

Abstract
Hypomethylating agent (HMA) failure myelodysplastic syndrome (MDS) patients have poor outcomes and urgent need for novel therapies. Hedgehog pathway signaling upregulation plays a central role in myeloid neoplasm pathogenesis and leukemia stem cell survival. We evaluated the efficacy and safety of the smoothened inhibitor glasdegib in HMA-failure MDS (n = 35, median age 73 years). According to the International Prognostic Scoring System and the MD Anderson Global Risk Model, 54% and 77% had higher risk disease, respectively. Overall response was 6% (n = 2), and best response was marrow complete remission with hematologic improvement in both patients. Median OS and median follow-up were 10.4 and 42.8 months, respectively. Drug response/stable disease (SD) resulted in better OS than treatment failure (20.6 [95% CI, 10.4-] vs 3.9 months [95% CI, 0.7–9.1]; P < .0001). Response/SD was confirmed to be an independent covariate for improved OS (P < .0001). Grade 3 or higher infections occurred in 11% of patients (n = 4); non-hematologic toxicities were rare. Early mortality (< 30 days) occurred in 11% of patients (n = 4). Glasdegib was well tolerated among HMA-failure MDS patients, although single-agent activity was limited. SD or better resulted in notably superior OS. These results support further investigation of glasdegib, potentially in novel drug combinations, in MDS patients.
Keywords: MDS, Hedgehog, PF-04449913, Glasdegib, Hypomethylating agent failure
1. Introduction
Myelodysplastic syndromes (MDS) encompass a heterogeneous group of clonal neoplastic hematopoietic stem-cell diseases, which are characterized by ineffective hematopoiesis, bone marrow (BM) dysplasia, and risk of transformation to acute myeloid leukemia (AML). Treatment with the hypomethylating agents (HMA) azacitidine and decitabine are the current standard of care for high-risk MDS patients, and HMA failure in high-risk MDS patients results in a median overall survival (OS) of less than 6 months and a 2-year OS probability of 15% [1]. Furthermore, patients with low-risk MDS who experience HMA failure have a median OS of only 17 months [2]. The low OS rates among both low- and high-risk patients demonstrate the clear need for novel therapeutic approaches to the treatment of MDS patients with HMA failure.
The hedgehog (HH) signaling pathway plays a critical role in hematopoietic stem-cell survival, mediating cell self-renewal and resistance to chemotherapy [3]. In myeloid leukemia models, HH activation is essential for the maintenance of cancer stem cells as well as intrinsic chemoresistance [4–6]. To cue the HH pathway, HH ligands, including the sonic, desert, and Indian varieties, bind to the trans-membrane receptor patched, which works to inhibit activity of the smoothened (SMO) protein. The binding of a ligand to patched leads to SMO derepression, resulting in SMO activation and HH signal transduction via zinc finger glioma-associated transcriptional regulators (e.g. GLI1, GLI2, and GLI3) [7–9]. GLI1 and GLI2 are activators that play an important role in AML pathogenesis. GLI1 and GLI2 expression is often a predictor of inferior event-free survival (EFS) and OS. GLI2 expression is associated with FLT3 mutational status in AML while GLI3 acts as a strong repressor of HH signaling [10,11]. BM stromal cells of MDS patients show HH ligand upregulation, as well as decreased expression of HH-interacting protein, a negative regulator of HH signaling [12–14]. Moreover, GLI1 expression directly correlates with increased DNA methyltransferase 1 expression, with targeted knockdown of this pathway inducing cell-cycle arrest and apoptosis in MDS cell lines.(13)
Glasdegib (PF-04449913) is a potent orally bioavailable small-molecule inhibitor of SMO that was investigated in a phase 1 study that involved patients with myeloid malignancies, demonstrating clinical activity in 49% of patients (n = 23). Results included 1 AML patient with complete remission and incomplete hematological recovery and 2 MDS patients with hematologic improvement (HI) [15]. Glasdegib was well tolerated, with the most common treatment-related adverse events being dysgeusia, decreased appetite, and alopecia. Additionally, glasdegib in combination with low dose cytarabine has recently been approved by the FDA for the frontline treatment of AML patients who are ≥ 75 years of age or who are ineligible for intensive chemotherapy due to comorbidities. On the basis of these encouraging results, we designed a phase 2 study to evaluate the efficacy and safety of glasdegib among patients with HMA-refractory MDS or chronic myelomonocytic leukemia (CMML). The recommended dose based on the phase 1 trial was 200 mg or lower once daily.
