

Abstract
Prophages are phages that are integrated into bacterial genomes and which are key to understanding many aspects of bacterial biology. Their extreme diversity means they are challenging to detect using sequence similarity, yet this remains the paradigm and thus many phages remain unidentified. We present a novel, fast and generalizing machine learning method based on feature space to facilitate novel prophage discovery. To validate the approach, we reanalyzed publicly available marine viromes and single-cell genomes using our feature-based approaches and found consistently more phages than were detected using current state-of-the-art tools while being notably faster. This demonstrates that our approach significantly enhances bacteriophage discovery and thus provides a new starting point for exploring new biologies.
INTRODUCTION
Prophages are bacteriophages integrated into bacterial genomes where they play an important role in the ecology, physiology and evolution of their bacterial hosts (1,2). Despite the many and varied examples of ways that prophages impact bacterial biology, our knowledge is based on a somewhat limited number of prophages in well-studied prokaryotes (3–5). Integrated prophages are known to impact the phenotype of their hosts in a number of different ways, that is collectively referred to as lysogenic conversion. The most well-known example of this is an increase in virulence, by the carriage of toxin genes where prophage encoded toxins can contribute directly to human diseases such as cholera, shigellosis, diphtheria and botulism. Increased virulence is not limited to toxins, with other virulence factors encoded on prophages such as vapE, that increases the virulence of Streptococcus pneumoniae (6). Beyond lysogenic conversion prophages also provide a mechanism of horizontal gene transfer (HGT) between different hosts via generalized, specialized and lateral transduction (7).
To comprehensively understand the importance of prophages and further interrogate their multitude of roles, it is first necessary to predict their presence within prokaryotic host genomes. Current bioinformatics prediction tools largely rely on sequence similarities and therefore struggle to identify novel motifs with no close analogs in the public searchable databases. The current most widely used and very useful prophage prediction tool is PHASTER (8), which carries out sensitive comparisons to existing phage genes by combining sequence similarity searches with gene presence and synteny. When it comes to extracting viral sequences from (meta)genomic data, there is a reliance on well-understood phages and the available tools are dependent on linking databases with sequence similarity approaches as is partly the case with VirSorter (9) and VIBRANT (10) or with features, such as transcription orientation, protein length and amino acid composition (Prophage Hunter (11)).
Although some feature-based approaches have been applied to predict prophages and phages from (meta)genomic datasets (11–13), they have either been trained on a limited set of biological features (e.g. only nucleotide frequencies), or an inadequate amount of training data. Nevertheless, when applied alongside careful curation of training data, such approaches yield valuable new information, as demonstrated through the recent characterization of inoviruses (14), and significantly reduce prediction times, improving the speed of experiments and scalability of such models to big data.
Previously, transcription strand directionality and gene length have been identified as crucial biological features differentiating between phages and bacteria (9,11–13). Marvel (13) has utilized additional features such as intergenic distances and gene density (13) and other features have been used as proxies for structural sequence properties such as single amino acid frequencies (Prophage Hunter) and AT/GC skew (PhiSpy). So far, the used amount of sequence data has remained limited: Marvel, PhiSpy and Prophage Hunter are trained using 1029, 547 and 718 prokaryotic genomes, respectively, while VIBRANT uses the information from 181 prokaryotic genomes (10–13). It has been previously noted that no single tool can identify all the prophages in all bacterial genomes suggesting that multiple features remain to be learnt from the data (12).
To overcome the current limitations and to increase our knowledge of phage space we present PhageBoost, a bioinformatics machine learning tool for fast, generalizable and explainable detection and discovery of prophage regions. PhageBoost extracts the viral signal from the host background by shifting from sequence space into biological feature space. As proteins with similar functions can share attributes, or features, despite being far apart in sequence space (15), this makes predictions less prone to sequence similarity limitations.
Our approach calculates and engineers biological features from both nucleotide and amino acid sequences for every gene, and then uses machine learning to predict which gene belongs to bacteria or phages. The resulting prediction probabilities are parsed to longer regions, which are considered viral if their probability distributions differ from the background. PhageBoost utilizes biological features such as GC-content, amino acid composition, gene length, gene direction, intergenic distances and codon adaptation index (CAI) (full list of features in Supplementary Table S1), and extreme gradient boosting (XGBoost) (16) to learn the differences between the host and phage genes in relation to the complete genome signal.
MATERIALS AND METHODS
PhageBoost workflow
For a (meta-)genome prediction, PhageBoost starts working after the gene calling by expecting nucleotides, amino acids and coordinates as inputs for each gene. We have currently implemented PhageBoost to start from a fasta-file and implemented gene prediction using cythonized Prodigal (17) from Pyrodigal v.0.2.1 (https://github.com/althonos/pyrodigal). However, any gene caller results can be used as input. Contrary to the predictions of free phages, our approach relies on the ‘learning’ of the difference in features space between the prophage and the bacterial host sequence where PhageBoost will calculate the biological features for each gene and transform them relative to the background of all the genes in the set of contigs before predicting. This is done by standardizing the features by subtracting the mean and scaling to unit variance before the probabilistic classification of each gene as phage or bacteria. Afterward, the genes above a probability threshold (default 0.9) are parsed to regions using multiple adjustable parameters that allow customizable pattern matching: length of a minimum number of genes (default 10), the neighboring genes required to have the same threshold (default 0) and the allowed gap between genes (default 5). We further smooth the predictions using Parzen rolling windows (18) of 20 periods and look at the smoothed probability distribution across the genome. We disregard regions having either a summed smoothed probability <0.5, or region less than one unit of standard deviation away from the negative predictions smoothed average, and finally, we reject those regions whose the probability distributions differ from the probability distribution generated from the negative predictions by using Kruskal–Wallis rank test (default, alpha: 0.001) as implemented in Scipy (19). The algorithm returns the predicted probabilities, smoothed probabilities and predicted regions for each gene. Ultimately, each phage region can then be filtered out from the input fasta-file using start and stop coordinates.
