

Abstract
Mastermind-like domain-containing 1 (MAMLD1) has been shown to play an important role in the process of sexual development and is associated with 46,XY disorders of sex development (DSDs). However, the causative role of MAMLD1 variations in DSDs remains disputable. In this study, we have described a clinical series on children from unrelated families with 46,XY DSD harbouring MAMLD1 variants. Whole exome sequencing (WES) was performed for each patient. WES data were filtered using common tools and disease customisation algorithms, including comparison against lists of known and candidate MAMLD1-related and DSD-related genes. Lastly, we investigated the hypothesis that MAMLD1-related DSD may follow an oligogenic mode of inheritance. Forty-three potentially deleterious/candidate variants of 18 genes (RET, CDH23, MYO7A, NOTCH2, MAML1, MAML2, CYP1A1, WNT9B, GLI2, GLI3, MAML3, WNT9A, FRAS1, PIK3R3, FREM2, PTPN11, EVC, and FLNA) were identified, which may have contributed to the patient phenotypes. MYO7A was the most commonly identified gene. Specific gene combinations were also identified. In the interactome analysis, MAMLD1 exhibited direct connection with MAML1/2/3 and NOTCH1/2. Through NOTCH1/2, the following eight genes were shown to be associated with MAMLD1:WNT9A/9B, GLI2/3, RET, FLNA, PTPN11, and EYA1. Our findings provide further evidence that individuals with MAMLD1-related 46,XY DSD could carry two or more variants of known DSD-related genes, and the phenotypic outcome of affected individuals might be determined by multiple genes.
Keywords: disorders of sexual development, MAMLD1, oligogenic inheritance, hypospadias, whole exome sequencing
Introduction
Disorders of sex development (DSDs) comprise a group of congenital diseases associated with the atypical development of internal and external genital structures. These conditions may be related to genetic variation, developmental programming, and hormone expression (1). Our understanding of DSDs has evolved significantly over the past several decades owing to the extensive research conducted on mammalian sex development and the genetic mechanisms underlying DSDs (2–5). Various underlying causes, such as mutations in the genes encoding proteins associated with sex determination and development as well as genital development, have been described (5). However, genotype-phenotype correlations are difficult to evaluate owing to the high phenotypic and genotypic diversity among individuals.
Mastermind-like domain-containing 1 gene (MAMLD1), also known as chromosome X open reading frame 6 (CXorf6) or F18 (online Mendelian inheritance in man (OMIM)# 300120), was first reported in two cases of myotubular myopathy and male hypogenitalism (6, 7). It was identified as a suitable candidate gene in patients with 46,XY DSD and was shown to be expressed in foetal Leydig cells at a time point close to the critical period for sex development (8, 9). In mouse Leydig tumour cells, the transient knockdown of Mamld1 mRNA expression led to a significant reduction in testosterone (T) production (10). MAMLD1 contains the target sequence of steroidogenic factor (SF-1), which is a modulator of gene transcription involved in testicular differentiation (11, 12). MAMLD1 transactivates the non-canonical Notch-targeted Hes 3 promoter (8, 12). Hes3 regulates cell differentiation and proliferation during embryonic development (13). Therefore, MAMLD1 appears to play an important role during sex development and is associated with 46,XY DSD.
To date, approximately 30 MAMLD1 sequence variations have been identified in 46,XY DSD patients and recorded in the human gene mutation database (14). Disease-causing MAMLD1 variants can carry nonsense, missense, or frameshift mutations; insertions; or deletions, and these may even include complex variants (14). They are found to be scattered throughout the gene sequence and are not restricted to significant hotspots, owing to which the genotype-phenotype correlations remain obscure (14). Patients with 46,XY DSDs share a wide variety of phenotypic features (8, 9). The most significant phenotypic feature observed is hypospadias. Other phenotypes include cryptorchidism, micropenis, complete female external genitalia, and primary amenorrhea (14–17).
However, even after investigation of the condition, the causative role of MAMLD1 variations in DSDs remains disputable for several reasons. First, some MAMLD1 variants (P359S, V505A, and N662S) have also been identified in normal individuals (9, 15, 16), while others (P359S and Q580R) have not been detected in all affected patients from a single family (9). Second, several MAMLD1 variations have been confirmed to be associated with wild-type activity in functional studies (15, 18), and the animal experiments have shown that Mamld1-KO male mice present with normal genitalia and reproduction (19). Therefore, the role of MAMLD1 in sex development requires further elucidation; the broad spectrum of phenotypes indicates the presence of various modifying factors, such that a single pathogenic variant may neither be necessary nor sufficient for pathogenesis.
Our understanding of the genetic architecture of sex development-related inherited disorders has increased considerably over the past few years. Initial discoveries pertained to the identification of genes encoding proteins associated with disorders with Mendelian (monogenic) inheritance. In recent years, gene discovery efforts have evolved to consider more complex inheritance patterns, such as oligogenic inheritance, in which the accumulation of inherited low-penetrance variants in multiple genes contributes to the disorder (20). Oligogenic inheritance has been noted in several disorders, such as in congenital hypogonadotropic hypogonadism (21), inherited cardiac disorders (20), and NR5A1-related DSDs (22), using high-throughput sequencing (HTS).
In the present study, we investigated the hypothesis that MAMLD1-related DSDs may follow an oligogenic mode of inheritance. Whole exome sequencing (WES) was performed for ten subjects with 46,XY DSD harbouring MAMLD1 variants. WES data were filtered using common tools and a disease-tailored algorithm including lists of MAMLD1-related and DSD-related known and candidate genes designed by Fluck et al. (23). Using this method, we attempted to provide evidence that phenotypic outcomes may be determined by multiple genes.
Materials and Methods
Subjects
Patients ranged in age from 2 months to 14 years and had been admitted to Beijing Children’s Hospital in the past 5 years. Ten patients (1–10) from unrelated families, the members of which were confirmed to harbour mutations in MAMLD1, were recruited in the study. Each of the patients were assigned the same number as in our previously reported study (14). Patients with abnormal liver or kidney function, or those with systemic diseases that affect physical development, were excluded.
