

Abstract
Aims:
Lewy body diseases (LBD) are characterized by alpha-synuclein (SYN) pathology, but co-morbid Alzheimer’s disease (AD) pathology is common and the relationship between these pathologies in microanatomic hippocampal subfields is understudied. Here, we use digital histological methods to test the association between hippocampal SYN pathology and the distribution of tau and amyloid-beta (Aβ) pathology in LBD and contrast with AD subjects. We also correlate pathologic burden with antemortem episodic memory testing.
Methods:
Hippocampal sections from 49 autopsy-confirmed LBD cases, 30 with no/low AD co-pathology (LBD-AD) and 19 with moderate/severe AD co-pathology (LBD+AD), and 30 AD patients were stained for SYN, tau, and Aβ. Sections underwent digital histological analysis of subfield pathological burden which was correlated with antemortem memory testing.
Results:
LBD-AD and LBD+AD had similar severity and distribution of SYN pathology (p>0.05), CA2/3 being the most affected subfield (p<0.02). In LBD, SYN correlated with tau across subfields (R=0.49, p<0.001). Tau burden was higher in AD than LBD+AD (p<0.001), CA1/subiculum and entorhinal cortex (ERC) being most affected regions (p=0.04 to <0.01). However, tau pathology in LBD-AD was greatest in CA2/3, which was equivalent to LBD+AD. Aβ severity and distribution was similar between LBD+AD and AD. Total hippocampal tau and CA2/3 tau was inversely correlated with memory performance in LBD (R=−0.52, −0.69, p=0.04, 0.009).
Conclusions:
Our findings suggest that tau burden in hippocampal subfields may map closely with the distribution of SYN pathology in subfield CA2/3 in LBD diverging from traditional AD and contribute to episodic memory dysfunction in LBD.
Keywords: synuclein, tau, neuropathology, Parkinson’s Disease, dementia with Lewy Bodies, Alzheimer’s disease, digital histology, hippocampus
Introduction
Lewy body diseases (LBD: Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies) are all pathologically defined by the presence of neuronal alpha-synuclein (SYN) Lewy body (LB) and neurite (LN) pathology [1]. Distinctive SYN-positive LNs in the cornu ammonis (CA) sectors 2 and 3 (CA2/3) have been characterized as a feature of DLB and advanced stages of PD [2–5]. Prior work using traditional histologic methods has implicated both SYN and co-occurring tau pathology in this subfield on cholinergic denervation and the biological basis of memory impairment in LBD [2, 6]. The hippocampus is a distinct neuroanatomical structure with intrinsic connectivity important for anterograde memory and is a likely epicentre for tau pathology in aging and Alzheimer’s disease (AD) dementia. Despite the well-established pathological findings of SYN CA2/3 pathology in LBD, there is limited data on the relationship between the distribution of SYN and tau pathology in the hippocampal formation in LBD compared to AD. Indeed, moderate to severe amounts of AD co-pathology are common, occurring in approximately 50% of autopsied LBD series [7–9]. AD co-pathology is associated with a worse prognosis [7–10] in LBD and preliminary clinicopathological and biomarker studies suggest specific associations with discrete clinical characteristics of cognitive impairment [11–17]. In PD and DLB, patients with AD co-pathology may perform worse on naming tasks and tests of episodic memory than those with a ‘pure’ synucleinopathy [16–18]. Therefore, AD tau co-pathology is likely a major influence on clinical heterogeneity within LBD (for a recent review see [15]). Recent in vitro work even suggests that certain strains of SYN pathology may be capable of cross-seeding tau pathology [19]. However, detailed studies of the neuropathological distribution of tau, Aβ, and SYN in the well-characterized microanatomic pathways of the hippocampal formation and their clinical correlates are few in LBD and AD, which is in part due to the subjective and semi-quantitative nature of traditional neuropathological methods and small number clinicopathological studies [20–22].
Our previous work using digital histopathologic methods in LBD found that the distribution of neocortical tau pathology associated with the severity of SYN pathology, deviated from the patterns observed in AD without SYN pathology and correlated with worse performance on domain-specific tasks [17] further suggesting a link between tau and SYN pathology. Here, we extend this work to a focused study examining tau, Aβ, and SYN pathology in subfields of the hippocampus, where the distribution of tau in AD has been shown to follow the performant pathway[23, 24], but is understudied in LBD[2]. Based on our previous work, we hypothesized that tau pathology in LBD may deviate from the patterns of pathology present across hippocampal subfields as seen in AD without SYN pathology and that this altered pattern of tau pathology will relate to antemortem memory function.
Materials and Methods
Participants
Patients and data were abstracted from the University of Pennsylvania Integrated Neurodegenerative Disease Database (INDD) [25]. Patients selected were clinically evaluated and followed at the University of Pennsylvania’s Parkinson’s disease and Movement Disorder Clinic, Frontotemporal Dementia Center, Alzheimer’s disease Core Center, or the Michael J. Crescenz VA Medical Center’s Parkinson’s disease Research, Education, and Clinical Center. Cases were selected from our previously reported Penn LBD autopsy cohort [17] of 55 patients who met clinical criteria for LBD (PDD or DLB) [26, 27], had autopsy-confirmed LB synucleinopathy (i.e. brainstem, limbic or neocortical stage)[21], had at least one documented cognitive test for study. Forty-nine of these LBD patients with dementia (32 PDD, 17 DLB) had sufficient hippocampal tissue available for this focused study of the hippocampal subfield distribution of pathology (Table 1). An age-matched disease reference cohort of 30 patients with typical amnestic AD and a primary neuropathological diagnosis of AD with an absence of neocortical or hippocampal SYN were selected to compare the distribution of tau pathology between LBD and AD. All autopsies were performed at the Penn Center for Neurodegenerative Disease Research (CNDR) using validated neuropathological criteria [20] and were analysed for the presence of co-pathologies as described [28] for neuropathological diagnosis as below. All procedures were performed with prior informed consent in accordance with Penn Institutional Review Board guidelines.
Table 1.
Patient Demographics
| LBD-AD N=30 |
LBD+AD N=19 |
AD N=30 |
|
|---|---|---|---|
| Clinical Characteristics | |||
| Clinical Phenotype | DLB: 6 PDD: 24 |
DLB: 11 PDD: 8† |
AD: 30 |
| Sex, count male (%) | 23 (77) | 14 (74) | 14 (47)* |
| Age at Deatha | 77.3 (8.7) | 78.6 (6.2) | 74.4 (10.7) |
| Disease Durationa | 15.8 (7.3) | 9.1 (6.6)† | 11.0 (4.8)*† |
| Neuropathology | |||
| Brain Weightb | 1325 (134) | N=18, 1331 (134) | 1081 (136)*†‡ |
| Post Mortem Intervalc | 13.7 (11.5) | N=18, 16.6 (8.6) | 10.7 (5.9) |
| McKeith Stagesd | |||
| Brainstem | 2 (7) | 0 (0) | 0 (0) |
| Limbic | 8 (27) | 2 (11) | 0 (0) |
| Neocortical | 20 (67) | 17 (89) | 0 (0) |
| AD Neuropathologic changee | |||
| No | 11 (37) | 0 (0) | 0 (0) |
| Low | 19 (63) | 0 (0) | 0 (0) |
| Intermediate | 0 (0) | 11 (58) | 3 (10) |
| High | 0 (0) | 8 (27) | 27 (30)*‡ |
| Other Co-Pathologiesf | |||
| PART | 3 (10) | 0 (0) | 0 (0) |
| TDP-43 | 7 (23) | 8 (42)† | 17(57)* |
| ARTAG | N=23, 8 (35) | N=17, 12 (71) | N=23, 17(74)* |
years (SD).
grams (SD).
hours (SD).
count (%) derived from McKeith et al. 2005.
count (%) from Montine et al. 2011.
count (%) Unless specified all values are derived from the whole cohort.
Abbreviations: PART: primary age related tauopathy, TDP-43: TAR DNA binding progrein-43. ARTAG: age related tau astrogliopathy
p<0.05 across groups
p<0.05 compared to LBD-AD
p<0.05 compared to LBD+AD
Neuropathologic Diagnosis
Fresh tissue samples obtained at autopsy were fixed over-night in 70% ethanol with 150 mM sodium chloride (referred to simply as EtOH) or 10% neutral-buffered formalin (NBF). Tissue samples were trimmed, placed into cassettes and processed through a series of alcohol, xylene and Surgipath EM-400 paraffin embedding media (Leica Microsystems; Buffalo Grove, IL) with incubations overnight in a Shandon tissue processor (Thermo Scientific; Waltham, MA). All incubations were done under vacuum and at ambient temperature except paraffin (62°C). Tissue was then embedded into paraffin blocks and 6-μm-thick sections were cut for analysis. All tissue was processed in an identical manner. Sections were stained using immunohistochemistry (IHC) with established antibodies as described [25]. Expert neuropathologists (EBL, JQT) applied current diagnostic criteria to assign Thal phases[29], Braak tau stages[23], CERAD neuritic plaque stages[30], α-synuclein LB stages[21], and the presence of TDP-43 and aging-related tau astrogliopathy (ARTAG) co-pathology[31]. Final neuropathology diagnosis for each case was rendered using standard semi-quantitative assessments for each pathology in each brain region [20]. Based on modern neuropathological criteria using NIA-AA criteria [20] we categorized LBD patients into those with an intermediate or high level of AD neuropathologic change (ADNPC) (LBD+AD: N=19) and no or low levels of ADNPC (LBD-AD: N=30) (Table 1).