Feature generation
We calculated 1587 different features (Supplementary Table S1) using the BioPython SeqUtils ProteinAnalysis module (20) and in-house scripts for each gene. These features consist of 400 dipeptide combinations using normal amino acid alphabets and 1000 tripeptide and 100 dipeptide combinations using simplified amino acid alphabets (21), and 87 more generic features. To avoid model bias and make the model more generalizable, we applied simple feature engineering and selection. We selected the 208 features (see Supplementary Table S1) that were always present and high variance throughout the training data genomes. We further transformed the feature values relative to the genome differences. We calculated the average gene length and average intergenic distances for a random subset of 10 982 prokaryote genomes utilized in PhageBoost training and 9435 phage genomes from GenBank (retrieved 1 April 2018).
Training dataset generation
For a machine learning model to be able to predict if a region in a bacterial genome is a prophage, the model needs to be trained on (i) a trusted, preferable experimentally verified, positive dataset of known prophage regions, (ii) a similar trusted negative dataset of strictly prokaryotic regions and (iii) further validated with a similar dataset that was not used in any part of the training. In essence, to make sure the dataset is valid, it should consist of independent and identically distributed random variables. As no gold standard dataset is currently available, we constructed a bespoke dataset in silico which should ideally be as close as possible to reality. For the training dataset generation, we used all completely sequenced bacterial and archaeal genomes from NCBI’s RefSeq (22) up to February 2019 and chose those chromosomes which had 300 genes or more, resulting in 13 994 genomes (Supplementary Table S2). In order to create a classification value for the training data, phage regions and phage-free regions were defined using labels generated from clusters of orthologous groups (COGs) (23), and prokaryotic virus orthologous groups (pVOGs) (24). The genomic regions that were designated as phage regions consisted of regions where only pVOGs exist throughout a stretch of 10 genes. In contrast, the phage-free regions were defined as regions where only COGs exist throughout the ten genes stretch without the presence of any pVOGs. From the 13 994 genomes used for the model training and test data, we extracted 31 973 phage regions and 101 747 phage-free regions totaling 4 007 643 genes. We further validated the training data using MMSeqs2 (25) to align the genes used in the training data against the GenBank phage genes (retrieved 1 April 2018). A total of 859 770 of 1 090 269 genes (78.9%) labeled as phage have a hit to 366 722 non-redundant phage genes whereas 325 439 of 2 564 993 genes (12.7%) labeled as bacteria have a hit to 34 625 non-redundant phage genes (Supplementary Figure S3). The vast majority of bacterial genes which had similarity to phage genes were ‘typical’ bacteria genes, for example the transcriptional regulator LysR or ABC transporters that were annotated as such and which are known to be found in albeit at low frequencies in phage genomes (Supplementary Table S7).
Training sample weights
We used the individual genes found in the regions to train the classifier model. As not all bacterial genomes were equally present in the training data, this can skew the machine learning algorithm to toward prophage regions from the most abundant bacteria. Additionally, this can cause the machine learning model to learn patterns driven by particular taxonomical lineage or gene homology. To give equal importance to less abundant bacteria, and in order to limit model bias caused by redundant genes, we calculated clusters to generate sample weights during training. MMseqs (25) and MCL (26) (inflation parameter set to 2) were used to assign the genes to the gene clusters in a similar fashion as previously described (27). We applied majority voting to eliminate model bias caused by having the same features assign to both phage and prokaryote. Thus, to limit the bias caused by gene clusters without the majority group, we removed the gene cluster when the dominant group proportion was <0.7 and pooled the gene clusters with <10 members for more even sample weight generation. We managed the potential bias caused by taxonomy grouping genes belonging to the same family if more than 5000 genes present for the family. From the 116 512 controlled gene clusters, we generated 23 329 unique labels for calculating sample weights (see ‘Training of the model’ section).
Training of the model
After filtering steps during the sample weight generation of the 4 007 643 genes, we trained the model using 3 672 101 genes as the training data. We used a test dataset of 23 329 genes for early stopping during training. These 23 329 genes were generated by random sampling of single genes from each gene cluster (see ‘Training sample weights’). After taking a sample from each group as test data for early stopping of model training, the sample weights used in the actual training were computed using the balanced mode from Scikit-learn v0.22.1 (28). The datasets are available as supplementary data. The final gradient boosting decision tree model using XGBoost v.1.0.2 (16) was trained on 3 672 101 genes until no further model improvement was observed for ten boosting rounds on the test data using classification error as the evaluation metric. The model hyperparameters were manually fine-tuned to avoid generating false positives while driving the log-likelihood score lower after getting the initial idea of parameters through a ten-fold cross-validated search with Bayesian optimization framework Optuna v. 0.9 (29).
