

Abstract
Bottom-up proteomics relies on identification of peptides from tandem mass spectra, usually via matching against sequence databases. Confidence in a peptide–spectrum match can be characterized by a score value given by the database search engines, and it depends on the information content and the quality of the spectrum. The latter are influenced by experimental parameters, of which the collision energy is the most important one in the case of collision-induced dissociation. We examined how the identification score of the Byonic and Andromeda (MaxQuant) engines varies with collision energy for more than a thousand individual peptides from a HeLa tryptic digest on a QTof instrument. We thereby extended our earlier study on Mascot scores and corroborated its findings on the potential bimodal nature of this energy dependence. Optimal energies as a function of m/z show comparable linear trends for the three engines. On the basis of peptide-level results, we designed methods with one or two liquid chromatography–tandem mass spectrometry (LC-MS/MS) runs and various collision energy settings and assessed their practical performance in peptide and protein identification from the HeLa standard sample. A 10–40% gain in various measures, such as the number of identified proteins or sequence coverage, was obtained over the factory default settings. Best performing methods differ for the three engines, suggesting that the experimental parameters should be fine-tuned to the choice of the engine. We also recommend a simple approach and provide reference data to ease the transfer of the optimized methods to other mass spectrometers relevant for proteomics. We demonstrate the utility of this approach on an Orbitrap instrument. Data sets can be accessed via the MassIVE repository (MSV000086379).
Keywords: mass spectrometry, bottom-up proteomics, peptide fragmentation, database search, identification score, collision energy optimization, transferability
Introduction
Over the past few decades, mass spectrometry (MS) coupled to (nano)-liquid chromatography (nano-LC) has become an indispensable analytical tool in identification, quantitation, and characterization of proteins.1−10 The most established method is the bottom-up approach, also called shotgun proteomics, where proteins in a complex sample are first digested to peptides, and the latter are then separated in one or more dimensions and identified via tandem MS. Most frequently, peptide ions are fragmented by collisions with inert gas molecules (collision-induced dissociation, CID).11 The conventional data acquisition strategy is the data-dependent analysis (DDA) involving mass selection and MS/MS measurement of the most abundant precursors present in a preceding MS scan. The resulting fragment ion spectra of individual peptides are then searched against a sequence database to identify the peptide sequence, and proteins are inferred from the identified peptides. The similarity between the experimental and theoretical spectra, and thereby the confidence of the peptide–spectrum match (PSM), is characterized by a score value.12,13 An increase in the confidence of the PSMs is attained if the given experimental MS/MS spectrum contains a large number of characteristic peptide fragment ions (e.g., b/y sequence ions) and only a small number of unidentifiable (noise) peaks. An overall high level of confidence in the PSMs is key to identify a large number of peptides and, ultimately, proteins.
Collision energy (CE) is one of the most important parameters for tandem MS/MS measurements because it determines relative ion abundance of the various fragments and thereby the information content of the taken spectra. Although manufacturers typically provide recommendations, different kinds of spectra may be optimal for different purposes, requiring different collision energy settings. Workflow-specific optimization therefore promises significant performance gain. Still, earlier literature studies did not directly target the most efficient shotgun proteomics identification. For example, several papers determined CE values corresponding to a given survival yield (e.g., 50%) for peptides,14−16 while others sought the highest intensity for given parent–fragment transitions.17−23 Other studies addressed the most efficient sequence validation of biosimilar drugs, e.g., monoclonal antibodies (mAbs), and it was found that a combination of multiple CEs (or other experimental parameters relevant for the fragmentation method) may have a beneficial effect.24,25 Recently, our laboratory thoroughly tested the energy dependence of sequence validation procedures for mAbs26 and leveraged the results to develop a simple workflow. Using two collision energies 6–10 eV apart and an automated data processing, we could obtain a sequence coverage ca. 25% higher than the straightforward method with factory settings.
As a rare example of studies targeting identification confidence, Hinneburg et al. carried out MS/MS fragmentation of synthetic glycopeptides on a QTof instrument and determined optimal energetics for the peptide part, as characterized by the intensity coverage score.27 However, this systematic investigation included only a few dozen peptides. Another study, from Diedrich et al., examined the number of identified peptides and proteins from the tryptic digest of HEK293T lysate at three distinct CEs, as well as with a stepped method combining the three, using a Q Exactive mass spectrometer.28 However, only fairly small differences in the number of identified peptides under the various conditions were reported, and the full body of peptide-level results remained undisclosed. Tholey and co-workers also investigated the effect of varying collision energies on peptide identification as part of a study focusing on phosphopeptide quantitation.29
To our knowledge, we were the first to systematically study energy dependence of peptide identification confidence on large sets of individual peptides.30 The MS/MS spectra of several thousands of tryptic peptides from a digest of Escherichia coli and HeLa cell lysate were recorded at 21 different CEs, spanning a range of 40 eV, on a Bruker QTof instrument. The data were analyzed with Mascot, one of the most frequently used search engines.31 Mascot score vs energy curves revealed that only less than half of the peptides have a single well-defined optimal CE yielding maximum score (unimodal behavior). More than half of the peptides, in contrast, showed either a broad plateau or two different maxima (bimodal behavior). Optimum CE as a function of m/z of the peptide was shown to follow separate, significantly different linear trends for the unimodal and bimodal cases. This observation may form the basis of the DDA experiment design, including the potential use of multiple CEs even for the identification of unmodified peptides.
In the present work, we extend and leverage our studies on CE dependence to suggest optimal workflows for the practical measurement of a complex sample in its entirety. First, we broaden our previous investigation on Mascot to other search engines.12,13 We chose Andromeda since it is the one integrated into the widely used MaxQuant quantitative proteomics platform.32,33 We also included Byonic, a widely used hybrid method, which uses a small amount of de novo sequencing to extract candidate peptides from the database34 and overcomes the limitation of pure database searching programs, which cannot deal efficiently with nonspecific cleavages or several peptide modifications.
To transform these peptide-level results showing complicated and diverse energy dependence into practical approaches, we create and test workflows using combinations of multiple collision energies, within the same run or in two separate runs. We demonstrate the benefit of this approach for proteomics measurements on a HeLa standard digest.
Experimental Section
Chemical Reagents
LC-MS-grade solvents and HeLa tryptic digest standard were purchased from Sigma-Aldrich Kft. (Budapest, Hungary) and Thermo Fisher Scientific (Waltham, MA), respectively. MassPREP E. coli digestion standard was from Waters (Milford, MA).
Mass Spectrometry Analysis
Nano-LC-MS/MS studies of the HeLa tryptic digest standard samples were performed using standard laboratory methods for proteomics investigation (see details in Supporting Information S1) with varying collision energy settings. Briefly, in each run, 50 ng of HeLa was subjected to nano-LC-MS/MS analysis using a Dionex Ultimate 3000 RSLC nano-LC coupled to a Bruker Maxis II ETD Q-TOF via a CaptiveSpray nanoBooster ionization source. Peptides were separated on an Acquity M-Class BEH130 C18 or an Acclaim PepMap RSCL C18 analytical column using gradient elution, following trapping on an Acclaim PepMap100 C18 trap column. Solvent A consisted of water + 0.1% formic acid, while Solvent B was acetonitrile + 0.1% formic acid. Spectra were collected using a fixed cycle time of 2.5 s and the following scan speeds: MS spectra at 3 Hz, CID on precursors at 16 Hz for abundant ones (intensity >40 000) and at 4 Hz for peaks of low abundance (7500 < intensity < 40 000). These intensity threshold values provide good-quality MS/MS spectra and are typically used in our laboratory in DDA proteomics measurements for protein identification. An active exclusion of 2 min after one spectrum was used, except if the intensity of the precursor was elevated threefold. We evaluated two separate QTof experimental data sets, as discussed in the forthcoming sections.
