

Abstract
Purpose:
Preclinical evidence indicates that the bone marrow microenvironment provides a protective niche for leukemic stem cells, allowing them to evade the effects of BCR-ABL tyrosine kinase inhibitors (TKIs), but that targeting of the JAK-STAT pathway with the JAK2 inhibitor ruxolitinib increases TKI-induced apoptosis. A phase I clinical trial (NCT01702064) investigated the tolerability and safety of treating chronic-phase chronic myeloid leukemia patients with ruxolitinib in combination with the BCR-ABL TKI nilotinib and explored initial efficacy evidence.
Experimental design:
Eleven patients already treated with single-agent nilotinib (300–400 mg twice daily) commenced combination therapy, and molecular responses were evaluated after 6 months. Three ruxolitinib dose cohorts were studied: 5 mg, 10 mg, and 15 mg twice daily.
Results:
One patient experienced a grade 3/4 adverse event (hypophosphatemia) and 36% of patients experienced grade 1/2 anemia. Of 10 patients who were evaluable for responses, 40% had undetectable BCR-ABL transcripts, as measured by quantitative RT-PCR after 6 months. Plasma inhibitory assay results revealed a decrease in phospho-STAT3 levels after treatment with ruxolitinib. The recommended phase 2 dose of ruxolitinib was 15 mg BID.
Conclusions:
Overall, this combination was safe and well-tolerated, and the molecular responses were encouraging, thereby warranting further investigation in a phase 2 trial.
Keywords: Chronic myeloid leukemia, Nilotinib, Ruxolitinib, Molecular disease
1. Introduction
Chronic myeloid leukemia (CML) is characterized by the presence of the Philadelphia chromosome t(9;22)(q34;q11.2), which ultimately forms the BCR-ABL oncoprotein [1]. Tyrosine kinase inhibitors (TKIs) directed against the tyrosine kinase activity of BCR-ABL are the primary therapeutic option in CML and have proven to be extremely effective at inducing hematologic, cytogenetic, and molecular remissions. Five TKIs are currently approved by health authorities for the treatment of CML: the first-generation TKI imatinib; the second-generation TKIs dasatinib, nilotinib, and bosutinib; and the third-generation TKI ponatinib [2–6]. Nevertheless, TKIs do not completely eradicate CML, tending to leave most patients in need of lifelong therapy.
In recent years, investigators have started to question whether the discontinuation of TKIs is feasible among chronic-phase CML (CP-CML) patients who have achieved durable deep molecular responses. The first clinical trial to address this was the Stop Imatinib (STIM1) study, in which imatinib-treated patients who had ≥2 years of undetectable levels of BCR-ABL transcripts, as measured by peripheral blood quantitative real-time PCR (qRT-PCR), discontinued their imatinib. With a median follow-up time of 77 months, 61% of patients experienced molecular relapses, defined as loss of major molecular response, or 2 consecutive positive PCR results with at least a 1-log increase in BCR-ABL transcripts; nevertheless, 38% remained in treatment-free remission (TFR) [7]. Many other TFR studies have been completed since the STIM1 study with remarkably similar results, with both first- and second-generation TKIs showing TFR rates ranging from 40% to 60% [8–13]. Data from these trials were convincing enough that in 2016 the National Comprehensive Cancer Network CML Panel opted to incorporate TKI discontinuation into guidelines for the management of CML [14].
Despite these promising results, TKIs do not completely eradicate CML. Even among patients with undetectable BCR-ABL transcripts by qRT-PCR, evidence of leukemia persists when more sensitive detection methods are used [15,16]. Minimal residual disease (MRD) and biological factors that influence CML stem cells can lead to molecular relapse after treatment discontinuation [17,18]. Therefore, only a small subset of CP-CML patients worldwide are ultimately eligible for a trial of TKI cessation, whereas others never achieve deep enough molecular responses to make stopping treatment a realistic option. In most instances, eligible patients must have maintained at least a 4- or 4.5-log reduction in BCR-ABL transcripts from baseline molecular responses (MRs) of MR4.0 and MR4.5, respectively, for a minimum of 24 months prior to stopping treatment [7,9,14]. Data from clinical trials using imatinib, dasatinib, or nilotinib as first-line treatments in CP-CML suggest that approximately 20% to 55% of patients will achieve MR4.5 after 5 years of treatment, meaning that the remaining patients may not have the opportunity to attempt TKI cessation [3,6].
Preclinical data suggest that MRD is the result of BCR-ABL-independent drug resistance [19,20]. Even with increasing doses of BCR-ABL TKIs, CML cells residing in sanctuary sites such as the bone marrow are protected. The bone marrow microenvironment contains a variety of cytokines and growth factors that are capable of inducing signal transducers and activators of transcription 3- (STAT3-) Y705 phosphorylation via the Janus kinase- (JAK-) STAT pathway. Constitutive activation of this pathway can contribute to BCR-ABL-independent CML-cell survival, thereby evading the apoptotic effects of BCR-ABL TKIs. Therefore, inhibition of the phosphorylation of STAT3 via alternative pathways is required to eliminate this protective mechanism and eradicate MRD [19,20].
By using cell lines derived from CML patients and CML patient cells grown in media conditioned with HS-5 bone marrow stromal cells, our group has demonstrated that the JAK-STAT3 pathway is a promising target for further therapeutic interventions. By knocking down JAK2 and TYK2 in CML cell lines or using pan-JAK inhibitors, ruxolitinib, or small interfering RNA technology, we and others have been able to demonstrate the reversal of drug resistance against BCR-ABL TKIs in CML [19–21]. These studies have led to the belief that pharmacological inhibition of the JAK2-TYK2-STAT3 pathway could overcome bone marrow microenvironment-mediated drug resistance and possibly lead to the eradication of MRD for patients with CML. Ruxolitinib is an oral inhibitor of JAK that selectively inhibits JAK1 and JAK2 with modest-to-marked selectivity against TYK2 [22]. Ruxolitinib has health authority approval for the treatment of intermediate and high-risk myelofibrosis and polycythemia vera in patients who have had an inadequate response or are intolerant of hydroxyurea.
On the basis of these promising preclinical data, we designed a phase I clinical trial using ruxolitinib in combination with nilotinib to treat CP-CML patients, to determine the maximum-tolerated dose (MTD) of ruxolitinib and establish a toxicity profile. We also assessed preliminary evidence regarding the impact of ruxolitinib on phospho-STAT3– (pSTAT3–) Y705 in CML cell lines and the rate of MR4.5 after 6 months that was experienced by patients on the trial.