Clinical information was obtained from the medical record of each patient. Two experienced paediatric endocrinologists performed physical examinations and assessments. The information included, but was not limited to, age at visit, social gender, chief complaint, family history, bone age, birth length, birth weight, gestational age, history of gestation, penile length, testis size, testis position, urethral meatus, scrotal appearance, electrolyte levels, and liver and kidney function. Hormone investigations involved measurement of the levels of luteinizing hormone, follicle-stimulating hormone, anti-Müllerian hormone, inhibin-B, testosterone, adrenocorticotropic hormone, cortisol, 17-hydroxyprogesterone, and dehydroepiandrosterone sulphate. A flow chart of the study design is presented in Figure 1 . The clinical evaluation algorithm of patients with DSD is presented in Figure 2 .
Figure 1.

Flow chart of the study design.
Figure 2.

The clinical evaluation algorithm of DSD patients. * abnormal puberty indicating that a girl with primary amenorrhoea with or without breast development, a girl who virilizes at puberty or a boy with pubertal delay; DSD, Disorders of Sex Development; FSH, Follicle Stimulating Hormone; LH, Luteinizing Hormone; T, Testosterone; DHEA, Dehydroepiandrosterone; E2, Estradiol; AMH, Anti-Mullerian Hormone; DHT, Dihydrotestosterone; 17-OHP, 17-OH-Progesterone; ACTH, Adrenocorticotrophic Hormone; FBG, Fast Blood Glucose; HCG, Human Chorionic Gonadotropin; HMG, Human Menopausal Gonadotropin; CHH, Congenital Hypogonadotropic Hypogonadism; Dysg, Gonadal Dysgenesis.
Molecular Analysis
WES was performed for all patients. Genomic DNA was extracted from the peripheral blood samples of probands and their parents, and was sent to Chigene Translational Medicine Research Center Co., Ltd (Beijing Kangso Medical Laboratory Zhongguancun Huakang Gene Institute) for commercial sequencing. Putative candidate variants were confirmed using Sanger sequencing.
Bioinformatics Analysis
WES data were filtered using a disease-tailored list of MAMLD1-related and DSD-related known and candidate genes (N=606), similar to the algorithm previously designed by Camats et al. (22, 23). A project-specific filter for DSD-related and MAMLD1-related genes was generated by conducting a search in published literature and databases. The DSD-related gene list included genes with (potentially) deleterious variants reported in patients with 46,XY and 46,XX DSDs; genes with (potentially) disease-causing variants reported in syndromic patients with involvement of sex development; genes “related” to DSD in KO/mutant animal models (mice and rats); and overexpressed, upregulated, or downregulated genes in rodent embryonic gonadal cells. The following bioinformatics software tools were used for the interpretation and classification of variants: InterVar (http://wintervar.wglab.org/; clinical interpretation of genetic variants using the ACMG/AMP 2015 guideline), VarSome (The Human Genomics Search Engine; https://varsome.com/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Alamut Visual 2.11 (https://www.interactive-biosoftware.com/es/alamut-visual/).
After annotation, variant analysis was performed according to the following steps. A) WES data of each patient were first filtered using the MAMLD1- and DSD-related known and candidate gene lists; B) Variants with minor allele frequency (MAF) <0.05, or those undetected in gnomAD, 1000 Genomes (China), and ExAC (East Asia), were retained; C) Variants that were considered irrelevant for our study, including 1) variants detected in more than two patients, 2) variants in repeat regions, 3) variants in genes or gene regions with high variability (MAF >0.05), and 4) variants with low coverage and/or low quality, were discarded.
A search was performed for reported (potentially) disease-causing variants using the Human Gene Mutation Database (HGMD® Professional 2018.2, http://www.biobase-international.com/product/hgmd; Biobase) and dbSNP (http://www.ncbi.nlm.nih.gov/snp/). The search tool for the retrieval of interacting genes/proteins (STRING, http://string-db.org/) was used to analyse interactions within gene carriers of notable variants (DSD-related and/or MAMLD1-related). A medium confidence of 0.400 was noted. STRING data were extracted from known interactions (curated databases, experimentally determined interactions), predicted interactions (gene neighbourhood, gene fusions, gene co-occurrence), and other inferred evidences such as text mining, co-expression, and protein homology.
Ethics Approval
The project was approved by the Institutional Medical Ethics Review Board of Beijing Children’s Hospital (ID: 2019-k-156). Written informed consent was obtained from all patients or their legal guardians. This study was conducted in accordance with the principles of the Declaration of Helsinki.
Results
Clinical Features
The clinical features, hormone profiles, and molecular results of the ten Chinese patients harbouring MAMLD1 mutations are summarised in a previously published article (14). The age at visit of each subjected was under 3 years. The salient phenotypic feature was hypospadias (8/10); other phenotypic features included cryptorchidism (3/10) and micropenis (7/10). Serum T, luteinizing hormone, and follicle-stimulating hormone levels were sufficiently high in patients #3 and #6, who were in the mini-puberty period. An adequate response of T levels to human chorionic gonadotropin (hCG) stimulation was observed in patients #1–3, #6, and #8. Overall, nine genetic variants were identified, including six missense variations (p.P334S, p.S662R, p.A421P, p.T992I, p.P542S, and p.R927L) and three nonsense variations (p.R356X, p.Q152X, and p.Q124X). All patients had inherited the variants from their mothers. Detailed information on the functional domains is presented in Figure 3 .
Figure 3.

Detailed variation information of functional domains of the nine MAMLD1 variants.
