

Abstract
Structure-based optimization was conducted to improve the potency, selectivity, and cell-based activities of β-catenin/B-cell lymphoma 9 (BCL9) inhibitors based on the 4′-fluoro-N-phenyl-[1,1′-biphenyl]-3-carboxamide scaffold, which was designed to mimic the side chains of the hydrophobic α-helical hot spots at positions i, i + 3, and i + 7. Compound 29 was found to disrupt the β-catenin/BCL9 protein–protein interaction (PPI) with a Ki of 0.47 μM and >1900-fold selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs. The proposed binding mode of new inhibitors was consistent with the results of site-directed mutagenesis and structure–activity relationship studies. Cell-based studies indicated that 29 disrupted the β-catenin/BCL9 interaction without affecting the β-catenin/E-cadherin interaction, selectively suppressed transactivation of Wnt/β-catenin signaling, downregulated expression of Wnt target genes, and inhibited viability of Wnt/β-catenin-dependent cancer cells in dose-dependent manners. A comparison of the biochemical and cell-based assay results offered the directions for future inhibitor optimization.
Graphical Abstract
INTRODUCTION
The Wnt/β-catenin signaling pathway plays a key role in directing cell proliferation, differentiation, and survival.1,2 β-Catenin is the central mediator of this signaling pathway. The loss-of-function mutations in the suppressor genes of the Wnt/β-catenin signaling pathway, such as adenomatous polyposis coli (APC) and Axin, and/or the activation mutations at the N-terminal phosphorylation sites of the β-catenin gene (CTNNB1), stabilize β-catenin into the dephosphorylated state and result in accumulation of β-catenin in the cytoplasm. β-Catenin is then translocated into the cell nucleus, where it displaces the repressor protein Groucho/TLE, binds with the T-cell factor (Tcf)/lymphoid enhancer-binding factor (Lef) family of transcription factors, and recruits coactivators, B-cell lymphoma 9 (BCL9), Pygo, CREB-binding protein (CBP), etc., to activate transcription of specific Wnt/β-catenin target genes, such as cyclin D1, c-myc, survivin, and LEF1. These Wnt target genes then cause the initiation and progression of cancers3–6 and fibroses.7–9 The other causes for the hyperactivation of Wnt/β-catenin signaling are the autocrine/paracrine activation of Wnt ligands, frizzled (Fzd) and dishevelled (Dvl), and the epigenetic silencing of Wnt antagonist genes. Wnt/β-catenin signaling is also hyperactive in cancer stem cells to maintain the self-renewal of cancer stem cells,10–14 seed cancer metastasis,15 and drive the resistance to current therapies.16–19 Further, tumor cell intrinsic activation of β-catenin signaling prevents the recruitment of CD103+ dendritic cells by suppression of chemokine CCL4 expression and excludes CD8+ T-cells from the tumor microenvironment.20,21 The activation of β-catenin signaling was also reported to promote intratumoral regulator T cell (Treg) survival and infiltration, suppressing T-cell immunity.22,23
Significant efforts have been made to discover inhibitors for the upstream effectors of the Wnt/β-catenin signaling pathway.24–31 However, the inhibition of the upstream Wnt effectors can potentially cause the cross-regulatory effects on the noncanonical Wnt signaling pathways. Second, these inhibitors cannot confer efficacy to the diseases that harbor the more downstream APC and Axin loss-of-function mutations and the β-catenin activation mutations. The formation of the β-catenin-containing transcriptional complex in the cell nucleus is the penultimate step of this pathway. The transcriptional overactivation of Wnt/β-catenin signaling is dependent on the formation of this complex, in which β-catenin interacts with BCL9.32 BCL9 functions as a scaffolding structure of the Wnt enhanceosome and brings β-catenin to Tcf/Lef to transcribe specific Wnt target genes that cause the diseases.33 The crystallographic analysis34,35 and the biochemical studies36–38 reveal that the homology domain 2 (HD2) of BCL9/B9L adopts an α-helical structure to interact with the first armadillo repeat of β-catenin. Mammalian genomes encode two BCL9 paralogues, BCL9 and B9L (BCL9-like).39,40 The use of siRNAs39–42 and shRNAs43 against BCL9/B9L markedly decreases β-catenin-dependent gene expression and diminishes the growth of cancer cells in vitro and in vivo. The dominant negative constructs of BCL9/B9L that lack the C-terminal domains greatly inhibit the activity of Wnt/β-catenin signaling.39,41 Hence, the inhibitors that can disrupt the β-catenin/BCL9 protein–protein interaction (PPI) not only provide new chemical probes to understand the biology regulated by β-catenin signaling but also offer important starting points for drug discovery to treat advanced cancers and fibroses.
Triazole-stapled44 and olefin-stapled45 BCL9 L351–F374 α-helical peptides have been reported. The olefin-stapled BCL9 peptide was reported to target β-catenin, disrupt the β-catenin/ BCL9 PPI, and suppress transcription of Wnt target genes. This stapled peptide also inhibited tumor cell growth, angiogenesis, and metastasis without overt damage to normal tissues in mouse xenograft models for colorectal carcinoma and multiple myeloma.45 Compound screening identified a small molecule, carnosic acid, that disrupted the β-catenin/BCL9 PPI and inhibited β-catenin dependent transcription.35,46 In the previous study, we designed 4′-fluoro-N-phenyl-[1,1′-biphenyl]-3-carboxamide as a generalizable scaffold that itself can directly mimic the side chains of α-helical hot spots at positions i, i + 3, and i + 7.47 The further derivatization generated 1 in Figure 1. The biochemical AlphaScreen assay indicated that 1 disrupted the β-catenin/BCL9 PPI with an inhibition constant (Ki) of 2.1 ± 0.41 μM and showed 125-fold selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs.47 The proposed binding mode of 1 was evaluated with the site-directed mutagenesis studies using β-catenin D145A, E155A, D145A/E155A, L159S, and L156S/L178S and the structure–activity relationship (SAR) studies of 29 derivatives. The cellular bioavailability studies indicated that 1 can easily be taken up by cancer cells.48 This compound also dissociated the β-catenin/BCL9 PPI in cells, selectively suppressed transactivation of Wnt/β-catenin signaling, downregulated expression of Wnt target genes, and inhibited growth of Wnt/β-catenin-dependent cancer cells. To further improve the potency and selectivity of this series of inhibitors, herein, we report the structure-based optimization, synthesis, and biological characterization of new derivatives.
Figure 1.
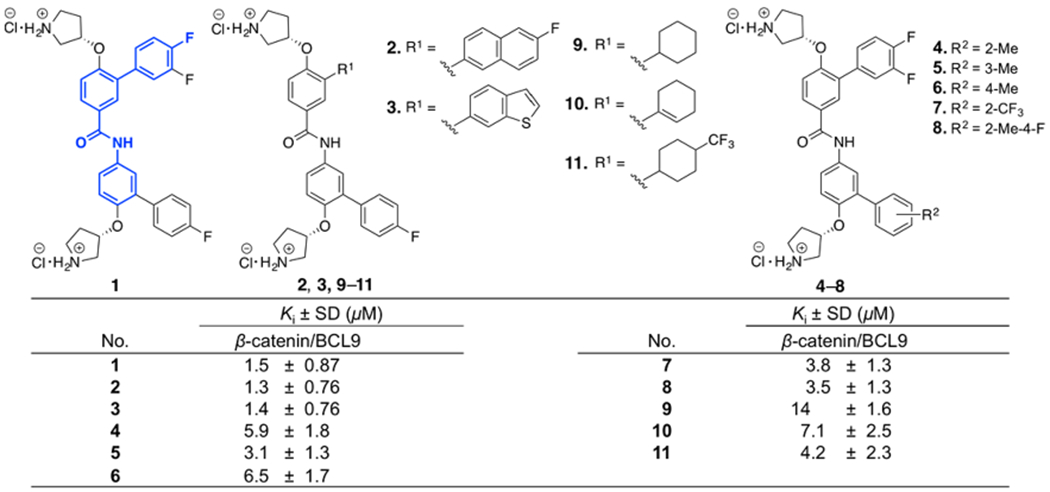
Chemical structures and the AlphaScreen Ki values of 1–11. Each set of data was expressed as mean ± standard deviation (n = 3).
RESULTS
Inhibitor Design and Synthesis.
Explore the Hydrophobic Interaction.
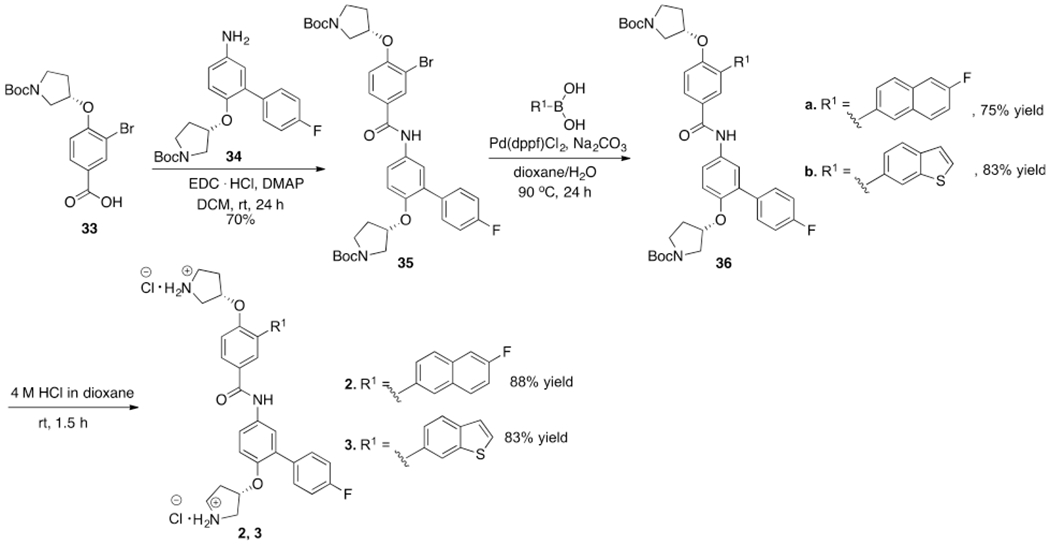
As described in previous studies, the β-catenin/BCL9 PPI interface has two hot regions.34,36–38,41,47,49 In hot region 1, residues D162, E163, and D164 of human β-catenin form an acidic knob to interact with H358 and R359 of human BCL9.34,36–38,41 This PPI interface is shallow and dominated by electrostatic interactions. In hot region 2, residues L366 (i), I369 (i + 3), and L373 (i + 7) of human BCL9 interact with a surface pocket that is lined with L159, V167, A171, M174, L178, L148, A149, A152, and L156 of human β-catenin, as shown in Supplementary Figure S1.34,41,44,47 This PPI interface is dominated by the hydrophobic interaction. The model of the interaction of 1 with β-catenin reported in the previous study47 was used as the starting point for inhibitor optimization. The first step of inhibitor design based on 1 was to increase its hydrophobic interactions with β-catenin. Compounds 2–8 in Figure 1 were designed and synthesized. The synthetic route for 2 and 3 is shown in Scheme 1. The synthetic route for 4–8 is shown in Supplementary Scheme S1. The AlphaScreen assay50 was used to evaluate the inhibitory activities of 2–8 in in vitro biochemical assays using full-length β-catenin and the BCL9 peptide (G350–P375). The results were expressed as the Ki in Figure 1. Compounds 2 and 3 displayed similar biochemical activities as 1 , indicating the limited gain of the potency by increasing the hydrophobic interaction.
Scheme 1.
The 3,4-difluorophenyl ring of 1 was also replaced with the aliphatic ring to explore the hydrophobic interaction. Compounds 9–11 were designed and synthesized. The synthetic route of 9–11 is shown in Supplementary Scheme S2. The AlphaScreen assay results in Figure 1 indicated that these three compounds were not as potent as 1.
Search for the Favorable Polar Interactions.
Although the hot region 2 is primarily hydrophobic, it does have the helical dipole and the polar functional groups. One strategy we used was the replacement of the CH group in the aromatic rings of 1 with the N atom.51 This method introduces the polarity to the compounds and reduces the lipophilicity. Compounds 12–16 were designed and synthesized. The synthetic routes for 12 and 13 are shown in Supplementary Scheme S3. The synthetic routes for 14–16 are shown in Supplementary Scheme S4. The biochemical assay results of 12–16 in Figure 2 indicated none of these compounds were as potent as 1.
Figure 2.
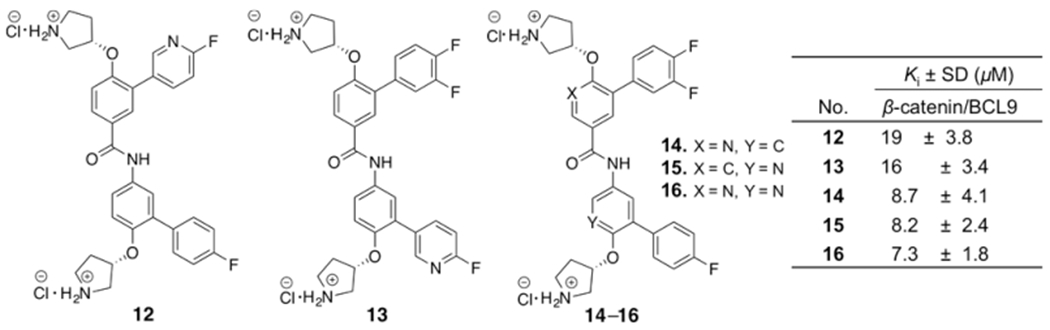
Chemical structures and the AlphaScreen Ki values of 12–16. Each set of data was expressed as mean ± standard deviation (n = 3).
Search for the Favorable H-Bonding and Salt Bridge Interactions.
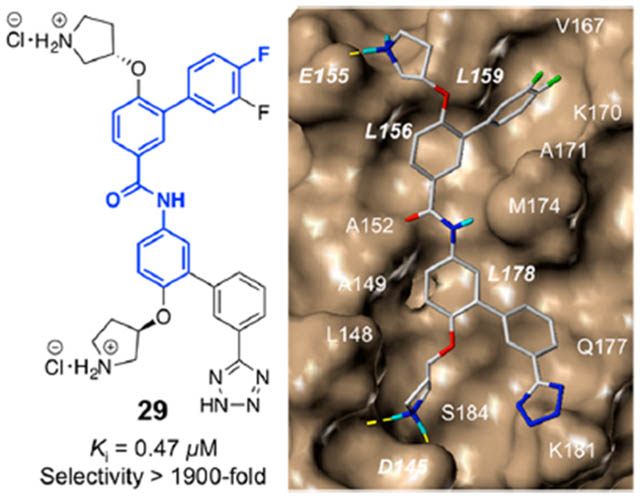
The crystal structures of β-catenin in complex with the BCL9 HD2 domain shows β-catenin Lys181 is adjacent to the hot region 2.34,35 Another strategy for inhibitor optimization was targeting this residue. Compounds 17–23 were designed to form H-bonding interactions with β-catenin Lys181. The synthetic route for 17–23 is shown in Supplementary Scheme S5. Carboxylic acid and bioisosteres were introduced to 1 to form salt bridge interactions with Lys181. Compounds 24–30 were designed and synthesized. The synthetic route for 24–26 is shown in Supplementary Scheme S6. The synthetic routes for 27–30 are shown in Scheme 2. The AlphaScreen assay results of 17–30 are shown in Figure 3. The biochemical inhibitory activities of 27 and 30 were comparable with that of 1. Compound 29 exhibited the Ki of 0.47 ± 0.14 μM for disruption of the β-catenin/BCL9 PPI. This Ki value is very close to the dissociation constant (KD) of 0.47 μM for the β-catenin/BCL9 interaction.34
Scheme 2.
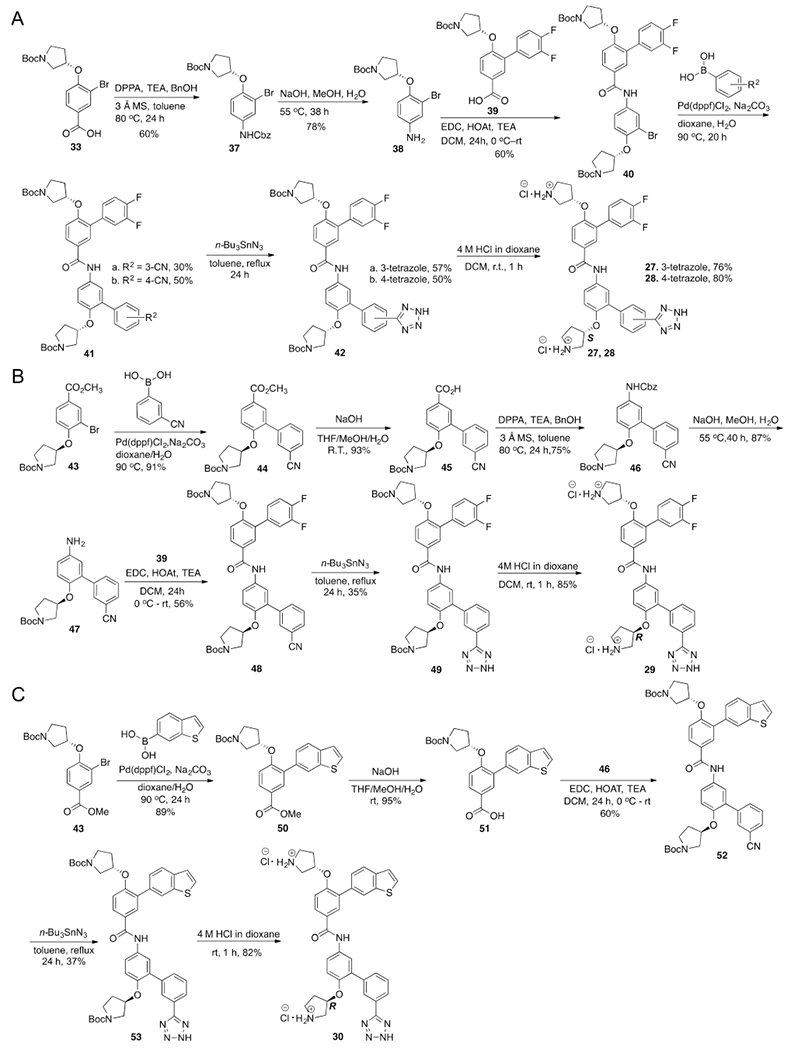
Figure 3.
