

Abstract
The E3 ubiquitin ligase Rad18 promotes a damage-tolerant and error-prone mode of DNA replication termed trans-lesion synthesis that is pathologically activated in cancer. However, the impact of vertebrate Rad18 on cancer genomes is not known. To determine how Rad18 affects mutagenesis in vivo, we have developed and implemented a novel computational pipeline to analyze genomes of carcinogen (7, 12-Dimethylbenz[a]anthracene, DMBA)-induced skin tumors from Rad18+/+ and Rad18−/− mice. We show that Rad18 mediates specific mutational signatures characterized by high levels of A(T)>T(A) single nucleotide variations (SNVs). In Rad18−/- tumors, an alternative mutation pattern arises, which is characterized by increased numbers of deletions >4 bp. Comparison with annotated human mutational signatures shows that COSMIC signature 22 predominates in Rad18+/+ tumors whereas Rad18−/− tumors are characterized by increased contribution of COSMIC signature 3 (a hallmark of BRCA-mutant tumors). Analysis of The Cancer Genome Atlas shows that RAD18 expression is strongly associated with high SNV burdens, suggesting RAD18 also promotes mutagenesis in human cancers. Taken together, our results show Rad18 promotes mutagenesis in vivo, modulates DNA repair pathway choice in neoplastic cells, and mediates specific mutational signatures that are present in human tumors.
INTRODUCTION
Genetic instability is a hallmark and enabling characteristic of cancer cells (1,2). During multi-step tumorigenesis mutations in key growth-regulatory genes (including oncogenes and tumor suppressors) allow cells to overcome normal barriers to proliferation, adapt to their environments, progress to malignancy and resist therapy. Because genetic change promotes tumorigenesis and enables cancers to adapt to therapy there is great interest in defining the molecular mechanisms of mutagenesis. Understanding mutagenic mechanisms employed by neoplastic cells may reveal new molecular vulnerabilities and provide new opportunities for preventing and treating cancer.
Cancer genome sequencing endeavors have cataloged the mutations that occur in thousands of human cancers. Nik-Zainal et al. applied mathematical models to extract five distinct ‘mutational signatures’ from 21 different human breast cancers (3). Those workers annotated mutational patterns based upon the frequencies of 96 different context-dependent single nucleotide variations (SNVs), each one occurring in a unique trinucleotide sequence context (with one nucleotide flanking both 5′ and 3′ positions of the mutated nucleotide). A study of ∼4.9 million mutations from 7042 different human cancers revealed over 20 distinct mutational patterns (4). Additional mutational signatures have been identified and curated in the ‘Catalogue Of Somatic Mutations In Cancer’ (COSMIC) v2 database which currently describes 30 distinct mutation signatures (5). The mutational signature has become a valuable tool for classifying mutations and for quantifying their contribution to the overall mutational spectra in cancer samples from humans (6–8) and mice (6,9).
Mutagenesis results from error-prone replication or repair of damaged DNA. Genotoxic exposures (from environmental, metabolic and pharmaceutical sources) can induce diverse mutagenic DNA lesions. A major challenge in cancer biology is to identify the DNA lesions and genome maintenance processes that generate the known mutational signatures documented in the COSMIC database. To definitively identify cancer-relevant mutagenic processes it is necessary to experimentally model defects in candidate DNA repair pathways and test whether these recapitulate mutational patterns observed in human cancer.
Trans-lesion synthesis (TLS) is an attractive candidate mechanism for mutagenesis in cancer. TLS is a DNA damage-tolerant and error-prone (mutagenic) form of DNA synthesis that employs the specialized Y-family DNA polymerases DNA Polymerase Eta (Polη), DNA Polymerase Kappa (Polκ), DNA Polymerase Iota (Polι) and REV1 (10). In contrast with the high-fidelity DNA polymerases Epsilon and Delta (Polϵ and Polδ) that replicate the bulk of the genome each cell cycle, Y-family DNA polymerases have very low fidelity and processivity on undamaged DNA templates. However, the Y-family DNA polymerases can replicate damaged DNA templates with relative accuracy. Moreover, each Y family TLS polymerase preferentially replicates DNA templates harboring specific cognate DNA lesions. Collectively the Y-family TLS polymerases allow cells to replicate genomes harboring diverse DNA lesions and confer resistance to a wide variety of genotoxic exposures (11). However, failure to select the most appropriate Y-family DNA polymerases for TLS of their preferred cognate lesions can lead to error-prone TLS and mutagenesis. For example, individuals with xeroderma pigmentosum variant (XPV, a sunlight-sensitivity and cancer propensity) lack functional Polη (12,13). Consequently compensatory and error-prone TLS of CPD by alternative Y-family DNA Polymerases (specifically Polκ and Polι) leads to hypermutability of ultraviolet (UV)-irradiated XPV cells thereby explaining the sunlight-induced skin cancer-susceptibility of XPV individuals (14,15).
Owing to their error-propensity on undamaged DNA and non-cognate DNA lesions, Y-family DNA polymerases must be stringently regulated to limit mutagenesis. Accordingly, there has been great interest in elucidating the mechanisms of TLS polymerase activation and selection. Recruitment of Y-family TLS polymerases to damaged DNA is promoted by the E3 ubiquitin ligase RAD18 which mono-ubiquitinates Lysine 164 of PCNA (a DNA polymerase processivity factor) at stalled replication forks (16–18). RAD18-deficient cells are compromised for PCNA mono-ubiquitination and often fail to activate TLS or to sustain damage-tolerant DNA synthesis following genotoxic injury. Persistent S-phase arrest and aberrant accumulation of single-stranded DNA (ssDNA) and DNA double-stranded breaks (DSB) following genotoxic injury are hallmarks of many RAD18- and TLS-deficient cells (17,19).
While RAD18 and TLS polymerases provide a potential mechanism for both DNA damage tolerance and mutagenesis in cancer, with the possible exception of sunlight-induced skin cancers in XPV patients, there is no direct evidence that the RAD18/TLS pathway shapes cancer genomes. In fact, although regulation of TLS polymerases by RAD18 has been studied extensively in tissue culture models, the impact of RAD18 on mutagenesis is unclear. For example, in different published studies RAD18 is reported to both stimulate and inhibit mutagenesis of ectopically-transfected plasmid DNAs (20–22) and the extent to which RAD18 impacts DNA replication fidelity in a natural chromosomal setting has never been studied. Moreover, there is considerable debate on whether RAD18 is even required for TLS. Many investigators have reported that Y-family DNA polymerases are activated independently of RAD18 and of mono-ubiquitinated PCNA in a variety of experimental settings including yeast (23,24), human cells and cell extracts (25–27), mouse embryonic fibroblasts (MEFs) (28,29) mouse pre-B cells (30) and chicken DT40 cells (31). Thus, the extent to which RAD18 affects mutagenesis remains controversial.
Increasingly it is appreciated that there are links between RAD18 and cancer. For example, RAD18 is pathologically overexpressed in many cancers (20,32) and RAD18-mediated TLS allows cells to withstand oncogene-induced DNA replication stress and DNA damage (33,34). Elevated RAD18 expression can promote PCNA ubiquitination and recruitment of Y-family polymerases to replication forks even in the absence of DNA damage (17). Therefore, the selective pressure for oncogene-expressing neoplastic cells to sustain high-level RAD18 expression and activity could provide a mechanism for error-prone replication and mutability in cancer.
Given recent findings that RAD18 is pathologically elevated in cancers and that TLS is required for tolerance of oncogenic stresses and cancer cell fitness (32,34), it is important to determine the impact of RAD18 on mutagenesis in cancer. Here we sought to resolve the debate regarding role of RAD18 in mutagenesis. To unambiguously test the contribution of RAD18 to mutagenesis in a physiological setting we have performed whole exome sequencing (WES) and compared genomes of tumors generated in Rad18+/+ and Rad18−/− mice. To analyze mouse WES data, we developed a new and rigorous bioinformatics pipeline. Using state-of-the-art computational tools, we have empirically determined the impact of Rad18 on different genetic alterations including SNVs and insertions/deletions (INDELs). Moreover, we have compared mutational patterns in mouse genomes with annotated human mutational signatures catalogued in the COSMIC database. Thus, our work defines for the first time how Rad18 impacts the cancer genome and determines the relative levels of different mutational signatures that are present in human cancer. In addition to advancing our knowledge of the RAD18 pathway and its impact on mutagenesis and carcinogenesis, our study validates the use of a new computational pipeline for analyzing WES data and interrogating mechanisms of genome maintenance in vivo.
The computational pipeline and tools we developed here are generally applicable to any tumorigenesis study with other genetically-modified mice (and other carcinogenic drivers): application of our methodologies will enable other investigators to analyze mouse tumor genomes and compare mutational events observed with human mutational signatures in the COSMIC database. Clearly such studies will be necessary if are to address an important knowledge gap and definitively identify the underlying mechanisms of the COSMIC signatures whose molecular etiologies are not yet known.
MATERIALS AND METHODS
Mice and breeding
Mice were maintained with a standard diet in a pathogen-free environment. Rad18−/− mice were described previously (35,36) and were back-crossed to C57BL/6J (Charles River Laboratories) to prevent genetic drift.
Genotoxin administration
7,12-Dimethylbenz[a]anthracene (DMBA, ACROS Organics™) was dissolved in olive oil to [6.66 mg/ml]. Mice were orally-gavaged with DMBA solution (versus olive oil control) to 50mg/kg as described previously (36).
Cells and culture
Rad18+/+ and Rad18−/− MEFs were obtained from day 13 embryos and cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, streptomycin sulfate (100 μg/ml) and penicillin (100 units/ml) as previously described (17). MEF were trypsinized and re-plated at a density of 1:3 after reaching 80% confluence. To induce DNA damage, Rad18+/+ and Rad18−/− MEF were treated with UV (20 J/m2) or DMBA (1 μM). Twenty-four hours later chromatin extracts were prepared as previously described (17,33).
SDS-PAGE and immunoblotting
Chromatin-enriched cell extracts were analyzed by sodium dodecyl sulphate-polyacrylamide gelelectrophoresis (SDS-PAGE) and immunoblotting as described previously (36). Primary antibodies used for immunoblot analysis were: p-ATM S1981 (SC-47739), mouse monoclonal PCNA clone PC10 (SC-56), RAD18 (A301-340A, Bethyl Laboratories Inc.; ATM (GTX70104, Gene Tex); and mouse monoclonal γH2AX S139 (05-636, EMD Millipore).
PCR analysis of Ras genes
To verify the mutational status of Ras genes in DMBA-induced tumors we used the primer pair GCAAGGGTGTAGGCTGGTTC (forward)/CTCACGGGCTAGCCATAGGTG (reverse) to polymerase chain reaction (PCR) amplify a 302 bp fragment of Hras spanning codon 61 and the primer pair ACTCTGTACATCTGTAGTCACT (forward) / TGGTTCCCTAACACCCAGTT (reverse) to amplify a 427 bp fragment of Kras spanning codon 12 from genomic DNA. PCR products were purified using a QIAquick PCR Purification Kit from Qiagen (Cat # 28104). Purified DNA samples were submitted to Eton Bioscience Inc. for Sanger sequencing. Proportions of SNV were determined using Thermo Fisher Sanger sequencing data analysis tool ‘Variant Analysis (VA)’ to measure the trace report of peaks.