Energy-Dependence Studies
As part of this work, we analyzed the score–collision energy relationship for the Byonic and Andromeda search engines on a previously recorded collision-energy-dependent mass spectrometric data set, the Mascot analysis of which had been published earlier.30 In this data set, collision energies applied to fragment the peptides were chosen to be the sum of an m/z-dependent preoptimized collision energy (for the equation, see Supporting Information S2) and a collision energy shift mapping the −20 to +20 eV range in 2 eV steps in 21 separate runs. Hence, 21 different collision energy values for each peptide were measured, centered at a peptide-specific default value and with a lower limit of 5 eV. All LC-MS/MS runs in this data set were recorded using an elevated threshold of 400 000 cps for MS/MS measurements to decrease the run-to-run variability of the set of actually analyzed parent ions in the DDA measurements.30 Actually two specific increased threshold values, namely, 100 000 and 400 000 cps, were tested in repetitions of identical runs. These were selected such that in a normal experiment ca. one-half and one-third of the precursor ions selected for MS/MS had intensities above these thresholds, respectively. Finally, we chose the latter for the energy-dependence studies because it provided variability statistics on the complex HeLa sample similar to those on the simple enolase, ensuring that (almost) the same peptides are measured at all energies. The increase in threshold could be done at the expense of the notably reduced number of identified peptides from the complex sample, about one-third of that with the default threshold.30
Performance Gain Study
The above energy-dependence studies allow optimum energies to be determined for each individual peptide, but obviously, we cannot directly apply these in practice since the identity of the peptides is not known at the time of the measurement. For the practical purposes set to be the goal of the present paper, we used these results to choose six different m/z-dependent collision energy settings for actual LC-MS/MS runs and carried out five repetitions of each (30 LC-MS/MS runs overall). First, we measured a reference, which was chosen to be the manufacturer’s recommendation for protein identification (“factory setting”). Next, we wanted to get close to the optimal collision energy with respect to the Mascot score for unimodal peptides and employed settings corresponding to the linear fit between the optimum collision energy and the m/z of the peptide.30 We refer to this setting as “100%” throughout the text. As a third and fourth setup, we uniformly increased the collision energy to 135% of the value coming from this fit and decreased it to 70% of the value coming from this fit. These settings practically coincide with the linear fits to the lower and higher energy optimum collision energies of the bimodal behavior peptides. Finally, two stepped methods were tested. In the first case, 50% of the time was used to acquire a spectrum with the collision energy set to 100%; then, in the other half of the time, the CE was reduced to 70% (step 100/70%). In the second stepped method, the time was split into three equal intervals, using 70, 100, and 135% collision energy (step 100/70/135%). (For a more detailed explanation of the chosen collision energy settings, see the Results and Discussion section.)
Further Test Measurements Addressing Transferability
Additional measurements to support transferability of our results were carried out using the above experimental setup on E. coli digest samples (300 ng per run) and on HeLa samples on a Thermo Orbitrap Fusion instrument (see Supporting Information S13 and S14).
Data Analysis
The raw QTof data were first recalibrated using Bruker Compass DataAnalysis software 4.3 (Bruker Daltonik GmbH, Bremen, Germany) for the internal calibrant. MS/MS spectra were searched against the human SwissProt database using three different search engines, namely, Mascot, Byonic, and Andromeda. The common parameters were set as follows: trypsin as the enzyme and maximum two missed cleavages allowed.
Mascot Search
MS/MS peak list generation was performed using ProteinScape software 3.1 (Bruker Daltonik GmbH, Bremen, Germany). Database search was performed using the Mascot search engine version v.2.5 (Matrix Science, London, U.K.). The mass tolerance was determined according to the recommended protocol35 and was set as 7 ppm for precursors and 0.05 Da for fragment peaks. The list of relevant variable modifications was obtained using error-tolerant search35 and included methionine oxidation and glutamine and asparagine deamidation, while cysteine carbamidomethylation was selected as fixed modification. The resulting Mascot output files (.dat) were subject to further analysis by Scaffold and Serac programs (see below).
Byonic Search
We employed Byonic v3.5.0 for our analyses (Protein Metrics, Cupertino, CA). Regarding mass tolerance values and the list of variable modifications, recommendations of the Preview module were used. These included 7 and 15 ppm for precursor and fragment mass tolerance, respectively, and the following variable modifications: oxidation (M) as common and pyro-glutamination (N-term Q and N-term E) and ammonia loss (N-term C) and acetyl loss (protein N-term) as rare modifications. Note that the hybrid characteristic of Byonic allows handling of more variable modifications compared to database searches. The mzIdentML output files were further analyzed by the Scaffold program, whereas the Excel reports were the input files for data aggregation carried out by Serac.
Andromeda Search
The raw files of the mass spectrometric experiments were searched by Andromeda as integrated into MaxQuant version 1.6.7;32,36 the peak lists were created by MaxQuant software itself. The default mass tolerances for Bruker QTof instrument were used (0.006 Da and 40 ppm for precursor and fragment tolerance), and the same modification list was applied as for Mascot database search. The results of the search were further analyzed by Scaffold and Serac programs.
Aggregation for Energy Dependence
For the study of the energy dependence of peptide fragmentation, we used our recently developed program called Serac.26 Analogous to our previous work on Mascot data,30 we collected identification scores as a function of collision energy from the energy-dependent mass spectrometric data series for the Byonic and Andromeda search engines and determined the optimal collision energy. (More details can be found in Supporting Information S3.) Briefly, we first extracted the data from Byonic Excel reports and the allPeptides.txt output file of the MaxQuant program. The score vs energy shift functions were then normalized by dividing all values with the maximum score for the given peptide ion. Byonic score values, Byonic log Prob values, and Andromeda scores were all investigated. To ensure that we draw conclusions on the basis of confident peptide identifications, only peptides meeting certain minimum requirements were taken into account. First, depending on the chosen measure of identification confidence, we only considered a peptide ion identified at a given collision energy
if its Byonic score exceeded 200, or
its log Prob value exceeded 1.3 (p < 0.05), or
its Andromeda score was above 50.
Further, a peptide ion was only included in the energy-dependence analysis if it was identified at least at three consecutive collision energy values and for at least one collision energy it was found to have
a Byonic score value above 400 (being a “very good” score),37 or
a log Prob value exceeding 2 (p < 0.01), or
an Andromeda score greater than 75.32
For each peptide ion, the optimum energy was determined from the normalized score vs collision energy shift data sets by fitting one or two Gaussian functions. The score cutoff, while important to avoid false identifications biasing our results, resulted in no data points at low scores; therefore, we decided to add two points with zero score at a shift of ±35 eV to avoid erroneously wide peaks to be fitted. Where there were less than 12 data points in the original data, we only attempted to fit one Gaussian. Data for peptide ions with at least 12 points were fitted using both one and two Gaussian function, and the two-peak fit was accepted if it provided significantly better fit. The nonlinear fits were carried out, and the corresponding plots were generated using the levmar38 and PGPLOT39 libraries through their Perl Data Language interfaces. The positions of the center of the Gaussian peaks were considered as optimal values: a single energy value for one-peak fits (unimodal peptides) and two energy values for each two-peak fit (lower and higher energy optimum of the bimodal peptides).
