

SUMMARY
Leukemia stem cells (LSCs) are believed to have more distinct vulnerabilities than the bulk acute myeloid leukemia (AML) cells, but their rarity and the lack of universal markers for their prospective isolation hamper their study. We report that genetically clonal induced pluripotent stem cells (iPSCs) derived from an AML patient and characterized by exceptionally high engraftment potential give rise, upon hematopoietic differentiation, to a phenotypic hierarchy. Through fate-tracking experiments, xenotransplantation, and single-cell transcriptomics, we identify a cell fraction (iLSC) that can be isolated prospectively by means of adherent in vitro growth that resides on the apex of this hierarchy and fulfills the hallmark features of LSCs. Through integrative genomic studies of the iLSC transcriptome and chromatin landscape, we derive an LSC gene signature that predicts patient survival and uncovers a dependency of LSCs, across AML genotypes, on the RUNX1 transcription factor. These findings can empower efforts to therapeutically target AML LSCs.
In Brief
Wesely et al. report that AML-iPSC derived hematopoietic cells are hierarchically organized and contain cells with hallmark features of LSCs (iLSCs). Through integrative genomic studies of bulk and single-cell transcriptomes and chromatin accessibility, they derive a LSC gene signature and identify RUNX1 as an AML LSC dependency with therapeutic implications.
Graphical abstract
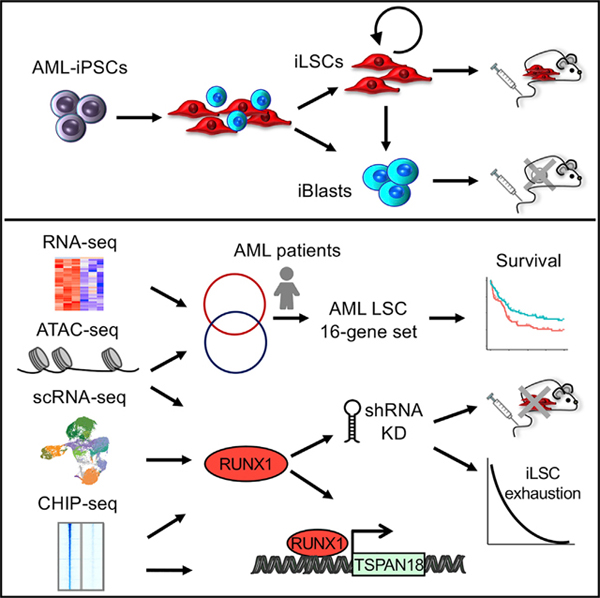
INTRODUCTION
The malignant cells in acute myeloid leukemia (AML) display hierarchical organization with leukemia stem cells (LSCs) residing on the apex (Kreso and Dick, 2014; Reinisch et al., 2015). AML LSCs share many properties with normal hematopoietic stem cells (HSCs), including self-renewal, and can thus initiate and maintain AML by giving rise to identical, as well as more differentiated, daughter cells that cannot propagate the disease. Human AML LSCs are classically studied in xenograft models whereby cells obtained from patients are transplanted into immunodeficient mice. The definition and properties of human LSCs are dictated by their observed properties in these assays: (1) self-renewal ability, defined as the ability to give rise to leukemic engraftment that can be maintained over serial transplantation; and (2) the ability to give rise to more differentiated progeny that are unable to engraft (Kreso and Dick, 2014; Thomas and Majeti, 2017).
Although there is substantial evidence for the existence of both murine and human LSCs, significant challenges for their study exist. Their similarities to normal HSCs, their rarity, and the unavailability of specific and universal immunophenotypic markers that distinguish LSCs from the rest of AML cells make their prospective isolation, study, and use in drug discovery challenging (Ho et al., 2016; Pollyea et al., 2014).
Here, we report that leukemia cells derived through in vitro differentiation of genetically clonal induced pluripotent stem cells (iPSCs) derived from an AML patient are hierarchically organized. A subpopulation of cells growing in adherence to plastic resides in the apex of this hierarchy and is highly enriched for cells with an HSC immunophenotype and leukemia-initiating potential. We characterize this hierarchical organization by cellular replating assays, mathematical modeling, and single-cell transcriptomics. We then use this human LSC model to characterize the transcriptome and chromatin landscape of AML LSCs and identify specific vulnerabilities. By integrative genomics analyses, we derive an LSC gene signature and reveal a role for the RUNX1 transcription factor (TF) in the maintenance of human LSCs.
RESULTS
Hematopoietic Cells Derived from Genetically Clonal AML-iPSC Lines Exhibit Morphologic and Phenotypic Heterogeneity
We and Chao et al. (2017) previously reported that iPSC lines derived from patients with AML (AML-iPSCs) re-establish a leukemic phenotype upon hematopoietic differentiation (Kotini et al., 2017). Although hematopoietic cells derived from normal human pluripotent stem cells (hPSCs; including iPSCs and embryonic stem cells [ESCs]) fail to engraft in immunodeficient mice—a limitation that constitutes a major roadblock to disease modeling and cell therapies (Rowe et al., 2016)—we showed that, in contrast, hematopoietic cells from AML-iPSC lines could engraft (Chao et al., 2017; Kotini et al., 2017). Of note, this is the only report of engraftment of hematopoietic cells derived from hPSCs through directed differentiation without transgenes. In particular, we found that all iPSC lines derived from one specific AML patient (patient #4)—harboring a complex translocation resulting in the loss of chromosome 7q—exhibited exceptionally high engraftment potential in NOD/SCID/IL-2Rγ−/− (NSG) mice, compared to other AML-iPSC lines; could serially transplant a lethal leukemia; and could be propagated long term in vitro as immature CD34+ myeloid cells (Kotini et al., 2017). This was observed in multiple iPSC lines derived from that patient, including iPSC lines derived from a subclone of the patient’s leukemia, harboring an additional KRAS G12D mutation in addition to the translocation (Kotini et al., 2017). In contrast, all other existing AML-iPSC lines, derived from 4 AML patients from different genetic subgroups (Chao et al., 2017; Kotini et al., 2017; unpublished data) showed low-level or no engraftment after intravenous administration into NSG mice. We, therefore, set to further investigate the unusually high engraftment and leukemia-initiating potential of these AML-iPSCs.
We observed that upon transfer of hematopoietic cells derived from these AML-iPSC lines into standard tissue-culture-treated plates on day 14 of hematopoietic differentiation, two cell populations with marked differences in their adherence properties are apparent: one population of cells that firmly attached to the plate and a population of cells growing in suspension. The first population consisted of moderately elongated cells that, when allowed to grow in confluency, formed a monolayer with cobblestone morphology, whereas the second contained round cells with typical hematopoietic progenitor cell morphology (Figure 1A). The same phenomenon was observed in all iPSC lines from this patient, but not in any other AML-iPSC lines (Chao et al., 2017; Kotini et al., 2017; unpublished data). Two lines, AML-4.10 and AML-4.24 (with and without the KRAS G12D mutation, respectively; both harbor the patient’s chromosomal translocation), were further characterized. The adherent cells expressed high levels of HSC and LSC cell surface markers, whereas suspension cells had a higher expression of markers of progenitor cells (Figures 1B, S1A, and S1B). Both fractions expressed comparable levels of the pan-hematopoietic marker CD45 and the myeloid lineage and AML marker CD33 (Figures 1B, S1A, and S1B). Most adherent cells had an HSC immunophenotype CD34+/CD38−/CD90+/CD45RA−/CD49f+ (Figures 1C and S1C). Notably, cells with this immunophenotype are only transiently detected at low levels (below 1%–2%) in hematopoietic differentiation cultures from normal hPSCs (Figure S1D). Both cell fractions could be expanded in culture for at least several weeks (Figure 1D). After manual separation and culture for 3 weeks, the adherent cells maintained the morphology of immature blast cells with a high nucleus-to-cytoplasm ratio, whereas the suspension fraction gave rise to cells with morphological features of more mature myeloid cells, macrophages, and neutrophils (Figures 1E and S1E). Cell cycle analysis showed that the adherent fraction contained more cells in G0/G1 phase, whereas suspension cells had a higher fraction of cells in S phase (Figures 1F and S1F). These observations led us to the hypothesis that the adherent cells may represent a LSC population (thereafter referred to as iLSC) and the suspension cells their less immature progeny (thereafter referred to as iBlasts).
Figure 1. Hematopoietic Cells Derived from AML-iPSCs Show Phenotypic Heterogeneity.
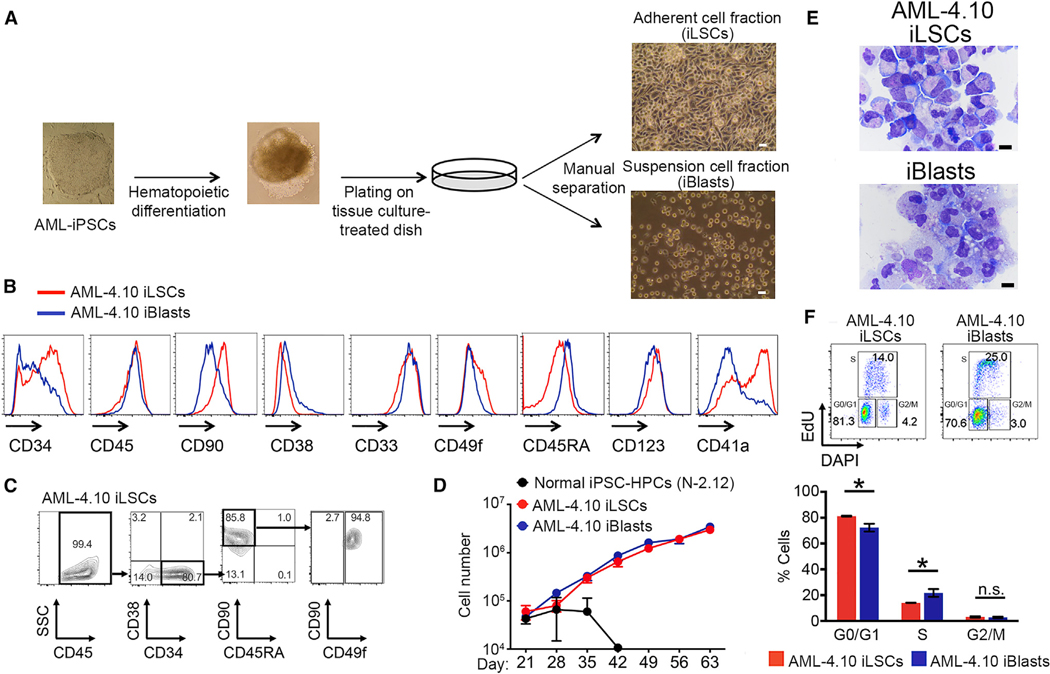
(A) Hematopoietic cells obtained from in-vitro-directed differentiation of AML-iPSCs segregate into an adherent (iLSC) and a suspension (iBIast) cell fraction upon plating on regular tissue-culture-treated dishes. Scale bar, 5 μm.
(B) Expression of hematopoietic markers by flow cytometry in iLSCs and iBlasts from the AML-4.10 iPSC line.
(C) HSC markers in iLSCs from the AML-4.10 iPSC line on day 16 of hematopoietic differentiation.
(D) Expansion of iLSCs and iBlasts from the AML-4.10 iPSC line in liquid culture compared to hematopoietic cells derived from a normal iPSC line, N-2.12 (Kotini et al., 2015, 2017). Mean and SEM of 6 independent experiments are shown.
(E) Wright-Giemsa-stained cytospins of manually separated cells. Scale bar, 10 μm.
(F) Cell cycle analysis of iLSCs and iBlasts from the AML-4.10 iPSC line. Representative flow cytometry plots (top) and cumulative data (bottom) are shown. Mean and SEM from 4 independent experiments are shown. *p < 0.05, t test. See also Figure S1.
The iLSC Fraction Contains the Engraftment Potential and Serially Transplants a Lethal Leukemia
To test this hypothesis, we first transplanted the iLSC and iBlast manually separated cell fractions on day 16 of hematopoietic differentiation into NSG mice (Figure 2A). These experiments showed that the engraftment potential was contained within the iLSC fraction, with the iBlast fraction only rarely giving detectable engraftment at very low levels (Figures 2B and S2A). In contrast, the iLSCs gave rise to robust high-level engraftment in the bone marrow (BM), spleen, and peripheral blood (Figures 2B and S2A). The engrafted cells were primarily CD33+ myeloid cells, consistent with a myeloid leukemia (Figures 2C, 2D, and S2B). The mice transplanted with iLSCs became sick and had enlarged spleens, and their BM was infiltrated with immature human CD45+ cells with iBlast morphology (Figures 2E, 2F, S2C, and S2D). The mouse leukemias originating from iLSCs contained a higher number of phenotypic LSCs (CD34+/CD38−) and were serially transplantable (Figures 2G–2I). Transplantation of BM cells from mice engrafted with iLSCs into secondary recipients resulted in robust engraftment and led to the death of the recipient mice within 3 weeks of transplantation (Figure 2I). The frequency of leukemia-initiating cells (LICs) was estimated by limiting dilution assay transplantation experiments to be approximately 1:5,600 in iLSCs, which is more than 35 times the calculated frequency in the iBlast fraction (−1:200,000) (Figure S2E).
Figure 2. The iLSC Fraction Contains the Engraftment Potential.