Whole exome sequencing (WES)
For tumors, whole-exome DNA libraries were created with the Agilent SureSelect XT2 mouse All Exon kit according to the manufacturer's instructions, and then they were sequenced on a HiSeq2000 (Illumina) using a V3 or V4 flow cell generating 125-bp paired-end reads. Library preparation and sequencing were performed by UNC HTS core facility. For normal tissues, sequencing (WES) was performed by Novogene INC using Novaseq PE150. (https://en.novogene.com/). Bioinformatic analysis of WES data and identification of single-nucleotide variants (SNVs) and INDELs—raw sequencing reads were mapped to the Mus musculus (house mouse) reference genome (assembly GRCm38/mm10) using BBMap package (version 37.00), (37) and the resulting sequence alignment map (SAM) files were converted to binary alignment map (BAM) files using SAMtools 1.3.1 (38). Picard 2.2. 4 (http://broadinstitute.github.io/picard) was used to validate/sort BAM files, build BAM indices and remove duplicates. The analytic tools RealignerTargetCreator and IndelRealigner in Genome Analysis Toolkit 3.4.36 (GATK) (39) were used to perform local realignments around INDELs.
To call single-point variations, a base quality score recalibration was performed by using GATK 3 BaseRecalibrator and AnalyzeCovariates. Known single-nucleotide polymorphisms (SNPs) (mgp.v3.snps.rsIDdbSNPv137) and INDELs (mgp.v3.INDELs.rsIDdbSNPv137) reported in the C57BL/6NJ background by the Mouse Genome Project of the Sanger Institute (40) were removed as one step in the base quality score recalibration. The BAM files prepared for substitution-calling were then generated, and they contained 109 to 121 million reads per sample (Supplementary Table S1). Finally, substitutions were called by using HyplotypeCaller in GATK 3 to generate variant calling format (VCF) files. Then VCF files were refined to only include exome regions using BEDTools 2.26.0 (41).
Several SNVs and INDELs residing in the vicinity (at chr6 100 000 000−130 000 000 bp) of the disrupted Rad18 locus (located at chr6, 112 619 850−112 696 686 bp) were resulted from the gene-targeting strategy used to generate the Rad18-null mice (35) and were therefore excluded from our analysis of all (Rad18+/+ and Rad18−/−) samples.
A normalization of the number of somatic SNVs and the number of somatic INDELs to the library size (total reads in a sample) was performed as follows to allow a convenient range of the normalized number (Supplementary Table S2).
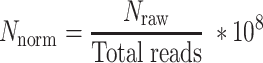 |
Here, to count the number of mutations and INDELs, we removed duplicated mutations/INDELs in VCF files and broke those reads corresponding to multi-allelic variants into multiple single-variant records.
Variants were annotated by ANNOVAR (v20180416) (42) and the corresponding gene information was derived from the Ensembl database (43). We first downloaded the GTF file and the genome FASTA file for the GRCm38.p6 mouse reference genome (GENCODE release M23) (44).
We then used the gtfToGenePred tool to convert the GTF file to a GenePred file, and we generated a transcript FASTA file following the instructions and scripts provided by ANNOVAR. In the meantime, using the getBM function of R package biomaRt, we extracted gene information from the Ensembl database (release 98) ‘mmusculus_gene_ensembl’ in mart ‘ENSEMBL_MART_ENSEMBL’. This database was also built for the GENCODE V23 track. Lastly, we joined the results from ANNOVAR and Ensembl by the Ensembl gene ID of each gene.
Annotation of mutation patterns in our mouse tumors and comparison with mutational signatures in human cancer
We used two strategies to analyze mutational signatures represented by trinucleotides with 1 bp flanking 5′ and 3′ of every SNV to generate 96 possible mutation types, as first described by Alexandrov et al. (4):
Our first strategy identified mutational signatures in mouse tumor samples using R package SomaticSignatures (45). Here we called the mutation patterns in our mouse WES data in each sample and in each sample group (by pooling samples within a group) according to the relative proportions of the 96 mutation types using trinucleotide frequencies. The number of mutational signatures in our mouse tumor samples was based on a selection criterion using residual sum of squares. We used the following procedure to estimate the relative contribution of each mutational signature to the total mutational burden in our tumor samples (Figure 5D): for SNVs embedded in trinucleotide sequences, we denoted the frequencies of all possible mutation types across multiple samples to a matrix
 . The non-negative matrix factorization method can be used to identify mutational signatures by decomposing
. The non-negative matrix factorization method can be used to identify mutational signatures by decomposing  into two matrices
into two matrices  and
and  with the constraint of positive components (3). For the
with the constraint of positive components (3). For the 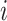 th mutation in the
th mutation in the 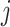 th sample, given a fixed number
th sample, given a fixed number 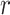 of signatures,
of signatures,  . Thus,
. Thus, 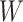 describes the composition of each signature in terms of the 96 mutational types, and
describes the composition of each signature in terms of the 96 mutational types, and  represents the relative contribution of each signature in each sample.
represents the relative contribution of each signature in each sample.Our second strategy assessed the relative contribution of 30 given human cancer mutational signatures defined in the COSMIC v2 to the total mutational burden in each of our mouse tumor samples (Figure 5F). We denoted the frequencies of all possible mutation types for a fixed sample to a vector
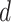 . We denoted the composition of 30 cancer mutational signatures in terms of the 96 trinucleotide contexts (mutational types) as a matrix
. We denoted the composition of 30 cancer mutational signatures in terms of the 96 trinucleotide contexts (mutational types) as a matrix  .
.  is known and available for downloading from the COSMIC website. By finding a non-negative vector
is known and available for downloading from the COSMIC website. By finding a non-negative vector 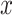 such that
such that  for each sample, we obtained the relative contribution matrix
for each sample, we obtained the relative contribution matrix 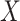 of known cancer mutational signatures. The
of known cancer mutational signatures. The 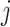 th column of
th column of 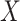 is the non-negative vector
is the non-negative vector 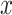 for the
for the 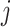 th sample. In particular, we solved for the
th sample. In particular, we solved for the 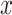 ’s using the non-negative least squares optimization though R package MutationalPatterns (46). Both above relative contributions of mutational signatures can be normalized within a sample by dividing the observed mutation-type contribution by the total contribution, to obtain a Normalized Relative Contribution (NRC, see Figure 5C and E).
’s using the non-negative least squares optimization though R package MutationalPatterns (46). Both above relative contributions of mutational signatures can be normalized within a sample by dividing the observed mutation-type contribution by the total contribution, to obtain a Normalized Relative Contribution (NRC, see Figure 5C and E).
Figure 5.

Annotating DMBA-induced mutational signatures in Rad18+/+ and Rad18−/− mouse tumors. (A) Mutation type probabilities (calculated as relative proportions in a sample) of different SNVs in all possible trinucleotide contexts in Rad18+/+ (WT) and Rad18−/− (KO) mouse tumors. (B) Identification of four distinct mutation signatures (designated as mS1-mS4) in DMBA-induced tumors from Rad18+/+ and Rad18−/− mice. (C) Heatmap showing the normalized relative contribution of mutation signatures mS1–mS4 to the total mutational burden of DMBA-induced tumors in Rad18+/+ and Rad18−/− mice (x-axis). (D) Stacked bar chart showing the relative contribution of mutation signatures mS1–mS4 to the total mutational burden of DMBA-induced tumors in Rad18+/+ and Rad18−/− mice (x-axis). (E) Heatmap showing the normalized relative contribution of mutational signatures from human tumors (hS1–hS30 on the x-axis) to the mutational portraits of DMBA-induced tumors in Rad18+/+ and Rad18−/− mice (y-axis). (F) Stacked bar chart showing the relative contribution of mutation signatures from human tumors (hS1–hS30) to the total mutational burden of DMBA-induced tumors in Rad18+/+ and Rad18−/− mice (x-axis). The relative contributions of the following COSMIC signatures were statistically different between Rad18+/+ and Rad18−/− mice: hS1 (P = 0.008), hS3 (P = 0.033), hS4 (P = 0.012), hS11 (P = 0.009), hS12 (P = 0.003), hS20 (P = 0.031), hS22 (P = 0.015), hS25 (0.011) and hS28 (P = 0.041).
Defining relationships between RAD18 expression and SNVs in human cancer
Lung adenocarcinoma (LUAD), bladder urothelial carcinoma (BLCA), lung squamous cell carcinoma (LUSC) and kidney renal clear cell carcinoma (KIRC) were selected for evaluation of relationships between RAD18 status and genome instability in human tumors. TCGA datasets containing RNA expression, mutation, genomic alteration (downloaded on 20 March 2019), and clinical information (downloaded in May 2019) for these tumors were from the TCGA data portal (https://portal.gdc.cancer.gov). Specific datasets used in this study include: (i) HTSeq-FPKM-UQ gene expression quantification (for those duplicated samples from the same patient, we choose the sample with higher gene expression for downstream analysis), (ii) somatic mutation aggregated and masked by Mutect2 and organized as MAF files publically available and (iii) copy Number Variation (CNV) files with −1 as a loss, 0 as neutral, 1 as gain of copies that are defined by numeric focal-level CNV values generated with ‘Masked Copy Number Segment’ files from tumor aliquots using GISTIC2, on a project level. Only protein-coding genes were retained, and their numeric CNV values were further thresholded by a noise cutoff of 0.3 in the Gene Level Copy Number Scores matrix generated by GISTIC2 (and take every individual replicate as a sample), (iv) smoking information is also available for most subjects in LUAD, LUSC and BLCA, but not available (N/A) for >80% KIRC patients. To avoid zeros when using log scale for display, log2(FPKM + 1) was used to display gene expression data obtained by RNAseq.
To evaluate the interaction between smoking and RAD18 gene expression, we defined two populations: ‘smokers’ (a group including both current smokers, and current reformed smokers who smoked >1 cigarettes per day) and ‘non-smokers’ (life-long non-smokers). Patients lacking ‘Tobacco Smoking History Indicator’ or records of ‘cigarettes per day’, were excluded from downstream analysis (Supplementary Figure S1). The smoking cutoff was determined by the distribution of number of cigarettes per day in LUAD and LUSC (Figure 6C and Supplementary Figure S5C). The numbers of patients for data analyzed in this study (including gene expression using RNAseq, CNV and SNV) are shown in Table 1. If there are technical replicates or biological replicates in data for one patient, we only kept mRNA files with the highest RAD18 expression and SNV files with the smallest data ID.
Figure 6.

