

Abstract
Gulf War Illness (GWI) is thought to be a chronic neuroimmune disorder caused by in-theater exposure during the 1990–1991 Gulf War. There is a consensus that the illness is caused by exposure to insecticides and nerve agent toxicants. However, the heterogeneity in both development of disease and clinical outcomes strongly suggests a genetic contribution. Here, we modeled GWI in 30 BXD recombinant inbred mouse strains with a combined treatment of corticosterone (CORT) and diisopropyl fluorophosphate (DFP). We quantified transcriptomes from 409 prefrontal cortex samples. Compared to the untreated and DFP treated controls, the combined treatment significantly activated pathways such as cytokine-cytokine receptor interaction and TNF signaling pathway. Protein-protein interaction analysis defined 6 subnetworks for CORT + DFP, with the key regulators being Cxcl1, Il6, Ccnb1, Tnf, Agt, and Itgam. We also identified 21 differentially expressed genes having significant QTLs related to CORT + DFP, but without evidence for untreated and DFP treated controls, suggesting regions of the genome specifically involved in the response to CORT + DFP. We identified Adamts9 as a potential contributor to response to CORT + DFP and found links to symptoms of GWI. Furthermore, we observed a significant effect of CORT + DFP treatment on the relative proportion of myelinating oligodendrocytes, with a QTL on Chromosome 5. We highlight three candidates, Magi2, Sema3c, and Gnai1, based on their high expression in the brain and oligodendrocyte. In summary, our results show significant genetic effects of the CORT + DFP treatment, which mirrors gene and protein expression changes seen in GWI sufferers, providing insight into the disease and a testbed for future interventions.
Keywords: BXD RI mice, Gulf War Illness, Neuroinflammation, Transcriptome, Genetics
1. Introduction
Gulf War Illness (GWI) is a medically unexplained chronic condition suffered by an estimated 25–32% of the 956,600 troops deployed during the 1990–1991 Gulf War, the majority of whom came from the USA (Kerr, 2015; National Academies of Sciences and Medicine, 2016). The disorder is characterized by fatigue, musculoskeletal pain, cognitive dysfunction, chemical sensitivities, loss of memory and sleep disruption, symptoms thought to result from neuroimmune activation and that resembles most features of ‘sickness behavior’ (Dantzer et al., 2008). Although these symptoms were first reported within 6 months of troops returning home (Gavaghan, 1994; Robinson, 1995), they have persisted for almost 30 years (Blanchard et al., 2019; Tillman et al., 2019).
There are three main questions that remain to be answered in respect to GWI. First, what caused the disorder? Second, why did only a subset those deployed develop the illness, and why is there such heterogeneity in symptoms? Third, why have the symptoms persisted so long?
A consensus has been reached on the first of these questions: that exposure to toxicants in-theater, combined with high circulating levels of stress hormone (cortisol) as might be expected in the physical environment of the Gulf War (White et al., 2016). The most likely causative combination of exposures is the high physical stress of the war theater environment (leading to high levels of the stress hormone, cortisol) and exposure to irreversible acetylcholinesterase (AChE) inhibitors (Abdullah et al., 2012; Golomb, 2008; Research Advisory Committee (RAC) on Gulf War Veterans’ Illnesses, 2014; Kerr, 2015; Koo et al., 2018; Locker et al., 2017; O’Callaghan et al., 2015a; Pierce et al., 2016; Steele et al., 2011; Sullivan et al., 2018; Winkenwerder, 2003; Zakirova et al., 2016). The troops deployed to the Gulf War were exposed to a wide range of AChE inhibitors (Binns et al., 2008; Kerr, 2015), including pyridostigmine bromide, sarin, organophosphate pesticides, as well as the insect repellant, DEET. Approximately 250,000 troops may have been exposed to low levels of nerve agents via destruction of storage depots (Tuite and Haley, 2013). In fact, there is a correlation between the number of nerve agent alarms heard and occurrence of GWI (Haley and Tuite, 2013).
Although some of the heterogeneity in symptoms of GWI may be explained by different exposures, much of it cannot be, as not all individuals within a given unit developed GWI, and those who did show markedly different symptoms. It is likely that genetics is a significant contributor to susceptibility and response to the exposures (Jones et al., 2020). The rodent model developed by O’Callaghan and colleagues (O’Callaghan et al., 2015a) and employed by Jones et al., (Jones et al., 2020), and here, combines corticosterone (CORT, the rodent analogue of cortisol) with diisopropyl fluorophosphate (DFP), an irreversible AChE inhibitor and sarin surrogate. This model has been repeatedly demonstrated to alter neuroimmune and oligodendrocyte function in the central nervous system (Ashbrook et al., 2018; Belgrad et al., 2019; Koo et al., 2018; O’Callaghan et al., 2015a; Zakirova et al., 2016).
We have previously shown significant variation in three important neuroinflammatory markers in response to CORT plus DFP across the BXD recombinant inbred (RI) mouse family and mapped a causative locus (Jones et al., 2020). The BXD family are a set of 152 RI strains, all of which constitute a unique mosaic of alleles derived from the parental C57BL/6J (B6) and DBA/2J (D2) strains (Ashbrook et al., 2019). These strains segregate for ~6 million variants, similar to most human populations, and therefore can be used to model the genetic variability among individuals. Furthermore, because all the strains are inbred, the same genome can be studied repeatedly under a variety of environmental conditions, allowing the collection of a large phenome of data which can be jointly studied (Roy et al., 2020; Wang et al., 2016).
In the current study, we examined whole-genome RNA-seq in the prefrontal cortex (PFC) of 30 strains of the BXD family, plus the parental strains, to identify common responses across strains, identify variants which medicate neuroinflammatory response to the treatment, and determine the extent to which genetic differences alter neuro-morphological responses.
2. Materials and methods
2.1. Mice
The subjects for this study were male and female mice from 30 BXD RI strains, and the parental B6 and D2 strains. The animals were between 2 and 4 months of age at testing and ~2 animals per strain, sex and treatment group were used. All animals were obtained from the breeding colony at the UTHSC vivarium. All procedures were approved by the UTHSC animal care and use committee.
2.2. Experimental treatment
All animals had access to water and food ad libitum and controlled climate at 20 ± 2 °C and 35% relative humidity. The animals were divided into 3 treatment groups as follows:
Control (CTL) group. These animals received plain tap water for fluid, saline injection, and were euthanized by cervical dislocation six hours after injection.
DFP group. These animals received plain tap water for fluid and were injected with DFP, 4 mg/kg, i.p. Six hours after injection the animals were euthanized by cervical dislocation followed by decapitation. The dose of DFP used was which based on its ability to produce the symptoms of cholinergic crisis [e.g., SLUD (salivation, lacrimation, urination, and defecation) and seizures] while displaying mortality below LD25 (Locker et al., 2017).
CORT + DFP group. These animals received tap water containing 20 mg% CORT dissolved in 0.6% (v/v) EtOH vehicle for 8 days. On the 8th day, the animals were injected with DFP, 4 mg/kg, i.p. Six hours after injection the animals were euthanized by cervical dislocation followed by decapitation.
We already tested these animals for CORT consumption and proinflammatory cytokine gene expression in a previous study. Those data are reported in Jones et al. (2020), and will not be included in this study.
2.3. Tissue harvest
After being freed from the skull, the whole brain was put immediately into the mouse brain matrices with the ventral side up at 4 °C. A 90° cut was made 1 mm from the posterior edge of olfactory bulb, and another 90° cut 2 mm from the first cut, with the brain tissue between the 1st and 2nd cut defining the PFC. The PFC was weighed, frozen in a dry ice bath with isopentane, and stored at −80 °C. The choice of the PFC is based on the work by O’Callaghan and colleagues (O’Callaghan et al., 2015a) in which they showed that CORT + DFP produced similar effects of proinflammatory cytokine gene expression. Also, the PFC is the only cortical area to receive direct projections from the spinal cord (Maule et al., 2018). The cortical regions involved in fatigue, pain, and hyperalgesia have been reported to be associated with diminished white matter integrity in GW veterans (Binns et al., 2008).
2.4. RNA extraction
Total RNA was extracted using Trizol reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions. Approximately 30 mg of PFC tissue was added into a 2 ml tube containing 700 μl QIAzol Lysis Reagent and one 5 mm stainless steel bead (Qiagen, Hilden, Germany). The tissue was homogenized for 2 min in a Tissue Lyser II (Qiagen, Hilden, Germany) with a speed frequency of 30 r followed by incubating for 5 min. 140 μl chloroform was added into the homogenate, shaken vigorously for 15 s, and centrifuged for 15 min at 12,000×g at 4 °C. 280 μl upper aqueous was then transferred into a new collection tube containing 500 μl 100% ethanol. The mixture was loaded into a RNeasy mini‐spin column (Qiagen, Valencia, CA, USA), once with Buffer RWT and twice with Buffer RPE purification. All RNA had been treated with DNase to avoid DNA contamination, and verified by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA with OD260/280 > 1.8 and RIN > 8.0 were used for library preparation.
2.5. Library preparation and sequencing
One microgram of RNA was used for cDNA library construction at Novogene using an NEBNextUltra RNA Library Prep Kit for Illumina (cat# E7420S, New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s protocol. Briefly, mRNA was enriched using oligo (dT) beads followed by two rounds of purification and fragmented randomly by adding fragmentation buffer. The first strand cDNA was synthesized using random hexamers primer, after which a custom second-strand synthesis buffer (Illumina, San Diego, CA, USA), dNTPs, RNase H and DNA polymerase I were added to generate the second strand (ds cDNA). After a series of terminal repair, poly-adenylation, and sequencing adaptor ligation, the double-stranded cDNA library was completed following size selection and PCR enrichment. The resulting 250–350 bp insert libraries were quantified using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and quantitative PCR. Size distribution was analyzed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Qualified libraries were sequenced on an Illumina Novaseq Platform (Illumina, San Diego, CA, USA) using a paired-end 150 run (2 × 150 bases). An average of 40 million raw reads were generated from each library.
2.6. Data quality control and filtering
The raw reads underwent the following filter to produce clean data: 1) Remove reads containing adaptors; 2) Remove reads containing N > 10%; and 3) Remove reads in which 50% bases have a Qscore (Quality value) <= 5.
2.7. Alignment to the reference genome
Mus musculus (mouse) reference genome (GRCm38) and gene model annotation files were downloaded from the Ensembl genome browser (https://useast.ensembl.org/). Indices of the reference genome were built using STAR v2.5.0a (Dobin et al., 2013) and paired-end clean reads were aligned to the reference genome. STAR used the method of Maximal Mappable Prefix which can generate a precise mapping result for junction reads.
2.8. Quantification of gene expression level
In RNA-seq experiments, gene expression is estimated by the abundance of transcripts (count of sequencing) that map to a gene or exon. Read counts are proportional to gene expression level, gene length and sequencing depth. FeatureCount v0.6.1 (Liao et al., 2013) was used to count the number of read mapped to each gene. Transcripts Per Million (TPM) was calculated for each gene based on the length of the gene and reads mapped to that gene. In this normalization, the sum of all TPMs (genes-level) are equal to 1,000,000. Since the sequencing data were generated with two batches, the batch effects were further corrected with ComBat (Johnson et al., 2007) based on the log2 (TPM + 1) transformed values.
2.9. Differential expression analysis
Differential expression analyses between two conditions (DFP vs. CTL or CORT + DFP vs. CTL) were performed using the R package DESeq2 v1.22.2 (Love et al., 2014). DESeq2 provide statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using the Benjamini and Hochberg’s approach (Benjamini and Hochberg, 1995) for controlling the False Discovery Rate (FDR). Genes with an FDR < 0.01 found by DESeq2 were defined as differentially expressed genes (DEGs).
2.10. Gene set enrichment analysis
Gene set enrichment analysis for Gene Ontology (GO, biological process), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, and Mammalian Phenotype Ontology (MPO) were analyzed with WebGestalt (http://www.webgestalt.org/) (Wang et al., 2017). All protein coding genes in the mouse genome were used as the reference gene set. The statistical significance of enrichment between DEGs and the members of known GO terms, KEGG pathways, and MPO categories was calculated based on the hypergeometric test. The Benjamini and Hochberg correction (Benjamini and Hochberg, 1995) was used for multiple test correcting. A minimum overlap of 5 genes and an FDR cutoffs of 0.05 was required to determine the significance.
2.11. Protein-protein interaction (PPI) analysis of DEGs
PPI analysis of DEGs (FDR < 0.01 & Fold Change (FC) > 1.3) was based on the STRING database (https://string-db.org/) (Szklarczyk et al., 2018), which contained known and predicted PPIs. In this study, we first constructed the PPI networks by extracting the target gene lists from the database with minimum required interaction score of 0.7. Then Markov Cluster Algorithm (MCL) clustering was used for subnetwork construction, in which inflation parameter was set to 3.