2. Materials and methods
2.1. Patients
This was a single-center, open-label, 2-stage phase II study of glasdegib among patients who had MDS or CMML, according to World Health Organization (WHO) criteria, or AML with 20%–30% myeloblasts (refractory anemia with excess blasts in transformation), according to French-American-British criteria. Patients must have experienced refractory disease, progression, or relapse following prior HMA therapy. Patients with any risk score, as calculated by the International Prognostic Scoring System (IPSS), were eligible [16]. Inclusion criteria included age ≥ 18 years, an Eastern Cooperative Oncology Group performance status of 0–2, and adequate organ function (eg, serum creatinine ≤ 1.5 times the upper limit of normal [ULN], serum bilirubin < 1.5 times the ULN, and an aspartate aminotransferase/alanine aminotransferase < 2 times the ULN). Patients were excluded if they had a corrected QT interval ≥ 480 ms, a second malignancy requiring active therapy, or concurrent uncontrolled systemic illness.
2.2. Study design
All patients were given a daily oral dose of 100 mg of glasdegib over 4 consecutive weeks (days 1–28) for up to 4 cycles, with an optional continuation phase. There was no dose interruption between cycles. Dose escalation to 200 mg was allowed for patients who did not achieve at least HI following 2 cycles. Dose reduction to 50 mg was permitted for patients who experienced significant toxicity. Responses were assessed after patients completed cycles 2 and 4 (on day 1 of cycles 3 and 5), using International Working Group (IWG) 2006 criteria [17]. Low-(i.e. low or intermediate-1) and high-risk (i.e. intermediate-2 or high) patients were allowed to continue treatment after 4 cycles if they achieved HI or stable disease (SD). SD was defined as failure to achieve response with no evidence of disease progression after > 8 weeks. After completion of study therapy, patients were followed-up monthly for survival. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Moffitt Cancer Center Scientific Review Committee and Institutional Review Board. This trial is registered with ClinicalTrials.gov (NCT01842646).
2.3. Study objectives
The primary objective was to estimate the overall response rate to glasdegib, as defined by IWG 2006 criteria (i.e. complete remission (CR), marrow CR, partial remission, and/or HI), among patients with MDS and CMML after HMA failure [17]. Secondary objectives included ascertaining duration of response, OS, EFS, safety, and biomarker evaluation of response based on expression of HH targets. Duration of response was measured from the time of initial response until the date of disease progression. OS was defined as the period from the therapy start date until date of death. EFS was defined as the period from the therapy start date until the date of progression or death, whichever occurred first.
2.4. Correlative studies
To assess the relationship between response and expression of HH pathway targets, BM mononuclear cells were isolated using the Ficoll-Paque method. Total RNA was isolated using the RNeasy kit (Qiagen, Germantown, MD). Complementary DNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Real-time quantitative polymerase chain reaction analysis for each gene was performed using Taqman Gene Expression Arrays (SMO, Hs01090242_m1, PTCH1, Hs00181117_m1, GLI1, Hs00171790_m1; CCND1, Hs00765553_m1, c-MYC; Hs00153408; Applied Biosystems) and normalized to GAPDH (Hs99999905_m1) using the ABI PRISM 7900 H T Sequence Detection System (Applied Biosystems). Relative gene expression was calculated using the 2-ΔΔCt method. Immunohistochemically (IHC) stained paraffin-embedded BM trephine biopsies and clot sections were evaluated for expression of HH targets before and glasdegib treatment, as previously described, using diaminobenzidine immunoperoxidase staining on paraffin sections (EnVision + System-HRP (DAB + substrate); Dako, Santa Clara, CA). Primary antibodies to sonic/Indian HH (IgG rabbit polyclonal; N-19; Santa Cruz Biotechnology, Dallas, TX) at a ratio of 1:50, desert HH (IgG goat polyclonal; H-85; Santa Cruz Biotechnology) at a ratio of 1:250, GLI1 (N-16; Santa Cruz Biotechnology) at a ratio of 1:100, and SMO at a ratio of 1:50 (H-300; Santa Cruz Biotechnology) were incubated overnight with mouse monoclonal BMI1 antibody (Abcam Bmi1 antibody [1.T.21], Cambridge, MA), followed by a 30-minute incubation time with secondary antibodies (EnVision + System-HRP [DAB + substrate]; Dako). Immunodetection was performed with 3,3′ diaminobencidin (EnVision + System-HRP (DAB + substrate); Dako). Staining intensity was scored by grade category (grade 0 = < 10%; 1 = 10%–20%; 2 = 20%–50%, and 3 = > 50% positive cells).