Identification of an Oligogenic DSD Aetiology in Individuals With 46,XY DSD Harbouring MAMLD1 Variants
Forty-three potentially deleterious/candidate variants of 18 genes (RET, CDH23, MYO7A, NOTCH2, MAML1, MAML2, CYP1A1, WNT9B, GLI2, GLI3, MAML3, WNT9A, FRAS1, PIK3R3, FREM2, PTPN11, EVC, and FLNA) were identified in the ten hemizygous MAMLD1 patients ( Table 1 ). These variants may have contributed to the phenotype of each patient.
Table 1.
Identified genes and variants per patient after specific filtering.
| Patient | Gene | Locus | Type | HGVSc,HGVSp | dbSNP ID | Mutation source | MAF Value | ACMG classification | ||
|---|---|---|---|---|---|---|---|---|---|---|
| GnomAD | 1000Genomes (China) | ExAC (East Asian) | ||||||||
| 1 | MAMLD1 | Xq28 | miss | c.1986C>G, p.S662R | mother | VUS[(PM2,BP1)] | ||||
| 1 | RET | 10q11.2 | syn | c.3120G>C, p.L1040L | mother | VUS(PM2,BP7) | ||||
| 1 | CDH23 | 10q22.1 | c.6712+37C>T | rs750966744 | father | 0.000032 | 0.0003 | VUS(PM2) | ||
| 1 | MYO7A | 11q13.5 | c.3504-39T>C | rs192929996 | mother | 0.0013 | 0.019 | 0.017 | VUS(BS1) | |
| 1 | MAML1 | 5q35 | syn | c.1893G>A, p.Q631Q | Denove | VUS(PM2,BP7) | ||||
| 2 | MAMLD1 | Xq28 | non | c.1066C>T, p.R356X | rs782347827 | mother | 0 | P(PVS1,PM2,PP3) | ||
| 2 | WNT9B | 17q21 | c.*158C>T | rs1233082195 | father | 0.000008 | VUS(PM2,PP5) | |||
| 2 | NOTCH2 | 1p13-p11 | c.2600-24C>A | rs149097658 | mother | 0.0034 | 0.024 | 0.027 | VUS(BS) | |
| 2 | GLI2 | 2q14 | c.845+10G>A | rs199673018 | father | 0.00024 | 0 | 0.0005 | VUS | |
| 2 | GLI3 | 7p13 | c.1028+15G>A | rs116842918 | father | 0.0014 | 0.012 | 0.017 | LB(BS1,BP6) | |
| 3 | MAMLD1 | Xq28 | non | c.1066C>T, p.R356X | rs782347827 | mother | 0 | P(PVS1,PM2,PP3) | ||
| 3 | MYO7A | 11q13.5 | c.1554+22C>T | mother | 0.0006 | 0.0024 | VUS(PM2,BP4) | |||
| 3 | MYO7A | 11q13.5 | miss | c.3124T>G, p.W1042G | mother | VUS(PM1,PM2,PP3) | ||||
| 4 | MAMLD1 | Xq28 | miss | c.1261G>C, p.A421P | mother | VUS(PM2,PP3,BP1) | ||||
| 4 | MAML3 | 4q28 | miss | c.1612A>G, p.M538V | rs148778901 | mother | 0.0074 | 0.0072 | 0.0078 | LB (PP3,BS1) |
| 4 | GLI3 | 7p13 | miss | c.1843A>T, p.T615S | rs200913720 | mother | 0.0006 | 0.0024 | 0.0005 | VUS (PP3,BP6) |
| 4 | GLI3 | 7p13 | miss | c.674C>T, p.T225I | rs753769482 | mother | 0.001 | VUS (PM2,PP3) | ||
| 5 | MAMLD1 | Xq28 | non | c.454C>T, p.Q152X | mother | P(PVS1,PM2,PP3) | ||||
| 5 | RET | 10q11.2 | miss | c.1465G>A, p.D489N | rs9282834 | mother | 0.0296 | 0.014 | 0.028 | LB (BS1,BP6,BP4) |
| 5 | NOTCH2 | 1p13-p11 | c.2600-24C>A | rs149097658 | father | 0.0191 | 0.024 | 0.027 | VUS (BS1) | |
| 5 | GLI2 | 2q14 | syn | c.4611C>T, p.L1537L | rs1335405295 | father | VUS (PM2,BP7) | |||
| 6 | MAMLD1 | Xq28 | miss | c.1624C>T, p.P542S | rs146443503 | mother | 0.0015 | 0.016 | 0.021 | LB (BS1,BP1) |
| 6 | MYO7A | 11q13.5 | c.6051+9C>T | rs747742075 | father | 0.0018 | VUS (BS4) | |||
| 6 | CYP1A1 | 15q24.1 | miss | c.518C>G, p.T173R | rs28399427 | father | 0.008 | 0.0072 | 0.0094 | VUS (BP4) |
| 6 | WNT9A | 1q42 | c.353-30G>A | rs141239747 | mother | 0.0314 | 0.039 | 0.028 | B (BA1,BS1,BS2) | |
| 6 | FRAS1 | 4q21.21 | syn | c.10930C>T, p.L3644L | rs183729151 | father | 0.0025 | 0.0048 | 0.0022 | LB (BS4,BP7) |
| 6 | GLI3 | 7p13 | syn | c.3015C>T, p.A1005A | rs200965852 | father | 0.0173 | 0.024 | 0.019 | LB (BS1,BP6,BP7) |
| 7 | MAMLD1 | Xq28 | miss | c.1000C>T, p. P334S | rs41313406 | mother | 0.08 | 0.0031 | 0.0002 | LP(PM1,PM2,PM5,PP3) |
| 7 | CYP1A1 | 15q24.1 | syn | c.927C>T, p.N309N | rs368742906 | unknown | 0.0031 | 0.0024 | 0.0032 | VUS (PM2,BP7,BP4) |
| 7 | PIK3R3 | 1p34.1 | miss | c.290A>G, p.Q97R | rs1225342856 | unknown | VUS (PM1,PM2) | |||
| 8 | MAMLD1 | Xq28 | non | c.370C>T, p.Q124X | mother | P(PVS1,PM2) | ||||
| 8 | CDH23 | 10q22.1 | miss | c.1282G>A, p.D428N | rs188376296 | father | 0.0031 | 0.0048 | 0.0066 | VUS (PP5,PP3,BS4) |
| 8 | MYO7A | 11q13.5 | miss | c.1133G>A, p.R378H | rs397516282 | father | LP (PM2,PM5,PP3,BS4) | |||