Relationship between RAD18 expression and overall SNV in LUAD. (A) Boxplot showing RAD18 expression in tumors and adjacent normal tissues from LUAD patients. (*: P < 0.05; **: P < 0.01, ***: P < 0.001, ****: P < 0.0001). ‘n’, number of samples. (B) Boxplot showing RAD18 CNV (x-axis) and RAD18 gene expression in tumors from LUAD patients. On the x-axis, −1, 0 and 1 indicate RAD18 gene loss, neutral and gain. (C) Distribution of the number of cigarettes smoked by LUAD patients per day. (D and E) Boxplots showing total SNV counts (D) or numbers of each of the six individual SNV types (E) in LUAD samples grouped by RAD18 mRNA expression and smoker/nonsmoker. RAD18 expression is indicated by ‘high’ (upper half) or ‘low’ (bottom half) in smokers and non-smokers. SNV type CA means C(G)>A(T), and the same label rule holds for other SNV types. (F and G) Scatter plots showing total SNV counts (F) and individual SNV types (G) in LUAD samples according to RAD18 mRNA expression (x-axis), in smokers and non-smokers as indicated. (H) Mutation spectrum of LUAD grouping by RAD18 mRNA expression level and smoker/nonsmoker.
Table 1.
Summary of number of patients to analyze mRNA expression, CNV, SNV and smoking history for each of the four TCGA tumor types
| Human tumor | mRNA | CNV | SNV | mRNA + SNV | CNV + SNV | Smoking history | mRNA + SNV + smoking history | CNV + SNV + smoking history |
|---|---|---|---|---|---|---|---|---|
| LUAD | 513 | 517 | 565 | 508 | 512 | 449 | 441 | 441 |
| LUSC | 501 | 503 | 491 | 488 | 490 | 453 | 442 | 442 |
| BLCA | 408 | 410 | 412 | 408 | 410 | 359 | 351 | 352 |
| KIRC | 530 | 532 | 336 | 332 | 334 | N/A | N/A | N/A |
To investigate the association of genome-wide somatic mutations, e.g. SNV, with RAD18 CNV (or RAD18 gene expression), samples were divided into groups based on RAD18. For mRNA, tumor samples were divided into ‘RAD18-high’ (>upper half for RAD18 mRNA expression) and ‘RAD18-low’ (<lower half for RAD18 mRNA expression). To assess CNV, samples were divided into three groups based on RAD18 gene copy number scores. Thus, ‘−1’ represents RAD18 gene loss, ‘0’ represents no change in RAD18 copy number (i.e. neutral) and ‘1’ represents RAD18 gene gain. R package SomaticSignatures was used for identifying mutational signatures of SNVs. Wilcoxon rank sum test was used for assessing differences in SNVs between sample groups. Pearson Correlation and the corresponding P-values were used for assessing correlations between RAD18 expression and the total number of SNVs.
RESULTS
Ingested DMBA induces skin cancer in mice
In previous studies to define roles of Rad18 in hematopoiesis we determined the effects of 7, 12-Dimethylbenz[a]anthracene (DMBA, a myelosuppressive genotoxic agent) on hematopoietic stem cell function in Rad18+/+ and Rad18−/− mice (36). DMBA is a synthetic polycyclic aromatic hydrocarbon (PAH) that is used widely to model environmental genotoxicity, mutagenesis and carcinogenesis. Genome maintenance mechanisms that mediate DMBA-induced mutagenesis are not known. Unexpectedly, we noticed that many of the mice administered DMBA by oral gavage developed large (up to 1–2 cm3) palpable skin lesions (Figure 1A). Although topically-administered DMBA is widely used as a carcinogen for initiating skin tumorigenesis (47), ingested DMBA has not previously been shown to induce solid tumors. Histopathological analysis revealed that the DMBA-induced tumors of orally gavaged mice included papillomas and squamous cell carcinomas (Figure 1A). There was no significant difference in the incidence of solid tumors between Rad18+/+ and Rad18−/− mice (Figure 1B), although the Rad18−/− mice showed increased incidence of leukemia compared to Rad18+/+ littermates. However, the availability of multiple large solid clonal tumors from Rad18+/+ and Rad18−/− mice provided an excellent opportunity to define how Rad18 impacts the cancer genome in a physiological setting.
Figure 1.

Histopathology of DMBA-induced skin tumors in Rad18+/+ and Rad18−/- mice. (A) Rad18+/+ and Rad18−/− mice were treated with DMBA by oral gavage as described under ‘Materials and Methods’ section. Palpable skin tumors from Rad18+/+ (WT) and Rad18−/− (KO) mice were sectioned, subject to H&E staining, and evaluated by a pathologist. KO15 is a soft tissue/spindle cell tumor; KO18 and WT23 are papillomas; KO18, WT1, WT6, WT8, WT22 are mostly squamous cell skin cancers. Every tumor was from a different mouse. (B) Incidence of skin tumors and other neoplasia in Rad18+/+ and Rad18−/− mice. It was computed as the proportion of mice with identified tumors among all available mice (for around 20 mice per genotype).
Rad18 determines levels of SNVs and INDELs during DMBA-induced carcinogenesis
Genomic DNA was extracted from five Rad18+/+ and six Rad18−/− tumors and subjected to WES. The sequencing data were analyzed computationally as described under ‘Methods’ and as summarized in Figure 2A. WES reads were aligned to the GRCm38/mm10 mouse genome. After recalibrations and removing known SNPs and INDELs, somatic SNVs and INDELs were identified in exomes. First we quantified the raw and the normalized numbers of different SNVs that were detected in DMBA-induced tumors from Rad18+/+ and Rad18−/− mice. As shown in Figure 2B and C the predominant SNVs in the DMBA-induced mouse tumors were A(T)>T(A), accounting for 35.3% of the mutations in all 11 tumor samples (Figure 2B). The numbers of A(T)>T(A) mutations also differed between the Rad18+/+ and Rad18−/− sample groups: in Rad18−/− mice, A(T)>T(A) mutations ranged from 16.4 to 28.7% of all SNVs (average 24.02%, SD = 5.18%). In the Rad18+/+ mice, A(T)>T(A) mutations were more abundant and ranged from 33.6 to 47.2% (average 42.41%, SD = 5.46%) (Figure 2B). Every Rad18+/+ tumor contained more observed A(T)>T(A) when compared with Rad18−/− samples (P = 0.0043 for normalized numbers) (Figure 2B and C). All other SNVs were also significantly more abundant in Rad18+/+ tumors when compared with Rad18−/− samples (with a range of P = 0.0043 to 0.03 for normalized numbers) (Figure 2C). It was important to determine whether the reduced SNV count of Rad18−/− tumors relative to Rad18+/+ was a specific result of carcinogen-treatment rather than a pre-existing difference between wild-type and Rad18 mutant mice. Accordingly, we performed WES of genomic DNA from normal tissues (including lung, kidney, tail) from age-matched Rad18+/+ and Rad18−/− mice that did not receive DMBA. Sequences from normal tissues were also analyzed using the computational pipeline used to interrogate tumor samples. As shown in Supplementary Figure S2A and B, there were no significant differences in SNV levels between normal tissue genomes from Rad18+/+ and Rad18−/− mice. We conclude that Rad18 promotes DMBA-induced A(T)>T(A) substitutions and all other types of SNVs specifically during DMBA-induced carcinogenesis but that Rad18 does not affect baseline mutation rates in developing mice.
Figure 2.

Rad18 mediates DMBA-induced point mutations while averting the formation of DBSs and INDELs. (A) Summary of workflow and computational pipeline used to identify mutations in DMBA-induced mouse tumors. (B) Histograms showing raw numbers of different SNV subtypes in the individual tumors from Rad18+/+ (blue bars) and Rad18−/− (red bars) mice. The incidence of A(T)>T(A) mutations expressed as a percentage of the total SNV burden for each sample is as follows: KO15, 25.1%; KO16, 18.7%; KO18, 28.7%; KO19B, 27.2%; KO19T, 27.9%; KO26, 16.4%; WT1, 42. 2%; WT22, 46.7%; WT23, 33.6%; WT6, 42.4%; WT8, 47.2%. (C) Box-plots showing normalized numbers of different SNVs in Rad18+/+ (blue boxes) and Rad18−/− (red boxes) mice. P-values were calculated in Wilcoxon Rank Sum tests. (D) Immunoblot analysis of DNA damage markers in DMBA-treated MEFs. Rad18+/+ and Rad18−/− MEF were treated with UV (5J/m2), DMBA (1μM), or were left untreated for controls. After 6 h, cells were collected and fractionated to prepare detergent-insoluble chromatin extracts. Chromatin-containing fractions were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies. For PCNA two different exposures of the same ECL membrane are shown to demonstrate equivalent protein loading and effects of treatments on PCNA monoubiquitination (PCNA-Ub). (E) Histograms showing raw numbers of insertions and deletions in the individual tumors from Rad18+/+ (blue bars) and Rad18−/− (red bars) mice. (F) Box-plots showing normalized numbers of different insertions and deletions in Rad18+/+ (blue boxes) and Rad18−/− (red boxes) mice. P-values were calculated using Wilcoxon Rank Sum tests. The normalized number of deletions 4+ bp is significantly different between the two sample groups (P = 0.0043).
We considered the potential consequences of DMBA genotoxicity on the genome in the absence of Rad18. We hypothesized that Rad18-deficient cells would fail to sustain replication of genomes harboring bulky DNA adducts and instead accumulate DSB due to collapse of stalled DNA replication forks. To test our hypothesis, we treated Rad18+/+ and Rad18−/− cells with DMBA and used immunoblotting to test for the presence of the DSB markers γH2AX and ATM (pS1981). As shown in Figure 2D, DMBA-induced PCNA mono-ubiquitination was detectable in WT but not Rad18−/− cells. Therefore, the Rad18-mediated TLS pathway is normally activated in response to DMBA genotoxicity. Consistent with our hypothesis, Rad18-deficient cells aberrantly accumulated elevated levels of the DSB markers γH2AX and ATM (pS1981) following DMBA treatment. Indeed, Rad18 was more important for suppressing accumulation of secondary DNA damage from DMBA than from UV radiation-induced lesions (Figure 2D) that are known to be tolerated via the Rad18 pathway (17). We conclude that DMBA genotoxicity leads to elevated levels of DSB in Rad18-null cells when TLS is compromised.
Next, we determined how DMBA-induced DSBs arising from Rad18-deficiency impact the genome in vivo. To investigate back-up mechanisms that compensate for Rad18-deficiency during DMBA-induced carcinogenesis we interrogated WES data for alternative genetic changes including insertions and deletions. Figure 2E and F show that Rad18−/− tumor contains more deletions (of >4bp) when compared with Rad18+/+ samples (P = 0.0043). In normal tissues from mice that did not receive carcinogen, there was no significant difference in numbers of indels in Rad18−/- mice when compared to Rad18+/+ (Supplementary Figure S2C and D). To ensure that tumor heterogeneity did not affect our conclusions we examined the variant allele frequency (VAF) in Rad18+/+ and Rad18−/- groups. Higher VAF values suggest higher homogeneity of tumor samples. In Supplementary Figure S3 we show that the average variant allele frequencies of SNVs and Indels are comparable between genotypes, indicating that Rad18 does not affect tumor heterogeneity. Taken together, the results of Figure 2 show that Rad18 mediates lesion bypass and mutagenesis at sites of carcinogen-induced DNA damage. In the absence of Rad18 (when replicative by-pass of carcinogen-adducted DNA is not possible), DSB are generated and processed via error-prone mechanism(s) leading to 4 bp+ deletions.