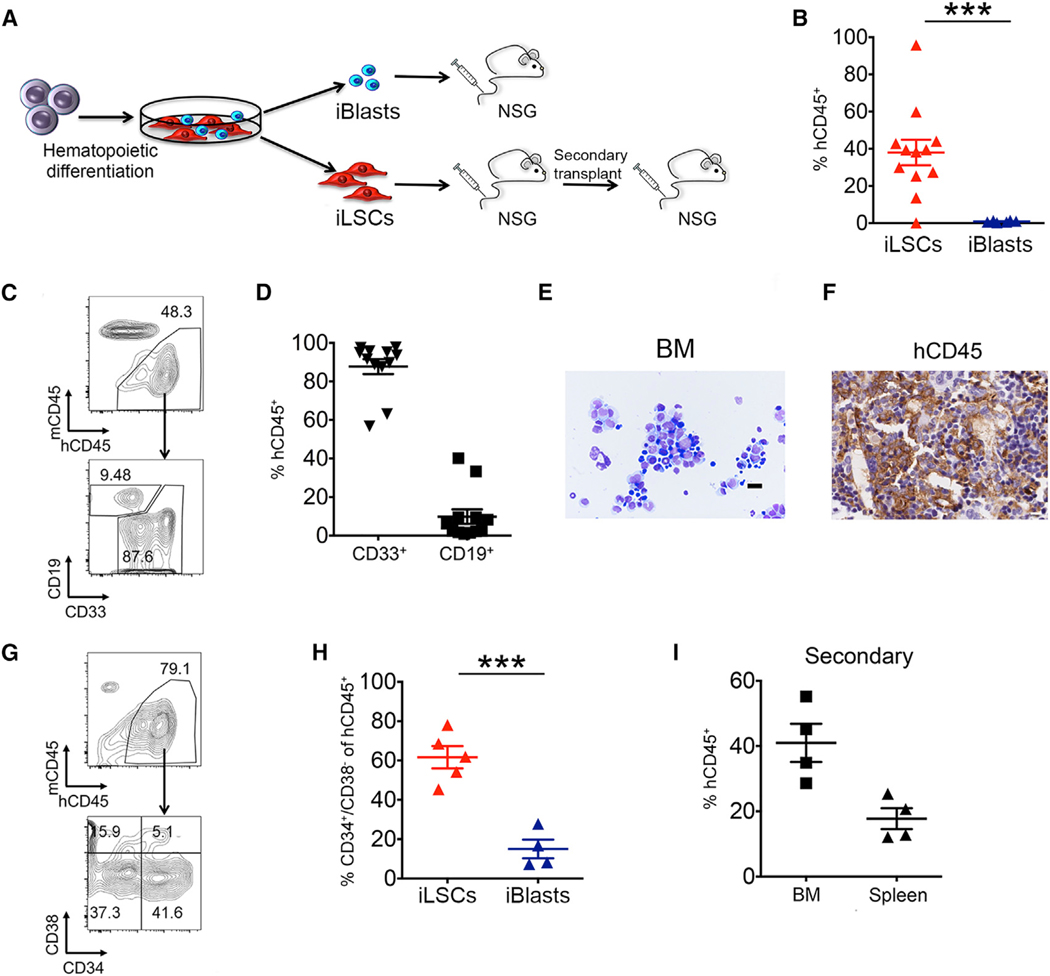
(A) Scheme of transplantation experiments. AML-4.10 and AML-4.24 iPSCs were differentiated along the hematopoietic lineage for 12–14 days and plated on tissue-culture-treated plates. iLSCs and iBlasts were manually separated 2 days later and intravenously injected into sublethally irradiated or busulfan-treated NSG mice. BM cells from mice injected with iLSCs were isolated and transplanted into secondary recipients.
(B) Levels of human engraftment in the BM of mice 8–12 weeks after transplantation. Each data point represents a unique mouse. The mean and SEM from 3 independent transplantation experiments, 2 with iPSC line AML-4.10 and 1 with AML-4.24, are shown. ***p < 0.001, t test.
(C) Representative flow cytometry panels showing human cell engraftment and lineage markers CD33 (myeloid) and CD19 (lymphoid) in the BM of recipient mice 8–12 weeks post-transplantation with iLSCs.
(D) Fraction of myeloid (CD33+) and lymphoid (CD19+) lineage cells within the hCD45+ population in the BM of mice transplanted with iLSCs. Each data point represents a unique mouse. Mean and SEM from 3 independent transplantation experiments are shown.
(E) Wright-Giemsa-stained cytospin of BM cells of a recipient mouse transplanted with iLSCs. Scale bar, 100 μm.
(F) Human CD45 detection by immunohistochemistry in the BM of a recipient mouse transplanted with iLSCs. Scale bar, 5 μm.
(G) Representative flow cytometry panels showing human cell engraftment and human phenotypic LSCs in the BM of recipient mice 8–12 weeks post-transplantation.
(H) Fraction of CD34+/CD38− cells within the hCD45+ population in the BM of mice injected with iLSCs or iBlasts. Each data point represents a unique mouse. Mean and SEM are shown. ***p < 0.001, t test.
(I) Human engraftment levels in the BM and spleen of secondary recipient mice. Each data point represents a unique mouse. Mean and SEM are shown. See also Figure S2.
To test whether the LSC activity of iLSCs is dependent on their adhesion properties, we compared the engraftment of iLSCs cultured for 1 week after manual separation under adherent conditions or under conditions preventing adhesion. No difference was observed in either the immunophenotype or the engraftment potential between the two culture conditions (Figure S2F). Furthermore, iLSCs and iBlasts had a comparable in vivo homing ability (Figure S2G). These results suggest that initial adherence is an LSC property in this model, but it does not per se confer LSC properties. These results argue against a direct link between adhesion and LSC activity in this model but firmly establish that adherent growth can be leveraged for easy prospective enrichment of the iLSC subset.
iLSCs Reside on the Apex of a Phenotypic Hierarchy
To test the hypothesis that iLSCs lie on the top of the phenotypic hierarchy, we first manually separated them from the bulk culture and immunophenotypically assessed the derivative cultures after 7 days. As expected, separation of the iLSCs resulted in enrichment for HSC markers CD34, CD90, CD49f, and CD41a (Figure 3A). After 7 days, the culture became more heterogeneous and immunophenotypically more similar to the starting bulk cells before separation (Figure 3A). This result shows that the iLSCs can reproduce the phenotypic heterogeneity of the bulk population. To further examine the interrelation between iLSCs and iBlasts, we separated iLSCs and iBlasts and cultured them separately for additional 4 weeks (Figure S3A). Each week, iLSCs and iBlasts from each plate were separated, counted, and plated back. The cultures initiated by iLSCs over time became populated with increasing numbers of iBlasts that reached approximately 70%–80% of the total cells by the end of the 4-week period (Figure S3A, left panels). In contrast, cultures initiated by iBlasts contained predominantly iBlasts (> 80%) throughout the culture period (Figure S3A, right panels). These results are consistent with culture dynamics whereby the iLSCs predominantly give rise to iBlasts, whereas movement in the opposite direction, i.e., iBlasts generating iLSCs, is much less frequent, if it happens at all.
Figure 3. The iLSCs Are on the Apex of the Phenotypic Hierarchy.
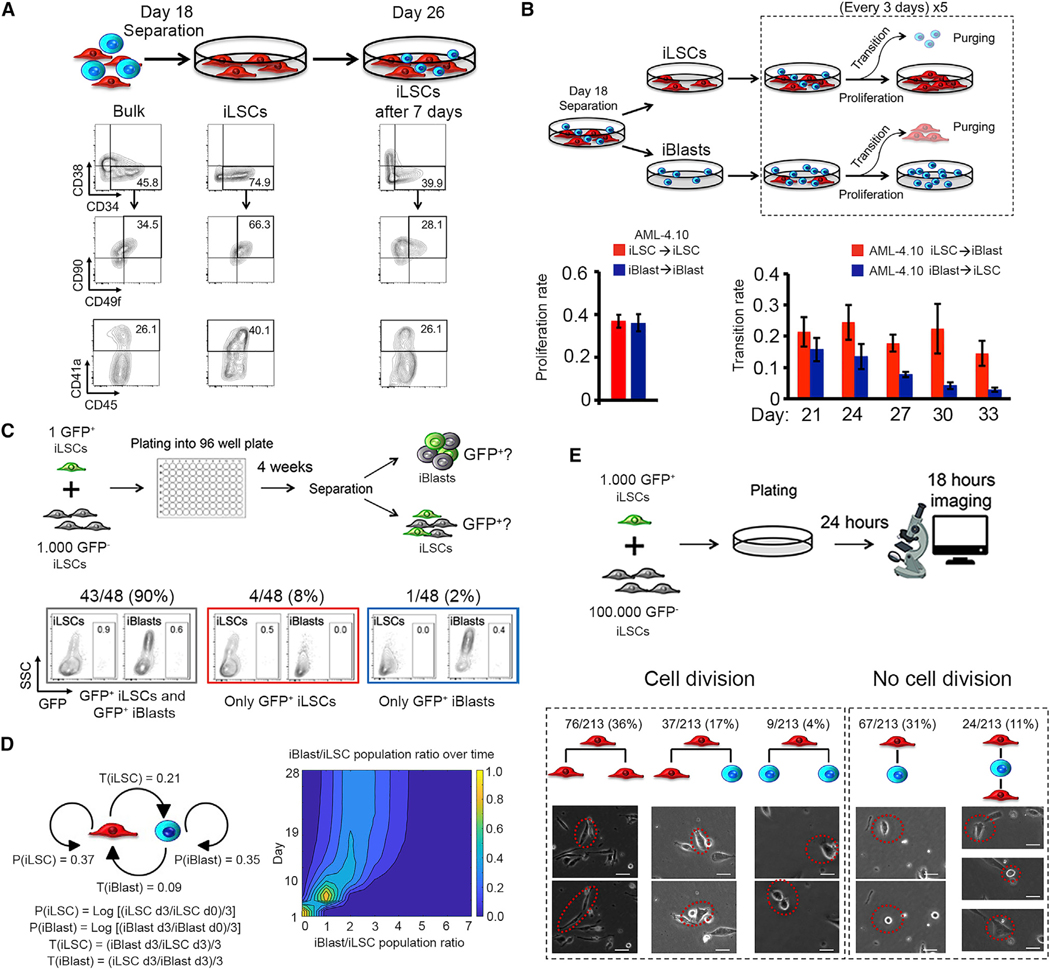
(A) iLSCs were manually separated from the bulk population. Bulk cells and iLSCs immediately after separation, as well as after 7 days in culture, were analyzed for HSC markers.
(B) Top panel: scheme of experimental setup for calculation of proliferation and transition rate of iLSCs and iBlasts. iLSCs and iBlasts, separated on day 18, were counted and purged from the other cell type every 3 days for a total of 15 days. Bottom panels: proliferation rate (left) and transition rate (right) of cells derived from line AML-4.10, calculated as shown in Table S1. Mean and SEM from 3 independent experiments are shown. Proliferation rate is averaged across all time points.
(C) Top panel: schematic over view of the experimental design. Single GFP+ iLSCs were mixed with 1,000 GFP− iLSCs, both derived from the AML-4.10 line, and plated in one well of a 96-well plate. The cells were allowed to grow for a sufficient amount of time (4 weeks) to allowe numeration of total cell numbers and GFP+ cell numbers by cell counts and flow cytometry. Bottom panels: representative flow cytometry panels and cumulative results from 48 wells that contained a GFP+ cell population detectable by flow cytometry.
(D) Scheme of Markov modeling parameters derived from the experiment shown in (B) and Figure S3B (left) and used for a Monte-Carlo simulation that recapitulates the single-cell experiment depicted in (C) (right). Distribution of iBlast/iLSC ratio values for days 0–28 across 1,000 random simulations starting with one single iLSC. For day 1, the iBlast/iLSC ratio is 0 for 100% of the models. Over time, the ratio shifts and converges to a steady-state value with some distribution between the 1,000 random models. The iBlast/iLSC ratio on day 28 shows a mean value of 2.5. P(iLSC): proliferation rate of iLSCs; P(iBlast): proliferation rate of iBlasts; T(iLSC): transition rate from iLSCs to iBlasts; T(iBlast): transition rate from iBlasts to iLSCs.
(E) Top panel: schematic of experimental setup. GFP+ iLSCs from the AML-4.10 line were mixed with unmarked iLSCs, allowed to adhere for 24 h, and imaged continuously for 18 h. Cumulative results and representative pictures of different cell division outcomes, in which an iLSC gives rise either to 2 iLSCs, to 1 iLSC and 1 iBlast, or to 2 iBlasts (from left to right). Representative pictures of cell transitions iLSC→iBlast and iBlast→iLSC without cell division are also shown (right panels). Scale bar, 20 μm. See also Figure S3, Tables S1 and S2, and Video S1.
To further investigate and quantify these population dynamics, we used mathematical modeling. To calculate a proliferation rate for each cell subset, iLSCs and iBlasts, and a transition rate, whereby each subset gives rise to the other (iLSC→iBlast and iBlast→iLSC), we used the following experimental design: separated iLSCs and iBlasts were enumerated every 3 days with simultaneous purging of the other cell subset for a total of 5 time points (Figure 3B). In agreement with the previous observations, iLSCs and iBlasts had similar proliferation rates, but the transition rate iLSC→iBlast was roughly double the iBlast→iLSC transition rate (average values of 0.21 and 0.09, respectively, for line AML-4.10; and 0.16 and 0.09, respectively, for line AML-4.24) (Figures 3B and S3B; Table S1). We next wanted to test the interrelationship between iLSCs and iBlasts at the single-cell level. Because iLSCs could not be grown at very low plating densities, we generated a GFP-marked AML-4.10 line and plated one single GFP+ iLSC per well of a 96-well plate together with 1,000 supporting unmarked iLSCs. After a period of time of 4 weeks (that we found to be sufficient for detection of GFP+ cells in single wells by flow cytometry), the iLSC and iBlast subsets of each individual well were separated and assessed for GFP+ cells. According to a Poisson distribution based on the number of GFP− wells, we confirmed that the majority of wells with GFP+ cells had a single GFP+ iLSC plated (33 out of 48 GFP+ wells) (Figure S3C). The vast majority of informative wells with a GFP+ cell population detectable by flow cytometry (43 out of 48) contained GFP+ cells in both the iLSC and iBlast fraction (Figure 3C). These results show that a single iLSC can give rise to iBlasts.
To further test if the population dynamics of iLSCs and iBlasts can be explained by a Markov model defined by the experimentally calculated proliferation and transition rates and their empirical variance (Figure 3D, left panel), we generated a Monte-Carlo simulation that recapitulates the single-cell experiment depicted in Figure 3C. Specifically, we generated 1,000 independent random simulations of the population dynamics starting with a single iLSC and monitored over 28 days. This showed rapid convergence to a steady-state iBlast/iLSC population ratio (mean = 2.5, standard deviation = 1) (Figure 3D, right panel). To empirically test this predicted long-term population ratio, the absolute iLSC and iBlast cell numbers from 26 wells of cultures originating from a single GFP+ iLSC were counted (Table S2). The empirical average iBlast/iLSC ratio was found to be 3.2, which is very similar to the value predicted by the model. Furthermore, in agreement with these results, the previous bulk culture experiments (Figure S3A) showed similar convergence to a steady-state iBlast/iLSC ratio of approximately 80% iBlasts and 20% iLSCs in both AML-4.10 and AML-4.24, which is close to the predicted steady-state iBlast/iLSC ratio of 2.5, corresponding to 72% iBlasts and 28% iLSCs. Bulk hematopoietic cells from day 24 of hematopoietic differentiation culture of the AML-4.10 line maintained engraftment potential, consistent with the long-term persistence of an LSC population (Figure S3D). Finally, time lapse imaging of cultures of GFP− marked and unmarked iLSCs revealed a predominance of symmetric (iLSC→iLSC→iLSC) divisions, which accounted for 76 of 122 tracked cell divisions (63%) (Figure 3E; Video S1). A derivation of iBlasts from iLSCs occurred through both asymmetric and symmetric cell divisions (iLSC→iLSC+iBlast and iLSC→iBlast+iBlast), with the former being approximately 4 times more frequent than the latter (37 of 122 versus 9 of 122 cell divisions) (Figure 3E; Video S1). Interestingly, a direct conversion of iLSCs to iBlasts and the reciprocal transition of iBlasts to iLSCs without cell division were also observed (Figure 3E; Video S1).
