

Abstract
Side population (SP) cells with stem-like properties, also known as cancer stem cells (CSC) have been recognized as drivers of the resistance phenotype in many cancers. Central to the characteristic stem-like phenotype of CSCs in cancer is the activity of the SOX2 transcription factor whose upregulation has been associated with enrichment of many oncogenes. This study outlines the fabrication of a lipoplex of SOX2 small interfering RNA (CL-siSOX2) for targeted treatment of SOX2-enriched, CSC-derived orthotopic and xenograft lung tumors in CB-17 SCID mice. CL-siSOX2 induced tumor contraction in cisplatin-naïve and cisplatin-treated groups by 85% and 94% respectively. Reduction in tumor weight and volume following treatment with CL-siSOX2 was associated with reduced protein expression of SOX2 and markers of tumor initiation, inflammation, invasion and metastasis in mice tumor xenografts. In addition, histological staining of lung tumor sections showed reduction in SOX2 expression was associated with inhibition markers of epithelial-to-mesenchymal transition.
Keywords: Nanoparticles, Cationic lipoplex, SOX2 siRNA, Gene delivery, Cancer stem cells, Lung cancer
Lung cancer is the leading cause of cancer-related deaths globally. In 2017, it was estimated that lung cancer diagnosis and deaths accounted for 13.2% and 25.9% respectively of the national cancer statistics in the United States (https://seer.cancer.gov/csr/1975_2014/). Effective treatment of lung cancer is hindered by severe adverse effects and acquired resistance due to lack of specificity of therapies, and up-regulation of and/or mutations in oncogenes respectively. Acquired tumor resistance is a complex process that is driven by the dynamic nature of cancer cells to sustain proliferation and evade cell death.1 Importantly, up-regulation and/or mutation of oncogenes have been shown to drive the resistance phenotype to more advanced intractable stages of tumor growth.1 The identification and therapeutic targeting of these oncogenes forms the basis of a new approach to cancer treatment known as targeted therapy.
A subset of tumor cells has been recognized as enabling replicative immortality in many tumor types including lung tumors. These side populations (SPs) are also known as cancer stem cells (CSCs) because they share similar properties such as self-renewal and pluripotency with embryonic stem cells.2 The CSC theory adduces a critical role for CSCs in promoting metastasis of cancer and enabling drug resistance.3 These CSCs are characterized by low cell cycling potentials, which enable them to escape most cytotoxic therapies that target rapidly dividing cells. CSCs from different types of cancer have been widely characterized to overexpress embryonic stemness factors including SOX2, OCT4, Nanog, and KLF4, as well as multidrug resistance markers including c-Myc, P-glycoprotein, MRP1–6 and ABCG2.4–6
SOX2 (SRY HMG-Box 2) is a transcription factor that regulates oncogenic signaling pathways in different types CSCs. In human lung CSCs, a positive feedback loop between SOX2 and EGFR sustained high tumorigenic properties, and upregulation of SOX2 enhanced self-renewability and resistance of cancer cells to EGFR inhibitors.2,7 Silencing of SOX2 in mice bearing A549 xenograft tumors decreased the expression of mediators of Wnt signaling in a subset of EGFR-mutant human lung adenocarcinoma cells constitutively expressing SOX2; treatment with PI3K/Akt inhibitors depleted SOX2 expression and hindered cell proliferation.8,9 And cooperation between SOX2, beta catenin and POU5F1 promoted insulin-like growth factor-1 (IGF-1)-mediated self-renewal and tumorigenicity whereas inhibition of SOX2 abrogated cancer cell growth and metastasis.6,8,10
Currently, no drug is approved for SOX2 targeted therapy in cancer. However, evidence of the therapeutic value of SOX2 targeting has been demonstrated with cancer stem cell (CSC)-selective compounds (e.g. Salinomycin and all-trans retinoic acid),11,12 mTOR allosteric inhibitor (e.g. Rapamycin),13 experimental SOX2 immunotherapies (e.g. SOX2 DNA and peptides),14,15 and gene therapies (e.g. microRNA; miRNA, and small interfering RNA; siRNA).16,17 Importantly, molecular tools such as RNA provide opportunities for demonstrating the potential clinical benefits of SOX2-specific targeted therapies. The usefulness of these molecular tools, however, is often offset by challenges to their stability, tumor targeting, and efficient cellular uptake in vivo. The ideal delivery system for macromolecule therapies (e.g. siRNA) in lung cancer must be able to evade the effects of degrading enzymes and antibodies, as well as promote the specific targeting to the tumor cells and subsequent extravasation of the therapy.
Lipid nanocarriers have been shown to provide the versatility for ensuring ideal delivery characteristics for macromolecules in vivo.18–20 We previously reported the successful formulation and delivery of liposomes containing short hairpin RNA (shRNA) targeting Annexin A2 (AnxA2) in a mouse orthotopic lung tumor model.21 In the current study, SOX2 siRNA was formulated in a cationic lipoplex nanocarrier for targeted treatment of SOX2-enriched, H1650 CSC-derived lung tumors in mice.
Materials and methods
Chemicals
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-mPEG [2000]) were obtained from Avanti Polar Lipids (Alabaster, AL). DMEM:F12 culture medium, fetal bovine serum (FBS), penicillin/streptomycin/neomycin (PSN) cocktail, nitrogen supplement (10×), and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-tricarbocyanine iodide (DiR) were obtained from Life Technologies (Grand Island, NY). Laminin, poly-D-Lysine, fibroblast growth factor (FGF) and epidermal growth factor (EGF) were from Sigma Aldrich (St. Louis, MO). Matrigel® extracellular matrix was procured from BD Biosciences (Bedford, MA). Lipidure®-coat plates were kindly donated by NOF Corporation (Japan). Primary and secondary antibodies were from Cell Signaling Technology (Danvers, MA). Control and SOX2 siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The SOX2 siRNA (sc-38,408) consisted of a triplex with the following sequences (5′ → 3′ orientation): (sc-38408A) sense: GAAUGGACCUUGUAUAGAUTT; antisense: AUCUAUACAAGGUCCAUUCTT, (sc-38408B) sense: GGACAGUUGCAAACGUGAATT; antisense: UUCACGUUUGCAACUGUCCTT, and (sc-38408C) sense: GAAUCAGUCUGCCGAGAAUTT; antisense: AUUCUCGGCAGACUGAUUCTT. Cultrex® cell migration assay kit was obtained from Trevigen (Gaithersburg, MD). All other chemicals and reagents were of cell culture or reagent grade.
Cell lines
Human A549 and H1650 cells were procured from the American Type Culture Collection (ATCC) and sorted into main population (MP) and side population (SP) cells as previously described.7 MP cells were maintained in DMEM:F12 media, supplemented with FBS (10% v/v) and a PSN cocktail (2% v/v). SP cells were cultured in an ultra-low attachment culture flask coated with a thin film of simulated basement membrane layer consisting of laminin-bound poly-D-Lysine. Cells were refreshed with serum-free selective base media containing FGF (10 μg/mL), EGF (10 μg/mL), nitrogen supplement (1X), and PSN cocktail (2% v/v). Cells were maintained under standard culture conditions of 95% air and 5% CO2 at 37 °C.
H1650 SP and H1650 MP sphere formation in suspension
Side population (SP) or main population (MP) cells from H1650 and A549 were plated in ultra-low attachment 96 well plates at a density of 10,000 cells/well in 100 μL medium (100,000 cells/mL) in serum free stem cell selective media and incubated for 10 days to form the spheres. The total number of spheres greater than 50 μm size was counted using a phase contrast microscope (Nikon Instruments). Results were presented as average number of spheres with standard deviation of triplicates of each experiment.
H1650 SP and H1650 MP cell migration assay
Cell migration assay was performed based on the modified Boyden chamber assay method using Trevigen’s Cultrex® cell migration 24-well assay kit according to manufacturer’s protocol, and a detailed description of the procedure is provided in the Supplementary Materials and Methods section.
H1650 SP and H1650 MP cell viability
Cell viability studies of H1650 SP and MP cells were performed with cisplatin treatment as previously described.21 In summary, cells (1 × 104 cells/well) were inoculated in a 96-well format for 16–18 h followed by treatment with different concentrations of cisplatin for 72 h. Cells were sequentially fixed in glutaraldehyde (0.025% w/v), stained with crystal violet solution (0.01% w/v), and resuspended in disodium hydrogen solution. Absorbance readings were taken at 540 nm to determine cell viability. Results representing 4–6 separate experiments were presented as average of percentage of cells inhibited vs. treatment with SEM.
SOX2 protein expression in H1650 SP and H1650 MP cells
SOX2 protein expression in H1650 SP or H1650 MP cell lysates was performed per standard protocol as previously described.21 A detailed outline of the procedure is provided in the Supplementary Materials and Methods section.
Preparation and characterization of cationic lipoplexes
A detailed description of the preparation and characterization of cationic lipoplexes (CL) from combining DOTAP, DPPC and DSPE-mPEG [2000] as adapted from our previous study,21 with a detailed description of the procedure provided in the Supplementary Materials and Methods section.