| 8 | MYO7A | 11q13.5 | c.4152+15A>G | rs1033447071 | father | VUS (PM2,BS4) | ||||
| 8 | FREM2 | 13q13.3 | miss | c.4916G>A, p.R1639K | rs77886481 | mother | 0.0197 | 0.014 | 0.022 | VUS (BS1) |
| 8 | FRAS1 | 4q21.21 | c.7372-24A>G | rs78365404 | mother | 0.013 | 0.0096 | 0.019 | B (BS1,BS2,BS4) | |
| 8 | FRAS1 | 4q21.21 | miss | c.8493C>G, p.F2831L | rs774409872 | mother | 0.0019 | – | – | VUS (BS4) |
| 9 | MAMLD1 | Xq28 | miss | c.2975C>T, p.T992I | mother | VUS(PM2,BP1,BP4) | ||||
| 9 | CDH23 | 10q22.1 | c.9077+7C>T | rs76114420 | mother | 0.0259 | 0.022 | 0.018 | LB (BS1,BP6) | |
| 9 | WNT9B | 17q21 | syn | c.1059C>T, p.Y353Y | rs537242221 | mother | 0.0006 | 0.0024 | 0 | VUS (BP7) |
| 9 | MAML3 | 4q28 | miss | c.2969C>T, p.P990L | rs185593153 | father | 0.0006 | 0.0048 | 0.0011 | VUS |
| 9 | FRAS1 | 4q21.21 | c.5857-434C>T | mother | VUS (PM2,BS4) | |||||
| 10 | MAMLD1 | Xq28 | miss | c.2780C>T, p.R927L | rs782511956 | mother | LB (PM2,BP1,BP4) | |||
| 10 | CDH23 | 10q22.1 | c.4210-16C>A | rs775928557 | father | VUS (PM2) | ||||
| 10 | CDH23 | 10q22.1 | c.9077+7C>T | rs76114420 | mother | 0.0259 | 0.022 | 0.018 | LB (BS1,BP6) | |
| 10 | MYO7A | 11q13.5 | syn | c.324C>T, p.Y108Y | rs116892396 | mother | 0.0161 | 0.026 | 0.021 | LB (BS1,BP6,BP7) |
| 10 | MAML2 | 11q21 | miss | c.385G>A, p.D129N | rs892433964 | mother | 0.0012 | VUS (PP3) | ||
| 10 | PTPN11 | 12q24 | c.1448-38G>T | mother | VUS (PM2) | |||||
| 10 | NOTCH2 | 1p13-p11 | syn | c.6783G>A, p.E2261E | rs759118563 | mother | 0.0001 | VUS (PM2,BP7) | ||
| 10 | PIK3R3 | 1p34.1 | miss | c.950A>G, p.N317S | rs114180250 | father | 0.0129 | 0.019 | 0.013 | VUS (BS1) |
| 10 | EVC | 4p16 | c.1316-20G>T | rs773461223 | mother | VUS (PM2) | ||||
| 10 | EVC | 4p16 | c.1564-6C>T | rs188245524 | mother | 0.0012 | 0.0024 | 0.0038 | VUS (BP6,BP4) | |
| 10 | FRAS1 | 4q21.21 | miss | c.6569C>T, p.S2190F | rs200166354 | father | 0.0031 | 0.0024 | 0.0033 | VUS (BS4) |
| 10 | FLNA | Xq28 | syn | c.3756G>A, p.A1252A | rs186619828 | mother | 0 | 0 | 0 | VUS (BP6) |
ACMG, the American College of Medical Genetics and Genomics; MAF, Minor Allele Frequency; VUS, Uncertain Significance; P, Pathogenic; B, Benign; LB, Likely Benign; LP, Likely Pathogenic.
Among the eighteen genes, four have been previously identified in patients with hypospadias (CYP1A1, FLNA, GLI3, and GLI2), three have been reported to be associated with cryptorchidism (FLNA, RET, and PTPN11), and one has been identified in patients with micropenis (EVC). In addition, eight genes were identified in patients with DSDs (FRAS1, FREM2, and NOTCH2) and/or were reported to be associated with other syndromes combined with DSDs (CYP1A1, EVC, FRAS1, PTPN11, and RET). Fourteen genes were previously shown to be associated with sexual or gonadal development (CDH23, EVC, FLNA, FRAS1, FREM2, GLI2, GLI3, MAML3, MYO7A, NOTCH2, PIK3R3, RET, WNT9A, and WNT9B). Based on information from OMIM, all patients, except patient #7, presented at least one variant in a gene with autosomal dominant (AD) inheritance (GLI2, FLNA, GLI3, NOTCH2, PTPN11, and RET); the other genes (CDH23 and MYO7A) exhibited either AD or autosomal recessive (AR) inheritance. FLNA undergoes X-linked recessive inheritance, while CYP1A1, FRAS1, FREM2, and EVC are known to undergo AR inheritance.
MYO7A was the most commonly identified gene. Seven MYO7A variants were identified in five patients (patients 1, 3, 6, 8, and 10); five CDH23 variants were identified in five patients (patient 1, 8, 9, and 10); five FRAS1 variants were identified in four patients (patient 6, 8, 9, and 10); four GLI3 variants were identified in three patients (patient 2, 4, and 6); NOTCH2 variants were identified in three patients (patient 2, 5, and 10). Variants of the following genes were detected in any two patients: CYP1A1 variants in patients 6 and 7, PIK3R3 variants in patients 7 and 10, RET variants in patients 1 and 5, MAML3 variants in patients 4 and 9, WNT9B variants in patients 2 and 9, and GLI2 variants in patients 2 and 5.