2. Patients and methods
2.1. Study population
Patients with a diagnosis of CP-CML and no history of progression to accelerated or blast-phase CML per the MD Anderson Cancer Center Criteria were enrolled in the study. Eligible patients were 18 years or older with an Eastern Cooperative Oncology Group performance status ≤ 2 and were receiving treatment with 300 or 400 mg oral nilotinib twice daily as first-, or second-line therapies. All patients had achieved complete cytogenetic responses or had been treated with nilotinib for a minimum of 6 months, and all patients had detectable BCR-ABL transcripts when tested using qRT-PCR with a sensitivity of at least 4.5 logs. At baseline, all patients had an absolute neutrophil count ≥1500 cells/dL, platelet count ≥100 000 cells/dL, total bilirubin ≤1.5 times the institutional upper limit of normal, aspartate aminotransferase and alanine aminotransferase ≤2.5 times the institutional upper limit of normal, and serum creatinine within the institutionally normal limits. This study was approved by the institutional review board and all investigators had Good Clinical Practice training and abided by the guidelines. The study was sponsored by Incyte Corporation and registered at clinicaltrials.gov (NCT01702064).
2.2. Study design
This was a phase I dose-escalation clinical trial that used a “3 + 3 design,” with 3 dose cohorts and a 2-patient expansion at the MTD. All patients remained on their prestudy doses of nilotinib. Twice daily ruxolitinib was added at 5, 10, and 15 mg in dose cohorts 1, 2, and 3, respectively. Patients remained on combination therapy for 6 months. After 6 months, ruxolitinib was discontinued, and patients continued treatment with nilotinib without any dose adjustments. Quantitative RT-PCR was used to measure BCR-ABL transcript levels in the peripheral blood and/or bone marrow every 3 months. The primary endpoint was the MTD of ruxolitinib. The period to assess for dose-limiting toxicities was the first 28 days. Secondary endpoints included toxicity assessments; rate of 4.5-log reductions in BCR-ABL transcript levels from a standardized baseline (MR4.5) at 6 months; changes in patient-reported fatigue, as assessed by the Fatigue Symptom Inventory (FSI); and pSTAT3 levels, as assessed using a plasma inhibitory assay (PIA).
2.3. Response criteria
Quantitative RT-PCR was used to measure BCR-ABL transcript levels in the blood and/or bone marrow to assess molecular responses. Complete blood counts with differentials were drawn every 2 weeks during cycles 1 and 2 and then monthly until completion of study treatment. Bone-marrow biopsies and aspirates were taken at baseline and at end of treatment to confirm complete cytogenetic response in the first 2 cohorts. End-of-treatment bone marrow biopsies were removed from the protocol by an amendment that was enacted prior to cohort 3. Adverse events were recorded at each patient visit. Patients were considered evaluable for safety if they had received ≥1 dose of ruxolitinib and for response if they had completed all 6 cycles of study treatment or if they had developed disease progression during study participation. Molecular responses were measured in log changes in BCR-ABL transcript levels measured by qRT-PCR, with a sensitivity of at least 4.5 logs. MR4.0 was defined as a 4-log reduction in BCR-ABL transcripts from a standardized baseline, which has a value of 0.01% using the International Scale (IS). MR4.5 was defined as a 4.5-log reduction in BCR-ABL transcripts from a standardized baseline, which has a value of 0.0032% on the IS.
2.4. Phospho-STAT3 analysis
Peripheral blood plasma samples were collected from 4 patients before and during treatment (on cycle 1, day 28), frozen, and later used to perform a PIA to measure pSTAT5 (Tyr694) and pSTAT3 (Ser727) activity in the K562 cell line and pSTAT5 (Tyr694) and pSTAT3 (Ser727 and Tyr705) activity in the KU812 cell [23]. For theses analyses, K562 and KU812 cells were plated at 100 000 cells per well in a 96-well plate. Each cell line was resuspended at 1 × 106 cells per mL in RPMI, 100 μL of which was added to each of 45 wells per cell line. Plasma was thawed and spun at 3000 rpm RPM for 5 min to pellet any particulates, and 75 μL of sera was added to the wells. Plates were incubated for 2 h at 37 °C, spun at 1280 RPM for 5 min, and washed twice with 100 μL of phosphate-buffered saline (PBS). The cells were then resuspended in 100 μL of 1.6% formaldehyde per well and incubated for 10 min and spun again at 1280 RPM for 5 min. Cells were washed twice with 100 μL of PBS and then resuspended in 200 μL of 95% methanol at 4 °C for 10 min. After incubation, they were spun at 1280 RPM for 5 min and washed twice more with 100 μL of PBS. This was followed by re-suspending well contents with 200 μL of flow cytometry staining buffer and storing them overnight. The following day, plates were spun at 1280 RPM for 5 min. The cells were then suspended with 50 μL of flow cytometry staining buffer (with 2 μL of Fc block per mL). Master mixes of each antibody were made, with 2 μL mL needed. Plates were incubated for 1 h, washed with 100 μL flow cytometry staining buffer, resuspended in 50 μL of flow cytometry staining buffer, and analyzed using Intellicyt iQueScreener Plus. The median fluorescent intensity (MFI) of all samples collected for isotype background values were subtracted from all samples. Percentage changes were calculated as % change = (MFI treatment/MFI pretreatment) × 100. The readout was the reduction from 100% STAT signaling. For the samples taken before and during treatment, paired t-tests were used to measure differences in pSTAT3 (Tyr705) in the KU812 cells after incubation, and paired 2-tailed t-tests were performed to determine significance values.
2.5. Fatigue symptom inventory (FSI)
Previous studies have validated the FSI as a reliable measure of fatigue in cancer patients, and the fatigue severity, interference, and duration subscales were used for this analysis. The fatigue severity subscale assesses the amount of fatigue experienced in the past week and the patient’s current level of fatigue [24–26], the fatigue interference subscale measures how fatigue has interfered in patients’ lives over the preceding week, and the fatigue duration subscale assesses the number of days patients experience fatigue per week and the amount of time they feel fatigued per day. Seven patients completed the FSI at baseline and during at least one of the 3- or 6-month follow-up visits. The 3- and 6-month follow-up visits were collapsed into a single follow-up for the purposes of our analyses. Paired samples t-tests were used to measure differences in fatigue severity, interference, and duration over time.
