner P. A. Identification of ultramodified proteins using top-down tandem mass spectra. J. Proteome Res. 2013, 12, 5830–5838. 10.1021/pr400849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W. X.; Guner H.; Gregorich Z. R.; Chen A. J.; Ayaz-Guner S.; Peng Y.; Valeja S. G.; Liu X. W.; Ge Y. MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol. Cell. Proteomics 2016, 15, 703–714. 10.1074/mcp.O115.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitzky A.; Golay M. J. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964, 36, 1627–1639. 10.1021/ac60214a047. [DOI] [Google Scholar]
- Eilers P. H. C. A perfect smoother. Anal. Chem. 2003, 75, 3631–3636. 10.1021/ac034173t. [DOI] [PubMed] [Google Scholar]
- Percival D. B.; Walden A. T.. Wavelet Methods for Time Series Analysis, Cambridge series in statistical and probabilistic mathematics; Cambridge University Press: Cambridge, 2000. [Google Scholar]
- Du P.; Kibbe W. A.; Lin S. M. Improved peak detection in mass spectrum by incorporating continuous wavelet transform-based pattern matching. Bioinformatics 2006, 22, 2059–2065. 10.1093/bioinformatics/btl355. [DOI] [PubMed] [Google Scholar]
- Stead D. A.; Preece A.; Brown A. J. P. Universal metrics for quality assessment of protein identifications by mass spectrometry. Mol. Cell. Proteomics. 2006, 5, 1205–1211. 10.1074/mcp.M500426-MCP200. [DOI] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J.; Liko I.; Landreh M.; Shutin D.; Bolla J. R.; Jefferies D.; Agasid M.; Yen H. Y.; Ladds M. J. G. W.; Lane D. P.; Khalid S.; Mullen C.; Remes P. M.; Huguet R.; McAlister G.; Goodwin M.; Viner R.; Syka J. E. P.; Robinson C. V. Combining native and ‘omics’ mass spectrometry to identify endogenous ligands bound to membrane proteins. Nat. Methods 2020, 17, 505–508. 10.1038/s41592-020-0821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris-Díaz M. D.; Richtera L.; Zitka O.; Krężel A.; Adam V. A chemometric-assisted voltammetric analysis of free and Zn(II)-loaded metallothionein-3 states. Bioelectrochemistry 2020, 134, 107501 10.1016/j.bioelechem.2020.107501. [DOI] [PubMed] [Google Scholar]
- Mantini D.; Petrucci F.; Pieragostino D.; Del Boccio P.; Di Nicola M.; Di Ilio C.; Federici G.; Sacchetta P.; Comani S.; Urbani A. LIMPIC: a computational method for the separation of protein MALDI–TOF–MS signals from noise. BMC Bioinformatics 2007, 8, 101. 10.1186/1471-2105-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellers R. T.; Greer J. B.; Early B. P.; Yu X.; LeDuc R. D.; Kelleher N. L.; Thomas P. M. ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15, 1235–1238. 10.1002/pmic.201400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study and an R script that follows the results section are deposited in https://github.com/ManuelPerisDiaz/Data_MetaOdysseus. The MetaOdysseus R package is deposited in GitHub and can be freely downloaded from the following link: https://github.com/ManuelPerisDiaz/MetaOdysseus.