Digital Pathology
In the Penn Digital Neuropathology lab, adjacent hippocampal sections were stained for tau (AT8, Thermo Fisher, 1:1000, no antigen retrieval), Aβ (NAB228, CNDR, 1:40,000, formic acid antigen retrieval), and SYN (SYN303, CNDR, 1:30,000 formic acid antigen retrieval) for analysis in this study. Briefly, 6μm sections were deparaffinized and rehydrated using graded ethanols, after which the appropriate antigen retrieval method described above was implemented. Slides then were placed in a 30% H202 in methanol solution for 30minutes, washed in 0.1M tris(hydroxymethyl)aminomethane (TRIS) at 7.6pH, blocked with 2% fetal bovine serum (FBS) in 0.1M TRIS, and then slides were incubated in primary antibody at 4°F overnight. The second day, after washing with 1% TRIS solution and blocking with 2% FBS, slides were incubated in biotinylated horse anti-mouse IgG or goat anti-rabbit IgG secondary antibody 1:1000 (Vector laboratories, Burlingame California) for 1 hour at room temperature, then for an additional 1 hour in avidin/biotin-based peroxidase (Vector laboratories, Burlingame California). The chromogen used was 3,3’-diaminobenzidine (DAB: Vector laboratories, Burlingame California) with haematoxylin as the counter stain. Slides were then dehydrated in graded ethanols, treated with xylene, and coverslipped. Digital images of histology slides at 20x magnification were obtained using a Lamina slide scanning system (Perkin Elmer, Waltham MA) with a pixel size of 6.5 μm2 (i.e. pixel resolution of 0.325 μm), camera resolution of 2560 × 2160, and a bit depth of 16 (.mrxs image file). Halo digital image software v1.90 (Indica Labs, Albuquerque NM) was used to calculate %area occupied (%AO) of reactivity for tau, Aβ and SYN pathology as previously published, which included inter-rater validation and comparisons to traditional ordinal scores[17, 32]. First, digital images of histology sections were manually segmented by trained investigators into hippocampal subfields based on cytoarchitectural features, as previously described [33] (Entorhinal cortex (ERC), CA2/3, CA1/Subiculum (CA1/SUB), and CA4/Dentate gyrus (CA4/DG)) (RI, CP, SM) (Figure 1). In brief, we used the cellular morphological features presented by Duvernoy in 2005 of the size, shape and density of neuronal cells to define subfields[34] as we have done previously for pathology-imaging correlation work (Adler et al 2015). Briefly, CA1 was defined by the presence of neuronal cells in the pyramidal layer that are ovoid and sparsely populated while the subiculum defining neurons are characterized by pyramidal cells similar in size and shape to CA1 with a more striated appearance; thus, CA1 and subiculum were combined into a single ROI. CA2 defining neuronal cells were defined by triangular shape and more densely packed compared to CA1 and CA3 defining neuronal cells are similar shape to CA2, but slightly more spaced out; thus, we combined CA2/3 subfields into a single ROI. The dentate gyrus DG is defined by small, round, very densely packed neurons with a granular layer in the hilum that comprised CA4 as per Amaral and Lavanex[35]. The entorhinal cortex was defined as the cortical region adjacent to the rhinal sulcus. We sought to avoid the white matter tracts from the stratum radiatum, stratum laconusem, stratum moleculare, and vestigial hippocampal sulcus and thus the lateral boundaries of our manual segmentation of hippocampal subfields was defined in part by grey-white border. We used conservative ROIs combining these subfield regions to ensure accurate identification using cellular features. We tested accuracy of this measure by performing blinded draw re-draw analysis performed by two separate investigators in a subset of regions (n=259 individual subfields from 37 images) and found high-level of reproducibility (R2=0.96, p<0.001).
Figure 1. Hippocampal Subfield Segmentation.
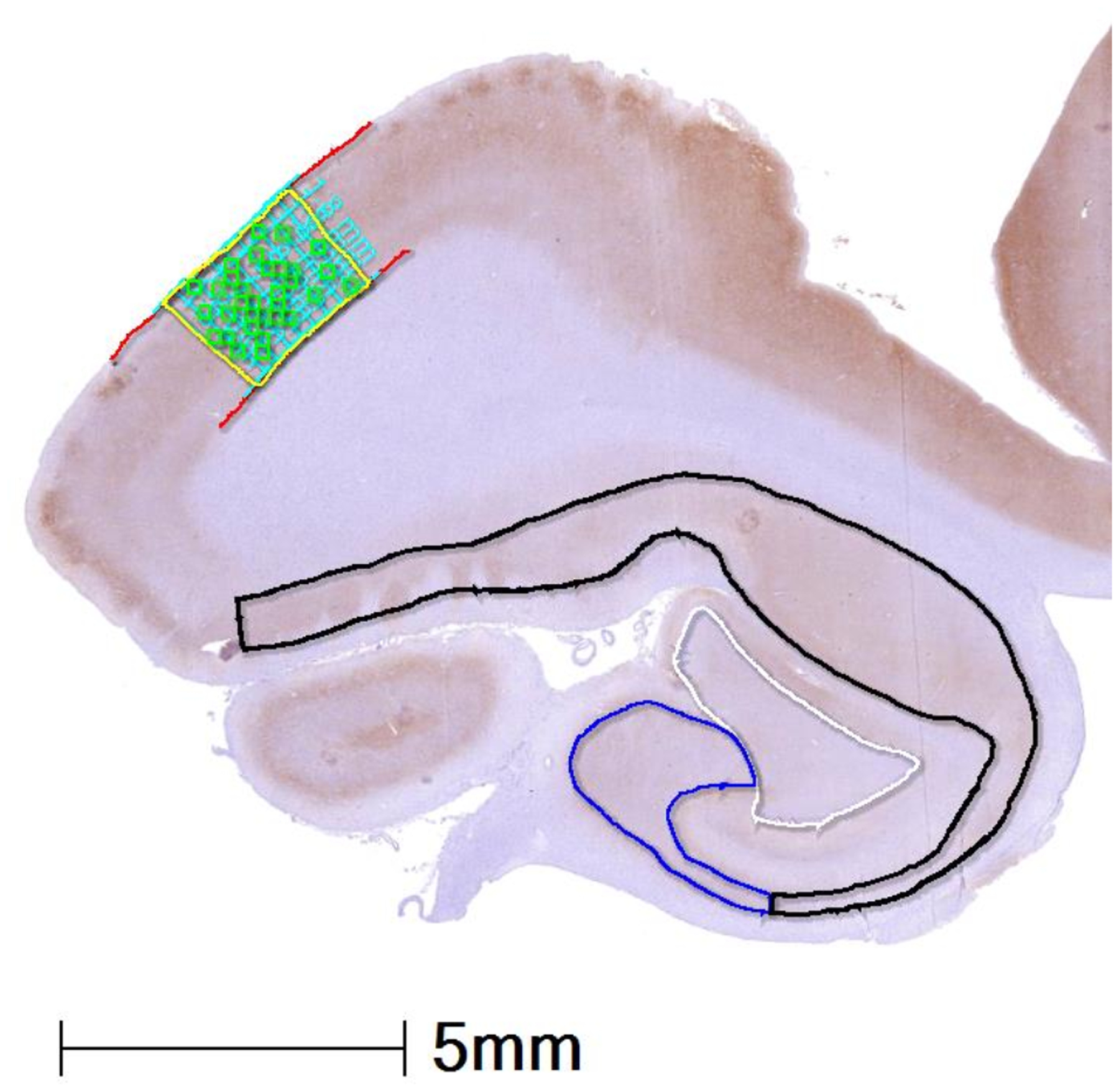
Representative image of hippocampal anatomy with example of manually segmented hippocampal subfields from an LBD+AD case stained for tau (AT8). CA4/DG: white, CA2/3: blue, CA1/SUB: black, ERC: yellow with grey matter outlined in red, orthogonal transecting lines in blue, and random tiles which were averaged for %AO measurement in green. Abbreviations: CA1: cornu ammonis field 1, SUB: subiculum, CA2/3 cornu ammonis field 2 and 3, CA4: cornu ammonis field 4, DG: dentate gyrus, ERC: entorhinal cortex.
Colour deconvolution intensity thresholds were optimized for each stain, by averaging values of red-blue-green colour vectors and minimal optical density values visually tuned from 5 representative slides per stain for tau, Aβ, and SYN (Table S1). Because SYN303 can detect normal monomeric SYN resulting in chromogen signal in the neuropil that can be difficult to separate from signal of Lewy pathology using colour deconvolution alone, we added an object detection algorithm, based on size and shape of positive signal in Lewy pathology, to further differentiate SYN pathology from background normal SYN reactivity as we have done previously (Figure 2 and Table S1)[17]. This additional step ensures specificity for Lewy bodies and large Lewy neurites. Percent area occupied values from the ERC were compared to traditional ordinal severity scores (0: none to 3: severe) in the same region assigned by blinded raters. Statistically significantly higher % area occupied values were associated with higher ordinal scores for each pathology type (tau: F(48)=115, Aβ: F(47)=123, SYN: F(48)=71.8, p<0.001 for each (Figure S1)). All LBD tissue was stained in a single staining batch for each stain, while AD cases were stained in a separate single staining batch for tau and Aβ. We used empirically defined digital image analysis algorithms for each staining run (Table S1) to mitigate batch effects as we have validated previously[36]. Appropriate tau, Aβ, and SYN algorithms were applied to CA1/SUB, CA2/3, CA4/DG subfields. In the case of the ERC which has laminar organization, we utilized an additional step previously validated and published, whereby we used a vertical transect method [37] to sample representative grey matter and used a random sampling derived from applying 175 μm2 tiles with a subsequent random 70% dropout to reduce sampling bias [17, 36, 38, 39]
Figure 2. Digital Histology Detection Overlay.