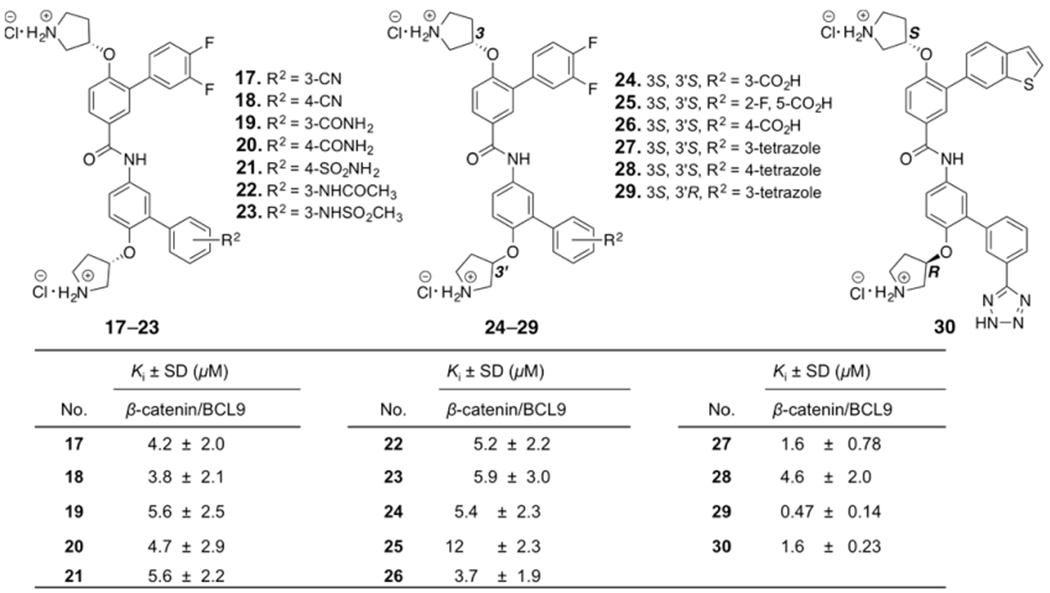
Chemical structures and the AlphaScreen Ki values of 17–30. Each set of data was expressed as mean ± standard deviation (n = 3).
Introduce Conformational Constraints.
Two compounds, 31 and 32, were designed and synthesized. The synthetic routes for 31 and 32 are shown in Scheme 3. The biochemical assay results of these two compounds are shown in Figure 4. Compound 32 exhibited the similar biochemical inhibitory activity as 1.
Scheme 3.
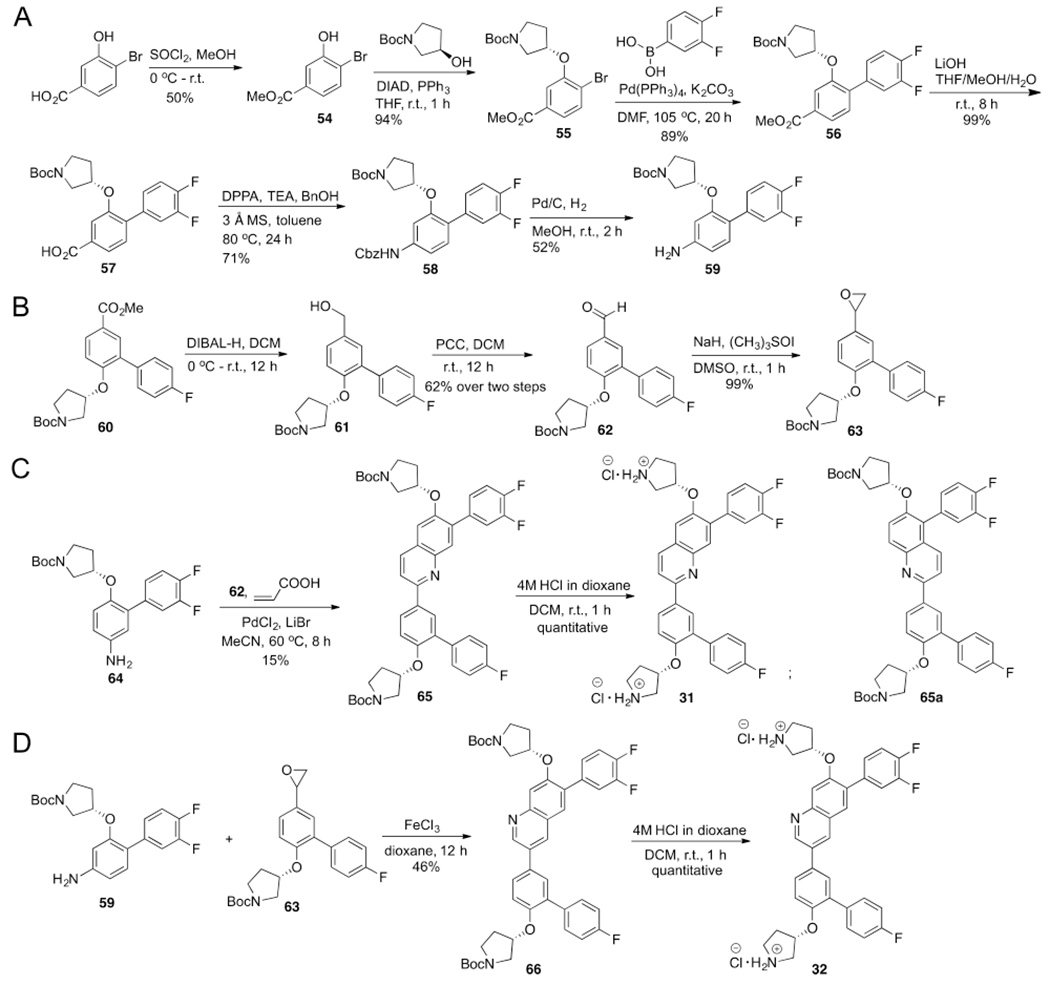
Figure 4.
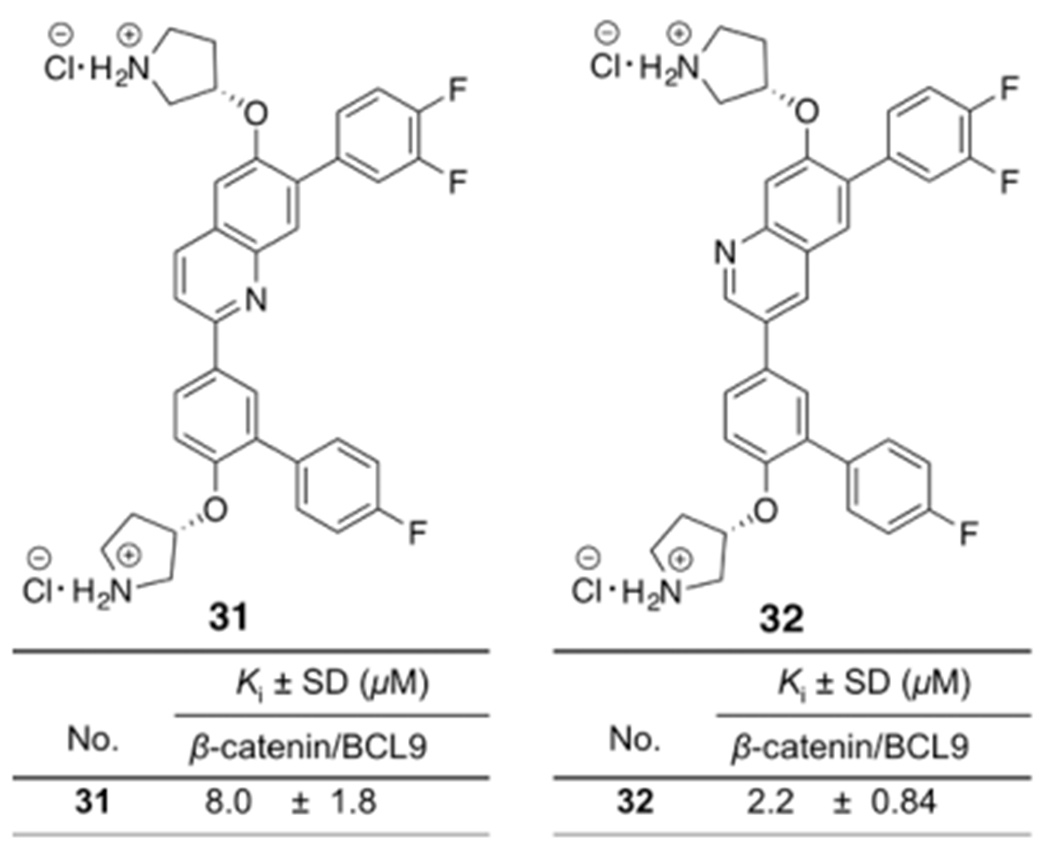
Chemical structures and the AlphaScreen Ki values of 31 and 32. Each set of data was expressed as mean ± standard deviation (n = 3).
Biochemical Characterizations.
Inhibitor Selectivity between β-Catenin/BCL9 and β-Catenin/E-Cadherin PPIs.
β-Catenin is at least associated with two functions in cells. One is that the nuclear β-catenin interacts with Tcf/Lef, BCL9/B9L, CBP/p300, etc., to culminate Wntβ-catenin signaling. The second is that the cytosolic β-catenin interacts with E-cadherin to act as a structural component of adherens junctions. The crystallographic analyses of β-catenin in complexes with BCL934,35 and E-cadherin52 indicated that the interface of β-catenin to interact with BCL9 was also used to bind region V of E-cadherin. Compounds 29 and 30 were assessed for their selectivity between β-catenin/BCL9 and β-catenin/E-cadherin PPIs using the AlphaScreen selectivity assay.50 As shown in Table 1, these two compounds exhibited high selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs with 29 of >1900-fold.
Table 1.
Selectivities of 29 and 30 between β-Catenin/BCL9 over β-Catenin/E-Cadherin Interactionsa
|
Ki ± SD (μM) |
|||
|---|---|---|---|
| compd | β-catenin/BCL9 | β-catenin/E-cadherin | selectivity of BCL9/cadherin |
| 29 | 0.47 ± 0.14 | >915 | >1900 |
| 30 | 1.6 ± 0.23 | >915 | >570 |
Each set of data was expressed as mean ± standard deviation (n = 3).
Site-Directed Mutagenesis Studies.
In previous studies, compound 1 was reported to bind with β-catenin but not the BCL9 HD2 domain in isothermal titration calorimetry (ITC) studies.47 The crystallographic studies indicated that D145, E155, and K181 were not involved in the β-catenin/BCL9 binding.34,35 Our AlphaScreen and fluorescence polarization (FP) experiments indicated that β-catenin D145A,47 E155A,47 D145A/E155A,47 and IK181A mutants had no effect on the β-catenin/BCL9 PPI, as shown in Figure 5A. This result allowed us to use the AlphaScreen competitive inhibition assays to evaluate the effects of these mutations on the Ki of 3, 27, and 29. As shown in Figure 5B, the Ki values of 3 for wild type β-catenin/BCL9, β-catenin D145A/BCL9, and β-catenin D145A and E155A/BCL9 interactions were 1.3 ± 0.80, 4.2 ± 1.1, and 12 ± 3.8 μM, respectively, indicating that the carboxylic side chains of both D145 and E155 of β-catenin were important for the inhibitory potency of 3. However, β-catenin K181 was not targeted by 3 because the Ki value of 3 for the β-catenin K181A/BCL9 interaction was 1.7 ± 0.26 μM. This result is consistent with the proposed binding mode of 3 with β-catenin in Figure 5C (the stick model of Figure 5C is shown in Supplementary Figure S2A). Compounds 27 and 29 were designed to form salt bridge interactions with β-catenin K181A. The AlphaScreen Ki of these two compounds for wild type β-catenin/BCL9, β-catenin D145A/BCL9, β-catenin D145A and E155A/BCL9, and β-catenin K181A/BCL9 PPIs are shown in Figure 5B. β-Catenin D145, E155, and K181 are important for 27 and 29 to bind with β-catenin and disrupt the β-catenin/BCL9 PPI. This result is consistent with the proposed binding mode in Figure 5D (the stick model of Figure 5D is shown in Supplementary Figure S2B).
Figure 5.
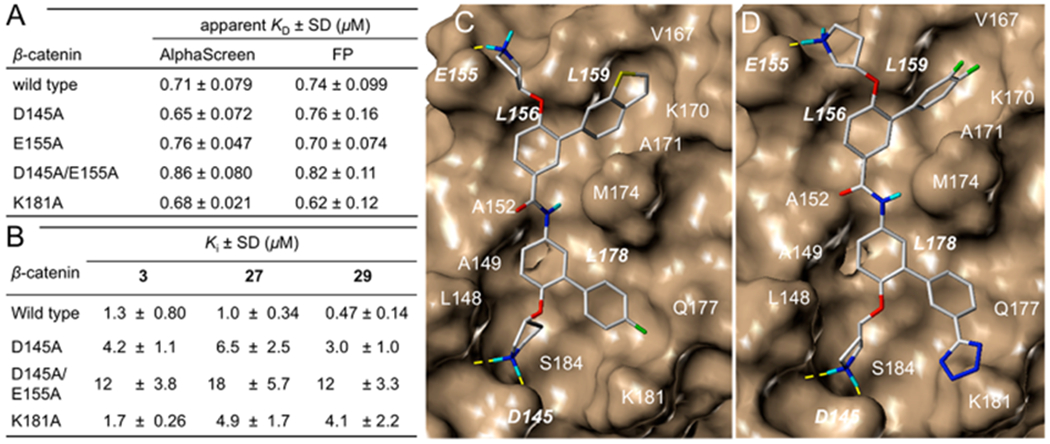
(A) Apparent KD values of wild-type and mutant β-catenin proteins with wild-type BCL9 HD2 domain. (B) AlphaScreen competitive inhibition assay of 3, 27, and 29 to disrupt wild-type β-catenin/wild-type BCL9 and mutant β-catenin/wild-type BCL9 PPIs. Each set of data was expressed as mean ± standard deviation (n = 3). (C) AutoDock docking result of 3 with β-catenin (PDB ID, 2GL7). (D) AutoDock docking result of 29 with β-catenin (PDB ID, 2GL7).
Cell-Based Studies.
Inhibition of Viability of Wnt/β-Catenin-Dependent Cancer Cells.
The MTS assay was conducted to determine the effects of 1–32 on inhibition of cancer cell growth. Three cancer cell lines with hyperactive Wnt/β-catenin signaling, SW480, HCT116, and MDA-MB-231, and one cancer cell line with the normal Wnt/β-catenin signaling pathway, A549, were used in this study. The tested compounds were incubated with cells for 72 h before the addition of the MTS reagent (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxyme-thoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) and the electron coupling reagent, phenazine methosulfate (PMS), for detection.53,54 The half maximal inhibitory concentrations (IC50) of these compounds are shown in Table 2. All of the compounds inhibited viability of cancer cells. Among them, compounds 4, 8, 14, 15, 27, 29, and 31 are selective for Wnt/β-catenin hyperactive cancer cells over the cancer cells that have normal Wnt signaling.
Table 2.
MTS Assay To Monitor the Inhibitory Activities of 1–32 on the Viability of Cancer Cells, and the Wnt-Responsive TOPFlash Luciferase Reporter Assay To Examine the Effect on Wnt/β-catenin Transactivationa
| MTs IC50 ± SD (μM) |
||||||
|---|---|---|---|---|---|---|
| Wnt/β-catenin hyperactive |
TOPFlash IC50 ± SD (μM) |
|||||
| compd | SW480 | HCT116 | MDA-MB-231 | normal Wnt A549 | β-catenin-activated HEK239 | SW480 |
| 1 | 3.5 ± 0.32 | 3.1 ± 0.83 | 2.3 ± 0.25 | 8.8 ± 0.62 | 4.5 ± 1.3 | 2.9 ± 0.33 |
| 2 | 1.8 ± 0.94 | 5.6 ± 2.2 | 2.1 ± 1.1 | 3.7 ± 1.4 | 10 ± 3.4 | 12 ± 3.5 |
| 3 | 2.0 ± 0.80 | 7.5 ± 2.0 | 2.8 ± 1.7 | 9.4 ± 3.3 | 6.2 ± 1.9 | 1.2 ± 0.27 |
| 4 | 1.2 ± 0.62 | 5.7 ± 1.9 | 3.1 ± 1.5 | 23 ± 5.0 | 5.2 ± 1.3 | |
| 5 | 2.2 ± 1.3 | 1.4 ± 0.6 | 1.4 ± 0.7 | 4.5 ± 2.0 | ||
| 6 | 1.6 ± 0.71 | |||||
| 7 | 2.6 ± 0.67 | |||||
| 8 | 0.90 ± 0.51 | 5.6 ± 2.0 | 4.1 ± 1.8 | 10 ± 2.4 | 2.0 ± 0.29 | |
| 9 | 6.6 ± 2.7 | 3.8 ± 1.5 | 3.8 ± 1.5 | |||
| 10 | 3.7 ± 2.0 | 6.9 ± 3.2 | 3.4 ± 1.6 | 8.9 ± 2.7 | 8.7 ± 2.3 | |
| 11 | 3.6 ± 2.7 | 5.6 ± 2.7 | 2.3 ± 1.0 | 4.8 ± 2.4 | 10 ± 3.2 | 10 ± 3.3 |
| 12 | 21 ± 8.0 | |||||
| 13 | 15 ± 4.1 | |||||
| 14 | 3.0 ± 1.4 | 8.7 ± 3.5 | 3.6 ± 1.2 | 26 ± 7.4 | 22 ± 4.5 | 5.9 ± 1.0 |
| 15 | 1.7 ± 0.99 | 7.8 ± 3.3 | 3.1 ± 1.3 | 29 ± 7.9 | 10 ± 1.2 | 7.7 ± 1.2 |
| 16 | 9.9 ± 3.7 | |||||
| 17 | 4.3 ± 0.74 | >25 | ||||
| 18 | 6.1 ± 2.9 | >25 | ||||
| 19 | 190 ± 15 | |||||
| 20 | 160 ± 19 | |||||
| 21 | 220 ± 58 | >200 | >200 | |||
| 22 | 160 ± 9.1 | |||||
| 23 | 95 ± 8.9 | |||||
| 24 | >400 | >400 | 368 ± 30 | |||
| 25 | >400 | >400 | >400 | |||
| 26 | >400 | >400 | 391 ± 36 | |||
| 27 | 140 ± 11 | 190 ± 7.8 | 92 ± 16 | 410 ± 42 | ||
| 28 | 86 ± 13 | |||||
| 29 | 41 ± 6.2 | 55 ± 7.4 | 73 ± 8.9 | >400 | 25 ± 4.1 | 25 ± 2.7 |
| 30 | 150 ± 21 | 240 ± 29 | 430 ± 30 | 50 ± 5.2 | 43 ± 3.4 | |
| 31 | 3.6 ± 1.9 | 9.6 ± 2.5 | 15 ± 6.1 | 23 ± 4.4 | 26 ± 3.1 | 12 ± 1.5 |
| 32 | 1.7 ± 0.90 | 2.2 ± 1.0 | 3.1 ± 1.2 | 7.5 ± 2.9 | 14 ± 1.5 | 7.9 ± 1.4 |
Each set of data was expressed as mean ± standard deviation (n = 3). The data for 1 was reported in a previous study.47
Lactate Dehydrogenase (LDH) Cytotoxicity Assay.