Patient 1 carried four variants among four genes: RET, CDH23, MYO7A, and MAML1. Patient 2 carried four variants among four genes: WNT9B, NOTCH2, GLI2, and GLI3. Patient 3 carried two variants of MYO7A. Patient 4 carried three variants in MAML3 and GLI3. Patient 5 carried three variants among three genes: RET, NOTCH2, and GLI2. Patient 6 carried five variants among five genes: MYO7A, CYP1A1, WNT9A, FRAS1, and GLI3. Patient 7 carried two variants among two genes: CYP1A1 and PIK3R3. Patient 8 carried six variants among four genes: CDH23, MYO7A, FREM2, and FRAS1. Patient 9 carried four variants among four genes: CDH23, WNT9B, MAML3, and FRAS1. Patient 10 carried 11 variants among nine genes: CDH23, MYO7A, MAML2, PTPN11, NOTCH2, PIK3R3, EVC, FRAS1, and FLNA. The details of these variants are summarised in Table 1 .
In addition, patients 1, 8, and 10 presented combination variants of two genes: MYO7A-CDH23. Patients 2 and 5 both presented combination variants of two genes: NOTCH2-GLI2; patients 6 and 8 both presented combination variants of two genes: MYO7A-FRAS1.
Interactome Analysis of the Identified DSD- and MAMLD1-Related Genes
Interactome analysis was performed for the identification of DSD-related genes using bioinformatics software to assess possible gene–protein interactions. The network including all genes identified is presented in Figure 4A . The core network constructed using the Cytoscape Molecular COmplex DEtection (MCODE) software is shown in Figure 4B . Overall, a connection was detected among the 18 genes and we report that MAMLD1 is directly connected to MAML1/2/3 and NOTCH1/2. Through NOTCH, eight genes (WNT9A/9B, GLI2/3, RET, FLNA, PTPN11, and EYA1) were associated with MAMLD1. Some of these genes also acted as central nodes for further connections; for example, GLI3 for EVC, FGF10, GLI2, NOTCH1/2, and EYA1; RET for ZBTB16, FGF10, PIK3R3, PTPN11, and NOTCH1; EYA1 for FRAS1, MYO7A, FGF10, NOTCH1, WNT9B, and GLI3; and FGF10 for EYA1, GLI2/3, NOTCH1, PTPN11, and RET. In addition, the isolated gene couple that was revealed in our analysis was CYP1A1-HSD3B2.
Figure 4.
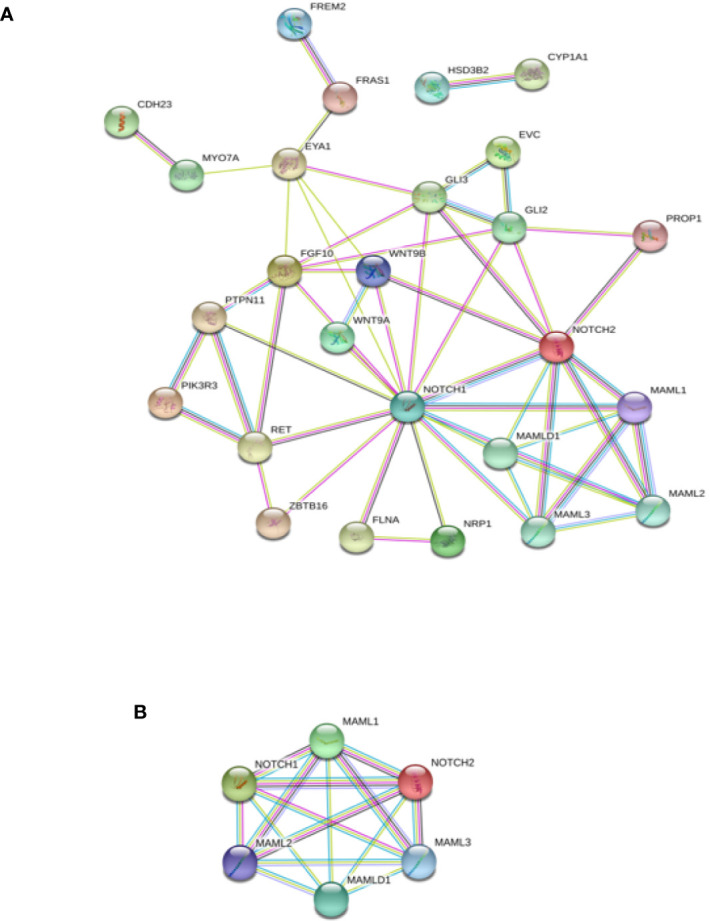
Interaction network of DSD- and MAMLD1-related genes identified in DSD individuals harbouring genetic variants in MAMLD1. (A) The scheme depicts an overview of detected genes and their interrelationship. (B) Core network of the detected genes and their interrelationship. Filled nodes show proteins with known or predicted 3D structure. Empty nodes depict proteins with unknown 3D structure. Candidate genes are underlined. Known interactions correspond to curated databases (turquoise lines) and experimentally determined interactions (pink lines). Predicted interactions correspond to gene neighbourhood (green lines), gene fusions (red lines) and gene co-occurrence (blue lines). Other interactions correspond to text mining (yellow lines), co-expression (black lines) and protein homology (violet lines).
The specific interactome of the genes identified in the patients is shown in Figure 5 . In patients 2, 5, and 10, MAMLD1 and MAMLD1-related genes (MAML1, MAML2, and MAML3) were directly related to NOTCH2 ( Figures 5B, E, J ). There were two networks each for patients 1 and 8: CHD23-MYO7A and MAML1-MAMLD1 for patient 1 ( Figure 5A ); CHD23-MYO7A and FREM2-FRAS1 for patient 8. In patient 2, NOTCH2 was shown to play a central role in its associated with the WNT9B, MAMLD1, and GLI2/3 network ( Figure 5B ). In patient 10, PTPN11, PIK3R3, CDH23, and MYO7A were directly related ( Figure 5J ). The CHD23-MYO7A network was identified in three patients (patients 1, 8, and 10).