In order to benchmark PhageBoost, we removed 54 genomes from the training data and recalculated the sample weights but used the same hyperparameters (see Benchmarking and comparisons using 54 genomes). This resulting dataset consisted of 3 655 262 genes, 23 278 clusters and 23 278 test genes for stopping the boosting (data available in Supplementary Data).
Model explanations
Explaining the model predictions was done by utilizing the Shapley additive explanations for ensembles of trees from shap v.0.35.0 (30) and the builtin method for current XGBoost versions for easy access to the same feature contributions and interactions for the predictions. To understand how the model learned during the training, we computed and visualized the feature contribution for the training data, as well as the link between the Shapley value and the feature value in Figure 1E and Supplementary Figure S1, including individual figures, and raw Shapley value data in supplementary data. We used the predicted feature contributions and extracted sorted order of the feature importance on models local output by using the standard approach taking the averages on the absolute values of the Shapley values. For Figure 1E, the feature value and Shapley value interaction were simplified using the Pearson correlation coefficient, while the figures in supplementary data have raw data visualized in a scatter plot. See Supplementary Figure S1 for the barplot of the impact of each feature during training. For Figure 1B–D, we smoothed the predictions by using Parzen (18) window rolling averages of twenty periods. We summed the raw contributions for each gene (Figure 1C), and visualized the smoothed ten features with the most contribution in the training dataset (with the highest absolute average) (Figure 1D), and summed total features followed by smoothing (Figure 1B). The workflow on how to create the images in Figure 1 is found in the Jupyter notebooks in GitHub: https://github.com/ku-cbd/PhageBoost/tree/master/notebooks.
Figure 1.

PhageBoost prophage predictions (in orange) along a bacterial chromosome (Haemophilus influenzae, NC_000907.1) (blue) (A) with total feature influence (B). The biological feature content varies among the prophage regions and from the bacterial genome (C). Individual feature contributions can be used to explain the model predictions (D) (30). This allows the extraction and validation of biological signals. Panel E shows the 10 most important features learned during the training phase that the model uses to discriminate between prophage and bacterial regions. Panels C and D show the influence of the same ten features along the predicted region. Colorbar: Pearson correlation coefficient between the feature values and Shapley values.
Benchmarking and comparisons using 54 genomes
We took the approach of using the previously reported 267 prophages from 54 genomes (31) that have been previously used to benchmark prophage prediction tools (8,9,32). We extracted the list of the validated prophages from the PHASTER website (Table 4, https://phaster.ca/statistics). For each genome in the 54 validation set for all the predictors, we used the regions begin and end coordinates (base pairs) to count the sensitivity (recall) and positive predicted values (PPV/precision) as previously suggested for phage dataset validation (8). We have further included the F1-score and false positive rate (FPR). The sensitivity, positive predictive value (PPV), FPR, accuracy and F1-score were defined as follows:
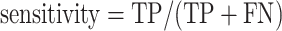 |
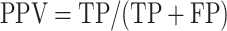 |
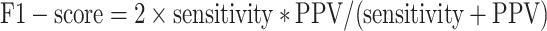 |
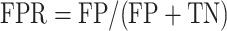 |
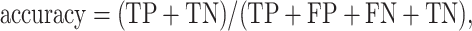 |
where TN = genome length (bp)—TP–FP–FN.
Where true positive (TP) equals the nucleotide found in both validation set and with the corresponding prophage prediction tool, false positive (FP) equals nucleotides found only in the predictions; false negative (FN) nucleotides found only from validation. However, we would like to point out, as already noted in 2016 by the developers of PHASTER, ‘[…] given that the ‘gold standard’ annotations used for evaluation are […] old, many prophages identified as ‘false positives’ relative to this standard are likely to be true prophages.’ (8). Thus, more than the reported prophages as in the genomes would negatively influence the PPV score (8,32). To link the prediction performance for each phage region and take into account the different sizes of the viral regions, we also calculated the proportion of region retained. We generated in-house python scripts to compute all three values.
We benchmarked PhageBoost against VirSorter (9), VIBRANT (10), Prophage Hunter (11) and PHASTER (8). We used the default settings for PhageBoost v.0.1.2, VIBRANT v.1.2.1 and VirSorter v.1.0.5 with the database db2 (9), while we manually submitted 54 genomes to the PHASTER web server using their URLAPI (https://phaster.ca/instructions) and chose not to use precomputed results. The genomes were uploaded to Prophage Hunter using their web user interface and annotated both with and without similarity matching. Results have been deposited to Supplementary Data.
Virome mapping
We mapped the short reads from 223 published marine viral metagenomic samples (33) to the 5539 single amplified genomes (34) Supplementary Table S4. These virome samples come from 65 Tara stations. We used the Anvi’o v.6.1 (35) platform for the work. We merged all the single-amplified genomes (SAGs) to a multi-fasta file, after which we mapped each short-read sample to the concatenated SAGs file using Bowtie2 v.2.3.5 (36) with the -a flag to return all the possible matches and otherwise default parameters to avoid signal dilution. The read recruitment varied from 0 to 5.22 percent (Supplementary Table S5). Afterward, by using the gene calling done by the original authors of the dataset (34), we extracted the coverage and detection information for each gene (Supplementary Data). Using the detection information, we extracted the regions found by the viral mapping with a custom in-house script. We defined a region found with a minimum number of five genes with detection of 1.0. We parsed the regions for each virome sample separately (supplementary data). We additionally extracted the prophage-like regions from the SAGs by selecting the regions that were at least 10 kb long and located 10 kb away from the edges.