Analysis of Performance Gain
The practical proteomics performance of LC-MS/MS runs using various collision energy settings in conjunction with various search engines was compared using Scaffold version 4.10 (Proteome Software, Portland, OR), using the following settings: LFDR-rescoring on, 95% peptide threshold, 1% FDR for proteins, and a minimum of two peptides was required for the identification of proteins.
Results and Discussion
Energy-Dependent Experiments
As a first part of this work, we wanted to broaden the scope of our earlier investigation on collision energy dependence of the identification score. We thus constructed energy-dependence curves of Byonic scores, Byonic log Prob values, and Andromeda scores, analogous to the Mascot score curves done earlier, for the peptides identified from the same experimental data set on a standard HeLa tryptic digest. It was found that Byonic score and log Prob values gave practically identical energy dependence, so we will discuss only the Byonic score here; results on log Prob can be found in Supporting Information S5. Overall, 21 different collision energies were examined for all peptides, mapping the energy dependence in 2 eV steps. When a given peptide was identified more than once in the same LC-MS/MS run, that is, measured several times at the same collision energy, the best scoring match was accepted. Overall, from these runs with an elevated MS/MS threshold, we identified 2188 and 1765 peptides using the Byonic and Andromeda search engines, respectively (see Table 1). Among these, 1378 and 1041 peptides, respectively, were considered sufficiently reliable to be included in the energy-dependence analysis (see the Data Analysis section). Using either search engine, approximately three-quarters of these peptides were doubly charged, and most of the rest were triply charged.
Table 1. Number of Peptides with Various Properties for All Search Engines Considered30 a.
| Mascot | Byonic | Andromeda | |
|---|---|---|---|
| identified from 21 runs | 2152 | 2188 | 1765 |
| considered for energy-dependence study | 1721 | 1378 | 1041 |
| from this, 2+ | 1405 | 1087 | 834 |
| from this, 3+ | 284 | 252 | 183 |
| from this, unimodal fit | 733 | 288 | 623 |
| from this, bimodal fit | 988 | 1090 | 418 |
Mascot results were taken from our earlier work.
Analogous to the previous Mascot results, normalized score vs collision energy curves revealed three qualitatively different curve shapes in all cases, namely, a single, well-defined maximum; a broad plateau; or two well-defined peaks. We therefore modeled them using one or two Gaussian functions, based on fit quality, and we refer to them as showing unimodal and bimodal behavior. In total, 79 and 40% of the peptides were found to be bimodal for Byonic and Andromeda, respectively, which points out that a large fraction of the curves shows either two maxima or a broadening due to overlapping peaks. As was discussed in our earlier paper,30 the phenomenon of bimodal curves can be associated with the different energy dependence of b- and y-type fragment intensities. Typically, y ions are more stable, less sensitive to the collision energy, and therefore more intense in the MS/MS spectra than b ions at all collision energies. Their optimum can be found ca. at the higher energy optimum of bimodal peptides. In contrast, b ions easily fragment to smaller ions and their intensity shows a relatively sharp maximum at the lower collision energy optimum. The heights of the two peaks in the score vs collision energy function are comparable, with the peak at the higher collision energy usually having a somewhat lower score. The data in our earlier paper30 shows that it is on average ∼93% of that at the lower energy peak for Mascot. The reason might be that although the contribution of y ions increases with the increase of collision energy, it is less energy-dependent, almost constant in a large collision energy range. The smaller effect of b ions is only present around the lower energy optimum. In addition, bimodal peptides have a fairly wide range of collision energies with relatively high scores (i.e., a plateau), as we found that even the minimum Mascot score between the two peaks is on average as much as 77% of the score at the lower energy peak (see Figure 1 for representative examples). Another interesting observation is that at higher m/z the bimodal behavior becomes more frequent.
Figure 1.
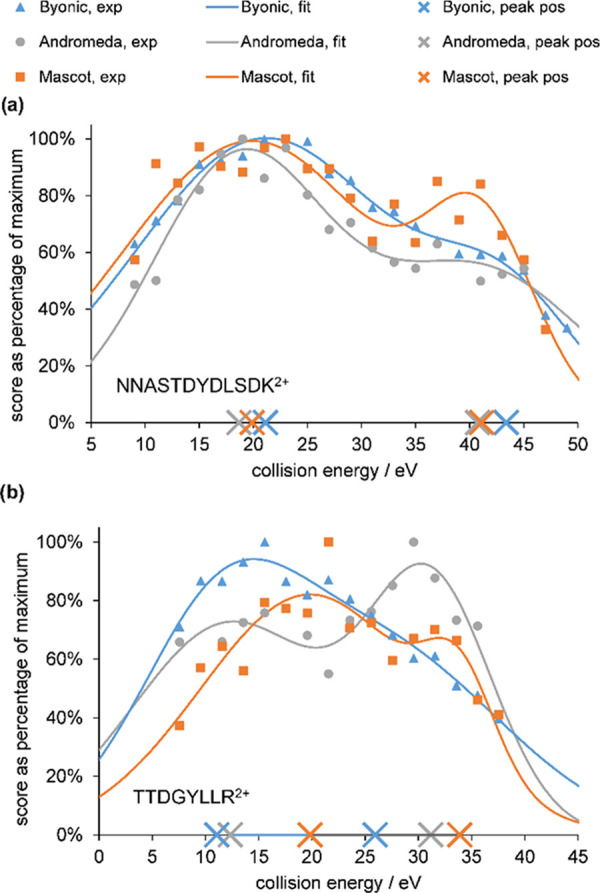
Result of fitting Gaussians to the energy-dependence data points (score as % of the maximum value vs collision energy in eV) of the bimodal example peptides (a) NNASTDYDLSDK2+ and (b) TTDGYLLR2+. Symbols denote measured data, while solid lines depict the two-Gaussian model functions. The peak positions of the latter are marked by crosses on the horizontal axis. Blue/triangles, gray/circles, and orange/squares depict Byonic, Andromeda, and Mascot results, respectively.
Figure 1a depicts example fitting results for the bimodal NNASTDYDLSDK2+ peptide (blue curve and triangles belong to Byonic, while gray curve and circles show Andromeda results) together with the earlier Mascot data as a comparison (indicated by orange curve and squares). The centers of the Gaussian functions were accepted as collision energy optimum values; they are denoted by crosses on the horizontal axis in Figure 1. In this particular example, the normalized score vs collision energy curves and the two Gaussian model functions obtained in the case of the three search engines have similar characteristics. The resulting optima fall close to each other; the lower optima are 21.1, 18.7, and 19.9 eV, while the determined higher energy optima are 43.4, 40.7, and 41.1 eV for Byonic, Andromeda, and Mascot search engines, respectively. Nevertheless, we also found numerous peptides that were identified by all three engines, but the behavior with them is not uniform. Figure 1b shows the representative example of a bimodal TTDGYLLR2+ peptide where the determined optimal collision energies for Byonic and Andromeda search engines are lower than for Mascot, especially for the lower energy optimum. A full explanation of all differences would require knowledge and analysis of details of the algorithms in the three engines (two of them being proprietary) and would not fit into the current study focusing on practical usability and design of protocols. We believe that key reasons include differences in what types of fragment ions the engines look for and how they utilize experimental intensity information.32,34,40 Regarding the former, b and y ions are always considered for all three engines. Further, Mascot and Byonic assume NH3 loss (K, N, Q, and R) and H2O loss (D, E, S, and T) from b/y ions, while Andromeda takes them into account only if the corresponding b/y ion is present. Finally, Byonic also includes a-type fragment ions as well. As an example, we included MS/MS spectra of the TTDGYLLR2+ peptide at Mascot’s and Byonic’s optimum energy, annotated by both engines, in Supporting Information S4. Byonic appears to take into account more of the low-intensity b fragment ions that favor low energies, which might be the reason for its optimum being lower. In addition, specifics of the peak list generation (Andromeda’s own algorithm vs MS vendor’s software for the other two engines) might also contribute to the observed differences. Again, as our ultimate goal is to design measurement workflows, we decided not to focus more on the differences of individual peptides but instead to move on to an analysis at an aggregated level.