In conclusion, our results so far establish that iLSCs possess both defining properties of LSCs: (1) the ability to serially engraft leukemia; and (2) the ability to give rise to more differentiated cells, the iBlasts, that cannot initiate and propagate leukemia.
Single-Cell Transcriptome Analysis Characterizes Cellular Heterogeneity and Hierarchy within the iLSC Population
To probe the degree of heterogeneity within the iLSCs and iBlasts, we performed single-cell transcriptome analyses in iLSCs, immediately after separation (day 16), as well as after 1 or 2 weeks in culture (day 23 and 30, respectively), in duplicate (Figures S4A–S4F). Unsupervised clustering revealed 13 distinct clusters (Figures 4A, 4B, and S4G). Based on the temporal changes in cluster representation over the 3 time points of the analysis (Figures 4B and S4H), 3 clusters were almost exclusively present at the earliest time point (day 16) and expressed typical HSC genes (Figure 4C). They were, thus, named stem cell I (SC I), SC II, and SC III. Three additional clusters were almost exclusively present at the latest time point (day 30) (Figures 4B and S4H). Based on marker gene expression, they were identified as mast cells (Mast), promonocytes/monocytes (Promono/Mono), and lymphoid progenitors (Lymph P) (Figures 4A, 4B, 4D, and S4G) (Athanasiadis et al., 2017; Pellin et al., 2019; Velten et al., 2017; Villani et al., 2017). The remaining 7 clusters consisted of a mixture of mainly day 23 and day 30 cells. Based on gene expression, they were annotated as follows: one cluster with shared expression of stem cell and megakaryocyte markers, also characterized by higher cell cycle (SC IV Meg/Cycling); one cluster of megakaryocyte progenitors (Mk Ps); one cluster of early progenitors characterized by high expression of high-mobility group (HMG) genes (early P HMG high); one cluster of granulocyte/monocyte progenitors (GMPs); two clusters of mast cell progenitors (Mono/Mast Ps) and immature mast cells (immature Mast); and one cluster of B cell progenitors (Pre/Pro Bs). The expression of cell cycle genes was generally higher in iBlast clusters and later time points, which is consistent with the more quiescent state of iLSCs (Figure 4E). To interrogate the cellular hierarchy within the iLSC population, we performed pseudotime analysis of the 4 clusters, namely, SC I, SC II, SC III, and SC IV Meg/Cycling. This analysis placed SC I on the apex of the inferred hierarchy (Figure 4F). SC I then gave rise to SC II, which subsequently branched into SC III and SC IV. This trajectory was consistent with increasing expression of cell cycle genes from SC I to SC IV (Figure 4E). By inspecting the most highly expressed genes that were not shared among the 4 SC clusters, we observed that SC I was characterized by a high expression of the genes PRDX1 and HSPB, encoding an antioxidant enzyme and a heat shock protein, respectively, which may confer protection against oxidative and other types of cellular stress (Figure S4I). SC II was characterized by the expression of MDM4 and PTPRS (Figure S4I). MDM4 is a negative regulator of TP53, which has been shown to regulate HSC quiescence and cell cycle entry (Liu et al., 2009), whereas PTPRS is a phosphatase that negatively regulates HSC self-renewal and repopulating activity (Quarmyne et al., 2015). SC III was characterized by higher DNMT1 expression, which has been shown to negatively regulate the cell cycle (Trowbridge et al., 2009), as well as by expression of GMNN and CENPF, two genes that regulate DNA replication and mitosis and whose expression is highest in the G2 phase of the cell cycle. SC IV showed predominant expression of megakaryocyte markers, such as PLEK, PF4, GP9, and CD41a (Figures 4C, 4D, and S4I) and showed the highest expression of cell cycle genes among all SC clusters. The presence of a CD41+ cell cluster with a concurrent expression of megakaryocyte lineage and HSC genes reflects the close relationship between HSCs and Mk Ps reported in several recent studies (Carrelha et al., 2018; Grinenko et al., 2014; Notta et al., 2016; Rodriguez-Fraticelli et al., 2018; Sanjuan-Pla et al., 2013; Shin et al., 2014; Yamamoto et al., 2018).
Figure 4. Single-Cell Transcriptome Analysis.
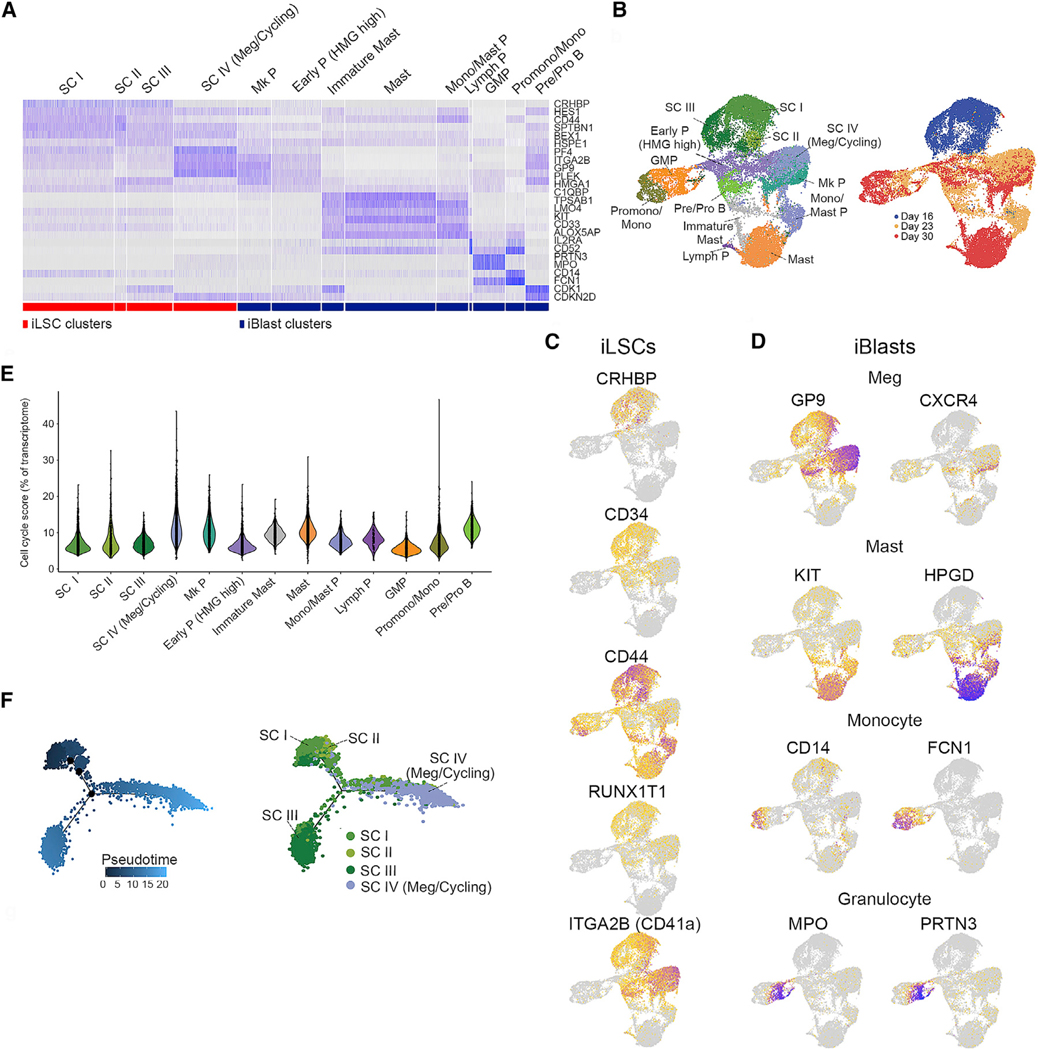
(A) Heatmap of the 13 clusters based on expression of representative genes (shown on the right). Cluster annotation (top) was based on manual curation of marker genes.
(B) UMAP plots of single cell transcriptome data colored by cluster (left) and colored by day of sample collection (right).
(C) Expression of selected HSC genes, as indicated, projected onto the UMAP plot from (B).
(D) Expression of selected hematopoietic lineage genes, as indicated, projected onto the UMAP plot from (B).
(E) Violin plots showing expression of cell cycle genes for each cluster.
(F) Pseudotime reconstruction of the hierarchy of clusters SC I-IV. See also Figure S4.
In summary, our single-cell transcriptome analyses further establish the “sternness” of iLSCs and characterize temporal changes in their expression profile. They show that iLSCs establish a hierarchy that resembles that of normal HSCs by giving rise to heterogeneous populations corresponding to immature and more mature progenitor cells of diverse hematopoietic lineages.
Integrated Genomics Analyses Identify a Human LSC Gene Signature
To probe the molecular properties of iLSCs and iBlasts, we performed transcriptome and chromatin accessibility analyses by RNA sequencing (RNA-seq) and ATAC-seq, respectively, in manually separated iLSCs and iBlasts. Differential gene expression analysis revealed a marked dominance of gene downregulation, in both the number of genes and effect size, in iLSCs compared to iBlasts (Figures 5A and 5B). Gene set enrichment analysis (GSEA) revealed HSC gene sets enriched in iLSCs and, conversely, myeloid lineage differentiation gene signatures enriched in iBlasts (Figure 5C). A comparison of genome-wide chromatin accessibility in iLSCs versus iBlasts also revealed a prominence of loss over a gain of accessible peaks (Figure 5D). Peaks more accessible in iLSCs were predominantly localized in intronic and intergenic regions, whereas peaks with higher accessibility in iBlasts were mostly found in promoters (Figure 5E). There was also enrichment for HSC, LSC, and self-renewal gene sets in genes more accessible in iLSCs and for myeloid differentiation gene sets in genes more accessible in iBlasts (Figure S5A).
Figure 5. Extraction of a Human LSC Gene Set through Integrated Genomics Analyses.
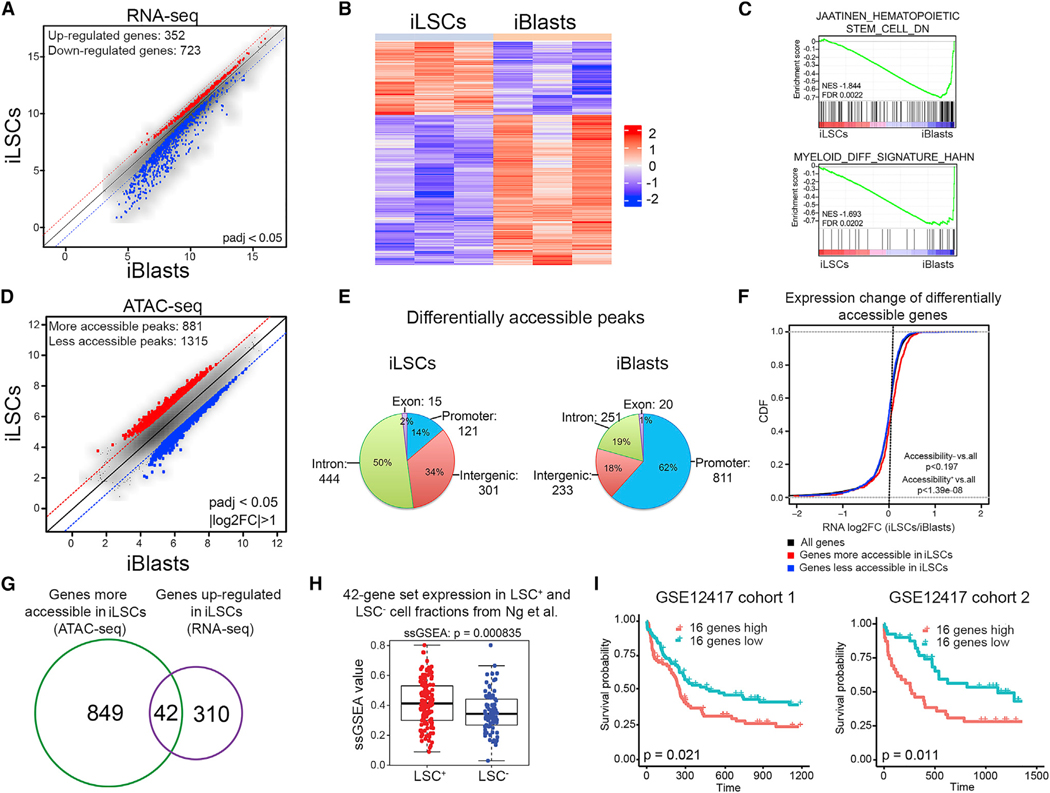
(A) Scatterplot of the average log2-normalized counts of differentially expressed genes in iLSCs compared to iBlasts. Genes with adjusted p < 0.05 were considered significantly differentially expressed.
(B) Row-normalized heatmap of differentially expressed genes between iLSCs and iBlasts.
(C) GSEA plots of the indicated gene sets against the rank list of differentially expressed genes between iLSCs and iBlasts. Enrichment for genes downregulated in HSCs (top) and for myeloid differentiation genes (bottom) in iBlasts compared to iLSCs indicates loss of HSC gene expression and gain of myeloid differentiation gene expression in iBlasts.
(D) Scatterplot of the average log2-normalized counts of differentially accessible peaks in iLSCs compared to iBlasts. Peaks with log2 fold change of >1 or log2 fold change of <—1 and Benjamini & Hochberg (BH)-adjusted p < 0.05 were considered significantly differentially accessible.
(E) Genomic distribution of differentially accessible peaks that are more accessible in iLSCs (left) or more accessible in iBlasts (right). Absolute numbers and percentages are shown.
(F) Cumulative distribution functions (CDFs) of the expression change (log2 fold change [log2FC]) for all genes (black), genes significantly more accessible in iLSCs (red) and genes significantly more accessible in iBlasts (blue). Genes more accessible in iLSCs are also upregulated in iLSCs (Kolmogorov-Smirnov [KS] test, p < 1.39e-08). Genes more accessible in iBlasts are not differentially expressed between iLSCs and iBlasts compared to background (KS test, p < 0.197).
(G) Venn diagram showing the overlap between differentially accessible genes (green) and differentially expressed genes (purple) in iLSCs compared to iBlasts.