We searched for similarity-based evidence by looking for resemblance of these regions to the GenBank phage genes, using MMSeqs2 (25) and retaining hits above 25% identity. We then calculated the total proportion of each region to any phage genomes. The average proportion was only 15% (Supplementary Figure S4). Since the prophages or temperate phages integrated to host genome could have a few lysogenic markers, we searched the following previously (11,37) suggested keywords for integration: ‘recombinase, excise, exciase, excisionase, recombination, transposase, lysogenic, prophage, temperate, integrase, repressor’. We found that only 102 regions (13.7%) had one or more of these gene annotations. However, this is a 62.8% increase from the original 22 659 virome regions where 1899 (8.4%) regions had a keyword hit or more. The overall low amount of keyword hits is not surprising given the low average proportion of regions mapped to phage genomes.
We further searched for evidence of attachment sites around the region by finding the longest string matches at the ends of the region extending it 5 kb outwards and 0.5 kb inwards. We then compared the generated distribution to random samples from the whole SAG dataset and found that the length of the string match is significantly (Z-test) higher in the prophage-like filtered region subset than random samples from the whole dataset. Repeats longer than 12 bp are commonly used to indicate the presence of the potential attachment sites (32,11). We found that 439 regions had a string size longer than or equal to 15 bp (Supplementary Figure S7).
Predicting viral signal from the SAGs
For the dataset of 5537 SAGs (34), we used the default settings for PhageBoost v.0.1.1 predictions. These are minimum region length 10, 5 allowed gaps and probability threshold 0.9. We used the default settings for VirSorter v.1.0.5 (9) predictions with the database db2 using all the phage hidden Markov models (HMMs) and curated HMMs. For the PHASTER (8) predictions, we submitted genomes to the PHASTER web server using their URLAPI (https://phaster.ca/instructions), and thus could not be benchmarked in the same way as other tools in terms of time. We chose and chose not to use pre-computed results together with a multi-fasta option. For VIBRANT v.1.2.1 (10), we used the default settings. To link the predictions to the regions found by viral read recruitment and to take into account the different sizes of the viral regions, we calculated the proportion of region retained.
Ecological significance
We further wanted to investigate the ecological significance of the filtered dataset regions found by linking this to the prediction tool. We took the metadata associated with the isolation and sampling locations for both the viromes and the SAGs and generated the data for each potential prophage region (Supplementary Table S4). Using kepler-gl v.2.2.0 (https://kepler.gl/), we visualized the sites by latitude and longitude metadata of the sampling spots of both viromes and SAGs for each prophage prediction tool (Figure 3). An interactive map is provided in supplementary data. For VIBRANT, the six SAGs came all from a single sampling location; these were mapped to 121 virome sampling sites. PHASTER found seven unique locations for SAGs and 67 virome locations, whereas VirSorter found five locations for SAGs and 124 locations for viromes, and PhageBoost had most considerably more ecological signals with 11 locations for SAGs and 125 locations for viromes (Supplementary Data). These results suggest that some of the prophages might be more globally present than previously understood using the current tools.
Figure 3.

The sampling locations of the SAGs with potential prophage regions found by different prophage predictors and confirmed by virome reads. The coordinates are jittered if multiple prophage predictors overlap. The size of the point is dependent on the number of SAGs found from those coordinates.
We used both GNU parallel v20200322 (38) and joblib v.0.14.1 to speed up the computation throughout the model preparation, parsing, and processing data for this paper. All the data visualization and figures were generated using either kepler-gl, Matplotlib (39) or Seaborn (40) and were manually fine-tuned for publication using Inkscape (http://www.inkscape.org/).
RESULTS AND DISCUSSION
Biological explanation of discriminative features
To evaluate the model behavior, explanations of the individual predictions of the machine learning model's outputs were created based on Shapley additive explanations (41). This approach allows an assessment of the general feature importance generated during model training and the subsequent exploration of which interactions inform predictions. Thus, we can then relate biological significance to the key feature contributions identified in our models.
To distinguish between prophage or bacterial origins, PhageBoost generates prediction probabilities for each gene across the bacterial genome or metagenomic contig (Figure 1). Of the many hundreds of biological features used, it is often smaller subsets drive the prophage prediction (Figure 1B–D). During the training phase, by explaining (30) the predictor local output, we can identify subsets of discriminating features which are shown in the order of relative importance in Figure 1E. A key strength of our approach is that the feature contribution can be related to the actual feature values in order to extract a biological signal that defines the prediction (Figure 1E).