From the analysis of individual peptides, we therefore zoomed out to the whole set of them and plotted the optimum collision energies as a function of the peptide ion m/z value. Results for doubly charged peptides using Byonic and Andromeda are shown in Figure 2a,b. Peak positions of the unimodal fits are represented by blue circles, while gray and orange circles belong to the lower energy and the higher energy optimum of bimodal peptides, respectively. Apparently, peak positions in each group follow linear trends with respect to m/z with relatively large R2 values (see dashed/dotted lines). We carried out a separate analysis for the triply charged peptides (see Supporting Information S6), which shows a broadly similar overall picture, with optimum energies being slightly lower as expected because of the higher charge. These findings are in line with the general observation that optimum collision energies are linear in m/z.15 Studies targeting 50% fragmentation,14 maximum fragment ion intensity for selected18,20 or multiple17,21 reactions, and scores of modified peptides27,41 all found and employ collision energies linear in m/z though the coefficient of determination and the actual parameters differ. In addition, Thermo has explicitly included the linear equation in their definition of “Normalized Collision Energy”.42
Figure 2.
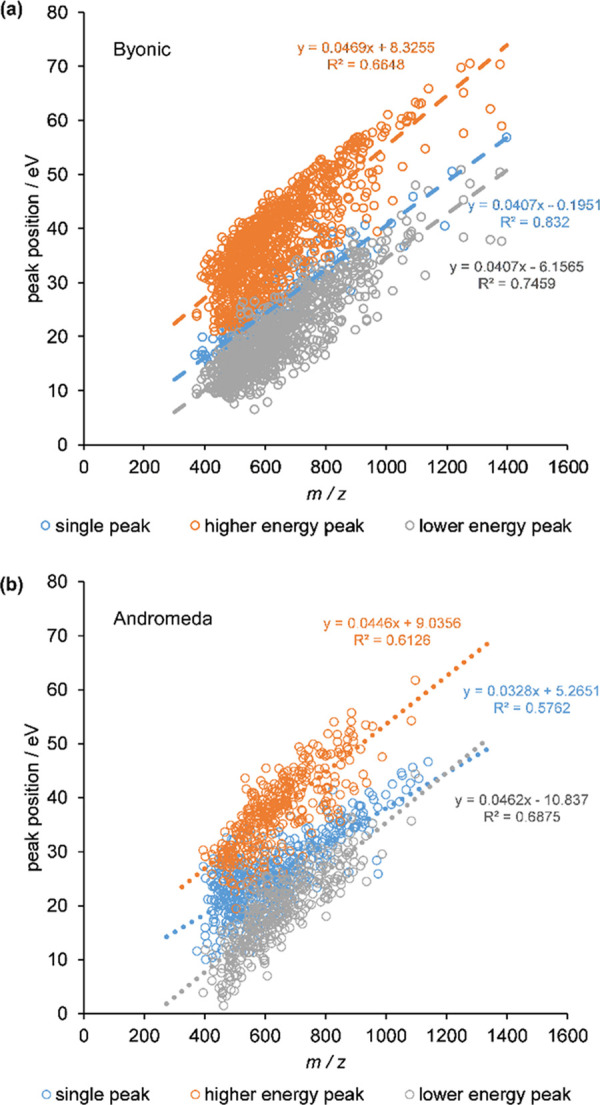
Peak positions in eV as a function of m/z for doubly charged peptides using the (a) Byonic search engine and (b) Andromeda search engine. Blue circles indicate the position of the sole peak for peptides having unimodal behavior, while orange and gray circles are the higher and the lower collision energies, respectively, for bimodal peptides. Dashed and dotted lines represent linear fits.
The large residual variance around the trend line points to further effects determining the optimum. As highlighted in our previous paper,30 the majority of the investigated peptides have +2 charge and only one basic residue (C-terminal R or K). This limits the extent to which the effect of factors like position and number of charged residues or potential presence of salt bridges can be assessed. At the same time, the variance of the +2 peptides with one basic residue is comparable to that of the full data set, suggesting that these factors alone cannot account for the observations and warranting further studies.
We also investigated the effect of precursor intensity on the score and on the optimal collision energy trend. In general, we found no strong correlation between the peptide intensity and the maximum achievable peptide score (see Supporting Information S7). Further, the typical run-to-run variations by ca. a factor of two in peptide precursor intensity do not translate into variations in score (see Supporting Information S7). Hence, the somewhat different timing of the precursor selection for a certain peptide between LC-MS/MS experiments does not affect the determination of optimal collision energy. Finally, optimal collision energies of peptides with higher intensity weakly tend to deviate less from the trend line, but there is no systematic relationship between the spread and the intensity (see Supporting Information S7).
To compare the three examined search engines, we displayed all of the obtained trend lines, together with those for Mascot from our earlier work, in Figure 3. The scoring functions are very different in the three search engines, which apparently gives rise to slightly different trends, particularly in the case of the lower energy peak of bimodal peptides. Apparently, Andromeda and Byonic favor somewhat lower optimum energies than Mascot in the m/z region of ca. 400–600, containing most of the analyzed peptides. On the other hand, considering the sizeable residual variance of the optimum energy around the trend lines (recall Figure 2), we can still conclude that the choice of search engine has only a minor effect on the notion of the “good spectrum” and, hence, on the optimal collision energy.
Figure 3.
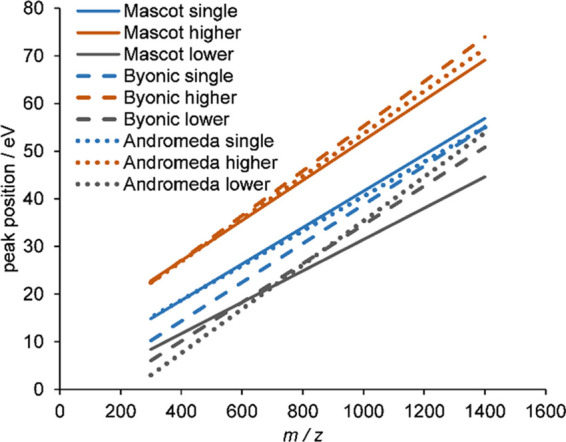
Linear fits to the optimal collision energy vs m/z data for doubly charged peptides. Solid lines represent the Mascot search engine results,30 while dashed and dotted lines belong to the Byonic and Andromeda search engine, respectively. Blue, single optimum for unimodal peptides; orange, higher energy optimum for bimodal peptides; gray, lower energy optimum for bimodal peptides.
Performance Gain over Factory Settings Using a Single LC-MS/MS Run
The results on individual peptides for all three search engines revealed reasonably good m/z-dependent linear trends for the optimum energies. These can form a basis for the selection of collision energies in a practical DDA measurement run, where only the m/z (but not the identity of the peptide) is known at the time the collision energy is chosen. To explore the potential proteomics performance gain via this approach, we used the results of the previous section to set up several m/z-to-collision energy mappings for a single LC-MS/MS run and tested the latter in actual measurements. Due to the close similarity of trend lines obtained for the three different search engines, we based the LC-MS/MS setups on the equations of the Mascot results, and we do not expect this choice to have any material impact on the conclusions.