Annotation of DMBA-induced SNVs and INDELs at the gene level in Rad18+/+ and Rad18−/− tumors
We sought to identify the target genes of DMBA-mediated mutagenesis in tumors from Rad18+/+ and Rad18−/− mice. We annotated SNVs and INDELs in all tumor samples (i.e. Rad18+/+ and Rad18−/−). As expected, all SNVs/INDELs from the final VCF file had corresponding annotated genes in the GRCm38.p6 mouse reference genome (GENCODE release M23). Of the 55335 genes annotated in the reference genome, 10428 genes contained SNVs in at least one tumor sample and 1404 genes contained INDELs in at least one tumor sample. One hundred and sixty-one genes contained SNVs in all 11 samples (i.e. from both Rad18+/+ and Rad18−/− tumors). One hundred and seventy-eight genes contained SNVs in all of the Rad18−/− samples (and three of these SNV-containing genes were specific to Rad18−/− tumors, namely Mcm7, Ly6g2 and Vcpkmt). Two hundred and six genes contained SNVs in all of the Rad18+/+ samples (and nine of these genes were specific to Rad18+/+ tumors: Acan, Cars, Cfap65, Col13a1, Col2a1, Fcgbp, Olfr617, Pkhd1 and Vmn2r8). One hundred and eighteen genes contained INDELs in all 11 of the Rad18+/+ and Rad18−/− samples. One hundred and fifty genes contained INDELs in all Rad18−/− tumors and of these INDEL-annotated genes, six were present exclusively in Rad18−/- tumors (Mcm7, Mtus1, Pkdrej, 1810009A15Rik, Gm49416 and Vcpkmt). One hundred and fifty-one genes contained INDELs in all the Rad18+/+ tumors, but none of these genes contained INDELs exclusively in the Rad18+/+ samples (Supplementary Table S5). Next, we asked whether the global (exome-wide) associations between Rad18 status and different mutation types (SNVs/INDELs) were also evident in defined SNV-annotated (or INDEL-annotated) subsets of genes. For these analyses, we focused on A(T)>T(A) substitutions and deletions >4bp which were more abundant in Rad18+/+ and Rad18−/− tumors, respectively (Figure 2). Figure 3A and B show that for those genes annotated with SNVs, the incidence of A(T)>T(A) mutations was significantly higher in Rad18+/+ tumors when compared to the Rad18−/− samples. The gene-level association between Rad18 and A(T)>T(A) SNVs is consistent with the exome-wide mutation patterns in Rad18+/+ and Rad18−/− groups (Figure 2). Therefore, A(T)>T(A) changes account for the Rad18+/+-specific SNV-containing genes. For the small number of group-specific genes (with no more than five G>A per sample) annotated with G(C)>A(T), the incidence of G(C)>A(T) mutations (higher in mS3/4) was significantly higher in Rad18-/- tumors when compared to the Rad18+/+ samples. However for the large number of group, common genes annotated with G(C)>A(T), there was no significant difference between Rad18+/+ and Rad18−/− groups. For those genes annotated with INDELs, the number of 4+ bp deletions was significantly higher in Rad18−/− tumors when compared to the Rad18+/+ samples (Figure 3C and D). Thus, the difference in number of INDEL-annotated genes between Rad18+/+ and Rad18−/− groups is consistent with the overall INDEL pattern for the two Rad18 genotypes (Figure 2). We conclude that deletions >4 bp account for the INDEL-annotated genes that are specific to the Rad18−/− tumors. Taken together, the results of Figure 3 show that the exome-wide associations between Rad18 and specific mutation types (SNVs and INDELs) were fully recapitulated at the individual annotated gene level.
Figure 3.

Comparison of numbers of gene-level annotated SNVs and 4+ bp deletions, between Rad18−/− (pink bars) and Rad18+/+(cyan bars) DMBA-induced mouse tumors. The left panels show boxplots of normalized SNV numbers while the right panels show bar plots of the raw numbers of SNV. (A) The number of A(T)>T(A) and G(C)>A(T) substitutions for SNV-annotated group-specific genes in each of the Rad18+/+ (cyan) and Rad18−/− (pink) samples. (B) The number of A(T)>T(A) and G(C)>A(T) substitutions for SNV-annotated common genes in each of the Rad18+/+ (cyan) and Rad18−/− (pink) samples. (C) The number of 4+ bp deletions for INDEL-annotated group-specific genes in the six Rad18−/− (pink) samples. Note: only data from Rad18−/− (pink) mice are shown because none of the Rad18+/+ samples contained genes unique to this genotype harboring 4+ bp deletions. (D) The number of 4+ bp deletions for INDEL-annotated common genes in each of the Rad18+/+ (cyan) and Rad18−/− (pink) samples. * ‘Group-specific genes’ refers to genes present exclusively in all samples from one group (i.e. Rad18 genotype) but not any of the samples in the other group. ‘Group-common genes’ refers to genes existing in all samples within one sample group (or Rad18 genotype), but not exclusively present in the other sample group.
Next, we sought to identify DMBA-induced oncogenic mutations in tumors from Rad18+/+ and Rad18−/− mice. The Ras family proto-oncogenes are known targets of DMBA-induced mutagenesis. DMBA-driven carcinogenesis in mice is often attributed to activating mutations in Kras and Hras genes (at codons 12 and 61, respectively). Therefore, we used our WES data to determine the mutational status of Hras and Kras genes in DMBA-induced tumors from Rad18+/+ and Rad18−/− mice. To independently verify the mutational status of Ras genes and validate our WES results, we individually PCR-amplified and sequenced the Kras and Hras genes from tumor DNA samples. Based on our data, Figure 4A, B, and Supplementary Table S3 show that, 5 out of 11 tumors collected in this study contained activating mutations in Ras genes. Mutated Kras and Hras were identified in two of the six Rad18−/− tumors and in three of the five Rad18+/+ tumors (Kras was mutated in KO26, WT22, WT6 while Hras was mutated in KO18 and WT1). The specific locations of mutations in Ras genes are shown in Supplementary Table S3. We conclude that DMBA-induced activating mutations in Ras family oncogenes are not dependent on the Rad18 pathway. Moreover, DMBA-induced carcinogenesis is not exclusively driven by Hras and Kras mutations.
Figure 4.

Identification of oncogenic drivers in DMBA-induced mouse skin tumors from Rad18+/+ and Rad18−/− mice. (A) Histogram showing the number of somatic SNVs for each of the 43 oncogenes that were altered in DMBA-induced mouse skin tumors from all Rad18+/+ and Rad18−/− mice. The identified SNVs are described according to locations (exonic, intronic, splice junction and UTR) and types (synonymous and non-synonymous) using different colors and transparencies as defined in the key. Black arrows point to Ras family genes. (B) Table showing the number of somatic SNVs identified in 43 different oncogenes for each individual Rad18+/+ and Rad18−/− tumor sample. (C) Histogram showing the number of somatic INDELs for each of the nine oncogenes that were altered in DMBA-induced mouse skin tumors from all Rad18+/+ and Rad18−/− mice. The identified INDELs are described according to locations (exonic, intronic and splice junction) and types (frameshift insertion or deletion) using different colors and transparencies as defined in the key. (D) Table showing the number of INDELs identified in 9 different oncogenes for each individual Rad18+/+ and Rad18−/− tumor sample.
Nassar and colleagues previously defined the mutational landscape of DMBA-induced skin tumors in wild-type C57BL6 mice and identified 48 candidate driver oncogenes harboring recurrent somatic mutations in those neoplasms (47). Together with these 48 previously identified driver genes encoding the Ras family proteins (Kras, Hras andRras) and genome maintenance factors (Trp53), we added eight other oncoproteins implicated in basal cell carcinomas such as components of the Sonic Hedgehog (SHH) pathway (Shh, Smo, Ptch1, Gli1, Gli2, Gli3, Foxm1 and Foxe1) to have a combined list of 56 oncogenic driver genes. We determined the mutational status of those 56 known oncogenes in the DMBA-induced tumors from Rad18+/+ and Rad18−/− mice. These 56 genes are all protein coding genes. By focusing on protein coding genes, based on the analysis of 21 902 protein-coding genes in the Ensembl database (release 98) SNVs in our study were annotated to 9543 genes. A total of 12 of the 48 candidate driver oncogenes identified by Nassar et al. (Alkbh3, Blm, Brpf1, Fbxw7, Gsto2, Nras, Rpl36, Sfrp2, Shh, Slit3, Sult1a1 and Dcaf1) and one gene (Foxm1) from the SHH pathway did not contain SNVs in our study.
Therefore, 43 out of the 56 oncogenic drivers were annotated with SNVs in at least 1 of the 11 Rad18+/+ and Rad18−/− tumor samples (Figure 4A and B), with an enrichment P-value of 1.04 × 10−6 (Pearson's Chi-squared test) and an odds ratio of 4.3. Therefore, driver oncogenes are significantly enriched in SNV-annotated genes from DMBA-induced tumors. Twenty-one of the 43 mutated oncogenes detected in this study harbored SNV in both Rad18+/+ and Rad18−/- samples, 4 oncogenes were mutated only in Rad18−/- tumors and 18 were mutated only in Rad18+/+ tumors.
We identified eight oncogenic drivers having G(C)>A(T) mutations (that are featured in signatures mS3 and mS4). within these eight genes, G>A substitutions occurred 13 times in Rad18−/− tumors and three times in Rad18+/+ samples. For the 59 total SNVs in Rad18−/− tumors and the 103 SNVs in Rad18+/+ tumors, G>A SNVs were significantly enriched in the Rad18−/− group (with a P-value of Fisher's exact test as 0.0001579 and odds ratio of 9.3). A(T)>T(A) SNVs occurred 25 times in Rad18−/− tumors and 69 times in the Rad18+/+ group (with an enrichment P-value for A(T)>T(A) in the Rad18+/+ of 0.0029 and an odds ratio of 2.7).
Of the 21 902 protein-coding genes in the Ensembl database, 1252 genes were annotated by INDELs in our study. Nine of those 56 oncogenic drivers were annotated with INDELs in at least 1 of the 11 Rad18+/+ and Rad18−/− tumor samples (Figure 4C and D), with an odds ratio of 3.2 and an enrichment P-value of 4.2 × 10−3 (Fisher's exact test). Six of the nine mutated oncogenes we identified contained INDELs in both Rad18+/+ and Rad18−/− samples, two oncogenes were mutated only in Rad18−/- tumors and one was mutated only in Rad18+/+ tumors.