2.5. Statistics
A Simon’s 2-stage design was used for this phase 2 trial and had an 87% power to evaluate the overall response rate (CR + partial remission + marrow CR + HI), with a null hypothesis of 10% or less versus the alternative hypothesis of an overall response rate of 30% or more. Twenty patients were to be enrolled in the first stage. If ≥ 2 patients achieved a response, the study could proceed to the second stage, at which an additional 15 patients could be enrolled. All time-to-event variables, including OS, EFS, and time to transformation to AML, were analyzed using the Kaplan-Meier method and log-rank test. Toxicity was graded according to Common Terminology Criteria for Adverse Events version 4.0, and data were reported as frequencies and percentages. Exploratory analyses were performed comparing responders to non-responders. Continuous and categorical variables were analyzed using the Wilcoxon Rank Sum and Kruskal-Wallis tests, respectively. All tests were 2 sided and considered statistically significant if P < .05. Multivariate Cox regression models were created to adjust for clinical and treatment characteristics. All analyses were performed using Statistical Analysis System 9.4 or R 3.5.1.
3. Results
3.1. Patient characteristics
From August 29, 2013, to September 2, 2015, 35 patients were accrued to the study. Patient characteristics are summarized in Table 1. The median age of the cohort was 73 years and predominantly male (69%). By WHO category, 74% of patients had MDS (n = 26), 16% had CMML (n = 5), and 13% (n = 4) had AML (20%–30% blasts). Of the MDS patients, 65% (n = 17) had excess blasts. The median BM blast percentage was 5%, with 33% of patients having poor-risk cytogenetics, according to their IPSS. By IPSS, 54% (n = 19) and 43% (n = 15) had high- and low-risk disease, respectively. According to the MD Anderson Global Risk Model, [18] 77% of patients had high-risk disease (n = 27). Ninety-seven percent of the cohort had received prior azacitidine, with 17% of patients having received prior decitabine. The median numbers of prior cycles of azacitidine and decitabine were 9 and 4, respectively.
Table 1.
Baseline patient characteristics of the study cohort.
| Characteristic | Patients (N = 35) |
|---|---|
| Sex, no. (%) | |
| Male | 24 (69) |
| Female | 11 (31) |
| Age, y | |
| Median | 73 |
| Range | 55–88 |
| WHO subtype, no. (%) | |
| RCMD | 9 (26) |
| RAEB-1 | 6 (17) |
| RAEB-2 | 11 (31) |
| CMML | 5 (16) |
| AML | 4 (13) |
| Cytopenias | |
| Grade 2 or 3, no. (%) | 27 (77) |
| ANC, mean (range), x109/L | 0.94 (0.06–7.65) |
| Hemoglobin, mean (range), g/dL | 9.3 (7.3–13.1) |
| Platelets, mean, (range), x109/L | 46 (9–270) |
| IPSS cytogenetic score, no. (%) | |
| Good | 13 (39) |
| Intermediate | 9 (27) |
| Poor | 11 (33) |
| IPSS Category, no. (%) | |
| Low/INT-1 | 15 (43) |
| INT-2/High | 19 (54) |
| Unknown | 1 (3) |
| MD Anderson Risk Category, no. (%) | |
| Low/INT-1 | 7 (20) |
| INT-2/High | 27 (77) |
| Unknown | 1 (3) |
| Baseline bone marrow blast percentage, % | |
| Median | 5 |
| Range | 0.2–23 |
| Prior HMA therapy | |
| Azacitidine, no. (%) | 34 (97) |
| Median no. of cycles/patient | 9 |
| Decitabine, no. (%) | 6 (17) |
| Median no. of cycles/patient | 4 |
Abbreviations: AML, acute myeloid leukemia; ANC, absolute neutrophil count; CMML, chronic myelomonocytic leukemia; HMA, hypomethylating agent; INT, intermediate; IPSS, international prognostic scoring system; RAEB, refractory anemia with excess blasts; RCMD, refractory cytopenia with multilineage dysplasia; WHO, World Health Organization.