(H) Single-sample GSEA (ssGSEA) score of the 42 genes from (G) in LSC+ and LSC− fractions from primary AML patient cells from Ng et al. (2016) (GEO: GSE76008), showing significant association with LSC+ status (Wilcoxon rank-sum test, p < 8.35e-4).
(I) Kaplan-Meier estimates of overall survival in two patient cohorts, as indicated, based on expression of the 16-gene ssGSEA score, showing negative association with patient survival in both cohorts. See also Figure S5 and Table S3.
There was a correlation between gene expression and chromatin accessibility changes for genes more accessible in iLSCs but not for genes more accessible in iBlasts (Figure 5F). Integration of the RNA-seq and ATAC-seq analyses identified a set of 42 genes that were both more highly expressed and more accessible in the iLSCs than the iBlasts (Figures 5G, S5B, and S5C; Table S3). This 42-gene signature was significantly upregulated in LSC+ cell fractions isolated from primary AML patient cells and was validated by xenotransplantation from a large published dataset by Dick and colleagues (Ng et al., 2016) (Figure 5H). To derive a gene set with potential predictive value for AML prognosis, we selected the genes among the 42-gene set that were individually significantly upregulated in the primary LSC+ compared to LSC− cell fractions (Wilcoxon rank-sum test, p < 0.05). This yielded a 16-gene set, which correlated with AML patient survival in two independent patient cohorts (Figure 5I).
The RUNX1 TF Is Critical for the Maintenance of Leukemia and for the Survival of iLSCs
To identify specific TFs as regulators of the AML LSC state, we first performed motif analysis of the ATAC-seq data. In two independent approaches, RUNX1 was among the top TFs whose motif was enriched in genomic regions more accessible in iLSCs (Figures 6A and S6A). RUNX1 motifs mainly localized in intronic and intergenic regions (Figure S6B), and the genes associated with RUNX1-motif-containing peaks were overall upregulated in iLSCs compared to iBlasts (Figure S6C). The RUNX1 motif was also one of the most enriched motifs in peaks associated with our LSC 42-gene set (Figure 6B). Additionally, we used the 42 gene set to derive an “iLSC score” to interrogate the single-cell transcriptome data (Aibar et al., 2017). The iLSC score correlated well with the distribution of the single-cell RNA-seq (scRNA-seq) clusters over the 3 time points of sample collection, validating this score as an effective signature of the iLSC versus iBlast state (Figure S6D). Variable TF “regulons” that positively correlated with the iLSC score across single cells involved TFs with known roles in hematopoiesis and leukemia and included RUNX1 (Figure 6C; Table S4).
Figure 6. The RUNX1 TF Is Critical for the Maintenance of LSCs.
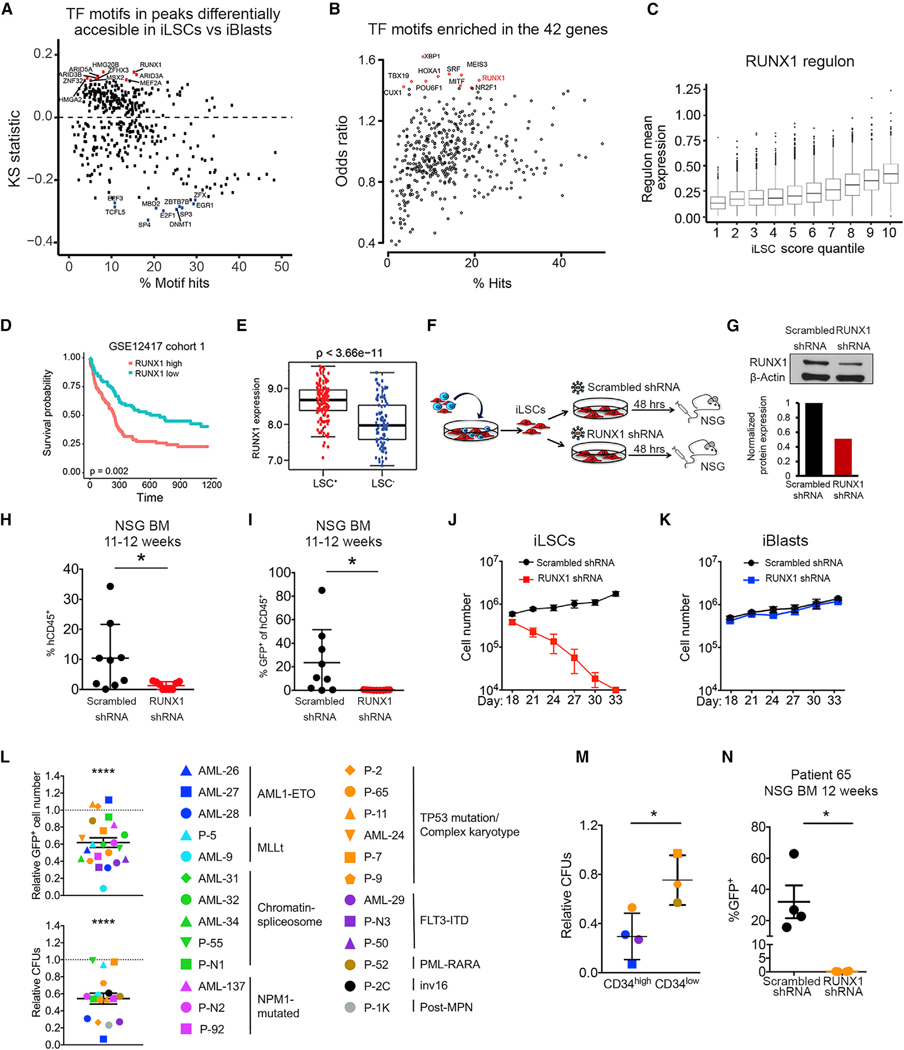
(A) TF motifs in peaks differentially accessible in iLSC sversus iBlasts.The x axis indicates the percentage of hits for eachTF motif in the atlas. They axis shows the KS statistic, indicating whether peaks containing a specific motif have increased (up) or decreased (down) accessibility log2FC in iLSCs versus iBlasts compared to the background (atlas). The top 10 motifs with increased accessibility in iLSCs are shown in red. The top 10 motifs with increased accessibility in iBlasts are shown in blue.
(B) TF motifs enriched in the 42 genes from Figure 5G, showing RUNX1 as one of the top enriched motifs. The x axis indicates the percentage of hits for each TF motif in peaks associated with the 42 genes. The y axis indicates motif enrichment for the peaks associated with the 42 genes compared to background (atlas), measured as odds ratio. The top 10 more enriched motifs are shown in red.
(C) Correlation of RUNX1 regulon mean expression with iLSC score. Mean regulon expression in single cells grouped in 10 quantiles based on iLSC score (1 corresponds to the quantile with the lowest iLSC score and 10 to the quantile with the highest iLSC score).
(D) Kaplan-Meier estimates of overall survival according to RUNX1 expression, showing that RUNX1 expression is negatively associated with AML patient survival.
(E) RUNX1 expression in LSC+ and LSC− cell fractions from primary AML cells from Ng et al. (2016), showing significantly higher expression in the LSC+ fraction. (F) Separated iLSCs were transduced with a lentiviral vector encoding an shRNA against RUNX1 orscrambled shRNA. At 48 h later, the cells were collected and intravenously injected into sublethally irradiated or busulfan-treated NSG mice.
(G) RUNX1 protein by western Blot in iLSCs transduced with lentiviral vectors encoding RUNX1 or scrambled shRNA 48 h after transduction.
(H) Engraftment levels inthe BM of mice 11–12 weeks after transplantation with iLSCs transduced with RUNX1 orscrambled shRNA. Each data point represents a unique mouse from two independent experiments. Mean and SEM are shown. *p < 0.05, t test.
(I) Assessment of GFP+ cells within the hCD45+ cells engrafted in the BM of mice 11–12 weeks after transplantation with iLSCs transduced with RUNX1 or scrambled shRNA. Each data point represents a unique mouse from two independent transplantation experiments. Mean and SEM are shown. *p < 0.05, t test.
(J) iLSCs were transduced with alentiviral vector encoding RUNX1 orscrambled shRNA.Total cells were counted every 3 days. Mean and SEM of 3 independent experiments are shown.
(K) iBlasts were transduced with a lentiviral vector encoding RUNX1 orscrambled shRNA.Total cells were counted every 3 days. Mean and SEM of 3 independent experiments are shown.
(L) Cell counts of transduced (GFP+) cells (top panel) and colony-forming units (CFUs; bottom panel) of primary AML samples from 9 different AML genetic groups, as indicated in the right, after RUNX1 KD relative to scrambled shRNA transduction. Mean and SEM are shown. ****p < 0.0001, t test.
(M) CFUs of primary AML cells after transduction with RUNX1 shRNA relative to scrambled shRNA stratified in CD34high (%CD34+ > 60%) and CD34low (% CD34+ < 40%). Mean and SEM are shown. *p < 0.05, t test.
(N) GFP+ engraftment of AML patient cells (patient 65) transduced with RUNX1 shRNA or scrambled shRNA in the BM of mice 12 weeks after transplantation. Each data point represents a unique mouse. *p < 0.05, t test. See also Figure S6 and Tables S4 and S5.
RUNX1 is a critical regulator of the hematopoietic system during development (Chen et al., 2009; Gao et al., 2018). RUNX1’s well-known roles in AML are due to a loss of function, as a partner in fusion genes generated by translocations and as a target of loss-of-function germline or somatic mutations (Chen et al., 2009; Sood et al., 2017). However, a previous study detected a correlation between RUNX1 expression and worse survival in AML patients (Fu et al., 2016). In agreement with this finding, we found that RUNX1 expression correlated with worse survival in a different AML patient cohort (Figure 6D).
These observations, taken together, led us to hypothesize that RUNX1 may play a role in sustaining LSCs and that this role may explain the negative correlation between RUNX1 expression and patient survival. Consistent with this hypothesis, RUNX1 expression was significantly higher in primary LSC+ cells from AML patients (Ng et al., 2016) (Figure 6E). To experimentally test if RUNX1 plays a role in the maintenance of LSCs, we first examined the effects of its depletion in iLSCs (Figure 6F). Reduction of the RUNX1 protein by approximately 50% in the iLSCs (through lentiviral short hairpin RNA (shRNA) transduction with over 90% efficiency) abolished their ability to engraft (Figures 6F–6I, S6E, and S6F). RUNX1 shRNA-transduced iLSCs had no detectable engraftment in 4 out of 9 mice, whereas 5 mice had modest levels of hCD45+ cell engraftment (1.7%–2.8%), consisting entirely of GFP− cells (Figures 6H and 6I). iLSCs with RUNX1 knockdown (KD) could also not be maintained in vitro and disappeared within 2 weeks of culture (Figure 6J). RUNX1 KD did not induce apoptosis or differentiation, and the kinetics of its effect were consistent with iLSC exhaustion (Figures S6G and S6H). In contrast, RUNX1 KD had no effect on the viability and growth of iBlasts (Figure 6K). Conversely, overexpression of RUNX1 in iBlasts conferred to them a more HSC-like phenotype (Figures S6I–S6K).
To test whether primary human AMLs are dependent on RUNX1 and whether such dependence is confined to any specific genetic groups or more generalizable to AML LSCs, we measured the effects of RUNX1 KD in 25 primary AML samples representative of common AML genetic groups: RUNX1 translocated (AML1-ETO), MLL translocated, AML with mutations in splicing and chromatin regulators (“chromatin-spliceosome”), NPM1 mutated, AML with TP53 mutations or complex karyotype, FLT3-ITD, and others (Table S5). RUNX1 KD decreased both bulk cell survival and colony formation (compared to scrambled shRNA transduction) in most samples across diverse genetic groups (Figures 6L and S6L; Table S5). The decrease in colony-forming units (CFUs) caused by RUNX1 KD correlated with the CD34+ cell fraction in the samples (for the CD34+ AML samples, which excluded NPM1-mutated AML that is typically CD34−) (Figure 6M). Furthermore, RUNX1 KD in AML patient cells abolished their engraftment (GFP+ cell engraftment) (Figures 6N and S6M–S6O). Taken together, these results support a role for RUNX1 in the maintenance of primary human AML LSCs across different genetic groups.
The AML-LSC RUNX1-Dependent Transcriptional Program
To further probe the role of RUNX1 in AML LSCs, we performed chromatin immunoprecipitation sequencing (ChIP-seq) analyses in iLSCs and iBlasts. In agreement with our chromatin accessibility and TF motif analyses, we found genome-wide RUNX1 binding to be dramatically higher in iLSCs than iBIasts (Figures 7A and 7B). Genes bound by RUNX1 preferentially in iLSCs versus iBlasts were enriched for RUNX1 deficiency gene signatures, as well as for HSC differentiation signatures, which may represent genes repressed by RUNX1 (Figure S7A). To identify RUNX1 target genes critical for the LSC state, we selected the genes among the 42 LSC genes (Figure 5G) that were more bound by RUNX1 in iLSCs than in iBlasts. This prioritization gave us 16 genes (Figure 7C). To further narrow these down, we interrogated the enrichment of RUNX1 motifs in their accessible chromatin (Figure S7B) and measured the effects of RUNX1 KD in the expression of the top 5 genes with more RUNX1 accessible motifs in iLSCs (Figure S7C). These analyses nominated TSPAN18, encoding tetraspanin 18, as the prominent candidate gene (Figure S7D). In support of TSPAN18 as a RUNX1 target gene critical for maintaining the RUNX1-dependent iLSC state, TSPAN18 overexpression partially reversed the effect of RUNX1 KD in iLSCs (Figures 7D and S7E). Furthermore, TSPAN18 expression was higher in the LSC-enriched cell fraction and decreased after RUNX1 KD in primary AML patient cells (Figures S7F and S7G). To interrogate the genome-wide LSC RUNX1-dependent program in an unbiased way, we performed RNA-seq and ATAC-seq following RUNX1 KD in iLSCs and iBlasts. These analyses, again, revealed a predominance of gene expression changes in iLSCs compared with that in iBlasts (Figure S7H). Integration of these data with the ChlP-seq analyses revealed 20 genes activated (down after RUNX1 KD) and 47 genes repressed by RUNX1 (up after RUNX1 KD) (Figures 7E and S7I). This gene signature was significantly enriched in LSC+ cell fractions from AML patients (Ng et al., 2016; Figure 7F) and negatively correlated with patient survival (Figure 7G). Furthermore, expression of the top 10 repressed and top 10 activated genes of this RUNX1-dependent LSC signature correlated with RUNX1 expression in AML patient cohorts (Figure 7H). Finally, expression of the top 3 repressed and top 3 activated RUNX1 target genes changed in the predicted direction in primary AML patient cells after RUNX1 KD (Figure S7J). Taken together, these results characterize a RUNX1-dependent gene program critical for sustaining AML LSCs and nominate TSPAN18 as an important mediator of the RUNX1 effects on AML LSC maintenance.