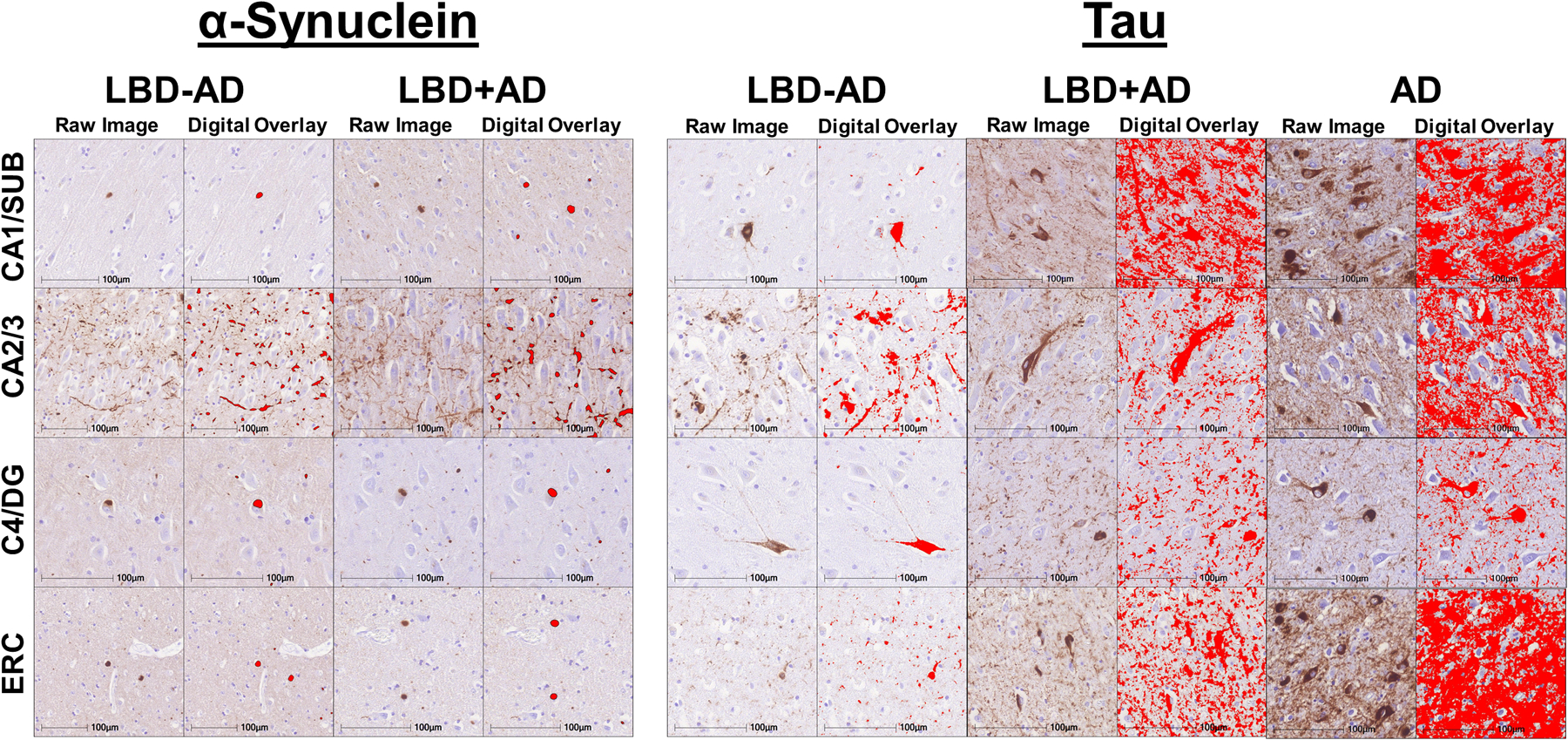
Representative photomicrographs in different hippocampal subfields in LBD-AD, LBD+AD, and AD cases stained for SYN (SYN303) and tau (AT8) The first column for each group shows raw images and the second column for each group depicts digital detection of pathology (%AO- red overlay).
Neuropsychological Testing
Neuropsychological testing was obtained at the first available research visit after the diagnosis of dementia was applied as defined by the diagnostic impression of the clinician from the clinical record. All patients had Mini-mental status exam testing (MMSE) [40] and a subset to patients (n=16. LBD-AD=12, LBD+AD=4) had detailed memory testing using the Hopkins Verbal Learning Test (HVLT) delayed recall scores (scores: 0–12) [41]. Neuropsychological testing was administered to participants by trained research personnel as described[42]. The median time between testing and death was 2.6 years with an interquartile range of 1.6–4.2 years.
Statistical Analysis
Differences in demographics were compared using fisher exact t-test, ANOVA, or independent sample t-test as appropriate. As %AO data were not normally distributed, a square-root transformation was used for all analyses. Between-group comparisons of pathological tau, Aβ, and SYN %AO in each subfield were performed using independent sample t-test. To test the regional distribution of tau, Aβ, and SYN pathology in specific hippocampal subfields within each group (i.e. LBD-AD, LBD+AD, and AD) we used linear mixed effects models, which can control for repeated measures of pathology within individual cases[43]. For the linear mixed effects models, we used pathology %AO measurement as the dependent variable and region as a fixed factor, with CA2/3 as reference region and controlling for sex, age at death, and disease duration with a random intercept for each case. Separate models were run for LBD-AD, LBD+AD, and AD. To directly compare the distribution of tau in hippocampal subfields of interest between groups, we also calculated the ratio of tau %AO in CA2/3 to CA1/Subiculum and compared between groups using independent sample t-tests. To test the association between %AO digital measures of tau and SYN in LBD, we used partial correlation with tau %AO as the dependent variable controlling for age at death, disease duration and sex in each hippocampal subfield. Finally, to test clinicopathological correlations of tau, Aβ, and SYN, neuropsychological performance and pathological burden (average %AO across subfields) were assessed using partial correlation controlling for the interval between testing and death, with exploratory models examining total tau and SYN burden as well as CA1/SUB and CA2/3 subfield burdens. All tests were two-tailed with a statistical threshold of 0.05 and performed using STATA v15.1 (College Station, TX).
Results
Demographics
Demographic information is detailed in Table 1. As expected, there were more patients with a DLB phenotype in the LBD+AD group as DLB is well described to have high rates of AD co-pathology[7, 44]. There were more female subjects in the AD group, which also had a lower brain weight than the LBD+AD group. Age at death was similar between groups.
Synuclein Hippocampal Subfield Burden in LBD
SYN pathology severity in each hippocampal subfield was similar in LBD+AD and LBD-AD (p>0.05 for each region) (Figures 2 and 3). We did not observe a significant difference in hippocampal subfield SYN burden between PDD and DLB clinical groups (p>0.05 for each region). As expected, the subfield with greatest SYN pathology in both groups was CA2/3 with significantly higher SYN pathology than all other regions in LBD-AD (p<0.001 for each) and higher than CA1/SUB and CA4/DG in LBD+AD (p=0.02 and <0.001 respectively) (Figure 2 and 3, and Table S2)
Figure 3. Hippocampal Subfield Synuclein Burden in LBD-AD and LBD+AD.
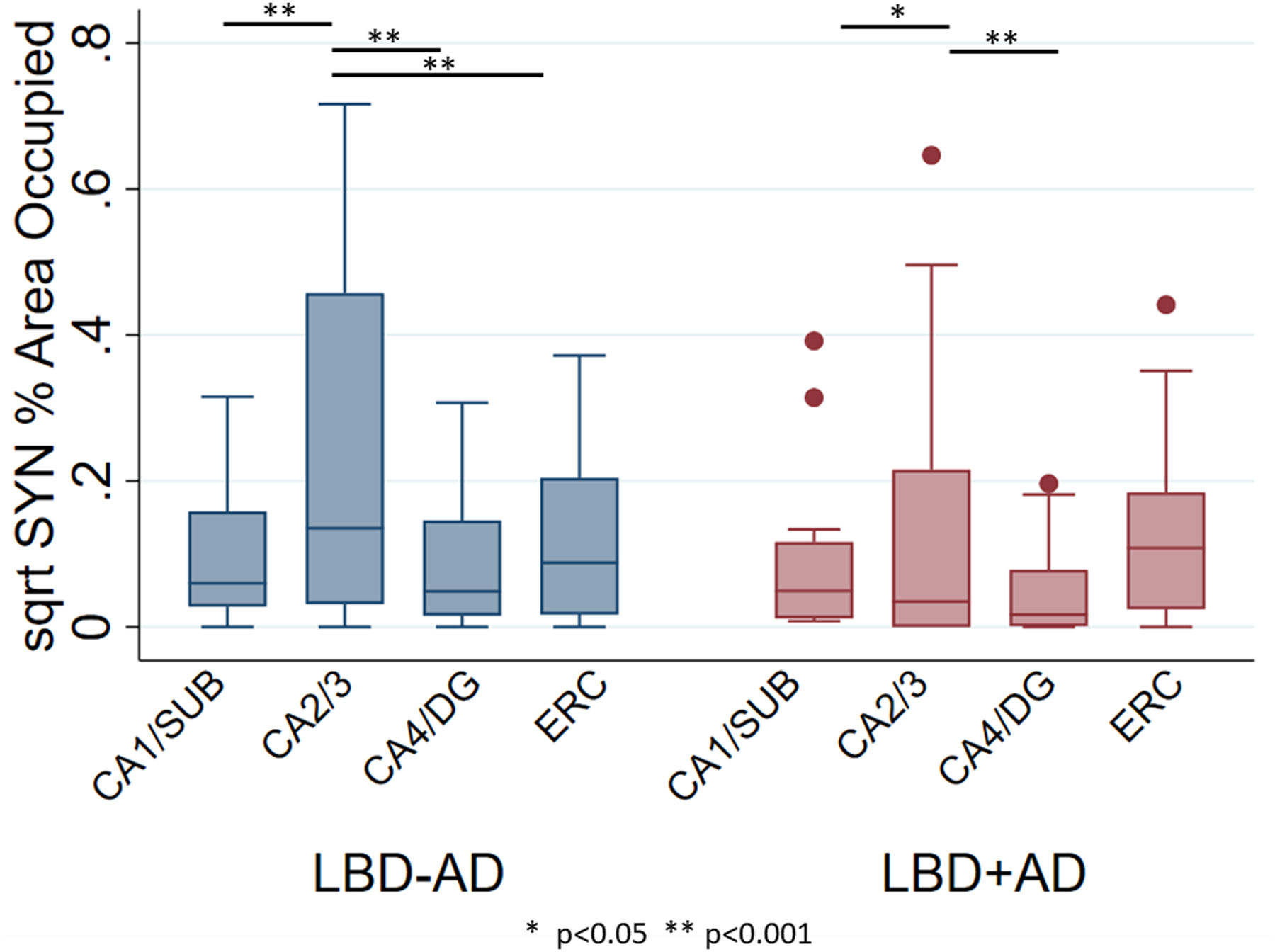
Box-plots depict median, interquartile range and range of square root of %AO of SYN pathology in hippocampal subfields of LBD-AD (blue) and LBD+AD (red) cases. Abbreviations: CA1/SUB: average cornu ammonis field 1 and subiculum, CA2/3 average cornu ammonis field 2 and 3, CA4/DG: average cornu ammonis field 4 and dentate gyrus, ERC: entorhinal cortex. Similar patterns of SYN deposition are observed across groups. * p<0.05, ** p<0.001 from linear mixed effects models comparing region.