We created five different setups (see Figure 4 and Supporting Information S8): (1) the first setup corresponded to the line fitted to the single Mascot score optimum of unimodal peptides vs m/z and was called 100%; (2) the second setup corresponded to an energy of 135% of the first setup (and was also called as such) and practically coincided with the fit to the higher energy Mascot score optimum of bimodal peptides; (3) 70% of the first setup, being an approximation of the lower energy Mascot score optimum of bimodal peptides; (4) a stepped setting, involving 70 and 100% in a 50–50% time distribution; and (5) another stepped method, combining the 70, 100, and 135% settings in equal times. The latter two methods were motivated by the observation that the unimodal optimum falls in between the optima for the bimodal peptides. Hence, a combination of these two or three collision energies within one LC-MS/MS run might provide favorable conditions for the measurement of both peptide classes simultaneously. As a reference, we also tested the manufacturer’s recommendation for protein identification, referred to as factory settings. Five repeated runs were recorded for each applied collision energy setup.
Figure 4.
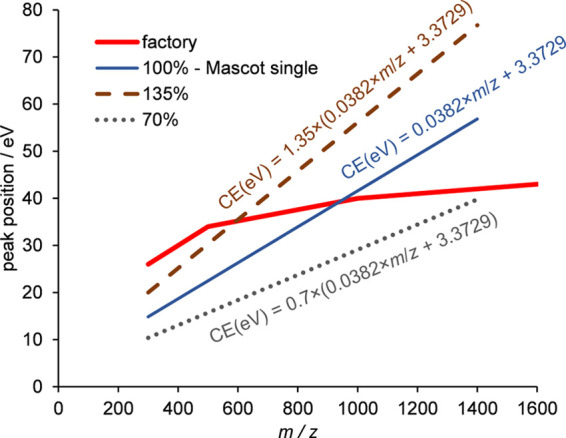
Compared energy settings in eV as a function of m/z for +2 peptides. (Results for +3 peptides can be found in Supporting Information S8.) Red line, factory setting (see Supporting Information S8 for equations). Blue line, 100% (the line fitted to the Mascot score optimum of unimodal peptides). Dashed brown line, 135% (practically the line fitted to the higher energy Mascot score optimum for bimodal peptides). Dotted gray line, 70% (practically the line fitted to the lower energy Mascot score optimum for bimodal peptides). The two applied stepped methods combine the 100 and 70% settings (blue and gray lines) and 100, 70, and 135% settings (blue, gray, and brown lines), respectively.
The performance of the various collision energy settings was characterized by the number of identified peptides and proteins. To bring the three studied search engines to the same platform, we determined the numbers by importing the raw results into Scaffold software and using Scaffold’s criteria for peptide and protein identification (see the section on Data Analysis). The values of the five repeats were averaged, and we then calculated the performance gain in % relative to the factory settings. All gains for all three search engines are summarized in Table 2, while Figure 5 illustrates the numbers for the Mascot search engine (similar bar charts for the other search engines can be found in Supporting Information S9).
Table 2. Performance Gain in Terms of Increase in the Number of Identified Peptides and Proteinsa.
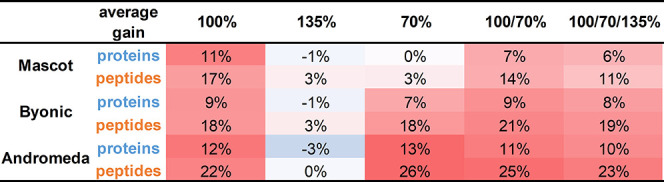
Averages over five repeats, obtained using various collision energy settings and analyzed by three different search engines, are shown, expressed as and color-coded on the basis of the gain in % over the factory setting (darker red means higher gains).
Figure 5.
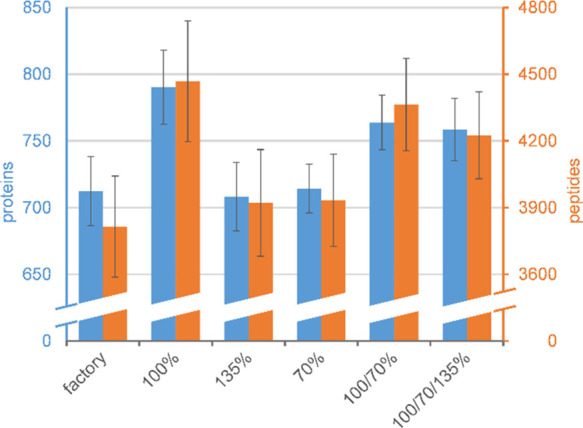
Number of identified peptides and proteins as average of five repeats at several collision energy settings analyzed by the Mascot search engine and Scaffold. Left-hand-side bars and scale (blue), number of proteins; right-hand-side bars and scale (orange), number of peptides. Error bars indicate ±1 standard deviation.
As can be seen, the collision energy setting of 100% means a significant gain for each search engine, raising the number of identified proteins from 712 to 790 for Mascot, from 878 to 957 for Byonic, and from 832 to 929 for Andromeda. In contrast, the high energy setting (135%) brings hardly any advantage over the manufacturer’s recommendations. The impact of the low energy setting (70%) is strikingly different among the three engines, performing best for Andromeda, on par with 100% for Byonic, and no better than the factory setting for Mascot. This difference might be related to the above-observed difference in the low-energy optima for the three engines.
Generally speaking, the stepped settings do not seem to show signs of synergism; the observed gains are usually between those of their constituents. Only in one case, for Byonic, was the combination of 100/70% marginally better than either 100 or 70%. In summary, 100% turned out to be the best setting for Mascot, 100/70% was the best performer for Byonic, and 70% was the most efficient for Andromeda, but several other settings yielded comparable results. In all cases, the best methods provided a gain of ∼10% in the number of proteins and ∼20% in the number of peptides (see Table 2).
Since better collision energy leads to improved spectrum quality, we expect not only a higher number of identified compounds but also higher confidence in them. To confirm this expectation, for each search engine, the best collision energy setting for a single LC-MS/MS run (as determined above) and the factory setting were also compared in terms of identification scores of the common peptides and sequence coverage of common proteins, as determined by Serac and Scaffold, respectively. The gains were determined as averages over three pairs of LC-MS/MS runs. As is apparent from Figure 6, the collision energy optimization process significantly improved the DDA analysis. Both the identification confidence of peptides and the sequence coverage of identified proteins increased significantly (on the order of 20%) for all three search engines applied. Andromeda profited most from the fine-tuning though it had slightly lower sequence coverage values at the factory setting to begin with. The larger sequence coverage values are in line with the increased number of peptides.
Figure 6.
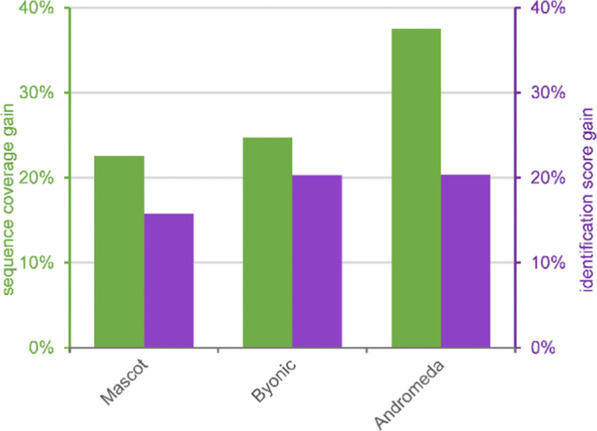
Performance gain of the best collision energy method for a single LC-MS/MS over factory setting in terms of identification scores and sequence coverage of proteins. Left-hand-side bars and scale (green), sequence coverage; right-hand-side bars and scale (purple), identification scores.