Figure 7. RUNX1-Dependent iLSC Transcriptional Program.
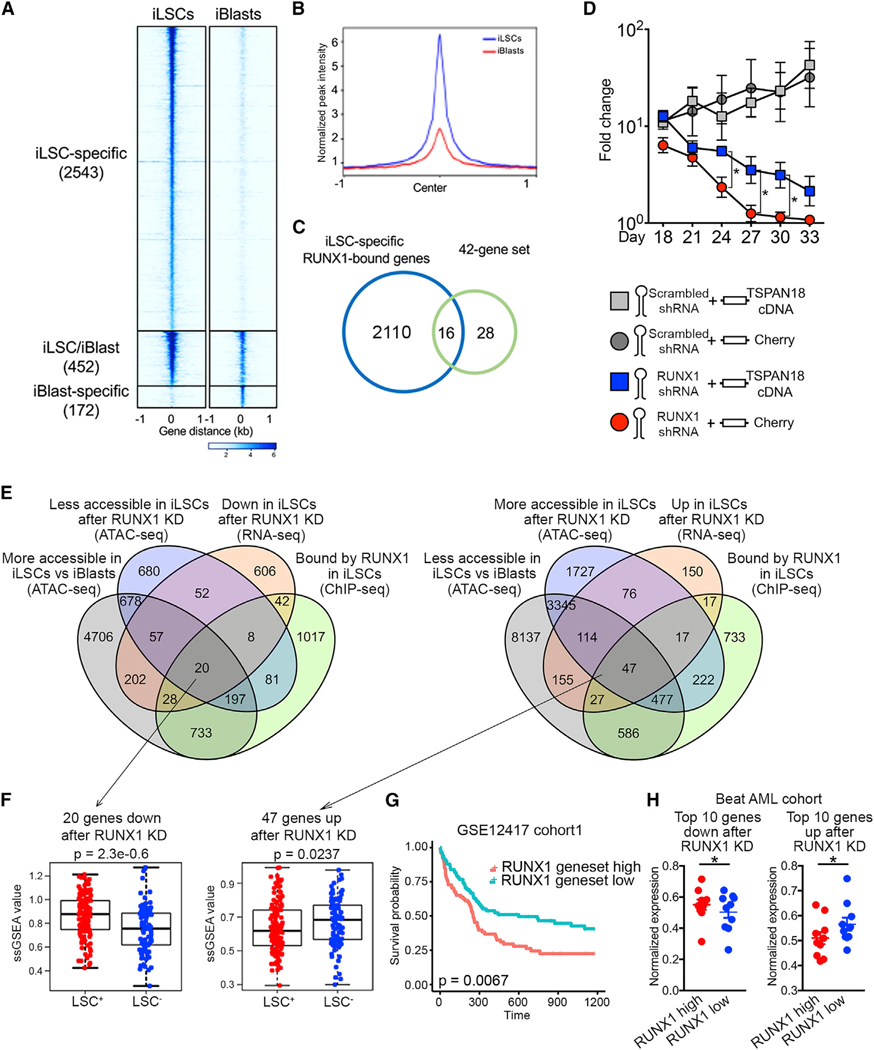
(A) Tornado plot of iLSC-specific, iBlast-specific, and iLSC/iBlast common RUNX1 ChIP-seq peaks. The number of peaks in each group is indicated in parentheses.
(B) iLSC-specific and iBlast-specific metapeaks from the tornado plot shown in (A).
(C) Venn diagram showing the overlap between iLSC-specific RUNX1-bound genes and the 42-gene set from Figure 5G (genes upregulated and more accessible in iLSCs versus iBlasts).
(D) iLSCs were co-transduced with a lentiviral vector encoding RUNX1 shRNA or scrambled shRNA and a lentiviral vector encoding TSPAN18 cDNA or Cherry only and counted every 3 days. Mean and SEM of 4 independent experiments are shown. *p < 0.05, t test.
(E) Left: Venn diagram showing overlap among genes more bound by RUNX1 in iLSCs than iBlasts (ChIP-seq, log2FC > 2), genes downregulated after RUNX1 KD in iLSCs (RNA-seq, adjusted pvalue [padj] < 0.05), genes less accessible after RUNX1 KD in iLSCs (ATAC-seq, padj < 0.05) and genes more accessible in iLSCs than iBlasts (ATAC-seq, padj < 0.05). Right: Venn diagram showing overlap among genes more bound by RUNX1 in iLSCs than iBlasts (ChIP-seq, log2FC > 2), genes upregulated after RUNX1 KD in iLSCs (RNA-seq, padj < 0.05), genes more accessible after RUNX1 KD in iLSCs (ATAC-seq, padj < 0.05), and genes less accessible in iLSCs than iBlasts (ATAC-seq, padj < 0.05).
(F) ssGSEA score of the 20 (down after RUNX1 KD) and 47 (up after RUNX1 KD) genes from E in LSC+ and LSC− fractions from primary AML patient cells from Ng et al. (2016) (GEO: GSE76008), showing significant association with LSC+ status (Wilcoxon rank-sum test).
(G) Kaplan-Meier estimates of overall survival based on the expression of the 20 and 47 genes as ssGSEA score, showing negative association with patient survival.
(H) Expression of the top 10 among the 20 and 47 genes, respectively, activated and repressed by RUNX1, in the Beat AML patient cohort stratified based on RUNX1 expression into RUNX1 high (above average) and RUNX1 low (below average). Min-max-normalized expression values are shown. *p < 0.05, t test. See also Figure S7.
DISCUSSION
We show here that hematopoietic cells derived from “exceptional engrafter” AML-iPSC lines exhibit phenotypic and functional heterogeneity and hierarchical organization, recapitulating an LSC model.
The study of human LSCs is currently only possible through retrospective identification of cell subsets from primary patient samples containing LSCs on the basis of engraftment potential in xenotransplantation, which severely restricts both the scale and ease of human LSC experimentation (Ng et al., 2016). Importantly, primary AML LSCs cannot be grown ex vivo for cellular assays, functional genetics, or pharmacologic testing. The model we report here enables easy prospective isolation by means of adherence to plastic of a cell fraction highly enriched for LSC activity. It, thus, offers unique opportunities for experimentation on the biology and therapeutic targeting of LSCs and CSCs more generally. The iPSC lines we present here appear unique in this phenotype and show exceptional engraftment potential among the very small number of AML-iPSC lines that currently exist. It is currently unclear whether an in vitro self-renewing LSC population will be found upon differentiation of other AML-iPSC lines and whether, if it isfound, it will similarly exhibit high in vitro adhesion properties. Although we did not find a requirement of in vitro adhesion for the maintenance of immunophenotypic iLSCs with repopulating potential, our data do not exclude a link between determinants of the increased adhesion properties and the iLSC state.
The LSC model posits that cancers are hierarchically organized as a result of non-genetic determinants and that cells with LSC properties exist at a distinct epigenetic state within a genetically homogeneous cancer cell population (Dick, 2008). The genetic and clonal composition of a given AML has now been shown to be a strong determinant of engraftment potential (Klco et al., 2014). Thus, genetic sources have become appreciated as another very prominent source of cancer heterogeneity that often confound claims of an LSC-type hierarchy (Kresoand Dick, 2014; Shackleton et al., 2009). Because both iLSC and iBlasts are derived from genetically clonal iPSC lines—originating from single cells of the original patient leukemia—this cellular system uniquely lends support to the LSC concept, as a definitive and unambiguous source of non-genetic heterogeneity.
LSCs are believed to have distinct properties from the bulk AML cells, which are hypothesized to, at least in part, account for AML relapse (Jones et al., 2018; Pollyea et al., 2014; Ye et al., 2018). Identifying unique dependencies of LSCs can, thus, open avenues for their specific therapeutic targeting in combination with standard chemotherapy to obtain lasting responses or even cures. Our genomics studies revealed a dramatically distinct transcriptome and chromatin landscape between LSCs and non-LSCs, suggesting that many opportunities for specific targeting exist. Accessible chromatin in iLSCs was found mostly in non-promoter regions, whereas iBlasts gained accessibility in promoter regions (Figure 5E). This finding, together with the pronounced gene upregulation underlying the iLSC-to-iBlast transition and ourscRNA-seq analyses, suggest that the iLSCs represent a relatively transcriptionally inactive state and that the transition to iBlasts is governed by transcriptional upregulation of diverse lineage genes (Velten et al., 2017).
Transcriptome profiling of LSCs has shown that LSC gene expression signatures are highly predictive of therapy failure (Eppert et al., 2011; Gentles etal., 2010; Ng et al., 2016). Thus, more recent efforts have been undertaken to derive molecular signatures that can predict patient outcomes as a means of translating research on LSCs into practical tests for patient prognostication in the clinic (Ng et al., 2016). Our study adds to these efforts datasets of transcriptomic and, importantly, chromatin accessibility data and a gene signature—non-overlapping with previously derived gene sets, including the one from Ng etal. (2016)—which correlates with patient survival. Unveiling mechanisms sustaining iLSCs may also enhance our understanding of the requirements for the generation of engraftable HSCs from normal hPSCs, as molecular pathways involved in normal HSCs are likely to overlap and intersect with those that sustain LSCs.
RUNX1 is a key transcriptional regulator of hematopoiesis, with a well-characterized role during the development of the hematopoietic system (Chen etal., 2009; Gao etal., 2018). RUNX1, also known as AML 1 protein (AML1), is often translocated or mutated in sporadic AML and is the target of germline mutations causing familial predisposition to AML (Hatlen et al., 2012; Song et al., 1999). Both somatic and inherited RUNX1 mutations are loss-of-function or dominant-negative mutations, supporting a tumor-suppressor function of RUNX1 (Sood et al., 2017). On the other hand, a requirement for wild-type RUNX1 for the growth and survival of certain types of AML (RUNX1 and MLL translocated) has been demonstrated (Goyama et al., 2013), and an oncogenic role of RUNX1 in cooperation with FLT3-ITD was also recently reported in a mouse model of AML (Behrens et al., 2017). Here, we report a role for RUNX1 in AML LSC maintenance.
RUNX1 expression significantly negatively correlated with survival in AML patients across genotypes (Figure 6D). We did not find evidence for preferential RUNX1 dependency or lack thereof in association with any specific genetic group. Rather, we detected a higher sensitivity to RUNX1 KD in patient samples enriched for LSCs (higher CD34+ fractions). Thus, it is the presence and frequency of LSCs rather than the leukemia genetics that is the main determinant of RUNX1 dependency, consistent with the data in our model showing a complete lack of RUNX1 dependence in iBlasts. Although we did not directly test the effect of RUNX1 KD in normal HSCs here, several lines of evidence suggest that RUNX1 complete deletion, depletion, or haploinsufficiency (as we mimic here by 50% shRNA KD) does not impair HSC function in adult mice or humans (Chen et al., 2009; Ichikawa et al., 2004). Furthermore, patients with inherited germline RUNX1 mutations that confer haploinsufficiency (RUNX1 familial platelet disorder) do not have any discernible HSC defects (Sood et al., 2017).
We, thus, propose that RUNX1 dependency represents a vulnerability of AML LSCs with implications for the therapeutic targeting of AML. Furthermore, the LSC-specific RUNX1 target genes that we characterize can provide additional nodes for targeting, with therapeutic implications. Among these, we show that TSPAN18 at least partially mediates the RUNX1 effects in LSCs. TSPAN18 is a particularly attractive target because, as a cell surface protein, it can be targeted with antibody or chimeric antigen receptor (CAR)-based approaches.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eirini P. Papapetrou (eirini.papapetrou@mssm.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
RNA-Seq data are accessible through GEO datasets: GSE124966. ATAC-Seq data are accessible through GEO datasets: GSE124967. Single-cell RNA-Seq data are accessible through GEO datasets: GSE124992. CHiP-Seq data are accessible through GEO datasets: GSE143679. RNA- and ATAC-Seq after RUNX1 KD data are accessible through GEO datasets: GSE143471.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strain
NOD/SCID/interleukin 2 receptor γ chain null (NOD/SCID/IL2rγ−/−, NSG) female mice, 6–8 weeks old, were purchased from the Jackson Laboratory. All mouse studies were performed in compliance with Icahn School of Medicine at Mount Sinai and Memorial Sloan Kettering Cancer Center laboratory animal care regulations.
Human iPSC lines
The generation of human iPSC lines AML-4.10 and AML-4.24, used in this study, was previously described (Kotini et al., 2017). The iPSCs were maintained on mitomycin-C treated embryonic fibroblasts (MEFs) at 37°C with 5% carbon dioxide and passaged twice a week with dispase solution, as described (Kotini etal., 2017). The AML-4.10, AML-4.24 and AML-4.10 GFPhiPSC lines were routinely examined for acquisition of chromosomal abnormalities, mycoplasma contamination or contamination from other cell lines.
Human Samples
Human bone marrow mononuclear cells and peripheral blood mononuclear cells from 25 AML patients were obtained with informed consent under protocols approved by a local Institutional Review Board at Memorial Sloan Kettering Cancer Center, the Icahn School of Medicine at Mount Sinai and the Sylvester Comprehensive Cancer Center, University of Miami. Additional information on the samples (gene mutations, genetic classification and cytogenetics) are available in Table S5.
METHOD DETAILS
Human iPSC lines and culture
Hematopoietic differentiation
Cytological analyses
Flow cytometry
Cell cycle analyses
Transplantation of iPSC-derived cells into NSG mice
Proliferation and transition rate analyses
Markov modeling
Single-cell plating experiments
Time-lapse imaging
Single-cell RNA sequencing
RNA sequencing
ATAC sequencing
RUNX1 knockdown and overexpression
Western Blot
qRT-PCR
Primary AML patient cell experiments
TSPAN18 rescue experiments
RUNX1 CHIP sequencing
Human iPSC lines and culture
The generation of iPSC lines AML-4.10 and AML-4.24 was previously described (Kotini et al., 2017). Briefly, these lines were derived with a Cre-excisable OKMS lentiviral vector from PBMCs from a female patient with secondary AML. The starting patient cells and all derived iPSC lines had a complex translocation involving chromosomes 1, 7, and 14, resulting in a deletion of 7q and additional material of unknown origin on chromosome 15 [46,XX,der(1)t(1;7;14)(q32;p11p22;p11.1),der(7)del(7)(p11p22)inv(7)(p11q31),der(14) t(1;14)(q32;p11.1),add(15)(p11.1)].