The cytotoxic compounds may cause cell death by damaging of the cell membrane through the nonspecific manner. The LDH release assay55,56 with the relatively short inhibitor incubation time57 (4 h in this study) was conducted to determine the cytotoxicity of the tested compounds. The result is shown in Supplementary Figure S3. Compounds 1–18 except 12 exhibited nonspecific cytotoxicity at high concentrations, such as 50 and 100 μM. Compounds 19–31 did not exhibit obvious cytotoxicity in the LDH assay even at the very high concentration of 800 μM.
Inhibition of β-Catenin/BCL9/Tcf Transcriptional Activity.
The TOPFlash luciferase reporter assay (in which the luciferase reporter has three wild-type Tcf4 binding sites) was performed with 1–4, 8, 10, 11, 14, 15, 17, 18, 21, 24–26, and 29–32. Their IC50 values are shown in Table 2. Among them, compounds 1–4, 8, 10, 11, 14, 15, and 29–32 inhibited the TOPFlash luciferase activities. However, all of these compounds, except 29 and 30, inhibited the FOPFlash luciferase reporter (with three mutant Tcf4 binding sites) activities at the higher concentrations. The results of 1–4, 8, 11, 14, 15, and 29–32 are shown in Figure 6. These results indicated that only 29 and 30 are specific for the Wnt/β-catenin signaling pathway. Compounds 1–4, 8, 11, 14, and 15 inhibited FOPFlash luciferase reporter activity because these compounds underwent the primary necrosis by damaging the cell membrane at high concentrations.
Figure 6.
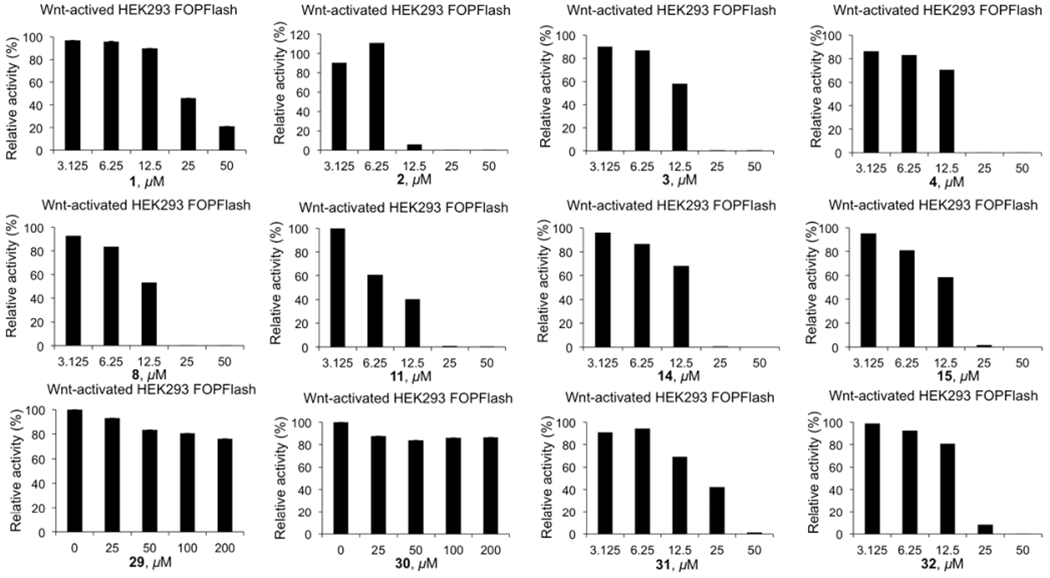
Wnt-responsive FOPFlash luciferase reporter assay results of inhibitors 1–4, 8, 11, 14, 15, and 29–32 in β-catenin activated HEK293 cells.
Inhibition of Expression of Wnt/β-catenin Target Genes.
Axin2 is a specific target gene of the Wnt/β-catenin signaling pathway.58 Cyclin D1, LEF1, and c-myc are upregulated in cancer cells with hyperactive Wnt/β-catenin signaling to promote tumor-igenesis. Quantitative real-time PCR (qPCR) studies were conducted for 3, 8, 29, and 31 (Figure 7). Similar to 1,47 compounds 3 and 29 did not inhibit house-keeper gene HPRT but dose-dependently inhibited Wnt target genes, Axin2, cyclin D1, and LEF1.
Figure 7.
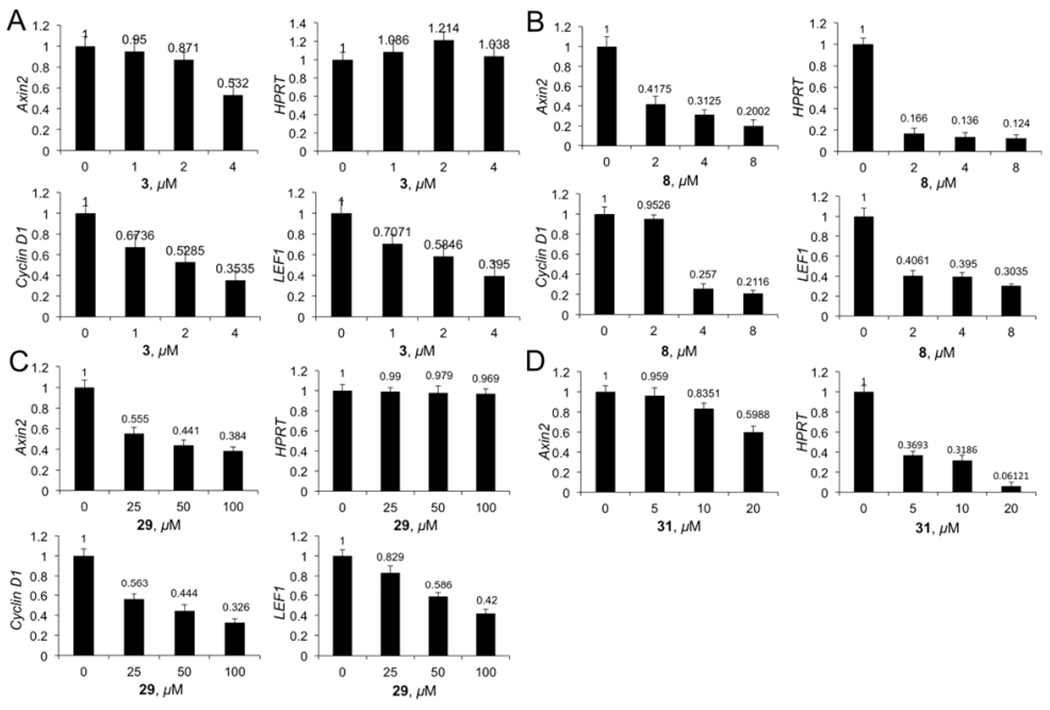
qPCR studies to determine the changes of mRNA expression of Axin2, LEF1, and cyclin D1 in SW480 cells in response to different concentrations of 3 (A), 8 (B), 29 (C), and 31 (D). House-keeper gene HPRT was used as the reference. Each set of data was expressed as mean ± standard deviation (n = 3).
As shown in Figure 8A,B, Western blot experiments indicated that the protein expression levels of cyclin D1 and c-myc were significantly decreased after treatment of 3 and 29, respectively. Both compounds can inhibit the level of the active form of β-catenin (ABC), which is nuclear β-catenin, in dose-dependent manners. These two compounds had no effect on the level of E-cadherin-bound β-catenin (i.e., total β-catenin), indicating these two compounds did not affect the upstream nodes of the Wnt pathway that regulate degradation of cytosolic β-catenin.
Figure 8.
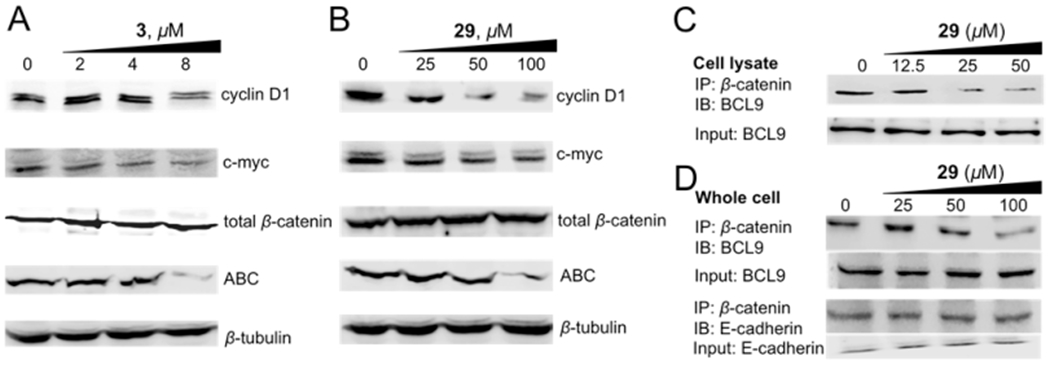
(A,B) Western blot analysis to monitor the change of protein expression of cyclin D1, c-myc, the active form of β-catenin (ABC), and the total β-catenin in response to different concentrations of 3 (A) and 29 (B) in SW480 cells. β-Tubulin was used as the internal reference. The quantitative data of Western blot analyses are shown in Supplementary Table S1. (C) Co-IP experiments to evaluate the disruption of the β-catenin/BCL9 PPI by 29 in HCT116 cell lysate. (D) Co-IP experiments to evaluate the disruption of the β-catenin/BCL9 PPI by 29 and the selectivity of 29 for β-catenin/BCL9 over β-catenin/E-cadherin PPIs in HCT116 cells. IP, immunoprecipitation; IB, immunoblotting; input, 10% of cell lysate. Each experiment was performed in triplicate. The quantitative data of co-IP experiments are shown in Supplementary Table S2.
Disruption of the β-Catenin/BCL9 PPI in Cells and Cell-Based Inhibitor Selectivity Studies.
The coimmunoprecipitation (co-IP) experiments were conducted to assess the effect of 29 for disruption of the interaction between full-length β-catenin and full-length BCL9 using HCT116 cell lysates. As shown in Figure 8C, compound 29 can dose-dependently disrupt the full-length β-catenin/full-length BCL9 PPI after 4 h incubation with HCT116 cell lysates. The co-IP experiments were also performed with HCT116 cells to evaluate the effect of 29 on the disruption of the β-catenin/BCL9 PPI and the selectivity over the β-catenin/E-cadherin PPI in the cellular context. As shown in Figure 8D, compound 29 inhibited the β-catenin/BCL9 PPI in a dose-dependent manner. A parallel experiment indicated that 29 did not affect the β-catenin/E-cadherin PPI at the concentrations that were sufficient to disrupt the β-catenin/BCL9 PPI.
Chemistry.
The synthetic route for 2 and 3 is shown in Scheme 1. The amide bond coupling reaction between the previously reported 3359 and 3447 generated key intermediate 35, which then underwent the Suzuki reaction with (6-fluoronaph-thalen-2-yl)boronic acid or (1-benzothiophen-6-yl)boronic acid to afford 36 in good yield. The Boc deprotection of 36 with 4 M HCl offered the final products 2 and 3.
The synthetic routes for 27–30 are shown in Scheme 2. Compound 33 was subjected to the Curtius rearrangement to generate 37. The deprotection of the Cbz protecting group gave 38. The amide bond coupling reaction of 38 with another previously reported key intermediate 3947 and then the Suzuki reaction with 3-cyanophenylboronic acid and 4-cyanophenylboronic acid generated 41a,b, respectively. The cycloaddition reaction of nitriles 41 with n-Bu3SnN3 and then the deprotection of the Boc protecting group with 4 M HCl offered 27 and 28. The Suzuki reaction between the previously reported 4347 and 3-cyanophenylboronic acid afforded 44. The hydrolysis of the methyl ester of 44 and the Curtius rearrangement reactions furnished 46. The deprotection of the Cbz group and the amide bond coupling reaction with 39 gave 48. The cycloaddition reaction of 48 with n-Bu3SnN3 resulted in 49, which offered the final product 29 after the deprotection of the Boc group. The Suzuki reaction between 43 and (1-benzothiophen-6-yl) boronic acid generated 50. The deprotection of the methyl ester of 50 and the amide coupling reaction with 46 afforded 52, which then underwent the cycloaddition reaction to furnish 53. The deprotection of the Boc group of 53 generated final product 30.
The synthetic routes for 31 and 32 are shown in Scheme 3. The esterification of 4-bromo-3-hydroxybenzoic acid and then the Mitsunobu reaction with (R)-N-Boc-3-pyrrolidinol offered 55. The Suzuki reaction of 55 with 3,4-difluorophenylboronic acid and the hydrolysis of the methyl ester of 56 afforded 57. The Curtius rearrangement of 57 and the deprotection of the Cbz group generated 59. The methyl ester group of the previously synthesized intermediate 6047 was reduced to a hydroxymethyl group by DIBAL-H. The alcohol 61 was then oxidized to aldehyde 62 by PCC. The epoxidation of 62 by trimeth-ylsulfoxonium iodide offered 63. The palladium-catalyzed sequential reaction60 using the previously reported aniline 64,47 aldehyde 62, and acrylic acid produced 65. The other regioisomer 65a was not observed in this reaction. This difference might be correlated with the steric hindrance of the 3,4-difluorophenyl ring. The deprotection of the Boc protecting group afforded the final product 31. The FeCl3 promoted tandem reaction of aniline 59 with styrene oxide 63 furnished 66.61 The deprotection of the Boc protecting group provided the final product 32.
DISCUSSION
Wnt/β-catenin signaling is frequently observed hyperactive in cancers. The most effective target to develop anticancer inhibitors for the Wnt/β-catenin signaling pathway is the β-catenin-containing transcriptional complex because this complex mediates the signaling at the downstream node in the pathway. However, despite numerous efforts, this target has proven to be challenging.62 To date, only one series of drug-like inhibitors targets this transcriptional complex, ICG-001 and its second-generation prodrug PRI-724.63 PRI-724 is in clinical trial phase II.64 ICG-001 and PRI-724 bind with transcription coactivator CBP to disrupt the β-catenin/CBP interaction.65 In the structure of the β-catenin-containing transcriptional complex, the β-catenin/BCL9 PPI represents a unique target for inhibitor design because (1) BCL9/B9L acts as an essential adaptor protein to recruit other cofactors and load β-catenin to the Wnt enhanceosome, facilitating the access of β-catenin to Tcf/Lef.33 The β-catenin/BCL9 PPI is required for transcription of Wnt responsive genes33,39,40,66–69 and tumorigenesis;41–43,70–72 (2) this PPI has a moderately strong binding affinity (KD = 0.47 μM); and (3) the β-catenin interface for binding with BCL9 is relatively small and has little overlap with those for the other β-catenin partners. Along with the efforts of the other groups to discover β-catenin/BCL9 inhibitors,35,44–46 we reported the design and synthesis of 1 as a new small-molecule inhibitor for this PPI.47
In the process of inhibitor optimization, we found that the gain of potency was limited by exploration of the hydrophobic interaction based on 1. Also, the more hydrophobic compounds cause cytotoxicity by damaging the cell membrane, as evident in the LDH release assay. The substitution of the CH group of the aromatic rings of 1 with a N atom made the positive impact on the molecular and physiological properties of 12–14, which displayed the lower cytotoxicity in the LDH release assay. However, none of them were more potent than 1. Compound 1 is a lipophilic base. The previous bioinformatic analyses indicated that the lipophilic bases were prone to causing target promiscuity.73–76 Hence, in the process of improving inhibitor potency and selectivity for the Wnt/β-catenin signaling pathway, we also aimed to introduce the polar groups; compound 29 was obtained. This compound exhibits a Ki of 0.47 μM for disruption of the β-catenin/BCL9 PPI and >1900-fold selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs in biochemical AlphaScreen assays. This compound dose-dependently inhibited TOPFlash luciferase reporter activity with an IC50 of 25 μM, did not exhibit cytotoxicity in the LDH release assay, and had no effect on FOPFlash luciferase reporter activity up to the concentration of 200 μM.
The Ki of 29 for disruption of the β-catenin/BCL9 PPI was 0.47 μM. The TOPFlash luciferase reporter (with three Tcf binding sites) activity of 29 was 25 μM. This difference could be caused by the zwitterionic nature of this compound. The intracellular concentration of 29 was determined by the method described in a previous study.48 The result is shown in the Supplementary Note. The cell-bound concentration of 29 at 37 °C was determined to be 6.6 nmol/million HCT116 cells for the 6 h incubation in 5 mL of DMEM media with 10% FBS, when the input concentration was 50 μM 29. The 4 °C incubation experiment indicated a significant amount (77%) of cell-bound 29 bound with the cell membrane through the nonspecific manner. The cell-bound concentration of 29 was 5.1 nmol/million HCT116 cells for 6 h incubation at 4 °C in 5 mL of DMEM media with 10% FBS. This result is different from that of 1.48 The cell-bound concentration of 1 at 37 °C was determined to be 6.7 nmol/million MDA-MB-231 cells for the 6 h incubation in 5 mL of DMEM media with 10% FBS, when the input concentration was 2 μM 1. At 4 °C, the cell-bound concentration of 1 was 1 nmol/million MDA-MB-231 cells for 6 h incubation at 4 °C in 5 mL of DMEM media with 10% FBS. Therefore, only 15% of 1 bound with the cell membrane by the nonspecific manner.