3. Results
3.1. Patient demographics
Between April 2013 and March 2016, 11 patients were enrolled at the H. Lee Moffitt Cancer Center and Research Institute. The median age was 41 years (range, 25–63 years) and 73% (n = 8) of patients were male. Thirty-six percent (n = 4) had received 1 TKI prior to nilotinib, while the remaining patients were taking nilotinib as first-line treatment. The nilotinib dose was 300 mg twice daily for 73% (n = 8) of patients and 400 mg twice daily for 27% (n = 3). Nilotinib was second-line therapy for all 3 patients taking 400 mg twice daily and for 1 patient taking 300 mg twice daily. The median time from diagnosis of CML to enrollment in the study was 11 months (range, 6–135 months). All patients were diagnosed in chronic phase, and none had a history of progression to advanced-phase CML (Table 1).
Table 1.
Patient characteristics.
| Patient Characteristics | N=11 |
|---|---|
| Sex, n (%) | |
| Male | 8 (73) |
| Female | 3 (27) |
| Median age, y (range) | 41 (25–63) |
| Median time from CML diagnosis, mo (range) | 11 (6–135) |
| Line of therapy | |
| First-line nilotinib, n (%) | 7 (64) |
| Second-line nilotinib, n (%) | 4 (36) |
| Nilotinib dose | |
| 300 mg twice daily, n (%) | 8 (73) |
| 400 mg twice daily, n(%) | 3 (27) |
| Evaluable for response, n (%) | 10 (91) |
| Baseline qRT-PCR (n=10), IS | 0.05% |
Abbreviations: CML, chronic myeloid leukemia; IS, International Scale; RT-PCR, real-time PCR.
3.2. Dose escalation
Ruxolitinib was given orally and concurrently with nilotinib, using the dose-expansion methodology planned. The MTD was not reached, and the 2 patients in the expansion cohort were treated with 15 mg of ruxolitinib twice daily, for a total of 5 patients treated at this dose. All 5 patients who received ruxolitinib 15 mg twice daily also took nilotinib 300 mg twice daily. As a result, the recommended phase 2 dose of ruxolitinib was established as being 15 mg twice daily.
3.3. Adverse events
All patients were eligible for safety and toxicity assessments. Treatment-emergent adverse events that investigators considered to be at least possibly related to the study combination are listed in Table 2. No dose-limiting toxicities were observed in study patients, and no dose reductions were required. The only grade 3/4 adverse event was grade 3 hypophosphatemia, which was observed in 1 patient taking nilotinib 300 mg twice daily who received ruxolitinib 15 mg twice daily. This was successfully corrected with oral potassium-phosphate supplementation. The most common adverse events across all dose levels were grades 1/2 hyperbilirubinemia (64%) and elevated alanine aminotransferase (45%). No dose reductions were required for nilotinib or ruxolitinib on the basis of these events.
Table 2.
Treatment-emergent adverse events, by ruxolitinib twice daily dose.
| Adverse Event | All doses, all grades (N=11) | Ruxolitinib 5 mg, grades 1/2 (n=3) | Ruxolitinib 10 mg, grades 1/2 (n=3) | Ruxolitinib, 15 mg (n=5) | |
|---|---|---|---|---|---|
| Grade 1/2 | Grade 3/4 | ||||
| Hyperbilirubinemia, n (%) | 7 (64) | 3 (100) | 1 (33) | 3 (60) | |
| Elevated ALT, n (%) | 5 (45) | 1 (33) | 2 (67) | 2 (40) | |
| Fatigue, n (%) | 4 (36) | 1 (33) | 1 (33) | 2 (40) | |
| Rash, n (%) | 4 (36) | 1 (33) | 3 (60) | ||
| Nausea, n (%) | 4 (36) | 2 (67) | 1 (33) | 1 (20) | |
| Anemia, n (%) | 4 (36) | 4 (80) | |||
| Dry eyes, n (%) | 3 (27) | 1(33) | 2 (40) | ||
| Elevated AST, n (%) | 3 (27) | 1 (33) | 1 (33) | 1 (20) | |
| Musculoskeletal pain, n (%) | 2 (18) | 2 (40) | |||
| Headache, n (%) | 2 (18) | 2 (66) | |||
| Hypophosphatemia, n (%) | 1 (9) | 1 (20) | |||
| Vomiting, n (%) | 1 (9) | 1 (33) | |||
| Diarrhea, n (%) | 1 (9) | 1 (33) | |||
| Abdominal bloating, n (%) | 1 (9) | 1 (33) | |||
| Hypertriglyceridemia, n (%) | 1 (9) | 1 (33) | |||
| Heartburn, n (%) | 1 (9) | 1 (20) | |||
| Dry mouth, n (%) | 1 (9) | 1 (20) | |||
| Hematochezia, n (%) | 1 (9) | 1 (20) | |||
| Constipation, n (%) | 1 (9) | 1 (20) | |||
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Grade 1/2 anemia was observed in 36% (n = 4) of patients, all of whom were in the cohort receiving 15 mg ruxolitinib twice daily (Table 3). In all cases, the investigators considered the anemia to be related to ruxolitinib, yet no patients described symptoms associated with anemia. One patient with baseline hemoglobin of 12.1 g/dL experienced a decline to 9.7 g/dL after 8 weeks on ruxolitinib; this was deemed to be treatment related, and ruxolitinib was discontinued. The progressive anemia occurred simultaneously with the start of lower gastrointestinal bleeding. After stopping ruxolitinib, the hemoglobin continued to trend down and reached a nadir of 8.7 g/dL at week 12. A colonoscopy was performed to assess the cause of the patient’s gastrointestinal bleeding and revealed mild diverticulitis, 1 non-bleeding polyp, and internal hemorrhoids that were thought to be the bleeding source. At week 13, the hemoglobin began trending up, and by week 17, it had returned to baseline. The mean change in hemoglobin from baseline to end of study in the 4 patients who developed anemia was 1.5 g/dL (range, 0.5–3.4 g/dL), and the mean change in hemoglobin from baseline to nadir during treatment was 1.7 g/dL (range, 1.2–2.4 g/dL). The median time to nadir during treatment was 10 weeks (range, 8–12 weeks).
Table 3.
Data for 4 patients who experienced adverse events of anemia.
| Patient ID | Baseline Hemoglobin, g/dL | On-treatment Hemoglobin Nadir, g/dL | End-of-study Hemoglobin, g/dL | Time to On-treatment Nadir, weeks |
|---|---|---|---|---|
| 8 | 14.7 | 13.3 | 13.9 | 12 |
| 9a | 14.5 | 13.3 | 14.0 | 8 |
| 11 | 13.7 | 11.8 | 12.3 | 12 |
| 12 | 12.1 | 9.7 | 8.7a | 8 |
Off ruxolitinib for 4 weeks prior to this end-of-study assessment.