We conclude that the SNV/INDEL-annotated genes in our DMBA experiment are enriched with known oncogenes in both Rad18−/− and Rad18+/+ tumors. However, the differences between those enrichment of SNV- or INDEL-annotated oncogenes when comparing Rad18−/− and Rad18+/+ tumors are unlikely to be statistically significant (data not shown). It remains to be determined which of the observed oncogenic mutations are carcinogenic drivers and which are passengers. Unexpectedly however, the absence of Ras mutations in some of the tumor samples demonstrates that DMBA-induced carcinogenesis can occur via Ras-independent mechanisms.
Defining DMBA-induced mutational signatures in Rad18+/+ and Rad18−/− mouse tumors
Having demonstrated that Rad18 status determines overall numbers of SNVs and INDELs, we sought to delineate ‘mutational signatures’ (i.e. the context-dependent mutation frequencies that consider two nucleotides in the flanking 5′ and 3′ positions of the SNV) as annotated by previous literature (3,4) and described in the COSMIC v2 database. We used R package SomaticSignatures (45) to identify mutational signatures based on SNVs in different trinucleotide settings in mouse WES data. As shown in Figure 5A, all tumor samples contained more T(A)>A(T) substitutions than the other five mutation types, regardless of Rad18 status. The high incidence of T(A)>A(T) mutations in our experimental model is consistent with dA residues as major targets of DMBA-induced genotoxicity, accounting for 80% of DMBA-adducted bases in vitro and in vivo (48,49). This result is also consistent with results of Figure 2B. The most frequent T(A)>A(T) mutation types occurred in the trinucleotide settings are CTG > CAG and GTG > GAG (Figure 5A). Rad18+/+ tumor genomes displayed a higher frequency of T(A)>A(T) mutations when compared with Rad18−/−. We identified four distinct mutational signatures which we designated as ‘mS1-mS4’ (Figure 5B—‘m’ indicates mouse). mS1 and mS2 are characterized by a preponderance of T(A)>A(T) mutations (58.2379 and 47.0344% T(A)>A(T) mutations, respectively), and are more dominant in Rad18+/+ than in Rad18−/− tumors. mS3 and mS4 display relatively more C(G)>T(A) and T(A)>C(G) mutations (Figure 5B). Therefore, in our DMBA carcinogenesis model, Rad18 modulates global DMBA-induced mutagenesis primarily via changes in T(A)>A(T) mutations.
The stacked bar chart in Figure 5D shows the relative contribution of each of the signatures mS1–mS4 to the total alterations present in each of the tumors generated in Rad18+/+ and Rad18−/− mice. After normalizing, mS3 and mS4 were found to contribute more than 70% of the mutations in Rad18−/− tumor samples KO15, KO16 and KO26, and more than 55% of the mutations in KO18, KO19B and KO19T (Figure 5C and Supplementary Table S4). On the other hand, mS1 and mS2 contributed more than 70% of the mutations in Rad18+/+ tumors WT1, WT22, WT6 and WT8, and more than 50% of the mutations in WT23 (Figure 5C). The decreased levels of signatures mS1 and mS2 (characterized by A(T)>T(A)) in the Rad18−/− tumors (when compared with Rad18+/+) are consistent with a major role for Rad18 in mediating error-prone replication of DMBA-addicted dA residues (Figure 5C and D).
Next, we determined the relationship between mutational patterns annotated in human tumors and DMBA-induced mutational signatures in mouse tumors. The COSMIC database catalogues 30 mutational signatures identified in human cancers and referred to here as ‘hS1–30’ (where ‘h’ indicates human). The heat-map in Figure 5E shows the contribution of each of the signatures hS1-hS30 (x-axis) to the mutation patterns of DMBA-induced tumors in Rad18+/+ and Rad18−/− mice (y-axis). As shown in Figure 5E, human mutational signatures 22 and 4 contribute more to the overall mutational patterns of Rad18+/+ tumors than Rad18−/- samples. However, mutational signatures 1, 3 and 12 contribute more to the overall mutational portrait of Rad18−/− tumor genomes when compared with Rad18+/+ samples.
The data shown in Figure 5E are also depicted in Figure 5F where stacked bar charts quantitatively illustrate the reduced levels of overall mutations in Rad18−/− tumors relative to Rad18+/+, as well as the differential contributions of human COSMIC mutational signatures to Rad18+/+ and Rad18−/− tumor genomes. Notably, contributions of COSMIC signatures hS4 (P = 0.012), hS22 (P = 0.0154) and hS25 (P = 0.012) were significantly reduced in Rad18−/− tumors when compared with Rad18+/+, while hS1 (P = 0.008), hS3 (P = 0.034), hS11 (P = 0.009) and hS12 (P = 0.003) were significantly increased in Rad18−/− relative to Rad18+/+ tumors (Figure 5F). P-values here were obtained using Student's t-test and Wilcoxon Rank Sum test of the Relative Contribution between two sample groups to get the minimum P-values between the two tests, then multiple testing adjusted (FDR procedure). COSMIC Signature 22 is present in urothelial carcinoma and liver cancer and has been associated with aristolochic acid exposure, Signature 4 is found in head and neck and lung cancers and has been attributed to tobacco smoke PAHs such as Benzo[a]pyrene. Therefore, the results of Figure 5 suggest the interesting possibility that the RAD18 pathway mediates mutagenesis in response to common human exposures such as aristolochic acid and tobacco smoke. Signature 3 (which is elevated in Rad18−/− tumors) is found in breast and ovarian cancers and is associated with error-prone repair of DNA DSB.
For comparison with tumors, we also attempted to annotate mutation signatures in genomes from normal tissues of Rad+/+ and Rad18−/− mice that did not receive carcinogen. As shown in Supplementary Figure S4A, normal mouse tissues did have SNV, consistent with published findings that mutations do arise in non-cancerous tissues (50). Supplementary Figure S4A shows that C>T mutations predominate in normal tissues, regardless of Rad18 status.
We also determined the contribution of COSMIC mutation signatures 1–30 to the genomes of normal tissues. All of the COSMIC signatures which contributed differentially to Rad18+/+ and Rad18−/− tumor genomes (hS1, hS3, hS4, hS11, hS12, hS20, hS22, hS25 and hS28) made similar relative contributions to normal tissue genomes of Rad18+/+ and Rad18−/− mice. As shown in Supplementary Figure S4B and C, with the exception of hS5, hS6 and hS24 there were no significant differences in the relative contributions of any human COSMIC signatures to the overall mutational portraits when comparing Rad18+/+ and Rad18−/- normal tissue samples.
We conclude that Rad18 specifically determines both the overall mutation frequency and the relative contribution of individual COSMIC signatures to the overall mutational portraits of tumors, but does not significantly affect genomes of normal tissues.
Correlations between RAD18 and mutational patterns in human cancer
Based on our finding that Rad18 impacts mutational patterns in a mouse carcinogenesis model we investigated associations between RAD18 status and mutations in human tumors. We analyzed relationships between RAD18 and mutagenesis in the settings of LUAD and LUSC because exposure to genotoxic chemicals in cigarette smoke is a clear risk factor for these diseases (51). Indeed, tobacco smoke-induced genotoxicity and mutagenesis in lung cancer are attributed largely to PAHs such as the DMBA-related compound Benzo[a]pyrene (52). Thus, stratification of patients as ‘nonsmokers’ and ‘smokers’ (see ‘Materials and Methods’ section, Figure 6C and Supplementary Figure S5C) based on patient smoking history provided an opportunity to investigate how interplay between RAD18 and tobacco smoke impacts mutagenesis. To understand how RAD18 gene might impact mutagenesis, we compared the number of SNVs and the mutational signatures between different tumor sample groups as defined by RAD18 mRNA expression levels or RAD18 CNV (as described in ‘Materials and Methods’ section).
We also selected BLCA and KIRC to investigate associations between RAD18 status, smoking and mutagenesis (Table 1) for two reasons: First, both LUSC and LUAD cohorts mostly have small numbers of samples with RAD18 CNV gain and loss, while BLCA tumors mainly have RAD18 gain and KIRC mainly have RAD18 loss (Table 2 and Supplementary Figure S8). Second, similar to LUAD, smoking is strongly associated with the etiology of both LUSC and BLCA. Therefore, these four datasets provided interesting tumor contexts in which to investigate the mutagenic consequences of differential RAD18 expression.
Table 2.
Number of TCGA tumor samples that have CNV (including gain, neutral and loss) in the RAD18 gene
| Human tumor | CNV data alone | CNV + SNV | CNV + SNV + mRNA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gain | Neutral | Loss | Gain | Neutral | Loss | Gain | Neutral | Loss | |
| LUAD | 16 | 524 | 15 | 16 | 519 | 15 | 16 | 513 | 15 |
| LUSC | 19 | 490 | 15 | 19 | 477 | 15 | 19 | 474 | 15 |
| BLCA | 72 | 337 | 6 | 72 | 337 | 6 | 71 | 334 | 6 |
| KIRC | 1 | 509 | 79 | 0 | 316 | 45 | 0 | 315 | 41 |
RAD18 mRNA was expressed at higher levels in tumors than in adjacent normal tissues for all four tumor types (Figure 6A; Supplementary Figures S5A, 6A and 7A). LUSC, LUAD, BLCA expressed higher levels of RAD18 than KIRC. As expected, RAD18 mRNA levels were positively correlated with high RAD18 copy numbers (Figure 6B; Supplementary Figures S5B, 6B and 7B) in all four tumor types. We infer that RAD18 expression is associated with mutations (SNV) regardless of whether RAD18 expression is determined based on its transcript levels or gene copy number.
From our analysis of LUAD data, we observed that SNVs were higher in smokers versus non-smokers (Figure 6D), as also described in previous studies. Interestingly however, we found that in the smokers, total numbers of SNVs were significantly higher in tumors expressing high RAD18 (‘RAD18-high’) when compared with the group expressing low RAD18 levels (‘RAD18-low’)—see Figure 6D. However, for non-smokers, the correlation between number of total SNVs and RAD18 gene expression was less significant (Figure 6D).
Next, we analyzed the relationship between RAD18 expression and individual SNVs in LUAD. As shown in Figure 6E, three types of SNVs, specifically C(G)>A(T), C(G)>G(C), T(A)>C(G), were significantly associated with RAD18 expression, particularly in the smokers. However, while the other three SNV types (C(G)>T(A), T(A)>A(T), T(A)>G(C)) showed higher medians in the RAD18-high group when compared to RAD18-low, this trend was not significant at a P-value of 0.05 (Figure 6E). We further tested the correlation between RAD18 and the number of SNVs using a continuous scale of RAD18 gene expression, as shown in Figure 6F and G. In those analyses, RAD18 and the number of SNVs showed a significant positive-correlation in LUAD, both for total SNVs (Figure 6F) and for all individual SNV types except T(A)>G(C) in LUAD nonsmokers (Figure 6G) where the P-value was marginal (P = 0.059).
Finally, to determine whether RAD18 is likely to impact mutations in a sequence context specific manner in human tumors, we analyzed relationships between RAD18 expression and smoking status on SNVs in different possible trinucleotide settings. The results of Figure 6H suggest no significant difference in mutational patterns (in a tri-nucleotide setting) between RAD18-high and RAD18-low groups for LUAD.