Four cancer cells were examined in cell-based studies. SW480 and HCT116 cells have hyperactive Wnt signaling. SW480 cells harbor the deletion of APC,77 whereas HCT116 cells harbor the heterozygous three-base deletion of CTNNB178 that blocks β-catenin phosphorylation and ubiquitination. Although the expression levels of β-catenin in MDA-MB-231 cells are low,79–81 MDA-MB-231 cells have high expression levels of upstream Wnt effectors, low-density lipoprotein-related proteins 5 and 6 (LRP 5/6),81,82 frizzled (Fzd),83,84 and dishevelled.79 MDA-MB-231 cells are susceptible to the activation by Wnt ligands to promote cancer cell growth in vitro and in vivo.83,85 β-Catenin siRNA knocks down β-catenin expression in MDA-MB-231 cells and blocks cancer cell migration and invasion.80,86 These three cancer cell lines were chosen based on their dependence on β-catenin for growth and survival. However, A549 cells do not contain active Wnt/β-catenin signaling.87,88
In this study, the tetrazolium MTS assay was used to examine the inhibitory effects of new compounds on cancer cell viability. The MTS assay, however, cannot distinguish the effects on cell death and cell growth inhibition. LDH release was used as the preferred biomarker to examine the effect on cell death because LDH is a soluble cytoplasmic enzyme that is present in all examined cells of this study. It is released to the extracellular space when the cytoplasmic membrane is damaged. Through the combinational use of MTS (compounds were incubated with cancer cells for 72 h) and LDH release (compounds were incubated with cancer cells for 4 h) assays, the effects of these compounds on cytotoxicity and on inhibition of the viability of cancer cells can be differentiated.
Compounds 2–18 displayed high activities in MTS assays. However, they also exhibited cytotoxicity in the LDH release assay. They are not suitable for the further inhibitor optimization. In contrast, compounds 24–30 inhibited growth of cancer cells in the MTS assays but had no effect on LDH release activity, indicating that the future inhibitor optimization should take into consideration decreasing compound hydrophobicity, and compound 29 can be used as the new starting point. Compound 29 also exhibits selectivity on inhibiting the viability of Wnt-activated cancer cells over the cancer cells that have normal Wnt signaling. It is interesting that the IC50 of 29 for the inhibition of TOPFlash luciferase reporter activity is lower than that for the MTS assay.
Similar to carnosic acid35 and 1,47 compounds 3 and 29 also selectively decrease the level of active form of β-catenin (i.e., nuclear β-catenin) without affecting the level of total β-catenin (i.e., E-cadherin-bound β-catenin). With the result that 29 disrupts the β-catenin/BCL9 interaction in co-IP experiments (Figures 8C,D), there are at least two possible mechanisms why 29 promotes the selective degradation of nuclear β-catenin. One is that 29 disrupts the interaction of β-catenin with BCL9, and inhibitor-bound β-catenin is susceptible to the H1 (the first α-helix of β-catenin armadillo repeats)-dependent proteasomal degradation.35 The second mechanism is that after the disruption of the PPI, compound 29 destabilizes β-catenin tertiary structure, and promotes β-catenin degradation. The further mechanistic studies are required to address this question.
Although 29 is more potent and selective than 1 in biochemical assays, it should be noted that 29 still has multiple carboaromatic rings, which are often considered to be the potential risk of the high serum albumin binding, and the inhibition of cytochrome P450 and human ether-a-go-go-related gene product (hERG), potentially lowering the developability of the compound.89–92 In addition to improving inhibitor potency and maintaining inhibitor selectivity, the further optimization will be centered on the replacement of the aromatic rings with the aliphatic alternatives and the increase of the three-dimensional features of the designed compounds.
CONCLUSION
The Wnt/β-catenin signaling pathway is frequently implicated in tumorigenesis of many cancers. The pathway is not only involved in the proliferation of cancer cells but more importantly responsible for cancer progression, metastasis, and drug resistance. The activation of β-catenin signaling also blocks recruitment of cytotoxic T cells, promotes Treg cell survival and infiltration, and excludes anticancer immunity. However, it has been proven difficult to discover inhibitors for the downstream effectors of the Wnt/β-catenin signaling pathway. The β-catenin/BCL9 PPI in the transcriptional complex is an interesting target for chemical probe development and drug discovery. In this study, structure-based design and synthesis was conducted to optimize the inhibitors based on the 4′-fluoro-N-phenyl-[1,1′-biphenyl]-3-carboxamide scaffold. Compound 29 was found to disrupt the β-catenin/BCL9 PPI with a Ki of 0.47 μM and exhibited high selectivity (>1900-fold) for β-catenin/BCL9 over β-catenin/E-cadherin PPIs in the biochemical AlphaScreen assays. The site-directed mutagenesis studies indicated that β-catenin D145, E155, and K181 are important for the inhibitors to disrupt the β-catenin/BCL9 PPI. The cell-based studies indicated that 29 can selectively inhibit growth of cancer cells that have hyperactive Wnt/β-catenin signaling and did not show cytotoxicity in the LDH release assays. The co-IP experiments indicated that this compound disrupted the full-length β-catenin/BCL9 PPI while leaving the β-catenin/E-cadherin PPIs unaffected. The TOPFlash/FOPFlash luciferase reporter assays indicated that 29 can selectively suppress transactivation of Wnt/β-catenin signaling. This compound also downregulated expression of Wnt target genes in dose-dependent manners at both mRNA and protein levels, and only inhibited the downstream nodes of the Wnt/β-catenin signaling pathway. This study offered the directions for future inhibitor optimization.
EXPERIMENTAL SECTION
Chemical Synthesis.
General Methods, Reagents, and Materials.
All reagents were purchased from commercial sources and used without further purification unless stated otherwise. 1H NMR and 13C NMR spectra were recorded on the Bruker AVANCEIIIHD 500 (500 MHz), Varian VXR-500 (500 MHz), Varian Inova-400 (400 MHz), or Varian Unity-300 (300 MHz) spectrometers (125.7, 100, and 75 MHz for 13C NMR spectra, respectively) in d6-DMSO, d6-acetone, d4-methanol, and CDCl3. Chemical shifts were reported as values in parts per million (ppm), and the reference resonance peaks were set at 7.26 ppm (CHCl3), 3.31 ppm (CD2HOD), 2.50 ppm [(CD2H)2SO], and 2.05 ppm [(CD2H)2CO] for the 1H NMR spectra and 77.23 ppm (CDCl3), 49.00 ppm (CD3OD), 39.52 ppm (d6-DMSO), and 29.84 ppm (d6-acetone) for the 13C NMR spectra. Low-resolution mass spectra were determined on the Agilent 6120 single quadrupole MS with 1220 infinity LC system (HPLC-MS) with an ESI source. High-resolution mass spectra were determined on the Agilent G6230BA TOF LCMS Mass Spectrometer with a TOF mass detector. Thin-layer chromatography was carried out on E. Merck precoated silica gel 60 F254 plates with visualization accomplished with phosphomolybdic acid and ninhydrin spray reagents, or with a UV–visible lamp. Column chromatography was performed with SilicaFlash F60 (230–400 mesh). The purity of final compounds 2–32 was determined by HPLC analysis with two different conditions. The instrument was an Agilent 1260 Infinity II HPLC system with a quaternary pump, a vial sampler, and a diode array detector (DAD). A Kromasil 300–5–C18 column (4.6 × 250 mm) was used. The DAD detector was set to 220, 254, and 280 nm. The purity of all tested compounds was ≥95%.
N-(4′-Fluoro-6-(((S)-pyrrolidin-3-yl)oxy)-[1,1′-biphenyl]-3-yl)-3-(6-fluoronaphthalen-2-yl)-4-(((S)-pyrrolidin-3-yl)oxy)benzamide Dihydrochloride (2).
To a solution of 36a (0.40 g, 0.50 mmol) in CH2Cl2 (2 mL) under anhydrous conditions was added 4 M HCl in dioxane (10 mL). The mixture was then stirred at room temperature for 1–1.5 h. The solvent was removed under the reduced pressure to yield 2 as a white solid (0.27 g, 88%). 1H NMR (500 MHz, CD3OD) δ 8.09–7.91 (m, 4H), 7.86 (d, J = 8.5 Hz, 1H), 7.76–7.52 (m, 6H), 7.38–6.96 (m, 5H), 5.26 (s, 1H), 5.02 (s, 1H), 3.86–3.37 (m, 6H), 3.19 (dq, J = 30.1, 10.7, 10.2 Hz, 2H), 2.53–2.03 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 166.60, 163.34, 162.01, 161.39, 160.06, 156.52, 150.25, 134.67, 134.65, 134.25, 134.22, 133.62, 133.55, 133.46, 131.46, 131.37, 131.32, 131.26, 131.19, 130.95, 130.89, 130.82, 130.70, 128.91, 128.80, 128.28, 128.22, 126.87, 126.83, 124.29, 123.13, 121.91, 120.75, 116.26, 116.06, 115.80, 115.58, 114.93, 114.76, 113.57, 110.41, 110.24, 77.33, 76.71, 50.61, 44.44, 44.27, 31.05, 30.82. HRMS (ESI) Calcd for C37H33F2N3O3 (M + H)+ 606.2568, found 606.2554.
3-(Benzo[b]thiophen-6-yl)-N-(4′-fluoro-6-(((S)-pyrrolidin-3-yl)-oxy)-[ 1,1′-biphenyl]-3-yl)-4-(((S)-pyrrolidin-3-yl)oxy)benzamide Dihydrochloride (3).
It was prepared through the same procedure for 2 to yield 3 as a white solid (83% yield). 1H NMR (300 MHz, CD3OD) δ 8.07 (d, J = 1.5 Hz, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.88 (d, J = 8.3 Hz, 1H), 7.74–7.65 (m, 2H), 7.67–7.45 (m, 4h), 7.39 (d, J = 5.5 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.13 (td, J = 9.7, 9.3, 5.7 Hz, 3H), 5.25 (s, 1H), 5.02 (d, J = 3.5 Hz, 1H), 3.49 (dddt, J = 35.8, 31.2, 11.3, 5.3 Hz, 6H), 3.18 (ddt, J = 20.0, 11.6, 8.3 Hz, 2H), 2.46–2.07 (m, 4H). 13C NMR (75 MHz, CD3OD) δ 166.67, 156.51, 150.25, 140.3, 139.19, 134.24, 133.79, 133.46, 131.78, 131.48, 131.32, 131.22, 130.96, 128.70, 128.24, 127.15, 126.03, 124.28, 123.53, 123.05, 123.00, 121.88, 115.57, 114.97, 114.69, 113.76, 77.36, 76.82, 50.62, 44.40, 44.26, 31.04, 30.82. HRMS (ESI) Calcd for C35H32FN3O3S (M + H)+ 594.2227, found 594.2217.
3′,4′-Difluoro-6-((S)-pyrrolidin-3-yloxy)-N-(6-((S)-pyrrolidin-3-yloxy)-3′-(2H-tetrazol-5-yl)-[1,1′-biphenyl]-3-yl)-[1,1′-biphenyl]-3-carboxamide Dihydrochloride (27).
It was prepared through the same procedure as 2 to afford 27 as a white solid (76% yield). 1H NMR (500 MHz, CD3OD): δ ppm 8.30 (s, 1H), 8.03 (dd, J = 2.5 Hz, 9.0 Hz, 1H), 7.99–7.97 (m, 2H), 7.80–7.72 (m, 3H), 7.65 (t, J = 8.0 Hz, 1H), 7.57–7.47 (m, 1H), 7.41–7.30 (m, 2H), 7.26 (d, J = 9.0 Hz, 1H), 7.19 (d, J = 9.0 Hz, 1H), 5.31 (s, 1H), 5.14 (s, 1h), 3.66–3.62 (m, 1H), 3.56–3.52 (m, 3H), 3.52–3.37 (m, 3H), 3.29–3.23 (m, 1H), 2.39–2.17 (m, 4H). 13C NMR (125 MHz, CD3OD): δ ppm 166.17, 156.00, 150.08, 139.11, 133.20, 131.94, 130.67, 130.39, 129.28, 129.11, 129.05, 128.00, 125.94, 125.91, 125.89, 125.46, 124.20, 124.02, 122.16, 118.26, 118.12, 116.76, 116.62, 114.84, 113.24, 109.99, 76.90, 76.52, 50.48, 50.36, 44.23, 44.10, 30.75, 30.66. HRMS (ESI) Calcd for C34H31F2N7O3 (M + H)+ 624.2535, found 624.2528.
3′,4′-Difluoro-6-((S)-pyrrolidin-3-yloxy)-N-(6-((S)-pyrrolidin-3-yloxy)-4′-(2H-tetrazol-5-yl)-[1,1′-biphenyl]-3-yl)-[1,1′-biphenyl]-3-carboxamide Dihydrochloride (28).
It was prepared through the same procedure as 2 to afford 28 as a white solid (80% yield). 1H NMR (500 MHz, CD3OD): δ ppm 8.09 (dd, J = 2.0 Hz, 8.5 Hz, 2H), 8.03 (dd, J = 2.0 Hz, 8.5 Hz, 1H), 8.00–7.97 (m, 1H), 7.76 (dd, J = 3.0 Hz, 9.0 Hz, 1H), 7.74–7.66 (m, 3H), 7.57–7.46 (m, 1H), 7.39–7.29 (m, 2H), 7.25 (d, J = 8.5 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 5.31 (s, 1H), 5.08 (s, 1H), 3.65–3.62 (m, 1H), 3.56–3.43 (m, 4H), 3.41–3.33 (m, 1H), 3.29–3.22 (m, 1H), 3.20–3.11 (m, 1H), 2.402.25 (m, 2H), 2.25–2.11 (m, 2H). 13C NMR (125 MHz, CD3OD): δ ppm 156.00, 150.20, 139.59, 133.38, 131.73, 130.38, 129.37, 129.05, 128.15, 126.40, 126.18, 125.82, 123.94, 121.99, 118.27, 118.12, 116.78, 116.64, 115.59, 113.26, 77.37, 76.53, 50.54, 50.41, 44.22, 44.04, 30.70, 30.48. HRMS (ESI) Calcd for C34H31F2N7O3 (M + H)+ 624.2535, found 624.253.
3′,4′-Difluoro-6-(((S)-pyrrolidin-3-yl)oxy)-N-(6-(((R)-pyrrolidin-3-yl)oxy)-3′-(2H-tetrazol-5-yl)-[1,1′-biphenyl]-3-yl)-[1,1′-biphenyl]-3-carboxamide Dihydrochloride (29).
It was prepared through the same procedure as 2 to afford 29 as a white solid (85% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.26 (s, 1H), 9.40 (d, J = 32.9 Hz, 4H), 8.29 (s, 1H), 8.06 (d, J = 7.4 Hz, 1H), 8.01 (d, J = 6.6 Hz, 2H), 7.84 (d, J = 11.8, Hz, 2H), 7.74 (d, J = 8.2 Hz, 2H), 7.66 (d, J = 7.6 Hz, 1H), 7.48 (q, J = 4.5, 3.3 Hz, 2H), 7.27 (d, J = 8.5 Hz, 1H), 7.18 (d, J = 8.6 Hz, 1H), 5.23 (s, 1H), 5.06 (s, 1H), 3.59–3.06 (m, 8H), 2.23–2.00 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 166.10, 156.13, 150.57, 150.11, 148.66, 139.12, 134.51, 133.13, 133.06, 130.94, 130.56, 130.45, 130.25, 129.33, 128.27, 127.93, 127.13, 125.94, 124.51, 123.79, 123.26, 119.02, 117.90, 117.41, 116.41, 78.56, 54.58, 54.13, 47.35, 46.93, 33.64, 33.14. HRMS (ESI) Calcd for C34H31F2N7O3 (M + H)+ 624.2535, found 624.253.
(R)-3-((5-(3-(Benzo[b]thiophen-6-yl)-4-(((S)-pyrrolidin-1-ium-3-yl)oxy)benzamido)-3′-(2H-tetrazol-5-yl)-[1,1′-biphenyl]-2-yl)oxy)-pyrrolidin-1-ium Chloride (30).
It was prepared through the same procedure as 2 to afford 30 as a white solid (82% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.29 (s, 1H), 9.53 (t, J = 59.7 Hz, 4H), 8.35 (t, J = 1.8 Hz, 1H), 8.29 (d, J =1.4 Hz, 1H), 8.17–8.08 (m, 2H), 8.05 (dd, J = 8.7, 2.4 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.91–7.85 (m, 2H), 7.83–7.74 (m, 2H), 7.71–7.63 (m, 2H), 7.50 (d, J = 5.5 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 7.21 (d, J = 9.0 Hz, 1H), 5.27 (t, J = 5.0 Hz, 1H), 5.17–5.00 (m, 1H), 3.65–3.46 (m, 2H), 3.34 (m, 4H), 3.13 (d, J = 9.8 Hz, 2H), 2.27–2.01 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.90, 156.35, 149.79, 139.70, 139.24, 138.99, 134.05, 133.82, 132.50, 131.08, 130.62, 129.94, 129.81, 129.34, 128.49, 128.19, 128.17, 126.67, 126.23, 124.17, 123.78, 123.71, 123.49, 121.80, 114.97, 113.76, 76.78, 76.53, 50.06, 50.01, 44.26, 44.12, 31.51, 31.40. HRMS (ESI) Calcd for C36H33N7O3S (M + H)+ 644.2444, found 644.2434.
7-(3,4-Difluorophenyl)-2-(4′-fluoro-6-((S)-pyrrolidin-3-yloxy)-[1,1′-biphenyl]-3-yl)-6-((S)-pyrrolidin-3-yloxy)quinoline Dihydrochloride (31).