3.2. Response to therapy and survival
Thirty-four of the 35 patients were evaluable for response (Table 2). One patient, who received only 1 dose of glasdegib immediately before being admitted to the hospital with cellulitis, declined further therapy, died 1 week later, and was subsequently deemed non-evaluable for response. The overall response rate was 6% (n = 2) with best overall response of marrow CR in 2 patients, both of whom were MDS patients with excess blasts. Additionally, both responding patients had HI and neutrophil response; one had platelet response. Both patients responded at the 100 mg dose level. The median time to response was 81 days (range, 28–134 days). SD was observed in 56% of patients (n = 19). Thirty-eight percent (n = 13) of patients had treatment failure, including 10 patients with progressive disease and 3 patients who died during study treatment secondary to disease/infection, before their responses could be assessed. The median number of cycles received was 3 (range, 0–11 cycles). One responding patient received 7 cycles of therapy and had a durable response that lasted an additional 9 months, with transfusion independence after drug discontinuation. The other responding patient received 4 cycles of therapy and electively discontinued, without evidence of disease progression. The median overall survival (OS) of the cohort was 10.4 months, with a median follow-up of 42.8 months (range, 0.3–47.7 months; Fig. 1A). The median EFS of the entire cohort was 6.4 months. Based on IPSS category, patients with low-risk disease (i.e. low/intermediate-1) had greater OS than patients with high-risk disease (i.e. intermediate-2/high; median OS 15.0 vs 7.7 months; P = .03; Fig. 1B). Patients who achieved drug response/SD had significantly better OS than patients who experienced treatment failure, which was defined as progressive disease or death during treatment (median OS 20.6 vs 3.9 months, respectively; P < .0001; Fig. 1C). In the cohort of patients with high-risk disease according to IPSS (n = 19), OS was also significantly better among patients who responded/had SD than among those who had treatment failure (median OS 19.5 months vs 3.6 months, respectively; P = .004; Fig. 1D). Response/SD was confirmed as an independent covariate for improved OS in Cox regression modeling that included age, gender, WHO, and IPSS risk category (HR 0.13; 95% CI, 0.05–0.34; P < .0001).
Table 2.
Overall Response Rates for Evaluable Patientsa.
| Response | Patients, no. (%) (n = 34) |
|---|---|
| Complete response/CRi | 0 (0) |
| Hematologic improvementb | 2 (6) |
| Stable disease | 19 (56) |
| Failure | 13 (38) |
Abbreviations: CRi, complete response with incomplete blood count recovery.
Median no. of cycles received = 3 (range, 0–11).
Both patients also achieved marrow complete remission.
Fig. 1.