Performance Gain Using a Combination of Multiple LC-MS/MS Runs
As noted before,
the investigation of individual peptides revealed
two types of behavior (unimodal and bimodal),30 and it seemed logical to use two different collision energy settings.
Besides combining them within one run (see above), we studied the
effect of using them in two separate runs and aggregating the results
via data processing. Specifically, we tested the number of identified
peptides and proteins from the joint processing of two LC-MS/MS measurements,
one run with the 100% method and another one collected at 70% collision
energy setting. Since repetition of runs may already increase the
number of identified peptides/proteins,43 the reference we took is the combination of two runs, both taken
at the best setting for single LC-MS/MS runs. Combined data analysis
was performed by Scaffold using identical criteria as above. The procedure
was carried out for all possible combinations of the repeated measurements.
Where identical runs were combined, we created all possible pairs
from the five repetitions, leading to 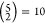 combinations. Where different runs were
combined, five repetitions from one setting and five repetitions from
the other setting meant 5 × 5 = 25 combinations. The results
over these 10 or 25 combinations were then averaged. Figure 7 depicts the results for the
number of proteins together with the single run at factory setting
and the optimum method for the given search engine as comparisons.
The number of identified peptides shows very similar trends and is
presented in Supporting Information S10. As can be seen, two measurements provided 10–20% more identified
proteins than a single LC-MS/MS run, but the use of two runs with
different collision energy settings could not provide a consistent
benefit over a repetition at the optimum setting. In fact, the latter
resulted in more identified proteins (by 4% for Mascot) or performed
similarly well (in the cases of Byonic and Andromeda) to the combination
of 100 and 70% methods.
combinations. Where different runs were
combined, five repetitions from one setting and five repetitions from
the other setting meant 5 × 5 = 25 combinations. The results
over these 10 or 25 combinations were then averaged. Figure 7 depicts the results for the
number of proteins together with the single run at factory setting
and the optimum method for the given search engine as comparisons.
The number of identified peptides shows very similar trends and is
presented in Supporting Information S10. As can be seen, two measurements provided 10–20% more identified
proteins than a single LC-MS/MS run, but the use of two runs with
different collision energy settings could not provide a consistent
benefit over a repetition at the optimum setting. In fact, the latter
resulted in more identified proteins (by 4% for Mascot) or performed
similarly well (in the cases of Byonic and Andromeda) to the combination
of 100 and 70% methods.
Figure 7.
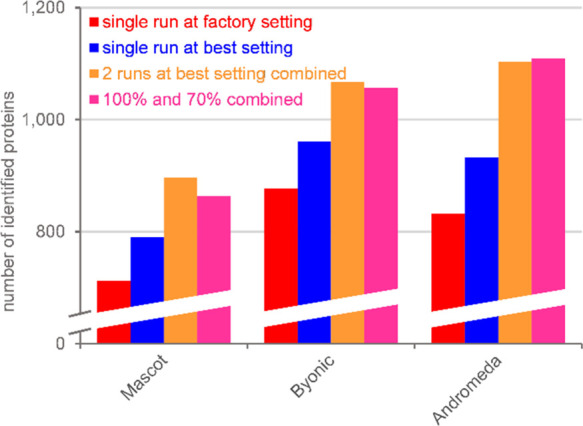
Number of identified proteins using several protocols analyzed by various search engines. Red, single run at factory setting; blue, single run at optimal setting; orange, two runs at optimal setting combined; and pink, two runs—one at 100% and another one at 70% collision energy setting combined.
Practical Approach to Collision Energy Optimization
In the above sections, we demonstrated the potential gain in performance of proteomics measurements from a collision energy optimization directly targeting identification scores. The steps we performed provided us with a general picture of energy-dependent peptide behavior, but the actual collision energy settings are specific to our instrument. Comparison of spectra of model compounds44 and of large sets of peptides45 across instruments relevant for proteomics (QTof, Orbitrap) indicates that energy dependence is broadly similar but the optimal energies may differ to some extent, hampering the direct transfer of the optimized settings across instruments. In line with this, a brief survey of the recent proteomics literature suggests some variation in the employed collision energy settings, but the authors typically did not provide any details regarding their choice, hinting that no targeted optimization was carried out. Here, we suggest a simple approach to transfer our optimization concept to any other instrument, which has the potential to bring about comparable performance gains. A key step of this optimization is to find the equivalent of the 100% trend line. While here it was obtained from hundreds of peptides, we propose that it can also be determined from a handful of carefully selected ones. We suggest carrying out energy-dependent measurements focusing only on a few peptides from the HeLa standard that are identifiable in a wide energy range, show unimodal behavior, and have their Mascot optimum energy very close to the 100% line. After scoring the results with Mascot and identifying the best energy for each peptide, the 100% line can be fitted for the alternative instrument. We provide a collection of peptides recommended for this purpose in Supporting Information S11.
To support and strengthen our conclusions and prove the utility of the above-presented collision energy optimization protocol, we designed and performed several further experiments. First, we carried out a performance check on E. coli digest in the above-described manner using our Bruker QTof instrument (see Supporting Information S12). A 300 ng sample was injected in each LC-MS/MS run, and four replicates were measured. Applying the optimized collision energy values, we could achieve gains of 5–13 and 8–18% over the factory settings in the numbers of identified proteins and peptides, respectively, depending on the search engine used. These findings corroborate the generality of our approach to the tryptic digest sample.
As a next step, we focused on the transferability to another mass spectrometer. To this end, we carried out LC-MS/MS measurements on HeLa tryptic digest using Thermo Fusion Orbitrap equipment with varying HCD collision energies from NCE = 13% to 45% (see Supporting Information S13). From these measurements, most of the reference peptides presented in Table S2 could be identified at several collision energy settings. Based on the well-defined energy-dependent behavior of the Mascot score, 20 peptides were chosen for benchmarking purposes covering the whole m/z range (see Table S2). Since NCE values of Thermo instruments already contain a charge factor, we deliberately refrained from separate calibration for different charge states. The optimal collision energy setting was determined by the fitting procedure of the Mascot score vs collision energy curves of the 20 peptides, and NCE = 28% was obtained as average. Comparison of the default—NCE = 35%—and the optimized method in two or three replicates resulted in 5–13% more proteins and 8–28% more peptides upon optimization (see Supporting Information S14). These results clearly show the applicability of the designed protocol to set up other instruments.
Finally, we repeated the experiments on the Thermo Obritrap Fusion mass spectrometer operating in the low-energy CID (ion trap—IT) fragmentation mode (see Supporting Information S13). Since in ITs the parent ion is selectively energized and the energy of the dissociating ion does not change with increasing excitation, typically the increase of collision energy has minimal effect on the mass spectra.46−48 Therefore, no significant collision energy dependence of the identification scores and number of identified proteins/peptides is expected. Indeed, despite the large investigated collision energy range (NCE from 19 to 43%), both the fragments itself and their relative intensity were the same in the MS/MS spectra and the performance of the LC-MS/MS runs remained constant.