2.12. Drug repositioning analysis
Drug repositioning analysis was conducted with GREP (v1.1.0) (Sakaue and Okada, 2019), a package used for quantifying a set of genes in clinically defined categories and capturing potential drugs that targeting the gene set. The input of the gene set is the DEGs (FDR < 0.01 & FC > 1.3) induced by DFP or CORT + DFP treatment, respectively. The International Statistical Classification of Diseases and Related Health Problems (ICD, v10) was used for the drug indication categorization. Drug-indicated disease category information was retrieved from Therapeutic Target Database (http://db.idrblab.net/ttd/).
2.13. Estimates of cell type composition changes
To estimate the proportions of different cell types within our samples, we used the R Bioconductor package DeconRNASeq (Du et al., 2019) with our RNA-seq data. To identify genes whose expression are characteristic of different frontal cortex cell types, we downloaded RNA-seq data for populations enriched for specific central nervous system cell types from the Gene Expression Omnibus (GEO) (Ashbrook et al., 2018; Edgar et al., 2002), Series GSE52564. This series contains data from the Mus musculus cerebral cortex (Zhang et al., 2014), which we used as a reference of cell-type-specific gene expression. These RNA-seq data were trimmed and aligned and gene expression was quantified identically to our own RNA-seq data, as described above. For each of the six cell types, (astrocytes, neurons, oligodendrocyte precursor cells (OPC), myelinating oligodendrocytes (MO), microglia and endothelial cells) we produced a gene expression signature by finding those genes with a five-fold difference in expression in one cell type, compared to each of the others. These gene expression signatures were then used to estimate the proportion of each cell type within each of our samples.
We then compared the effect of treatment on each of the cell types using ANOVA, with Tukey Honestly Significant Differences post-hoc. Analysis was carried out using the stats R package (Team, 2013), and figures were drawn using ggplot2 (Wickham, 2016). The mean estimate for the proportion of each cell type per strain was then calculated, with CTL or CORT + DFP treatment, and these two differences between the estimates. These estimates were uploaded to GeneNetwork (http://www.genenetwork.org/; GN IDs 21497–21517) (Mulligan et al., 2017; Parker et al., 2017; Sloan et al., 2016), and quantitative trait loci (QTL) were mapped.
2.14. QTL mapping
QTL mapping allows the identification of linkage between any region of the genome, and a phenotype of interest. The fast linear regression equations of Haley and Knott (Haley and Knott, 1992) were used for initial QTL mapping. Using 5000 permutations of the phenotypes, genome-wide significant (p < 0.05), and suggestive (p < 0.63) thresholds were calculated within GeneNetwork. The suggestive threshold corresponds to approximately one false QTL per genomescan. Likelihood ratio statistic (LRS) scores were converted to log of the odds (LOD) scores by dividing by 4.61, and confidence intervals were defined by a LOD drop of 1.5 on either side of the peak LOD value (Manichaikul et al., 2006). We considered QTL intervals that achieved genome-wide significance for one phenotype, and genome-wide suggestive for others, as highest priority for candidate gene analysis. We used the January 2017 BXD genotype file (www.genenetwork.org/webqtl/main.py?FormID=sharinginfo&GN_AccessionId=600).
There are now updated linear mixed model mapping algorithms, such as GEMMA (Zhou and Stephens, 2012), available on GeneNetwork (Sloan et al., 2016), and these account for kinship among strains. Traits with significant and suggestive QTLs detected using Haley-Knott mapping were remapped using GEMMA to confirm LRS scores and locations of QTLs even in the presence of kinship structure.
2.15. Candidate gene identification
We examined genes within the QTL 1.5 LOD confidence intervals focusing on genes with non-synonymous single nucleotide polymorphism SNPs (nsSNPs) or insertions/deletions (InDels). These genes are high priority candidates, although we acknowledge that causal variants may lie in non-coding regions. For each of these high priority candidates we then examined which GO:biological processes (Consortium, 2015) and KEGG pathways (Kanehisa et al., 2012) the gene was annotated as being part of, and highlighted those which may relate to our phenotypes. We also reviewed known effects of mutations using the Mouse Genome Informatics (MGI) Phenotypes, Alleles and Disease Models Search (www.informatics.jax.org/allele) (Bello et al., 2015). To cover more recent findings we also checked the NCBI Entrez information (www.ncbi.nlm.nih.gov/gene) about the gene and its human homolog, particularly related articles in PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) and GeneRIFs (https://www.ncbi.nlm.nih.gov/gene/about-generif).
To get data specific to the BXD family, transcriptome data for many different organs and tissues are available in GeneNetwork, including some tissues with data at different ages. We used them to find which of these transcripts have cis-eQTLs, and the expression of which correlate with proportion of cell types.
3. Results
3.1. Summary statistics of RNA-seq
RNA sequencing of the 409 samples yielded a total of 19.5 billion reads (paired-end 300 bp). Each sample, on average, received 47.8 million reads and 90% of those (42.9 million reads) was uniquely mapped onto the mouse reference genome (GRCm38) with STAR aligner (Dobin et al., 2013). In addition, approximately 36.8 million reads per sample were properly assigned to the Ensembl mouse gene models. Detailed RNA-seq summary statistics are listed in Supplementary Data 1.
3.2. Differentially expressed genes
In order to investigate the effects of the DFP and CORT + DFP treatments on PFC mRNA expression levels, we performed differential expression analysis using DESeq2 and gene set enrichment analysis with WebGestalt. We identified a total of 3736 DEGs (Supplementary Data 2) between DFP and CTL (Fig. 1A), of which 2203 genes were upregulated in the DFP group. Enrichment analysis demonstrated that those DEGs are significantly involved in immune system related biological processes (Fig. 1B) and KEGG pathways (Fig. 1C). Furthermore, hundreds of genes are functionally related to immune system physiology (374 genes), adaptive immunity (226 genes), and cell-mediated immunity (252 genes) (Fig. 1D).
Fig. 1.
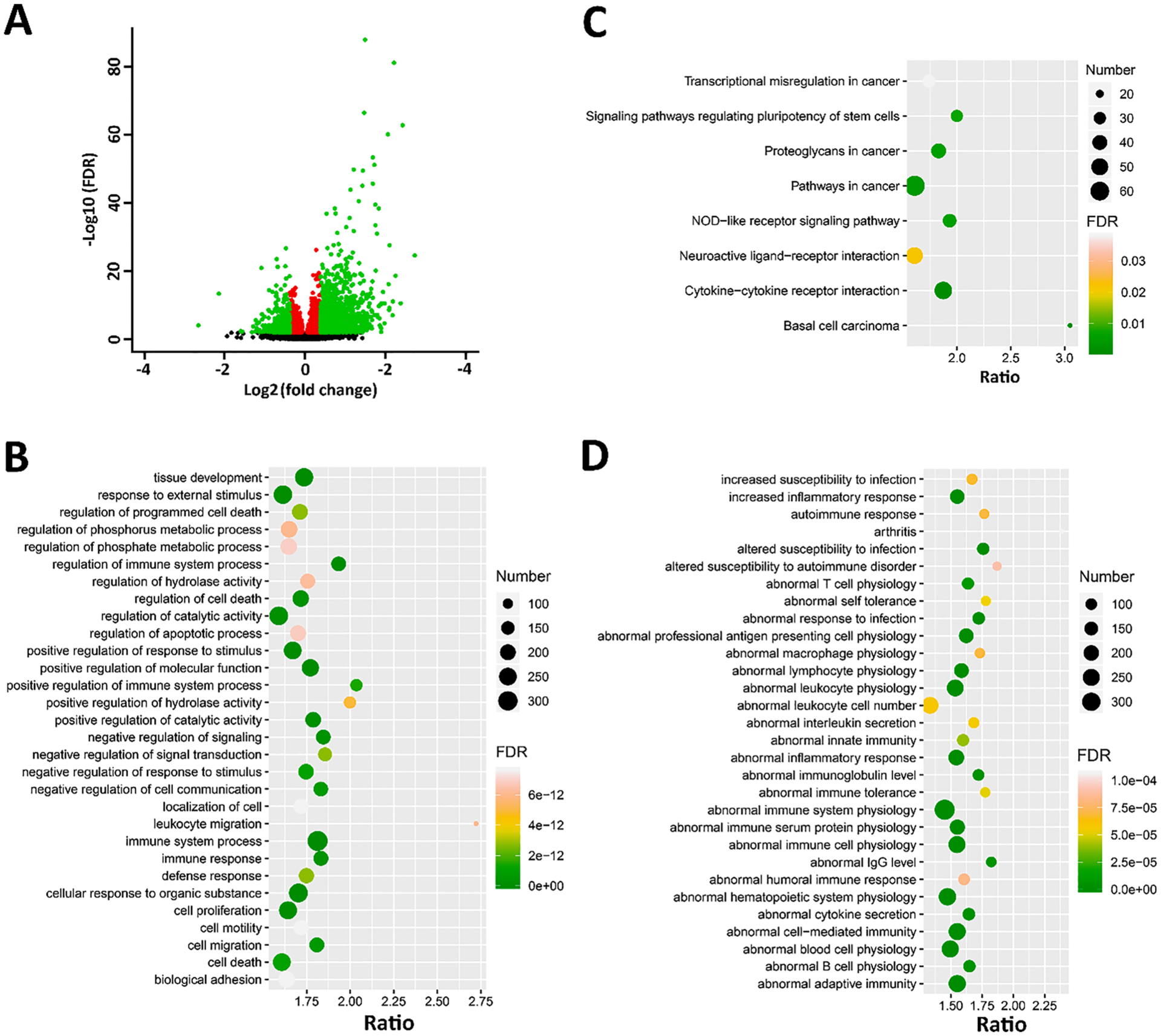
DEGs between DFP and CTL. (A) Volcano plot for the DEGs. The x-axis is the Log2(FC) for DFP treatment vs. CTL group, and the y-axis is the Log10 (FDR). Red dots are genes with FDR < 0.01, while green dots are genes with FDR < 0.01 and FC > 1.3. (B-D) Gene set enrichment analysis for GO (biological process), KEGG pathways, and MPO of the DEGs. All three analysis were performed by WebGestalt (http://www.webgestalt.org/) and plotted with ggplot2. For GO and MPO, top 30 enriched terms were shown. For KEGG, all the significant enriched terms were shown here. The x-axis represents the enrichment ratio; the size of the dots represent the number of genes; and the color indicated the FDR value.
We identified 2408 DEGs (Supplementary Data 2) between CORT + DFP and CTL group (Fig. 2A). Among those, 1070 genes are upregulated with the CORT + DFP treatment. GO enrichment showed that they participate in the immune system (Fig. 2B). Enrichment of KEGG pathways not only identified 3 common pathways with DFP vs. CTL group, including basal cell carcinoma, cytokine-cytokine receptor interaction, and pathways in cancer, but also highlighted several novel terms, such as inflammatory bowel disease (IBD), NF-kappa B signaling pathway, Th17 cell differentiation, and TNF signaling pathway (Fig. 2C). The phenotype enriched terms also align well with the terms over-represented for the DEGs induced by the DFP treatment (Fig. 2D and Fig. 1D). These results indicate that the treatment is having the expected effect on immune-related genes, as observed in previous studies using only a single strain (Ashbrook et al., 2018; Locker et al., 2017; Miller et al., 2018; O’Callaghan et al., 2015a).
Fig. 2.