Tau and Aβ Hippocampal Subfield Burden in LBD and AD
We compared the regional patterns of tau %AO in LBD groups and to a reference cohort of age-matched AD without SYN pathology. Tau %AO was higher in LBD+AD than LBD-AD in CA1/SUB and ERC (t(40, 47)=3.1–5.5, p=0.003, <0.001 respectively). In LBD-AD, CA2/3 had the highest tau burden, with significant elevations over CA4/DG and ERC (p<0.001 and p=0.02 respectively). CA2/3 tau burden was similar between LBD-AD and LBD+AD (p=0.6). In both LBD+AD and AD, CA1/SUB and ERC were higher than CA2/3 (LBD+AD and AD p<0.001 for both) and CA4/DG was lower (LBD+AD and AD P<0.001 for both) suggesting the pattern of early tau pathology in LBD (LBD-AD) was divergent from LBD+AD and AD (Figure 4, Table S3). To test the patterns of subfield tau pathology directly between groups, we calculated a ratio of the square root of CA2/3 to CA1/SUB tau %AO and found LBD-AD had a higher ratio of CA2/3 to CA1/SUB (1.49±0.92) tau burden than both LBD+AD (0.68±0.32, t(34)=3.3 p=0.003) and AD (0.74±0.17, t(39)=3.6 p=0.001). In contrast, LBD+AD and AD had similar CA2/3 to CA1/SUB ratios (p=0.4). As Braak tau stage increases in LBD, the CA2/3 to CA1/Sub ratios assume more AD like values (i.e. lower ratio values across increasing Braak stages) (F(3,33)=5.9 p=0.006) (Figure 5).
Figure 4. Hippocampal Subfield Tau and Aβ Burden in LBD-AD, LBD+AD, and AD.
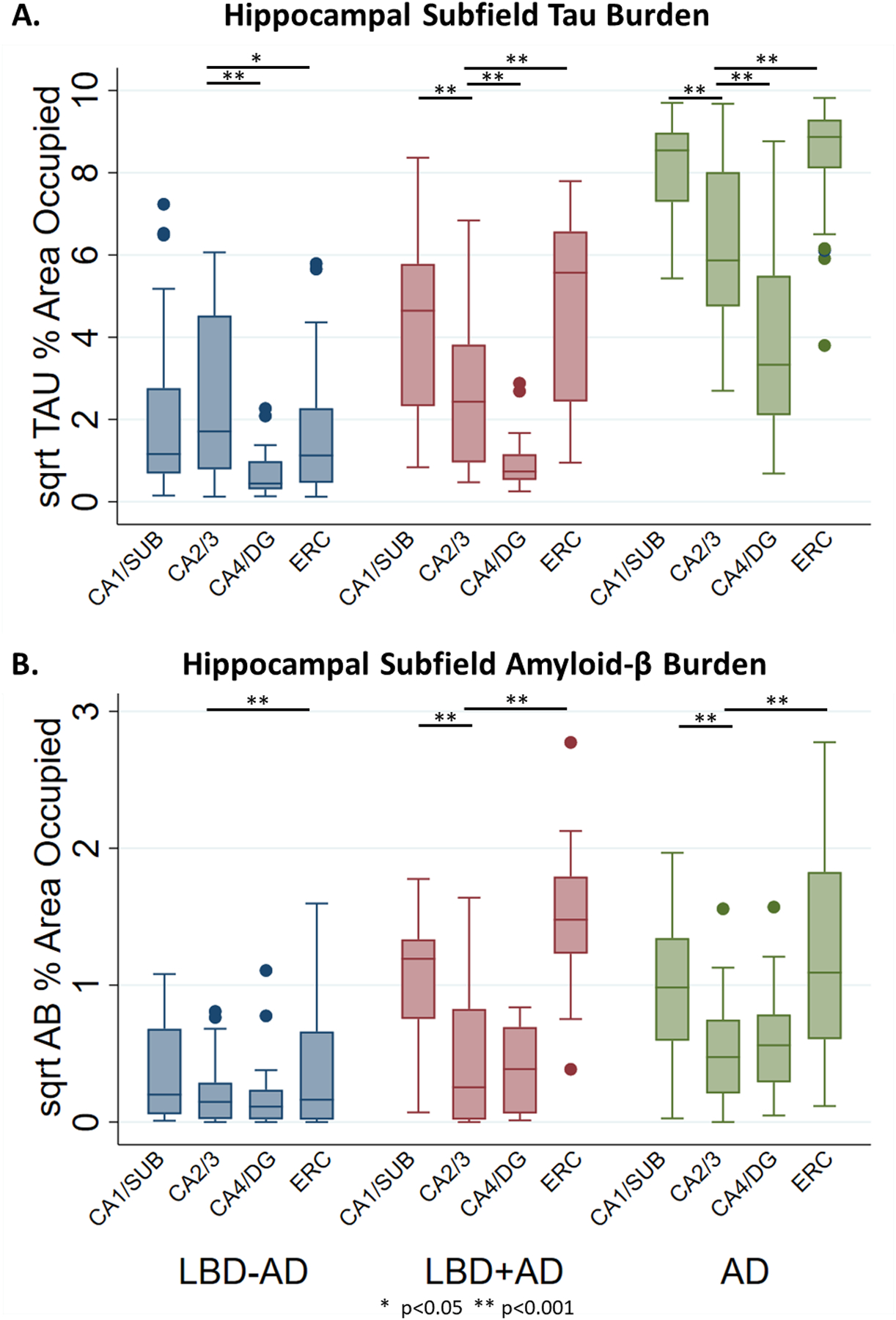
Box-plots depict median, interquartile range and range of square root of %AO of pathology in hippocampal subfields of LBD-AD (blue), LBD+AD (red), and AD (green) cases for A) Tau %AO in LBD-AD, LBD+AD, and AD and B) Aβ %AO in LBD-AD, LBD+AD, and AD. Similar patterns of tau deposition are observed between LBD+AD and AD although AD consistently has higher degree of tau deposition. LBD-AD shows a pattern of tau deposition similar to SYN deposition. LBD+AD and AD have similar distribution and severity of Aβ pathology
Abbreviations: CA1/SUB: average cornu ammonis field 1 and subiculum, CA2/3 average cornu ammonis field 2 and 3, CA4/DG: average cornu ammonis field 4 and dentate gyrus, ERC: entorhinal cortex. * p<0.05, ** p<0.001 from linear mixed effects models.
Figure 5. Ratio of CA2/3 Tau to CA1/SUB Tau.
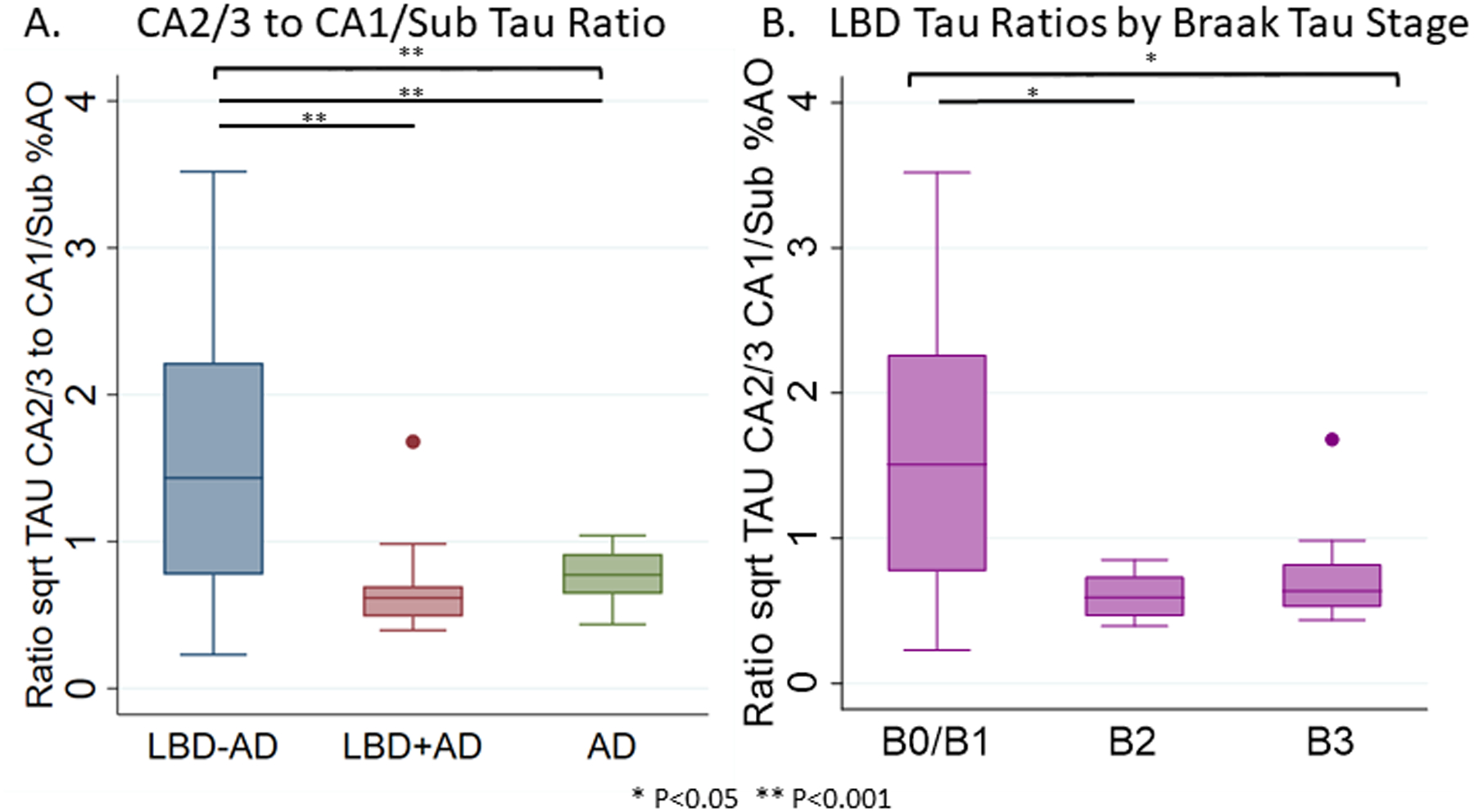
Box-plots depict median, interquartile range and range of the ratio of the square root of tau %AO in CA2/3 to CA1/SUB in A) LBD-AD (blue), LBD+AD (red), and AD (green) and B) in the entirety of the LBD cohort by Braak tau stage. Brackets indicate ANOVA differences across all groups, while lines indicate significant differences between groups. * p<0.05 ** p<0.001.