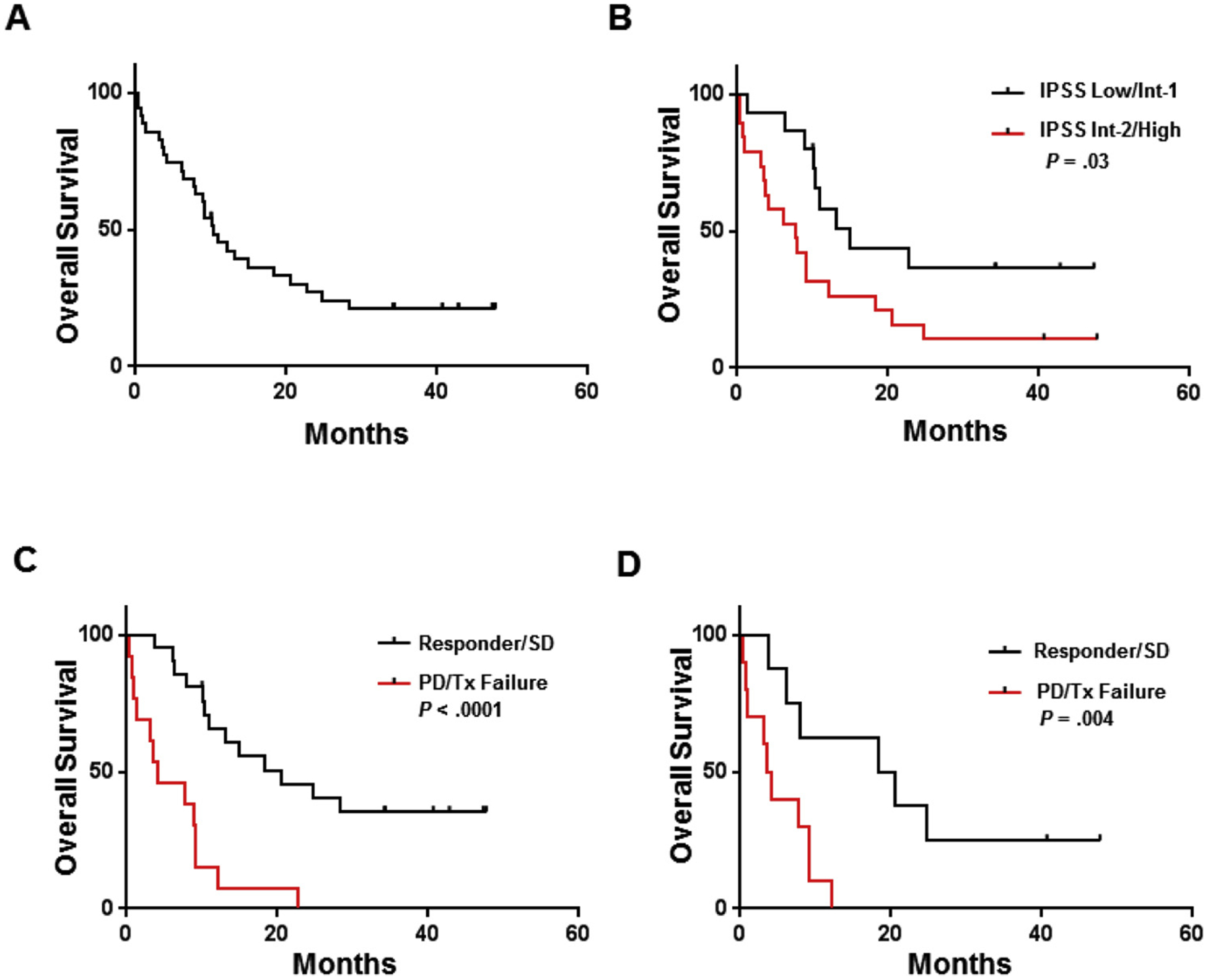
Overall survival of patients treated with glasdegib. Kaplan-Meier curves of OS (A) in the entire cohort, (B) stratified by IPSS classification, and stratified by response (ie, responder/stable disease versus progressive disease/treatment failure) in (C) the overall cohort and (D) the high-risk cohort. Abbreviations: INT, intermediate; IPSS, international prognostic scoring system; PD, progressive disease; SD, stable disease; Tx, treatment.
3.3. Safety
Treatment-emergent grades 1/2 adverse events (AEs) that occurred in ≥ 10% of patients are shown in Table 3. The most common grade 1/2 AEs included pain (n = 22 [63%]), dysgeusia (n = 21 [60%]), nausea (n = 11 [31%]), and anorexia (n = 11 [31%]). Other than infections (n = 4), grade 3 and higher non-hematologic toxicities were rare and included muscle pain (n = 1), rash (n = 1), and dyspnea (n = 1), of which only muscle pain was attributed to the study drug. Fourteen percent of patients (n = 5) had dose reductions to 50 mg because of toxicities after a median of 3 cycles (range, 2–4 cycles). Additionally, 3 patients (9%) had their dose escalated to 200 mg orally daily. Four patients (11%) died within 30 days of treatment initiation.
Table 3.
Treatment-emergent adverse events for all patients treated with glasdegib.
| Toxicity | Grades 1–2, no. (%) | Grades 3–5, no. (%) | Total, no. (%) |
|---|---|---|---|
| Pain | 22 (63) | 1 (3) | 23 (66) |
| Dysgeusia | 21 (60) | 21 (60) | |
| Nausea | 11 (31) | 11 (31) | |
| Anorexia | 11 (31) | 11 (31) | |
| Skin/rash | 8 (23) | 1 (3) | 9 (26) |
| Oral mucositis | 9 (26) | 9 (26) | |
| AST elevated | 8 (23) | 8 (23) | |
| Alopecia | 8 (23) | 8 (23) | |
| ALT elevated | 6 (17) | 6 (17) | |
| Dizziness | 6 (17) | 6 (17) | |
| Dyspnea | 5 (14) | 1 (3) | 6 (17) |
| Infections | 2 (6) | 4 (11) | 6 (17) |
| Fatigue | 6 (17) | 6 (17) | |
| Diarrhea | 6 (17) | 6 (17) | |
| Constipation | 5 (14) | 5 (14) | |
| Fever | 5 (14) | 5 (14) | |
| Hyperkalemia | 5 (14) | 5 (14) | |
| Hyperglycemia | 4 (11) | 4 (11) | |
| Thrombocytopenia | 2 (6) | 2 (6) | 4 (11) |
| Headache | 4 (11) | 4 (11) |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
3.4. HH pathway correlatives
There were no response associated differences in baseline expression levels of HH pathway components or downstream signaling intermediates that could be observed via messenger RNA (mRNA) expression analysis or IHC (Supplementary Table 1). However, in comparison to baseline expression of HH pathway targets, patients with progressive disease after cycle 2 had a trend for increased PTCH expression (P = .13; Fig. 2A), with significantly increased mRNA expression of GLI1 as well as downstream signaling components, including MYC and Cyclin D1 (Fig. 2B–D). However, there was no difference in protein expression on serial assessment by IHC (Supplementary Table 1).