Although regression tree boosting models are more complex than linear univariate models, biological insights can be gained from the ranking of the feature importances. The strongest feature, which PhageBoost uses to discriminate between prophage and bacterial regions, is the CAI, which measures the synonymous codon usage bias for genes with respect to a set of reference genes. Originally, the CAI is based only on highly expressed genes, but here we used the genome data as we did not have expression data (42). Temperate phages generally adapt their codon usage to be concordant with their hosts with time, as they utilize the host translational machinery. There is evidence for this in a small set of coliphages that have been examined, with greater adaptation in temperate phages compared to virulent phages (43). However, adaptation is further complicated by the phage carriage of tRNAs that may reduce the need to evolve codon usage. Why a low CAI is such a strong feature is not immediately obvious, it may be that the identified phages are very recent acquisitions and did not have time to evolve. Other relevant features are the length of genes and the lengths of the regions in between the genes in the observed region—which is not surprising as phage genes tend to be shorter (640 ± 132 bp) than prokaryotic genes (936 ± 67 bp) and phage genomes are more compact, thereby having shorter intergenic distances (prokaryotes: 511 ± 493 bp, phages: 78 ± 14 bp). Other important features consist of the GC content at the third position in the codon, the molecular weight and the percentages of threonine, histidine and cysteine, where more cysteine and histidine residues are signatures of prophages, and higher contents of threonine are attributes of bacterial regions. The Shannon entropy has already been shown to be discriminative between bacterial genomes and phage genomes (44) and has previously been used for detecting prophages in bacterial genomes (12). The grand average index of hydropathy (GRAVY) which is essentially a measure of hydrophobicity of proteins, and it had been shown that phage proteins used in phage display may be more hydrophobic (45). For a full set of feature importances see Supplementary Figure S1 and Table S3.
Benchmarking and validation of novel predictions
We chose to benchmark PhageBoost in two ways (Figure 2)—by comparing its performance to existing state-of-the-art methods and by discovering previously unknown viral signals by mapping marine viral fractions on to single amplified marine microbial genomes.
Figure 2.

Software benchmarking. Validation against a previously reported dataset of prophages from 54 prokaryotes (31)# with a total found regions, positive predictive value (PPV), and sensitivity measured for single genomes as well as phage proportion (in bp) found per prophage. (A) Virome (33) mapping validated phage signals marine SAGs (34) and a filtered subset with a prophage-like pattern. (B)# Prophage Hunter failed to run NC_003155 both with and without sequence similarity (SM).
Experimentally verified prophages from 54 prokaryotic genomes
We used the genomes of 54 prokaryotes that have at least the previously reported 267 prophages (31) as the validation set and retrained the PhageBoost model after omitting these genomes from the training data. We benchmarked this model against four prediction tools VIBRANT (10), VirSorter (9), Prophage Hunter (11) and PHASTER (8) using three predefined metrics: number of regions found, sensitivity and positive predictive value (PPV) (8,32) (Figure 2A), and other common machine learning metrics such as F1-Score (Supplementary Figure S8), false positive rate (Supplementary Figure S9) and accuracy (Supplementary Figure S10, averaged results in Supplementary Table S10). VIBRANT, PHASTER and Prophage Hunter were more conservative when making predictions and had the highest PPV values of 0.83, 0.69 and 0.57, respectively, but they also found the least amount of the validated phages. PhageBoost with a PPV of 0.44 was higher than VirSorter with a PPV of 0.31. Given that the validation genomes could have more than the reported prophages (8), negative conditions (true negatives and false positives) are impossible to accurately verify. Thus, identifying these prophages would negatively influence the PPV. PhageBoost identified the most validated prophage regions with 231, whereas PHASTER identified 224, VirSorter 221, VIBRANT 192 and Prophage Hunter 173. Only eight prophage regions were uniquely found by PhageBoost, however the shared amounts of two tools or more showcase the accumulation of prophage regions to the PhageBoost side (Supplementary Figure S2). Only 13 prophages were found by two tools but not by PhageBoost. VirSorter and PHASTER, which found the second most regions these were 18 and 22 prophages respectively. We believe PhageBoost results in the future could be improved by generating a more comprehensive training dataset. While PhageBoost reached a higher sensitivity (0.82) than Prophage Hunter (0.66), PHASTER (0.78) and VIBRANT (0.73), VirSorter (0.85) had the highest sensitivity finding larger proportions of the validated regions (Figure 2A). This is evident by manually investigating the prediction regions for each tool (Supplementary Table S6). We note that while PhageBoost predicts the most prophages, the region borders could be further finetuned by looking at the potential attachment sites and in general the regions should be verified by other means such as virome coverage data or looking at the functional annotations if any.
We further calculated the nucleotide-level accuracy (Supplementary Figure S9) and false positive rate (FPR) (Supplementary Figure S10). We found that VIBRANT was the best performing (0.987) with the least false predicted base pairs (0.007), while overall, the accuracy was over 90% regardless of the predictor (See Supplementary Table S10 for averaged set of metrics). PhageBoost had the lowest minimum accuracy score for each genome (Supplementary Figure S9). Regarding false positive predictions, PhageBoost barely fitted under 5% margin (0.049), while VirSorter had the highest (0.079) (Supplementary Figure S10 and Table S10). The compactness of score distributions of accuracy and FPR for PHASTER (0.016), Prophage Hunter (0.023), and VIBRANT was surprisingly dense. This might suggest the utilization of the 54 prokaryotic genomes and their prophages during their final model development. VIBRANT includes at least seven of these genomes in the training data (10). For PHASTER and Prophage Hunter, the used genomes are not evident through literature search (8,11,32). While this is making predictor comparison biased with these genomes as benchmarking using these genomes, we see that utilizing all the information available for the deployed predictor is justified. However, external evaluation sets are thus needed.
Superimposition of TARA Ocean viral samples on single-cell genomic marine microbes
As there are no universal conserved marker genes for phages, we utilized recently published marine datasets (33,34) to demonstrate that PhageBoost can discover previously unseen prophage signals. We reason that prophage regions in marine bacteria will have similarities to marine phages and by superimposing marine phage sequences on marine bacterial genomes, we will be able to enrich for prophage regions without sequence similarities to existing databases.