In contrast Aβ %AO was similar in all regions between LBD+AD and AD (t(38–42)=0.2–1.8, p=0.07–0.8) with ERC and CA1/SUB Aβ having greater Aβ %AO than CA2/3 (p<0.001 for all) (Figure 4, Table S4). As expected Aβ %AO was higher in LBD+AD than LBD-AD in all regions (t(41–46)=2.1–7.5, p=0.04-<0.001).
Next we tested the association between tau and SYN across hippocampal subfields in the total LBD cohort and found that average total SYN %AO correlated with average total tau %AO when controlling for age at death, sex, and disease duration (R=0.49, p<0.001). This was the case in both the LBD+AD and LBD-AD groups individually (LBD-AD: R=0.51, p=0.007. LBD+AD: R=0.57, p=0.02). In individual subfields across the entire LBD cohort, there were significant associations between SYN and tau with the strongest association found in CA2/3 (R=0.67, p<0.001), then CA1/SUB (R=0.62, p<0.001) and ERC (R=0.31, p=0.03). The scant tau and SYN in CA4/DG appeared correlated graphically but did not reach statistical significance (R=0.28, p=0.06) (Figure 6).
Figure 6. Regional Correlation between SYN and tau Burdens in LBD.
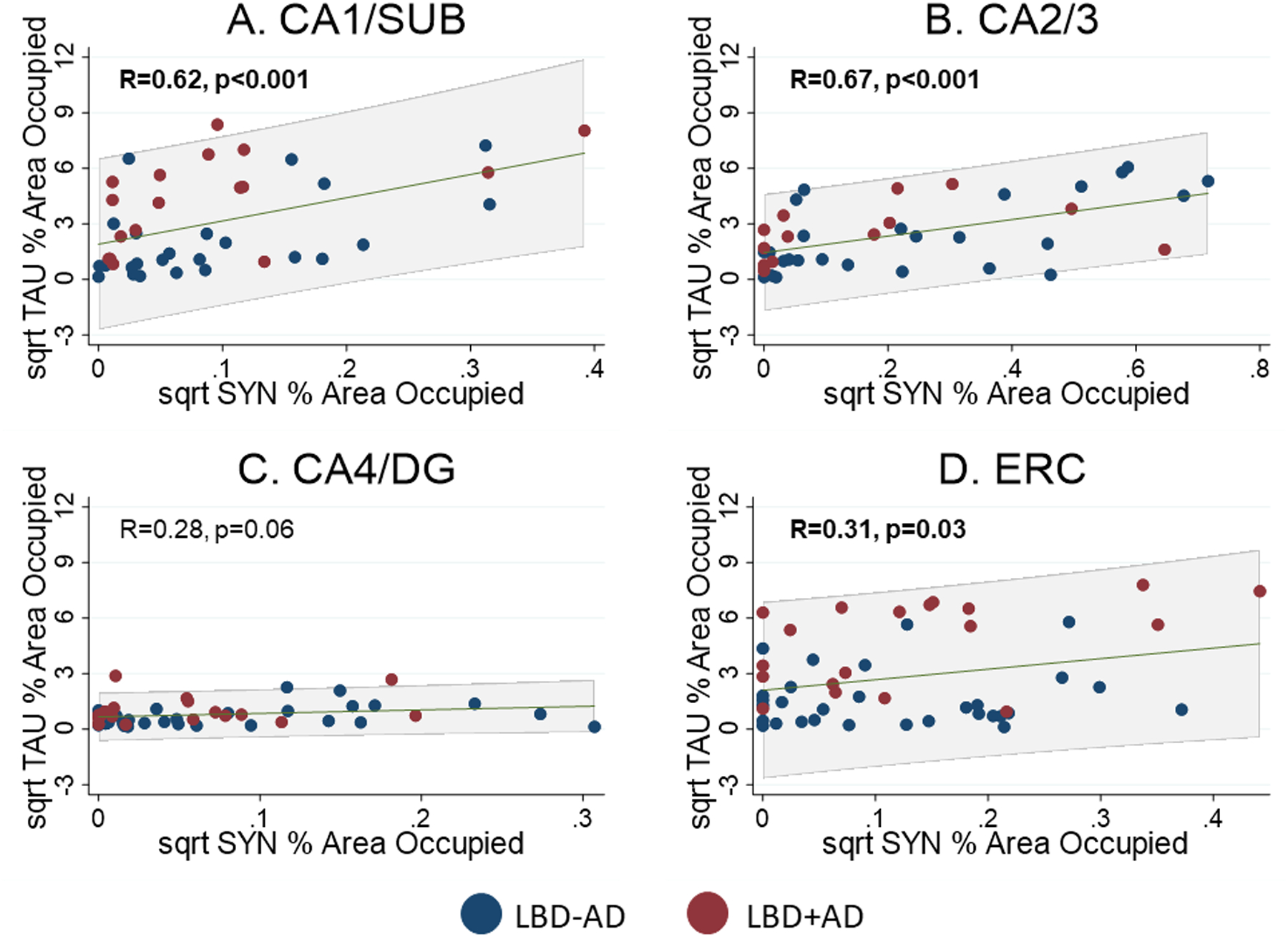
Scatterplots showing relationship between square root SYN %AO and tau %AO in A) CA1/SUB, B) CA2/3, C) CA4/DG, and D) ERC. Blue points are from LBD-AD cases and Red points indicated LBD+AD cases. Linear prediction is shown in green with 95% confidence intervals shown in grey shaded region. Significant correlations were observed in CA1/SUB and CA2/3 and ERC. Abbreviations: CA1/SUB: average cornu ammonis field 1 and subiculum, CA2/3 average cornu ammonis field 2 and 3, CA4/DG: average cornu ammonis field 4 and dentate gyrus, ERC: entorhinal cortex.
Exploratory Clinicopathological Correlation
LBD+AD patients performed slightly worse than LBD-AD on both MMSE (22.4±4.2 versus 21.5±7.6) and HVLT delayed recall testing (2.4±2.3 versus 1.3±1.9) but did not reach statistical significance (p=0.6 and 0.4 respectively). We did not observe a significant association for tau %AO with MMSE scores (p>0.1 for each) but in the subset of patients with detailed memory testing data available, after controlling for interval between testing and death, we observed a significant negative correlation between total hippocampal tau %AO and performance on HVLT delayed recall testing (R=−0.52, p=0.04) but no such correlations with total SYN %AO or Aβ%AO (R=−0.29,−0.04, p=0.30, 0.87 respectively). In an exploratory subfield analysis restricted to CA2/3 and CA1/SUB, HVLT delayed recall scores were associated with tau %AO in CA2/3 (R=−0.69, p=0.009) but not SYN or Aβ %AO (R=−0.31, 0.04, p=0.28, 0.89 respectively) (Figure 7). We did not observe any significant associations between tau, Aβ, or SYN %AO in CA1/SUB and memory performance (p>0.15 for both).
Figure 7. HVLT Delayed Recall Scores and Tau, Aβ, and SYN %AO Burden.