Animals study assurances
Four- to six-week old male SCID-beige mice with mean weight of 21 gram (n = 45) (C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7) were procured from Taconic Biosciences (Cambridge City, IN). All animals were maintained on a standard animal chow and water ad libitum. Criteria for excluding mice from study, or early termination of treatment included significant loss (>25%) in body weight of mice from baseline and/or visual observation of general distress. Animals were randomized into 9 groups (n = 5) and allowed to acclimate for 6 days. All animal study protocols were approved by the Florida A&M University Institutional Animal Care and Use Committee (IACUC). Procedures involving animals were carried out in an AAALAC-approved facility located at Florida A&M University-College of Pharmacy and Pharmaceutical Sciences’ F.H. Humphies Science Research Building.
Tissue kinetics of fluorescent cationic lipoplexes in mice
SCID-beige mice (n = 5) were injected (intraperitoneal, i.p.) with 100 μL of fCL. Animals were sacrificed at hourly intervals under isoflurane exposure followed by resectioning of the lungs. The kinetics of the fCL on the basis of lung tissue perfusion was investigated by digital imaging using the Carestream Molecular Imaging In-Vivo MS FX PRO (Bruker, Billerica, MA). Results representing three different readings were taken and presented as total flux (perfusion per second) vs. time (h).
Animal model of xenograft and orthotopic tumor and treatment
SCID-beige mouse modeling of orthotopic and xenograft lung tumors are as previously described,21–23 and treatment are detailed in the Supplementary Materials and Methods section.
Efficacy studies
Efficacy assessment of treatment on xenograft tumor growth progression over the treatment duration was performed on the basis of body weight variations from baseline (gram), tumor volume (mm3), tumor weight (mg), immunohistochemistry (IHC) staining and hematoxylin–eosin (H&E) staining. Xenograft tumor measurements were performed using an electronic caliper on day 0, 4, 6, 8, 11 and 13, and the tumor volume estimated according to:
where, W = width of tumor, and L = length of tumor.
IHC staining for SOX2, E-cadherin and N-cadherin, and H&E staining of resected lung tissue were performed as previously described.22 Results for body weight, tumor volume and tumor weight were presented as graphs of means with standard deviation (n = 5).
Immunoblotting of tumor lysates
Assessment of protein expression of xenograft tumors was performed by western blot as previously described,24 with detailed description provided in the Supplementary Materials and Methods section.
Statistical analysis
Where applicable, results were analyzed using the GraphPad Prism version 5.0 software (GraphPad Software, Inc.; La Jolla, CA). Statistically significant differences between groups were determined by two-tailed unpaired student t test at 95% confidence interval where P < 0.05 is considered significant.
Results
H1650 SP cells are upregulated for SOX2 and exhibit resistance phenotypes to chemotherapy
A549 MP and SP cells produced 9.33 ± 1.57 and 26.33 ± 1.63 spheres (>50 μm3) respectively and H1650 MP and SP produced 17.33 ± 2.65 and 36.33 ± 2.30 spheres respectively (Figure 1, A). H1650 SP exhibited a 1.5-fold increase in number of spheroid formed (Figure 1, B), as well as a 1.4-fold increase in migratory potential (Figure 1, C) compared to H1650 MP cells. Treatment of H1650 MP and SP cells with cisplatin yielded significant differences in mean cell viabilities with IC50 values of 11.03 ± 3.65 μMand 136.12 ± 7.32 μM respectively (Figure 1, D). H1650 SP cells had a 1.7-fold increase in SOX2 protein expression compared to H1650 MP cells (0.87 ± 0.05 vs. 0.52 ± 0.04 respectively) (Figure 1, E).
Figure 1.

H1650 SP cells have high tumorigenic and migratory potentials, which drive resistance to chemotherapy. (A) H1650 SP suspension cultures were more tumorigenic compared to H1650 MP cells, as well A549 SP and MP cells. (B) H1650 SP adherent cultures readily propagate as spheroids on Lipidure®-coat plates; (C) H1650 SP cells have higher migration rates compared to main population cells; (D) H1650 SP cells exhibit higher resistance potential to 72-h treatment with cisplatin compared to H1650 MP cells; and (E) SOX2 is significantly upregulated in H1650 SP cells compared H1650 MP cells. Statistical analysis: sphere (****P < 0.0001); cell viability, IC50 (**P = 0.0073); cell migration (**P = 0.0024); and SOX2 expression (****P < 0.0001).
Preparation and characterization of cationic lipoplexes loaded with siSOX2
Table 1 shows parameters for characterization of cationic lipoplexes (CL) loaded with or without control siRNA (CL-siScr) and SOX2 siRNA (CL-siSOX2). Results represent means with standard deviation.
Table 1.
Characterization of siRNA-loaded cationic lipoplexes.
| Formulation | Hydrodynamic Diameter (nm) | Polydispersity Index | Zeta Potential (mV) | Entrapment Efficiency (%) |
|---|---|---|---|---|
| CL | 78.50 ± 2.12 | 0.30 ± 0.12 | 18.72 ± 1.26 | — |
| CL-siScr | 93.50 ± 0.71 | 0.30 ± 0.03 | 16.76 ± 0.97 | 94.22 ± 5.36 |
| CL-siSOX2 | 93.50 ± 0.71 | 0.20 ± 0.12 | 15.33 ± 1.06 | 92.59 ± 3.12 |
Lipoplexes were prepared with the phospholipids DPPC, DOTAP, and DSPE-PEG (2000) at an optimized lipid/siRNA (N/P) ratio of 2.6:1. Lipoplexes were dispersed in HEPES-buffered deionized water (pH 7–7.4) and characterized on the basis of size (nm), polydispersity, surface charge (mV) and entrapment efficiency (%). Results represent mean with standard deviation.
Lung and tumor kinetics of cationic lipoplexes in mice
Figure 2 represents the kinetics of fluorescent labeled cationic lipoplexes (fCL) in xenograft tumor (Figure 2, A) and organs (Figure 2, B) at different exposure times. Total flux of fCL in tumor tissue at 1, 2, 3, and 4 h were 25.38 ± 6.82, 54.81 ± 8.52, 84.80 ± 12.26, and 100 ± 15.18 perfusions per second (p/sec) respectively. Significant differences in tumor tissue uptake were observed at 1 h vs. 2 h and 2 h vs. 3 h. The total flux of fCL in lung estimated hourly from 1 to 5 h were 7698 ± 945, 9325 ± 1620, 5984 ± 893, 6256 ± 1104, and 9012 ± 1836 (p/sec) respectively (Figure 2, B).
Figure 2.
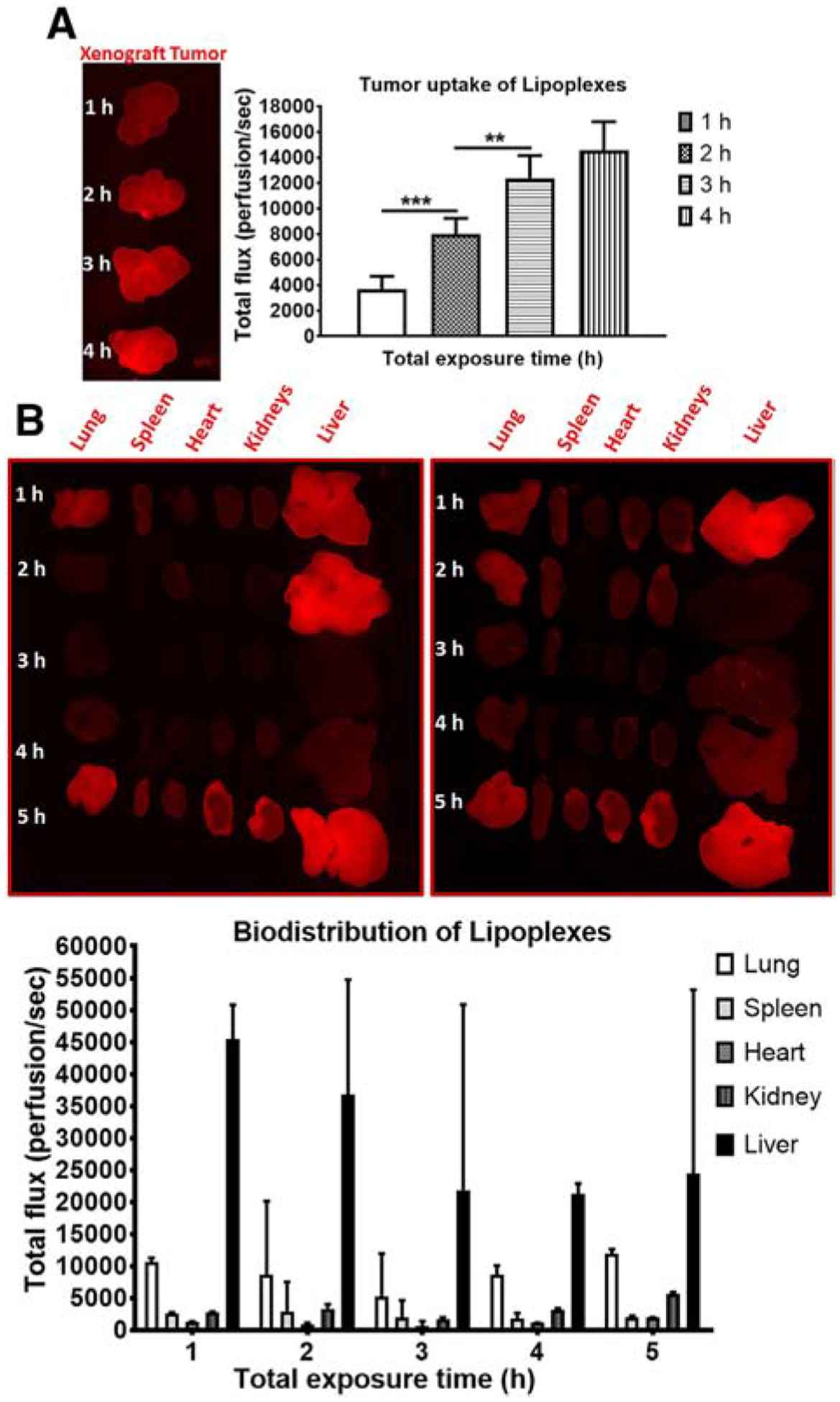
Fluorescent cationic lipoplexes (fCL) are efficient in trafficking to lung and xenograft tumor tissue in mice. DiR-labeled lipoplexes (fCL) were administered into C.B.17 SCID mice bearing H1650 SP as xenograft tumors. Distribution kinetics (A) tumor and (B) organs were determined on the basis of fluorescence intensity (perfusion/sec) using the Carestream Molecular Imaging In-Vivo MS FX PRO (Bruker). Statistical analysis: **P = 0.0020 and ***P = 0.0003.