Conclusions
For tandem mass spectrometry, collision energy is of key importance in determining spectrum characteristics. Bottom-up proteomics relies on identification of peptides via matching their MS/MS spectra against a database. In this work, we examined how the confidence of peptide identification (the “score”) depends on the collision energy and what the optimum value might be, for two widely used search engines, Byonic and Andromeda (as part of the MaxQuant tool). We used our findings, together with our earlier results30 on Mascot, to design optimized proteomics workflows and assessed the performance of the latter on an actual proteomics sample. The main conclusions we could draw from our work are as follows.
As a function of collision energy, Byonic and Andromeda scores for identification of a given peptide may simply show a maximum (“unimodal behavior”). But for a significant fraction of peptides, the dependency is more sophisticated and can be characterized either by two separate maxima or by a broad plateau (“bimodal behavior”). These observations are in line with our earlier findings on Mascot and corroborate that the common cause might be the different energy dependences of b- and y-type fragment ions.30
Across all peptides, the optimum collision energy depends linearly on the peptide ion m/z with significant residual variance. Different charges and the different classes (unimodal and bimodal) follow separate trend lines. On the other hand, different search engines display very similar trends for identical categories.
Knowledge of the various m/z-dependent trends in optimum collision energies allowed us to design experimental workflows with different collision energy settings that can be used in the LC-MS/MS measurements of a complex sample in its entirety. We assessed the performance of single-run and two-run workflows on a tryptic HeLa digest standard.
For all search engines, the optimized workflows outperformed the factory settings according to several key measures of proteomics. The optimization increased the number of identified peptides and proteins, sequence coverage, and identification score of the individual peptides. Gains in the various measures ranged from 10 to 40% for single-run workflows. Dedicated two-run workflows did not provide additional improvement over that expected by simply repeating measurements.
Even though differences between search engines were minor in peptide-level trends, the best performing workflows were different for them. Mascot was found to yield optimal results only in a narrow energy range. Byonic and Andromeda showed less clear preferences, but they, Andromeda in particular, tend to work better with lower energies.
Confidence in the identification of a peptide from an MS/MS spectrum stems from characteristics that are formed in a complex interplay of many energy-dependent fragmentation processes. Our investigations highlight the importance of optimizing the collision energy directly with the database search scores as target and also suggest that the specific search engine should be taken into account when the experimental settings are fine-tuned. We recommended a simple protocol and provided reference data to carry out this fine-tuning on other mass spectrometers widely used in proteomics. As an example, we demonstrated the usefulness of this protocol on an Orbitrap instrument.
Acknowledgments
Á.R. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. Funding from the National Research, Development and Innovation Office (NKFIH PD-132135 and K 131762) is gratefully acknowledged. The research was supported by GINOP-2.3.3-15-2016-00020.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00518.
S1, details of nano-LC-MS/MS measurements; S2, collision energy settings; S3, determination of the optimal collision energy; S4, annotated MS/MS spectra at the Byonic and Mascot collision energy optima; S5, results of Byonic log Prob values; S6, results of triply charged peptides for Byonic and Andromeda score values; S7, effect of precursor intensity; S8, tested energy settings for +3 peptides; S9, number of identified peptides and proteins using various energy settings for Byonic and Andromeda search engines; S10, number of identified peptides using several protocols analyzed by various search engines; S11, peptides recommended for determination of “100%” trend line on other mass spectrometers; S12, obtained performance gain on Bruker Maxis II ETD QTof instrument via collision energy optimization from E. coli tryptic digest standard; S13, details of nano-LC-MS/MS measurements and benchmarking of Thermo Orbitrap Fusion instrument; S14, obtained performance gain on Thermo Orbitrap Fusion instrument with HCD fragmentation via collision energy optimization on HeLa tryptic digest standard (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Han X.; Aslanian A.; Yates J. R. Mass Spectrometry for Proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490. 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez P.; Müller M.; Appel R. D. Automated Protein Identification by Tandem Mass Spectrometry: Issues and Strategies. Mass Spectrom. Rev. 2006, 25, 235–254. 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- Domon B.; Aebersold R. Mass Spectrometry and Protein Analysis. Science 2006, 312, 212–217. 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Vitek O.; Aebersold R. Analysis and Validation of Proteomic Data Generated by Tandem Mass Spectrometry. Nat. Methods 2007, 4, 787–797. 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Yates J. R.; Ruse C. I.; Nakorchevsky A. Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- Holl S.; Mohammed Y.; Zimmermann O.; Palmblad M. Scientific Workflow Optimization for Improved Peptide and Protein Identification. BMC Bioinformatics 2015, 16, 284–296. 10.1186/s12859-015-0714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M. L.; Savitski M. M.; Zubarev R. A. Improving Protein Identification Using Complementary Fragmentation Techniques in Fourier Transform Mass Spectrometry. Mol. Cell. Proteomics 2005, 4, 835–845. 10.1074/mcp.T400022-MCP200. [DOI] [PubMed] [Google Scholar]
- Baldwin M. A. Protein Identification by Mass Spectrometry. Mol. Cell. Proteomics 2004, 3, 1–9. 10.1074/mcp.R300012-MCP200. [DOI] [PubMed] [Google Scholar]
- Szabo Z.; Janaky T. Challenges and Developments in Protein Identification Using Mass Spectrometry. TrAC, Trends Anal. Chem. 2015, 69, 76–87. 10.1016/j.trac.2015.03.007. [DOI] [Google Scholar]
- Olsen J. V.; Mann M. Status of Large-Scale Analysis of Post-Translational Modifications by Mass Spectrometry. Mol. Cell. Proteomics 2013, 12, 3444–3452. 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias L. A.; Santos I. C.; Brodbelt J. S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2020, 92, 227–251. 10.1021/acs.analchem.9b04859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A. I. A Survey of Computational Methods and Error Rate Estimation Procedures for Peptide and Protein Identification in Shotgun Proteomics. J. Proteomics 2010, 73, 2092–2123. 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J. C.; Choudhary J. S.. PSM Scoring and Validation. In Proteome Informatics; Bessant C., Ed.; The Royal Society of Chemistry, 2017; pp 69–92. [Google Scholar]
- Neta P.; Simon-Manso Y.; Yang X.; Stein S. E. Collisional Energy Dependence of Peptide Ion Fragmentation. J. Am. Soc. Mass Spectrom. 2009, 20, 469–476. 10.1016/j.jasms.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Haller I.; Mirza U. A.; Chait B. T. Collision Induced Decomposition of Peptides. Choice of Collision Parameters. J. Am. Soc. Mass Spectrom. 1996, 7, 677–681. 10.1016/1044-0305(96)85613-3. [DOI] [PubMed] [Google Scholar]