3.4. Molecular responses
Ten patients were evaluable for responses. The patient who stopped ruxolitinib following the advent of anemia and lower gastrointestinal bleeding was removed from study treatment prior to the first BCR-ABL measurement and, therefore, was not included in the response assessment. Quantitative RT-PCR was performed to measure BCR-ABL transcript levels at baseline, after 3 months, and at the end of the study. All 11 patients had baseline molecular testing and 27% had less than MMR, 45% have values between MMR and MR4.0, and 27% had values between MR4.0 and MR4.5. Ten patients had end-of-study molecular testing and 10% had less than MMR, 40% had values between MMR and MR4.0, 10% were between MR4.0 and MR4.5, and 40% had MR4.5 (summarized in Table 4). When comparing baseline values to end-of-study values, 40% (n = 4) had ≤0.5-log change in BCR-ABL transcripts. Twenty percent (n = 2) had reductions of 0.5 to < 1 logs, 30% (n = 3) had ≥1-log reductions, and 10% (n = 1) had progressive disease.
Table 4.
Change in molecular response from baseline to end of study.
| Molecular Response | No. of Patients | |
|---|---|---|
| Baseline, n=11 | End-of-Study, n=10 | |
| > 0.1% | 3 | 1 |
| 0.01–0.1% | 5 | 4 |
| 0.0032–0.01% | 3 | 1 |
| < 0.0032% | 0 | 4 |
The median time from diagnosis to study enrollment for the 4 patients with MR4.5 was 8 months (range, 6–135 months). Three (75%) of those with MR4.5 received nilotinib as their first TKI at a dose of 300 mg twice daily and 1 (25%) had been treated with 1 prior TKI and was taking nilotinib 400 mg twice daily. Of the 6 evaluable patients who received nilotinib as their first-line TKI, 3 (50%) achieved MR4.5. The median time from diagnosis to study enrollment for these 6 patients was 9 months (range, 6–25 months).
The median qRT-PCR value on cycle 1 day 1 of study treatment was 0.05% (range, 0.002%–0.8%) reported on the IS. One patient (ID #9) had PCR > 0.0032% IS when he was screened for enrollment, making him eligible for the study; however, PCR drawn on cycle 1 day 1 was 0.0025% IS, suggesting that he had MR4.5 prior to beginning ruxolitinib. Nine patients had qRT-PCR checked after completing 6 months of study participation, and the median value was 0.0052% IS (range, 0%–0.1%). The median change in BCR-ABL transcript levels after 6 months of ruxolitinib was a 1-log reduction. All PCR results are listed and presented longitudinally in Fig. 1.
Fig. 1.
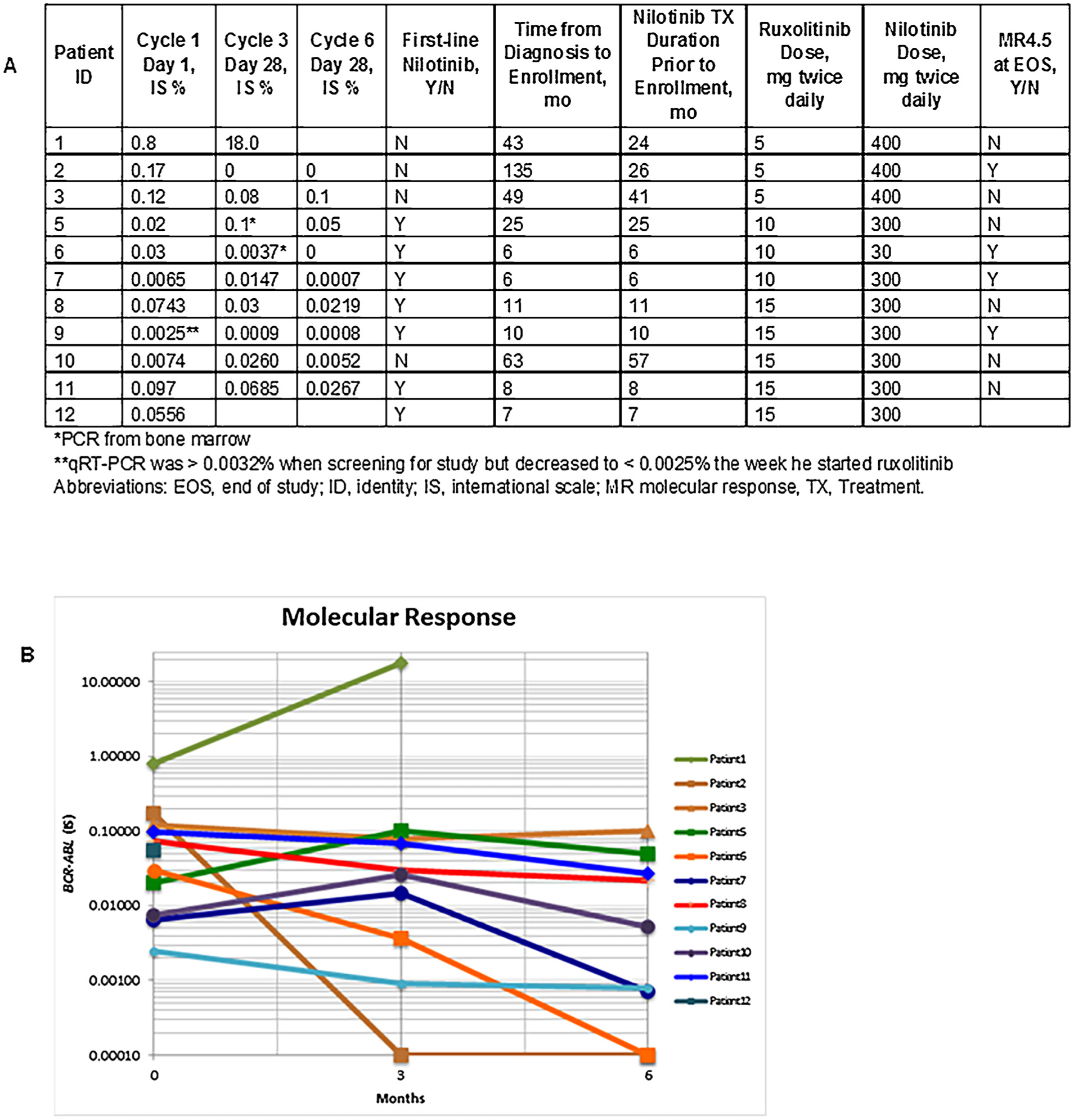
Patient responses over time. (A) Quantitative real-time PCR results, using international scale. (B) Patients’ molecular responses over the 6-month period of the clinical trial are shown according to the international scale (IS).