Cationic lipoplexes loaded with SOX2 siRNA (CL-siSOX2) reverse tumor growth in mice
Cationic lipoplexes encapsulating SOX2 siRNA (CL-siSOX2) were effective in inducing tumor regression in both orthotopic and xenograft tumors in mice after single treatment or combination with cisplatin (Figure 3, A). Figure 3, B shows mice body weight measurements over the total treatment course with a significant decrease in body weight observed in cisplatin group compared to CL-siScr and CL-siSOX2 groups at day 8; all mice in the cisplatin group were sacrificed on day 8 due to significant loss in body weight (25.88%) and a general observation of emaciation and distress. Although, body weight in the CL-siSOX2 + cisplatin group decreased by 12.94% (day 8), 22.35% (day 13) and 25.88% (day 15), animals did not exhibit the distress observed in the cisplatin group. Notwithstanding, tumor volume reduction (% baseline) in the cisplatin group at day 8 (38.31 ± 29.92%) was comparable to that observed in CL-siSOX2 and CL-siSOX2 + cisplatin at day 8 (35.97 ± 25.97% and 31.28 ± 22.47% respectively), with the tumor in the latter two groups decreasing further on day 11 (38.90 ± 26.08% and 29.86 ± 8.46% respectively) and day 13 (37.00 ± 23.77% and 25.35 ± 12.99% respectively) (Figure 3, C). Tumor weight measurements (mg) as an endpoint measure of efficacy of treatment with CL-siScr, cisplatin, CL-siSOX2 and CL-siSOX2 + cisplatin were 579.53 ± 13.74, 75.37 ± 8.56, 73.60 ± 17.11 and 29.45 ± 7.15 respectively (Figure 3, D). The decrease in tumor weight in the cisplatin, CL-siSOX2 and CL-siSOX2 + cisplatin groups were significant compared to CL-siScr.
Figure 3.
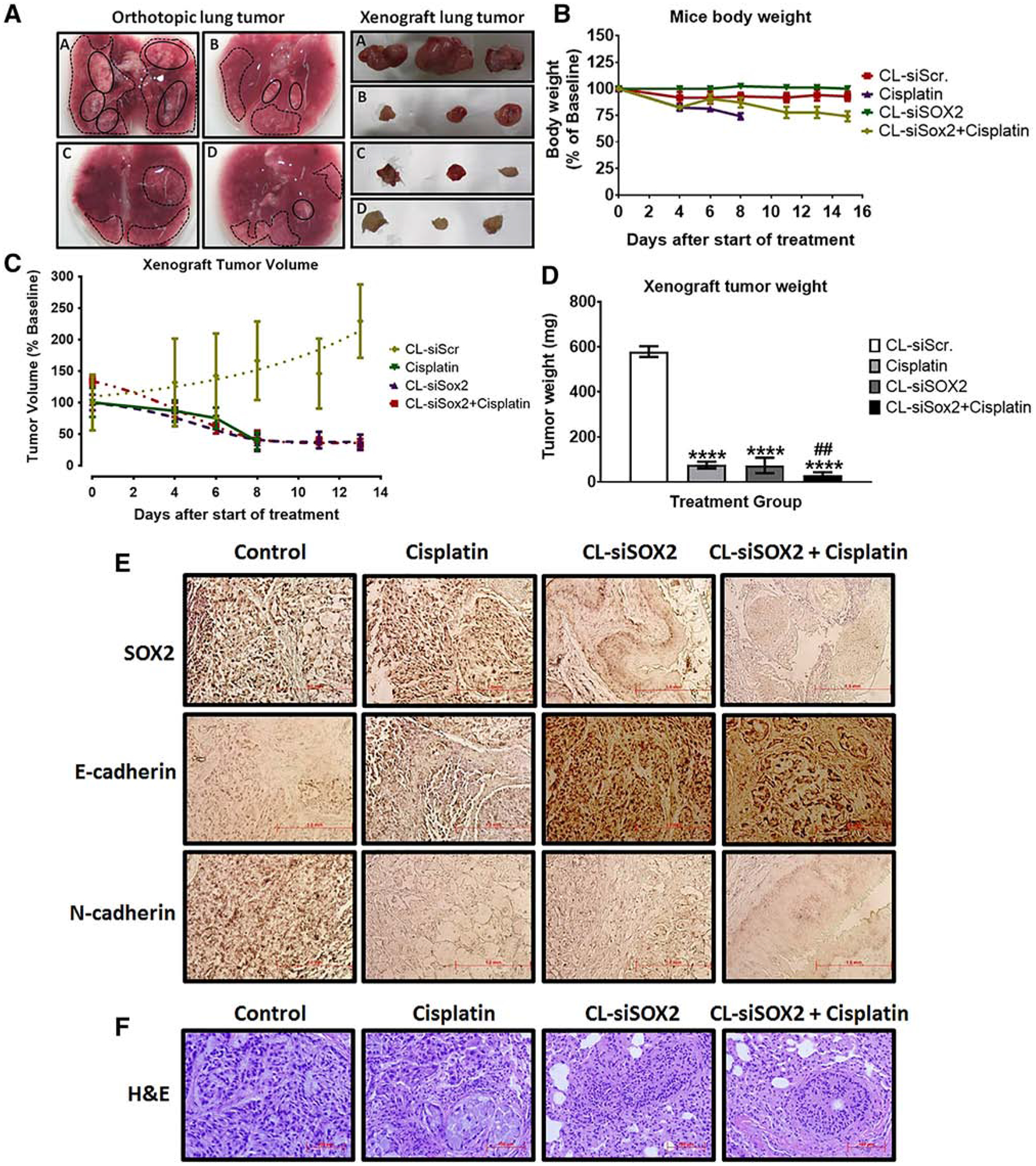
Cationic lipoplexes encapsulating SOX2 siRNA (CL-siSOX2) inhibit lung tumor growth in orthotopic and xenograft mice models. C.B.17 SCID-beige mice were injected (A) orthotopically (left panel) or subcutaneously (right panel) with H1650 SP cells as described under materials and methods. Animals received (a) CL-siScr, (b) cisplatin (2 mg/kg; i.p., on day 1 and 6), (c) CL-siSOX2 (22 nmol/100 μL; i.p. for 6 days on alternate days) and (d) CL-siSOX2 + cisplatin. Mice were sacrificed and orthotopic and xenograft lung tumors resected. Micrographs of orthotopic tumors are shown as either nodular (thick black outlines) or areas of sparsely disseminated (broken black lines) groups. (B-D) H1659 SP-derived xenograft tumors in C.B.17 SCID-beige mice were allowed to grow to approximately 150 mm3 in diameter followed by treatment as described above. Mice body weight (B), tumor volume (C) and tumor weight (D) were determined and presented as mean with standard deviation. (D) represents endpoint data at day 15 of treatment except for the cisplatin group; animals in the cisplatin group were sacrificed at day 8 due to increased distress. (E) Immunohistochemical staining for SOX2, E-cadherin and N-cadherin, and (F) H&E staining were performed on resected lung tissue and images were captured by light microscopy. Statistical analysis: student t test: (treatment vs. siScr: *P < 0.05; **P < 0.01; ****P < 0.0001, and treatment vs. cisplatin: ##P < 0.01).