Fig. 2.
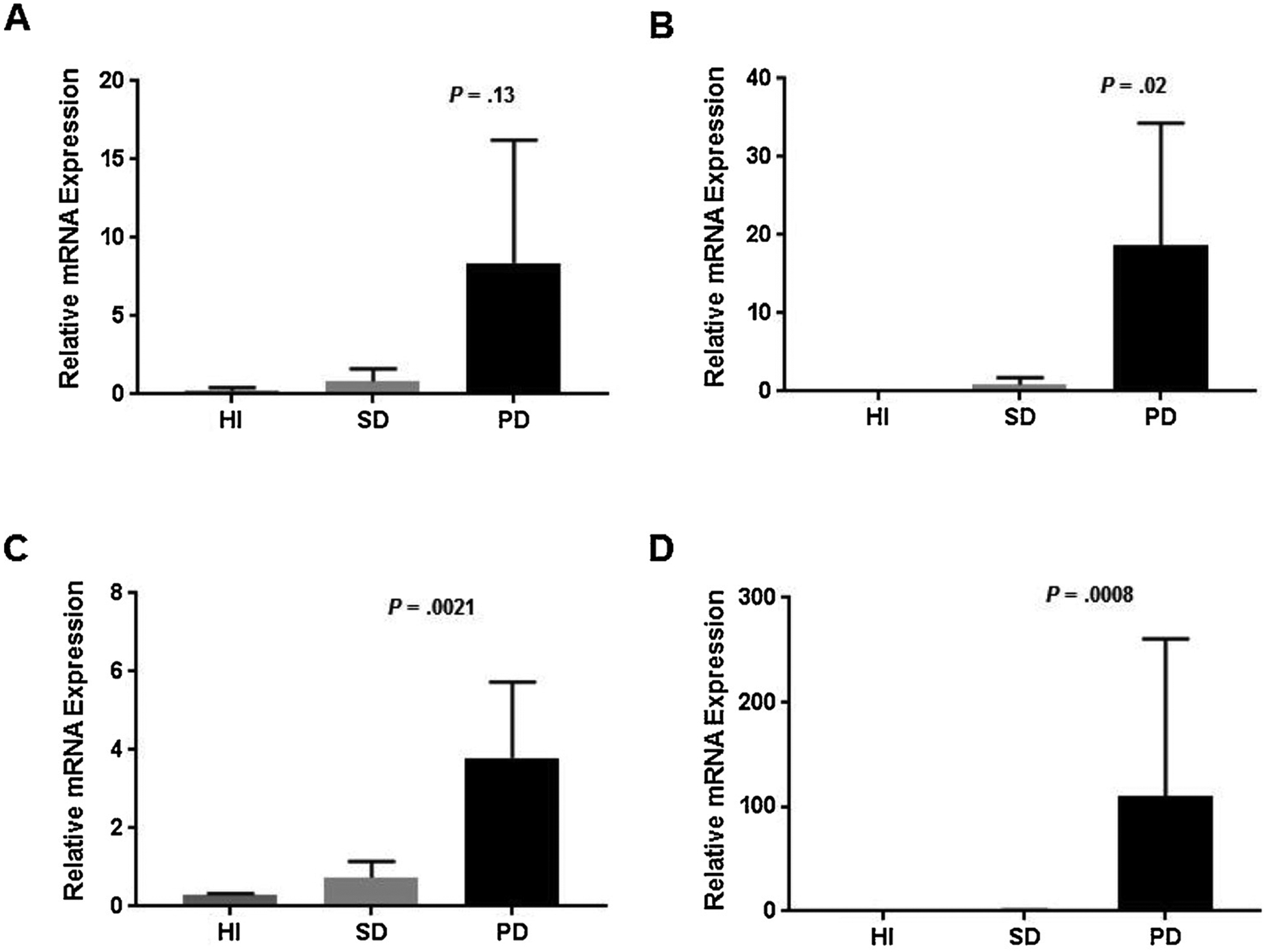
Serial mRNA expression of Hedgehog pathway components. The relative gene expression of (A) PTCH, (B) GLI1, (C) MYC, and (D) CCND1 were shown by serial assessment of patients with hematologic improvement, stable disease, and progressive disease. This is a two-column image. Abbreviations: HI, hematologic improvement; PD, progressive disease; SD, stable disease.