The 3 patients who achieved ≥ 1-log reductions in BCR-ABL transcript levels were treated with either ruxolitinib 5 mg twice daily (n = 1) or 10 mg twice daily (n = 2). One patient in cohort 1 had progressive disease after 3 months of study participation, with a loss of complete cytogenetic response. Notably, the fluorescence in situ hybridization for BCR-ABL that was ordered for this patient on cycle 1 day 1 of study treatment was positive in 18% of cells (sensitivity was 0.5%), although cytogenetics in the bone marrow remained diploid at that time. These data suggest that the disease was likely progressing prior to study enrollment. A kinase domain mutation analysis was done at the time this patient discontinued the study and no mutations were identified.
3.5. Phospho-STAT3 analyses
We used PIA to assess the impact of ruxolitinib treatment on the pSTAT3 levels of 4 patients (1 from cohort 2 and 3 from cohort 3; Table 5). Peripheral blood samples were taken and frozen at baseline and on day 28 of cycle 1. Day 28 peripheral blood samples were drawn prior to the administration of that day’s ruxolitinib. The samples had low levels of pSTAT3 (Y705) in K562 cells at baseline, making them suboptimal for assessing true changes in pSTAT3 (Y705) over time, however these cells were adequate for measuring changes in pSTAT3 (S727) and pSTAT5 (Y694). KU812 cells had higher baseline pSTAT3 (Y705) levels and were deemed a more accurate cell line for PIA analysis. We measured changes in pSTAT3 (S727) and pSTAT5 (Y694) using the baseline and ruxolitinib-treated samples. There was a statistically significant decrease in pSTAT3 (Y705) in the KU812 cells when exposed to the patient plasma samples taken during treatment, as compared to the samples taken at baseline (p = .035), suggesting an on-target effect of ruxolitinib in these instances. In the K562 cells, there was no significant change in pSTAT3 (S727); however, there was a statistically significant decrease in pSTAT5 (Y694) when the K562 cells were exposed to the ruxolitinib-treated plasma samples (p = .047) Fig. 2 shows the flow cytometry plot from patient number 11, indicating a reduction in pSTAT3 (Y705) and pSTAT5 (Y694) in the D28 sample (notated as EOS [end of study]).
Table 5.
Percentage change from baseline for peripheral blood samples taken on day 28 of cycle 1 and assessed for pSTAT3, using plasma inhibitory assay.
| Patient ID | Ruxolitinib Dose Cohort, mg | K562 cells | KU812 cells | |
|---|---|---|---|---|
| pSTAT3 (Ser727), % change | pSTAT5 (Tyr694), % change | pSTAT3 (Tyr705), % change | ||
| 7 | 10 | +12 | −55 | −4 |
| 9 | 15 | +12 | −14 | −4 |
| 10 | 15 | +12 | −62 | −7 |
| 11 | 15 | +12 | −23 | −12 |
Fig. 2.
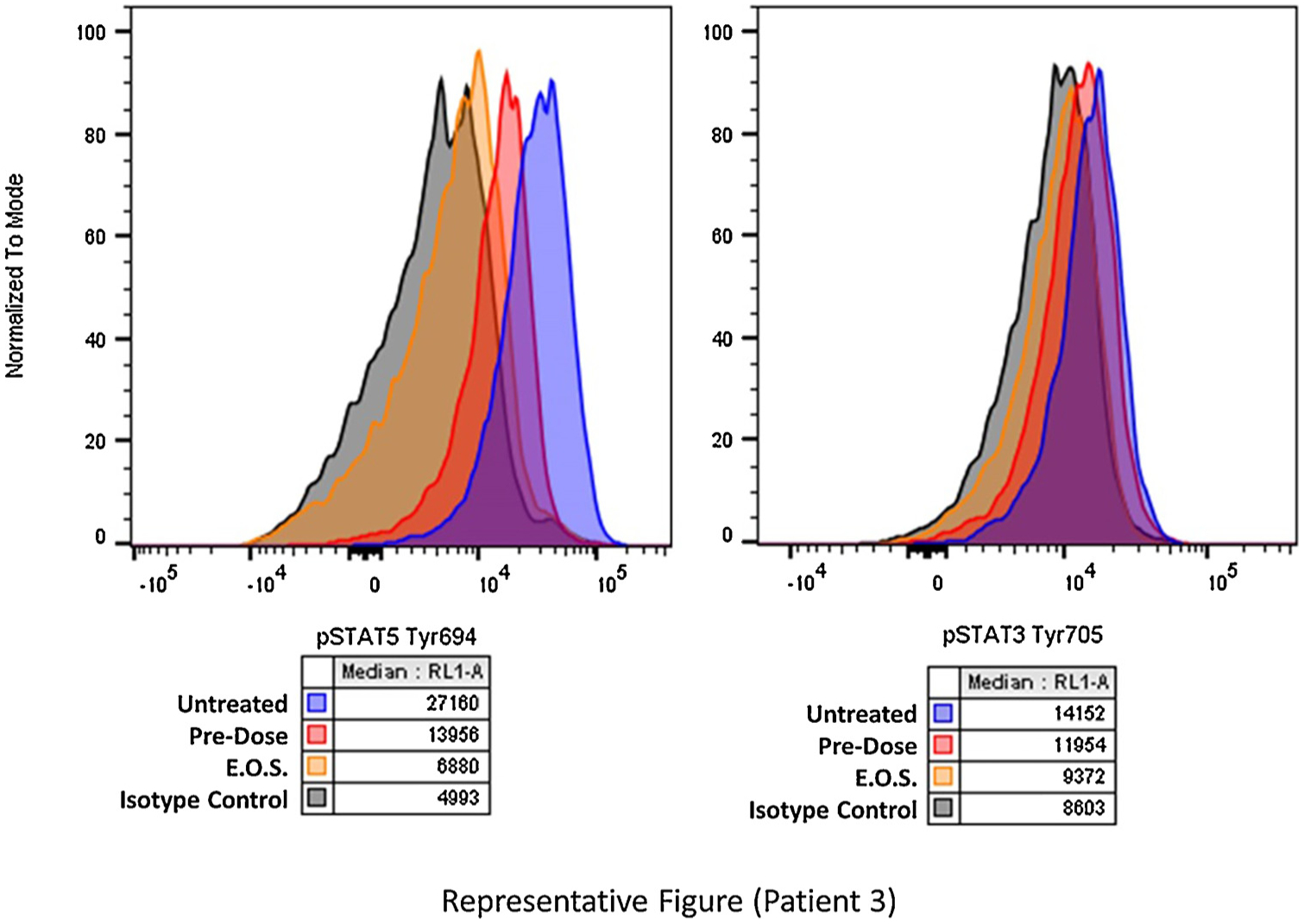
Flow cytometry plot of plasma inhibitory assay for patient number 11, indicating that pSTAT5 (Y694) and pSTAT3 (Y705) are less in the day-28 samples (notated as EOS [end of study]) than in the pre-dose samples.