In fact, smoking had greater impact on mutational patterns than RAD18, very similar to our mouse experiment in which DMBA treatment induced greater deviation from the reference genome than Rad18 status.
In BLCA, RAD18 expression was elevated in tumors when compared with normal healthy adjacent tissue (Supplementary Figure S5A), exactly as we observed in LUAD. RAD18 expression was positively associated with RAD18 CNV in BLCA (Supplementary Figure S5B). Moreover, when using RAD18 CNV to group BLCA samples, the numbers of all six SNV types were significantly higher in the ‘RAD18 CNV gain’ groups when compared with ‘RAD18-loss’ and ‘RAD18-neutral’ groups (Supplementary Figure S5C and D). As observed in LUAD, numbers of each of the six SNVs and the overall numbers of SNVs across the six sub-types were higher in RAD18-high BLCA groups for both non-smokers and smokers (Supplementary Figure S5E and F). The significant positive correlation between the number of overall SNVs and RAD18 gene expression was also evident using the continuous gene expression scale (Supplementary Figure S5G and H) in each SNV type and across six SNV types. Indeed, the association between RAD18 and SNVs was more significant in BLCA than in LUAD particularly for non-smokers.
Moreover in LUSC, as with LUAD, we observed a significant positive correlation between RAD18 (as measured by mRNA levels) and overall SNV, as well as SNV subtypes (specifically C(G)>A(T), C(G)>G(C), C(G)>T(A)—see Supplementary Figure S6D–G), when separating tumors into RAD18-high and RAD18-low groups, and by continuous RAD18 gene expression. However, in contrast with LUAD, RAD18 expression was not significantly associated with SNV in LUSC non-smokers (Supplementary Figure S6). After stratification of LUSC patients by smoking status, we observed no significant correlation between RAD18 expression or RAD18 CNV and mutational patterns in a tri-nucleotide setting (Figure 6H; Supplementary Figures S5I, J and 6H). We also confirmed previous indications that both LUAD and LUSC have prominent C→ A SNVs that are less abundant in non-smokers than smokers (53).
Taken together, the results of Figure 6, Supplementary Figures S5 and 6 are consistent with the hypothesis that RAD18 expression contributes to smoking-induced SNVs (and less significantly to basal SNVs) in multiple cancer types. Therefore, consistent with our analysis of mutational patterns of DMBA-induced tumors in mice, RAD18 is likely to promote mutagenesis in lung tumors and perhaps other cancers that are driven by environmental genotoxic exposures. To complement our analysis of how RAD18 impacts genome stability in human cancers we also examined KIRC, in which the RAD18 gene is lost in 13.4% of tumor samples—see Table 2). The unusually high incidence of RAD18 loss in KIRC is most likely due to proximity of RAD18 gene to the VHL tumor suppressor (which is frequently deleted in KIRC but not typically altered in LUAD, LUSCC or BLCA). The apparent lack of strong relationship between RAD18 CNV and mRNA expression in KIRC (Supplementary Figure S7B) is probably because RAD18 mRNA is expressed at very low levels in the kidney (https://www.proteinatlas.org/ENSG00000070950-RAD18/tissue) and is therefore largely unaffected by RAD18 gene copy number. Therefore, we conclude that the impact of RAD18 on mutagenesis and genome stability is tissue-specific and of greatest relevance to those cancers that express high levels of RAD18 mRNA.
Based on our analyses of mouse tumors showing increased numbers of Indels in Rad18−/− tumors relative to Rad18+/+ we also investigated the relationship between RAD18 expression and deletions >4bp in TCGA data from human tumors. We examined the ‘masked TCGA WES data’ in the four tumor types studied here but found that the number of Indels per tumor sample is extremely low (typically less than 10). Therefore, there is insufficient power for detecting the relationship between the number of deletions >4bp and RAD18 expression, and instead we focused our analysis on the relationship between RAD18 and SNV load in human tumors.
DISCUSSION
The extent to which TLS pathway activation depends on Rad18 has been controversial, with some investigators suggesting that activation of TLS polymerases is Rad18-independent (28,54–55). Moreover, despite extensive study, the roles of Rad18 in error-prone DNA synthesis and mutagenesis have only been studied using artificial reporter systems such the SupF assay, often yielding contradictory results (21,22). Our study is the first to test roles for Rad18 in mutagenesis in a physiological chromosomal setting. We show that Rad18-deficient mice show reduced rates of DMBA-induced mutagenesis compared to WT animals, thereby demonstrating that Rad18 is a physiologically relevant determinant of error-prone DNA synthesis on damaged DNA templates. We infer that in DMBA-treated cells, leading strand DNA synthesis (indicated by the dashed arrow in Figure 7) at sites of DNA damage (indicated by red explosion in Figure 7) is sustained in a Rad18-dependent and error-prone manner.
Figure 7.

Hypothetical Role of Rad18 in PAH-induced mutagenesis. Leading strand DNA synthesis (dashed arrow) stalls at sites of PAH-induced DNA damage such as bulky adducts and AP sites (indicated by the red explosion). In Rad18+/+ cells, the Y-family TLS DNA polymerases (Polη, Polκ, Polι, REV1) are recruited to stalled DNA replication forks and promote error-prone replicative bypass of DNA lesions, resulting in mutagenesis (SNV). In Rad18−/− cells, TLS polymerases are not efficiently recruited to sites of PAH-induced DNA damage leading to persistent replication fork stalling and eventually to fork collapse. The DBSs resulting from collapsed replication forks are re-ligated via error-prone mechanisms such as TMEJ leading to insertions and deletions. See ‘Discussion’ section for details.
DMBA genotoxicity has been used extensively for modeling carcinogenesis in experimental animals. Interestingly however, the genome maintenance mechanisms underlying DMBA-induced mutagenesis have not been defined. DMBA is known to induce DNA damage primarily (∼80%) at dA and to a lesser extent (∼20%) at dG residues, leading to G(C)>T(A) and A(T)>T(A) transversions due to dA misincorporation (56). Therefore, the most abundant Rad18-dependent substitutions, namely A(T)>T(A) and G(C)>T(A) detected in our experiments most likely result from error-prone replication and repair of DMBA-adducted dA and dG residues. Further work is necessary to identify the Y-family DNA polymerase(s) that mediate Rad18-dependent bypass at sites of DMBA-induced DNA damage. Based on past studies, Polκ and Polη are good candidate effectors of Rad18 in DMBA-induced mutagenesis: Polκ mediates bypass at B[a]P-adducted dG bases (57) and may similarly perform bypass of the structurally-related DMBA-adducted dG residues. Additionally, the mechanism of DMBA mutagenesis is thought to involve depurination of the adducted base followed by error-prone replication at the resulting apurinic/apyrimidinic (AP) non-coding site. Polη is implicated in error-prone TLS at AP sites (58) and is therefore a strong candidate mediator of Rad18-mediated mutagenesis at DMBA-induced AP sites. We also note that DMBA-induced A(T)>T(A) and G(C)>T(A) substitutions also occur (albeit to a lesser extent) in the absence of Rad18. Interestingly, in some instances Polη is recruited to stalled replication in a Rad18-independent manner (25,28,54). Therefore, Polη might also contribute to the DMBA-induced mutations that we detected in Rad18−/− mice.
What then is the source of INDELs that arise in Rad18−/− mice? Rad18-loss and TLS-deficiency lead to persistent DNA replication fork stalling and fork ‘collapse’ resulting in DSBs (see Figure 7). The DSB arising during DNA replication can be repaired via error-prone or error-free mechanisms. In a previous study, Tjisterman and colleagues clonally propagated Caenorhabditis elegans lacking TLS polymerases (Polκ and Polη) for 50 generations and identified a class of deletions that they attributed to the A-family DNA polymerase theta (Polθ, also termed PolQ) (59). PolQ mediates an error-prone mechanism of DSB repair that acts on DNA ends containing long 3′ single-stranded overhangs (60) and accounts for repairs associated with microhomologies (61). Similar to Polκ and Polη deficient worms, Rad18-deficient mice are TLS-compromised. We hypothesize therefore that the deletions detected in Rad18-deficient mouse tumors arise via the same Polq-mediated mechanism that generates deletions in Polκ and Polη-mutant C. elegans (Figure 7).
The extent to which RAD18 contributes to mutational signatures of human cancers is not known. Therefore, it was of great interest to determine relationships between whether Rad18-sensitive mutation patterns identified in our study (mS1–mS4) and human cancer-associated mutational signatures 1–30 described in the COSMIC database. We show that COSMIC signatures 22 and 4 contribute most to mutational patterns of DMBA-induced tumors and show a Rad18-dependency. Signature 22 has been found in urothelial (renal pelvis) carcinoma and liver cancers and is specifically associated with exposure to Aristolochic Acid (AA). AA is a genotoxic plant alkaloid and, similar to DMBA, primarily induces dA-linked bulky adducts leading to A(T)>T(A) transversions (62,63). Mutational signature 4 is found in smoking-associated cancers (including head and neck, LUAD, lung squamous carcinoma, small cell lung carcinoma and oesophageal cancer) and is attributed to B[a]P and other carcinogenic PAH in tobacco smoke. Based on our finding that DMBA-induced mutation patterns resembling signatures 22 and 4 are Rad18-dependent in mice, it seems very likely that the RAD18 pathway in humans contributes significantly to COSMIC mutational signatures 22 and 4 that are induced by adduct-forming chemical exposures.
Mutational signature 22 is also present in DMBA-induced tumors from Rad18−/- mice (indicating that this mutation pattern can also be generated via Rad18-independent mechanisms). However, in contrast with Rad18+/+ tumors, Signature 22 does not dominate the overall mutational profile of DMBA-induced Rad18−/− tumor genomes. Moreover, Signatures 3, 1 and 12 contribute more significantly to the overall mutational portrait of Rad18−/− genomes when compared with Rad18+/+ tumors. This observation is consistent with our interpretation that Rad18-deficient cells rely on back-up genome maintenance pathways to process the products of DMBA genotoxicity, including stalled and collapsed replication forks (Figure 7).
Signature 3 is found in BRCA1/2-mutated breast, ovarian and pancreatic cancers and is associated with defective homologous recombination (HR, an error-free DSB repair process) and channeling of DSB into error-prone TMEJ which generates INDELs with overlapping microhomology (64). Signature 1 is present in all cancer types and is associated with small INDELs, possibly indicating a mutagenic mechanism involving DSB intermediates. RAD18 contributes to activation of the HR pathway (65,66) which, similar to TLS, facilitates S-phase progression in cells harboring damaged genomes. Therefore, defects in RAD18-mediated HR and/or TLS could explain the formation of DSB and INDEL that constitute mutational signatures 3 and 1. The etiology of signature 12 is unknown and it is interesting to speculate that this signature arises due to compensatory activity by a mutagenic pathway that serves as backup when S-phase progression is impaired by HR or TLS defects (a mechanism that also explains how Signature 3 arises).