Figure 5.

Interaction networks of DSD- and MAMLD1-related genes identified per MAMLD1 individual. (A) to (J) correspond to the interaction networks per patient. Filled nodes show proteins with known or predicted 3D structure. Empty nodes depict proteins with unknown 3D structure. Candidate genes are underlined. Known interactions correspond to curated databases (turquoise lines) and experimentally determined interactions (pink lines). Predicted interactions correspond to gene neighborhood (green lines), gene fusions (red lines) and gene co-occurrence (blue lines). Other interactions correspond to text mining (yellow lines), co-expression (black lines) and protein homology (violet lines).
Discussion
The genetic architecture of human inheritance has traditionally been divided into two major types. Typically, complex traits exhibit polygenic architectures resulting from the presence of several common variants with low effect, whereas rare traits are usually associated with monogenic determinants with high effect (24). There is growing evidence that suggests that these two classes of phenotypes might not be as biologically distinct as previously considered, and genetic structure of a lineage is present, rather than a dichotomy (25). Mendelian disorders have also been found to be influenced by multiple or common genetic variants (26–28).
Sex development is a highly complex biological event that requires the expression and regulation of a large number of genes with spatial and temporal precision. Although there is considerable information regarding sex development in individuals with monogenic DSDs, the broad spectrum of phenotypes in numerous DSD cases remains less understood. In this study, we investigated the hypothesis that MAMLD1-related DSD may follow an oligogenic mode of inheritance.
All enrolled patients harboured MAMLD1 variants, and all of them shared a broad spectrum of phenotypes. The most prevalent phenotype was hypospadias and others included cryptorchidism, bifid scrotum, and/or micropenis. The T levels in patients were within the normal range, and were indicative of the mini-puberty stage. An appropriate T response after hCG stimulation was observed in all patients for whom data were available. Additionally, we detected 43 potential disease-causing variants of 18 genes with reported MAMLD1 interaction. Interactome analysis of identified DSD-related genes was performed to evaluate the possible gene–protein interactions. Using the obtained information, we constructed a genetic map of potential oligogenic hits identified in the patients with 46,XY DSD harbouring heterozygous MAMLD1, keeping in mind the existing view of genetic interactions in male sex determination and development. Our findings provide further evidence that individuals with MAMLD1-related 46,XY DSD could harbour two or more variants of known DSD-related genes. The phenotypic outcome might be determined by multiple genes.
A series of studies have been conducted to elucidate the role of oligogenic inheritance in DSDs. A recent study suggested that the expanded DSD phenotypes associated with NR5A1 mutations resulted from the oligogenic inheritance of other genes related to testicular development, such as MAP3K1, POR, CHD7, and AKR1C3 (22, 29). Similarly, Eggers et al. observed a storage effect in a cohort of patients with severe hypospadias. In three patients, they observed the oligogenic inheritance of variants of testis development-related genes (MAP3K1 and ZFPM2) in combination with VUS. Another patient with severe hypospadias was observed to carry two disease-causing variants of HSD3B2 and GNRHR (30, 31). In addition, among patients with 46,XY DSD of unknown aetiology, five patients were observed to carry a mutation in AR, besides carrying other variants in genes encoding proteins participating in androgen action or gonadal development (31). Recently, Flück et al. investigated additional genetic hits in patients with MAMLD1-related DSDs. Using HTS and a custom-tailored algorithm, they identified 55 potentially deleterious genetic variants of 41 additional genes (23). The above information indicates that oligogenic inheritance may contribute to a broader DSD phenotype than previously reported.
In the present study, seven MYO7A variants were identified in five patients (patients 1, 3, 6, 8, and 10), five CDH23 variants were identified in five patients (patients 1, 8, 9, and 10), and five FRAS1 variants were identified in four patients (patients 6, 8, 9, and 10). Several gene combinations were identified: MYO7A-CDH23 in three patients, NOTCH2-GLI2 in two patients, and MYO7A-FRAS1 in two patients. MYO7A, CDH23, and FRAS1 were the most frequently identified genes, which suggested a shared genetic basis.
Overall, some of the genes identified in the ten patients harbouring MAMLD1 variants were previously reported to be associated with specific syndromes in patients with genitourinary anomalies: RET was shown to be associated with congenital anomalies of the kidney and the urinary tract (CAKUT) syndrome, EVC with Ellis-van Creveld syndrome, FRAS1 and FREM2 with Fraser syndrome, PTPN11 with Noonan syndrome, and WNT9B with Mayer-Rokitansky-Küster-Hauser syndrome. However, none of the patients enrolled in this study presented with the complete set of phenotypic features typical to these syndromes, possibly because none of these variants induced the complete impairment of gene expression and protein function. On the contrary, advances in sequencing technology have greatly expanded and challenged the validity of the established phenotypes of known syndromes. In this respect, extensive studies on a greater number of cases should be conducted to obtain better evidence.
Interactome analysis was performed to identify DSD-related genes using bioinformatics software to assess possible gene–protein interactions ( Figures 4 and 5 ). Overall, a connection was observed among all 18 genes. MAMLD1 directly connected to MAML1/2/3 and NOTCH1/2. Through NOTCH1/2, eight genes (WNT9A/9B, GLI2/3, RET, FLNA, PTPN11, and EYA1) were associated with MAMLD1. Flück et al. have reported the interaction network of genes identified in DSD patients with MAMLD1 variants (23). However, there is the discrepancy between the present and the previous studies in the scheme of the interaction network. The presence of interactions between GLI2 and NOTCH2, MYO7A, and EYA1 have been displayed in the present study, and a known interaction between WNT9A and NOTCH2 disappeared in the present study. This phenomenon may caused by the version difference of STRING software, and the medium confidence, in the present study, the medium confidence was set to 0.4, and when it was set to be lower, the interaction between WNT9A and NOTCH2 were displayed, which means that the screening criteria in our study were higher than the reported article.