3.6. Fatigue assessment
Of the 7 patients who completed FSI questionnaires, 7 were included in the analysis of fatigue severity, but only 6 were included in the analyses of fatigue interference and duration. The data, analyzed by paired-samples t-tests, showed a decline in average fatigue severity from baseline (mean 2.00, standard deviation [SD] 1.12) to follow-up (mean 1.89, SD 1.23), although this was not statistically significant at p = .29. A slight increase in fatigue interference occurred from baseline (mean 0.67, SD 0.96) to follow-up (mean 0.93, SD 1.02), which also lacked statistical significance (p = .33). In regard to fatigue duration, no statistical difference was seen in the number of days patients felt fatigued per week (baseline mean 1.67, SD 0.52 versus follow-up mean 2.00, SD 1.67; p = .68). However, there was a significant increase in the amount of time patients felt fatigued per day from baseline (mean 1.17, SD 0.98) to follow-up (mean 2.00, SD 1.55; p = .04).
4. Discussion
With the availability of BCR-ABL TKIs, the long-term outcomes for patients with CP-CML have significantly improved, so that the life expectancy for most is similar to that of the general population [27]. In spite of this, TKIs do not completely eradicate CML stem cells. Indefinite TKI therapy has been the standard of care since the development of imatinib, although many patients may experience bothersome side effects and financial hardships associated with lifelong therapy [28]. A subset of patients achieves deep molecular remissions, affording those patients the opportunity to attempt TKI cessation; nevertheless, approximately 50% relapse and must restart therapy [7–13]. To date, it has been unclear as to why TKI therapy can be stopped, if hematopoietic stem cells are inherently resistant to TKIs. Results of the CML8 study support the idea of quiescent residual stem cells. In this study, DNA-PCR for BCR-ABL was performed for 26 patients at the time of treatment discontinuation. Thirteen of these patients maintained durable treatment-free remissions despite having positive DNA-PCR results [15]. These results should be regarded with some caution, as it remains possible that years after the cessation of therapy, a leukemic stem cell with genetic features of more advanced and resistant CML could emerge among some patients. This possibility is supported by the fact that very late relapses occur after allogeneic hematopoietic stem cell transplant in some rare cases [29]. As such, the focus of many CML investigators has become the identification of ways in which to target leukemic stem cells to completely eradicate the CML clone. By targeting the JAK-STAT pathway using ruxolitinib, we have postulated that we may alter the bone marrow microenvironment and allow for BCR-ABL TKI-induced apoptosis of leukemic stem cells [19,20].
Supporting the idea that dual inhibition of BCR-ABL and the JAK-STAT pathway could eradicate leukemic stem cells, Gallipoli et al have postulated that the role of JAK2 and phospho-STAT5 in the propagation of CD34 + CML cells is more pronounced when BCR-ABL is maximally inhibited by nilotinib. Thus, the combination of BCR-ABL inhibition and STAT inhibition are crucial to the eradication of leukemic stem cells. However, some evidence supports the idea that STAT3 may compensate for the suppression of STAT5, thus making the solitary inhibition of STAT3 or STAT5 alone less effective. Targeting JAK2, which is upstream of STAT3 and STAT5, is likely a more effective approach to inhibiting both STATs [30,31].
We studied the combination of the JAK2 inhibitor ruxolitinib with the second-generation BCR-ABL TKI nilotinib to determine the MTD of ruxolitinib and establish a safety profile for this combination. Our data suggest that the combination is safe and well tolerated and does not significantly worsen patient-experienced fatigue, which is the most common side effect of TKI treatment [28]. The sample size for fatigue assessments was too small to draw meaningful conclusions. The decline in fatigue severity was encouraging, although not statistically significant, whereas the patient-reported amount of time feeling fatigued per day increased over time. These results warrant further investigation into fatigue and health-related quality of life to better elucidate the patient experience while on this treatment combination.
Only one patient experienced a grade 3/4 adverse event, which was reversible hypophosphatemia. Although 80% of patients treated with ruxolitinib 15 mg twice daily developed grades 1 or 2 anemia, most of these patients did not experience symptoms. Only one patient experienced a severe drop in hemoglobin that coincided with the advent of a lower gastrointestinal bleed. In this case, ruxolitinib was stopped after 4 weeks and by week 17 hemoglobin had returned to baseline.
Molecular responses with this combination treatment were encouraging. Although we lack the ideal historical control for this study population, 40% of patients achieving MR4.5 over a 6-month period is unexpected. When looking only at those patients who had been receiving first-line nilotinib 300 mg twice daily prior to study therapy, 50% achieved MR4.5 after 6 months on combination therapy. In contrast, the cumulative incidence curve for rates of MR4.5 in the ENESTnd trial indicate that approximately 5% to 10% of patients receiving first-line nilotinib as a single agent may achieve this response over any 6-month time span [32].
Although our sample size for PIA analyses was small, our data provide proof of the principle that the addition of ruxolitinib limits phosphorylation of STAT3 (Y705) and STAT5 (Y694), indicating an on-target effect of the drug. We believe that it is the inhibition of pSTAT3 that alters the bone marrow microenvironment, thereby lifting resistance to TKI-induced apoptosis and potentially eradicating MRD [19,20]. A significant decrease in pSTAT5 was also seen in our PIA, and this dual pSTAT3 and pSTAT5 inhibition could have more impact than the solitary inhibition of STAT3 or STAT5 alone [30,31].