The patient also harbored subclonal KRAS and NRAS mutations. Line AML-4.10 has the KRAS G12D mutation. AML-4.24 is WT for both NRAS and KRAS. The iPSCs were maintained on mitotically inactivated MEFs, as described (Kotini et al., 2017).
Hematopoietic differentiation
Spin embryoid bodies (spin-EBs) were prepared and cultured in APEL2 medium, as described (Ng et al., 2008). Briefly, cells were dissociated into single cells with accutase and plated at 3,500 cells per well in round-bottom low-attachment 96-well plates in APEL2 medium containing 5% protein-free hybridoma medium (PFHM-II), 30 ng/ml bone morphogenetic protein 4 (BMP4) and 10 μM Y-27632. The plates were centrifuged at 800 rpm for 5 min to induce EB aggregation. After 24 hours, the medium was replaced by APEL2 medium containing 5% PFHM-II, 30 ng/mL BMP4 and 50 ng/mL FGF2. After 2 days, the cytokine cocktail was changed to 5% PFHM-II, 20 ng/ml vascular endothelial growth factor (VEGF), 10 ng/ml FGF2, 100 ng/ml stem cell factor (SCF), 20 ng/ml Flt3 ligand (FL), 20 ng/ml thrombopoietin (TPO), 40 ng/ml IL-3. On day 8, EBs were collected and resuspended in Stem Pro34 SFM medium with 1% nonessential amino acids (NEAA), 1 mM L-glutamine and 0.1 mM β-mercaptoethanol (2ME), supplemented with 100 ng/ml SCF, 20 ng/ml Flt3L, 20 ng/ml TPO, 40 ng/ml IL-3. The medium was thereafter replaced every two days.
In most experiments unless otherwise specified, iLSCs and iBlasts were manually separated as follows. Cells from bulk liquid hematopoietic culture in low adherence plates were transferred into tissue culture-treated plates on day 14–16 of hematopoietic differentiation. Every two days the supernatant containing the iBlasts was collected and transferred into new tissue culture-treated plates. iBlasts from all collection days were pooled together. Each collection of supernatant with iBlasts was followed by one wash with PBS which was discarded. The adherent cells remaining in the initial tissue culture-treated plates were maintained as iLSCs. iLSCs were collected and passaged using accutase.
Cytological analyses
Approximately 200,000 cells from liquid hematopoietic differentiation cultures were washed twice with PBS containing 2% FBS and resuspended in PBS. Cytospins were prepared on slides using a Shandon CytoSpin III cytocentrifuge (Thermo Electron Corporation). Slides were then air-dried for 30 mins and stained with the Hema 3 staining kit (Fisher Scientific Company LLC). The slides were read on a Nikon Eclipse Ci microscope and digital images were taken with a Nikon DS-Ri2 camera and NIS-Elements D4.40.00 software.
Flow cytometry
Following hematopoietic differentiation, the cells were dissociated with accutase into single cells. The following flow cytometry antibodies were used: CD34-PE (clone 563, BD PharMingen), CD45-BV510 (clone HI30, BD Horizon), CD90-PerCP/Cy5.5 (clone 5E10, BioLegend), CD38-PE-CF594 (clone HIT2, BD Horizon), CD33-PE-CF594 (clone WM53, BD Horizon), CD49f-BV421 (clone GoH3, BD Horizon), CD45RA-APC (clone MEM56, Life Technologies), CD123-BV711 (clone 9F5, BD Horizon), CD41a- PE-Cy7 (clone HIP8, BD PharMingen), mCD45-PE-Cy7 (clone 30-F11, BD Biosciences), CD45-APC (clone HI30, BD PharMingen), CD19-PerCP-Cy5.5 (clone HIB19, BD PharMingen). Cell viability was assessed with DAPI (Life Technologies). Intracellular staining of Patient 65 cells was performed with a biotin-conjugated TSPAN18 antibody (Lifespan Bioscience) and a secondary ani-biotin APC antibody (Miltenyi Biotec) using the Cytofix/Cytoperm Plus kit (BD Biosciences). Cells were assayed on a BD Fortessa and data were analyzed with FlowJo software (Tree Star).
Cell cycle analyses
For cell cycle analyses, iLSCs and iBlastsfrom hematopoietic differentiation day 18 were manually separated and 500,000 cells were plated into a 6-well plate. After 24 hours EdU was added to the media. After 4 hours the cells were harvested and analyzed with the Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit.
Transplantation of iPSC-derived cells into NSG mice
NSG mice were purchased from Jackson Laboratories and housed at the Center for Comparative Medicine and Surgery at Icahn School of Medicine at Mount Sinai or at the Center of Comparative Medicine and Pathology of Memorial Sloan Kettering Cancer Center. One day before transplantation, the mice were either injected intraperitoneally with 30mg/kg Busulfan solution or sublethally irradiated (250 rad). The latter also received IVIG one hour prior to irradiation and one hour prior to transplantation. Bulk cells, iLSCs and iBlasts derived from AML-iPSC lines after 16 or 24 days of hematopoietic differentiation were resuspended in StemPro-34 at 1×106 single cells per 100μL and injected into the mice via the tail-vein or retro-orbital route using a 25G needle. The injected mice were monitored daily and engraftment was assessed 8–12 weeks post transplantation or earlier if recipients appeared moribund. For limiting dilution assay experiments, cohorts of 5 NSG mice received 50, 100, 500, 1,000, 5,000, 10,000, 50,000, 100,000, 500,000 and 1,000,000 iLSCs or 50,000, 100,000, 500,000, 1,000,000 iBlasts. LSC frequency was calculated based on the number of non-engrafted mice using Poisson statistics 14 weeks after transplantation. All mouse studies were performed in compliance with Icahn School of Medicine at Mount Sinai and Memorial Sloan Kettering Cancer Center laboratory animal care regulations.
Proliferation and transition rate analyses
iLSCs and iBlasts were manually separated on hematopoietic differentiation day 18 and plated separately in 6-well plates. Every three days, iLSCs and iBlastsfrom duplicate wells initiated from iLSCs and duplicate wells initiated from iBlasts were separately harvested and counted. The proliferation rate was calculated using the formula: P(iLSC) = Log [(iLSC d3/iLSC d0)/3] and P(iBlast) = Log [(iBlast d3/iBlast d0)/3]. The transition rate was calculated using the formula: T(iLSC) = (iBlast d3/iLSC d3)/3 and T(iBlast) = (iLSC d3/iBlast d3)/3.
Markov modeling
The proliferation and transition dynamics of the iLSC and iBlast populations were modeled with a Markov model and population dynamics over time were simulated using the following dynamic equations:
To simulate the population behavior in a more realistic scenario where the proliferation and transition rates are noisy, we generated 1000 diversified random sets of the Markov model parameters (Gibbs sampling) that were analyzed as a Monte-Carlo simulation using the dynamic equations listed above. Specifically, for each initial Markov chain simulation the following parameters were randomized:
where the mean and standard deviation of each parameter were empirically estimated, and N(0,1) represent random Gaussian sampling with zero mean and unit standard deviation.
These 1000 random Markov models were then set to simulate the empirical experiment shown in Figure 3C, initiated with a single iLSC and monitored over 28 days with simulation of the iLSC and iBlast population growth according to the dynamic equations listed above. The ratio between the iBlast and iLSC population (iBlast/iLSC) was calculated for each day and for each of the 1000 random models. The distribution of iBlast/iLSC ratio values for each day across the 1000 random models was then calculated (Figure 3D, right).
Single-cell plating experiment
To generate a GFP-labeled AML-4.10 line, the AML-4.10 iPSC line was transduced with a lentiviral plasmid expressing GFP. The AML-4.10 iPSCs were dissociated with accutase into single cells and plated on Matrigel. Two days later the cells were transduced with the GFP lentiviral vector supernatant. After two days single cells were dissociated with accutase and replated at clonal density (500 cells per 60-mm dish). After 7–10 days, single colonies were picked in separate wells of a 6-well plate and allowed to grow for approximately 3–6 days. Colonies from each clone were dissociated into single cells with accutase and analyzed by flow cytometry for the expression of GFP. One clone with uniform and stable GFP expression in all cells over time was selected.
GFP+ and GFP− (parental line) AML-4.10 iPSCs were differentiated along the hematopoietic lineage and plated into tissue culture-treated plates on day 14 of hematopoietic differentiation. iLSCsfrom each line were manually separated on day 16 of hematopoietic differentiation, counted and mixed at a 1:1,000 ratio of GFP+ to GFP− cells. The cells were plated at 1,000 cells per well in 96-well plates in 100 μL of hematopoietic differentiation media. Every 5 days, 50 μL of hematopoietic media with 2x concentration of cytokines (200 ng/ml SCF, 40 ng/ml Flt3L, 40 ng/ml TPO, 80 ng/ml IL-3) was added in each well. After 28 days, iLSCs and iBlasts were separately harvested and analyzed for GFP+ cells by flow cytometry. In a subset of 26 wells, absolute iLSC and iBlast numbers were counted and the numbers of GFP+ cells calculated as shown in Table S2.
Time-lapse imaging
GFP+ and GFP− AML-4.10 hematopoietic cells were plated on tissue culture-treated plates on day 14 of hematopoietic differentiation. iLSCs from both lines were manually separated on day 16 of hematopoietic differentiation, counted and mixed at a 1:10 ratio of GFP+ to GFP− cells. The cells were plated at 10,000 cells per well in 6-well plates. After 24 hours the cells were washed with PBS and fresh medium was added. The 6-well plate was placed into the humidity chamber (37°C, 5% CO2) of an Olympus IX-70 Microscope (20x). Brightfield and GFP-fluorescence images were captured with a Q Imaging Retiga 2000R cooled CCD camera every 20 minutes for a total of 18 hours.
Single-cell RNA sequencing
Hematopoietic cells from the AML-4.10 iPSC line were plated in tissue culture-treated 6-well plates on day 14 of hematopoietic differentiation. After 2 days the iBlasts were removed from all wells by washing. iLSCs were collected with accutase on day 16. Total cells were harvested after 1 week (day 23) and after two weeks (day 30). Two duplicate wells were independently collected for each time point. All samples were collected at the same time from experiments initiated one week apart. The cells were sorted for live (DAPI−) CD45+ cells on a BD FACS Aria II and further processed using the Chromium10x Genomics® protocol (v.2). Samples were sequenced at an average of 50,000 reads per cell.
RNA sequencing
Bulk hematopoietic cells from the AML-4.10 iPSC line were plated on tissue culture-treated dishes on day 14 of hematopoietic differentiation. After 2 days iLSCs and iBlasts were manually separated and harvested. Triplicate samples were collected from 3 independent differentiation experiments. For RUNX1 knockdown, iLSCs and iBlasts from 3 independent differentiation experiments were separated on day 16 of hematopoietic differentiation, as above, and transduced with lentiviral vectors expressing either RUNX1 or scrambled shRNA. The cells were collected after 48 hours and transduction efficiency was assessed by flow cytometry (GFP). Total RNA was extracted with the RNeasy mini kit (QIAGEN). PolyA-tailed mRNA was selected with beads from 1 mg total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). cDNAs were generated using random hexamers and ligated to barcoded Illumina adaptors with the NEXTflex Rapid Directional RNA-Seq Library Prep Kit (Bioo Scientific). Sequencing of 75 nucleotide-long single-end reads was performed in a NextSeq-500 (Illumina).
ATAC sequencing
iLSCs and iBlasts from 3 independent differentiation experiments were separated on day 16 of hematopoietic differentiation and cells of each fraction were harvested. For RUNX1 knockdown, iLSCs and iBlasts from 3 independent differentiation experiments were separated on day 16 of hematopoietic differentiation and transduced with lentiviral vectors expressing either RUNX1 or scrambled shRNA. The cells were collected after 48 hours and transduction efficiency was assessed by flow cytometry (GFP). Samples were processed as follows: nuclei were isolated by washing with ATAC buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2) and lysing with 50 ul of ATAC lysis buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40). 1ml of ATAC buffer was added to dilute lysis buffer. Cell lysates were spun to obtain nuclear pellets, which were subjected to transposase reaction using the Illumina Nextera DNA Sample Preparation Kit according to the manufacturer’s instructions. The reaction was stopped by adding SDS to a final concentration of 0.2%. The final libraries were quantified using the Agilent BioAnalyzer. Sequencing of 75 nucleotide-long paired-end reads was performed in a NextSeq-500 (Illumina).