The specific interactome of the identified genes in all the patients studied is shown in Figure 5 . In patients 2, 5, and 10, MAMLD1 and MAMLD1-related genes (MAML1, MAML2, and MAML3) were directly related to NOTCH2 ( Figures 5B, E, J ). The core network is illustrated in Figure 4B , and shows that NOTCH 1/2 and MAMLD1-related genes constitute the core genes, which was in line with the reported literature (23). NOTCH signalling is a highly conserved signalling pathway and involves the participation of four transmembrane receptors (32). There is growing evidence that mis-regulation of NOTCH signalling may lead to common disorders, ranging from neuropsychiatric to metabolic disorders (33, 34). Furthermore, somatic mutations in genes encoding specific components of the pathway and/or mis-regulation of NOTCH signalling activity have also been linked to oncogenesis and tumour progression in different cancer types (35, 36). The efficient regulation of this pathway was also shown to be essential for the regulation of embryonic development in multiple organ systems, including the gonadal system (37). NOTCH signalling is implicated in Leydig cell differentiation in an inhibitory regulatory manner (37). Further investigations that focus on the functional impacts of each pathogenic mutation will likely provide a better mechanistic understanding of how specific phenotypes may be linked to defects in the NOTCH signalling pathway.
This study has several limitations. The sample size was considerably small to establish a relationship between the observed genotypes and phenotypes, and due to the small sample size, the impact of mutation or variant types had to be excluded from a genotype-phenotype correlation analysis, which reduced the significance of the observations. In addition, functional studies are necessary to further clarify the exact disease-causing effect in patients with 46,XY DSD harbouring MAMLD1 variations; however, when multiple variants are being searched for, which may contribute only partially, this testing method cannot be considered feasible. Therefore, in future studies, we intend to increase the sample size and also extend the follow-up period. Additionally, we intend to employ next-generation statistical analyses of genetic data to identify associations between a group of variants and complex traits in sex development. Moreover, recent improvements in gene editing enabled by advancements in the CRISPR-Cas 9 technology may provide an opportunity for testing the hypotheses on the potential of oligogenic inheritance in DSDs in the near future (38).
In conclusion, we believe our findings provide evidence that individuals with MAMLD1-related 46,XY DSDs could harbour two or more variants of known DSD-related genes. The phenotypic outcomes might be determined by multiple genes. A more extensive study involving other DSD cohorts is necessary to assess whether the genetic variants identified in this study are truly related to DSDs, and that the inclusion of these variants might help establish a better genotype–phenotype correlation.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to lilele2006@163.com.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Medical Ethics Review Board of Beijing Children’s Hospital (ID: 2019-k-156). Written informed consent was obtained from all patients or their legal guardians. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
CG examined and recruited the patients, conceived and designed the study, provided critical comments, and edited the manuscript. LL and FG collected and analysed the data and drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Special Fund of the Pediatric Medical Coordinated Development Center of Beijing Hospitals Authority (No.XTYB201808) and the Beijing Natural Science Foundation (No.7204260).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
We would like to thank the Chigene (Beijing) Translational Medical Research Center Co., Ltd. for the bioinformatics analysis. We would like to thank the family members of the participants and the medical staff involved in this study. Last, we are thankful to Editage (www.editage.cn) for English language editing.
References
- 1. Witchel SF. Disorders of Sex Development. Best Pract Res Clin Obstet Gynaecol (2018) 48:90–102. 10.1016/j.bpobgyn.2017.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cools M, Nordenstrom A, Robeva R, Hall J, Westerveld P, Fluck C, et al. COST Action BM1303 working group 1. Caring for individuals with a difference of sex development (DSD) : a consensus statement. Nat Rev Endocrinol (2018) 14:415–29. 10.1038/s41574-018-0010-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eggers S, Ohnesorg T, Sinclair A. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol (2014) 10:1673–83. 10.1038/nrendo.2014.163 [DOI] [PubMed] [Google Scholar]
- 4. Bashamboo A, McElreavey K. Gene mutations associated with anomalies of human gonad formation. Sex Dev (2013) 7:126–46. 10.1159/000342188 [DOI] [PubMed] [Google Scholar]
- 5. Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol (2013) 9:79–91. 10.1038/nrendo.2012.235 [DOI] [PubMed] [Google Scholar]
- 6. Hu LJ, Laporte J, Kress W, Kioschis P, Siebenhaar R, Poustka A, et al. Deletions in Xq28 in two boys with myotubular myopathy and abnormal genital development define a new contiguous gene syndrome in a 430 kb region. Hum Mol Genet (1996) 5:139–43. 10.1093/hmg/5.1.139 [DOI] [PubMed] [Google Scholar]
- 7. Laporte J, Guiraud-Chaumeil C, Vincent MC, Mandel JL, Tanner SM, Liechti-Gallati S, et al. Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC International Consortium on Myotubular Myopathy. European Neuro-Muscular Center. Hum Mol Genet (1997) 6:1505–11. 10.1093/hmg/6.9.1505 [DOI] [PubMed] [Google Scholar]
- 8. Ogata T, Laporte J, Fukami M. MAMLD1 (CXorf6): A New Gene Involved in Hypospadias. Horm Res (2009) 71:245–52. 10.1159/000208797 [DOI] [PubMed] [Google Scholar]
- 9. Fukami M, Wada Y, Miyabayashi K, Nishino I, Hasegawa T, Nordenskjold A, et al. CXorf6 is a causative gene for hypospadias. Nat Genet (2006) 38:1369–71. 10.1038/ng1900 [DOI] [PubMed] [Google Scholar]