The same procedure to prepare 2 was used to prepare 31 as a yellow solid (quantitative yield). 1H NMR (500 MHz, CD3OD): δ ppm 8.76 (d, J = 10.5 Hz, 1H), 8.25 (d, J = 8.5 Hz, 1H), 8.22–8.15 (m, 3H), 7.76 (s, 1H), 7.68–7.61 (m, 3H), 7.51–7.46 (m, 1H), 7.457.38 (m, 2H), 7.21 (t, J = 9.0 Hz, 2H), 5.46 (s, 1H), 5.35 (s, 1H), 3.773.71 (m, 1H), 3.68–3.43 (m, 6H), 3.28–3.24 (m, 1H), 2.49–2.29 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 163.46, 161.50, 156.87, 153.61, 152.91, 151.41, 149.53, 143.38, 138.19, 135.57, 133.16, 132.98, 131.91, 131.42, 131.32, 131.25, 129.79, 128.51, 126.36, 125.73, 123.61, 121.30, 118.64, 118.51, 117.23, 117.10, 114.85, 114.68, 114.48, 108.75, 102.74, 77.09, 76.76, 50.42, 50.21, 44.32, 44.25, 30.77, 30.65. HRMS (ESI) Calcd for C35MH30F3N3O2 (M + H)+ 582.2368, found 582.2345.
6-(3,4-Difluorophenyl)-3-(4′-fluoro-6-((S)-pyrrolidin-3-yloxy)-[1,1′-biphenyl]-3-yl)-7-((S)-pyrrolidin-3-yloxy)quinoline Dihydrochloride (32).
The same procedure for 2 was used to prepare 32 as a yellow solid (quantitative yield). 1H NMR (500 MHz, CD3OD): δ ppm 9.41 (s, 1H), 9.12 (brs, 1H), 8.23 (s, 1H), 7.90 (dd, J = 2.5, 8.5 Hz, 1H), 7.86 (d, J = 2.0 Hz, 1H), 7.68 (s, 1H), 7.67–7.60 (m, 3H), 7.49–7.39 (m, 2H), 7.36 (d, J = 8.5 Hz, 1H), 7.20 (t, J = 9.0 Hz, 2H), 5.55 (s, 1H), 5.26 (s, 1H), 3.80 (dd, J = 5.0 Hz, 13.0 Hz, 1H), 3.693.58 (m, 2H), 3.57–3.50 (m, 2H), 3.48–3.42 (m, 1H), 3.38–3.33 (m, 1H), 3.27–3.21 (m, 1H), 2.57–2.40 (m, 2H), 2.31–2.26 (m, 2H). 13C NMR (500 MHz, CD3OD): 163.35, 161.39, 157.33, 154.25, 144.86, 134.06, 133.70, 133.68, 132.10, 131.20, 131.13, 130.99, 129.83, 127.57, 126.24, 126.21, 126.19, 118.59, 118.44, 117.06, 116.92, 115.16, 114.75, 114.58, 77.27, 76.78, 50.50, 50.21, 44.36, 44.17, 30.67, 30.56. HRMS (ESI) Calcd for C35H30F3N3O2 (M + H)+ 582.2368, found 582.2348.
tert-Butyl (S)-3-((5-(3-Bromo-4-(((S)-1-(tert-butoxycarbonyl)-pyrrolidin-3-yl)oxy)benzamido)-4’-fluoro-[1,1’-biphenyl]-2-yl)oxy)-pyrrolidine-1-carboxylate (35).
To a solution of 3359 (1.0 g, 2.6 mmol) in CH2Cl2 (30 mL) was added 3447 (0.97 g, 2.6 mmol), EDC'HCl (0.75 g, 3.9 mmol), and DMAP (0.32 g, 2.6 mmol). The mixture was then stirred at room temperature for 24 h. The reaction mixture was diluted with CH2Cl2 (20 mL), washed with water (30 mL) and brine (30 mL), and dried over MgSO4. The solid was filtered, and the solvent was removed under reduced pressure. The residue was then purified by column chromatography to yield 35 as a white solid (1.3 g, 70%). 1H NMR (500 MHz, acetone-d6): δ ppm 8.91 (s, 1H), 7.62 (s, 1H), 7.41 (dd, J = 3.0, 9.0 Hz, 1H), 7.20 (s, 2H), 6.97–6.87 (m, 2h), 6.64 (d, J = 9.0 Hz, 1H), 6.57–6.52 (m, 3H), 4.63 (s, 1H), 4.38 (s, 1H), 3.12–2.73 (m, 8H), 2.70–2.54 (m, 1H), 1.72–1.52 (m, 2H), 0.82 (m, 18H). 13C NMR (125 MHz, acetone-d6): δ ppm 168.5, 163.53, 163.12, 161.18, 156.50, 154.08, 150.44, 134.78, 133.64, 132.81, 131.41, 131.18, 129.40, 128.75, 123.18, 120.90, 115.52, 114.91, 114.40, 112.48, 78.69, 78.50, 77.85, 77.05, 51.65, 51.54, 51.33, 51.17, 44.21, 43.95, 31.53, 31.37, 30.69, 30.36, 27.99.
tert-Butyl (S)-3-((5-(4-(((S)-1-(tert-Butoxycarbonyl)pyrrolidin-3-yl)oxy)-3-(6-fluoronaphthalen-2-yl)benzamido)-4′-fluoro-[l,l′-bi-phenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (36a).
To a solution of 35 (0.50 g, 0.67 mmol) in dioxane/water (3:1 (v:v)) was added (6-fluoronaphthalen-2-yl)boronic acid (0.15 g, 0.80 mmol), Pd(dppf)Cl2 (0.025 g, 0.034 mmol), and Na2CO3 (0.14 g, 1.3 mmol). The reaction mixture was heated to 90 °C under argon and stirred for 24 h. The reaction mixture was cooled to room temperature, diluted with ethyl acetate, washed with water and brine, and dried over Na2SO4. The solid was then filtered, and the solution was concentrated under vacuum. The residue was purified by column chromatography to yield 36a as a white solid (0.40 g, 75% yield). 1H NMR (500 MHz, acetone-d6) δ 9.64 (s, 1H), 8.15 (dd, J = 13.7, 2.4 Hz, 1H), 8.07–7.95 (m, 3H), 7.97–7.82 (m, 3H), 7.72 (d, J = 8.5 Hz, 1H), 7.66–7.58 (m, 1H), 7.52 (tt, J = 5.6, 2.6 Hz, 2H), 7.36 (td, J = 8.9, 2.6 Hz, 1H), 7.29–7.20 (m, 1H), 7.12 (td, J = 11.1, 10.0, 7.6 Hz, 3H), 5.19 (d, J = 14.3 Hz, 1H), 4.96 (d, J = 9.5 Hz, 1H), 3.74–3.14 (m, 8H), 2.30–1.92 (m, 4H), 1.75–0.96 (m, 18H). 13C NMR (126 MHz, acetone-d6) δ 164.91, 163.09, 161.93, 161.14, 159.98, 156.89, 154.21, 154.16, 154.5, 150.32, 150.26, 135.16, 135.04, 134.80, 133.88, 133.54, 133.47, 131.42, 131.37, 131.25, 131.15, 131.07, 130.92, 130.75, 130.62, 129.11, 129.5, 128.44 (d, J = 1.2 Hz), 128.34, 127.00, 126.86, 123.15, 122.07, 120.95, 116.49, 116.29, 115.51, 115.31, 114.90, 114.78, 114.73, 113.73, 113.59, 110.79, 110.76, 110.63, 78.74, 78.56, 77.85, 77.31, 77.04, 76.55, 51.66, 51.58, 51.21, 44.34, 44.24, 44.10, 43.99, 31.45, 31.38, 30.61, 30.56, 29.99, 28.03, 28.01
tert-Butyl (S)-3-((5-(3-(Benzo[B]thiophen-6-yl)-4-(((S)-1-(tert-butoxycarbonyl)pyrrolidin-3-yl)oxy)benzamido)-4′-fluoro-[1,1 ′-bi-phenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (36b).
It was prepared through the same procedure as 36a to afford 36b as a white solid (83% yield). 1H NMR (500 MHz, acetone-d6) δ 9.63 (s, 1H), 8.40–7.97 (m, 3H), 7.99–7.78 (m, 3H), 7.67 (t, J = 5.2 Hz, 1H), 7.61–7.50 (m, 3H), 7.45 (d, J = 5.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.15–6.98 (m, 3H), 5.16 (d, J = 6.4 Hz, 1H), 5.03–4.84 (m, 1H), 3.79–3.12 (m, 8H), 2.40–1.94 (m, 4H), 1.58–1.31 (m, 18H). 13C NMR (75 MHz, acetone-d6) δ 165.00, 163.72, 160.48, 156.90, 154.26, 154.06, 150.26, 139.93, 139.03, 134.83, 134.28, 133.91, 131.45, 131.35, 131.18, 130.65, 128.95, 128.33, 127.41, 126.31, 123.96, 123.24, 121.00, 115.49, 115.29, 114.99, 114.71, 113.72, 78.76, 78.58, 77.82, 77.41, 77.01, 76.45, 51.60, 51.20, 44.36, 44.25, 44.05, 31.48, 31.37, 30.61, 28.04.
(S)-tert-Butyl-3-(4-(((benzyloxy)carbonyl)amino)-2-bromophenoxy)pyrrolidine-1-carboxylate (37).
To a solution of 33 (0.39 g, 1.01 mmol) in dry toluene (20 mL) under anhydrous conditions was added diphenylphosphoryl azide (DPPA) (0.28 g, 1.01 mmol), Et3N (0.20 g, 2.02 mmol), benzyl alcohol (1.00 mL), and the activated molecular sieves (2 g). The mixture was stirred at room temperature for 10 min and then heated to 80 °C under nitrogen for 24 h. Upon completion, the molecule sieves were filtered, and the solution was diluted with EtOAc (50 mL). The organic layer was washed with water (50 mL) and brine (50 mL) and dried over MgSO4. The solid was filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography to yield 37 as a white solid (0.30 g, 60% yield). 1H NMR (500 MHz, CDCl3): δ ppm 7.63 (s, 1H), 7.41–7.26 (m, 5H), 6.81 (d, J = 8.5 Hz, 2H), 5.18 (s, 2H), 4.82 (s, 1H), 3.64–3.50 (m, 4H), 2.24–2.15 (br.m, 1H), 2.08–1.99 (m, 1H), 1.46 (s, 9H). 13C NMR (500 MHz, CDCl3): δ ppm 154.89, 154.76, 153.73, 150.12, 136.21, 133.18, 128.81, 128.50, 124.58, 119.25, 116.68, 114.37, 79.19, 78.31, 67.28, 51.69, 44.04, 31.92, 28.74.
(S)-tert-Butyl-3-(4-amino-2-bromophenoxy)pyrrolidine-1-carbox-ylate (38).
Compound 37 (1.69 g, 3.44 mmol) was dissolved in MeOH, and an aqueous solution of NaOH (3.6 g, 89.44 mmol) in 15 mL of water was added to the stirring mixture. The reaction was heated to 55 °C for 38 h. Upon completion, the reaction mixture was neutralized with HCl, and the organic solvent was removed. To the remaining aqueous layer was added saturated NaHCO3. The organic layer was washed with EtOAc three times. After the removal of the organic solvent, the dark brown solid was obtained (0.96 g, 78%). 1H NMR (500 MHz, CDCl3): δ ppm 6.89 (s, 1H), 6.74 (d, J = 14.0 Hz, 1H), 6.59–6.52 (m, 1H), 4.73 (s, 1H), 3.69–3.44 (m, 4H), 2.25–2.13 (m, 1H), 2.08–1.89 (m, 1H), 1.45 (s, 9H). 13C NMR (500 MHz, CDCl3): δ ppm 154.90, 146.45, 142.83, 128.63, 127.13, 120.5, 119.38, 115.21, 80.03, 79.16, 51.67, 44.08, 31.05, 28.75.
(S)-tert-Butyl-3-((5-((3-bromo-4-(((S)-1-(tert-butoxycarbonyl)-pyrrolidin-3-yl)oxy)phenyl)carbamoyl)-3′,4′-difluoro-[1,1′-biphen-yl]-2-yl)oxy)pyrrolidine-1-carboxylate (40).
Compound 3947 (0.17 g, 0.41 mmol) was dissolved in anhydrous CH2Cl2 at 0 °C. EDC'HCl (0.16 g, 0.82 mmol) and HOAt (0.062 g, 0.41 mmol) were added. The reaction mixture was allowed to stir at 0 °C for 30 min before 38 (0.15 g, 0.41 mmol) and Et3N (0.20 mL, 1.3 mmol) were added. The reaction was stirred overnight while warming up to room temperature. The reaction was quenched after 24 h with 1 M HCl. The aqueous layer was washed twice with EtOAc, and the solvent of the combined organic layers was removed under reduced pressure. Flash column chromatography was used to purify 40 as a white solid (0.18 g, 60% yield). 1H NMR (500 MHz, CDCl3): δ ppm 8.98–8.80 (m, 1H), 7.87–7.78 (m, 3H), 7.71–7.47 (m, 1H), 7.19 (t, J = 9.0 Hz, 1H), 7.12–7.04 (m, 2H), 7.00–6.72 (m, 2H), 4.91–4.80 (m, 2H), 3.693.40 (m, 7H), 3.37–3.23 (m, 1H), 2.23–1.99 (m, 4H), 1.42 (s, 18H). 13C NMR (500 MHz, CDCl3): δ ppm 165.19, 156.63, 156.38, 154.81, 150.77 150.49, 148.81, 134.26, 133.49, 130.26, 129.63, 128.82, 127.73, 125.98, 125.61, 120.90, 118.55, 116.94, 116.07, 113.87, 113.22, 80.00, 79.69, 51.58, 51.01, 44.29, 43.80, 31.66, 30.94, 28.60, 28.51.
(S)-tert-Butyl-3-((5-(6-(((S)-1-(tert-butoxycarbonyl)pyrrolidin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-3-ylcarboxamido)-3′-cyano-[1,1′-biphenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (41a).
It was prepared through the same procedure as 36 to afford 41a as a white solid (30% yield). 1H NMR (500 MHz, acetone-d6): δ ppm 9.64 (s, 1H), 8.5–8.03 (m, 2H), 7.87–7.79 (m, 4H), 7.71 (d, J = 7.5 Hz, 1h), 7.59 (t, J = 7.5 Hz, 1H), 7.51–7.44 (m, 1H), 7.35–7.31 (m, 2H), 7.26 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 9.0 Hz, 1H), 5.22–5.17 (m, 1H), 5.5–5.01 (m, 1H), 3.61–3.27 (m, 8H), 2.19–2.13 (m, 4H), 1.42 (s, 9H), 1.40 (s, 9H).
(S)-tert-Butyl-3-((5-(6-(((S)-1-(tert-butoxycarbonyl)pyrrolidin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-3-ylcarboxamido)-4′-cyano-[1,1′-biphenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (41b).
It was prepared through the same procedure as 36 to afford 41b as a white solid (50% yield). 1H NMR (500 MHz, acetone-d6): δ ppm 9.60 (s, 1H), 8.07–8.04 (m, 2H), 7.89–7.87 (m, 2H), 7.80 (d, J = 9.0 Hz, 2H), 7.72 (d, J = 8.5 Hz, 1H), 7.55–7.51 (m, 1H), 7.30 (d, J = 8.5 Hz, 1H), 7.19 (d, J = 8.0 Hz, 1H), 5.25–5.21 (m, 1H), 5.08–5.04 (m, 1H), 3.63–3.20 (m, 8H), 2.20–2.08 (m, 4H), 1.43 (s, 9H), 1.40 (s, 9H).
(S)-tert-Butyl-3-((5-(6-(((S)-1-(tert-butoxycarbonyl)pyrrolidin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-3-ylcarboxamido)-3′-(2H-tet-razol-5-yl)-[1,1′-biphenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (42a).
To a stirred solution of 41a (0.05 g, 0.064 mmol) in anhydrous toluene (5 mL) was added n-Bu3SnN3 (0.064 g, 0.19 mmol). The resulting solution was refluxed for 24 h. Upon completion, the reaction mixture was diluted with EtOAc (50 mL), washed with brine (20 mL × 3), and dried over Na2SO4. The solid was filtered, and the solvent was removed under the reduced pressure. The residue was purified by column chromatography (DCM/MeOH = 15:1–10:1) to yield 42a as a pale yellow solid (0.03 g, 57% yield). 1H NMR (300 MHz, acetone-d6): δ ppm 9.75 (s, 1H), 8.31–8.06 (m, 3H), 7.98 (s, 1H), 7.94–7.90 (m, 1H), 7.86 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.53–7.337.28 (d, J = 7.8 Hz, 1H), 7.15 (d, J = 9.0 Hz, 1H), 5.24–5.19 (m, 1H), 5.05–4.98 (m, 1H), 3.66–3.24 (m, 8H), 2.30–2.10 (m, 4H), 1.43–1.35 (m, 18H). MS (ESI) m/z = 846.4 [M + Na]+.
(S)-tert-Butyl-3-((5-(6-(((S)-1-(tert-butoxycarbonyl)pyiroliclin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-3-ylcarboxamido)-4′-(2H-tetrazol-5-yl)-[1,1′-biphenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (42b).
The procedure for 42a was used to prepare 42b as a white solid (50% yield). 1H NMR (500 MHz, acetone-d6): δ ppm 9.67 (s, 1H), 8.14 (d, J = 8.0 Hz, 2H), 8.06–8.04 (m, 2H), 7.88–7.83 (m, 2H), 7.68 ( d, J = 8.0 Hz, 2H), 7.52–7.44 (m, 1H), 7.35–7.30 (m, 2H), 7.26 (s, 1H), 7.14 (d, J = 8.0 Hz, 1H), 5.22–5.18 (m, 1H), 5.04–5.00 (m, 1H), 3.63–3.23 (m, 8H), 2.21–2.08 (m, 4H), 1.45–1.37 (m, 18H). MS (ESI) m/z = 846.4 [M + Na]+.
tert-Butyl (R)-3-((3′-Cyano-5-(methoxycarbonyl)-[1,1′-biphenyl]-2-yl)oxy)pyrroliCine-1-carboxylate (44).