We mapped the sequence reads from 223 metagenomic marine viral fractions samples (viromes) from 65 different TARA oceans stations (33) to 5537 marine SAGs (34). This gave a total of 22 659 unique regions with a signal out of 236 405 contigs (Supplementary Tables S4 and 5) that were consistently detected by at least 1× coverage. We then filtered regions that were at least 10 kb long and located 10 kb away from the edges, resulting in 746 regions which we hypothesize could be prophage regions within the marine microbial genomes.
Thereafter, we predicted viral signals from all 5537 SAGs using PhageBoost and three other leading prediction tools—PHASTER, VirSorter and VIBRANT (Table 1). The run times varied between the different programs where PhageBoost was clearly the fastest. PhageBoost finished the prediction in ∼33.5 h and was 9× faster than VIBRANT, and ∼19× faster than VirSorter with PHASTER taking more than two weeks to get results back from the server (Table 1). The number of predictions also varied widely, with PhageBoost predicting the highest numbers of virome mapped regions overall and for the potential prophage regions, with VirSorter second, and VIBRANT and PHASTER predicting notably fewer hits mostly finding some of the regions that were found in multiple virome samples (Figure 2B), which could indicate that these regions have already been deposited to virome databases. VirSorter was often found assigning the whole contig as phage, whereas PhageBoost classified smaller regions.
Table 1.
Prediction software comparisons for the marine SAGs
| Viral regions | Contigs | SAGsc | Time per SAG (mins) | Total time (h) | |
|---|---|---|---|---|---|
| PhageBoost | 5757 | 5081 | 2539 | 0.36 ± 0.2 | 33.37 |
| PHASTER | 2402 | 2303 | 1905 | -a | 401 |
| VirSorter | 3889 | 3872 | 2552 | 6.8 ± 4.9 | 627.21 |
| VIBRANT | 943 | 942 | 715 | 3.3 ± 1.5 | 301.8b |
| Total | 234593 | 5537 |
aJobs to web server submissions took 2 h 10 February 2020 20:02–22:00, results received 13 February 2020–27 February 2020 13:20; b the elapsed time for 5515 genomes, the remaining 242 failed to finish; c Single-amplified genomes.
We further used MMSeqs2 to search for in silico validation through similarity-based evidence from the phage genes. We analyzed all 22 659 unique regions found by the marine virome read recruitment of the SAGs that were annotated to SAR11-clade or Prochlorococcus by the original authors of dataset (34). The regions found from both lineages give hits to the phages. The most frequent phage references for both are Synechococcus phages (Supplementary Tables S8 and 9).
For the Prochlorococcus regions, PhageBoost and VirSorter find a comparable number of regions, whereas PHASTER and VIBRANT find relatively few. Evidently multiple regions do not share resemblance to the phage genes (Supplementary Figure S5), and those which do are mostly annotated as hypothetical protein (31%) (Supplementary Table S8). For the SAR11 regions, PhageBoost finds most hits while VirSorter is the clear second, whereas PHASTER and VIBRANT find few. Multiple regions share less resemblance to the phage genes than the Prochlorococcus regions (Supplementary Figure S6), and 30% of the genes are annotated as hypothetical protein in the reference (Supplementary Table S9).
By investigating the sampling locations of the SAGs where a potential prophage region was found, we further observed a substantial increase in predictions making it ecologically significant. The predicted prophage fragments are spread around to multiple locations in the ocean thereby increasing the ecological phage space. (Figure 3 and online methods). The regions that were most similar to the known phages in the GenBank databases were also the regions where most tools often agreed (Supplementary Figure S4). As the homology-based approaches cannot go beyond the current known sequence space, our results show that by utilizing the biological feature space and machine learning, PhageBoost is able to generalize and detect previously unknown viral signals for novel hosts such as Prochlorococcus and SAR11. Furthermore, as the viromes’ ecological signal doesn’t get saturated with positive predictions, this suggests that a repository of new prophages awaits to be discovered.
Strengths and limitations of PhageBoost
PhageBoost is a new tool for predicting prophages, where the aim is not to replace other available tools but to add a high-speed and accurate prophage detection, which makes it suitable for high-throughput sequencing projects. As PhageBoost is feature-based, it does not rely on sequence similarities or hallmark genes for the prediction, making it able to generalize and identify novel prophages that would otherwise have been missed by sequence similarity-based techniques. However, PhageBoost needs the bacterial background to identify the prophages, it will not detect prophages if they are present independently without a background. Also, as it is a gene-based workflow, it will not determine the exact prophage boundaries. PhageBoost is, in general, returning a prediction, which is a combination of what other currently available tools are predicting, including additional novel prophage genomes. Some of these could be overpredictions originating from the training data (Supplementary Figures S11–13) but without clear biological bias (Supplementary Figure S13 and Table S11). In the case of prophages, the overpredictions are very hard to assess as not even in the commonly used 54 genome validation dataset, it is guaranteed that all prophages have been identified. Therefore, it is challenging to verify positive predictions, both computationally and experimentally (31). For the validation, we excluded the 54 genomes from the PhageBoost training dataset, but as we used the public versions of the other prophage predictors to compare against, we cannot be sure that they have not been trained with the 54 validation genomes included in their training dataset.