The primary limitation of this phase I clinical trial is the small sample size; however, both our safety and exploratory efficacy data justify additional testing of this treatment combination. The recommended phase 2 dose of ruxolitinib is 15 mg twice daily. We cannot definitively say whether the molecular responses seen in patients enrolled on this single-arm clinical trial were truly because of the addition of ruxolitinib or whether they would have occurred with nilotinib alone. To better address this question, a randomized, phase II clinical trial of ruxolitinib plus nilotinib, or dasatinib versus nilotinib, or dasatinib alone is in development and expected to open to enrollment in 2018. A second phase 2 study in CP-CML patients who have failed a TFR attempt will use a combination of ruxolitinib and TKIs for 12 months prior to second TFR attempts and is also expected to open to enrollment in 2018. If the addition of ruxolitinib successfully alters the bone marrow microenvironment in a way that allows for TKI-induced killing of leukemic stem cells, it should follow that more patients will achieve deeper molecular responses, affording them the opportunity to attempt TKI discontinuation. If the leukemic clone is successfully eradicated with this treatment combination, this will signify the potential cure of CP-CML for some patients.
Statement of translational relevance.
Chronic myeloid leukemia stem cells are inherently resistant to tyrosine kinase inhibitors (TKIs); thus, complete eradication of this disease will likely require a combination of therapeutic agents to alter the bone marrow microenvironment and sensitize the leukemic stem cells to TKI-induced apoptosis. This phase 1 clinical trial describes safety and early efficacy data for a combination of therapies that are thought to potentially sensitize leukemic stem cells to TKIs, with the goal of fully eradicating the disease.
Acknowledgements
The authors would like to thank Sonya J. Smyk, Moffitt Cancer Center, for editorial support. She was not compensated beyond her regular salary.
Role of the funding source
This clinical trial was funded by Incyte Corporation. They provided funds and ruxolitinib for all patients on the trial, and provided early input on the design of this clinical study. Incyte was not involved in any data collection, analysis, or interpretation. Personnel at Incyte have reviewed a draft of this manuscript, but they did not participate in the writing of this report or decision to submit for publication. This work has been supported in part by the Biostatistics Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Declarations of interest
KS serves on Speakers Bureau and consults for Novartis; JPI consults for and serves on advisory boards for Novarits; LN also declares Novartis. LH, ES, FK, KH, and AN have no conflicts of interest to declare.
References
- [1].Rowley JD, Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining, Nature 243 (5405) (1973) 290–293. [DOI] [PubMed] [Google Scholar]
- [2].Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F, Fujihara S, Ortmann CE, Menssen HD, Kantarjian H, O’Brien SG, Druker BJ, Investigators I, Long-term outcomes of imatinib treatment for chronic myeloid leukemia, N. Engl. J. Med 376 (10) (2017) 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boque C, Shah NP, Chuah C, Casanova L, Bradley-Garelik B, Manos G, Hochhaus A, Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-naive chronic myeloid leukemia patients trial, J. Clin. Oncol 34 (20) (2016) 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gambacorti-Passerini C, Brummendorf TH, Kim DW, Turkina AG, Masszi T, Assouline S, Durrant S, Kantarjian HM, Khoury HJ, Zaritskey A, Shen ZX, Jin J, Vellenga E, Pasquini R, Mathews V, Cervantes F, Besson N, Turnbull K, Leip E, Kelly V, Cortes JE, Bosutinib efficacy and safety in chronic phase chronic myeloid leukemia after imatinib resistance or intolerance: minimum 24-month follow-up, Am. J. Hematol 89 (7) (2014) 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, DiPersio J, DeAngelo DJ, Abruzzese E, Rea D, Baccarani M, Muller MC, Gambacorti-Passerini C, Wong S, Lustgarten S, Rivera VM, Clackson T, Turner CD, Haluska FG, Guilhot F, Deininger MW, Hochhaus A, Hughes T, Goldman JM, Shah NP, Kantarjian H, Investigators P, A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias, N. Engl. J. Med 369 (19) (2013) 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hochhaus A, Saglio G, Hughes TP, Larson RA, Kim DW, Issaragrisil S, le Coutre PD, Etienne G, Dorlhiac-Llacer PE, Clark RE, Flinn IW, Nakamae H, Donohue B, Deng W, Dalal D, Menssen HD, Kantarjian HM, Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial, Leukemia 30 (5) (2016) 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Etienne G, Guilhot J, Rea D, Rigal-Huguet F, Nicolini F, Charbonnier A, Guerci-Bresler A, Legros L, Varet B, Gardembas M, Dubruille V, Tulliez M, Noel MP, Ianotto JC, Villemagne B, Carre M, Guilhot F, Rousselot P, Mahon FX, Long-term follow-up of the French stop imatinib (STIM1) study in patients with chronic myeloid leukemia, J. Clin. Oncol 35 (3) (2017) 298–305. [DOI] [PubMed] [Google Scholar]
- [8].Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, Dang P, Goyne JM, Slader C, Filshie RJ, Mills AK, Melo JV, White DL, Grigg AP, Hughes TP, Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study, Blood 122 (4) (2013) 515–522. [DOI] [PubMed] [Google Scholar]
- [9].Hochhaus A, Masszi T, Giles FJ, Radich JP, Ross DM, Gomez Casares MT, Hellmann A, Stentoft J, Conneally E, Garcia-Gutierrez V, Gattermann N, Wiktor-Jedrzejczak W, le Coutre PD, Martino B, Saussele S, Menssen HD, Deng W, Krunic N, Bedoucha V, Saglio G, Treatment-free remission following frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: results from the ENESTfreedom study, Leukemia 31 (July (7)) (2017) 1525–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mahon F.-x., Richter J, Guilhot J, Hjorth-Hansen H, Almeida A, Janssen JJWMJ, Mayer J, Porkka K, Panayiotidis P, Stromberg U, Berger MG, Diamond J, Ehrencrona H, Kairisto V, Machova Polakova K, Mueller MC, Mustjoki S, Hochhaus A, Pfirrmann M, Saussele S, Cessation of tyrosine kinase inhibitors treatment in chronic myeloid leukemia patients with deep molecular response: results of the euro-ski trial, Blood 128 (22) (2016) 787–787. [Google Scholar]
- [11].Kadowaki N, Kawaguchi T, Kuroda J, Nakamae H, Matsumura I, Miyamoto T, Ishikawa J, Nagafuji K, Imamura Y, Yamazaki H, Shimokawa M, Akashi K, Kanakura Y, Discontinuation of nilotinib in patients with chronic myeloid leukemia who have maintained deep molecular responses for at least 2 years: a multicenter phase 2 stop nilotinib (nilst) trial, Blood 128 (22) (2016) 790–790. [Google Scholar]
- [12].Kumagai T, Nakaseko C, Nishiwaki K, Yoshida C, Ohashi K, Takezako N, Takano H, Kouzai Y, Murase T, Matsue K, Morita S, Sakamoto J, Wakita H, Sakamaki H, Inokuchi K, Discontinuation of dasatinib after deep molecular response for over 2 years in patients with chronic myelogenous leukemia and the unique profiles of lymphocyte subsets for successful discontinuation: a prospective, multicenter Japanese trial (D-STOP trial), Blood 128 (22) (2016) 791–791. [Google Scholar]
- [13].Hughes TP, Boquimpani CM, Takahashi N, Benyamini N, Clementino NCD, Shuvaev V, Ailawadhi S, Lipton JH, Turkina AG, Moiraghi EB, Nicolini FE, Dengler J, Sacha T, Kim D-W, Fellague-Chebra R, Acharya S, Krunic N, Jin Y, Mahon F-X, Treatment-free remission in patients with chronic myeloid leukemia in chronic phase according to reasons for switching from imatinib to nilotinib: sub-group analysis from ENESTop, Blood 128 (22) (2016) 792–792. [Google Scholar]
- [14].NCCN, Clinical Practice Guidelines in Oncology Chronic Myeloid Leukemia, (Version 1.2018), (2017) https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf.