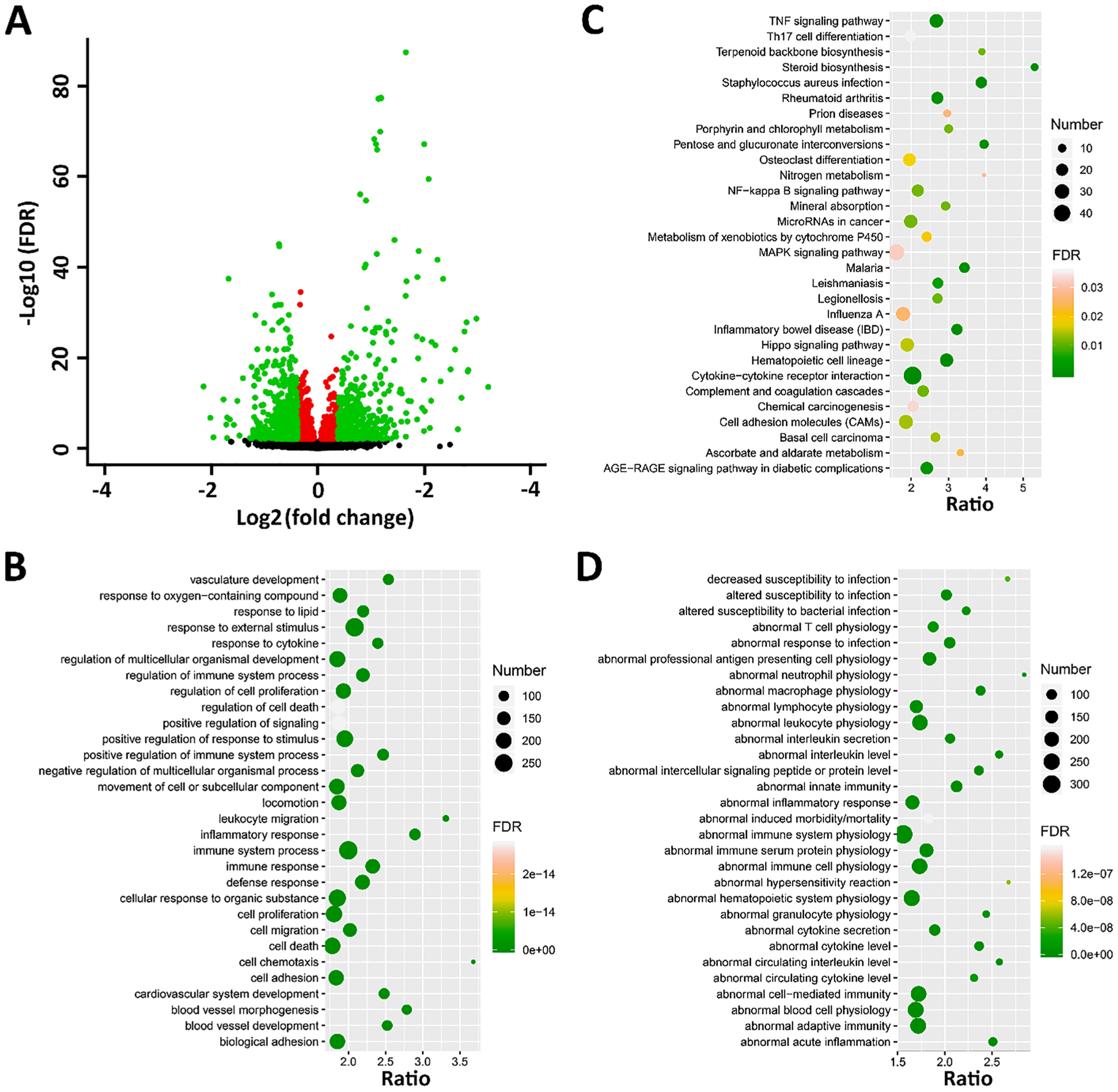
DEGs between CORT + DFP and CTL. (A) Volcano plot for the DEGs. The x-axis is the Log2(FC) for DFP treatment vs. CTL group, and the y-axis is the Log10(FDR). Red dots are genes with FDR < 0.01, while green dots are genes with FDR < 0.01 and FC > 1.3. (B-D) Gene set enrichment analysis for GO (biological process), KEGG pathways, and MPO of the DEGs. All three analysis were performed by WebGestalt (http://www.webgestalt.org/) and plotted with ggplot2. top 30 enriched terms were shown. The x-axis represents the gene ratio; the size of the dots represents the number of genes; and the color indicated the FDR value.
3.3. Comparisons of the DEGs between DFP vs. CTL and CORT + DFP vs. CTL
To investigate the different impacts of DFP or CORT + DFP treatment on mRNA expression levels, we directly compared the DEGs between DFP vs. CTL and CORT + DFP vs. CTL. Results showed 901 common genes between those two DEG sets (Fig. 3). In addition, 2835 and 1508 genes are specifically induced by DFP and CORT + DFP treatment, respectively (Fig. 3). Enrichment analysis highlighted that those genes are involved in distinct KEGG pathways, especially for the genes specifically induced by the combined treatment of CORT + DFP. This combination significantly affected pathways like cytokine-cytokine receptor interaction, inflammatory bowel disease (IBD), NF-kappa B signaling pathway, Th17 cell differentiation, and TNF signaling pathway (Fig. 3). Again, in agreement with previous findings, the combined treatment has an enhanced effect on immune related pathways when compared with the DFP treatment alone.
Fig. 3.
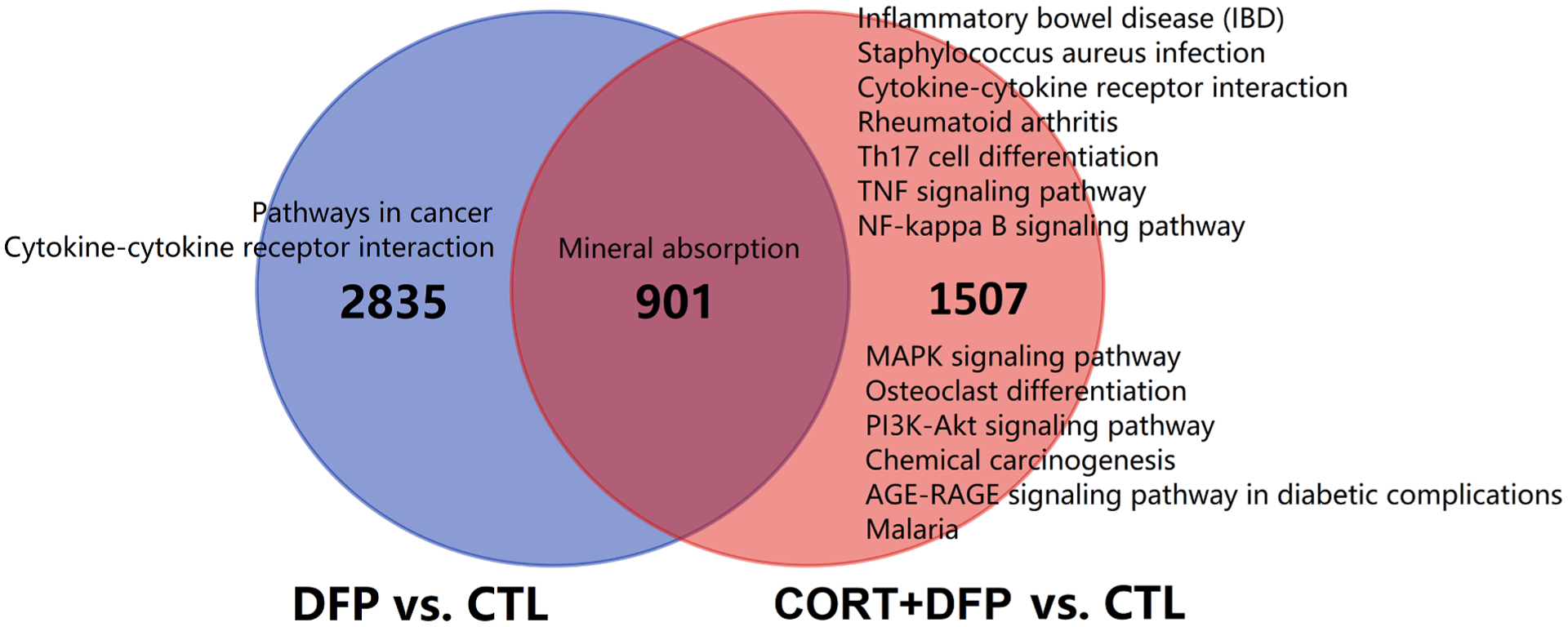
Venn diagrams showing the overlaps for the DEGs between DFP vs. CTL and CORT + DFP vs. CTL. The numbers indicated common or unique genes for those two groups. For each gene set, KEGG pathway enrichment analysis was conducted with WebGestalt (http://www.webgestalt.org/) and the significant enriched terms (FDR < 0.05) was listed alongside the diagrams.
3.4. Protein-protein interaction (PPI) subnetworks for DEGs
In order to further dissect the potential interactions of the DEGs and identify their key regulators, we uploaded the DEGs (FDR < 0.01 and FC > 1.3) into STRING (https://string-db.org/) to search for PPIs. By performing MCL clustering, we identified 4 and 6 subnetworks (# genes > 10) for DFP and CORT + DFP, respectively. Genes in each subnetwork are highly inter-connected (interaction score ≥ 0.7). For the DFP treatment, the key regulators are: Gng11, Cdc20, Igfbp3, and Iigp1 (Fig. 4). Those subnetworks function in neuroactive ligand-receptor interaction (subnetwork 1), protein ubiquitination (subnetwork 2), insulin-like growth factor binding (subnetwork 3), and innate immune response (subnetwork 4). For the CORT + DFP treatment, the key regulators are: Cxcl1, Il6, Ccnb1, Tnf, Agt, and Itgam (Fig. 5). Those subnetworks function in cytokine-cytokine receptor interaction (subnetwork 1, 2, and 4), p53 signaling pathway (subnetwork 3), neuroactive ligand-receptor interaction (subnetwork 5), and complement and coagulation cascades (subnetwork 6). Interestingly, none of these six hub genes have significant eQTL, suggesting that they are integrating influences from a number of other genes, rather than being under the control of a single variant of large effect.
Fig. 4.
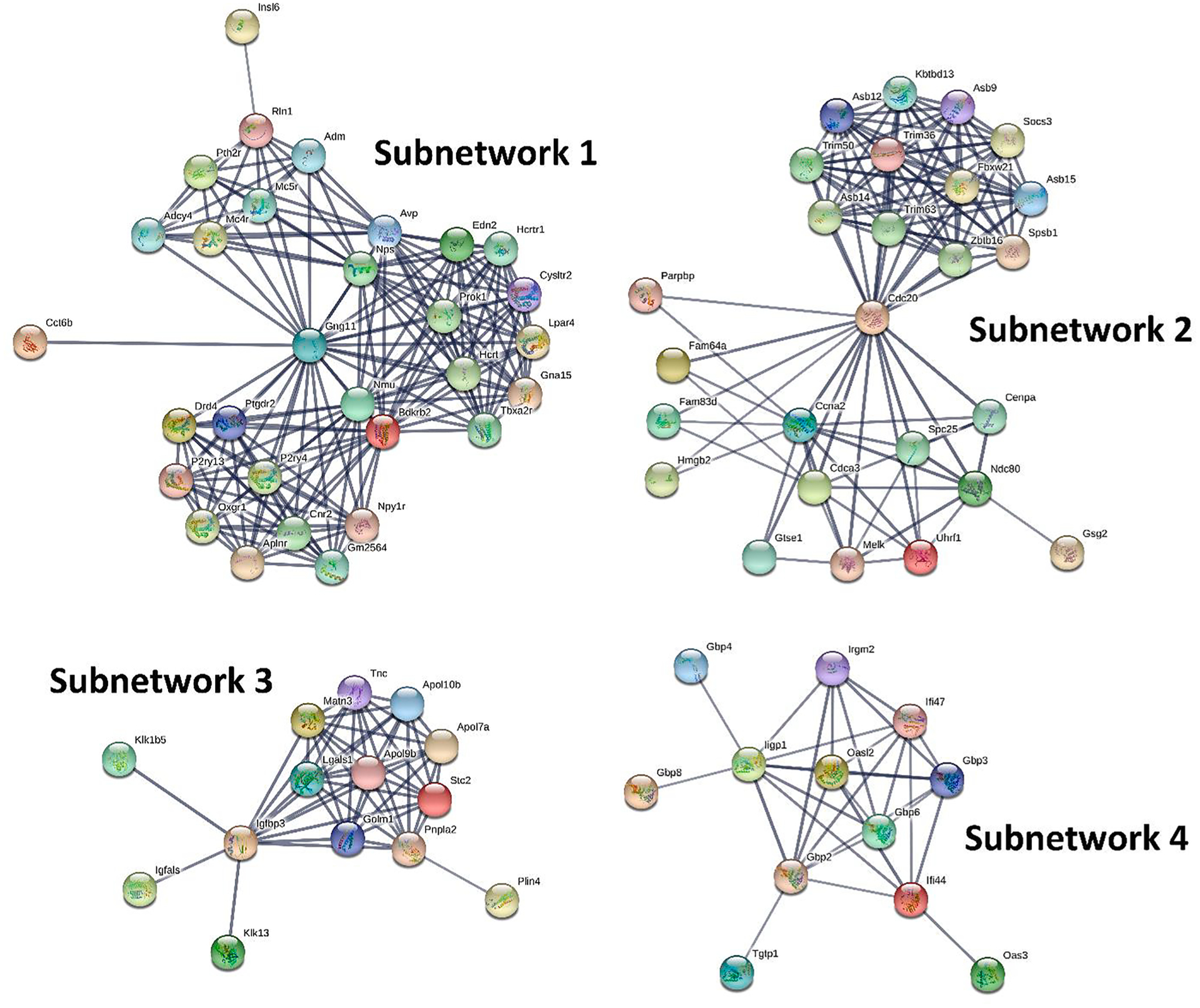
PPI subnetworks for the DEGs between DFP and CTL. The most salient DEGs were submitted to STRING (https://string-db.org/) for searching PPIs. Then followed by MCL subnetwork clustering with a minimum required interaction score of 0.7 and inflation parameter of 3. In each subnetwork, nodes represent genes while edges represent PPIs between two genes.