PhageBoost seeks to explain what features have been important for the decision if a given region is a prophage or not. Some high-ranking features, like the CAI have earlier been demonstrated as indicators of HGT and genomic islands (46–48) and there is the possibility that part of the overpredictions could be due to recent HGT. As prophage integration is one of the main mechanisms of HGT (7,46), by investigating the local feature importance interactions of predictions, we show that the combination of features together with CAI is tuned for selecting prophages rather than only just HGTs or genomic islands (Figure 1D).
In conclusion, PhageBoost is a fast prophage predictor, which is independent of sequence similarity, able to generalize and therefore is able to predict from new unseen data to facilitate the discovery of previously unknown prophages. We have applied PhageBoost on 5537 single-cell genomics data and have found consistently more viral regions and considerably faster than with the current state-of-art tools. This finding was validated with available marine virome data. In order to support larger sequencing projects, Phageboost can work on multiple file formats, including compressed files, and is freely available as an interactive online prediction server and a command-line tool. This allows future work for generating and inferring insights from large datasets which could help for example solve the state of lysogenic activity as well as providing new approaches to study the phylogeny of phages and host-phage interactions.
DATA AVAILABILITY
The PhageBoost predictor is available as an online prediction server at http://www.phageboost.dk and freely available to academic users at GitHub: https://github.com/ku-cbd/PhageBoost. The PB13994 training datasets are available at a frozen archive as https://doi.org/10.17894/ucph.64136536–6353-430b-96ca-701ce89921c4.
The PhageBoost source code is available at GitHub: https://github.com/ku-cbd/PhageBoost.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Tom Delmont for his valuable advice leading to the choice of the marine validation dataset, and Antonio Fernandez Guerra for helpful discussions in taming the possible model bias arising from taxonomy.
Contributor Information
Kimmo Sirén, Section for Evolutionary Genomics, The GLOBE Institute, University of Copenhagen, Copenhagen,1353 Denmark.
Andrew Millard, Department of Genetics and Genome Biology, University of Leicester, LE1 7RH Leicester, UK.
Bent Petersen, Section for Evolutionary Genomics, The GLOBE Institute, University of Copenhagen, Copenhagen,1353 Denmark; Centre of Excellence for Omics-Driven Computational Biodiscovery, AIMST University,08100 Kedah, Malaysia.
M Thomas P Gilbert, Section for Evolutionary Genomics, The GLOBE Institute, University of Copenhagen, Copenhagen,1353 Denmark; Center for Evolutionary Hologenomics, The GLOBE Institute, University of Copenhagen,1353 Copenhagen, Denmark; University Museum, NTNU, 7012 Trondheim, Norway.
Martha R J Clokie, Department of Genetics and Genome Biology, University of Leicester, LE1 7RH Leicester, UK.
Thomas Sicheritz-Pontén, Section for Evolutionary Genomics, The GLOBE Institute, University of Copenhagen, Copenhagen,1353 Denmark; Centre of Excellence for Omics-Driven Computational Biodiscovery, AIMST University,08100 Kedah, Malaysia.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Mælkeafgiftsfonden project ‘Metacheese’; Innovation Fund Denmark project ‘Bac4Crop’.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cohen D., Melamed S., Millman A., Shulman G., Oppenheimer-Shaanan Y., Kacen A., Doron S., Amitai G., Sorek R. Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature. 2019; 574:691–695. [DOI] [PubMed] [Google Scholar]
- 2. Bernheim A., Sorek R. Viruses cooperate to defeat bacteria. Nature. 2018; 559:482–484. [DOI] [PubMed] [Google Scholar]
- 3. Kupczok A., Neve H., Huang K.D., Hoeppner M.P., Heller K.J., Franz C.M.A.P., Dagan T Rates of mutation and recombination in siphoviridae phage genome evolution over three decades. Mol. Biol. Evol. 2018; 35:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gentile G.M., Wetzel K.S., Dedrick R.M., Montgomery M.T., Garlena R.A., Jacobs-Sera D., Hatfull G.F. More evidence of Collusion: a new Prophage-Mediated viral defense system encoded by Mycobacteriophage Sbash. Mbio. 2019; 10:e00196-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chatterjee A., Duerkop B.A. Sugar and fatty acids Ack-celerate prophage induction. Cell Host Microbe. 2019; 25:175–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rezaei Javan R., Ramos-Sevillano E., Akter A., Brown J., Brueggemann A.B. Prophages and satellite prophages are widespread in Streptococcus and may play a role in pneumococcal pathogenesis. Nat. Commun. 2019; 10:4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramisetty B.C.M., Sudhakari P.A. Bacterial ‘Grounded’ Prophages: Hotspots for genetic renovation and innovation. Front. Genet. 2019; 10:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roux S., Enault F., Hurwitz B.L., Sullivan M.B. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015; 3:e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kieft K., Zhou Z., Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020; 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wenchen S., Sun H.X., Zhang C., Cheng L., Peng Y., Deng Z., Wang D., Wang Y., Hu M., Liu W. et al. Prophage Hunter: an integrative hunting tool for active prophages. Nucleic Acids Res. 2019; 47:W74–W80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akhter S., Aziz R.K., Edwards R.A. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012; 40:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amgarten D., Braga L.P.P., da Silva A.M., Setubal J.C MARVEL, a tool for prediction of bacteriophage sequences in metagenomic bins. Front. Genet. 2018; 9:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roux S., Krupovic M., Daly R.A., Borges A.L., Nayfach S., Schulz F., Sharrar A., Matheus Carnevali P.B., Cheng J.-F., Ivanova N.N. et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth's biomes. Nat. Microbiol. 2019; 4:1895–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jensen L.J., Gupta R., Blom N., Devos D., Tamames J., Kesmir C., Nielsen H., Staerfeldt H.H., Rapacki K., Workman C. et al. Prediction of human protein function from post-translational modifications and localization features. J. Mol. Biol. 2002; 319:1257–1265. [DOI] [PubMed] [Google Scholar]