- 10. Nakamura M, Fukami M, Sugawa F, Miyado M, Nonomura K, Ogate T. Mamld1 knockdown reduces testosterone production and Cyp17a1 expression in mouse Leydig tumor cells. PloS One (2011) 6:e19123. 10.1371/journal.pone.0019123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab (2015) 29(4):607–19. 10.1016/j.beem.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fakami M, Wada Y, Okada M, Kato E, Katsumata N, Baba T, et al. Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem (2008) 283:5525–32. 10.1074/jbc.M703289200 [DOI] [PubMed] [Google Scholar]
- 13. Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development (2007) 134:1243–51. 10.1242/dev.000786 [DOI] [PubMed] [Google Scholar]
- 14. Li L, Su C, Fan L, Gao F, Liang X, Gong C, et al. Clinical and molecular spectrum of 46,XY disorders of sex development that harbour MAMLD1 variations: case series and review of literature. Orphanet J Rare Dis (2020) 15(1):188. 10.1186/s13023-020-01459-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalfa N, Cassorla F, Audran F, Oulad AI, Philibert P, Beroud C, et al. Polymorphisms of MAMLD1 gene in hypospadias. J Pediatr Urol (2011) 7:585–91. 10.1016/j.jpurol.2011.09.005 [DOI] [PubMed] [Google Scholar]
- 16. Chen Y, Thai HT, Lundin J, Lagerstedt-Robinson K, Zhao S, Markljung E, et al. Mutational study of the MAMLD1 gene in hypospadias. Eur J Med Genet (2010) 53:122–6. 10.1016/j.ejmg.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 17. Ruiz-Arana IL, Hubner A, Cetingdag C, Krude H, Gruter A, Fukami M, et al. A novel hemizygous mutation of MAMLD1 in a patient with 46, XY complete gonadal dysgenesis. Sex Dev (2015) 9:80–5. 10.1159/000371603 [DOI] [PubMed] [Google Scholar]
- 18. Camats N, Fernanez-Cancio M, Audi L, Mullis PE, Moreno F, Gonzalez CI, et al. Human MAMLD1 Gene variations seems not sufficient to explain a 46, XY DSD phenotype. PloS One (2015) 10:e0142831. 10.1371/journal.pone.0142831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyado M, Nakamura M, Miyado K, Morohashi K, Sano S, Nagata E, et al. Mamld1 deficiency significantly reduces mRNA expression levels of multiple genes expressed in mouse fetal Leydig cells but permits normal genital and reproductive development. Endocrinology (2012) 153:6033–40. 10.1210/en.2012-1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cerrone M, Remme AC, Tadros R, Bezzina C, Delmar M. Beyond the one gene-one disease paradigm complex genetics and pleiotropy in inheritable cardiac disorders. Circulation (2019) 140:595–610. 10.1161/CIRCULATIONAHA.118.035954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert Consensus Document: European Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. Nat Rev Endocrinol (2015) 11:547–64. 10.1038/nrendo.2015.112 [DOI] [PubMed] [Google Scholar]
- 22. Camats N, Fernandez-Cancio M, Audi L, Schaller A, Fluck CE. Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: an oligogenic origin? Eur J Hum Genet (2018) 26:1329–38. 10.1038/s41431-018-0202-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fluck CE, Audi L, Fernandez-Cancio M, Sauter KS, Martinez de LI, Castano L, et al. Broad phenotypes of disorders/differences of sex development in MAMLD1 patients through oligogenic disease. Front Genet (2019) 10:746. 10.3389/fgene.2019.00746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs rare allele hypotheses for complex diseases. Curr Opin Genet Dev (2009) 19:212–9. 10.1016/j.gde.2009.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katsanis N. The continuum of causality in human genetic disorders. Genome Bio (2016) 17:233. 10.1186/s13059-016-1107-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med (2017) 376:21–31. 10.1056/NEJMoa1516767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR, et al. Genome-wide association metaanalysis identififies fifive modififier loci of lung disease severity in cystic fifibrosis. Nat Commun (2015) 6:8382. 10.1038/ncomms9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Freund MK, Burch KS, Shi H, Mancuso N, Kichaev G, Garske KM, et al. Phenotype-Specifific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits. Am J Hum Genet (2018) 103:535–52. 10.1016/j.ajhg.2018.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mazen I, Abdel-Hamid M, Mekkawy M, Bignon-Topalovic J, Boudjenah R, El Gammal M, et al. Identification of NR5A1 mutations and possible digenic inheritance in 46,XY gonadal dysgenesis. Sex Dev (2016) 10:147–51. 10.1159/000445983 [DOI] [PubMed] [Google Scholar]
- 30. Kon M, Suzuki E, Dung VC, Hasegawa Y, Mitsui T, Muroya K, et al. Molecular basis of non-syndromic hypospadias: systematic mutation screening and genomewide copy-number analysis of 62 patients. Hum Reprod (2015) 30:499–506. 10.1093/humrep/deu364 [DOI] [PubMed] [Google Scholar]
- 31. Eggers S, Sadedin S, van den Bergen JA, Robevska G, Ohnesorg T, Hewitt J, et al. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol (2016) 17(1):243. 10.1186/s13059-016-1105-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol (2012) 23:450–7. 10.1016/j.semcdb.2012.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab (2015) 26:248–55. 10.1016/j.tem.2015.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology (2015) 61:382–92. 10.1002/hep.27268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koch U, Radtke F. Notch signaling in solid tumors. Curr Top Dev Biol (2010) 92:411–55. 10.1016/S0070-2153(10)92013-9 [DOI] [PubMed] [Google Scholar]
- 36. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all time . Nat Rev Cancer (2011) 11:338–51. 10.1038/nrc3035 [DOI] [PubMed] [Google Scholar]
- 37. Windley SP, Wilhelm D. Signaling pathways involved in mammalian sex determination and gonad development. Sex Dev (2016) 9:297–317. 10.1159/000444065 [DOI] [PubMed] [Google Scholar]
- 38. Gifford CA, Ranade SS, Samarakoon R, Salunga HT, de Soysa TY, Huang Y, et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science (2019) 364:865–70. 10.1126/science.aat5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to lilele2006@163.com.