Fig. 5.
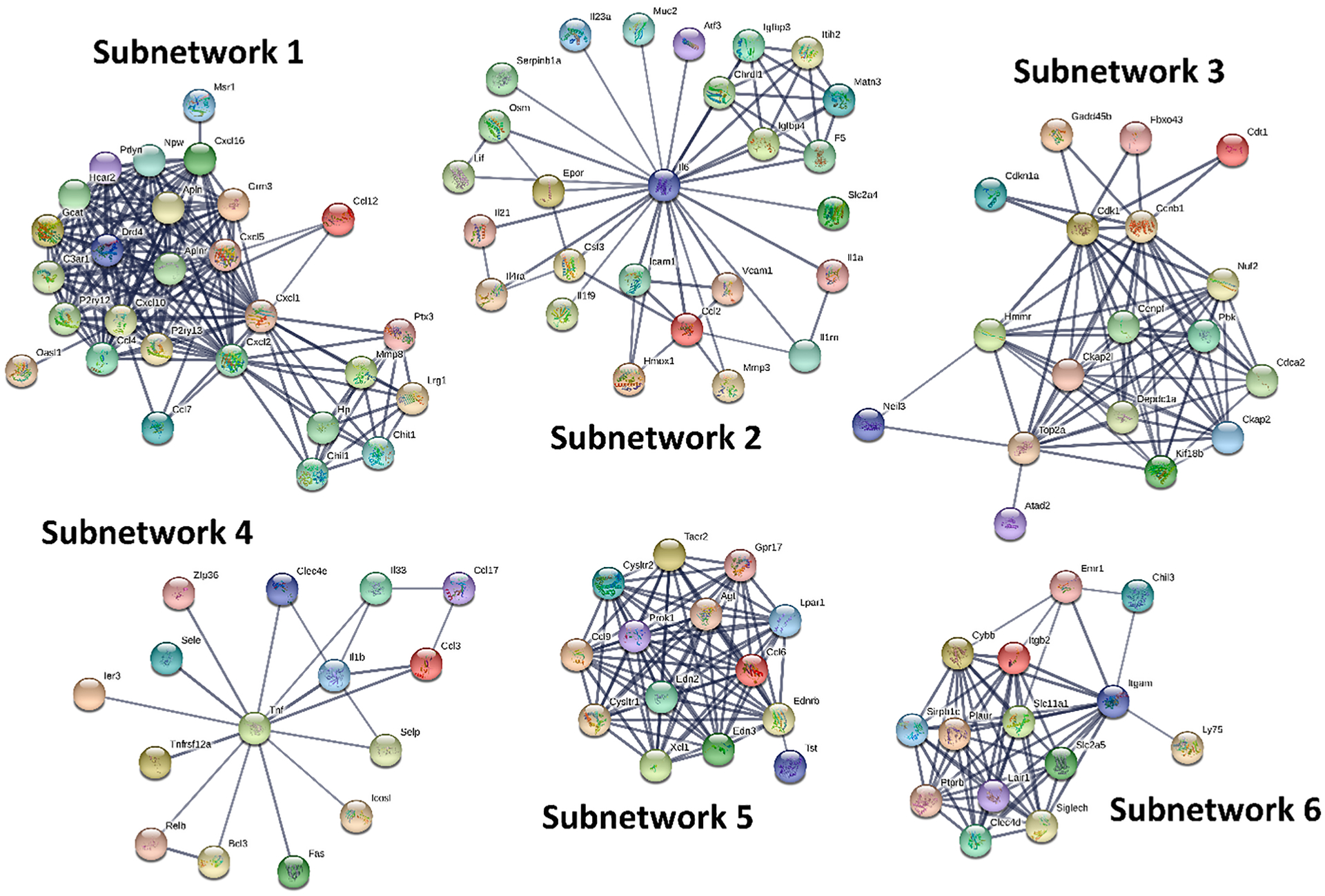
Protein-protein interaction (PPI) subnetworks for DEGs between CORT + DFP and CTL. The most salient DEGs were submitted to STRING (https://string-db.org/) for identification of PPIs. These PPIs were then clustered using MCL subnetwork clustering with a minimum required interaction score of 0.7 and inflation parameter of 3. In each subnetwork, nodes represent genes while edges represent PPIs between two genes.
3.5. Drug repositioning analysis
In addition to the discovery of new uses of existing drugs, drug repositioning analysis also allow us gain insight into a disease’s similarities with others. We performed drug repositioning analysis using GREP package (Sakaue and Okada, 2019) for the DEGs (FDR < 0.05 and FC > 1.3). As shown in Table 1. The genes differentially expressed with CORT + DFP treatment are significantly over-represented in chronic lower respiratory diseases, haemolytic anaemias, diseases of esophagus, noninfective enteritis and colitis, and inflammatory polyarthropathies. A total of 69 genes was covered in those terms and 30 of them are inflammatory response related genes, including Adora2b, Alox5, C5ar1, Ccl2, Ccr1, Cd14, Cxcl10, Cxcr2, Cysltr1, Ednrb, Fas, Hp, Icam1, Il1a, Il1b, Il21, Il23a, Il33, Il4r, Il6, Nr1h4, Osm, Ptgs2, Reg3a, Sele, Selp, Serpine1, Tek, Tlr2, and Tnf. There are two terms, chronic lower respiratory diseases and digestive system disorders of fetus and newborn, which are enriched in both CORT + DFP group and DFP group. The other DFP-specific enriched terms include complications predominantly related to the puerperium, certain early complications of trauma, and pulmonary heart disease and diseases of pulmonary circulation (Table 1). Those terms totally included 36 genes and 12 of them are inflammatory response related genes, including Ada, Ager, Bdkrb2, Ccl2, Cnr2, Nos2, Ppara, Serpine1, Tbxa2r, Tek, Tlr7, and Trpv1. Detailed gene-drug pairs are listed in Supplementary Data 3.
Table 1.
Lists of significant enriched ICD terms for the DEGs. Fisher Exact P < 0.05 are bolded.
| Group | Group Name | DFP vs. CTL | CORT + DFP vs.CTL | ||
|---|---|---|---|---|---|
| Odds Ratio | Fisher Exact P | Odds Ratio | Fisher Exact P | ||
| J40-J47 | Chronic lower respiratory diseases | 1.97 | 0.0165 | 2.75 | 0.0001 |
| D55-D59 | Haemolytic anaemias | 0.90 | 0.6572 | 4.03 | 0.0024 |
| D60-D64 | Aplastic and other anaemias | 1.83 | 0.1992 | 3.76 | 0.0035 |
| K50-K52 | Noninfective enteritis and colitis | 0.73 | 0.7917 | 2.51 | 0.0055 |
| P75-P78 | Digestive system disorders of fetus and newborn | 7.79 | 0.0473 | 13.77 | 0.0055 |
| D50-D53 | Nutritional anaemias | 0.88 | 0.6721 | 3.26 | 0.0114 |
| B50-B64 | Protozoal diseases | 1.79 | 0.0891 | 2.21 | 0.0169 |
| M15-M19 | Arthrosis | 1.27 | 0.2916 | 1.95 | 0.0177 |
| M05-M14 | Inflammatory polyarthropathies | 1.58 | 0.0679 | 1.78 | 0.0183 |
| K20-K31 | Diseases of oesophagus, stomach and duodenum | 0.98 | 0.5993 | 2.67 | 0.0190 |
| M45-M49 | Spondylopathies | 1.63 | 0.2199 | 2.48 | 0.0263 |
| T66-T78 | Other and unspecified effects of external causes | 1.44 | 0.2205 | 2.04 | 0.0272 |
| 170–179 | Diseases of arteries, arterioles and capillaries | 1.61 | 0.0725 | 1.75 | 0.0305 |
| T80-T88 | Complications of surgical and medical care, not elsewhere classified | 0.00 | 1.0000 | 2.38 | 0.0321 |
| M00-M03 | Infectious arthropathies | 1.37 | 0.2416 | 1.85 | 0.0406 |
| E10-E14 | Diabetes mellitus | 1.53 | 0.0677 | 1.57 | 0.0435 |
| A30-A49 | Other bacterial diseases | 1.48 | 0.2223 | 1.97 | 0.0483 |
| 085–092 | Complications predominantly related to the puerperium | 12.67 | 0.0013 | 3.90 | 0.1237 |
| J80-J84 | Other respiratory diseases principally affecting the interstitium | 2.80 | 0.0319 | 1.96 | 0.1372 |
| 195–199 | Other and unspecified disorders of the circulatory system | 2.37 | 0.0038 | 1.43 | 0.1667 |
| T79-T79 | Certain early complications of trauma | 4.50 | 0.0208 | 1.70 | 0.3558 |
| 126–128 | Pulmonary heart disease and diseases of pulmonary circulation | 2.21 | 0.0133 | 1.07 | 0.4830 |
| 130–152 | Other forms of heart disease | 1.80 | 0.0245 | 0.95 | 0.6144 |
3.6. Proinflammatory cytokines expression level distribution across BXD RI strains
Several studies have shown that GWI is associated with proinflammatory cytokines such as TNFα, IL1β, and IL6 (O’Callaghan et al., 2015a, 2016; Khaiboullina et al., 2015). The PPI subnetwork analysis also revealed two subnetworks of Il6 and Tnf for CORT + DFP treatment (Fig. 5, subnetwork 2 and 4). Because our study included 32 strains, this allows us to detect the strain and treatment effects for the expression variances of proinflammatory cytokines genes. As shown in Fig. 6, all three genes, Il6, Tnf, and Il1b, showed large variance in expression across the BXD strains. Analysis of variance revealed significant effects for both strain and treatment (Jones et al., 2020). For example, BXD98 shows very little difference in expression of Il6 between DFP and CORT + DFP, and BXD66 actually shows a down-regulation in expression with CORT + DFP treatment compared to DFP. This gives a clear indication that the genome is significantly mediating the effect of our treatments.
Fig. 6.
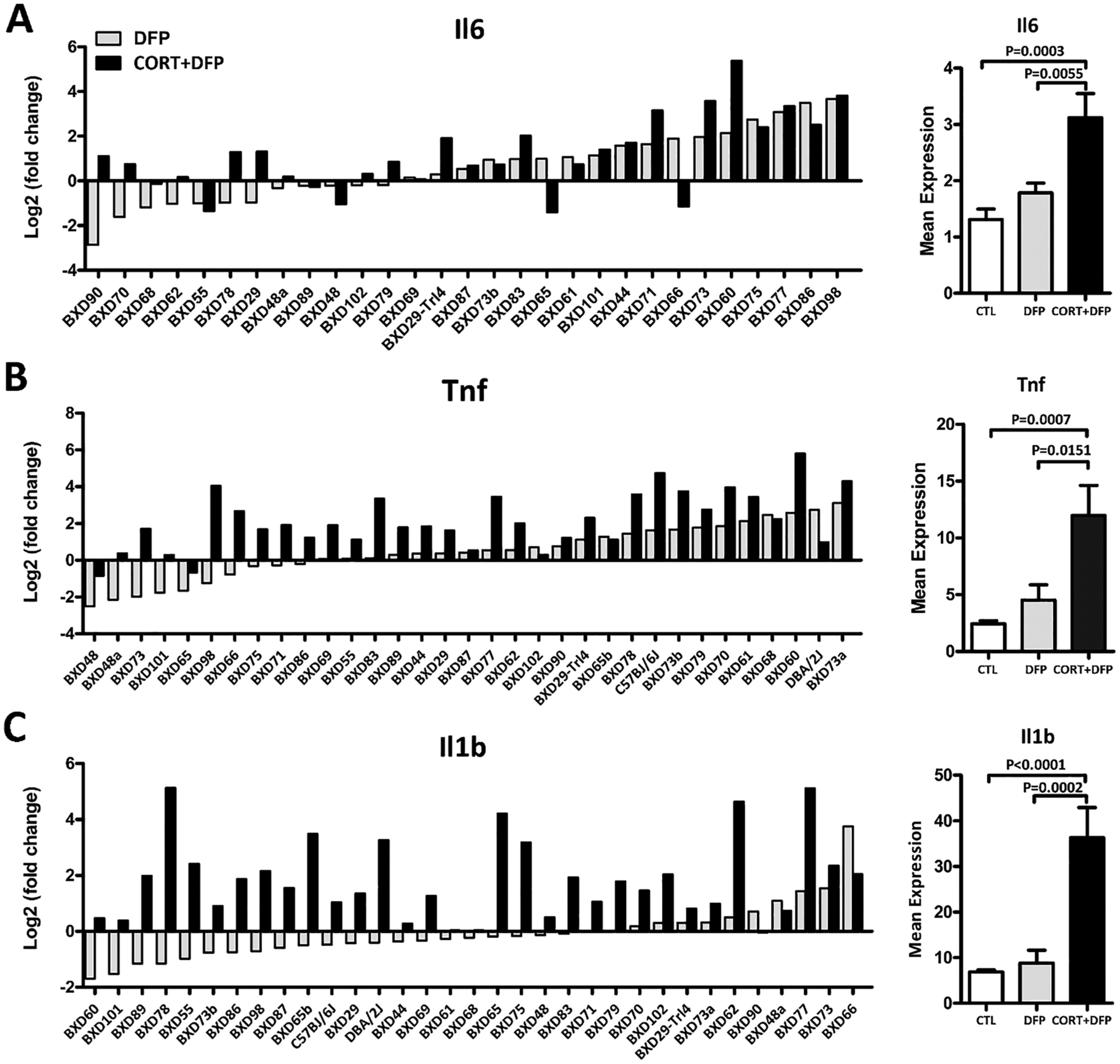
Proinflammatory cytokines gene Il6 (A), Tnf (B), and Il1b (C) expression distribution across the BXD RI strains. Left panel shown the relative fold change of Il6, Tnf, and Il1b in the DFP and CORT + DFP groups compared to the CTL across the BXD RI strains. Right panel shown the overall expression of the three genes in the three groups.