- Baker E. S.; Tang K.; Danielson W. F.; Prior D. C.; Smith R. D. Simultaneous Fragmentation of Multiple Ions Using IMS Drift Time Dependent Collision Energies. J. Am. Soc. Mass Spectrom. 2008, 19, 411–419. 10.1016/j.jasms.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein C. A.; Gafken P. R.; Martin D. B. Collision Energy Optimization of B-and y-Ions for Multiple Reaction Monitoring Mass Spectrometry. J. Proteome Res. 2011, 10, 231–240. 10.1021/pr1004289. [DOI] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Abbatiello S. E.; Zhang S.; Whiteaker J. R.; Paulovich A. G.; Carr S. A.; MacCoss M. J. Effect of Collision Energy Optimization on the Measurement of Peptides by Selected Reaction Monitoring (SRM) Mass Spectrometry. Anal. Chem. 2010, 82, 10116–10124. 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Graaf E. L.; Altelaar M. A. F.; Van Breukelen B.; Mohammed S.; Heck A. J. R. Improving SRM Assay Development: A Global Comparison between Triple Quadrupole, Ion Trap, and Higher Energy CID Peptide Fragmentation Spectra. J. Proteome Res. 2011, 10, 4334–4341. 10.1021/pr200156b. [DOI] [PubMed] [Google Scholar]
- Wu C.; Shi T.; Brown J. N.; He J.; Gao Y.; Fillmore T. L.; Shukla A. K.; Moore R. J.; Camp D. G.; Rodland K. D.; et al. Expediting SRM Assay Development for Large-Scale Targeted Proteomics Experiments. J. Proteome Res. 2014, 13, 4479–4487. 10.1021/pr500500d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood C. A.; Eastham A.; Lee L. W.; Risler J.; Mirzaei H.; Falkner J. A.; Martin D. B. Rapid Optimization of MRM-MS Instrument Parameters by Subtle Alteration of Precursor and Product m/z Targets. J. Proteome Res. 2009, 8, 3746–3751. 10.1021/pr801122b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P.; Aebersold R. Selected Reaction Monitoring–Based Proteomics: Workflows, Potential, Pitfalls and Future Directions. Nat. Methods 2012, 9, 555–566. 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Holman S. W.; Sims P. F. G.; Eyers C. E. The Use of Selected Reaction Monitoring in Quantitative Proteomics. Bioanalysis 2012, 4, 1763–1786. 10.4155/bio.12.126. [DOI] [PubMed] [Google Scholar]
- He L.; Anderson L. C.; Barnidge D. R.; Murray D. L.; Hendrickson C. L.; Marshall A. G. Analysis of Monoclonal Antibodies in Human Serum as a Model for Clinical Monoclonal Gammopathy by Use of 21 Tesla FT-ICR Top-Down and Middle-Down MS/MS. J. Am. Soc. Mass Spectrom. 2017, 28, 827–838. 10.1007/s13361-017-1602-6. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. Sequencing Larger Intact Proteins (30-70 KDa) with Activated Ion Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2018, 29, 140–149. 10.1007/s13361-017-1808-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Révész Á.; Rokob T. A.; Jeanne Dit Fouque D.; Hüse D.; Háda V.; Turiák L.; Memboeuf A.; Vékey K.; Drahos L. Optimal Collision Energies and Bioinformatics Tools for Efficient Bottom-up Sequence Validation of Monoclonal Antibodies. Anal. Chem. 2019, 91, 13128–13135. 10.1021/acs.analchem.9b03362. [DOI] [PubMed] [Google Scholar]
- Hinneburg H.; Stavenhagen K.; Schweiger-Hufnagel U.; Pengelley S.; Jabs W.; Seeberger P. H.; Silva D. V.; Wuhrer M.; Kolarich D. The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. J. Am. Soc. Mass Spectrom. 2016, 27, 507–519. 10.1007/s13361-015-1308-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich J. K.; Pinto A. F. M.; Yates J. R. Energy Dependence of HCD on Peptide Fragmentation: Stepped Collisional Energy Finds the Sweet Spot. J. Am. Soc. Mass Spectrom. 2013, 24, 1690–1699. 10.1007/s13361-013-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke D.; Hung C. W.; Cassidy L.; Tholey A. Optimized Fragmentation Conditions for ITRAQ-Labeled Phosphopeptides. J. Proteome Res. 2013, 12, 2755–2763. 10.1021/pr400113n. [DOI] [PubMed] [Google Scholar]
- Révész Á.; Rokob T. A.; Jeanne Dit Fouque D.; Turiák L.; Memboeuf A.; Vékey K.; Drahos L. Selection of Collision Energies in Proteomics Mass Spectrometry Experiments for Best Peptide Identification: Study of Mascot Score Energy Dependence Reveals Double Optimum. J. Proteome Res. 2018, 17, 1898–1906. 10.1021/acs.jproteome.7b00912. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J. C.; Creasy D. M.; Cottrell J. S. Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data. Electrophoresis 1999, 20, 3551–3567. . [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Cox J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Bern M.; Cai Y.; Goldberg D. Lookup Peaks: A Hybrid of de Novo Sequencing and Database Search for Protein Identification by Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 1393–1400. 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- Matrix Science, Mascot Search. http://www.matrixscience.com/blog/back-to-basics-optimize-your-search-parameters.html#comments. July 16, 2018.
- MaxQuant. https://www.maxquant.org/ (accessed January 2019).
- Bern M.; Kil Y. J.; Becker C. Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinf. 2012, 40, 13.20.1–13.20.14. 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourakis M. I. A.Levmar: Levenberg-Marquardt Nonlinear Least Squares Algorithms in C/C++, Version 2.6; Institute of Computer Science, Foundation for Research and Technology: Hellas, Heraklion, Greece. http://www.ics.forth.gr/~lourakis/levmar/ (accessed October 2017).
- Pearson T.PGPLOT 5.2.2; California Institute of Technology. http://www.astro.caltech.edu/~tjp/pgplot/ (accessed October 2017).
- Matrix Science, Search Paramters. http://www.matrixscience.com/help/search_field_help.html (accessed January 2019).
- Zhang Y.; Ficarro S. B.; Li S.; Marto J. A. Optimized Orbitrap HCD for Quantitative Analysis of Phosphopeptides. J. Am. Soc. Mass Spectrom. 2009, 20, 1425–1434. 10.1016/j.jasms.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Thermo Fisher Scientific Product Support Bulletin 104. Normalized Collision Energy Technology. http://tools.thermofisher.com/content/sfs/brochures/PSB104-Normalized-Collision-Energy-Technology-EN.pdf (accessed January 2019).
- Tabb D. L.; Vega-Montoto L.; Rudnick P. A.; Variyath A. M.; Ham A. J. L.; Bunk D. M.; Kilpatrick L. E.; Billheimer D. D.; Blackman R. K.; Cardasis H. L.; et al. Repeatability and Reproducibility in Proteomic Identifications by Liquid Chromatography-Tandem Mass Spectrometry. J. Proteome Res. 2010, 9, 761–776. 10.1021/pr9006365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazsó F. L.; Ozohanics O.; Schlosser G.; Ludányi K.; Vékey K.; Drahos L. Quantitative Comparison of Tandem Mass Spectra Obtained on Various Instruments. J. Am. Soc. Mass Spectrom. 2016, 27, 1357–1365. 10.1007/s13361-016-1408-y. [DOI] [PubMed] [Google Scholar]
- Szabo D.; Schlosser G.; Vékey K.; Drahos L.; Révész Á. Collision Energies on QTof and Orbitrap Instruments: How to Make Proteomics Measurements Comparable?. J. Mass Spectrom. 2020, (in press) 10.1002/jms.4693. [DOI] [PubMed] [Google Scholar]
- March R. E. An Introduction to Quadrupole Ion Trap Mass Spectrometry. J. Mass Spectrom. 1997, 32, 351–369. . [DOI] [Google Scholar]
- McLuckey S. A.; Goeringer D. E. Slow Heating Methods in Tandem Mass Spectrometry. J. Mass Spectrom. 1997, 32, 461–474. . [DOI] [Google Scholar]
- Cooks R. G.; Kaiser R. E. Jr. Quadrupole Ion Trap Mass Spectrometry. Acc. Chem. Res. 1990, 23, 213–219. 10.1021/ar00175a002. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.