We have found no evidence that human tumors with Signatures 12, 3 or 1 are deficient in RAD18. It is formally possible that tumors containing these signatures have defects in other genes that compromise the RAD18 pathway. However, Rad18 is very important for preventing DNA replication-associated DNA damage. Therefore, we favor the hypothesis that Rad18-loss leads to a secondary effect such as DNA replication fork collapse and that subsequent error-prone repair recapitulates mutational mechanism(s) responsible for these signatures in human cancers.
Our mouse carcinogenesis experiments showing that Rad18 deficiency leads to decreased incidence of SNV provides the first demonstration that the Rad18 pathway is an important determinant of mutagenesis in cancer. Interestingly however, RAD18 is not typically deficient in human tumors (Supplementary Figure S8). Instead, RAD18 protein is pathologically stabilized and activated in cancer cells (due to its association with the Cancer/Testes Antigen MAGE-A4—which is expressed broadly in many cancer types including 48% of non-small cell lung carcinomas (67), 38% of bladder cancers (68) and 13% of breast cancers (69). Moreover, our analysis of TCGA gene expression data show that RAD18 mRNA is often overexpressed in many tumors including LUAD, LUSC and BLCA. From our past cell culture studies, even slight increases in RAD18 expression lead too DNA damage-independent induction of PCNA mono-ubiquitination and inappropriate recruitment of the error-prone Y-family DNA polymerases to DNA replication fork (17). Therefore, the aberrant high level expression of RAD18 mRNA and RAD18 protein in cancer cells is likely to be of great consequence. Our analyses show that RAD18 expression is strongly associated with increased incidence of diverse SNV in many tumors (LUAD, LUSC and BLCA) and a causal relationship between elevated RAD18 and mutagenesis is highly likely. RAD18 can activate four different Y-family TLS polymerases, all of which are mutagenic on undamaged DNA templates, while having a different subset of preferred cognate lesions and each resulting in distinct mutagenic outcomes. Therefore, RAD18 might have broad impact on mutagenesis both basally and in the face of diverse carcinogenic exposures. In addition to the new role we have demonstrated here for Rad18 as a mutagenic driver in cancer, our past work has shown that RAD18 allows neoplastic cells to tolerate intrinsic oncogene-induced DNA damage and DNA replication stress (33). The tolerance of oncogene-induced genotoxicity conferred by the RAD18 pathway also helps explain how RAD18 provides neoplastic cells with a selective advantage and facilitates tumorigenesis. The selection for RAD18 expression and RAD18 pathway activity that occurs during tumorigenesis is also likely to impact cancer therapy: RAD18-mediated genome maintenance allows cancer cells to tolerate chemotherapeutic drugs such as cisplatin (70) and camptothecin (66). Moreover, high RAD18 activity is likely to promote therapy-induced mutations that allow tumors to adapt, evolve and evade treatment.
In summary RAD18 endows neoplastic cells with important enabling characteristics (DNA damage tolerance, mutability) that contribute to multi-step tumorigenesis and chemoresistance. Therefore, RAD18 may serve as a useful biomarker of susceptibility to specific therapeutic agents. Moreover, the pathological activation of RAD18 in cancer (e.g. by MAGE-A4) may represent a new vulnerability and provide opportunities for new treatments that ameliorate DNA damage tolerance and mutability specifically in cancer cells.
Finally, our newly developed mouse genome-specific computational pipeline is a novel and innovative aspect of this study that enabled unambiguous analysis of WES data and interpretation of mutagenic mechanism using experimental mice. The smoothness and versatility of our WES analytical pipeline stems from three fundamental components: (i) SNV/indel pipeline for mouse tumor WES. (ii) Mutational signature-calling and TEMJ detection. (iii) Specific gene-associated mutation profiles in TCGA. Regarding the first component, most available bioinformatics tools for WES data are based on human genomes. Our SNV/indel pipeline for mouse tumor WES has improved the older murine-based methodologies used by Nassar et al. (47) and surpasses the current state-of-the-art mouse DNAseq application (71). Our second component serves to evaluate the genome instability in mouse WES data (e.g. by comparing WT and Rad18-null genomes in our study). Our third component uses RAD18 gene expression or CNV to define sample groups and detect the RAD18-associated mutational signatures in related human tumors. This three-component pipeline constitutes a valuable method that can be utilized for defining the mutagenic consequences of any candidate DNA repair (or other) gene, and any carcinogenic driver (including genotoxins or oncogenes) using experimental mice (and perhaps using clonal cell lines).
Overall, the concept of the mutational signature has advanced our understanding of human cancer genetics. However, the underlying mutagenic mechanisms of many COSMIC signatures remain unknown. Moreover, in many cases mechanistic underpinnings of COSMIC signatures have been inferred but not demonstrated through experimentation. Clearly to understand the mutagenic processes that drive cancer, there is a need to experimentally test the contributions of specific genes and pathways to individual mutational signatures and overall mutational portraits of tumors, exactly as we have done for Rad18. The important goal of identifying the molecular etiology of COSMIC signatures will be facilitated by the methodologies that we have described and validated in this study.
DATA AVAILABILITY
The raw WES data of the 11 mouse samples and the corresponding sample annotation file generated in this study have been submitted to the SRA database (www.ncbi.nlm.nih.gov/sra) under accession number BioProject PRJNA626010 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA626010). Source codes can be accessed via https://github.com/jitonglou/RAD18_project_data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Xiaobei Zhao, Neil Hayes, Yun Li and Leonard McMillan for valuable discussion and advice for the analysis of WES data.
Contributor Information
Jitong Lou, Department of Biostatistics, University of North Carolina at Chapel Hill, 135 Dauer Drive, 3101 McGavran-Greenberg Hall, Chapel Hill, NC 27599, USA.
Yang Yang, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, 614 Brinkhous-Bullitt Building, Chapel Hill, NC 27599, USA.
Qisheng Gu, Division of Oral and Craniofacial Health Sciences, Adam School of Dentistry, University of North Carolina at Chapel Hill, 385 S. Columbia Street, Chapel Hill, NC 27599, USA.
Brandon A Price, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Chapel Hill, NC 27599, USA; Curriculum in Bioinformatics and Computational Biology, University of North Carolina at Chapel Hill, 120 Mason Farm Road, Chapel Hill, NC 27599, USA.
Yuheng Qiu, Department of Statistics, Purdue University, 250 N. University St, West Lafayette, IN 47907, USA.
Yuri Fedoriw, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, 614 Brinkhous-Bullitt Building, Chapel Hill, NC 27599, USA.
Siddhi Desai, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, 614 Brinkhous-Bullitt Building, Chapel Hill, NC 27599, USA.
Lisle E Mose, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Chapel Hill, NC 27599, USA.
Brian Chen, Department of Biostatistics, University of North Carolina at Chapel Hill, 135 Dauer Drive, 3101 McGavran-Greenberg Hall, Chapel Hill, NC 27599, USA.
Satoshi Tateishi, Department of Cell Maintenance, Institute of Molecular Embryology and Genetics, Kumamoto University, 2-2-1 Honjo Chuoku, Kumamoto 860-0811, Japan.
Joel S Parker, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Chapel Hill, NC 27599, USA; Department of Genetics, University of North Carolina at Chapel Hill, 120 Mason Farm Road, Chapel Hill, NC 27599, USA.
Cyrus Vaziri, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, 614 Brinkhous-Bullitt Building, Chapel Hill, NC 27599, USA.
Di Wu, Department of Biostatistics, University of North Carolina at Chapel Hill, 135 Dauer Drive, 3101 McGavran-Greenberg Hall, Chapel Hill, NC 27599, USA; Division of Oral and Craniofacial Health Sciences, Adam School of Dentistry, University of North Carolina at Chapel Hill, 385 S. Columbia Street, Chapel Hill, NC 27599, USA; Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Chapel Hill, NC 27599, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health [R01 ES029079, CA215347 to C.V., D.W.]; UNC Lineberger Comprehensive Cancer Center, Developmental Award (to Y.Y.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000; 100:57–70. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 3. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A. et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rogozin I.B., Pavlov Y.I., Goncearenco A., De S., Lada A.G., Poliakov E., Panchenko A.R., Cooper D.N. Mutational signatures and mutable motifs in cancer genomes. Brief. Bioinform. 2018; 19:1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rogozin I.B., Goncearenco A., Lada A.G., De S., Yurchenko V., Nudelman G., Panchenko A.R., Cooper D.N., Pavlov Y.I. DNA polymerase eta mutational signatures are found in a variety of different types of cancer. Cell Cycle. 2018; 17:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jager M., Blokzijl F., Kuijk E., Bertl J., Vougioukalaki M., Janssen R., Besselink N., Boymans S., de Ligt J., Pedersen J.S. et al. Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer. Genome Res. 2019; 29:1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang M.N., Yu W., Teoh W.W., Ardin M., Jusakul A., Ng A.W.T., Boot A., Abedi-Ardekani B., Villar S., Myint S.S. et al. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res. 2017; 27:1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohmori H., Friedberg E.C., Fuchs R.P., Goodman M.F., Hanaoka F., Hinkle D., Kunkel T.A., Lawrence C.W., Livneh Z., Nohmi T. et al. The Y-family of DNA polymerases. Mol. Cell. 2001; 8:7–8. [DOI] [PubMed] [Google Scholar]
- 11. Prakash S., Johnson R.E., Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005; 74:317–353. [DOI] [PubMed] [Google Scholar]
- 12. Masutani C., Kusumoto R., Yamada A., Dohmae N., Yokoi M., Yuasa M., Araki M., Iwai S., Takio K., Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999; 399:700–704. [DOI] [PubMed] [Google Scholar]
- 13. Johnson R.E., Kondratick C.M., Prakash S., Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999; 285:263–265. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y., Woodgate R., McManus T.P., Mead S., McCormick J.J., Maher V.M. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007; 67:3018–3026. [DOI] [PubMed] [Google Scholar]
- 15. Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reissner T., Chaney S., Friedberg E.C., Wang Z., Carell T. et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009; 28:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kannouche P.L., Wing J., Lehmann A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004; 14:491–500. [DOI] [PubMed] [Google Scholar]
- 17. Bi X., Barkley L.R., Slater D.M., Tateishi S., Yamaizumi M., Ohmori H., Vaziri C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell. Biol. 2006; 26:3527–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hedglin M., Benkovic S.J. Regulation of Rad6/Rad18 activity during DNA damage tolerance. Annu. Rev. Biophys. 2015; 44:207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bi X., Slater D.M., Ohmori H., Vaziri C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem. 2005; 280:22343–22355. [DOI] [PubMed] [Google Scholar]