CL-siSOX2 inhibits expression of stemness markers in xenograft tumors
Protein expression for the stemness markers SOX2, OCT4, Nanog, c-Myc and KLF4 are presented in Figure 4. CL-siSOX2 significant decreased expression of SOX2 (10.34 ± 1.26%), Nanog (14.26 ± 7.43%), c-Myc (6.16 ± 0.51%) and KLF4 (33.34 ± 2.87%) compared to CL-siScr. However, CL-siSOX2 upregulated expression of OCT4 (124.59 ± 9.96) compared to CL-siScr. CL-siSOX2 + cisplatin significantly decreased expression of SOX2 (0.93 ± 0.05%), OCT4 (2.73 ± 0.34%), Nanog (0.75 ± 0.05%), c-Myc (1.25 ± 0.06%), and KLF4 (8.86 ± 0.07%) compared to individual treatments.
Figure 4.
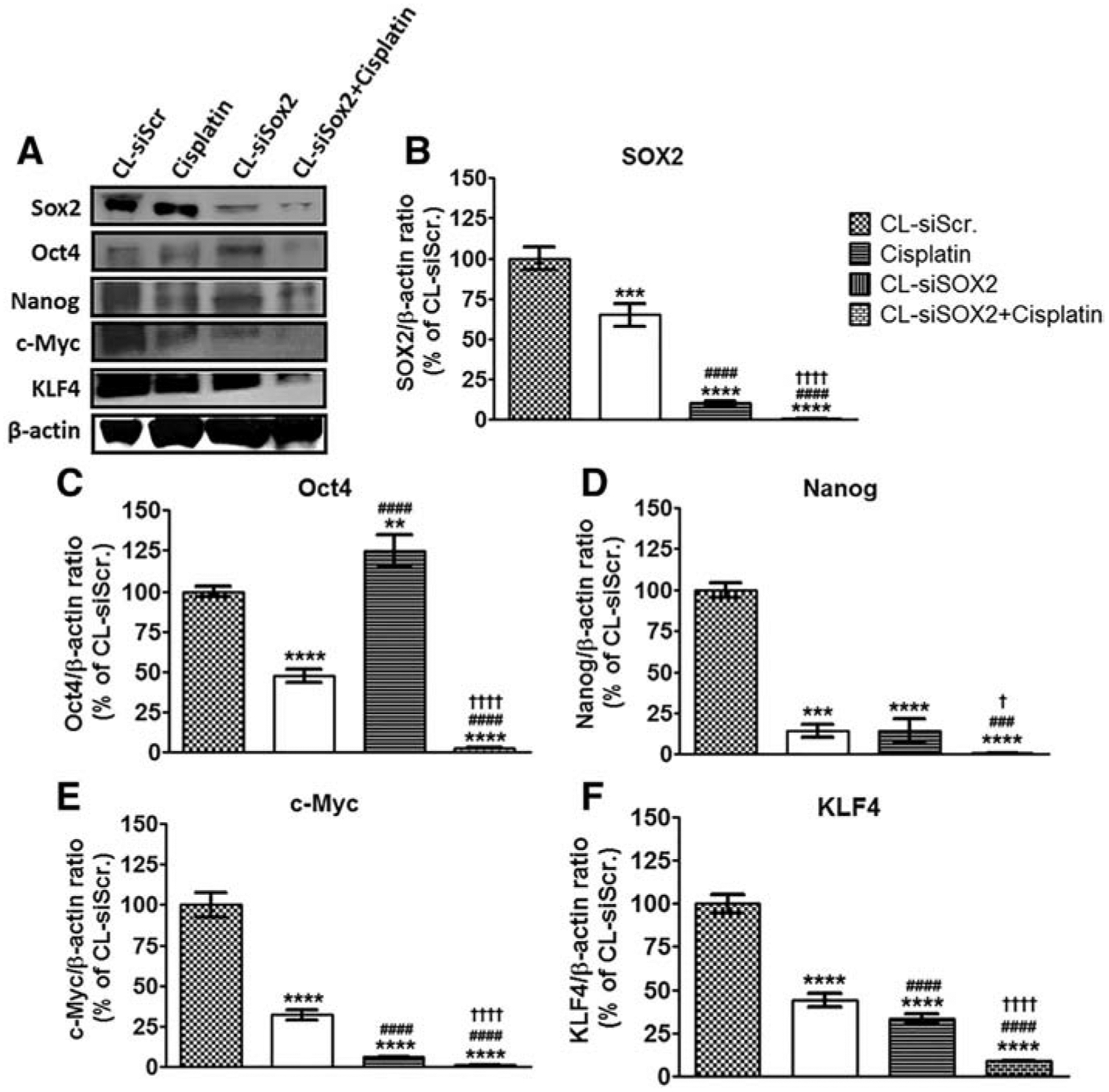
Cationic lipoplex loaded with siSOX2 (CL-siSOX2) inhibit expression of stemness markers in mice xenograft tumors. C.B.17 SCID mice with H1650 SP cells as tumor xenografts received treatment as described. (A) Immunoblotting of tumor lysates show knockdown of (B) SOX2 and associated stemness factors including (C) OCT4, (D) Nanog, (F) KLF4, as well as the oncogene (E) c-Myc. Results were calculated as protein/β-actin ratio and presented as mean percent of the CL-siScr with SD. Statistical analysis: student t test: (treatment vs. siScr: **P < 0.01; ***P < 0.001; ****P < 0.0001, treatment vs. cisplatin: ###P < 0.001; ####P < 0.0001, and treatment vs. CL-siSOX2: †P < 0.05; ††††P < 0.0001).
CL-siSOX2 inhibits expression of markers of multidrug resistance in xenograft tumors
Treatment with CL-siSOX2 resulted in significant reduction in expression of markers of tumor resistance including Wnt3a (53.36 ± 3.32%), Wnt5a/b (49.22 ± 4.22%), phospho-β-catenin (32.37 ± 1.67%), Dvl2 (45.10 ± 2.52%) and ABCG2 (56.69 ± 2.99%) compared to CL-siScr (Figure 5). Likewise, cisplatin decreased expression of the respective proteins to 44.88 ± 4.01%, 33.60 ± 2.40%, 21.86 ± 1.72%, 26.18 ± 1.71% and 47.24 ± 3.09% compared to CL-siScr. CL-siSOX2 + cisplatin resulted in enhanced reduction in the expression of the respective protein to 0.81 ± 0.01%, 6.18 ± 0.55%, 0.55 ± 0.51%, 0.41 ± 0.02% and 18.47 ± 1.25% in a significant manner compared individual treatments.
Figure 5.
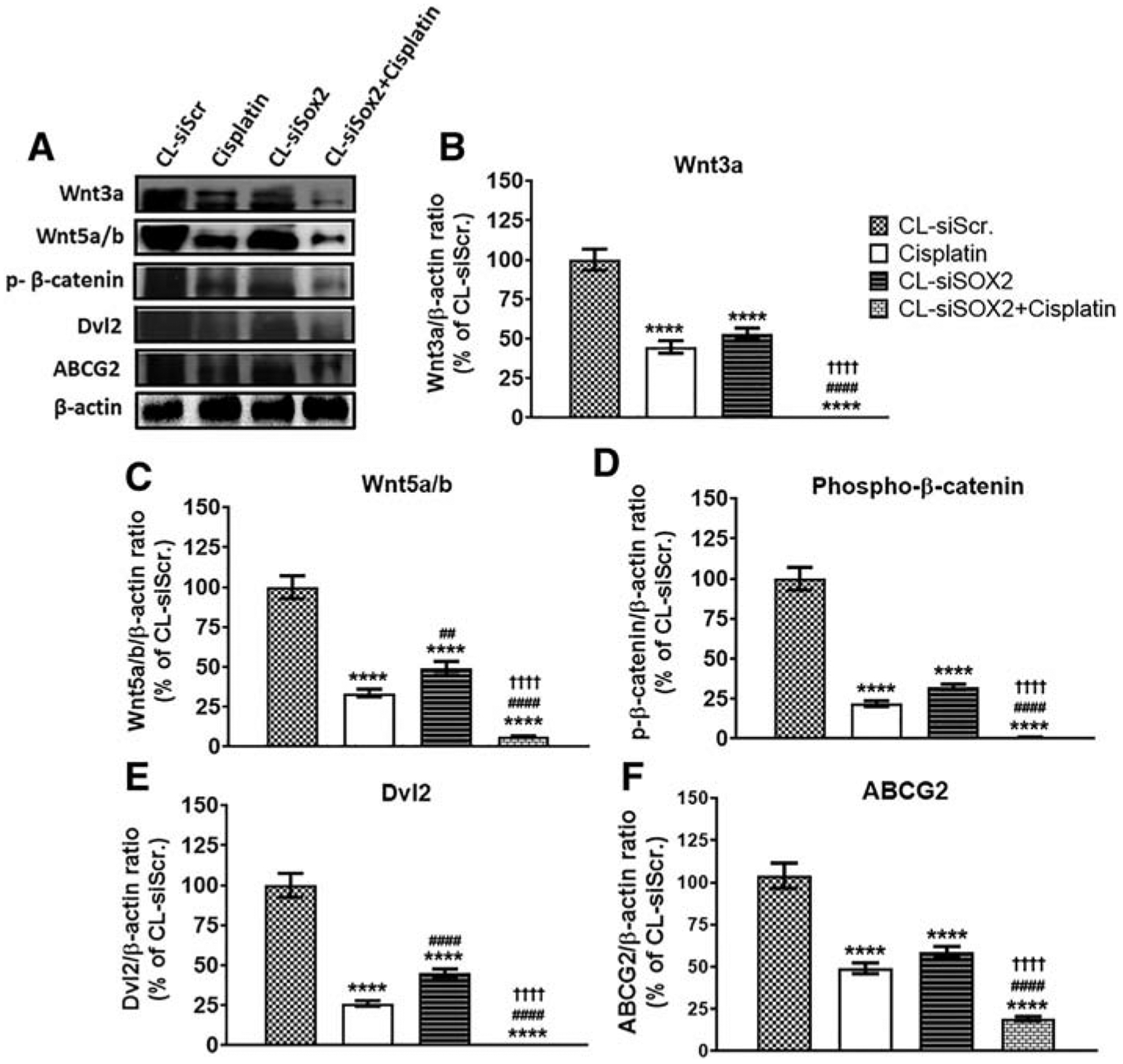
Cationic lipoplex loaded with siSOX2 (CL-siSOX2) inhibits expression of markers of tumor resistance in mice xenograft tumors. C.B.17 SCID mice with H1650 SP cells as tumor xenografts received treatment as described. (A) Immunoblotting of tumor lysates shows protein expression of (B) Wnt3a, (C) Wnt5a/b, (D) phospho-β-catenin, (E) Dvl2 and (F) ABCG2. Results were calculated as protein/β-actin ratio and presented as mean percent of the CL-siScr with SD. Statistical analysis: student t test: (treatment vs. siScr: ****P < 0.0001, treatment vs. cisplatin: ##P < 0.01; ####P < 0.0001, and treatment vs. CL-siSOX2: ††††P < 0.0001).