- [15].Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA, Slader C, Field C, Dang P, Filshie RJ, Mills AK, Grigg AP, Melo JV, Hughes TP, Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR, Leukemia 24 (10) (2010) 1719–1724. [DOI] [PubMed] [Google Scholar]
- [16].Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, Arber DA, Slovak ML, Forman SJ, Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment, Blood 101 (12) (2003) 4701–4707. [DOI] [PubMed] [Google Scholar]
- [17].Cortes J, O’Brien S, Kantarjian H, Discontinuation of imatinib therapy after achieving a molecular response, Blood 104 (7) (2004) 2204–2205. [DOI] [PubMed] [Google Scholar]
- [18].Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O, Blanchet O, Marit G, Gluckman E, Reiffers J, Gardembas M, Mahon FX, Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years, Blood 109 (1) (2007) 58–60. [DOI] [PubMed] [Google Scholar]
- [19].Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA, Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance, Mol. Cancer Ther 7 (10) (2008) 3169–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nair RR, Tolentino JH, Argilagos RF, Zhang L, Pinilla-Ibarz J, Hazlehurst LA, Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML, Leuk. Res 36 (6) (2012) 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Quintarelli C, De Angelis B, Errichiello S, Caruso S, Esposito N, Colavita I, Raia M, Pagliuca S, Pugliese N, Risitano AM, Picardi M, Luciano L, Saglio G, Martinelli G, Pane F, Selective strong synergism of Ruxolitinib and second generation tyrosine kinase inhibitors to overcome bone marrow stroma related drug resistance in chronic myelogenous leukemia, Leuk. Res 38 (2) (2014) 236–242. [DOI] [PubMed] [Google Scholar]
- [22].Chiappori AA, Soliman H, Janssen WE, Antonia SJ, Gabrilovich DI, INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect, Expert Opin. Biol. Ther 10 (6) (2010) 983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, DeAngelo D, Galinsky I, Giles F, Estey E, Kantarjian H, Cohen P, Wang Y, Roesel J, Karp JE, Small D, Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors, Blood 108 (10) (2006) 3477–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hann DM, Jacobsen PB, Azzarello LM, Martin SC, Curran SL, Fields KK, Greenberg H, Lyman G, Measurement of fatigue in cancer patients: development and validation of the fatigue symptom inventory, Qual. Life Res 7 (4) (1998) 301–310. [DOI] [PubMed] [Google Scholar]
- [25].Broeckel JA, Jacobsen PB, Horton J, Balducci L, Lyman GH, Characteristics and correlates of fatigue after adjuvant chemotherapy for breast cancer, J. Clin. Oncol 16 (5) (1998) 1689–1696. [DOI] [PubMed] [Google Scholar]
- [26].Donovan KA, Jacobsen PB, Small BJ, Munster PN, Andrykowski MA, Identifying clinically meaningful fatigue with the fatigue symptom inventory, J. Pain Symptom Manage 36 (5) (2008) 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gambacorti-Passerini C, Antolini L, Mahon FX, Guilhot F, Deininger M, Fava C, Nagler A, Della Casa CM, Morra E, Abruzzese E, D’Emilio A, Stagno F, le Coutre P, Hurtado-Monroy R, Santini V, Martino B, Pane F, Piccin A, Giraldo P, Assouline S, Durosinmi MA, Leeksma O, Pogliani EM, Puttini M, Jang E, Reiffers J, Piazza R, Valsecchi MG, Kim DW, Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib, J. Natl. Cancer Inst 103 (7) (2011) 553–561. [DOI] [PubMed] [Google Scholar]
- [28].Efficace F, Baccarani M, Breccia M, Alimena G, Rosti G, Cottone F, Deliliers GL, Barate C, Rossi AR, Fioritoni G, Luciano L, Turri D, Martino B, Di Raimondo F, Dabusti M, Bergamaschi M, Leoni P, Simula MP, Levato L, Ulisciani S, Veneri D, Sica S, Rambaldi A, Vignetti M, Mandelli F, Gimema, health-related quality of life in chronic myeloid leukemia patients receiving long-term therapy with imatinib compared with the general population, Blood 118 (17) (2011) 4554–4560. [DOI] [PubMed] [Google Scholar]
- [29].Socie G, Stone JV, Wingard JR, Weisdorf D, Henslee-Downey PJ, Bredeson C, Cahn JY, Passweg JR, Rowlings PA, Schouten HC, Kolb HJ, Klein JP, Long-term survival and late deaths after allogeneic bone marrow transplantation. Late effects working committee of the International bone marrow transplant registry, N. Engl. J. Med 341 (1) (1999) 14–21. [DOI] [PubMed] [Google Scholar]
- [30].Gallipoli P, Cook A, Rhodes S, Hopcroft L, Wheadon H, Whetton AD, Jørgensen HG, Bhatia R, Holyoake TL, JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo, Blood 124 (9) (2014) 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin H, Chen M, Rothe K, Lorenzi MV, Woolfson A, Jiang X, Selective JAK2/ABL dual inhibition therapy effectively eliminates TKI-insensitive CML stem/progenitor cells, Oncotarget 5 (18) (2014) 8637–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Larson RA, Kim D-W, Issaragrilsil S, le Coutre P, Dorlhiac Llacer PE, Etienne G, Clark RE, Flinn I, Nakamae H, Hochhaus A, Saglio G, Kantarjian HM, Donohue B, Deng W, Menssen HD, Hughes TP, Efficacy and safety of nilotinib (NIL) vs imatinib (IM) in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): long-term follow-up (f/u) of ENESTnd, Blood 124 (21) (2014) 4541–4541. [Google Scholar]