RUNX1 knockdown and overexpression
RUNX1 and Scrambled shRNA sequences were inserted into the 3′ UTR of the G-U6 lentiviral vector (Kotini et al., 2015). The shRNA sequences are shown in the key resources table. Vectors were packaged as described (Kotini et al., 2015). 5 million iLSCs separated on day 18 of hematopoietic differentiation, were transduced with either RUNX1 or scrambled shRNA. Two days after transduction the cells were harvested and injected into sublethally irradiated or busulfan-treated NSG mice via the tail-vein route. For overexpression of RUNX1, 250,000 iBlasts, separated on day 18 of hematopoietic differentiation, were transduced with the mP2A-RUNX1c lentiviral vector (Chang et al., 2018) or an empty GFP vector. Seven days after transduction the cells were harvested and assessed by flow cytometry.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD134-PE | BD PharMingen | Cat# 550761; RRID: AB_393871 |
| CD41a- PE-Cy7 | BD PharMingen | Cat# 561424; RRID: AB_10642584 |
| CD90-PerCP/Cy5.5 | Biolegend | Cat# 328118; RRID: AB_2303335 |
| CD45-APC | BD PharMingen | Cat# 555485; RRID: AB_398600 |
| CD45-BV510 | BD Horizon | Cat# 563204; RRID: AB_2738067 |
| CD49f-BV421 | BD Horizon | Cat# 562582; RRID: AB_2737666 |
| CD33-PE-CF594 | BD Horizon | Cat# 562492, RRID: AB_2713912 |
| CD38-PE-CF594 | BD Horizon | Cat# 562288, RRID: AB_11153122 |
| CD123-BV711 | BD Horizon | Cat# 563161; RRID: AB_2738038 |
| CD19-PerCP-Cy5.5 | BD PharMingen | Cat# 561295; RRID: AB_10644017 |
| mCD45-PeCy7 | BD Biosciences | Cat# 552848; RRID: AB_394489 |
| RUNX1 | Abnova | Cat# H00000861-M05; RRID:AB_565542 |
| β-Actin | Cell Signaling | Cat# 4970; RRID:AB_2223172) |
| TSPAN18 | Lifespan Bioscience | Cat# LS-C157288 |
| Anti-Biotin APC | Miltenyi Biotec | Cat# 130–111-069; RRID:AB_2661379 |
| RUNX1 | Abcam | Cat# ab23980; RRID:AB_2184205 |
| Rabbit IgG | Santa Cruz Biotech | Cat# sc-2027; RRID:AB_737197 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SCF | R&D Systems | Cat# 255-SC |
| Flt3 ligand | R&D Systems | Cat# 308-FK |
| TPO | R&D Systems | Cat# 288-TP |
| IL-3 | R&D Systems | Cat# 203-IL |
| APEL2 | StemCell Technologies | Cat# 05275 |
| BMP4 | R&D Systems | Cat# 314-BP |
| Y-27632 | Tocris | Cat# 1254; CAS: 129830–38-2 |
| FGF2 | R&D Systems | Cat# 233-FB |
| PFHM-II | Fisher Scientific Company LLC | Cat# 12–040-077 |
| VEGF | Peprotech | Cat# 100–20 |
| StemPro-34 SFM | Life technologies | Cat# 10639011 |
| MethoCult GF+ | StemCell Technologies | Cat# H4435 |
| X-VIVO 15 | Lonza | Cat# 04–418Q |
| BIT 9500 | StemCell Technologies | Cat# 09500 |
| Busulfan | Sigma | Cat# B2635; CAS: 55–98-1 |
| Matrigel | Corning | Cat# 354277 |
| Formaldehyde | Sigma | Cat# F8775; CAS: 50–00-0 |
| Glycine | Sigma | Cat# G8898; CAS: 56–40-6 |
| Proteasome inhibitor cocktail | Millipore | Cat# 535140 |
| Protein G Magnetic Beads | New England Biolabs | Cat# S1430S |
| Proteinase K | Invitrogen | Cat# AM2546 |
| Critical Commercial Assays | ||
| TaqMan Universal master mix | Applied Biosystems | Cat# 4304437 |
| Trizol | Life technologies | Cat# 15596018 |
| Invitrogen SuperScript III First-Strand Synthesis System | Life technologies | Cat# 18080051 |
| SsoFast EvaGreen Supermix | Bio-Rad | Cat# 172–52G1 |
| Hema 3 staining kit | Fisher Scientific Company LLC | Cat# 23123869 |
| RNeasy mini kit | QIAGEN | Cat# 741G4 |
| NEXTflex Rapid Directional RNA-Seq Library Prep Kit | Bio Scientific Corporation | Cat# 5138 |
| Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit | Fisher Scientific Company LLC | Cat# C1G632 |
| Pierce BCA Protein Assay Kit | Fisher Scientific Company LLC | Cat# 23227 |
| Novex NuPAGE 4–12% Bis-Tris Protein Gels, 1.5mm, 10 well | Invitrogen | Cat# NPG335BOX |
| ECL Western Blotting Detection Reagents | GE Healthcare | Cat# 45-GGG-878 |
| Cytofix/Cytoperm Plus | BD Biosciences | Cat# 555G28 |
| Nextera DNA Flex Library Prep Kit | Illumina | Cat# 15G28212 |
| AMPure XP for PCR Purification | Beckman Coulter Inc | Cat# A6388G |
| Deposited Data | ||
| Raw RNA-seq data | GEO dataset browser | GSE124966 |
| Raw ATAC-seq data | GEO dataset browser | GSE124967 |
| Raw scRNA-seq data | GEO dataset browser | GSE124992 |
| Raw ChIP-seq data | GEO dataset browser | GSE143679 |
| Raw RNA-seq data (RUNX1 KD) | GEO dataset browser | GSE143471 |
| Raw ATAC-seq data (RUNX1 KD) | GEO dataset browser | GSE143471 |
| Experimental Models: Organisms/Strains | ||
| NOD/SCID/interleukin 2 receptor g chain null mice | Jackson Laboratory | GG5557 |
| Recombinant DNA | ||
| mP2A-RUNX1c | Chang et al., 2018 | N/A |
| G-U6 | Papapetrou et al., 2010 | N/A |
| mP2A-mCherry-mCherry | This paper | N/A |
| mP2A-mCherry-TSPAN18 | This paper | NM_130783 |
| Sequence-Based Reagents | ||
| shRNA RUNX1 | This paper | https://portals.broadinstitute.org/gpp/public/trans/details?transName=NM_GG1GG189G.2 |
| Scrambled shRNA | Kotini et al., 2015 | N/A |
| Primers used for qRT-PCR analyses, see Table S6 | This paper | N/A |
| Software and Algorithms | ||
| FlowJo software | TreeStar, Inc. | N/A |
| STAR alignment tool | PMID: 23104886 | https://github.com/aIexdobin/STAR |
| Bowtie 2 alignment tool | PMID: 22388286 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad Software, Inc. | N/A |
| R-package | N/A | http://www.R-project.org |
| NIS-Elements BR | Nikon Instruments Inc. | N/A |
| NIS-Elements D4.40.00 | Nikon Instruments Inc. | N/A |
| FIMO | Grant et al., 2011 | http://meme-suite.org/db/motifs |
| HOMER | PMID: 20513432 | http://homer.ucsd.edu/homer/ |
| GSEA | PMID: 16199517 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Cell Ranger | 10X Genomics | https://www.1Gxgenomics.com/ |
| Seurat package | Satija et al., 2015 | N/A |
| Monocle | Trapnell et al., 2014 | N/A |
| SCENIC | Aibar et al., 2017 | N/A |
| GENIE3 | Huynh-Thu et al., 2010 | N/A |
| RcisTarget | Aibar et al., 2017 | N/A |
| UCSC Genome browser | University of California, Santa Cruz | https://genome.ucsc.edu/ |
| Other | ||
| 7500 Fast Real-Time PCR System | Applied Biosystems | N/A |
| BD Fortessa | BD Biosciences | N/A |
| Evos XL Core Cell Imaging System | Thermo Fisher Scientific Inc. | N/A |
| Nikon DS-U3 | Nikon Instruments Inc. | N/A |
| Shandon CytoSpin III | Thermo Electron Corporation | N/A |
| Nikon Eclipse Ci | Nikon Instruments Inc. | N/A |
| Nikon DS-Ri2 camera | Nikon Instruments Inc. | N/A |
| NextSeq-500 | Illumina Inc | N/A |
| HiSeq 2500 | Illumina Inc | N/A |
| Hi-seq 2000 | Illumina Inc | N/A |
| Q Imaging Retiga 2000R cooled CCD | Q Imaging | N/A |
| Olympus IX-70 Microscope | Olympus | N/A |
| LSC+ and LSC- cell fractions data | Ng etal., 2016 | GSE76008 |
| Cohort 1 (163 samples analyzed on U133A&B arrays) | GEO dataset browser | GSE12417 |
| Cohort 2 (79 samples analyzed on U133 Plus 2.0 array) | GEO dataset browser | GSE12417 |
| Beat cohort | dbGaP and Genomic Data Commons | phs001657.v1.p1. |
Western Blot
iLSCs transduced with a lentiviral vector encoding either RUNX1 or scrambled shRNA, were lysed two days after transduction with high salt buffer (0.3 M KCl) supplemented with protease inhibitor. Protein concentrations were determined by bicinchoninic acid assay (Pierce Biotechnology Inc.) and 20 mg of protein from each extract were diluted in Laemmli SDS sample buffer and resolved by electrophoresis on Bolt 4% to 12% Bis-Tris precast gels (Invitrogen) and blotted on nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk diluted inTris-buffered saline and incubated with primary antibody RUNX1 (clone 4E7, Abnova) and β-Actin (Cell Signaling). After washing, blots were incubated with HRP-conjugated secondary antibody and developed using ECL Western Blotting Detection Reagents (Amersham GE Healthcare). Band intensity was quantified by ImageJ.
qRT-PCR
RNA was isolated from iLSCs two days after transduction with RUNX1 or scrambled shRNA with Trizol (Life Technologies). Reverse transcription was performed with Superscript III (Life Technologies) and qPCR was performed with the SsoFast EvaGreen Supermix (Bio-Rad) using primers shown in the key resources table. Reactions were performed in triplicate in a 7500 Fast Real-Time PCR System (Applied Biosystems).
Primary AML patient cell experiments
Patient samples were obtained with informed consent under protocols approved by a local Institutional Review Board at Memorial Sloan Kettering Cancer Center, the Icahn School of Medicine at Mount Sinai and the Sylvester Comprehensive Cancer Center, University of Miami. Cryopreserved primary AML patient cells were thawed and cultured in X-VIVO 15 containing 20% BIT 9500, 1% nonessential amino acids (NEAA), 1 mM L-glutamine and 0.1 mM β-mercaptoethanol (2ME), supplemented with 100 ng/ml SCF, 50 ng/ml Flt3L, 50 ng/ml TPO and 20 ng/ml IL-3. After 1–3 days of prestimulation (depending on cell viability), 300,000 to 1,000,000 cells were transduced with either RUNX1 or scrambled shRNA lentiviral vectors. 48 hours later the cells were harvested and transduction efficiency (%GFP+ cells) was assessed by flow cytometry. 5,000 cells were plated in duplicate in semi-solid methylcellulose media containing SCF, IL3, IL6, EPO, G-CSF, GM-CSF and colonies were scored after 10–14 days. In parallel, 200,000 – 5000,000 cells were plated in liquid culture (X-VIVO 15 with 20% BIT 9500,1% NEAA, 1 mM L-glutamine, 0.1 mM 2ME, 100 ng/ml SCF, 50 ng/ml Flt3L, 50 ng/ml TPO and 20 ng/ml IL-3) and counted 5 days later. Absolute cell number values were multipled by the fraction of GFP+ cells in the beginning of the counts (48 hours post-transduction) and at the final time point (day 5 or day 3 in experiments with limiting numbers of surviving cells), as detailed in Table S5. For xenotransplantation, patient cells transduced with RUNX1 or scrambled shRNA were collected 48 hours after transduction and injected i.v. into busulfan-treated NSG mice at 1×106 cells per mouse.
TSPAN18 rescue experiments
mCherry and the TSPAN18 cDNA (NM_130783) linked by a P2A peptide were cloned into the mP2A lentiviral vector (Kotini et al.,2015). Vector production was performed as described (Kotini et al., 2015). iLSCs were separated from iBlasts on day 16 of hematopoietic differentiation. 300,000 iLSCs were simultaneously transduced with a lentiviral vector expressing RUNX1 shRNA or scrambled shRNA and GFP and a lentiviral vector expressing TSPAN18 and mCherry or mCherry alone. Transduction efficiency (GFP and Cherry) was assessed by flow cytometry 48 hours after transduction.
RUNX1 ChIP sequencing
iLSCs and iBlasts from 2 independent differentiation experiments were separated on day 16 of hematopoietic differentiation. Formaldehyde-fixed cell pellets were suspended into 0.5 mL sonication buffer (0.1% SDS, 1% Triton, 1 mM EDTA, 16.7 mM Tris pH 8, 167 mM NaCl) with 1x proteasome inhibitor cocktail (Millipore, #535140). The samples were sonicated using Bioruptor Pico (Diage- node) for 12 cycles with 30 s on/off. ChIP was performed at 4°C O/N with 1 μg of chromatin DNA sample, 1.5 μg of RUNX1 antibody (Abcam, ab23980) or Rabbit IgG (Santa Cruz Biotech, sc-2027) and 20 μL of magnetic Protein A/G beads (NEB). The beads were washed twice with the following buffers: high salt buffer (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris pH 8,500 mM NaCl); Li buffer (0.25 M LiCl, 1% NP40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris pH 8); Tris (pH 8). The beads were then suspended in 13 μLof H20,15μ of TD and 2 μL of TDE1 (Illumina, 15028212) and incubated at 37°C with constant shaking for 1 h. The beads were washed twice with TE (pH 8), resupsended in 20 μL buffer (100 mM Tris pH 8, 1mM EDTA, 0.1% SDS, 250 mM NaCl) with 1 mL of Proteinase K (Invitrogen, AM2546) and incubated at 65°C for 4 h. The DNA in the supernatant was purified with 2x Ampure beads (Beckman). The purified DNA was PCR-amplified for 12 cycles with Illumina index primers and sequenced on an Illumina Nextseq 500 platform.
QUANTIFICATION AND STATISTICAL ANALYSIS
scRNA-seq analysis
Differential gene expression analysis of RNA-seq data
GSEA analysis
Association of LSC gene sets with primary AML samples
Survival analyses
Differential accessibility analysis of ATAC-seq data
Motif enrichment analysis
CHIP-seq analysis
Statistical analysis
scRNA-seq analysis
Reads were aligned to the human reference genome hg19. Chromium 10x data was processed using Cell Ranger (v.2.1.0) with default parameters. The Seurat package (v.2.3.4) was used to perform unbiased clustering of single cells (Satija et al., 2015). Briefly, cells with unique molecular identifier (UMI) count lower than 1,000 or mitochondrial UMIs higher than 40% were filtered out. To account for potential doublets, cells with UMI count higher than 40,000 were also filtered out. The data was log normalized using a scale factor of 10,000. Potential confounders of clustering (number of UMIs, percentage of mitochondrial reads and cell cycle) were regressed out. Cell clustering was performed using the following parameters: reduction type = pca, dim.use 1:20, resolution = 0.5. Cluster marker expression analysis was performed using the FindAllMarkers function (min.pct = 0.1, thres.use = 0.3). UMAP was used to visualize the clusters and gene expression. Clusters were manually annotated based on the top differentially expressed genes. Cell cycle and iLSC score analyses were performed by calculating the percentage of the transcriptome mapping to the corresponding gene set. Pseudotime ordering was performed using Monocle (Trapnell et al., 2014), setting the origin of pseudotime in the cell state containing the highest fraction of cells from the first time point (day 16). We identified regulons using SCENIC (v.1.0.1.1) (Aibar et al., 2017) with default parameters, using 10,000 randomly sampled cells from our dataset. Briefly, GENIE3 (v.3.5) was used to calculate the co-expression of transcription factors and target genes (Huynh-Thu et al., 2010), keeping genes with 3 molecules detected in at least 1% of the cells and also present in the RcisTarget (v.3.8) database (Aibar etal., 2017). For each regulon, each cell was scored by the mean of the normalized expression of all genes associated with the regulon. Regulons whose scores were highly variable across all cells were selected by calculating modified z-scores of the variances using the formula: z = (x - median(x)) / MAD(x), where MAD(x) = 1.4826 * median(|x - median(x)|). Regulons with z scores > 10 were selected as regulons of interest and are shown in Table S4.