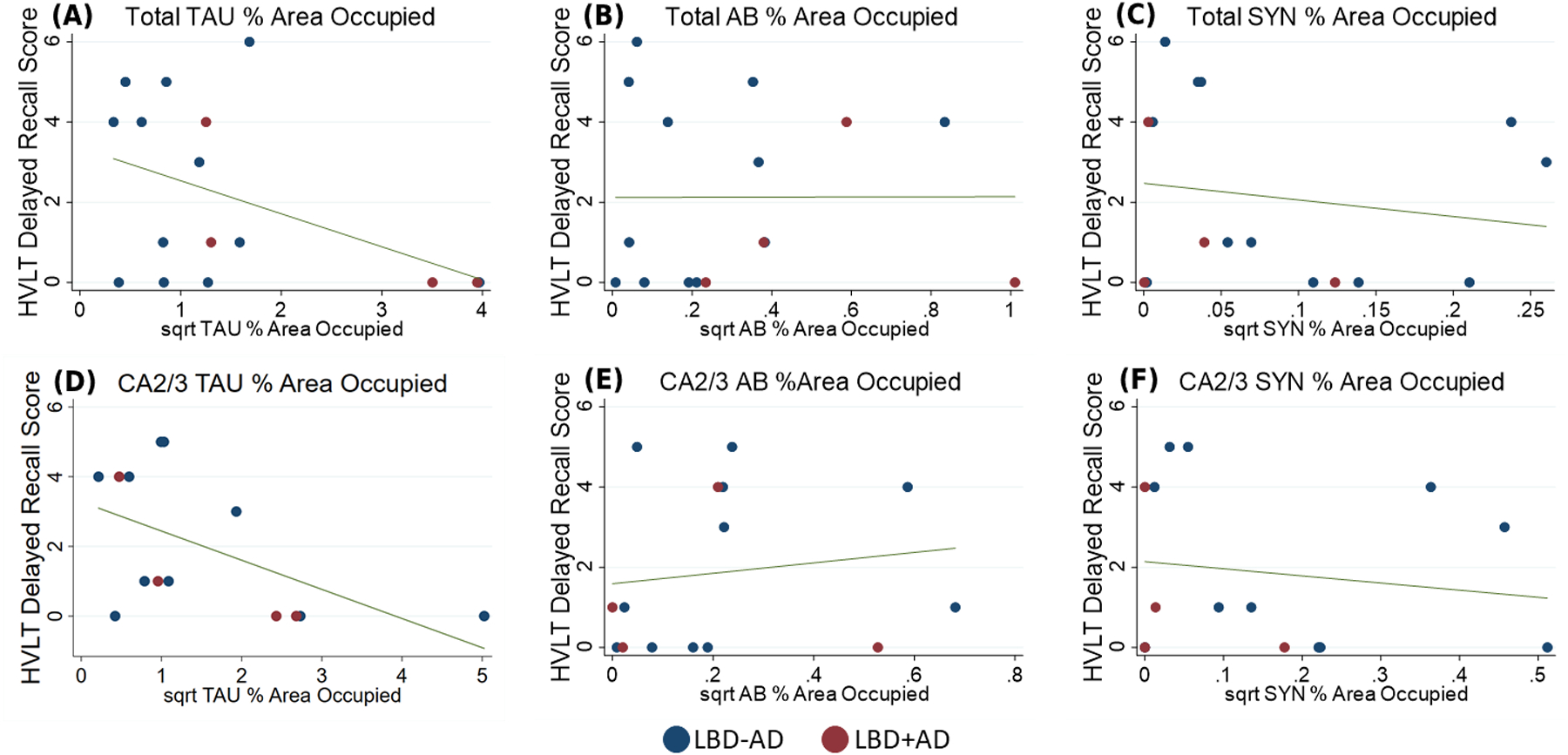
Hopkins Verbal Learning Task Delayed Recall Score plotted against a) square root total tau %AO and b) square root total Aβ %AO c) square root total SYN %AO d) square root CA2/3 tau %AO and e) square root CA2/3 Aβ %AO f) square root CA2/3 SYN %AO Blue: LBD-AD, Red: LBD+AD, Line: predicted linear fit.
Discussion
The hippocampal formation is a well-studied anatomical structure in regards to connectivity in non-human primate and the hypothesized propagation and spread of tau and Aβ amyloid pathology in AD [23, 29, 45–49]. In LBD, the presence of prominent SYN pathology CA2/3 and the ERC suggests a possible distinct pathway of propagation of LBD pathology between these regions that has been demonstrated in mice and non-human primates where deep layers of the ERC have reciprocal connections with this region[50–53]. Less is known regarding the patterns of SYN and AD co-pathology in LBD and how they may interact. Here we used digital histologic methods to perform a detailed examination of hippocampal subfield pathology to determine the distribution of association of tau, Aβ, and SYN pathology in LBD-AD, LBD+AD, and AD and the ramifications of these pathologies on antemortem memory testing in LBD. We find that overall hippocampal SYN deposition severity and distribution is similar between LBD-AD and LBD+AD in this dementia cohort with prominent CA2/3 subfield SYN pathology (Figure 3). We also find that SYN pathology correlated with tau pathology across subfields (Figure 6). Interestingly, LBD patients with lower levels of AD co-pathology (LBD-AD) appeared to have a pattern of tau pathology mirroring the distribution of SYN, with greatest tau levels seen in CA 2/3 which was equivalent to LBD+AD. However, hippocampal tau pathology was overall much more severe in AD than LBD+AD even when controlling for Braak tau stage but had a similar distribution across subfields. Lastly, tau pathology had a negative correlation with antemortem memory performance, suggesting tau pathology may influence clinical phenotype of dementia in LBD. These novel data on tau distribution in the LBD hippocampus reinforce previous experimental model data suggesting a synergistic effect of SYN and tau pathology in LBD and have important clinical implications.
Moderate to severe AD co-pathology [20], is found in approximately 10% of non-demented PD patients, about 35% of PDD patients, and 70% of DLB patients at autopsy[1, 8, 9, 54]. AD co-pathology exhibits a robust influence on clinical heterogeneity in LBD (for review see [15]), shortening time to dementia in PD [8, 14, 55] and overall survival in PD and DLB [7, 8]. PD patients with high degrees of AD co-pathology are more likely to have an akinetic-rigid motor phenotype [9, 13, 14] and DLB patients are less likely to have cognitive fluctuations or visual hallucinations[11, 12, 27]. In terms of memory impairment specifically, LBD patients with co-occurring AD pathology are more likely to exhibit episodic memory impairments or impairments in naming as opposed to the attentional and executive dysfunction that are hallmarks of Lewy pathology [16–18, 26].
Hippocampal subfields have distinct cytoarchitecture and are selectively vulnerable to different types of pathologies. Specifically, tau pathology in AD likely begins in the trans entorhinal cortex and spreads to other regions of the entorhinal cortex and CA1, whereas SYN pathology in PD preferentially affects CA2/3 initially [4, 24]. Aβ plaques first affect the hippocampal formation in the ERC and CA1/Sub, with later involvement of CA 2/3 and CA4/DG [29, 45]. Histopathological staging schemes are developed through comparison of the presence or absence of pathology in regions across serial autopsies and our digital data measuring pathology severity recapitulates these staging data with increasing %AO of tau and Aβ in AD and SYN in LBD from earlier to later stage regions (Figures 3 and 4). These data suggest that objective digital measures of disease severity may aid in histopathological staging efforts and our continuous measures of pathology facilitated statistical modelling of regional comparisons to account for demographics that otherwise would be challenging with standard ratings.
As expected, we observed that CA2/3 was the hippocampal subfield most affected by SYN pathology in LBD-AD and LBD+AD [2, 4, 56]. We did not observe regional differences of SYN inclusions between pathological (LBD-AD vs LBD+AD) or clinical subgroups (PDD vs DLB) in this LBD cohort of demented patients [17]. SYN pathology in PD and DLB is thought to ascend rostrally, affecting the hippocampus as part of the limbic stage of the disease [4, 27]. As limbic and neocortical stage Lewy pathology is felt to be sufficient to cause dementia, our cohort of demented patients may have already reached a ceiling effect of SYN pathology that may obscure differences between the subgroups that may have occurred earlier in their disease course [27]. Future work will examine more detailed sampling across the spectrum of non-demented PD and more advanced PDD and DLB studied here.
We find novel evidence across the LBD cohort that SYN and tau correlated with each other across subfields of the hippocampus and within both LBD-AD and LBD+AD. Interestingly, we found that the tau burden deviated from classic histopathological staging in the LBD-AD group, and that these patients had prominent CA2/3 tau pathology that was equivalent to the LBD+AD group, suggesting that likely early tau pathology in LBD-AD may share similar predilection to CA2/3 as SYN pathology, rather than rather than the ERC or CA1/SUB, areas linked to traditional Braak tau staging in AD. We found the relative amount of tau in the CA1/Subiculum increased with increasing Braak tau stage (Figure 5) and it is interesting to speculate that there may be overlapping progression of AD-related tau and a possible separate process of SYN-mediated tau propagation exemplified in LBD in CA 2/3. We previously found divergent neocortical patterns of tau in LBD+AD compared to AD where LBD+AD had relatively higher concentrations of tau in the temporal neocortex [17]. Here we see a more AD-like pattern of tau in the hippocampus in LBD+AD and a potentially divergent pattern in LBD-AD. These observations could be related to alterations in early tau deposition in CA2/3 that are later partially obscured when higher degrees of tau pathology occur in LBD where the tau pathology adopts a more AD-like pattern with greater CA1/Subiculum tau pathology that is equivalent to CA2/3 tau. While these results represent cross-sectional data, they provide novel indirect support for model data for an interaction of these proteins. Indeed, new in vitro studies suggest that SYN may exhibit strain like properties and be capable of cross-seeding tau[19, 57, 58] and neuropathological and in vivo PET imaging studies suggest that neocortical tau pathology in LBD may diverge from what is seen in AD [17, 59–61]. These studies would suggest the possibility that pathological SYN and tau interact in a clinically relevant way in LBD. Indeed, we found a correlation of tau in CA 2/3 with memory function during life, suggesting that even in mildly effected patients, hippocampal tau could contribute to clinical features. While tau pathology in LBD has identical biochemical structure of paired helical filaments in LBD[62], future work using strain-specific[58] and conformational epitopes of tau and SYN may elucidate specific AD and SYN related forms of tau pathology.