3.7. QTL mapping of differentially expressed genes
By using a genetic reference population, we are able to map QTL which influence the expression of any gene across all three of our treatment groups. Therefore, we looked for QTL in our 1507 DEGs (FDR < 0.01 and FC > 1.3) which are strong and significant in CORT + DFP (LRS > 20), but have no evidence for a QTL in DFP or CTL (LRS < 12, ± 10 Mb). This would allow us to identify loci which are only important in the combined CORT + DFP treatment. Only 21 genes met this criterion, and there were no enriched annotations among these genes (Table 2). Interestingly, four of these genes all had trans-eQTL mapping to Chromosome (Chr) 6:91–95 Mb, suggesting that a variant in this region is influencing the expression of genes uniquely in the CORT + DFP treatment. We then examined if any genes in this region were found to be differentially expressed in CORT + DFP, and found three genes, Fgd5 (Log2(FC) = −0.18), Adamts9 (Log2(FC) = 0.55) and A730049H05Rik (Log2(FC) = 0.70). Interestingly, Adamts9 has a suggestive association with chronic fatigue syndrome (p = 2.8e-7) and with malaise and fatigue (p = 9.8e-6; http://pheweb.sph.umich.edu/ SAIGE-UKB/variant/3–64647592-C-G) in the UK BioBank. As this is a relatively poorly annotated gene, we next looked at the 20 genes which were most highly correlated with Adamts9 expression in our CORT + DFP data (r > 0.8) and carried out enrichment analysis. This showed a significant enrichment for response to cytokine (FDR = 0.002, 7 genes) and cellular response to cytokine stimulus (FDR = 0.01, 6 genes).
Table 2.
Table of transcripts with a strong, significant eQTL (LRS>20) in CORT+DFP treated animals, but no evidence in control or DFP treated animals (LRS<10). The four transcripts with a trans-eQTL on Chr 6 at 91–95 Mb are bolded.
| Symbol | Location | CTL | DFP | CORT + DFP | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean Expression | Max LRS at Peak | Max LRS Location | Mean Expression | Max LRS at Peak | Max LRS Location | Mean Expression | Max LRS at Peak | Max LRS Location | ||
| Sgk2 | Chr2: 162.987330 | 0.986 | 13.8 | Chr4: 58.363486 | 1.034 | 10.9 | Chr6: 54.998546 | 0.837 | 25.2 | Chrl5: 8.299029 |
| TrpS3ill | Chr2: 93.187548 | 5.711 | 15 | Chr9: 111.455945 | 5.794 | 10.1 | Chrl3: 83.781116 | 5.99 | 21 | Chr6: 93.599797 |
| Chil3 | Chr3: 106.147554 | 0.164 | 14.7 | Chr3: 109.660551 | 0.173 | 11.4 | Chrl5: 97.614562 | 0.575 | 20.4 | ChrX: 133.603916 |
| Gm43666 | Chr3: 33.703355 | 0.154 | 9.7 | Chrl9: 58.686202 | 0.148 | 11.1 | Chr6: 115.858131 | 0.116 | 25.8 | Chrl5: 72.500609 |
| A830029E22Rik | Chr3: 81.802025 | 0.681 | 17.2 | ChrlO: 27.031021 | 0.651 | 14.6 | Chr8: 15.921720 | 0.521 | 21.5 | Chr2: 139.724059 |
| Bglap2 | Chr3: 88.377736 | 0.297 | 12.4 | Chr4: 131.999242 | 0.335 | 12 | Chrl9: 32.049009 | 0.196 | 23.8 | Chr6: 76.542612 |
| Gml2724 | Chr4: 106.241216 | 0.231 | 15.5 | Chrll: 98.915692 | 0.175 | 11 | Chrl: 170.949421 | 0.137 | 23.1 | Chr6: 18.110893 |
| Ermap | Chr4: 119.175457 | 0.368 | 16.9 | Chrll: 35.140181 | 0.351 | 11 | Chr2: 70.716531 | 0.308 | 20.5 | Chrl5: 3.767744 |
| Retsat | Chr6: 72.598475 | 3.208 | 9.9 | Chr2: 173.426500 | 3.321 | 9.6 | Chr8: 75.212318 | 3.553 | 20.6 | Chr6: 91.793995 |
| Nat8f3 | Chr6: 85.732513 | 1.419 | 15.9 | Chr2: 138.500000 | 1.374 | 10.7 | Chr3: 105.059546 | 1.182 | 20.2 | ChrX: 100.000000 |
| Gsgll | Chr7: 125.878420 | 6.031 | 11.6 | Chrl5: 96.171491 | 6.128 | 12.6 | Chrl3: 116.536749 | 6.241 | 22.2 | Chr6: 92.602487 |
| Adgral | Chr7: 139.834174 | 4.831 | 12.8 | Chrl5: 96.186877 | 4.971 | 9.6 | Chrl5: 97.614562 | 5.056 | 20.2 | Chr6: 5.301296 |
| Spred3 | Chr7: 29.158829 | 4.554 | 10.2 | Chrl5: 96.186877 | 4.617 | 11.5 | Chr3: 76.579840 | 4.724 | 23.7 | Chr4: 57.500003 |
| 0ca2 | Chr7: 56.239760 | 0.295 | 11 | Chr9: 57.350996 | 0.27 | 7.8 | Chr9: 43.304228 | 0.234 | 26.3 | Chr6: 115.858131 |
| Krtl3 | Chrll: 100.117327 | 0.109 | 11.9 | Chrl4: 78.286799 | 0.033 | 17 | ChrlO: 113.399853 | 0.055 | 35.6 | Chrll: 114.671594 |
| Gml2185 | Chrll: 48.904656 | 1.161 | 12.8 | Chr3: 141.827741 | 1.077 | 17.4 | Chr9: 43.304228 | 1.004 | 23.6 | Chrl3: 54.842814 |
| Gm49123 | Chrl4: 45.551838 | 0.24 | 8.5 | Chrl9: 28.403278 | 0.203 | 9.9 | ChrlO: 3.180582 | 0.138 | 20.1 | Chr4: 41.176874 |
| Ptk2b | Chrl4: 66.153257 | 7.13 | 11.3 | Chrll: 98.136016 | 7.225 | 9 | Chrl6: 86.457592 | 7.515 | 23 | Chr6: 91.706864 |
| Book | Chrl5: 38.933144 | 6.34 | 16.6 | Chrl9: 27.531578 | 6.343 | 20.7 | Chrl5: 38.986548 | 6.252 | 20.9 | ChrX: 114.159505 |
| Upklb | Chrl6: 38.773184 | 1.058 | 39.8 | Chrl6: 36.946690 | 0.942 | 42.6 | Chrl6: 36.946690 | 0.698 | 22.4 | Chrl7: 5.991544 |
| AC117629.1 | Chrl9: 26.246024 | 0.727 | 9.2 | Chrl9: 27.531578 | 0.714 | 9 | Chrll: 64.680151 | 0.576 | 21 | Chr7: 136.435575 |
3.8. Estimates of cell type composition changes
Previous studies have shown that the treatment employed here alters the proportion of different cell types within the brain (Ashbrook et al., 2018). We observed a similar pattern, with a significant effect of CORT + DFP treatment on the relative proportion of all cell types, except for microglia (Fig. 7). We saw understandably larger variation than observed in the previous study, due to using 32 strains rather than a single strain. Using a range of related strains allowed us to map loci which may be causing this response to the treatment. To investigate this, we calculated the mean proportion of each cell type in each strain, and preformed QTL mapping using GEMMA (Zhou and Stephens, 2012), within GeneNetwork (Sloan et al., 2016).
Fig. 7.
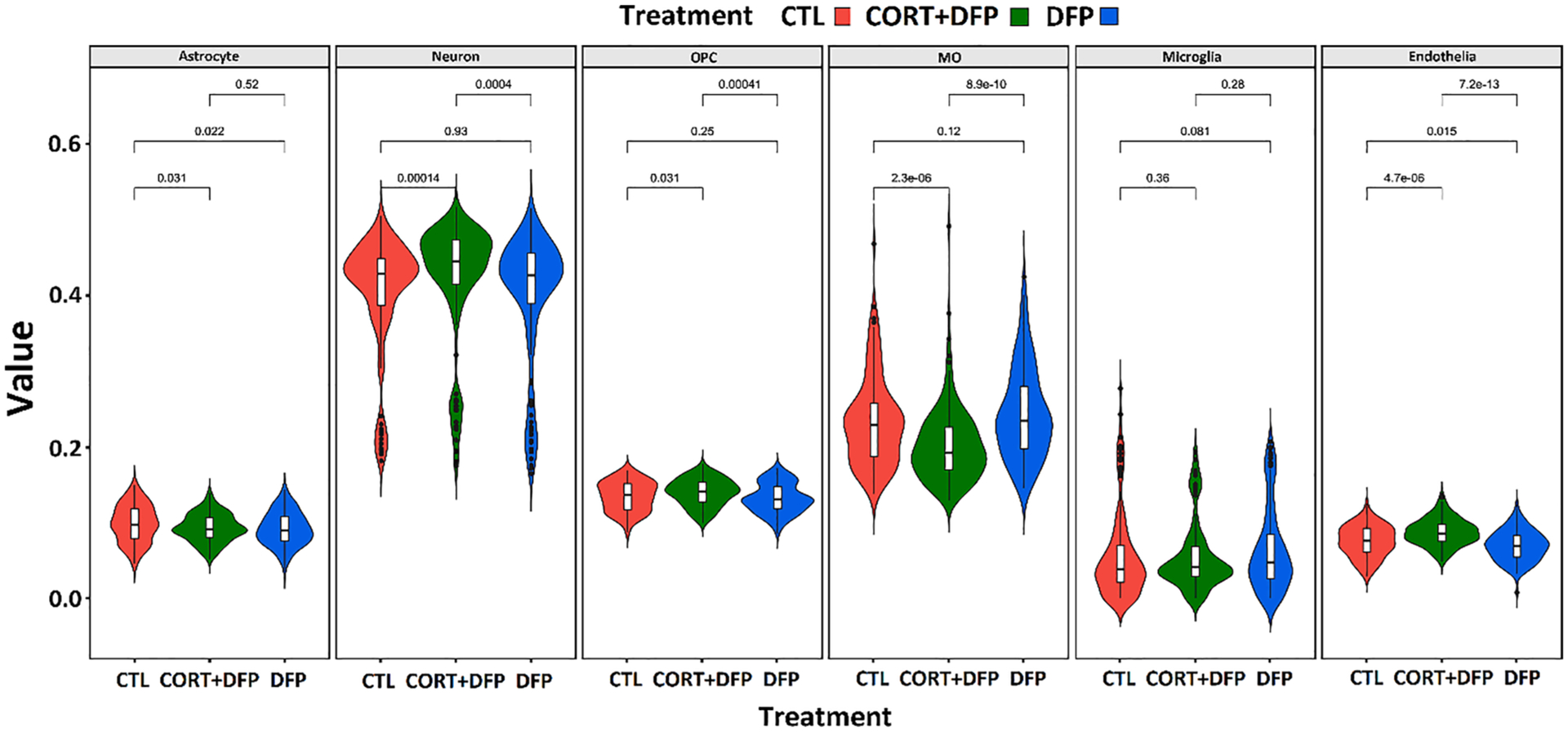
Violin plot of estimated relative proportion of six cell types (astrocytes, neurons, oligodendrocyte precursor cells (OPCs), oligodendrocytes, microglia and endothelial cells) in frontal cortex of treated (CORT + DFP, or DFP) and control animals. Treatment differences were detected using ANOVA, and p-values were calculated between treatment using Tukey HSD test.