- 16. Chen T., Guestrin C. XGBoost: a scalable tree boosting system. Proceedings of the 22Nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’16. 2016; NY: ACM; 785–794. [Google Scholar]
- 17. Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harris F.J. On the use of windows for harmonic analysis with the discrete Fourier transform. Proc. IEEE. 1978; 66:51–83. [Google Scholar]
- 19. Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020; 17:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murphy L.R., Wallqvist A., Levy R.M. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein. Eng. 2000; 13:149–152. [DOI] [PubMed] [Google Scholar]
- 22. Pruitt K.D., Tatusova T., Maglott D.R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005; 33:D501–D504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000; 28:33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grazziotin A.L., Koonin E.V., Kristensen D.M. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017; 45:D491–D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Steinegger M., Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 26. van Dongen S., Abreu-Goodger C. Using MCL to extract clusters from networks. Methods Mol. Biol. 2012; 804:281–295. [DOI] [PubMed] [Google Scholar]
- 27. Delmont T.O., Eren A.M. Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ. 2018; 6:e4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 29. Akiba T., Sano S., Yanase T., Ohta T., Koyama M. Optuna: a next-generation hyperparameter optimization framework. Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, KDD ’19. 2019; NY: ACM; 2623–2631. [Google Scholar]
- 30. Lundberg S.M., Nair B., Vavilala M.S., Horibe M., Eisses M.J., Adams T., Liston D.E., King-Wai Low D., Newman S.-F., Kim J. et al. Explainable machine-learning predictions for the prevention of hypoxaemia during surgery. Nat. Biomed. Eng. 2018; 2:749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casjens S. Prophages and bacterial genomics: what have we learned so far?. Mol. Microbiol. 2003; 49:277–300. [DOI] [PubMed] [Google Scholar]
- 32. Zhou Y., Liang Y., Lynch K.H., Dennis J.J., Wishart D.S. PHAST: a fast phage search tool. Nucleic Acids Res. 2011; 39:W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gregory A.C., Zayed A.A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C. et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell. 2019; 177:1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pachiadaki M.G., Brown J.M., Brown J., Bezuidt O., Berube P.M., Biller S.J., Poulton N.J., Burkart M.D., La Clair J.J., Chisholm S.W. et al. Charting the complexity of the marine microbiome through Single-Cell genomics. Cell. 2019; 179:1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eren A.M., Esen Ö.C., Quince C., Vineis J.H., Morrison H.G., Sogin M.L., Delmont T.O. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015; 3:e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clokie M.R.J., Blasdel B.G., Demars B.O.L., Sicheritz-Pontén T Rethinking phage Ecology by rooting it within an established plant framework. PHAGE. 2020; 1:121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tange O. GNU Parallel 20200522 ('Kraftwerk'). 2020; 10.5281/zenodo.3841377. [DOI]
- 39. Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007; 9:90–95. [Google Scholar]
- 40. Waskom M., Botvinnik O., O’Kane D., Hobson P., Lukauskas S., Gemperline D.C., Augspurger T., Halchenko Y., Cole J.B., Warmenhoven J. et al.. 2017; mwaskom/seaborn: v0.8.1 (September 2017) 10.5281/zenodo.883859. [DOI]
- 41. Lundberg S.M., Erion G.G., Lee S.-I. Consistent individualized feature attribution for tree ensembles. 2018; arXiv doi:07 March 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1802.03888.
- 42. Sharp P.M., Li W.H. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987; 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chithambaram S., Prabhakaran R., Xia X. Differential codon adaptation between dsDNA and ssDNA phages in Escherichia coli. Mol. Biol. Evol. 2014; 31:1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akhter S., Bailey B.A., Salamon P., Aziz R.K., Edwards R.A. Applying Shannon's information theory to bacterial and phage genomes and metagenomes. Sci. Rep. 2013; 3:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luck K., Travé G. Phage display can select over-hydrophobic sequences that may impair prediction of natural domain–peptide interactions. Bioinformatics. 2011; 27:899–902. [DOI] [PubMed] [Google Scholar]
- 46. Lawrence J.G., Ochman H. Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:9413–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garcia-Vallvé S., Palau J., Romeu A. Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Mol. Biol. Evol. 1999; 16:1125–1134. [DOI] [PubMed] [Google Scholar]
- 48. Garcia-Vallve S., Guzman E., Montero M.A., Romeu A. HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003; 31:187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PhageBoost predictor is available as an online prediction server at http://www.phageboost.dk and freely available to academic users at GitHub: https://github.com/ku-cbd/PhageBoost. The PB13994 training datasets are available at a frozen archive as https://doi.org/10.17894/ucph.64136536–6353-430b-96ca-701ce89921c4.
The PhageBoost source code is available at GitHub: https://github.com/ku-cbd/PhageBoost.