4. Discussion
In a cohort of HMA-refractory MDS, AML, and CMML patients, we demonstrated that glasdegib is safe and well tolerated. The most common treatment-emergent AEs were pain/muscle spasms, dysgeusia, nausea, and anorexia. Treatment-emergent AEs were similar to those observed in previous studies examining this patient population, with the exception of an increased risk of rash, oral mucositis, and transaminitis, which were all grades 1/2, with the exception a grade 3 rash that was experienced by one patient but considered to be unrelated to the study drug [15,19]. Dose reductions to 50 mg daily were uncommon, and the 3 patients whose dose was escalated to 200 mg daily tolerated it well.
There is increasing awareness of the dismal OS, typically less than 6 months, that HMA-refractory, high-risk MDS carries [1]. A recent phase 2 trial (NCT01546038) of glasdegib that compared low-dose cytarabine combination treatment with cytarabine alone showed an increased CR rate (15%) for the combination treatment but, more impressively, a significantly greater OS that was independent of cytogenetic risk [19]. Similarly, the combination of the SMO inhibitor sonidegib with azacitidine has been evaluated in a phase 1/1b study (NCT02129101) that resulted in an SD rate of 76% in relapsed/refractory AML patients, most of whom were HMA refractory [20]. In the current trial of heavily pretreated, primarily high-risk MDS patients, objective response rates were low, with only 6% of patients (n = 2) achieving response by IWG criteria; however, 56% of patients had SD. Notably, patients who had SD or better after treatment had a median OS of 20.6 months, which was significantly longer than patients with treatment failure, independent of other clinical variables. Greater OS was also observed in the high-risk cohort of patients who achieved SD/response. Importantly, achievement of SD or better was an independent covariate for improved OS.
Elucidation of potential response biomarkers and evaluation of synergism with additional agents will be critical to the advancement of HH-pathway inhibitors in myeloid malignancies. In this regard, a recent study identified GLI3 repressor (GLI3R) expression to be paramount to SMO inhibitor activity [11]. Specifically, GLI3R is required for SMO inhibitor response, which is epigenetically silenced in most of the cases in which low response rates have correlated to treatment with glasdegib as a single agent. Supporting this hypothesis, combination with the HMA decitabine restored GLI3 expression leading to SMO sensitivity [11]. However, increased response rates of sonidegib with azacitidine were not observed in the aforementioned clinical trial NCT02129101 (CR rate of 23% in frontline setting) [20]. Further evaluation of GLI3R expression as a biomarker of response in glasdegib-treated MDS patients would be of interest for future study.
Although we did not observe differential gene expression of HH pathway targets at baseline, patients with progressive disease had significant upregulation of downstream HH pathway targets on serial assessment, suggesting a potential resistance mechanism via SMO independent activation of GLI. Specifically, the HH pathway has significant crosstalk with other signaling pathways, including the Ras/Raf/MEK/ERK, PI3K/AKT/mTOR, epidermal growth factor receptor, and transforming growth factor beta pathways [21–23]. Furthermore, combination of SMO inhibitor with FLT3 inhibitors was found to be efficacious in both in vitro and in vivo AML models [24]. As these other pathways are actionable with approved therapies, potential combination with glasdegib should be evaluated in future studies.
In conclusion, glasdegib in HMA-refractory MDS and CMML patients was well tolerated with limited single-agent activity. The evidence of prolonged survival in patients who achieved SD was encouraging. As combination studies of glasdegib with low-dose cytarabine have highlighted, improved OS in treatment-naïve patients with MDS/AML is possible, [19] and future investigations should focus on identifying optimal glasdegib combination treatments for patients with myeloid malignancies.
Supplementary Material
Acknowledgements
We thank Paul Fletcher and Daley Drucker (Moffitt Cancer Center) for editorial support. They were not compensated beyond their regular salaries.