CL-siSOX2 inhibits expression of markers invasion and metastasis in xenograft tumors
Figure 6 shows western blot results after treatment with CL-siSOX2 with significant reduction in expression of Slug (29.80 ± 0.20%), and N-cadherin (4.73 ± 0.52%) while inducing the expression of E-cadherin (794.23 ± 50.59%) compared to CL-siScr. Notably, whereas cisplatin produced comparable expressions of Slug (24.01 ± 1.40%), E-cadherin (594.24 ± 30.98%) and N-cadherin (3.10 ± 0.11%) as CL-siSOX2, combination of both treatments yielded significant reductions in Slug (0.33 ± 0.12%) and an increase in E-cadherin (714.39 ± 15.49%) expression compared to the individual treatments. N-cadherin expression was significantly decreased in all treatment groups compared to CL-siScr, and significant differences were observed between cisplatin and CL-siScr, cisplatin and CL-siSOX2 + cisplatin and CL-siSOX2 and CL-siSOX2 + cisplatin.
Figure 6.
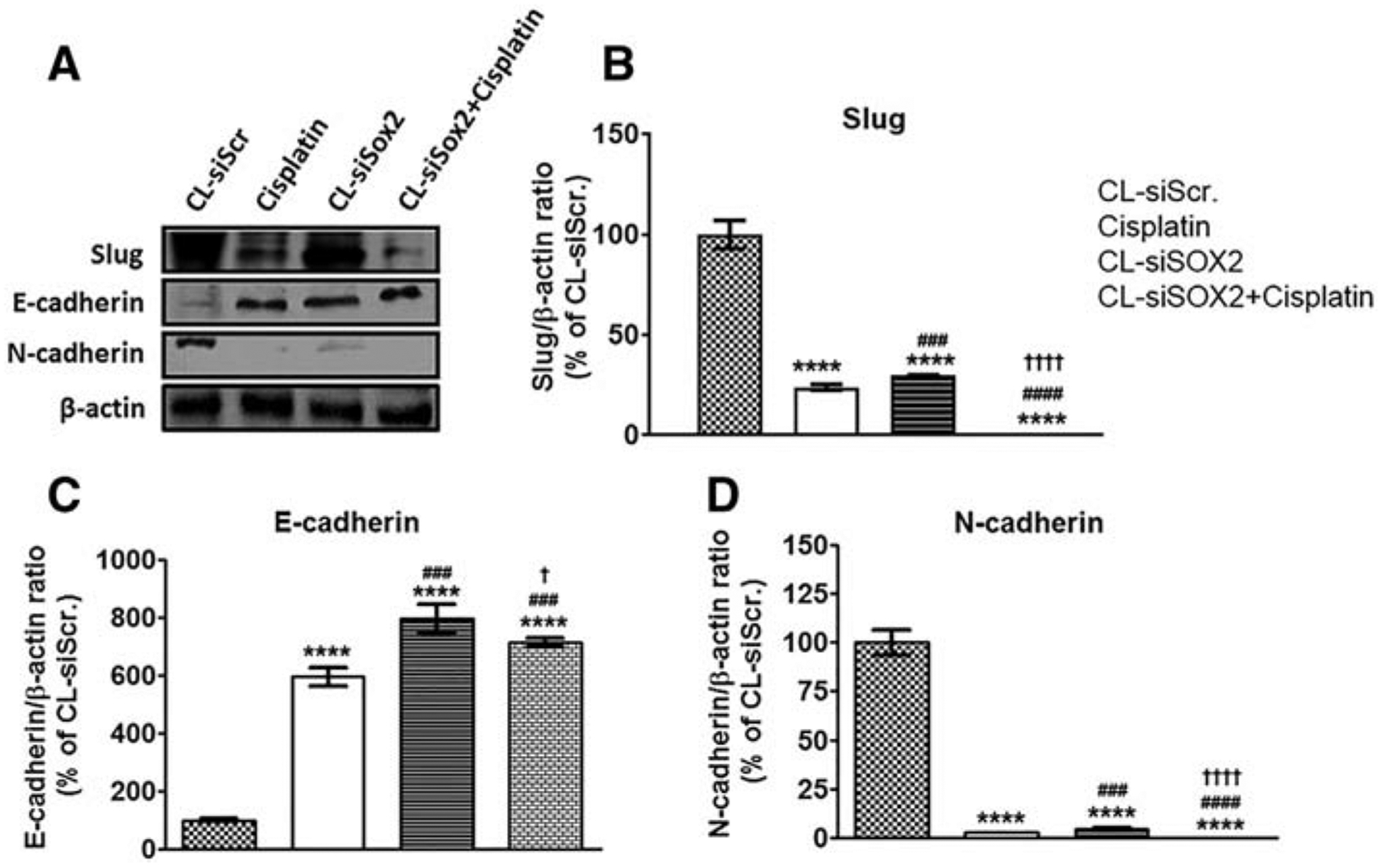
Cationic lipoplex loaded with siSOX2 (CL-siSOX2) inhibits markers of tumor invasion and metastasis in mice xenograft tumors. C.B.17 SCID mice with H1650 SP cells as tumor xenografts received treatment as described. (A) Immunoblotting of tumor lysates shows expression of (B) Slug, (C) E-cadherin and (D) N-cadherin. CL-siSOX2 and CL-siSOX2 + cisplatin downregulated the pro-invasion and metastatic Slug and N-cadherin, and induced the expression of anti-EMT (epithelial-to-mesenchymal transition) marker, E-cadherin. Results were calculated as protein/β-actin ratio and presented as mean percent of the CL-siScr with SD. Statistical analysis: student t test: (treatment vs. siScr: ****P < 0.0001, treatment vs. cisplatin: ###P < 0.001; ####P < 0.0001, and treatment vs. CL-siSOX2: †P < 0.05; ††††P < 0.0001).
CL-siSOX2 inhibits expression markers of inflammation in xenograft tumors
Figure 7 shows significant reduction in the expression of pro-inflammatory tumor markers TLR9, TLR1 and IKKγ with CL-siSOX2 (25.24 ± 1.71%, 35.72 ± 1.73% and 35.41 ± 2.72% respectively) and cisplatin (9.22 ± 0.28%, 34.34 ± 2.56% and 44.53 ± 3.30% respectively) compared to control. TLR9 and TLR1 expression were significant in CL-siSOX2 + cisplatin (9.22 ± 0.28%, 0.56 ± 0.03% and 0.12 ± 0.01% respectively) compared to single treatments.
Figure 7.

Cationic lipoplex loaded with siSOX2 (CL-siSOX2) inhibits expression of inflammatory markers in mice xenograft tumors. C.B.17 SCID mice with H1650 SP cells as tumor xenografts received treatment as described. (A) Immunoblotting of tumor lysates shows protein expression of (B) TLR9, (C) TLR1 and (D) IKKγ. Results were calculated as protein/β-actin ratio and presented as mean percent of the CL-siScr with SD. Statistical analysis: student t test: (treatment vs. siScr: ****P < 0.0001, treatment vs. cisplatin: ##P < 0.01; ###P < 0.001; ####P < 0.0001, and treatment vs. CL-siSOX2: ††††P < 0.0001).