In the LBD+AD group, we observed that hippocampal tau pathology overall was lower than in AD cases, even when controlling for Braak tau stage, mirroring our previous findings in the neocortex[17] and another previous digital pathology study that found lower tau pathology in DLB hippocampus compared to AD[63]. Similar to previous work, we found equivalent levels of Aβ plaque between LBD+AD and AD groups. This aligns with PET Aβ amyloid data in LBD showing similar severity of Aβ amyloidosis to matched AD group [17, 63]. While we cannot determine the timing of SYN, tau and Aβ pathology in LBD, it appears that at end-stage disease Aβ amyloidosis in LBD+AD is equivalent to AD, despite lower tau levels which may be associated in part to SYN-related degeneration in CA2/3 in a manner distinct from AD. Moreover, tau pathology appears to correlate more closely with memory symptoms that Aβ which is similar to AD and suggests that despite lower levels than those seen in AD tau pathology, these levels in LBD are clinically relevant.
Finally, we find novel evidence that hippocampal tau in CA2/3 relates to antemortem episodic memory performance. While episodic memory impairments are a core feature of typical amnestic AD [64], classic cognitive deficits in LBD include impairments in attention, working memory, and visuospatial function[26, 65]. Episodic memory deficits are only present in a subset of LBD patients during life and tend to be less severe than those found in AD, and previous studies have associated worse memory impairments in LBD with the presence of co-occurring AD pathology[16, 18, 27]. One detailed study of hippocampal subfield pathology in DLB was performed by Adamowicz et al. [2] where they studied a large autopsy confirmed series with detailed antemortem cognitive testing to detail the distribution of synuclein pathology in hippocampal subfields and how this and AD co-pathology may influence neuropsychological testing. We find several similarities to their previous conclusions using digital methods and add additional new data focused on comparing pathologic distributions between pathology-driven subgroups of LBD compared to AD without SYN co-pathology. They found that CA1 synuclein burden inversely correlated with memory performance, but that tau pathology consistently exhibited a larger effect. In the exploratory analysis in our study, we did not observe that CA1 synuclein burden correlated with memory impairments, and while we also found that tau pathology was a more significant contributor to memory impairment, this association was limited largely to the CA2/3 region in univariate and multivariate models. The reason for the discrepancies could be due to methodological differences in tissue preparation, patient characteristics or sample size, but nonetheless both studies suggest tau and SYN pathologies are intimately involved in the clinical manifestations of episodic memory in LBD.
There are limitations to this study. We used cellular morphology to identify hippocampal subfields using an established atlas-based approach [66], nonetheless these data represent a relative limited view (i.e. 6 um section) within the anterior-posterior axis of the hippocampus which may have distinct patterns of tau in AD [67]. Given challenges with anatomical segmentation, and to help facilitate the development of future MRI imaging biomarkers which often combine adjacent subfields due to the resolution of antemortem imaging [68, 69], we took the conservative approach of combining anatomically similar or adjacent subfields for analysis (CA1 and SUB, CA2 and CA3, DG and CA4) which could obscure more finely grained associations. Moreover, we included draw/re-draw validation of our manual segmentation of subfields and found high reproducibility (R2>0.95). The digital histologic method employed here detects a regional %AO of a pathology that is stained by immunohistochemistry and does not distinguish between different cellular pathologies. Thus, it is not fully able to exclude the contributions of other less-common co-morbid phosphorylated-tau pathologies labelled by AT8, such as ARTAG. Furthermore, there are currently no ARTAG tau specific antibodies. However, the likely contribution of ARTAG to these results is minimal since we restricted our subfield analysis to grey matter areas, avoiding the white matter subpial and subependymal regions that are more likely to harbour ARTAG pathology in these cases. Additionally, when present in grey matter, ARTAG pathology tends to affect CA4 and DG in the hippocampus which were the subfields where we consistently observed the least amount of tau %AO [30, 70, 71]. We also only had a subset of patients with detailed memory testing which precluded more sophisticated statistical modelling to interrogate the relationship between pathologic burden and memory performance. As such, the results of the exploratory analysis here should be interpreted cautiously, but we also had a relatively short time interval from testing and autopsy (median 2 years) which strengthens our clinicopathological correlation. We used a cross-section of neuropsychological data from these patients that was obtained as close to the onset of dementia as possible. This approach was used to minimize the length of time between testing and death but also to avoid floor effects of neuropsychological assessments at end-stage dementia, but future studies using longitudinal assessments may better elucidate these relationships between pathology and cognition, especially at early stage disease.
Despite these limitations, these novel data suggest that clinically-relevant tau pathology may originate in the hippocampus in LBD in association with prominent SYN in CA2/3 subfield. Moreover, this work adds to the growing literature of the strong influence of AD tau co-pathology on clinical heterogeneity of cognitive symptoms in LBD [2, 11, 12, 15–18].
Supplementary Material
Acknowledgements:
This work was supported by grants from the American Academy of Neurology/American Brain Foundation/Parkinson’s Foundation (2059), the Alzheimer's Association Research Foundation (AARF-16-443681), TL1TR001880, NIA AG061277, AG043503, AG062429 and NINDS NS088341. Data were contributed to this study by the Center on Alpha-synuclein Strains in Alzheimer Disease & Related Dementias at the University of Pennsylvania Perelman School of Medicine (U19 AG062418, Trojanowski JQ-PI) and the former Morris K. Udall Center at the University of Pennsylvania Perelman School of Medicine (P50 NS053488, Trojanowski JQ-PI). The authors declare that they have no conflicts of interest.
Abbreviations:
- LBD
Lewy body diseases
- LBD-AD
Lewy body disease with no/low degree of AD co-pathology
- LBD+AD
Lewy body disease with moderate/high degree of AD co-pathology
- SYN
alpha-synuclein
- Aβ
amyloid-beta
- CA1
cornu ammonis field 1
- SUB
subiculum
- CA2/3
cornu ammonis field 2 and 3
- CA4
cornu ammonis field 4
- DG
dentate gyrus
- ERC
entorhinal cortex
Footnotes
Ethical approval: All research was performed in accordance with the 1964 Declaration of Helsinki with procedures approved by the University of Pennsylvania Institutional Review Board. Informed consent was obtained from all individual participants in this study which included consent to publish.
Data Availability: The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Irwin DJ, Lee VM-Y, Trojanowski JQ. Parkinson’s disease dementia: convergence of [alpha]-synuclein, tau and amyloid-[beta] pathologies. Nat Rev Neurosci 2013; 14: 626–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, Gage FH. Hippocampal alpha-Synuclein in Dementia with Lewy Bodies Contributes to Memory Impairment and Is Consistent with Spread of Pathology. J Neurosci 2017; 37: 1675–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickson DW, Ruan D, Crystal H, Mark M, Davies P, Kress Y, Yen S-H. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: light and electron microscopic immunocytochemistry of CA2–3 neurites specific to DLBD. Neurology 1991; 41: 1402- [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24: 197–211 [DOI] [PubMed] [Google Scholar]
- 5.Galvin JE, Uryu K, Lee VM-Y, Trojanowski JQ. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proceedings of the National Academy of Sciences 1999; 96: 13450–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu AKL, Chau TW, Lim EJ, Ahmed I, Chang RC-C, Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta neuropathologica communications 2019; 7: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wakisaka Y, Furuta A, Tanizaki Y, Kiyohara Y, Iida M, Iwaki T. Age-associated prevalence and risk factors of Lewy body pathology in a general population: the Hisayama study. Acta Neuropathol 2003; 106: 374–82 [DOI] [PubMed] [Google Scholar]
- 8.Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, Keene CD, Montine TJ, Zabetian CP, Trojanowski JQ. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 2017; 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jellinger K, Seppi K, Wenning G, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm 2002; 109: 329–39 [DOI] [PubMed] [Google Scholar]
- 10.Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol 2008; 115: 409–15 [DOI] [PubMed] [Google Scholar]
- 11.Merdes A, Hansen L, Jeste D, Galasko D, Hofstetter C, Ho G, Thal L, Corey-Bloom J. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 2003; 60: 1586–90 [DOI] [PubMed] [Google Scholar]
- 12.Del Ser T, Hachinski V, Merskey H, Munoz DG. Clinical and pathologic features of two groups of patients with dementia with Lewy bodies: effect of coexisting Alzheimer-type lesion load. Alzheimer Dis Assoc Disord 2001; 15: 31–44 [DOI] [PubMed] [Google Scholar]
- 13.Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson’s disease. Brain 2009; 132: 2947–57 [DOI] [PubMed] [Google Scholar]
- 14.Compta Y, Parkkinen L, Kempster P, Selikhova M, Lashley T, Holton JL, Lees AJ, Revesz T. The significance of α-synuclein, amyloid-β and tau pathologies in Parkinson’s disease progression and related dementia. Neurodegener Dis 2014; 13: 154–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coughlin DG, Hurtig HI, Irwin DJ. Pathological Influences on Clinical Heterogeneity in Lewy Body Diseases. Mov Disord 2019: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peavy GM, Edland SD, Toole BM, Hansen LA, Galasko DR, Mayo AM. Phenotypic differences based on staging of Alzheimer’s neuropathology in autopsy-confirmed dementia with Lewy bodies. Parkinsonism Relat Disord 2016; 31: 72–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coughlin D, Xie SX, Liang M, Williams A, Peterson C, Weintraub D, McMillan CT, Wolk DA, Akhtar RS, Hurtig HI, Branch Coslett H, Hamilton RH, Siderowf AD, Duda JE, Rascovsky K, Lee EB, Lee VM, Grossman M, Trojanowski JQ, Irwin DJ. Cognitive and Pathological Influences of Tau Pathology in Lewy Body Disorders. Ann Neurol 2019; 85: 259–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraybill ML, Larson EB, Tsuang D, Teri L, McCormick W, Bowen J, Kukull W, Leverenz J, Cherrier M. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 2005; 64: 2069–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM-Y. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003; 300: 636–40 [DOI] [PubMed] [Google Scholar]