This analysis showed that for oligodendrocytess, but not other cell types, there was a significant locus affecting the difference in estimated cell type proportion in CORT + DFP treated animals vs control (Fig. 8A). Nor did any of the CORT + DFP cell proportions produce significant QTL if the control cell proportions were not accounted for. Since the proportion of each cell type is dependent upon the other cell types (i.e. because it is a proportion rather than an absolute value), we produced an eigentrait (Carter, 2013) from the first principal component (PC1) of all six phenotypes, and mapped this eigentrait. This identified the same QTL, but with a slightly longer 1.5 LOD drop confidence interval, and lower peak LOD (Fig. 8B). This suggests that the increase in oligodendrocytes is the driver behind the total change in cell proportion. The 1.5 LOD drop confidence interval spans Chr 5:16.86–20.07 Mb (Fig. 8C) and contains only 10 genes (Table 3).
Fig. 8.
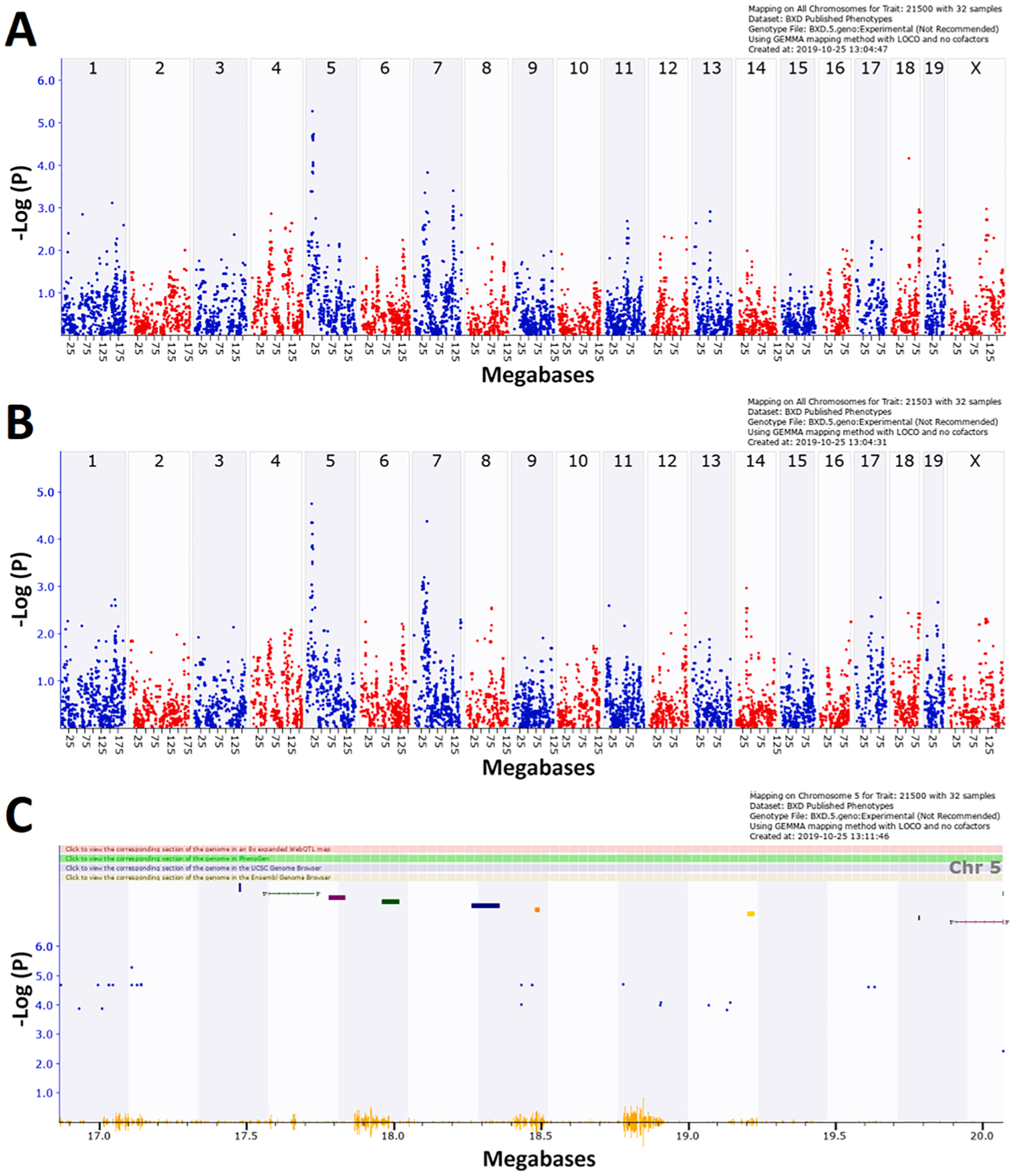
QTL mapping of difference in cell proportions between CORT + DFP treated and control animals for (A) myelinating oligodendrocytes (MOs), (B) eigentrait of all six cell types. (C) 1.5 LOD drop confidence interval of Chr 5 QTL. All figures were produced in GeneNetwork, using GEMMA. Each blue or red dot represents the −log(p) at a specific marker. In (C) bars at the top of the figure represent the location of genes. The orange ‘waves’ at the bottom of the figure represent SNP density.
Table 3.
Genes within the 1.5 LOD drop interval for the Chr5 QTL mediating proportion of myelinating oligodendrocytes (MOs) after CORT + DFP treatment.
| Symbol | Mb Start | Length (Kb) | SNP Count | SNP Density | Human Chr | Human Position (bp) |
|---|---|---|---|---|---|---|
| Speer4f | 17.47612 | 4.815 | 1 | 0.207684 | – | – |
| Speer4fl | 17.47612 | 4.815 | 1 | 0.207684 | – | – |
| Sema3c | 17.57482 | 155.452 | 67 | 0.431001 | 7 | 80742536–80922389 |
| Cd36 | 17.78169 | 54.207 | 5 | 0.092239 | 7 | 80602207–80679277 |
| Gnat3 | 17.96257 | 57.099 | 45 | 0.788105 | 7 | 80457291–80512503 |
| Gnail | 18.26513 | 95.279 | 12 | 0.125946 | 7 | 80134831–80226181 |
| AK076902 | 18.4817 | 13.556 | 50 | 3.688404 | – | – |
| 4921504A21Rik | 19.20237 | 24.188 | 22 | 0.909542 | – | – |
| AK051452 | 19.78453 | 2.62 | 0 | 0 | – | – |
| Magi2 | 19.90752 | 797.275 | 15 | 0.018814 | 7 | 78017055–79453667 |
Gene symbols and the gene position in mm10 are shown, along with the SNP count and density. The chromosome and bp position of human homologues are also indicated.
We next performed a systems genetics analysis to identify which of these 10 genes were high-likelihood candidates to be underlying the phenotype. None of the genes within the QTL interval have cis-eQTL, suggesting that differences in their regulation are not causing the phenotype. Nor do any protein coding genes have a trans-eQTL in this region. This suggests that the effect of this region is due to changes in protein function. There are 3304 variants in this region, and these were annotated with the variant effect predictor. Only one variant, in the long non-coding RNA Gm31752 (rs31737927), was predicted to be high impact, and three were predicted to be low impact, two in Sema3c (rs47333154, rs46955609) and one in Gnat3 (rs36506283)
To further investigate these 10 genes, we examined if they were known to be expressed in the brain. We collated data from several web services (Supplementary Data 4), which suggests that Sema3c, Cd36, Gnai1, and Magi2 are expressed in brain in both mouse and human. There is also evidence that Magi2, Sema3c, and Gnai1 are expressed in oligodendrocytes in at least one of the two species (http://celltypes.brain-map.org/rnaseq/mouse/cortex-and-hippocampus). These three genes are therefore high priority candidates.
To build a broader picture of how this locus might be influencing cell proportion, we also looked for genes which correlate with the PC1 eigentrait, to establish “Guilt by association”, e.g. genes which correlate with the eigentrait will point towards the pathways which are altered by our gene of interest. We identified 443 genes in the control gene expression data that significantly correlated with our PC1 eigentrait (p ≤ 0.05, r ≥ 0.5 or ≤ −0.5). Enrichment analysis using WebGestalt (Wang et al., 2017) identified many over represented pathways and annotations, especially those related to oxidative phosphorylation, the electron transport chain, and mitochondria (Fig. 9). This suggests that strains with higher baseline expression of genes related to the mitochondria have higher sensitivity to changes in cell proportions caused by the CORT + DFP treatment. To confirm this, we then took 14 genes within these categories (Mrps10, Mrpl27, Mrpl13, Mrpl52, Mrpl14, Mrpl21, Mrps16, Ndufa5, Ndufb5, Ndufa2, Ndufa1, Ndufb3, Ndufa6 and Ndufb10), and performed individual correlations between these genes and the cell proportion phenotypes. This confirmed that these genes positively correlated with both the PC1 eigengene, and the difference in proportion of MOs (Supplementary Data 5).
Fig. 9.
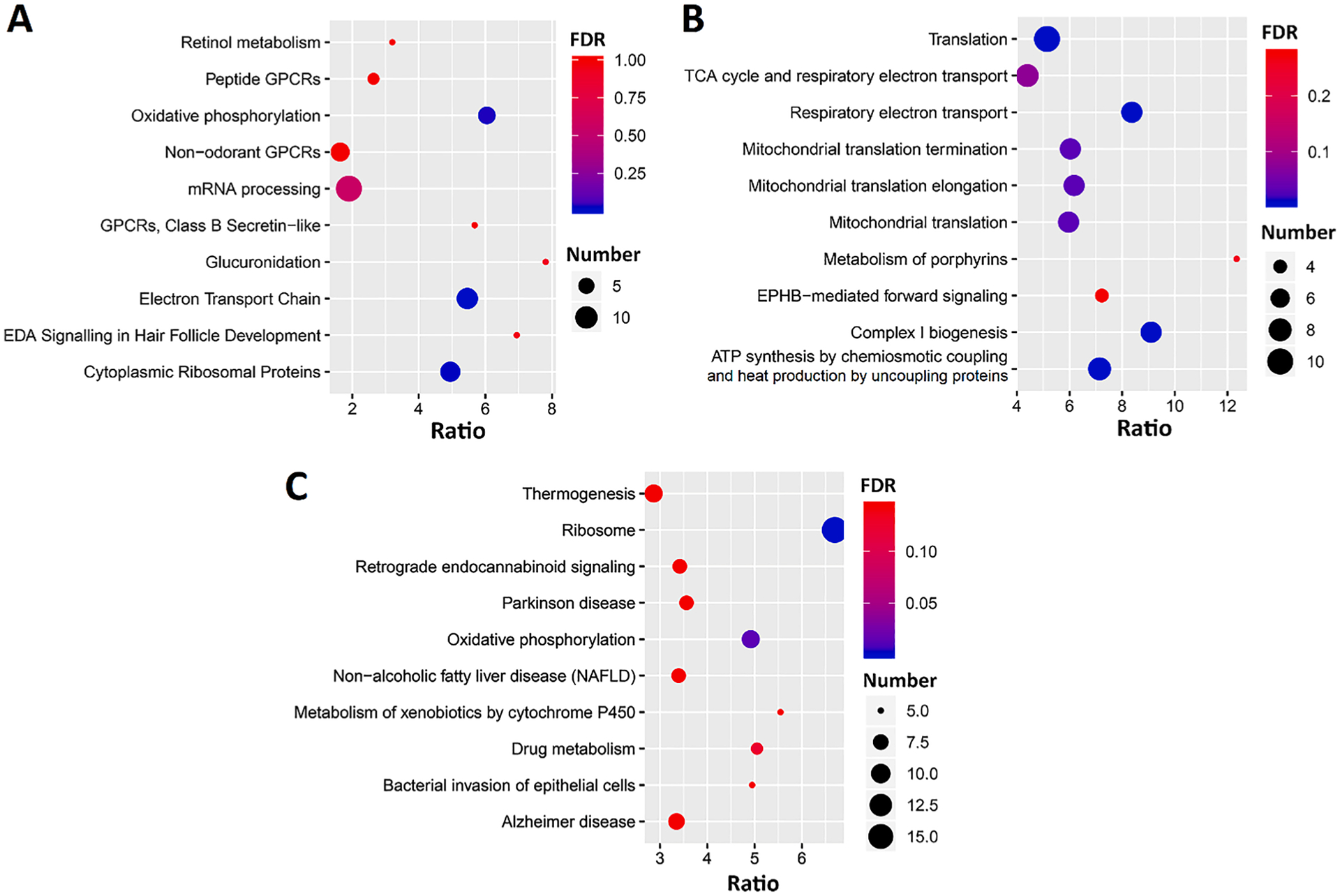
Pathways enriched in 2660 genes which correlate with our PC1 eigentrait, in the control RNA-seq data. Analysis for KEGG pathways (A), Reactome pathways (B), and Wiki pathways (C) were performed by WebGestalt (http://www.webgestalt.org/) and plotted with ggplot2. The x-axis represents the gene ratio; the size of the dots represents the number of genes; and the color indicated the FDR value.