CL-siSOX2 inhibits expression markers of tumor growth
Compared to CL-siScr, CL-siSOX2 reduced expression of Smad5, TGFβ, Bcl-2 and survivin to 44.48 ± 3.40%, 33.16 ± 2.94%, 4.15 ± 0.20% and 15.13 ± 1.01% respectively; whereas cisplatin reduced expression to 50.04 ± 3.53%, 49.37 ± 4.23%, 38.05 ± 1.81% and 24.46 ± 1.19% respectively (Figure 8). Compared to the individual treatments, however, the combined effect of CL-siSOX2 + cisplatin reduced Smad5, TGFβ, Bcl-2 and Survivin expression significantly to 0.27 ± 0.16%, 2.64 ± 0.21%, 0.00 ± 0.00% and 3.59 ± 0.09% respectively. Significant differences in the effects of cisplatin and CL-siSOX2 were observed for TGFβ, Bcl-2 and Survivin expression.
Figure 8.
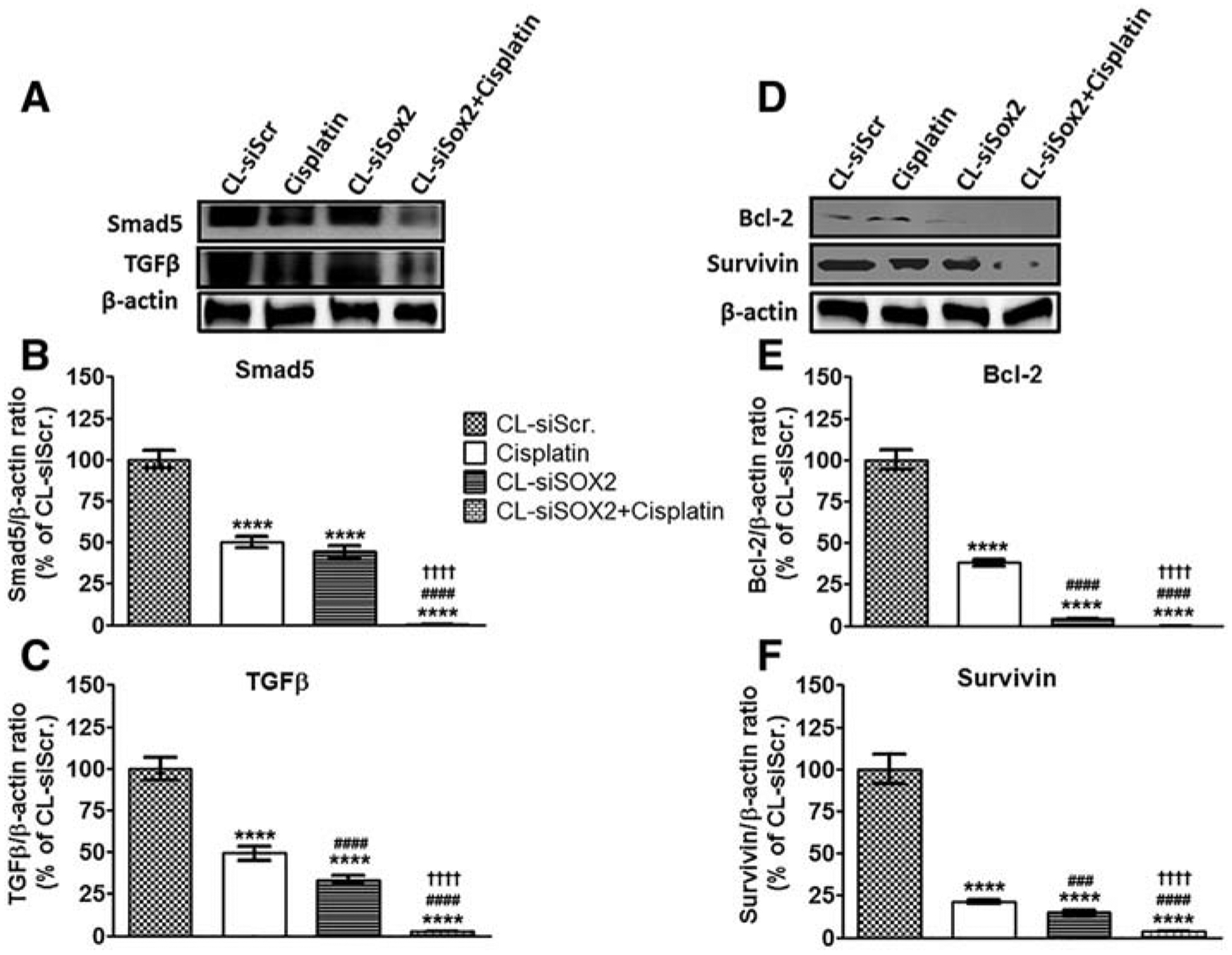
Cationic lipoplex loaded with siSOX2 (CL-siSOX2) inhibits expression of markers of tumor growth in mice xenograft tumors. C.B.17 SCID mice with H1650 SP cells as tumor xenografts received treatment as described. (A) Immunoblotting of tumor lysates shows protein expression of (B) Smad5, (C) TGFβ, (D) Bcl-2 and (E) Survivin. Results were calculated as protein/β-actin ratio and presented as mean percent of the CL-siScr with SD. Statistical analysis: student t test: (treatment vs. siScr: ****P < 0.0001, treatment vs. cisplatin: ###P < 0.001; ####P < 0.0001, and treatment vs. CL-siSOX2: ††††P < 0.0001).
Discussion
The SOX2 embryonic stem cell marker has gained attention for its wide-ranging transcriptional effects on enabling tumor growth and resistance in different types of cancer; for example, SOX2 promoted tumor growth and acquired resistance to radiation and chemotherapy in oral cancer and transcriptionally modulated tumor growth in gastric cancer.25,26 In lung cancer cells, SOX2 in cooperation with beta-catenin and POU5F1 was associated with increased IGF-R1 signaling and poor prognosis.10 Further, a positive feedback loop between SOX2 and EGFR promoted self-renewal and chemo-resistance in lung cancer.25 Central to the role of SOX2 in promoting the resistance phenotype in cancer is the existence of SOX2-enriched side populations (SPs) of cancer cells (cancer stem cells, CSCs) that are reposed with a functional capacity for promoting self-renewal and resistance in cancer.27 Therefore, for treatment of cancer to be effective over the long term, the spectrum of therapies must include targeting of these quiescent, residual CSCs.8,28 We proposed the use of a therapeutic small interfering RNA (siRNA) targeting SOX2 formulated in a cationic lipoplex (CL-siSOX2) for passive targeting and treatment of CSC-derived murine tumors.
Sustained proliferation is elemental to the survival, growth and metastatic dissemination of tumors. Indeed, cancer cells have long been associated with a higher potential for proliferation compared to normal cells, and inhibition of cell proliferation is an empirical measure of the potential clinical efficacy of anticancer therapy. SOX2-enriched H1650 SP cells were associated with higher proliferative and metastatic potential, which is evidenced by increased rates of sphere and spheroid formation, as well as increased migration and resistance to cisplatin compared to H1650 MP cells. As with other types of anticancer therapies (i.e. radiation and chemotherapy), targeted therapies also suffer from the fate of rapid progression towards acquired tumor resistance facilitated via extrinsic factors (e.g. secretion of enzymes and/or growth factors by stromal cells of the tumor microenvironment that deactivate drugs and/or activate primary and secondary resistance inside cancer cells respectively), and intrinsic factors (e.g. mutation of oncogenic signaling pathways, upregulation of efflux transporters, etc.).29–31 Nanoparticle drug formulation strategies provide opportunities for both active and passive targeted delivery of drugs to tumors while circumventing the effects of deactivating and/or inhibitory intrinsic and extrinsic resistance factors to improve cellular internalization and enhanced therapeutic efficacy, while limiting adverse effects due to improved targeting. Nanoparticles employing specific targeting, versatile drug loading, and imaging capabilities have been used in targeted delivery, imaging and treatment of different types of cancer.32–34 We proposed the use of fluorescent lipoplexes for targeted delivery of siRNA targeting SOX2 for imaging and treatment of lung tumors of stem cell origin.