It was prepared through the same procedure as 36 to afford 44 as a white solid (91% yield). 1H NMR (500 MHz, CDCl3) δ 8.05 (dd, J = 8.6, 2.2 Hz, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.77 (t, J = 1.8 Hz, 1H), 7.72–7.67 (m, 1H), 7.62–7.58 (m, 1H), 7.49 (d, J = 7.8 Hz, 1H), 6.97 (d, J = 8.6 Hz, 1H), 5.10–4.91 (m, 1H), 3.90 (s, 3H), 3.64–3.49 (m, 3H), 3.34–3.26 (m, 1H), 2.17–2.06 (m, 2H), 1.44 (s, 9H). 13C NMR (126 MHz, CDCl3) S 166.33, 157.42, 154.41, 138.58, 133.83, 132.91, 132.58, 131.53, 130.87, 129.15, 128.87, 123.52, 118.74, 112.75, 112.31, 79.78, 76.93 (d, J = 31.9 Hz), 52.13, 51.04, 44.07, 31.64, 28.45. MS (ESI) m/z = 445.2 [M + Na]+.
tert-Butyl (R)-3-((5-Amino-3′-cyano-[1,1′-biphenyl]-2-yl)oxy)-pyrrolidine-1-carboxylate (47).
Compound 44 was hydrolyzed under the basic condition to yield 45 (93% yield), which was used in next step without further purification. MS (ESI) m/z = 431.2 [M + Na]+.
The procedure to prepare 47 from 45 was the same as that of 38 from 33 to afford 47 as a dark brown solid (65% yield in two steps). 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H), 6.71 (t, J = 8.6 Hz, 2H), 4.62 (s, 1H), 4.05 (s, 2H), 3.63–3.30 (m, 3H), 3.19 (d, J = 8.6 Hz, 1H), 2.05–1.83 (m, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, CDCl3) S 154.46, 146.88, 140.48, 139.59, 133.83, 132.84, 131.04, 130.40, 128.66, 118.87, 117.78, 117.34, 116.44, 79.35, 78.46, 50.99 (d, J = 49.7 Hz), 43.84 (d, J = 44.0 Hz), 30.88, 28.43. MS (ESI) m/z = 402.2 [M + Na]+.
tert-Butyl (R)-3-((5-(6-(((S)-1-(tert-Butoxycarbonyl)pyrrolidin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-3-carboxamido)-3′-cyano-[1,1′-biphenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (48).
It was prepared through the same procedure as 40 to afford 48 as a white solid (56% yield). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J =21.3 Hz, 2H), 7.69 (d, J = 7.6 Hz, 3H), 7.56 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 7.7 Hz, 1H), 7.25 (s, 1H), 7.14 (s, 2H), 6.91 (s, 2H), 4.94 (s, 1H), 4.81 (s, 1H), 3.90–3.04 (m, 8H), 2.11 (dd, J = 25.8, 17.1 Hz, 4H), 1.42 (d, J = 1.8 Hz, 18H). MS (ESI) m/z = 803.3 [M + Na]+.
tert-Butyl (R)-3-((5-(6-(((S)-1-(tert-Butoxycarbonyl) pyrrolidin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-3-carboxamido)-3′-(2H-tetra-zol-5-yl)-[1,1′-biphenyl]-2-yl)oxy)pyrroliCine-1-carboxylate (49).
It was prepared through the same procedure as 42a to afford 49 as a white solid (35%). 1H NMR (400 MHz, acetone-d6) δ 8.47–8.26 (m, 1H), 8.17–8.05 (s, 2H), 7.85–7.05 (m, 10H), 5.25 (d, J = 33.5 Hz, 1H), 4.32 (dd, J = 5.6, 2.3 Hz, 1H), 3.77–3.25 (m, 8H), 2.45–2.03 (m, 4H), 1.46 (d, J = 11.4 Hz, 18H). MS (ESI) m/z = 846.4 [M + Na]+, MS (ESI) m/z = 822.4 [M − H]−.
tert-Butyl (S)-3-(2-(Benzo[B]thiophen-6-yl)-4-(methoxycarbonyl)-phenoxy)pyrrolidine-1-carboxylate (50).
The procedure to prepare 50 was the same as that for 36 to afford 50 as a white solid (89% yield). 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 4.9 Hz, 1H), 8.01 (t, J = 7.9 Hz, 1H), 7.97 (dt, J = 1.5, 0.8 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.52–7.41 (m, 2H), 7.35 (dd, J = 5.4, 0.9 Hz, 1h), 6.97 (d, J = 8.7 Hz, 1H), 4.97 (tt, J = 4.4, 2.2 Hz, 1H), 3.90 (s, 3H), 3.82–3.25 (m, 4H), 2.22–1.95 (m, 2H), 1.43 (d, J = 11.9 Hz, 9H). 13C NMR (126 MHz, CDCl3) δ 166.70, 157.70, 154.52, 154.35, 139.80, 139.69, 138.63, 133.59, 133.48, 133.04, 131.58, 130.49, 126.89, 126.78, 125.97, 123.62, 123.39, 123.14, 123.03, 122.90, 113.05, 112.93, 79.59, 76.28, 74.98, 52.01, 51.48, 51.05, 44.19, 43.83, 31.65, 30.88, 28.48.
tert-Butyl (R)-3-((5-(3-(Benzo[B]thiophen-6-yl)-4-(((S)-1-(tert-butoxycarbonyl)pyrroliCin-3-yl)oxy)benzamiCo)-3′-cyano-[1,1′-bi-phenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (52).
The procedure for the hydrolysis of 50 to yield 51 the same as that for the reparation of 38 (95% yield). Compound 51 was used directly in the next step without further purification.
The procedure to prepare 52 was the same as that to prepare 40 and afforded 52 as a white solid (60% yield). 1H NMR (500 MHz, CDCl3) δ 8.74 (dd, J = 102.4, 33.0 Hz, 1H), 8.00–7.79 (m, 3H), 7.79–7.58 (m, 5H), 7.50 (d, J = 7.7 Hz, 1H), 7.46–7.36 (m, 3H), 7.31 (d, J = 5.4 Hz, 1H), 7.14–6.67 (m, 2H), 4.85 (s, 1H), 4.77 (s, 1H), 3.61–2.98 (m, 8H), 2.11–1.83 (m, 4H), 1.41 (s, 18H). 13C NMR (126 MHz, CDCl3) δ 165.51, 156.87, 156.50, 154.74, 154.59, 154.37, 139.68, 139.13, 138.60, 133.94, 133.55, 132.84, 132.71, 131.59, 130.54, 130.45, 129.71, 128.67, 128.35, 128.08, 127.90, 127.68, 126.86, 125.93, 123.61. 123.07, 122.93, 121.82, 118.87, 114.85, 113.47, 113.29, 112.11, 111.96, 79.78, 79.60, 77.68, 76.18, 53.85, 53.46, 51.42, 50.94, 50.79, 44.27, 44.19, 43.79, 43.71, 31.59, 30.79, 28.46. MS (ESI) m/z = 823.3 [M + Na]>+.
tert-Butyl (R)-3-((5-(3-(Benzo[B]thiophen-6-yl)-4-(((S)-1-(tert-butoxycarbonyl)pyrroliCin-3-yl)oxy)benzamiCo)-3′-(2H-tetrazol-5-yl)-[1,1′-biphenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (53).
It was prepared by the same procedure as that for 42 to afford 53 as white solid (37% yield). 1H NMR (500 MHz, CDCl3) δ 9.00 (t, J = 47.7 Hz, 1H), 8.23–7.53 (m, 6H), 7.46–7.17 (m, 6H), 6.73 (dt, J = 82.1, 29.1 Hz, 2H), 4.95–4.43 (m, 2H), 3.72–3.04 (m, 8H), 2.03–1.71 (m, 4H), 1.52–1.22 (m, 18H). 13C NMR (126 MHz, CDCl3) δ 156.87, 155.49, 155.03, 154.49, 150.59, 150.26, 139.60, 138.79, 138.58, 133.46, 132.30, 131.62, 130.88, 128.89, 128.42, 128.13, 127.56, 126.85, 126.36, 125.90, 124.13, 123.59, 123.08, 122.85, 122.22, 114.45, 113.32, 80.17, 79.80, 76.09, 53.82, 53.43, 51.54, 51.32, 51.08, 50.83, 44.36, 44.00, 43.86, 31.74, 31.56, 30.74, 29.70, 28.49, 28.40. MS (ESI) m/z = 866.3 [M + Na]+.
Methyl 4-Bromo-3-hydroxybenzoate (54).
To a solution of 4-bromo-3-hydroxybenzoic acid (5.00 g, 13.80 mmol) in MeOH (30 mL) was added SOCl2 (2.50 g, 20.70 mmol) dropwise over 10 min at 0–5 °C. The mixture was stirred for 3 h at room temperature. The solvent was then removed under the reduced pressure, and the residue was taken into EtOAc (150 mL). The solution was washed with aqueous Na2CO3 (50 mL and brine (50 mL × 3), and dried over Na2SO4. The inorganic solid was removed by filtration, and the organic solvent was removed under the reduced pressure to yield 54 as a white solid (1.61 g, 50%). 1H NMR (300 MHz, DMSO-d6): δ ppm 10.74 (s, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 2.1 Hz, 1H), 7.28 (dd, J = 2.1, 8.1 Hz, 1H), 3.81 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ ppm 166.33, 154.99, 134.02, 130.70, 121.61, 116.98, 115.77, 52.99.
(S)-tert-Butyl 3-(2-Bromo-5-(methoxycarbonyl)phenoxy)-pyrrolidine-1-carboxylate (55).
The synthesis of 55 follows the same procedure as that previously described47 to afford 55 as a white solid (94%). 1H NMR (300 MHz, CDCl3): δ ppm 7.59 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 9.0 Hz, 2H), 4.99 (s, 1H), 3.89 (s, 3H), 3.63–3.58 (br.s, 4H), 2.24–2.10 (br.m, 2H), 1.44 (s, 9H). 13C NMR (75 MHz, CDCl3): δ ppm 166.43, 154.73, 153.94, 133.92, 130.64, 123.68, 119.53, 115.40, 79.78, 78.57, 52.63, 51.59, 44.34, 31.93, 31.15, 28.72.
(S)-tert-Butyl 3-((3′,4′-difluoro-4-(methoxycarbonyl)-[1,1′-bi-phenyl]-2-yl)oxy)pyrrolidine-1-carboxylate (56).
To a solution of 55 (1 g, 2.50 mmol) in dry DMF (25 mL) under anhydrous condition was added (3,4-difluorophenyl) boronic acid (0.47 g, 3.00 mmol), Pd(PPh3)4 (0.14 g, 0.13 mmol), and K2CO3 (0.52 g, 3.75 mmol). The mixture was heated to 105 °C under argon and stirred for 20 h. Then, the reaction mixture was cooled to room temperature, diluted with ethyl acetate, washed with water and brine, dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography to yield 56 as a yellow solid (0.96 g, 89% yield). 1H NMR (300 MHz, CDCl3): δ ppm 7.72 (dd, J = 1.8 Hz, 7.8 Hz, 1H), 7.59 (s, 1H), 7.36 (d, J = 8.1 Hz), 7.34–7.28 (m, 1H), 7.22–7.12 (m, 2H), 5.00–4.97 (m, 1H), 3.94 (s, 3H), 3.62–3.43 (br.m, 3H), 3.39–3.23 (m, 1H), 2.14–2.04 (m, 2H), 1.44 (s, 9H).
(S)-2-((1-(tert-Butoxycarbonyl)pyrrolidin-3-yl)oxy)-3′,4′-difluoro-[1,1′-biphenyl]-4-carboxylic Acid (57).
To the solution of 56 (0.33 g, 0.75 mmol) in a solvent mixture (14 mL, THF/MeOH/H2O = 4:2:1) was added LiOH (0.14 g, 6.00 mmol). The mixture was stirred for 8 h at room temperature. Then, the pH value was adJusted to 4–5 with HCl (1 M), diluted with water (50 mL), and extracted with EtOAc (50 mL× 3). The combined organic phase was dried over Na2SO4 and concentrated under vacuum to afford 57 as white solid (0.31 g, 99% yield). It was used directly in the next step without further purification. 1H NMR (300 MHz, CDCl3): δ ppm 7.78 (d, J = 7.8 Hz, 1H), 7.64 (s, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.35–7.30 (m, 1H), 7.22–7.17 (m, 2H), 5.01 (s, 1H), 3.76–3.43 (br.m, 3H), 3.41–3.23 (m, 1H), 2.21– 2.01 (m, 2H), 1.46 (s, 9H). 13C NMR (75 MHz, CDĈ): δ ppm 154.92, 153.87, 151.78, 135.31, 134.27, 131.22, 130.23, 125.77, 123.78, 118.83, 118.59, 117.26, 117.03, 115.14, 80.16, 51.40, 44.27, 28.67.
(S)-tert-Butyl 3-((4-Amino-3′,4′-difluoro-[1,1′-biphenyl]-2-yl)oxy)-pyrrolidine-1-carboxylate (59).
The procedure to prepare 58 was the same as that used to prepare 37. To a solution of 58 (0.75 g, 1.42 mmol) in MeOH (20 mL) was added 10% Pd on activated carbon (0.07 g, 10% by weight). The air was evacuated and exchanged with the H2 gas three times. The reaction mixture was allowed to stir under H2 for 2 h and then filtered through Celite. The solvent was removed under the reduced pressure to yield 59 (0.29 g, 52% yield). 1H NMR (500 MHz, CDCl3): δ ppm 7.08 (d, J = 8.0 Hz, 1H), 6.37 (d, J = 8.0 Hz, 1H), 6.25 (s, 1H), 4.79–4.76 (m, 1H), 3.78 (brs, 2H), 3.64–3.25 (m, 4H), 2.22–1.97 (m, 2H), 1.45 (s, 9H).
(S)-tert-Butyl 3-((4′-Fluoro-5-formyl-[1,1′-biphenyl]-2-yl)oxy)-pyrrolidine-1-carboxylate (62).
To a solution of the previously reported 6047 (1.00 g, 2.41 mmol) in CH2Cl2 (30 mL) was added DIBAL-H (3.61 mL, 1.0 M in THF) dropwise at −78 °C. The reaction mixture was allowed to rise to room temperature and stirred overnight. Then, the reaction mixture was quenched with NH4Cl (15 mL), extracted with CH2Cl2 (50 mL × 3), and dried over Na2SO4. After the removal of the inorganic solid and the solvent, the residue was purified by column chromatography (hexanes/acetone = 5:1–3:1) to yield 61 as a yellow oil (62% yield over two steps) (MS (ESI) m/z = 410.2 [M + Na]+).
To the solution of 61 (0.60 g, 1.55 mmol) in CH2Cl2 (20 mL) was added PCC (1.00 g, 4.65 mmol). The resulting mixture was stirred at room temperature overnight, diluted with DCM (80 mL), washed with brine (20 mL × 3), and dried over Na2SO4. After the removal of the inorganic solid and the solvent, the residue was purified by column chromatography (hexane/acetone = 6:1–5:1) to yield 62 (0.58 g, 99% yield) as a pale yellow oil. 1H NMR (300 MHz, CDCl3): δ ppm 9.96 (s, 1H), 7.89–7.85 (m, 2H), 7.45–7.39 (m, 2H), 7.12–7.02 (m, 3H), 5.04–4.99 (m, 1H), 3.70–3.23 (m, 4H), 2.15–2.09 (m, 2H), 1.45 (s, 9H). 13C NMR (75 MHz, CDCl3): δ ppm 190.97, 164.14, 160.87, 159.3 154.65, 133.08, 132.77, 131.64, 131.37, 131.26, 131.15, 130.50, 115.42, 115.13, 113.35, 79.92, 51.52, 44.17, 28.68. MS (ESI) m/z = 408.2 [M + Na]+.
(3S)-tert-Butyl-3-((4′-fluoro-5-(oxiran-2-yl)-[1,1′-biphenyl]-2-yl)-oxy)pyrrolidine-1-carboxylate (63).
To a solution of NaH (60% dispensed in mineral oil) (0.094 g, 2.34 mmol) in DMSO (15 mL) was added (CH3)3SOI. The resulting mixture was stirred at room temperature for 5 min. Then, the solution of 62 (0.30 g, 0.78 mmol) in DMSO (5 mL) was added slowly. The reaction mixture was stirred for another 1 h, and poured into ice water (50 mL), extracted with EtOAc (30 mL × 3), and dried over Na2SO4. The removal of the inorganic solid and the solvent yields 63 as a colorless oil (0.32 g, >99% yield). 1H NMR (500 MHz, CDCl3): δ ppm 7.43–7.40 (m, 2H), 7.22–7.18 (m, 2H), 7.07–7.04 (m, 2H), 6.92 (d, J = 8.0 Hz, 1H), 4.80–4.84 (m, 1H), 3.87–3.84 (m, 1H), 3.60–3.19 (m, 4H), 3.16–3.13 (m, 1H), 2.85–2.80 (m, 1H), 2.07–1.99 (m, 2H), 1.44 (s, 9H).
(S)-tert-Butyl-3-((2-(6-(((S)-1-(tert-butoxycarbonyl)pyrrolidin-3-yl)oxy)-4′-fluoro-[1,1′-biphenyl]-3-yl)-7-(3,4-difluorophenyl)-quinolin-6-yl)oxy)pyrrolidine-1-carboxylate (65).
A25 mL round-bottom flask was charged with a solution of 64 (0.050 g, 0.13 mmol) and 62 (0.049 g, 0.13 mmol) in MeCN (5 mL). Then, LiBr (0.011 g, 0.13 mmol), PdCl2 (0.0020 g, 0.013 mmol), and acrylic acid (0.019 g, 0.13 mmol) were added under the magnetic stirring. The resulting mixture was heated to 60 °C for 8 h, diluted with EtOAc (60 mL), washed with brine (20 mL × 3), and dried over Na2SO4. After the removal of the inorganic solid and the solvent, the residue was purified by column chromatography (hexanes/EtOAc = 2:1–1:1) to yield 65 as a yellow solid (0.015 g, 15% yield). 1H NMR (500 MHz, CDCl3): δ ppm 8.12– 8.08 (m, 4H), 7.85 (d, J = 8.5 Hz, 1H), 7.52 (dd, J = 5.5, 8.0 Hz, 2H), 7.47–7.40 (m, 1H), 7.32–7.30 (m, 1H), 7.24–7.19 (m, 1H), 7.`12– 7.06 (m, 4H), 5.06–5.03 (m, 1H), 4.96–4.93 (m, 1H), 3.783.23 (m, 8H), 2.23–2.04 (m, 4H), 1.46 (s, 9H), 1.44 (s, 9H). MS (ESI) m/z = 782.4 [M + H]+, MS (ESI) m/z = 804.4 [M + Na]+.