- 20.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on A, Alzheimer’s A. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica 2012; 123: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65: 1863–72 [DOI] [PubMed] [Google Scholar]
- 22.Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, Lee EB, Arnold SE, Duda JE, Hurtig H, Lee VM, Adler CH, Beach TG, Trojanowski JQ. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol 2016; 131: 393–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112: 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59 [DOI] [PubMed] [Google Scholar]
- 25.Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, Xie SX, McBride J, Wood EM, Schuck T, Irwin DJ, Gross RG, Hurtig H, McCluskey L, Elman L, Karlawish J, Schellenberg G, Chen-Plotkin A, Wolk D, Grossman M, Arnold SE, Shaw LM, Lee VM, Trojanowski JQ. A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2014; 10: 477–84 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007; 22: 1689–707 [DOI] [PubMed] [Google Scholar]
- 27.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017; 89: 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Hurtig HI, Siderowf A, Grossman M, McMillan CT, Miller B, Duda JE, Irwin DJ, Wolk D, Elman L, McCluskey L, Chen-Plotkin A, Weintraub D, Arnold SE, Brettschneider J, Lee VM, Trojanowski JQ. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018; 141: 2181–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800 [DOI] [PubMed] [Google Scholar]
- 30.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41: 479–86 [DOI] [PubMed] [Google Scholar]
- 31.Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M, Wolk DA, Irwin DJ, Weintraub D, Kim CF, Schuck T. Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. Journal of Neuropatholgy & Experimental Neurology 2017; 76: 270–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irwin DJ, Byrne MD, McMillan CT, Cooper F, Arnold SE, Lee EB, Van Deerlin VM, Xie SX, Lee VM, Grossman M, Trojanowski JQ. Semi-Automated Digital Image Analysis of Pick’s Disease and TDP-43 Proteinopathy. J Histochem Cytochem 2016; 64: 54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adler DH, Liu AY, Pluta J, Kadivar S, Orozco S, Wang H, Gee JC, Avants BB, Yushkevich PA. Reconstruction of the human hippocampus in 3D from histology and high-resolution ex-vivo MRI. In 2012 9th IEEE International Symposium on Biomedical Imaging (ISBI): IEEE. 2012: 294–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duvernoy HM. The human hippocampus: functional anatomy, vascularization and serial sections with MRI: Springer Science & Business Media. 2005 [Google Scholar]
- 35.Amaral D, Lavenex P. Hippocampal neuroanatomy In The hippocampus book Ed. RM In Andersen P, Amaral D (Eds.) & Bliss T& O’Keefe J(Ed.): Oxford University Press; 2007: 37–114 [Google Scholar]
- 36.Giannini LA, Xie SX, McMillan CT, Liang M, Williams A, Jester C, Rascovsky K, Wolk DA, Ash S, Lee EB. Divergent patterns of TDP‐43 and tau pathologies in primary progressive aphasia. Ann Neurol 2019; 85: 630–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armstrong RA. Quantifying the pathology of neurodegenerative disorders: quantitative measurements, sampling strategies and data analysis. Histopathology 2003; 42: 521–9 [DOI] [PubMed] [Google Scholar]
- 38.McMillan CT, Irwin DJ, Nasrallah I, Phillips JS, Spindler M, Rascovsky K, Ternes K, Jester C, Wolk DA, Kwong LK, Lee VM, Lee EB, Trojanowski JQ, Grossman M. Multimodal evaluation demonstrates in vivo (18)F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016; 132: 935–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, Van Deerlin VM, Seeley WW, Miller BL, Lee EB, Lee VM, Grossman M, Trojanowski JQ. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 2016; 79: 272–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–98 [DOI] [PubMed] [Google Scholar]
- 41.Benedict RH, Schretlen D, Groninger L, Brandt J. Hopkins Verbal Learning Test–Revised: Normative data and analysis of inter-form and test-retest reliability. The Clinical Neuropsychologist 1998; 12: 43–55 [Google Scholar]
- 42.Watson GS, Cholerton BA, Gross RG, Weintraub D, Zabetian CP, Trojanowski JQ, Montine TJ, Siderowf A, Leverenz JB. Neuropsychologic assessment in collaborative Parkinson’s disease research: A proposal from the National Institute of Neurological Disorders and Stroke Morris K. Udall Centers of Excellence for Parkinson’s Disease Research at the University of Pennsylvania and the University of Washington. Alzheimer’s & Dementia 2013; 9: 609–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics 1982; 38: 963–74 [PubMed] [Google Scholar]
- 44.Marui W, Iseki E, Kato M, Akatsu H, Kosaka K. Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer’s disease. Acta Neuropathol 2004; 108: 121–8 [DOI] [PubMed] [Google Scholar]
- 45.Van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer’s disease. In Prog Brain Res: Elsevier. 1990: 445–57 [DOI] [PubMed] [Google Scholar]
- 46.Insausti R, Amaral DG. Hippocampal formation In The Human Nervous System: Second Edition: Elsevier Inc; 2003: 871–914 [Google Scholar]
- 47.Insausti R, Munoz M. Cortical projections of the non‐entorhinal hippocampal formation in the cynomolgus monkey (Macaca fascicularis). Eur J Neurosci 2001; 14: 435–51 [DOI] [PubMed] [Google Scholar]
- 48.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 1984; 225: 1168–70 [DOI] [PubMed] [Google Scholar]
- 49.Braak H, Braak E, Ohm T, Bohl J. Alzheimer’s disease: mismatch between amyloid plaques and neuritic plaques. Neurosci Lett 1989; 103: 24–8 [DOI] [PubMed] [Google Scholar]
- 50.Rowland DC, Weible AP, Wickersham IR, Wu H, Mayford M, Witter MP, Kentros CG. Transgenically targeted rabies virus demonstrates a major monosynaptic projection from hippocampal area CA2 to medial entorhinal layer II neurons. J Neurosci 2013; 33: 14889–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witter MP, Amaral DG. Entorhinal cortex of the monkey: V. Projections to the dentate gyrus, hippocampus, and subicular complex. J Comp Neurol 1991; 307: 437–59 [DOI] [PubMed] [Google Scholar]
- 52.Ding S-L, Haber SN, Van Hoesen GW. Stratum radiatum in CA2 of the hippocampus is a unique target of the perforant path in human and nonhuman primates. Neuroreport 2010; 21: 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chevaleyre V, Siegelbaum SA. Strong CA2 pyramidal neuron synapses define a powerful disynaptic cortico-hippocampal loop. Neuron 2010; 66: 560–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med 2018; 16: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sabbagh MN, Adler CH, Lahti TJ, Connor DJ, Vedders L, Peterson LK, Caviness JN, Shill HA, Sue LI, Ziabreva I, Perry E, Ballard CG, Aarsland D, Walker DG, Beach TG. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord 2009; 23: 295–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dickson DW, Schmidt ML, Lee VM, Zhao ML, Yen SH, Trojanowski JQ. Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol 1994; 87: 269–76 [DOI] [PubMed] [Google Scholar]
- 57.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013; 154: 103–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Covell DJ, Robinson JL, Akhtar RS, Grossman M, Weintraub D, Bucklin HM, Pitkin RM, Riddle D, Yousef A, Trojanowski JQ, Lee VM. Novel conformation-selective alpha-synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson’s disease. Neuropathol Appl Neurobiol 2017; 43: 604–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kantarci K, Lowe VJ, Boeve BF, Senjem ML, Tosakulwong N, Lesnick TG, Spychalla AJ, Gunter JL, Fields JA, Graff-Radford J. AV-1451 tau and β-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 2017; 81: 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N, Sperling R, Growdon JH, Dickerson BC, Johnson K. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 2016; 73: 1334–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee SH, Cho H, Choi JY, Lee JH, Ryu YH, Lee MS, Lyoo CH. Distinct patterns of amyloid-dependent tau accumulation in Lewy body diseases. Mov Disord 2018; 33: 262–72 [DOI] [PubMed] [Google Scholar]
- 62.Iseki E, Togo T, Suzuki K, Katsuse O, Marui W, de Silva R, Lees A, Yamamoto T, Kosaka K. Dementia with Lewy bodies from the perspective of tauopathy. Acta Neuropathol 2003; 105: 265–70 [DOI] [PubMed] [Google Scholar]
- 63.Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, Colloby SJ, Jellinger K, Attems J. Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 2015; 129: 729–48 [DOI] [PubMed] [Google Scholar]
- 64.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 2011; 7: 263–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Litvan I, Aarsland D, Adler CH, Goldman JG, Kulisevsky J, Mollenhauer B, Rodriguez‐Oroz MC, Tröster AI, Weintraub D. MDS task force on mild cognitive impairment in Parkinson’s disease: Critical review of PD‐MCI. Mov Disord 2011; 26: 1814–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adler DH, Wisse LE, Ittyerah R, Pluta JB, Ding S-L, Xie L, Wang J, Kadivar S, Robinson JL, Schuck T. Characterizing the human hippocampus in aging and Alzheimer’s disease using a computational atlas derived from ex vivo MRI and histology. Proceedings of the National Academy of Sciences 2018; 115: 4252–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Phillips JS, Das SR, McMillan CT, Irwin DJ, Roll EE, Da Re F, Nasrallah IM, Wolk DA, Grossman M. Tau PET imaging predicts cognition in atypical variants of Alzheimer’s disease. Hum Brain Mapp 2018; 39: 691–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yushkevich PA, Amaral RS, Augustinack JC, Bender AR, Bernstein JD, Boccardi M, Bocchetta M, Burggren AC, Carr VA, Chakravarty MM. Quantitative comparison of 21 protocols for labeling hippocampal subfields and parahippocampal subregions in in vivo MRI: towards a harmonized segmentation protocol. Neuroimage 2015; 111: 526–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wisse LE, Daugherty AM, Olsen RK, Berron D, Carr VA, Stark CE, Amaral RS, Amunts K, Augustinack JC, Bender AR. A harmonized segmentation protocol for hippocampal and parahippocampal subregions: Why do we need one and what are the key goals? Hippocampus 2017; 27: 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.