We successfully, formulated siSOX2 in a cationic lipoplex with high drug loading. The formulation parameters were optimized to ensure nano-sized lipoplexes, which we predicted will facilitate lung tumor targeting will be sufficiently aided by an enhanced targeting of tumor vasculature resulting in increased extravasation into tumor tissue aided by the cationic charge of the lipoplexes; our predication was based on the effect of size and charge characteristics (<100 nm and ~15 mV respectively) of the lipoplexes. The precedent for this assumption is based on related studies that have demonstrated that cationic nanoparticles through electrostatic interactions are 15–30 times more efficient in targeting the luminal tumor vasculature subsequently resulting in extravasation compared to normal tissues.35–37 Moreover, surface functionalization of the lipoplex by pegylation was incorporated in the formulation process as a safeguard against potential degradation during plasma transit. Subsequently, the capacity of the lipoplexes to ensure lung exposure for subsequent tumor uptake was demonstrated by ex vivo fluorescence imaging (Figure 2). Tissue uptake of lipoplexes were observed in lung, spleen, heart, kidney and liver. Notably, uptake was higher in the liver compared to other tissues, which is supported by previous observations that initial uptake of nanoparticles, and subsequent saturation of phagocytic cells in the liver precede uptake by other tissues/organs.38 We also observed variable exposure of the lipoplexes to the lung; an initial increase in lung exposure (1 h) was followed by a decrease over the course of 2 h and 3 h, followed by an increase at 4 h and 5 h. This variable exposure kinetics may be explained by models of in vitro cellular uptake of nanoparticles that show initial saturation at the cell surface due to adsorption, and preceding endocytosis, where adsorption has been shown to occur at rates greater than three orders of magnitude faster than endocytosis.38 Subsequent increase in the rate of accessibility and recycling of surface receptors may have contributed to the clearance of surface adsorbed lipoplexes at 2 h and 3 h. Thus, the increased exposure at 5 h may be interpreted as intracellular accumulation rather than surface saturation by lipoplexes.
The H1650 lung cancer cell line has been validated in many studies as exhibiting resistance phenotypes to different treatments including small molecule chemo- and targeted therapies, as well as large molecules such as monoclonal antibodies.39–41 H1650 has also been characterized to demonstrate resistance to epidermal growth factor receptor (EGFR)-targeted therapies which is attributed to the presence of activating somatic mutations including EGFR-T790 M, KRAS and PTEN loss.42–44 Further complicating the therapeutic targeting of EGFR-enriched H1650 cells is the inherent heterogeneity of tumor cells typified by such subpopulations as the highly resistant, SOX2-overexpressing H1650 CSCs.7 Altogether, these characteristics present H1650 CSCs as a suitable cancer model for investigating the efficacy of a targeted therapy approach for sensitizing and/or treating resistant cancers.1,45,46
Many cancer treatment protocols comprise a combination treatment regimen as a strategy for enhancing efficacy, overcoming acquired resistance and improving the safety profile of treatment. For example, the monoclonal antibody cetuximab used in combination with cisplatin/vinorelbine as first-line therapy demonstrated superior efficacy compared to cisplatin/vinorelbine alone in a phase II clinical trial of patients with EGFR-positive advanced NSCLC.47 However, the incidence of hematologic, dermatologic, and respiratory toxicities was higher in the combination group compared to the chemotherapy group. Our study, while precluding the investigation of these toxicities showed evidence of distress of mice to cisplatin treatment, resulting in drastic loss in body weight, which necessitated the termination of the cisplatin group at day 8. The animals were, however, tolerant to CL-siSOX2 and CL-siSOX2 + cisplatin. Importantly, significant reduction in (xenograft) tumor volume and weight, and decreased number of (orthotopic) tumor nodules and areas of dissemination were observed with treatment with cisplatin, CL-siSOX2 and CL-siSOX2 + cisplatin. Additionally, semi-quantitative assessment of tissue sections showed decreased expression of SOX2 and reversal of EMT in the CL-siSOX2 and CL-siSOX2 + cisplatin groups; and histologic staining of tissue sections showed less tissue remodeling in the treatment groups.
SOX2, OCT4, and KLF4 have been shown to cooperate in promoting self-renewal and maintaining the pluripotent state of embryonic stem cells (ESCs).48 This tightly-controlled gene circuitry regulates developmental processes through spatial and temporal activation or repression of various downstream targets including transcription factors.49,50 However, in cancer cells this circuitry may be dysregulated resulting in aberrant transcriptional products and associated pathologic signal transduction effects.51 While downregulation of KLF4 may occur in tandem with SOX2, the reverse has been shown with SOX2 and OCT4, the result of a negative-feedback loop.52,53 Although, upregulation of OCT4 in the CL-siSOX2 group may prove problematic to the long-term suppression of the pluripotent state of any residual tumor, it is refreshing to observe that CL-siSOX2 + cisplatin completely abolished OCT4; thereby, supporting the rationale for the use of CL-siSOX2 in adjunctive therapy instead of as a stand-alone treatment.
Upregulation of SOX2 is associated with activation of mediators of tumor resistance.54–56 We observed that SOX2 overexpression promoted activation of Wnt/Catenin oncogenic signaling. However, CL-siSOX2 abrogated Wnt/Catenin-mediated resistance by down-regulation of Wnt3a, Wnt5a/b, phosphorylated β-catenin, and downstream targets, ABCG2 and Dvl2. We observed enhanced down-regulation of ABCG2 in the CL-siSOX2 + cisplatin group compared to individual treatments, which further supports a mutually-exclusive mechanism for the delayed progression of cisplatin-induced loss of body weight. Activation of β-catenin by SOX2 is through inhibition of its interaction with E-cadherin whose expression is mutually inhibitory to N-cadherin. SOX2 binds to the promoter region of Slug with resultant loss of E-cadherin function and enhancement of epithelial-to-mesenchymal transition (EMT).45,57 CL-siSOX2 facilitated reversal of EMT by upregulating E-cadherin, while downregulating Slug and N-cadherin. Toll-like receptors (TLR) activate pro-inflammatory NF-κB signaling resulting in enhanced tumor proliferation. Inhibition of canonical IKK/NF-κB activation has been shown to sensitize cancer cells to therapy.58 SOX2 down-regulation was associated with decreased expression of TLR1, TLR9 and IKKγ. Additionally, CL-siSOX2 disrupted the TGFβ/SMAD/SOX2 regulatory network by diminishing their expression together with Bcl-2 and Survivin.59
In conclusion, this study demonstrates the successful engineering of an efficient siRNA-loaded cationic lipoplex, for the therapeutic interference of SOX2 oncogenic activity in a mouse xenograft model of H1650 CSC/SP lung tumor. The study also defines the molecular events involved in attenuating tumor growth though SOX2 interference, resulting in the inhibition of markers of tumor growth, metastasis, invasion and chemoresistance. Consistent with other studies using specific siRNA targeting alone or in combination with other therapies such as radiation, chemotherapy, and/or targeted therapy,44,60 our results demonstrate the effectiveness of SOX2 siRNA in shrinking tumor volume in mice, either alone or in combination with cisplatin.
Supplementary Material
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nano.2019.02.007.
Conflict of Interest: Authors have no conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 2.Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia 2015;17(7):538–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg SB, Oxnard GR, Digumarthy S, Muzikansky A, Jackman DM, Lennes IT, et al. Chemotherapy With Erlotinib or Chemotherapy Alone in Advanced Non-Small Cell Lung Cancer With Acquired Resistance to EGFR Tyrosine Kinase Inhibitors. Oncologist 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuo D, Zhao WD, Wright FA, Yang HY, Wang JP, Sears R, et al. Assembly, annotation, and integration of UNIGENE clusters into the human genome draft. Genome Res 2001;11(5):904–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A 2007;104(3):973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiang R, Liao D, Cheng T, Zhou H, Shi Q, Chuang TS, et al. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br J Cancer 2011;104 (9):1410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, et al. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer 2012;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Xu Y, Chen Y, Li X, Mou W, Wang L, et al. SOX2 gene regulates the transcriptional network of oncogenes and affects tumorigenesis of human lung cancer cells. PLoS One 2012;7(5):e36326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dogan I, Kawabata S, Bergbower E, Gills JJ, Ekmekci A, Wilson III W, et al. SOX2 expression is an early event in a murine model of EGFR mutant lung cancer and promotes proliferation of a subset of EGFR mutant lung adenocarcinoma cell lines. Lung Cancer 2014;85(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu C, Xie D, Yu SC, Yang XJ, He LR. Yang J, et al. beta-Catenin/POU5F1/SOX2 transcription factor complex mediates IGF-I receptor signaling and predicts poor prognosis in lung adenocarcinoma. Cancer Res 2013;73(10):3181–9. [DOI] [PubMed] [Google Scholar]
- 11.Tang QL, Zhao ZQ, Li JC, Liang Y, Yin JQ, Zou CY, et al. Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett 2011;311(1):113–21. [DOI] [PubMed] [Google Scholar]