(S)-tert-Butyl-3-((3-(6-(((S)-1-(tert-butoxycarbonyl)pyrrolidin-3-yl)oxy)-4′-fluoro-[ 1,1′-biphenyl]-3-yl)-6-(3,4-difluorophenyl)-quinolin-7-yl)oxy)pyrrolidine-1-carboxylate (66).
A 25 mL round-bottom flask equipped with a magnetic stirrer bar was charged with 59 (0.020 g, 0.050 mmol), 63 (0.039 g, 0.10 mmol), FeCl3 (0.0024 g, 0.015 mmol), and dioxane (10 mL). The resulting mixture was heated to reflux overnight. After 12 h, the reactions mixture was diluted with EtOAc (60 mL), washed with brine (20 mL × 3), and dried over Na2SO4. After the removal of the inorganic solid and the solvent, the residue was purified by column chromatography (DCM/MeOH = 80:1–70:1) to yield 66 as a yellow solid (0.018 g, 46%). 1H NMR (500 MHz, acetone-d6): δ ppm 9.23 (s, 1H), 8.54 (s, 1H), 7.99 (s, 1H), 7.83–7.81 (m, 2H), 7.66–7.64 (m, 2H), 7.59–7.57 (m, 2H), 7.46–7.33 (m, 3H), 7.19 (t, J = 8.5 Hz, 2H), 5.37–5.34 (m, 1H), 5.19–5.14 (m, 1H), 3.73–3.78 (m, 8H), 2.38–2.10 (m, 4H), 1.441.40 (m, 18H). MS (ESI) m/z = 782.4 [m + H]+.
Ligand Docking Using AutoDock 4.2.
The same Autodock procedure described in a previous study59 was used to dock 3 and 29.
Protein Expression and Purification.
Full-length β-catenin (residues 1–781) were cloned into a pET-28b vector carrying a C-terminal 6 × histidine (Novagen) and transformed into E. coli BL21 DE3 (Novagen). Cells were cultured in LB medium with 30 μg/mL kanamycin until the OD600 was approximately 0.8, and then protein expression was induced with 400 μM IPTG at 20 °C overnight. Cells were lysed by sonication. The proteins were purified by three steps of chromatography, including Ni-NTA affinity chromatography (30210, Qiagen), HiTrap Q HP anion exchange chromatography (17–1154-01, GE Healthcare Life Science), and size-exclusion chromatography with a HiLoad 26/600 Superdex 200 pg column (28–9893-36, GE Healthcare Life Science) using an AKTA Pure FPLC system (GE Healthcare Life Science). Protein was eluted in a buffer containing 20 mM Tris (pH 8.5), 100 mM NaCl, and 2 mM DTT. The purity of β-catenin was greater than 95% as determined by SDS-PAGE gel analysis. Thermal-shift assay was performed on an CFX96 Real Time System (Bio-Rad) to monitor protein stability and detect protein aggregation. Protein unfolding was evaluated through measuring the fluorescence changes of fluorescent dye Sypro Orange when interacting with wild-type or mutant β-catenin proteins. A temperature increment of 1°/min was applied. All proteins were stable, and no aggregation was observed under storage or assay conditions. Proteins were aliquoted and stored at −80 °C.
BCL9 Peptide Synthesis and Purification.
Human BCL9 (residues 350–375), N-terminally biotinylated human BCL9 (residues 350–375), human E-cadherin (residues 824–877), and N-terminally biotinylated human E-cadherin (residues 824–877) were synthesized by InnoPep, Inc. (San Diego, CA, www.innopep.com). All synthesized peptides were purified by HPLC with purity >95%. The structures were validated by LC/MS. The sequences are as follows (Ahx, 6-aminohexanoic acid):
| Peptide | Sequence |
| BCL9 26-mer | H-350GLSQEQLEHRERSLQTLRDIQRMLFP375-NH2 |
| biotinylated BCL9 26-mer | biotin-Ahx-350GLSQEQLEHRERSLQTLRDIQRMLFP375-NH2 |
| E-cadherin 54-mer | H-824APPYDSLLVFDYEGSGSEAASLSSLNSSESDKDQDYDYLNEWGNRFKKLADMYG877-NH2 |
| biotinylated E-cadherin 54-mer | biotin-824APPYDSLLVFDYEGSGSEAASLSSLNSSESDKDQDYDYLNEWGNRFKKLADMYG877-NH2 |
AlphaScreen Competitive Inhibition Assays.
For the competitive inhibition assays of β-catenin/BCL9 PPI, the negative control (equivalent to 0% inhibition) refers to 5.0 nM biotinylated BCL9, 40 nM His6-tagged β-catenin, and 10 μg/mL of donor and acceptor beads in a final volume of 25 μL of assay buffer, but no tested inhibitor present. The positive control (equivalent to 100% inhibition) refers to 5.0 nM biotinylated BCL9 and 10 μg/mL of donor and acceptor beads in a final volume of 25 μL of assay buffer. For the competitive inhibition assays of β-catenin/E-cadherin interactions, the negative control (equivalent to 0% inhibition) refers to 10 nM biotinylated E-cadherin, 40 nM His6-tagged β-catenin, and 10 μg/mL of donor and acceptor beads in a final volume of 25 μL of assay buffer. The positive control (equivalent to 100% inhibition) of β-catenin/E-cadherin interactions refers to 10 nM biotinylated E-cadherin and 10 μg/mL of donor and acceptor beads in a final volume of 25 μL of assay buffer.
For the β-catenin/BCL9 assay, 5 nM biotinylated BCL9 and 40 nM His6-tagged β-catenin were incubated in assay buffer at 4 °C for 30 min. For the β-catenin/E-cadherin assay, 10 nM biotinylated human E-cadherin, and 40 nM His6-tagged human β-catenin were added and incubated in assay buffer at 4 °C for 30 min. Different concentrations of the tested inhibitor were added and incubated in 20 μL of assay buffer at 4 °C for another 1 h. All of the above assay plates were covered and gently mixed on an orbital shaker. The donor and acceptor beads were then added to the plates to a final concentration of 10 μg/mL in 25 μL of assay buffer. The mixture was incubated for 1 h at 4 °C before detection. The IC50 value was determined by nonlinear least-squares analysis of GraphPad Prism 5.0. The K values were derived from the IC50 values using a method reported by Nikolovska-Coleska et al.93 The assays were conducted under the conditions required by Nikolovska-Coleska et al.’s equation for determining the K values. All of the experiments were performed in triplicate. The results were expressed as mean ± standard deviation. The inhibitor selectivity for β-catenin/BCL9 over β-catenin/E-cahderin interactions was defined as the ratio of the respective Ki value of β-catenin/E-cadherin interactions over that of β-catenin/BCL9 interactions.
MTs Cell Viability Assay.
Colorectal cancer cell lines SW480 and HCT116, triple-negative breast cancer cell line MDA-MB-231, and lung cancer cell line A549 were seeded in 96-well plates at 5 × 103 cells/well, maintained overnight at 37 °C, and incubated in the presence of inhibitors at various concentrations. Cell viability was monitored after 72 h using a freshly prepared mixture of one part phenazine methosulfate (PMS, Sigma) solution (0.92 mg/mL) and 19 parts MTs agent (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt, Promega) solution (2 mg/mL). Cells were incubated in 10 μL of this solution at 37 °C for 3 h, and A490 was measured. The effect of each compound is expressed as the concentration required to reduce A490 by 50% (IC50) relative to vehicle-treated cells. Experiments were performed in triplicate.
LDH Assay.
The cytotoxic effects of the inhibitors on the cell membrane integrity were determined by measuring the activity of LDH using CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). SW480 cells were seeded in 96-well plates at 2 × 104 cells/well, maintained overnight at 37 °C, and incubated in the presence of inhibitors at various concentrations. After 4 h of incubation, the cell culture supernatant was incubated with diagnostic reagents in the LDH kit according to the manufacturer’s instructions. The activity of LDH was calculated as the following equation after detection at 490 nm using Synergy 2 plate reader (Biotek): percent cytotoxicity = 100 × (experimental release – background group)/(maximum release × background group). Experiments were performed in triplicate.
Cell Transfection and Luciferase Assay.
FuGENE6 (E269A, Promega) 96-well plate format was used for the transfection of HEK293 and SW480 cells according to the manufacturer’s instruction. HEK293 cells were cotransfected with 45 ng of TOPFlash or FOPFlash reporter gene, 135 ng of pcDNA3.1—β-catenin, and 20 ng of pCMV-RL normalization reporter gene. SW480 cells were cotransfected with 60 ng of the TOPFlash or FOPFlash reporter gene and 40 ng of pCMV-RL normalization reporter. Cells were cultured in DMEM and 10% FBS at 37 °C for 24 h, and different concentrations of inhibitors or DMSO were added. After 24 h, the luciferase reporter activity was measured using the Dual-Glo system (E2940, Promega). Normalized luciferase activity in response to the treatment with inhibitors was compared with that obtained from the cells treated with DMSO. Experiments were performed in triplicate.
qPCR Analysis.
SW480 cells at 1 × 106/mL were treated with inhibitors at different concentrations for 24 h. Total RNAs were extracted with TRIzol (15596026, Life Technologies), and the cDNA was synthesized with the superscript III first-strand kit (18080–051, Invitrogen). qPCR experiments were performed using the iQ SYBR green supermix kit (170–8880, BIO-RAD) on an CFX96 Real Time System (BIO-RAD). The threshold cycle (CT) values were normalized to that of internal reference GAPDH. Experiments were performed in triplicate. The primer pairs are shown below.
| human GAPDH | forward | 5′-GAAGGTGAAGGTCGGAGTC-3′ |
| revrese | 5′-GAAGATGGTGATGGGATTTC-3′ | |
| human HPRT | forward | 5′-GCTATAAATTCTTTGCTGACCTGCTG-3′ |
| reverse | 5′-AATTACTTTTATGTCCCCTGTTGACTGG-3′ | |
| human Axin2 | forward | 5′-AGTGTGAGGTCCACGGAAAC-3′ |
| reverse | 5′-CTTCACACTGCGATGCATTT-3′ | |
| human LEF1 | forward | 5′-GACGAGATGATCCCCTTCAA-3′ |
| reverse | 5′-AGGGCTCCTGAGAGGTTTGT-3′ | |
| human cyclin D1 | forward | 5′-ACAAACAGATCATCCGCAAACAC-3′ |
| reverse | 5′-TGTTGGGGCTCCTCAGGTTC-3′ |
Western Blotting.
SW480 cells at 1 × 106 cells/mL were treated with different concentrations of inhibitors for 24 h. Cells were lysed in buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitors. After centrifugation at 12,000 rpm for 20 min at 4 °C, the supernatant was loaded onto an 8% SDS polyacrylamide gel for electrophoretic analysis. Separated proteins were transferred onto nitrocellulose membranes for immunoblot analysis. The antibodies against total β-catenin (610153, BD Biosciences, most of which is phosphorylated β-catenin and represents the E-cadherin bound pool), the active form of β-catenin (ABC, 05–665, EMD Millipore, dephosphorylated at positions S37 and T41 of β-catenin), cyclin D1 (sc-853, Santa Cruz Biotechnology, Inc.), c-myc (D84C12, Cell Signaling), and β-tubulin (sc-55529, Santa Cruz Biotechnology, Inc.) were incubated with the membranes overnight at 4 °C, respectively. IRDye 680LT goat antimouse IgG (827–11080, LiCOR) or IRDye 800CW goat antirabbit IgG (827–08365, LiCOR) was used as the secondary antibody. The images were detected by the Odyssey Infrared Imaging System (LiCOR). Experiments were performed in duplicate.
Co-IP Assays.
Two sets of co-IP experiments were conducted: one, the inhibitor was added onto the cell lysates, and second, the inhibitor was added onto the live cells. For the cell lysate co-IP experiments, HCT116 cells were lysed first in buffer A containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 2 mM EDTA, and protease inhibitors. Different concentrations of the inhibitor were incubated with the HCT116 cell lysates at 4 °C for 4 h. For the whole cell co-IP experiments, HCT116 cells at 1 × 106/mL were treated with different concentrations of the inhibitor for 24 h. Cells were then lysed in buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 2 mM EDTA, and protease inhibitors. For both cell lysate and whole cell co-IP experiments, the lysates were then preadsorbed to A/G plus agarose (sc-2003, Santa Cruz Biotechnology) at 4 °C for 1 h. Preadsorbed lysates were incubated with a specific primary antibody against β-catenin (610153, BD Biosciences) overnight at 4 °C. A/G plus agarose was then added to the lysate mixture and incubated for 3 h. The beads were washed five times with the lysis buffer at 4 °C. The bound protein was eluted by boiling in the SDS sample buffer and loaded onto 8% SDS polyacrylamide gel for electrophoretic analysis. Separated proteins were transferred onto nitrocellulose membranes for immunoblot analysis. The antibodies against BCL9 (ab37305, Abcam) and E-cadherin (610404, BD Biosciences) were incubated with the membranes, respectively. IRDye 680LT goat antimouse IgG (827–11080, LiCOR) was used as the secondary antibody. The images were detected by the Odyssey Infrared Imaging System (LiCOR). Experiments were performed in duplicate.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Department of Defense CDMRP BCRP breakthrough award W81XWH-14-1-0083. We thank John A. Wisniewski, Daniel Powell, and Jinya Yin for the initial synthesis of some compounds. The H. Lee Moffitt Cancer Center & Research Institute is an NCI-designated Comprehensive Cancer Center, supported under NIH grant P30-CA76292.
ABBREVIATIONS USED
- ABC
active form of β-catenin
- APC
adenomatous polyposis coli
- BCL9
B-cell lymphoma 9
- B9L
BCL9-like
- CBP
CREB-binding protein
- co-IP
coimmunoprecipitation
- DAD
diode array detector
- DPPA
diphenylphosphoryl azide
- Dvl
disheveled
- Fzd
frizzled
- HD2
homology domain 2
- KD
dissociation constant
- LDH
lactate dehydrogenase
- Lef
lymphoid enhancer-binding factor
- LRP5/6
low-density lipoprotein-related proteins 5 and 6
- PMS
phenazine methosulfat
- PPI
protein-protein interaction
- qPCR
quantitative real-time PCR
- sFRP1
secreted Fzd-related protein 1
- Tcf
T-cell factor
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00068.