Funding
This work has been supported in part by the Biostatistics Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References
- [1].Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. , Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure, J. Clin. Oncol 29 (24) (2011) 3322–3327, 10.1200/jco.2011.35.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jabbour EJ, Garcia-Manero G, Strati P, Mishra A, Al Ali NH, Padron E, et al. , Outcome of patients with low-risk and intermediate-1-risk myelodysplastic syndrome after hypomethylating agent failure: a report on behalf of the MDS Clinical Research Consortium, Cancer 121 (6) (2015) 876–882, 10.1002/cncr.29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Takebe N, Harris PJ, Warren RQ, Ivy SP, Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways, Nat. Rev. Clin. Oncol 8 (2) (2011) 97–106, 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- [4].Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. , Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia, Nature 458 (7239) (2009) 776–779, 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. , Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation, Cancer Cell 14 (3) (2008) 238–249, 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- [6].Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, Aberson HL, Ferreira CV, Peppelenbosch MP, et al. , Hedgehog signaling maintains chemoresistance in myeloid leukemic cells, Oncogene 29 (48) (2010) 6314–6322, 10.1038/onc.2010.375. [DOI] [PubMed] [Google Scholar]
- [7].Taipale J, Beachy PA, The Hedgehog and Wnt signalling pathways in cancer, Nature 411 (6835) (2001) 349–354, 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- [8].Irvine DA, Copland M, Targeting hedgehog in hematologic malignancy, Blood 119 (10) (2012) 2196–2204, 10.1182/blood-2011-10-383752. [DOI] [PubMed] [Google Scholar]
- [9].Kalderon D, Transducing the hedgehog signal, Cell 103 (3) (2000) 371–374. [DOI] [PubMed] [Google Scholar]
- [10].Wellbrock J, Latuske E, Kohler J, Wagner K, Stamm H, Vettorazzi E, et al. , Expression of hedgehog pathway mediator GLI represents a negative prognostic marker in human acute myeloid leukemia and its inhibition exerts antileukemic effects, Clin. Cancer Res 21 (10) (2015) 2388–2398, 10.1158/1078-0432.CCR-14-1059. [DOI] [PubMed] [Google Scholar]
- [11].Chaudhry P, Singh M, Triche TJ, Guzman M, Merchant AA, GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML, Blood 129 (26) (2017) 3465–3475, 10.1182/blood-2016-05-718585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zou J, Hong Y, Tong Y, Wei J, Qin Y, Shao S, et al. , Sonic hedgehog produced by bone marrow-derived mesenchymal stromal cells supports cell survival in myelodysplastic syndrome, Stem Cells Int. 2015 (2015) 957502, , 10.1155/2015/957502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zou J, Zhou Z, Wan L, Tong Y, Qin Y, Wang C, et al. , Targeting the sonic Hedgehog-Gli1 pathway as a potential new therapeutic strategy for myelodysplastic syndromes, PLoS One 10 (8) (2015) e0136843, , 10.1371/journal.pone.0136843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kobune M, Iyama S, Kikuchi S, Horiguchi H, Sato T, Murase K, et al. , Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms, Blood Cancer J. 2 (2012) e87, 10.1038/bcj.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, et al. , Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study, Lancet Haematol. 2 (8) (2015), 10.1016/S2352-3026(15)00096-4 e339–346. [DOI] [PubMed] [Google Scholar]
- [16].Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. , International scoring system for evaluating prognosis in myelodysplastic syndromes, Blood 89 (6) (1997) 2079–2088. [PubMed] [Google Scholar]
- [17].Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, et al. , Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia, Blood 108 (2) (2006) 419–425, 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- [18].Kantarjian H, O’Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. , Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System, Cancer 113 (6) (2008) 1351–1361, 10.1002/cncr.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cortes JE, Heidel FH, Heuser M, Fiedler W, Smith BD, Robak T, et al. , A phase 2 randomized study of low dose Ara-C with or without glasdegib (PF-04449913) in untreated patients with acute myeloid leukemia or high-risk myelodysplastic syndrome, Blood 128 (22) (2016) 99-. [Google Scholar]
- [20].Tibes R, Kosiorek HE, Dueck A, Palmer J, Slack JL, Knight EA, et al. , Phase I/ IB study of Azacitidine and hedgehog pathway inhibition with sonidegib (LDE225) in myeloid malignancies, Blood 130 (Suppl 1) (2017) 2629-. [Google Scholar]
- [21].Brechbiel J, Miller-Moslin K, Adjei AA, Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer, Cancer Treat. Rev 40 (6) (2014) 750–759, 10.1016/j.ctrv.2014.02.003. [DOI] [PubMed] [Google Scholar]
- [22].Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. , Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways, Proc. Natl. Acad. Sci. U. S. A 104 (14) (2007) 5895–5900, 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Parascandolo A, Laukkanen MO, De Rosa N, Ugolini C, Cantisani MC, Cirafici AM, et al. , A dual mechanism of activation of the Sonic Hedgehog pathway in anaplastic thyroid cancer: crosstalk with RAS-BRAF-MEK pathway and ligand secretion by tumor stroma, Oncotarget 9 (4) (2018) doi: 10.10.18632/onco-target.23388. 4496–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lim Y, Gondek L, Li L, Wang Q, Ma H, Chang E, et al. , Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia, Sci. Transl. Med 7 (291) (2015) 291ra96, , 10.1126/scitranslmed.aaa5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.