- 20. Gao Y., Mutter-Rottmayer E., Greenwalt A.M., Goldfarb D., Yan F., Yang Y., Martinez-Chacin R.C., Pearce K.H., Tateishi S., Major M.B. et al. A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat. Commun. 2016; 7:12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim H., Dejsuphong D., Adelmant G., Ceccaldi R., Yang K., Marto J.A., D’Andrea A.D. Transcriptional repressor ZBTB1 promotes chromatin remodeling and translesion DNA synthesis. Mol. Cell. 2014; 54:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeman M.K., Lin J.R., Freire R., Cimprich K.A. DNA damage-specific deubiquitination regulates Rad18 functions to suppress mutagenesis. J. Cell Biol. 2014; 206:183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roche H., Gietz R.D., Kunz B.A. Specificities of the Saccharomyces cerevisiae rad6, rad18, and rad52 mutators exhibit different degrees of dependence on the REV3 gene product, a putative nonessential DNA polymerase. Genetics. 1995; 140:443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Minesinger B.K., Jinks-Robertson S. Roles of RAD6 epistasis group members in spontaneous polzeta-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics. 2005; 169:1939–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Acharya N., Yoon J.H., Gali H., Unk I., Haracska L., Johnson R.E., Hurwitz J., Prakash L., Prakash S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:17724–17729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nikolaishvili-Feinberg N., Jenkins G.S., Nevis K.R., Staus D.P., Scarlett C.O., Unsal-Kacmaz K., Kaufmann W.K., Cordeiro-Stone M. Ubiquitylation of proliferating cell nuclear antigen and recruitment of human DNA polymerase eta. Biochemistry. 2008; 47:4141–4150. [DOI] [PubMed] [Google Scholar]
- 27. Schmutz V., Janel-Bintz R., Wagner J., Biard D., Shiomi N., Fuchs R.P., Cordonnier A.M. Role of the ubiquitin-binding domain of Poleta in Rad18-independent translesion DNA synthesis in human cell extracts. Nucleic Acids Res. 2010; 38:6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hendel A., Krijger P.H., Diamant N., Goren Z., Langerak P., Kim J., Reissner T., Lee K.Y., Geacintov N.E., Carell T. et al. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLos Genet. 2011; 7:e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wit N., Buoninfante O.A., van den Berk P.C., Jansen J.G., Hogenbirk M.A., de Wind N., Jacobs H. Roles of PCNA ubiquitination and TLS polymerases kappa and eta in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015; 43:282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krijger P.H., van den Berk P.C., Wit N., Langerak P., Jansen J.G., Reynaud C.A., de Wind N., Jacobs H. PCNA ubiquitination-independent activation of polymerase eta during somatic hypermutation and DNA damage tolerance. DNA Repair (Amst.). 2011; 10:1051–1059. [DOI] [PubMed] [Google Scholar]
- 31. Okada T., Sonoda E., Yamashita Y.M., Koyoshi S., Tateishi S., Yamaizumi M., Takata M., Ogawa O., Takeda S. Involvement of vertebrate polkappa in Rad18-independent postreplication repair of UV damage. J. Biol. Chem. 2002; 277:48690–48695. [DOI] [PubMed] [Google Scholar]
- 32. Gao Y., Tateishi S., Vaziri C. Pathological trans-lesion synthesis in cancer. Cell Cycle. 2016; 15:3005–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y., Gao Y., Mutter-Rottmayer L., Zlatanou A., Durando M., Ding W., Wyatt D., Ramsden D., Tanoue Y., Tateishi S. et al. DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J. Cell Biol. 2017; 216:3097–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nayak S., Calvo J.A., Cong K., Peng M., Berthiaume E., Jackson J., Zaino A.M., Vindigni A., Hadden M.K., Cantor S.B. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020; 6:eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun J., Yomogida K., Sakao S., Yamamoto H., Yoshida K., Watanabe K., Morita T., Araki K., Yamamura K., Tateishi S. Rad18 is required for long-term maintenance of spermatogenesis in mouse testes. Mech. Dev. 2009; 126:173–183. [DOI] [PubMed] [Google Scholar]
- 36. Yang Y., Poe J.C., Yang L., Fedoriw A., Desai S., Magnuson T., Li Z., Fedoriw Y., Araki K., Gao Y. et al. Rad18 confers hematopoietic progenitor cell DNA damage tolerance independently of the Fanconi Anemia pathway in vivo. Nucleic Acids Res. 2016; 44:4174–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bushnell B. BBMap: A Fast, Accurate, Splice-Aware Aligner. 2014; https://escholarship.org/uc/item/1h3515gn. [Google Scholar]
- 38. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keane T.M., Goodstadt L., Danecek P., White M.A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011; 477:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hunt S.E., McLaren W., Gil L., Thormann A., Schuilenburg H., Sheppard D., Parton A., Armean I.M., Trevanion S.J., Flicek P. et al. Ensembl variation resources. Database. 2018; 2018:bay119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gehring J.S., Fischer B., Lawrence M., Huber W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015; 31:3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blokzijl F., Janssen R., van Boxtel R., Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018; 10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nassar D., Latil M., Boeckx B., Lambrechts D., Blanpain C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 2015; 21:946–954. [DOI] [PubMed] [Google Scholar]
- 48. Devanesan P.D., RamaKrishna N.V., Padmavathi N.S., Higginbotham S., Rogan E.G., Cavalieri E.L., Marsch G.A., Jankowiak R., Small G.J. Identification and quantitation of 7,12-dimethylbenz[a]anthracene-DNA adducts formed in mouse skin. Chem. Res. Toxicol. 1993; 6:364–371. [DOI] [PubMed] [Google Scholar]
- 49. Cheng S.C., Prakash A.S., Pigott M.A., Hilton B.D., Lee H., Harvey R.G., Dipple A. A metabolite of the carcinogen 7,12-dimethylbenz[a]anthracene that reacts predominantly with adenine residues in DNA. Carcinogenesis. 1988; 9:1721–1723. [DOI] [PubMed] [Google Scholar]
- 50. Yizhak K., Aguet F., Kim J., Hess J.M., Kubler K., Grimsby J., Frazer R., Zhang H., Haradhvala N.J., Rosebrock D. et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019; 364:eaaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T. et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016; 354:618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pleasance E.D., Stephens P.J., O’Meara S., McBride D.J., Meynert A., Jones D., Lin M.L., Beare D., Lau K.W., Greenman C. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010; 463:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hsu Y.C., Chang Y.H., Chang G.C., Ho B.C., Yuan S.S., Li Y.C., Zeng J.W., Yu S.L., Li K.C., Yang P.C. et al. Tumor mutation burden and recurrent tumors in hereditary lung cancer. Cancer Med. 2019; 8:2179–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haracska L., Unk I., Prakash L., Prakash S. Ubiquitylation of yeast proliferating cell nuclear antigen and its implications for translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:6477–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Acharya N., Yoon J.H., Hurwitz J., Prakash L., Prakash S. DNA polymerase eta lacking the ubiquitin-binding domain promotes replicative lesion bypass in humans cells. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:10401–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chakravarti D., Pelling J.C., Cavalieri E.L., Rogan E.G. Relating aromatic hydrocarbon-induced DNA adducts and c-H-ras mutations in mouse skin papillomas: the role of apurinic sites. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:10422–10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suzuki N., Ohashi E., Kolbanovskiy A., Geacintov N.E., Grollman A.P., Ohmori H., Shibutani S. Translesion synthesis by human DNA polymerase kappa on a DNA template containing a single stereoisomer of dG-(+)- or dG-(-)-anti-N(2)-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene). Biochemistry. 2002; 41:6100–6106. [DOI] [PubMed] [Google Scholar]
- 58. Kokoska R.J., McCulloch S.D., Kunkel T.A. The efficiency and specificity of apurinic/apyrimidinic site bypass by human DNA polymerase eta and Sulfolobus solfataricus Dpo4. J. Biol. Chem. 2003; 278:50537–50545. [DOI] [PubMed] [Google Scholar]
- 59. Roerink S.F., van Schendel R., Tijsterman M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014; 24:954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yousefzadeh M.J., Wyatt D.W., Takata K., Mu Y., Hensley S.C., Tomida J., Bylund G.O., Doublie S., Johansson E., Ramsden D.A. et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLos Genet. 2014; 10:e1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wyatt D.W., Feng W., Conlin M.P., Yousefzadeh M.J., Roberts S.A., Mieczkowski P., Wood R.D., Gupta G.P., Ramsden D.A. Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks. Mol. Cell. 2016; 63:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stiborova M., Arlt V.M., Schmeiser H.H. DNA adducts formed by aristolochic acid are unique biomarkers of exposure and explain the initiation phase of upper urothelial cancer. Int. J. Mol. Sci. 2017; 18:2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hoang M.L., Chen C.H., Chen P.C., Roberts N.J., Dickman K.G., Yun B.H., Turesky R.J., Pu Y.S., Vogelstein B., Papadopoulos N. et al. Aristolochic acid in the etiology of renal cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 2016; 25:1600–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Morganella S., Alexandrov L.B., Glodzik D., Zou X., Davies H., Staaf J., Sieuwerts A.M., Brinkman A.B., Martin S., Ramakrishna M. et al. The topography of mutational processes in breast cancer genomes. Nat. Commun. 2016; 7:11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang J., Huen M.S., Kim H., Leung C.C., Glover J.N., Yu X., Chen J. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 2009; 11:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Palle K., Vaziri C. Rad18 E3 ubiquitin ligase activity mediates Fanconi anemia pathway activation and cell survival following DNA Topoisomerase 1 inhibition. Cell Cycle. 2011; 10:1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peikert T., Specks U., Farver C., Erzurum S.C., Comhair S.A. Melanoma antigen A4 is expressed in non-small cell lung cancers and promotes apoptosis. Cancer Res. 2006; 66:4693–4700. [DOI] [PubMed] [Google Scholar]
- 68. Bergeron A., Picard V., LaRue H., Harel F., Hovington H., Lacombe L., Fradet Y. High frequency of MAGE-A4 and MAGE-A9 expression in high-risk bladder cancer. Int. J. Cancer. 2009; 125:1365–1371. [DOI] [PubMed] [Google Scholar]
- 69. Otte M., Zafrakas M., Riethdorf L., Pichlmeier U., Loning T., Janicke F., Pantel K. MAGE-A gene expression pattern in primary breast cancer. Cancer Res. 2001; 61:6682–6687. [PubMed] [Google Scholar]
- 70. Nojima K., Hochegger H., Saberi A., Fukushima T., Kikuchi K., Yoshimura M., Orelli B.J., Bishop D.K., Hirano S., Ohzeki M. et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005; 65:11704–11711. [DOI] [PubMed] [Google Scholar]
- 71. Lange S., Engleitner T., Mueller S., Maresch R., Zwiebel M., Gonzalez-Silva L., Schneider G., Banerjee R., Yang F., Vassiliou G.S. et al. Analysis pipelines for canc 37er genome sequencing in mice. Nat. Protoc. 2020; 15:266–315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw WES data of the 11 mouse samples and the corresponding sample annotation file generated in this study have been submitted to the SRA database (www.ncbi.nlm.nih.gov/sra) under accession number BioProject PRJNA626010 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA626010). Source codes can be accessed via https://github.com/jitonglou/RAD18_project_data.