Determination of the intracellular concentration of 29, description of the hot region 2 for the β-catenin/BCL9 PPI, stick models of AutoDock docking results of 3 and 29 with β-catenin, LDH cytotoxicity assay results of 1–9 and 11–31, quantitative data for Western blot and co-IP experiments, synthesis and structural characterizations of 4–26, HPLC conditions and traces, and NMR spectra of 2–32 (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Nusse R; Clevers H Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [DOI] [PubMed] [Google Scholar]
- (2).Anastas JN; Moon RT WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [DOI] [PubMed] [Google Scholar]
- (3).van de Wetering M; Sancho E; Verweij C; de Lau W; Oving I; Hurlstone A; van der Horn K; Batlle E; Coudreuse D; Haramis A-P; Tjon-Pon-Fong M; Moerer P; van den Born M; Soete G; Pals S; Eilers M; Medema R; Clevers H The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [DOI] [PubMed] [Google Scholar]
- (4).Lu D; Zhao Y; Tawatao R; Cottam HB; Sen M; Leoni LM; Kipps TJ; Corr M; Carson DA Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 3118–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sukhdeo K; Mani M; Zhang Y; Dutta J; Yasui H; Rooney MD; Carrasco DE; Zheng M; He H; Tai Y-T; Mitsiades C; Anderson KC; Carrasco DR Targeting the β-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc. Natl. Acad. Sci U. S. A 2007, 104, 7516–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dow LE; O’Rourke KP; Simon J; Tschaharganeh DF; van Es JH; Clevers H; Lowe SW Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015, 161, 1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brack AS; Conboy MJ; Roy S; Lee M; Kuo CJ; Keller C ; Rando TA Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [DOI] [PubMed] [Google Scholar]
- (8).Lancaster MA; Louie CM; Silhavy JL; Sintasath L; Decambre M; Nigam SK; Willert K; Gleeson JG Impaired Wnt-β-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat. Med 2009, 15, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Akhmetshina A; Palumbo K; Dees C; Bergmann C; Venalis P; Zerr P; Horn A; Kireva T; Beyer C; Zwerina J; Schneider H; Sadowski A; Riener M-O; MacDougald OA; Distler O; Schett G; Distler JHW Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun 2012, 3, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Malanchi I; Peinado H; Kassen D; Hussenet T; Metzger D; Chambon P; Huber M; Hohl D; Cano A; Birchmeier W; Huelsken J Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 2008, 452, 650–653. [DOI] [PubMed] [Google Scholar]
- (11).Barker N; Ridgway RA; van Es JH; van de Wetering M; Begthel H; van den Born M; Danenberg E; Clarke AR; Sansom OJ; Clevers H Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [DOI] [PubMed] [Google Scholar]
- (12).Wang Y; Krivtsov AV; Sinha AU; North TE; Goessling W; Feng Z; Zon LI; Armstrong SA The Wnt/β-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Huebschman ML; Lane NL; Liu H; Sarode VR; Devlin JL; Frenkel EP Molecular heterogeneity in adjacent cells in triple-negative breast cancer. Breast Cancer: Targets Ther. 2015, 7, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tammela T; Sanchez-Rivera FJ; Cetinbas NM; Wu K; Joshi NS; Helenius K; Park Y; Azimi R; Kerper NR; Wesselhoeft RA; Gu X; Schmidt L; Cornwall-Brady M; Yilmaz ÖH; Xue W; Katajisto P; Bhutkar A; Jacks T A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Malanchi I; Santamaria-Martïnez A; Susanto E; Peng H; Lehr H-A; Delaloye J-F; Huelsken J Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [DOI] [PubMed] [Google Scholar]
- (16).Creighton CJ; Li X; Landis M; Dixon JM; Neumeister VM; Sjolund A; Rimm DL; Wong H; Rodriguez A; Herschkowitz JI; Fan C; Zhang X; He X; Pavlick A; Gutierrez MC; Renshaw L; Larionov AA; Faratian D; Hilsenbeck SG; Perou CM; Lewis MT; Rosen JM; Chang JC Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yeung J; Esposito MT; Gandillet A; Zeisig BB; Griessinger E; Bonnet D; So CWE β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [DOI] [PubMed] [Google Scholar]
- (18).Fong CY; Gilan O; Lam EYN; Rubin AF; Ftouni S; Tyler D; Stanley K; Sinha D; Yeh P; Morison J; Giotopoulos G; Lugo D; Jeffrey P; Lee SC-W; Carpenter C; Gregory R; Ramsay RG; Lane SW; Abdel-Wahab O; Kouzarides T; Johnstone RW; Dawson S-J; Huntly BJP; Prinjha RK; Papenfuss AT; Dawson MA BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Milanovic M; Fan DNY; Belenki D; Däbritz JHM; Zhao Z; Yu Y; Dörr JR; Dimitrova L; Lenze D; Monteiro Barbosa IA; Mendoza-Parra MA; Kanashova T; Metzner M; Pardon K; Reimann M; Trumpp A; Dörken B; Zuber J; Gronemeyer H; Hummel M; Dittmar G; Lee S; Schmitt CA Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [DOI] [PubMed] [Google Scholar]
- (20).Spranger S; Bao R; Gajewski TF Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [DOI] [PubMed] [Google Scholar]
- (21).Sweis RF; Spranger S; Bao R; Paner GP; Stadler WM; Steinberg G; Gajewski TF Molecular drivers of the non-T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol. Res. 2016, 4, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ding Y; Shen S; Lino AC; Curotto de Lafaille MA; Lafaille JJ β-Catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat. Med. 2008, 14, 162–169. [DOI] [PubMed] [Google Scholar]
- (23).Keerthivasan S; Aghajani K; Dose M; Molinero L; Khan MW; Venkateswaran V; Weber C; Emmanuel AO; Sun T; Bentrem DJ; Mulcahy M; Keshavarzian A; Ramos EM; Blatner N; Khazaie K; Gounari F β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl. Med. 2014, 6, 225ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen B; Dodge ME; Tang W; Lu J; Ma Z; Fan C-W; Wei S; Hao W; Kilgore J; Williams NS; Roth MG; Amatruda JF; Chen C; Lum L Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Proffitt KD; Madan B; Ke Z; Pendharkar V; Ding L; Lee MA; Hannoush RN; Virshup DM Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [DOI] [PubMed] [Google Scholar]
- (26).Lum L; Clevers H Cell biology. The unusual case of Porcupine. Science 2012, 337, 922–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lu D; Choi MY; Yu J; Castro JE; Kipps TJ; Carson DA Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13253–13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fujii N; You L; Xu Z; Uematsu K; Shan J; He B; Mikami I; Edmondson LR; Neale G; Zheng J; Guy RK; Jablons DM An antagonist of dishevelled protein-protein interaction suppresses β-catenin-dependent tumor cell growth. Cancer Res. 2007, 67, 573–579. [DOI] [PubMed] [Google Scholar]
- (29).Thorne CA; Hanson AJ; Schneider J; Tahinci E; Orton D; Cselenyi CS; Jernigan KK; Meyers KC; Hang BI; Waterson AG; Kim K; Melancon B; Ghidu VP; Sulikowski GA; LaFleur B; Salic A; Lee LA; Miller DM III.; Lee E. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Huang S-M; Mishina YM; Liu S; Cheung A; Stegmeier F; Michaud GA; Charlat O; Wiellette E; Zhang Y; Wiessner S; Hild M; Shi X; Wilson CJ; Mickanin C; Myer V; Fazal A; Tomlinson R; Serluca F; Shao W; Cheng H; Shultz M; Rau C; Schirle M; Schlegl J; Ghidelli S; Fawell S; Lu C; Curtis D; Kirschner MW; Lengauer C; Finan PM; Tallarico JA; Bouwmeester T; Porter JA; Bauer A; Cong F Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [DOI] [PubMed] [Google Scholar]
- (31).Waaler J; Machon O; Tumova L; Dinh H; Korinek V; Wilson SR; Paulsen JE; Pedersen NM; Eide TJ; Machonova O; Gradl D; Voronkov A; von Kries JP; Krauss S A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [DOI] [PubMed] [Google Scholar]
- (32).Kramps T; Peter O; Brunner E; Nellen D; Froesch B; Chatterjee S; Murone M; Züllig S; Basler K Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell 2002, 109, 47–60. [DOI] [PubMed] [Google Scholar]
- (33).van Tienen LM; Mieszczanek J; Fiedler M; Rutherford TJ; Bienz M Constitutive scaffolding of multiple Wnt enhanceosome components by Legless/BCL9. eLife 2017, 6, e20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sampietro J; Dahlberg CL; Cho US; Hinds TR; Kimelman D; Xu W Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 2006, 24, 293–300. [DOI] [PubMed] [Google Scholar]
- (35).de la Roche M; Rutherford TJ; Gupta D; Veprintsev DB; Saxty B; Freund SM; Bienz M An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 2012, 3, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hoffmans R; Basler K Identification and in vivo role of the Armadillo-Legless interaction. Development 2004, 131, 4393–4400. [DOI] [PubMed] [Google Scholar]
- (37).Hoffmans R; Basler K BCL9–2 binds Arm/β-catenin in a Tyr142-independent manner and requires Pygopus for its function in Wg/Wnt signaling. Mech. Dev. 2007, 124, 59–67. [DOI] [PubMed] [Google Scholar]
- (38).Valenta T; Gay M; Steiner S; Draganova K; Zemke M; Hoffmans R; Cinelli P; Aguet M; Sommer L; Basler K Probing transcription-specific outputs of β-catenin in vivo. Genes Dev. 2011, 25, 2631–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Adachi S; Jigami T; Yasui T; Nakano T; Ohwada S; Omori Y; Sugano S; Ohkawara B; Shibuya H; Nakamura T; Akiyama T Role of a BCL9-related β-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 2004, 64, 8496–8501. [DOI] [PubMed] [Google Scholar]
- (40).Brembeck FH; Schwarz-Romond T; Bakkers J; Wilhelm S; Hammerschmidt M; Birchmeier W Essential role of BCL9–2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004, 18, 2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).de la Roche M; Worm J; Bienz M The function of BCL9 in Wntβ-catenin signaling and colorectal cancer cells. BMC Cancer 2008, 8, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Brembeck FH; Wiese M; Zatula N; Grigoryan T; Dai Y; Fritzmann J; Birchmeier W BCL9–2 promotes early stages of intestinal tumor progression. Gastroenterology 2011, 141, 1359–1370. [DOI] [PubMed] [Google Scholar]
- (43).Mani M; Carrasco DE; Zhang Y; Takada K; Gatt ME; Dutta-Simmons J; Ikeda H; Diaz-Griffero F; Pena-Cruz V; Bertagnolli M; Myeroff LL; Markowitz SD; Anderson KC; Carrasco DR BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 2009, 69, 7577–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kawamoto SA; Coleska A; Ran X; Yi H; Yang C-Y; Wang S Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem. 2012, 55, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Takada K; Zhu D; Bird GH; Sukhdeo K; Zhao J-J; Mani M; Lemieux M; Carrasco DE; Ryan J; Horst D; Fulciniti M; Munshi NC; Xu W; Kung AL; Shivdasani RA; Walensky LD; Carrasco DR Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 2012, 4, 148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).de la Roche M; Ibrahim AE; Mieszczanek J; Bienz M LEF1 and B9L shield β-catenin from inactivation by Axin, desensitizing colorectal cancer cells to tankyrase inhibitors. Cancer Res. 2014, 74, 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hoggard LR; Zhang Y; Zhang M; Panic V; Wisniewski JA; Ji H Rational design of selective small-molecule inhibitors for β-catenin/B-cell lymphoma 9 protein-protein interactions. J. Am. Chem. Soc 2015, 137, 12249–12260. [DOI] [PubMed] [Google Scholar]
- (48).Teuscher KB; Zhang M; Ji H A versatile method to determine the cellular bioavailability of small-molecule inhibitors. J. Med. Chem 2017, 60, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kawamoto SA; Thompson AD; Coleska A; Nikolovska-Coleska Z; Yi H; Wang S Analysis of the interaction of BCL9 with β-catenin and development of fluorescence polarization and surface plasmon resonance binding assays for this interaction. Biochemistry 2009, 48, 9534–9541. [DOI] [PubMed] [Google Scholar]
- (50).Zhang M; Wisniewski JA; Ji H AlphaScreen selectivity assay for β-catenin/B-cell lymphoma 9 inhibitors. Anal. Biochem 2015, 469, 43–53. [DOI] [PubMed] [Google Scholar]
- (51).Pennington LD; Moustakas DT The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. J. Med. Chem 2017, 60, 3552–3579. [DOI] [PubMed] [Google Scholar]
- (52).Huber AH; Weis WI The structure of the β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 2001, 105, 391–402. [DOI] [PubMed] [Google Scholar]
- (53).Barltrop JA; Owen TC; Cory AH; Cory JG 5-(3-Carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl) tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans as cell-viability indicators. Bioorg. Med. Chem. Lett 1991, 1, 611–614. [Google Scholar]
- (54).Cory AH; Owen TC; Barltrop JA; Cory JG Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [DOI] [PubMed] [Google Scholar]
- (55).Nachlas MM; Margulies SI; Goldberg JD; Seligman AM The determination of lactic dehydrogenase with a tetrazolium salt. Anal. Biochem. 1960, 1, 317–326. [DOI] [PubMed] [Google Scholar]
- (56).Decker T; Lohmann-Matthes M-L A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [DOI] [PubMed] [Google Scholar]
- (57).Niles AL; Moravec RA; Riss TL In vitro viability and cytotoxicity testing and same-well multi-parametric combinations for high throughput screening. Curr. Chem. Genomics 2009, 3, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Yan D; Wiesmann M; Rohan M; Chan V; Jefferson AB; Guo L; Sakamoto D; Caothien RH; Fuller JH; Reinhard C; Garcia PD; Randazzo FM; Escobedo J; Fantl WJ; Williams LT Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/β-catenin signaling is activated in human colon tumors. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 14973–14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wisniewski JA; Yin J; Teuscher KB; Zhang M; Ji H Structure-based design of 1,4-dibenzoylpiperazines as β-catenin/B-cell lymphoma 9 protein–protein interaction inhibitors. ACS Med. Chem. Lett. 2016, 7, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ji X; Huang H; Li Y; Chen H; Jiang H Palladium-catalyzed sequential formation of C–C bonds: efficient assembly of 2-substituted and 2,3-disubstituted quinolones. Angew. Chem., Int. Ed 2012, 51, 7292–7296. [DOI] [PubMed] [Google Scholar]
- (61).Zhang Y; Wang M; Li P; Wang L Iron-promoted tandem reaction of anilines with styrene oxides via C-C cleavage for the synthesis of quinolines. Org. Lett. 2012, 14, 2206–2209. [DOI] [PubMed] [Google Scholar]
- (62).Kahn M Can we safely target the WNT pathway? Nat. Rev. Drug Discovery 2014, 13, 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Emami KH; Nguyen C; Ma H; Kim DH; Jeong KW; Eguchi M; Moon RT; Teo J-L; Kim HY; Moon SH; Ha JR; Kahn M A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kimura K; Ikoma A; Shibakawa M; Shimoda S; Harada K; Saio M; Imamura J; Osawa Y; Kimura M; Nishikawa K; Okusaka T; Morita S; Inoue K; Kanto T; Todaka K; Nakanishi Y; Kohara M; Mizokami M Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/β-catenin inhibitor, in patients with hepatitis C virus-related cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine 2017, 23, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kim Y-M; Ma H; Oehler VG; Gang EJ; Nguyen C; Masiello D; Liu H; Zhao Y; Radich J; Kahn M The γ-catenin/ CBP complex maintains survivin transcription in β-catenin deficient/ depleted cancer cells. Curr. Cancer Drug Targets 2011, 11, 213–225. [DOI] [PubMed] [Google Scholar]
- (66).Townsley FM; Thompson B; Bienz M Pygopus residues required for its binding to Legless are critical for transcription and development. J. Biol. Chem 2004, 279, 5177–5183. [DOI] [PubMed] [Google Scholar]
- (67).Thompson BJ A complex of Armadillo, Legless, and Pygopus coactivates dTCF to activate wingless target genes. Curr. Biol. 2004, 14, 458–466. [DOI] [PubMed] [Google Scholar]
- (68).Townsley FM; Cliffe A; Bienz M Pygopus and Legless target Armadillo/β-catenin to the nucleus to enable its transcriptional co-activator function. Nat. Cell Biol. 2004, 6, 626–633. [DOI] [PubMed] [Google Scholar]
- (69).Hoffmans R; Städeli R; Basler K Pygopus and legless provide essential transcriptional coactivator functions to armadillo/β-catenin. Curr. Biol. 2005, 15, 1207–1211. [DOI] [PubMed] [Google Scholar]
- (70).Sakamoto I; Ohwada S; Toya H; Togo N; Kashiwabara K; Oyama T; Nakajima T; Ito H; Adachi S; Jigami T; Akiyama T Up-regulation of a BCL9-related β-catenin-binding protein, B9L, in different stages of sporadic colorectal adenoma. Cancer Sci. 2007, 98, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Deka J; Wiedemann N; Anderle P; Murphy-Seiler F; Bultinck J; Eyckerman S; Stehle J-C; André S; Vilain N; Zilian O; Robine S; Delorenzi M; Basler K; Aguet M Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 2010, 70, 6619–6628. [DOI] [PubMed] [Google Scholar]
- (72).Moor AE; Anderle P; Cantù C; Rodriguez P; Wiedemann N; Baruthio F; Deka J; André S; Valenta T; Moor MB; Győrffy B; Barras D; Delorenzi M; Basler K; Aguet M BCL9/9L-β-catenin signaling is associated with poor outcome in colorectal cancer. EBioMedicine 2015, 2, 1932–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Leeson PD; Springthorpe B The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- (74).Azzaoui K; Hamon J; Faller B; Whitebread S; Jacoby E; Bender A; Jenkins JL; Urban L Modeling promiscuity based on in vitro safety pharmacology profiling data. ChemMedChem 2007, 2, 874–880. [DOI] [PubMed] [Google Scholar]
- (75).Peters J-U; Schnider P; Mattei P; Kansy M Pharmacological promiscuity: dependence on compound properties and target specificity in a set of recent Roche compounds. ChemMedChem 2009, 4, 680–686. [DOI] [PubMed] [Google Scholar]
- (76).Peters J-U; Hert J; Bissantz C; Hillebrecht A; Gerebtzoff G ; Bendels S; Tillier F; Migeon J; Fischer H; Guba W; Kansy M Can. we discover pharmacological promiscuity early in the drug discovery process? Drug Discovery Today 2012, 17, 325–335. [DOI] [PubMed] [Google Scholar]
- (77).Nishisho I; Nakamura Y; Miyoshi Y; Miki Y; Ando H; Horii A; Koyama K; Utsunomiya J; Baba S; Hedge P Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [DOI] [PubMed] [Google Scholar]
- (78).Ilyas M; Tomlinson IPM; Rowan A; Pignatelli M; Bodmer WF β-Catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 10330–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Bilir B; Kucuk O; Moreno CS Wnt signaling blockage inhibits cell proliferation and migration, and induces apoptosis in triple-negative breast cancer cells. J. Transl Med. 2013, 11, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Xu J; Prosperi JR; Choudhury N; Olopade OI; Goss KH β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One 2015, 10, e0117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Maubant S; Tesson B; Maire V; Ye M; Rigaill G; Gentien D; Cruzalegui F; Tucker GC; Roman-Roman S; Dubois T Transcriptome analysis of Wnt3a-treated triple-negative breast cancer cells. PLoS One 2015, 10, e0122333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Liu C-C; Prior J; Piwnica-Worms D; Bu G LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 5136–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Yang L; Wu X; Wang Y; Zhang K; Wu J; Yuan Y-C; Deng X; Chen L; Kim CCH; Lau S; Somlo G; Yen Y FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [DOI] [PubMed] [Google Scholar]
- (84).Corda G; Sala G; Lattanzio R; Iezzi M; Sallese M; Fragassi G; Lamolinara A; Mirza H; Barcaroli D; Ermler S; Silva E; Yasaei H; Newbold RF; Vagnarelli P; Mottolese M; Natali PG; Perracchio L; Quist J; Grigoriadis A; Marra P; Tutt AN; Piantelli M; Iacobelli S; De Laurenzi V; Sala A Functional and prognostic significance of the genomic amplification of frizzled 6 (FZD6) in breast cancer. J. Pathol. 2017, 241, 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Wend P; Runke S; Wend K; Anchondo B; Yesayan M; Jardon M; Hardie N; Loddenkemper C; Ulasov I; Lesniak MS; Wolsky R; Bentolila LA; Grant SG; Elashoff D; Lehr S; Latimer JJ; Bose S; Sattar H; Krum SA; Miranda-Carboni GA WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol. Med. 2013, 5, 264–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Dey N; Barwick BG; Moreno CS; Ordanic-Kodani M; Chen Z; Oprea-Ilies G; Tang W; Catzavelos C; Kerstann KF; Sledge GW Jr.; Abramovitz M; Bouzyk M; De P; Leyland-Jones BR Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Uematsu K; He B; You L; Xu Z; McCormick F; Jablons DM Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene 2003, 22, 7218–7221. [DOI] [PubMed] [Google Scholar]
- (88).Grossmann TN; Yeh JT-H; Bowman BR; Chu Q; Moellering RE; Verdine GL Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Ritchie TJ; Macdonald SJF The impact of aromatic ring count on compound developability–are too many aromatic rings a liability in drug design? Drug Discovery Today 2009, 14, 1011–1020. [DOI] [PubMed] [Google Scholar]
- (90).Ritchie TJ; Macdonald SJF; Young RJ; Pickett SD The impact of aromatic ring count on compound developability: further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discovery Today 2011, 16, 164–171. [DOI] [PubMed] [Google Scholar]
- (91).Young RJ; Green DVS; Luscombe CN; Hill AP Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16, 822–830. [DOI] [PubMed] [Google Scholar]
- (92).Ritchie TJ; Macdonald SJF Physicochemical descriptors of aromatic character and their use in drug discovery. J. Med. Chem. 2014, 57, 7206–7215. [DOI] [PubMed] [Google Scholar]
- (93).Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004, 332, 261–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.