Differential gene expression analysis of RNA-seq data
Fastq files were aligned to GRCh37.75 (hg19) using STAR alignment tool. Aligned bam files were filtered for uniquely aligned reads with MAQ >10. Subsequently, RNA-seq reads were counted using GRCh37.75 annotation with tRNA and rRNA removed. Read counting was done using SummarizeOverlaps function from R-package “GenomicAlignments” with IntersectionNotEmpty mode. There were 10–20 million reads for each sample. We filtered genes by keeping genes with RPKM > 0.5 in at least three samples and count > 10 in at least three samples. There were 16,800 genes in the count table after filtering. Differential expression analysis was performed using R-package “DESeq2.” Genes with Benjamini & Hochberg (BH) adjusted P value <0.05 were considered significantly differentially expressed.
GSEA analysis
GSEA analysis was performed on all 4,733 curated gene sets in the Molecular Signatures Database (MSigDB, https://www.broadinstitute.org/msigdb) combined with 92 additional relevant gene sets from our experimentally derived or published hematopoietic self-renewal and differentiation signatures (PMID: 16199517, 24395885). Significantly enriched gene sets are defined by cutoff FDR < 0.05.
Association of LSC gene sets with primary AML samples
We generated per sample ssGSEA score for the different gene sets using R-package “GSVA” and tested the association of the ssGSEA score with LSC+ status, using the GSE76008 dataset, which includes 138 LSC+ samples and 89 LSC−samples, by Wilcoxon rank-sum test. The association of genes individually with LSC status was also determined by Wilcoxon rank-sum test.
Survival analyses
For survival analyses, we used public clinical data from GSE12417, which includes two cohorts. Cohort 1 includes 163 samples analyzed on U133A&B arrays and cohort 2 includes 79 samples analyzed on U133 Plus 2.0 array. To calculate the impact of RUNX1 expression and of the RUNX1-dependency gene signature on survival, we considered samples with expression above median as “high” and samples with expression below median as “low.” Survival curves were plotted using R package “survival” and p values were computed by log-rank test.
Differential accessibility analysis of ATAC-seq data
Fastq files were trimmed with Trimmomaticto remove adaptor sequences and aligned to GRCh37.75 (hg19) using Bowtie2 alignment tool. Subsequently, low quality PE reads with MAP < 20 were removed using samtools and duplicate reads were removed using Picard. All aligned reads where adjusted for shifts in Tn5 binding. We then called peaks for each replicate using macs2 with the following parameters (-p 1e-1-nomodel-shift −37-extsize 73). For each pair of replicates, we performed irreproducibility rate (IDR) analysis and considered a peak to be reproducible if IDR < 0.05. Finally, we created an ATAC-seq atlas of 65,339 peaks by combining all reproducible peaks (merging 2 peaks if they overlap by 75%). A peak was annotated as “promoter” if it localized within 2 kb of a transcription start site (TSS), as “exon” if it localized in an exon region, as “intron” if it localized in an intronic region, and as “intergenic” if it localized outside a transcript region and 2 kb away from a TSS. Each peak was assigned to the nearest gene within 50 kb.
Differential accessibility analysis was performed using R-package “DESeq2.” Peaks with log2fold change > 1 orlog2 old change < −1 and Benjamini & Hochberg (BH) adjust P value < 0.05 were considered significantly differentially accessible.
To determine accessibility changes at the gene level, we considered a gene to be of significant accessibility changes, if: (1) There was at least 1 peak within 50 kb with significant accessibility change; or (2) The distribution of accessibility change (log2FC) of all peaks within 50 kb was significantly shifted (p < 0.05) as compared to global background distribution by a KS test. Genome tracks were generated with the University of California, Santa Cruz (UCSC) genome browser.
Motif enrichment analysis
We used two independent approaches for motif analysis: (1) We first used FIMO (Grant et al., 2011) to search for motif hits in the ATAC-seq atlas with default parameters using CIS-BP Single Species DNA Homo_sapiens motif database (http://meme-suite.org/db/motifs), restricted to expressed TFs only, based on our RNA-seq filtering thresholds. For each motif, we compared accessibility log2FC distribution of peaks containing the motif with the accessibility log2FC of the atlas by KStest. The KS statistic was then plotted against the percentage of motif hits in the atlas. Positive/negative sign of KS statistic indicates the direction of change with positive indicating increased accessibility and negative decreased accessibility in iLSCs. (2) We used HOMER to search for top motif enrichment in the peaks with significant accessibility changes using known human motifs in the HOMER default database.
CHIP-seq analysis
The DNA sequencing reads were aligned to hg19 using Bowtie2 (version 2.2.6) with default parameters. MACS2 (version 2.1.1) was used to identify the enriched regions for RUNX1 binding relative to the IgG control with the shifting model and q-value 0.01, followed by the subtraction of known dark regions. Deeptools (version 3.3.1) was used to average profiles for RUNX1 binding. Genome tracks were generated with the University of California, Santa Cruz (UCSC) genome browser.
Statistical analysis
Statistical analysis was performed with GraphPad Prism software. Pairwise comparisons between different groups were performed using a two-sided unpaired unequal variance t test. For all analyses, p < 0.05 was considered statistically significant. Investigators were not blinded to the different groups.
Supplementary Material
Highlights.
AML-iPSC-derived hematopoietic cells recapitulate a LSC hierarchy
iLSCs can be easily prospectively isolated
A LSC 16-gene set correlates with AML patient survival
AML-iPSC-derived hematopoietic cells recapitulate a leukemia stem cell (LSC) hierarchy
ACKNOWLEDGMENTS
This work was supported by the US National Institutes of Health (NIH) grants R01HL121570 (E.P.P.), R01HL132071 (E.P.P.), and R01CA225231 (E.P.P. and M.G.K.); the New York State Stem Cell Board (E.P.P. and M.G.K.); the Pershing Square Sohn Cancer Research Alliance (E.P.P.); the Edward P. Evans Foundation, a Scholar award from the Leukemia and Lymphoma Society (LLS); and a RUNX1 Research Program (RRP)/Alex’s Lemonade Stand Foundation (ALSF) grant (E.P.P.). M.G.K. was supported by NIH grants R01DK101989, R01CA193842, R01HL135564, and R01CA186702; Center Support Grant/Core Grant (P30 CA008748); the Starr Cancer Consortium; the LLS Translational Research Program; the Susan and Peter Solomon Fund; the Tri-Institutional Stem Cell Initiative 2016–014; a Kimmel Scholar Award, a V-Scholar Award; a Geoffrey Beene Award; a LLS Career Development Award; and an ALSF A Award. S.D.N. was supported by NIH grants R01CA166835 and P30CA240139 and by a RRP/LLS grant. D.A.L. was supported by a Burroughs Wellcome Fund Career Award for Medical Scientists, a Pershing Square Sohn Prize for Young Investigators in Cancer Research, a National Institutes of Health Director’s New Innovator Award (DP2-CA239065), the LLS Translational Research Program, the NIH grant R01HL145283, and a Stand Up To Cancer Innovative Research Grant (SU2C-AACR-IRG-0616). J.W. was supported by a postdoctoral fellowship from the German Research Foundation (DFG; WE 6233). H.L. was supported by a Training Program in Stem Cell Biology fellowship from the New York State Department of Health (NYSTEM). A.Z. was supported by an EMBO long-term fellowship (ALTF 140–2016). N.D. was supported by a Medical Scientist Training Program grant (T32GM007739) from the National Institute of General Medical Sciences of the National Institutes of Health to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD/PhD Program. The authors thank Ravi Majeti, Mark Chao, and Florian Heidel for sharing research materials.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107688.
DECLARATION OF INTERESTS
E.P.P. has received honoraria from Celgene and Merck and research support from Incyte. M.G.K. has received consultant fees from Acent Therapeutics and research support from 28–7. These disclosures are not directly related to the present study.
REFERENCES
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadis EI, Botthof JG, Andres H, Ferreira L, Lio P, and Cvejic A.(2017). Single-cell RNA-sequencing uncovers transcriptional states and fate decisions in haematopoiesis. Nat. Commun 8, 2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens K, Maul K, Tekin N, Kriebitzsch N, Indenbirken D, Prassolov V, Müller U, Serve H, Cammenga J, and Stocking C. (2017). RUNX1 cooperates with FLT3-ITD to induce leukemia. J. Exp. Med 214, 737–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrelha J, Meng Y, Kettyle LM, Luis TC, Norfo R, Alcolea V, Boukarabila H, Grasso F, Gambardella A, Grover A, et al. (2018). Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 554, 106–111. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Kotini AG, Olszewska M, Georgomanoli M, Teruya-Feldstein J, Sperber H, Sanchez R, DeVita R, Martins TJ, Abdel-Wahab O, et al. (2018). Dissecting the Contributions of Cooperating Gene Mutations to Cancer Phenotypes and Drug Responses with Patient-Derived iPSCs. Stem Cell Reports 10, 1610–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MP, Gentles AJ, Chatterjee S, Lan F, Reinisch A, Corces MR, Xavy S, Shen J, Haag D, Chanda S, et al. (2017). Human AML-iPSCs Reacquire Leukemic Properties after Differentiation and Model Clonal Variation of Disease. Cell Stem Cell 20, 329–344.e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, and Speck NA (2009). Runxl is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457, 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick JE (2008). Stem cell concepts renewcancerresearch. Blood 112,4793–4807. [DOI] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. (2011). Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med 17, 1086–1093. [DOI] [PubMed] [Google Scholar]
- Fu L, Fu H, Tian L, Xu K, Hu K, Wang J, Wang J, Jing H, Shi J, and Ke X. (2016). High expression of RUNX1 is associated with poorer outcomes in cytogenetically normal acute myeloid leukemia. Oncotarget 7, 15828–15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Tober J, Gao P, Chen C, Tan K, and Speck NA (2018). RUNX1 and the endothelial origin of blood. Exp. Hematol 68, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles AJ, Plevritis SK, Majeti R, and Alizadeh AA (2010). Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA 304, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, Nakagawa M, Olsson A, Wunderlich M, Link KA, et al. (2013). Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Invest 123, 3876–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinenko T, Arndt K, Portz M, Mende N, Günther M, Cosgun KN, Alex- opoulou D, Lakshmanaperumal N, Henry I, Dahl A, and Waskow C.(2014). Clonal expansion capacity defines two consecutive developmental stages of long-term hematopoietic stem cells. J. Exp. Med 211, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatlen MA, Wang L, and Nimer SD (2012). AML1-ETO driven acute leukemia: insights into pathogenesis and potential therapeutic approaches. Front. Med 6, 248–262. [DOI] [PubMed] [Google Scholar]
- Ho TC, LaMere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, Liesveld JL, Mendler JH, Guzman M, Morrissette JD, et al. (2016). Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 128, 1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A,Wehenkel L, and Geurts P. (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS One 5, e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T, Mitani K, Chiba S, Ogawa S, Kurokawa M, and Hirai H. (2004). AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat. Med 10, 299–304. [DOI] [PubMed] [Google Scholar]
- Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nem- kov T, Pei S, Khan N, Adane B, Ye H, et al. (2018). Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 34, 724–740.e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, Fronick C, Magrini V, Demeter RT, Fulton RS, et al. (2014). Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 25, 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, Chang CJ, Boussaad I, Delrow JJ, Dolezal EK, Nagula- pally AB, Perna F, Fishbein GA, Klimek VM, Hawkins RD, et al. (2015). Functional analysis of a chromosomal deletion associated with myelo-dysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol 33, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, Chang CJ, Chow A, Yuan H, Ho TC, Wang T, Vora S, Solovyov A, Husser C, Olszewska M, et al. (2017). Stage-Specific Human Induced Pluripotent Stem Cells Map the Progression of Myeloid Transformation to Transplantable Leukemia. Cell Stem Cell 20, 315–328.e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, and Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. [DOI] [PubMed] [Google Scholar]
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, et al. (2009). p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ES, Davis R, Stanley EG, and Elefanty AG (2008). A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat. Protoc 3, 768–776. [DOI] [PubMed] [Google Scholar]
- Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD, et al. (2016). A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–37. [DOI] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. (2016). Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou EP, Korkola JE, and Sadelain M. (2010). A genetic strategy for single and combinatorial analysis of miRNA function in mammalian hematopoietic stem cells. Stem Cells 28, 287–296. [DOI] [PubMed] [Google Scholar]
- Pellin D, Loperfido M, Baricordi C, Wolock SL, Montepeloso A, Weinberg OK, Biffi A, Klein AM, and Biasco L. (2019). A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun 10, 2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollyea DA, Gutman JA, Gore L, Smith CA, and Jordan CT (2014). Targeting acute myeloid leukemia stem cells: a review and principles for the development of clinical trials. Haematologica 99, 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarmyne M, Doan PL, Himburg HA, Yan X, Nakamura M, Zhao L, Chao NJ, and Chute JP (2015). Protein tyrosine phosphatase-s regulates hematopoietic stem cell-repopulating capacity. J. Clin. Invest 125, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch A, Chan SM, Thomas D, and Majeti R. (2015). Biology and Clinical Relevance of Acute Myeloid Leukemia Stem Cells. Semin. Hematol 52, 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, and Camargo FD (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553,212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RG, Mandelbaum J, Zon LI, and Daley GQ (2016). Engineering Hematopoietic Stem Cells: Lessons from Development. Cell Stem Cell 18, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Bouriez Jones T, et al. (2013). Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502, 232–236. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M, Quintana E, Fearon ER, and Morrison SJ (2009). Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 138,822–829. [DOI] [PubMed] [Google Scholar]
- Shin JY, Hu W, Naramura M, and Park CY (2014). High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med 211, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, et al. (1999). Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet 23, 166–175. [DOI] [PubMed] [Google Scholar]
- Sood R, Kamikubo Y, and Liu P. (2017). Role of RUNX1 in hematological malignancies. Blood 129, 2070–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, and Majeti R. (2017). Biology and relevance of human acute myeloid leukemia stem cells. Blood 129, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge JJ, Snow JW, Kim J, and Orkin SH (2009). DNA methyl-transferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5, 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, Hirche C, Lutz C, Buss EC, Nowak D, et al. (2017). Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol 79, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Wilkinson AC, Ooehara J, Lan X, Lai CY, Nakauchi Y, Pritchard JK, and Nakauchi H. (2018). Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment. Cell Stem Cell 22, 600–607.e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Adane B, Khan N, Alexeev E, Nusbacher N, Minhajuddin M, Stevens BM, Winters AC, Lin X, Ashton JM, et al. (2018).). Subversion of Systemic Glucose Metabolism as a Mechanism to Support the Growth of Leukemia Cells. Cancer Cell 34, 659–673.e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data are accessible through GEO datasets: GSE124966. ATAC-Seq data are accessible through GEO datasets: GSE124967. Single-cell RNA-Seq data are accessible through GEO datasets: GSE124992. CHiP-Seq data are accessible through GEO datasets: GSE143679. RNA- and ATAC-Seq after RUNX1 KD data are accessible through GEO datasets: GSE143471.