- 12.Bhat-Nakshatri P, Goswami CP, Badve S, Sledge GW Jr, Nakshatri H. Identification of FDA-approved Drugs Targeting Breast Cancer Stem Cells Along With Biomarkers of Sensitivity. Sci Rep 2013;3:2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garros-Regulez L, Aldaz P, Arrizabalaga O, Moncho-Amor V, Carrasco-Garcia E, Manterola L, et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin Ther Targets 2016;20(4):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Favaro R, Appolloni I, Pellegatta S, Sanga AB, Pagella P, Gambini E, et al. Sox2 is required to maintain cancer stem cells in a mouse model of high-grade oligodendroglioma. Cancer Res 2014;74 (6):1833–44. [DOI] [PubMed] [Google Scholar]
- 15.Polakova I, Duskova M, Smahel M. Antitumor DNA vaccination against the Sox2 transcription factor. Int J Oncol 2014;45(1):139–46. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee P, Gupta A, Chattopadhyay D, Chatterji U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Sci Rep 2017;7(1):9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 2012;33(5):1462–76. [DOI] [PubMed] [Google Scholar]
- 18.Singhania A, Wu SY, McMillan NA. Effective Delivery of PEGylated siRNA-Containing Lipoplexes to Extraperitoneal Tumours following Intraperitoneal Administration. J Drug Deliv 2011;2011192562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Remaud S, Lopez-Juarez SA, Bolcato-Bellemin AL, Neuberg P, Stock F, Bonnet ME, et al. Inhibition of Sox2 Expression in the Adult Neural Stem Cell Niche In Vivo by Monocationic-based siRNA Delivery. Mol Ther Nucleic Acids 2013;e89:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landen CN, Merritt WM, Mangala LS, Sanguino AM, Bucana C, Lu C, et al. Intraperitoneal delivery of liposomal siRNA for therapy of advanced ovarian cancer. Cancer Biol Ther 2006;5(12):1708–13. [DOI] [PubMed] [Google Scholar]
- 21.Andey T, Marepally S, Patel A, Jackson T, Sarkar S, O’Connell M, et al. Cationic lipid guided short-hairpin RNA interference of annexin A2 attenuates tumor growth and metastasis in a mouse lung cancer stem cell model. J Control Release 2014;184:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andey T, Patel A, Jackson T, Safe S, Singh M. 1,1-Bis (3’-indolyl)-1-(p-substitutedphenyl)methane compounds inhibit lung cancer cell and tumor growth in a metastasis model. Eur J Pharm Sci 2013;50(2):227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SO, Andey T, Jin UH, Kim K, Singh M, Safe S. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 2012;31(27):3265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andey T, Sudhakar G, Marepally S, Patel A, Banerjee R, Singh M. Lipid nanocarriers of a lipid-conjugated estrogenic derivative inhibit tumor growth and enhance cisplatin activity against triple-negative breast cancer: pharmacokinetic and efficacy evaluation. Mol Pharm 2015;12 (4):1105–20. [DOI] [PubMed] [Google Scholar]
- 25.Chou YT, Lee CC, Hsiao SH, Lin SE, Lin SC, Chung CH, et al. The emerging role of SOX2 in cell proliferation and survival and its crosstalk with oncogenic signaling in lung cancer. Stem Cells 2013. [DOI] [PubMed] [Google Scholar]
- 26.Hutz K, Mejias-Luque R, Farsakova K, Ogris M, Krebs S, Anton M, et al. The stem cell factor SOX2 regulates the tumorigenic potential in human gastric cancer cells. Carcinogenesis 2014;35(4):942–50. [DOI] [PubMed] [Google Scholar]
- 27.Karachaliou N, Rosell R, Viteri S. The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl Lung Cancer Res 2013;2(3):172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao C, He J, Cheng H, Zhu Z, Xu H. Enhanced therapeutic effect of an antiangiogenesis peptide on lung cancer in vivo combined with salmonella VNP20009 carrying a Sox2 shRNA construct. J Exp Clin Cancer Res 2016;35(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487(7408):500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012;487(7408):505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krayem M, Journe F, Wiedig M, Morandini R, Najem A, Sales F, et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K) BRAF melanoma to vemurafenib. Eur J Cancer 2016;55:98–110. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Leonard M, Shu Y, Yang Y, Shu D, Guo P, et al. Overcoming Tamoxifen Resistance of Human Breast Cancer by Targeted Gene Silencing Using Multifunctional pRNA Nanoparticles. ACS Nano 2017;11(1):335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nascimento AV, Singh A, Bousbaa H, Ferreira D, Sarmento B, Amiji MM. Overcoming cisplatin resistance in non-small cell lung cancer with Mad2 silencing siRNA delivered systemically using EGFR-targeted chitosan nanoparticles. Acta Biomater 2017;47:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo Y, Cai X, Li H, Lin Y, Du D. Hyaluronic Acid-Modified Multifunctional Q-Graphene for Targeted Killing of Drug-Resistant Lung Cancer Cells. ACS Appl Mater Interfaces 2016;8(6):4048–55. [DOI] [PubMed] [Google Scholar]
- 35.Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest 1998;101(7):1401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bode C, Trojan L, Weiss C, Kraenzlin B, Michaelis U, Teifel M, et al. Paclitaxel encapsulated in cationic liposomes: a new option for neovascular targeting for the treatment of prostate cancer. Oncol Rep 2009;22(2):321–6. [PubMed] [Google Scholar]
- 37.Kalra AV, Campbell RB. Development of 5-FU and doxorubicin-loaded cationic liposomes against human pancreatic cancer: Implications for tumor vascular targeting. Pharm Res 2006;23(12):2809–17. [DOI] [PubMed] [Google Scholar]
- 38.Carlander U, Li D, Jolliet O, Emond C, Johanson G. Toward a general physiologically-based pharmacokinetic model for intravenously injected nanoparticles. Int J Nanomedicine 2016;11:625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu Y, Dong XZ, Liu X, Liu P, Chen YB. Enhanced Antitumor Activity of Cetuximab in Combination with the Jak Inhibitor CYT387 against Non-Small-Cell Lung Cancer with Various Genotypes. Mol Pharm 2016;13(2):689–97. [DOI] [PubMed] [Google Scholar]
- 40.Zhang FQ, Yang WT, Duan SZ, Xia YC, Zhu RY, Chen YB. JAK2 inhibitor TG101348 overcomes erlotinib-resistance in non-small cell lung carcinoma cells with mutated EGF receptor. Oncotarget 2015;6 (16):14329–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minami D, Takigawa N, Takeda H, Takata M, Ochi N, Ichihara E, et al. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol Cancer Res 2013;11(2):140–8. [DOI] [PubMed] [Google Scholar]
- 42.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7(3):169–81. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh G, Lian X, Kron SJ, Palecek SP. Properties of resistant cells generated from lung cancer cell lines treated with EGFR inhibitors. BMC Cancer 2012;12:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen G, Kronenberger P, Teugels E, Umelo IA, De Greve J. Targeting the epidermal growth factor receptor in non-small cell lung cancer cells: the effect of combining RNA interference with tyrosine kinase inhibitors or cetuximab. BMC Med 2012;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Casal R, Bhattacharya C, Ganesh N, Bailey L, Basse P, Gibson M, et al. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol Cancer 2013;12(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh S, Bora-Singhal N, Kroeger J, Laklai H. Chellappan SP. betaArrestin-1 and Mcl-1 modulate self-renewal growth of cancer stem-like side-population cells in non-small cell lung cancer. PLoS One 2013;8(2):e55982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosell R, Robinet G, Szczesna A, Ramlau R, Constenla M, Mennecier BC, et al. Randomized phase II study of cetuximab plus cisplatin/vinorelbine compared with cisplatin/vinorelbine alone as first-line therapy in EGFR-expressing advanced non-small-cell lung cancer. Ann Oncol 2008;19(2):362–9. [DOI] [PubMed] [Google Scholar]
- 48.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005;122(6):947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep 2011;12(7):720–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papapetrou EP, Tomishima MJ, Chambers SM, Mica Y, Reed E, Menon J, et al. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc Natl Acad Sci U S A 2009;106(31):12759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hattermann K, Fluh C, Engel D, Mehdorn HM, Synowitz M, Mentlein R, et al. Stem cell markers in glioma progression and recurrence. Int J Oncol 2016;49(5):1899–910. [DOI] [PubMed] [Google Scholar]
- 52.Rizzino A, Wuebben EL. Sox2/Oct4: A delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim Biophys Acta 2016;1859(6):780–91. [DOI] [PubMed] [Google Scholar]
- 53.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 2009;137(4):647–58. [DOI] [PubMed] [Google Scholar]
- 54.Li Y, Chen K, Li L, Li R, Zhang J, Ren W. Overexpression of SOX2 is involved in paclitaxel resistance of ovarian cancer via the PI3K/Akt pathway. Tumour Biol 2015;36(12):9823–8. [DOI] [PubMed] [Google Scholar]
- 55.Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med 2014;6(1):66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355(6320):84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC, Bamlet WR, et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogene 2013;2:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tapia MA, Gonzalez-Navarrete I, Dalmases A, Bosch M, Rodriguez-Fanjul V, Rolfe M, et al. Inhibition of the canonical IKK/NF kappa B pathway sensitizes human cancer cells to doxorubicin. Cell Cycle 2007;6 (18):2284–92. [DOI] [PubMed] [Google Scholar]
- 59.Ischenko I, Liu J, Petrenko O, Hayman MJ. Transforming growth factor-beta signaling network regulates plasticity and lineage commitment of lung cancer cells. Cell Death Differ 2014;21(8):1218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen G, Kronenberger P, Teugels E, Umelo IA, De Greve J. Effect of siRNAs targeting the EGFR T790M mutation in a non-small cell lung cancer cell line resistant to EGFR tyrosine kinase inhibitors and combination with various agents. Biochem Biophys Res Commun 2013;431(3):623–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.