Interestingly, far fewer genes (136) correlated between the PC1 eigengene and the post CORT + DFP treatment RNA-seq (p ≤ 0.05, r ≥ 0.5 or ≤ −0.5), and there were no significantly enriched annotations. This further suggests that it is baseline differences between the strains which are altering the proportions of cell types.
4. Discussion
There is huge variability in both the symptoms and susceptibility to GWI among veterans who were deployed to the 1990–1991 Gulf War. Ours is the first manuscript to take a genome-wide approach in a genetically diverse population, to more accurately model the genetic changes that occurred to those personnel deployed in the Persian Gulf.
Previous studies showed that exposure to DFP in mice could lead to a brain-wide neuroinflammatory response and pretreating with CORT augments this inflammation effect, which is consistent with a neuroimmune basis of GWI (O’Callaghan et al., 2015b). In this study, we treated a cohort of 30 BXD family members and their two parents (B6 and D2) with DFP or the combination of CORT + DFP. By comparing the PFC transcriptomes between the treatment and normal controls, thousands of DEGs were identified. The enrichment analysis showed that those DEGs significantly participated in immune system or inflammatory-related biological processes (Figs. 2 and 3), in good concordance with previous models and with human data. It is worth noting that several KEGG terms were significantly enriched in the CORT + DFP treatment condition compared to the DFP treatment alone, such as inflammatory bowel disease (IBD), NF-kappa B signaling pathway, Th17 cell differentiation, and TNF signaling pathway.
GWI is characterized by the persistence of inflammatory bowel disease (IBD). A new study showed that increased viral richness and alpha diversity correlated positively with gut bacterial dysbiosis and proinflammatory cytokines in the GWI mouse model (Seth et al., 2019). Broderick et al. (Broderick et al., 2011a) studied blood samples from GWI participants and suggested that chronic NF-kB activation as a potentially key component of GWI. In addition, depressive-like behaviors caused by exposure to chronic stress could be mediated by NF-κB signaling. Blockade of NF-κB could inhibit the actions of other proinflammatory cytokines (e.g. IL-6 and TNF-α) implicated in stress and depression (Koo et al., 2010). Multiple studies have demonstrated that T helper 17 (Th17) cells regulate inflammatory and autoimmune diseases. Th17 cells are involved in the pathogenesis of IBD (inflammatory bowel disease) and higher frequency of Th17 cells in patients with rheumatoid arthritis (RA) than in healthy controls (Singh et al., 2014). Studies also showed IL-6 cytokine and TGF-β together induce the differentiation of pathogenic TH17 cells from naive T cells (Bettelli et al., 2006). Cytokine IL-6 secretion was also reported critical in intestinal Th17 responses and promotes IL-22 expression in IBD (Li et al., 2014). Tumor Necrosis Factor (TNF) is one of the most potent physiological inducers of the nuclear transcription factor NF-kappa B (Schütze et al., 1995). Higher responsiveness of TNF-α have previously been identified in GWI subjects suggesting an autoimmune component to this illness (Broderick et al., 2011b; Warnatz and Voll, 2012).
Cytokines, secreted by a variety of cells, are extracellular proteins or glycoproteins that can regulate cell growth and differentiation, immunity, and participate in inflammation. Consistent with the findings that cytokines levels, such as IL-7, IL-4, TNF-α, IL-13, and IL-17F, in serum are significantly changed in GWI patients (Khaiboullina et al., 2015; Parkitny et al., 2015), our results also identified tens of differentially expressed cytokine genes, they are mainly involved into three cytokine-cytokine receptor interaction subnetworks (Fig. 5, subnetwork 1, 2, and 4), with the hub genes of Cxcl1, Il6, Tnf, respectively. In addition, those proinflammatory cytokine mRNA levels showed large variations across the BXD RI strains (Fig. 6), demonstrating both genetics and treatment modulated their expressions. This could explain part of the differential susceptibility to GWI, i.e. that genetics are a significant contributor to susceptibility and response to the exposures. Indeed, the Institute of Medicine (2010) concluded that GWI results from an interplay of genetic and environmental factors and the former may play a larger role for some affected. A case in point is rare variants in the BChE gene which confer reduced activity in the enzyme, are associated with increased susceptibility to GWI (Steele et al., 2015).
By using 32 inbred strains, we were able to demonstrate that the response to CORT + DFP is under genetic control, with differential response among strains. We were able to apply QTL mapping to identify DEGs that were under strong genetic control in the BXD, specifically under the CORT + DFP condition. We identified 21 genes that appear to be under differential genetic control in the CORT + DFP treatment, and found that a locus on Chr 6 was altering the expression of four of these genes. Within this locus, the gene Adamts9 is a good candidate for causality: it has been associated with chronic fatigue syndrome in UKBioBank data, and in our data is highly correlated with genes related to response to cytokines. Therefore, Adamts9 may be a gene which specifically responds to CORT + DFP, altering expression of a number of cytokine related genes, and resulting in fatigue related phenotypes. These results fit in well with previous studies of Adamts9, which has been linked with inflammation (García-Faroldi et al., 2013; Uysal et al., 2013; Yaykasli et al., 2009), and significantly associated with cognitive aging (Lin et al., 2017), which may be related to the cognitive disfunction seen in GWI sufferers.
We also identified a significant change in the proportion of oligodendrocytes under CORT + DFP treatment, in agreement with our previous work (Ashbrook et al., 2018). However, with the genetic variation that our new system adds, we were able to map a specific genetic locus altering oligodendrocyte cell proportion. Of the 10 genes we identified in this locus, only five have human homologues (Sema3c, Cd36, Gnat3, Gnai1, Magi2). Four of these genes have been associated with biological processes involving oligodendrocytes previously. The semaphorins, of which Sema3c is a member, are involved in oligodendrocyte guidance (Cohen et al., 2003; Spassky et al., 2002) and the control of the ingrowth of the septohippocampal projection (Steup et al., 2000). The innate immunity receptor Cd36 is believed to be associated with myelination and microglia (Eto et al., 2003; Li et al., 2015), microglial cell physiology (Eto et al., 2003), and peripheral nervous system regeneration (Su et al., 2008), and have been implicated in a wide variety of brain diseases, ranging from stroke to neurodegeneration (Garcia-Bonilla et al., 2014). Magi2 is expressed in Schwann cells (Poliak et al., 2002) and have been associated with several “behavior/neurological” and “nervous system” related phenotypes (Zhang et al., 2015), such as increased anxiety-related response, abnormal spatial working memory, abnormal brain interneuron morphology, abnormal neuron morphology, and abnormal dendritic spine morphology. Gnai1 may play a role in oligodendrocyte differentiation (Simon et al., 2016). Evidence from various sources also supports that these are the only of our ten genes which are expressed in the brain (Supplementary Data 5). Our results support previous work that CORT + DFP treatment decreases myelinating oligodendrocytes in response to the treatment in the frontal cortex of both mouse (Ashbrook et al., 2018) and rat (Belgrad et al., 2019) models. However, Belgrad et al. demonstrated that different brain regions may respond differently (Belgrad et al., 2019), which suggests that different gene variants may underlie the response of these different regions to CORT + DFP treatment.
Furthermore, it appears that strains with higher expression of mitochondrial related genes at baseline (i.e. in control treatment) have a larger increase in the proportion of oligodendrocytes following CORT + DFP exposure. This may indicate a role of the oxidative stress in the action of the exposure. Resent work has shown increased mitochondrial DNA copy number variants in veterans with Gulf War illness, compared to the non-afflicted (Ashbrook et al., 2018; Chen et al., 2017). Although Chen et al. suggested this could be a symptom of GWI, our data may suggest that those with higher mitochondrial DNA copy numbers were actually at increased risk of developing GWI. Alternatively, it could be that the baseline high levels of mitochondrial gene expression in these strains is an adaptive response to reduced mitochondrial function, making these strains more vulnerable to mitochondrial copy number changes caused by the CORT + DFP treatment.
In conclusion, we have shown significant genetic effects in response to CORT + DFP treatment, which mirrors gene and protein expression changes seen in GWI sufferers. By using BXD RI strains, we were able to identify hub genes, and map loci influencing these outcomes. RI strains provide an excellent resource for future work to look at mechanisms of action, as well as providing a testbed for interventions which model the genetic variability seen in the human population.
Supplementary Material
Acknowledgement
This research was supported in part by CDMRP grant W81XWH-17-10472.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The high-throughput sequencing data from this study have been submitted to the GeneNetwork under the GN Accession IDs GN880, GN881, and GN882 for CORT + DFP, CTL, and DFP, respectively.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbi.2020.06.018.
References
- Abdullah L, Evans JE, Bishop A, Reed JM, Crynen G, Phillips J, Pelot R, Mullan MA, Ferro A, Mullan CM, 2012. Lipidomic profiling of phosphocholine containing brain lipids in mice with sensorimotor deficits and anxiety-like features after exposure to Gulf War agents. NeuroMol. Med 14, 349–361. [DOI] [PubMed] [Google Scholar]
- Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, Lutz CM, Valenzuela A, Bohl CJ, Ingels JF, 2019. The expanded BXD family of mice: a cohort for experimental systems genetics and precision medicine. bioRxiv, 672097. [Google Scholar]
- Ashbrook DG, Hing B, Michalovicz LT, Kelly KA, Miller JV, de Vega WC, Miller DB, G. Broderick, J.P. O’Callaghan, P.O. McGowan, 2018. Epigenetic impacts of stress priming of the neuroinflammatory response to sarin surrogate in mice: a model of Gulf War illness. J. Neuroinflamm 15, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgrad J, Dutta DJ, Bromley-Coolidge S, Kelly KA, Michalovicz LT, Sullivan KA, O’Callaghan JP, Fields RD, 2019. Oligodendrocyte involvement in Gulf War Illness. Glia 67, 2107–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello SM, Smith CL, Eppig JT, 2015. Allele, phenotype and disease data at Mouse Genome Informatics: improving access and analysis. Mamm. Genome 26, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc.: Ser. B (Methodol.) 57, 289–300. [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK, 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. [DOI] [PubMed] [Google Scholar]
- Binns JH, Barlow C, Bloom FE, Clauw DJ, Golomb BA, Graves JC, Hardie A, Knox ML, Meggs WJ, Nettleman MD, 2008. Gulf war illness and the health of gulf war veterans. Department Of Veterans Affairs Washington Dc Research Advisory Committee On; …. [Google Scholar]
- Blanchard M, Molina-Vicenty HD, Stein PK, Li X, Karlinsky J, Alpern R, Reda DJ, Toomey R, 2019. Medical correlates of chronic multisymptom illness in Gulf War Veterans. Am. J. Med 132, 510–518. [DOI] [PubMed] [Google Scholar]
- Broderick G, Kreitz A, Fuite J, Fletcher MA, Vernon SD, Klimas N, 2011. A pilot study of immune network remodeling under challenge in Gulf War Illness. Brain Behav. Immun 25, 302–313. [DOI] [PubMed] [Google Scholar]
- Carter GW, 2013. Inferring gene function and network organization in Drosophila signaling by combined analysis of pleiotropy and epistasis. G3: Genes Genomes, Genetics 3, 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Meyer JN, Hill HZ, Lange G, Condon MR, Klein JC, Ndirangu D, Falvo MJ, 2017. Role of mitochondrial DNA damage and dysfunction in veterans with Gulf. War Illness. PloS one 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RI, Rottkamp DM, Maric D, Barker JL, Hudson LD, 2003. A role for semaphorins and neuropilins in oligodendrocyte guidance. J. Neurochem 85, 1262–1278. [DOI] [PubMed] [Google Scholar]
- Consortium GO, 2015. Gene ontology consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW, 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci 9, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Carey V, Weiss S, 2019. deconvSeq: Deconvolution of Cell Mixture Distribution in Sequencing Data. Bioinformatics. [DOI] [PubMed]
- Edgar R, Domrachev M, Lash AE, 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Yoshikawa H, Fujimura H, Naba I, Sumi-Akamaru H, Takayasu S, Itabe H, Sakoda S, 2003. The role of CD36 in peripheral nerve remyelination after crush injury. Eur. J. Neurosci 17, 2659–2666. [DOI] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Park L, Iadecola C, 2014. Commentary on Myers et al.: growing role of the innate immunity receptor CD36 in central nervous system diseases. Experim. Neurol 261, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Faroldi G, Rönnberg E, Orro A, Calounova G, Guss B, Lundequist A, Pejler G, 2013. ADAMTS: novel proteases expressed by activated mast cells. Biol. Chem 394, 291–305. [DOI] [PubMed] [Google Scholar]
- Gavaghan H, 1994. NIH panel rejects Persian Gulf syndrome. Nature 369, 8. [DOI] [PubMed] [Google Scholar]
- Golomb BA, 2008. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc. Natl. Acad. Sci 105, 4295–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley CS, Knott SA, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69, 315. [DOI] [PubMed] [Google Scholar]
- Haley RW, Tuite JJ, 2013. Epidemiologic evidence of health effects from long-distance transit of chemical weapons fallout from bombing early in the 1991 Persian Gulf War. Neuroepidemiology 40, 178–189. [DOI] [PubMed] [Google Scholar]
- Research Advisory Committee (RAC) on Gulf War Veterans’ Illnesses (2008). Gulf War Illness and the Health of Gulf War Veterans: research Update and Recommendations, 2009–2013. U.S. Government Printing Office, Washington, DC. [Google Scholar]
- Johnson WE, Li C, Rabinovic A, 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jones BC, Miller DB, Lu L, Zhao W, Ashbrook DG, Xu F, Mulligan MK, Williams RW, Zhuang D, Torres-Rojas C, 2020. Modeling the genetic basis of individual differences in susceptibility to Gulf War Illness. Brain Sci. 10, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M, 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr KJ, 2015. Gulf War illness: an overview of events, most prevalent health outcomes, exposures, and clues as to pathogenesis. Rev. Environ. Health 30, 273–286. [DOI] [PubMed] [Google Scholar]
- Khaiboullina SF, DeMeirleir KL, Rawat S, Berk GS, Gaynor-Berk RS, Mijatovic T, Blatt N, Rizvanov AA, Young SG, Lombardi VC, 2015. Cytokine expression provides clues to the pathophysiology of Gulf War illness and myalgic encephalomyelitis. Cytokine 72, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B-B, Michalovicz LT, Calderazzo S, Kelly KA, Sullivan K, Killiany RJ, O’Callaghan JP, 2018. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War Illness. Brain Behav. Immun 67, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS, 2010. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U.S.A 107, 2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Faustino J, Woo MS, Derugin N, Vexler ZS, 2015. Lack of the scavenger receptor CD 36 alters microglial phenotypes after neonatal stroke. J. Neurochem 135, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi QG, Lin F, Liang YG, Sun LJ, Mu JS, Wang YG, Su HB, Xu B, Ji CC, Huang HH, Li K, Wang HF, 2014. Cytokine IL-6 is required in Citrobacter rodentium infection-induced intestinal Th17 responses and promotes IL-22 expression in inflammatory bowel disease. Mol. Med. Rep 9, 831–836. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W, 2013. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin E, Tsai S-J, Kuo P-H, Liu Y-L, Yang AC, Kao C-F, Yang C-H, 2017. The ADAMTS9 gene is associated with cognitive aging in the elderly in a Taiwanese population. PLoS ONE 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locker AR, Michalovicz LT, Kelly KA, Miller JV, Miller DB, O’Callaghan JP, 2017. Corticosterone primes the neuroinflammatory response to Gulf War Illness-relevant organophosphates independently of acetylcholinesterase inhibition. J. Neurochem 142, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Dupuis J, Sen Ś, Broman KW, 2006. Poor performance of bootstrap confidence intervals for the location of a quantitative trait locus. Genetics 174, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maule AL, Janulewicz PA, Sullivan KA, Krengel MH, Yee MK, McClean M, White RF, 2018. Meta-analysis of self-reported health symptoms in 1990–1991 Gulf War and Gulf War-era veterans. BMJ Open 8, e016086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JV, LeBouf RF, Kelly KA, Michalovicz LT, Ranpara A, Locker AR, Miller DB, O’Callaghan JP, 2018. The neuroinflammatory phenotype in a mouse model of Gulf War illness is unrelated to brain regional levels of acetylcholine as measured by quantitative HILIC-UPLC-MS/MS. Toxicol. Sci 165, 302–313. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Mozhui K, Prins P, Williams RW, 2017. GeneNetwork: a toolbox for systems genetics In: Systems Genetics. Springer, pp. 75–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine, 2016. Gulf War and Health: Volume 10: Update of Health Effects of Serving in the Gulf War, 2016. National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- O’Callaghan JP, Kelly KA, Locker AR, Miller DB, Lasley SM, 2015. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J. Neurochem 133, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan JP, Michalovicz LT, Kelly KA, 2016. Supporting a neuroimmune basis of Gulf war illness. EBioMedicine 13, 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CC, Dickson PE, Philip VM, Thomas M, Chesler EJ, 2017. Systems genetic analysis in GeneNetwork.org. Curr. Protocols Neurosci 79, 8.39. 31–38.39. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkitny L, Middleton S, Baker K, Younger J, 2015. Evidence for abnormal cytokine expression in Gulf War Illness: a preliminary analysis of daily immune monitoring data. BMC Immunol. 16, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce LM, Kurata WE, Matsumoto KW, Clark ME, Farmer DM, 2016. Long-term epigenetic alterations in a rat model of Gulf War Illness. Neurotoxicology 55, 20–32. [DOI] [PubMed] [Google Scholar]
- Poliak S, Matlis S, Ullmer C, Scherer SS, Peles E, 2002. Distinct claudins and associated PDZ proteins form different autotypic tight junctions in myelinating Schwann cells. J. Cell Biol 159, 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, 1995. Veterans worry that unexplained medical problems a legacy of service during Gulf. War. CMAJ: Canadian Med. Assoc. J 152, 944. [PMC free article] [PubMed] [Google Scholar]
- Roy S, Sleiman MB, Jha P, Williams EG, Ingels JF, Chapman CJ, McCarty MS, Hook M, Sun A, Zhao W, 2020. Gene-by-environmental modulation of longevity and weight gain in the murine BXD family. bioRxiv, 776559. [Google Scholar]
- Sakaue S, Okada Y, 2019. GREP: genome for REPositioning drugs. Bioinformatics. [DOI] [PMC free article] [PubMed]
- Schütze S, Wiegmann K, Machleidt T, Krönke M, 1995. TNF-induced activation of NF-κB. Immunobiology 193, 193–203. [DOI] [PubMed] [Google Scholar]
- Seth RK, Maqsood R, Mondal A, Bose D, Kimono D, Holland LA, Janulewicz Lloyd P, Klimas N, Horner RD, Sullivan K, Lim ES, Chatterjee S, 2019. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon K, Hennen S, Merten N, Blättermann S, Gillard M, Kostenis E, Gomeza J, 2016. The orphan G protein-coupled receptor GPR17 negatively regulates oligodendrocyte differentiation via Gαi/o and its downstream effector molecules. J. Biol. Chem 291, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RP, Hasan S, Sharma S, Nagra S, Yamaguchi DT, Wong DT, Hahn BH, Hossain A, 2014. Th17 cells in inflammation and autoimmunity. Autoimmun Rev 13, 1174–1181. [DOI] [PubMed] [Google Scholar]
- Sloan Z, Arends D, Broman KW, Centeno A, Furlotte N, Nijveen H, Yan L, Zhou X, Williams RW, Prins P, 2016. GeneNetwork: framework for web-based genetics. J. Open Source Software 1. [Google Scholar]
- Spassky N, De Castro F, Le Bras B, Heydon K, Quéraud-LeSaux F, Bloch-Gallego E, Chédotal A, Zalc B, Thomas J-L, 2002. Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J. Neurosci 22, 5992–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele L, Lockridge O, Gerkovich MM, Cook MR, Sastre A, 2015. Butyrylcholinesterase genotype and enzyme activity in relation to Gulf War illness: preliminary evidence of gene-exposure interaction from a case–control study of 1991 Gulf War veterans. Environ. Health 14, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele L, Sastre A, Gerkovich MM, Cook MR, 2011. Complex factors in the etiology of Gulf War illness: wartime exposures and risk factors in veteran subgroups. Environ. Health Perspect 120, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steup A, Lohrum M, Hamscho N, Savaskan NE, Ninnemann O, Nitsch R, Fujisawa H, Püschel AW, Skutella T, 2000. Sema3C and netrin-1 differentially affect axon growth in the hippocampal formation. Mol. Cell. Neurosci 15, 141–155. [DOI] [PubMed] [Google Scholar]
- Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ, 2008. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 29, 1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K, Krengel M, Bradford W, Stone C, Thompson TA, Heeren T, White RF, 2018. Neuropsychological functioning in military pesticide applicators from the Gulf War: Effects on information processing speed, attention and visual memory. Neurotoxicol. Teratol 65, 1–13. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, 2018. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC, 2013. R: A language and environment for statistical computing.
- Tillman GD, Spence JS, Briggs RW, Haley RW, Hart Jr. J, Kraut MA, 2019. Gulf War illness associated with abnormal auditory P1 event-related potential: evidence of impaired cholinergic processing replicated in a national sample. Psychiatry Res.: Neuroimaging 283, 7–15. [DOI] [PubMed] [Google Scholar]
- Tuite JJ, Haley RW, 2013. Meteorological and intelligence evidence of long-distance transit of chemical weapons fallout from bombing early in the 1991 Persian Gulf War. Neuroepidemiology 40, 160–177. [DOI] [PubMed] [Google Scholar]
- Uysal S, Ünal ZN, Erdoğan S, Akyol S, Ramazan Yiğitoğlu M, Hirohata S, Işık B, Demircan K, 2013. Augmentation of ADAMTS9 gene expression by IL-1β is reversed by NFκB and MAPK inhibitors, but not PI3 kinase inhibitors. Cell Biochem. Funct 31, 539–544. [DOI] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, Zhang B, 2017. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pandey AK, Mulligan MK, Williams EG, Mozhui K, Li Z, Jovaisaite V, Quarles LD, Xiao Z, Huang J, 2016. Joint mouse–human phenome-wide association to test gene function and disease risk. Nat. Commun 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatz K, Voll RE, 2012. Pathogenesis of autoimmunity in common variable immunodeficiency. Front. Immunol 3, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RF, Steele L, O’Callaghan JP, Sullivan K, Binns JH, Golomb BA, Bloom FE, Bunker JA, Crawford F, Graves JC, 2016. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex 74, 449–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, 2016. ggplot2: Elegant Graphics for Data Analysis. Springer. [Google Scholar]
- Winkenwerder W, 2003. Environmental exposure report: pesticides final report. U.S. Department of Defense, Office of the Special Assistant to the Undersecretary of Defense (Personnel and Readiness) for Gulf War Illnesses Medical Readiness and Military Deployments, Washington, D.C. [Google Scholar]
- Yaykasli KO, Oohashi T, Hirohata S, Hatipoglu OF, Inagawa K, Demircan K, Ninomiya Y, 2009. ADAMTS9 activation by interleukin 1β via NFATc1 in OUMS-27 chondrosarcoma cells and in human chondrocytes. Mol. Cell. Biochem 323, 69–79. [DOI] [PubMed] [Google Scholar]
- Zakirova Z, Crynen G, Hassan S, Abdullah L, Horne L, Mathura V, Crawford F, Ait-Ghezala G, 2016. A chronic longitudinal characterization of neurobehavioral and neuropathological cognitive impairment in a mouse model of Gulf War agent exposure. Front. Integr. Neurosci 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Zhong P, Shin SM, Metallo J, Danielson E, Olsen CM, Liu Q-S, Lee SH, 2015. S-SCAM, a rare copy number variation gene, induces schizophrenia-related endophenotypes in transgenic mouse model. J. Neurosci 35, 1892–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M, 2012. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.