

Abstract
Metagenomics is the study of genomic DNA recovered from a microbial community. Both assembly-based and mapping-based methods have been used to analyze metagenomic data. When appropriate gene catalogs are available, mapping-based methods are preferred over assembly based approaches, especially for analyzing the data at the functional level. In this study, we introduce CAMAMED as a composition-aware mapping-based metagenomic data analysis pipeline. This pipeline can analyze metagenomic samples at both taxonomic and functional profiling levels. Using this pipeline, metagenome sequences can be mapped to non-redundant gene catalogs and the gene frequency in the samples are obtained. Due to the highly compositional nature of metagenomic data, the cumulative sum-scaling method is used at both taxa and gene levels for compositional data analysis in our pipeline. Additionally, by mapping the genes to the KEGG database, annotations related to each gene can be extracted at different functional levels such as KEGG ortholog groups, enzyme commission numbers and reactions. Furthermore, the pipeline enables the user to identify potential biomarkers in case-control metagenomic samples by investigating functional differences. The source code for this software is available from https://github.com/mhnb/camamed. Also, the ready to use Docker images are available at https://hub.docker.com.
INTRODUCTION
Metagenomics is an interdisciplinary research field which lies in the intersection of molecular genomics, microbial ecology and data analysis. The main subject of this field is metagenome, which refers to as the total genomic content of microorganisms present in a certain environment. Metagenomics is based on microbial culture-independent methods, including high-throughput genome-sequencing techniques. In this way, DNA from the inhabiting microorganisms (i.e. the microbiome) are extracted from a certain environment, e.g. intestine, and are studied using various computational techniques (1).
There are two main approaches for analyzing metagenomic data: (i) taxonomic profiling, which describes the phylogenetic diversity of microorganisms in samples; and (ii) functional profiling, which includes computational strategies for mapping genomic sequences to ‘functional’ groups, to gain an insight on the functional capacities of microbiota in samples (2).
Comprehensive functional analysis of microbiome can significantly improve our understanding of biochemical capabilities of the microbial community (3). A possible strategy for functional profiling of microbiome is to assemble reads to larger contigs, and then, predict the gene content for functional profiling (4). For poorly unexplored microbiomes, assembly based strategies are inevitable, despite the fact that most conventional assemblies are of relatively low accuracy (5). In contrast, for those environments that have already been studied extensively, like the human gut, good microbiome gene catalogs have previously been compiled (6). In such cases, mapping-based analysis can be used for functional analysis of metagenomes.
In shotgun metagenomics studies, the collection of microorganisms is studied through direct DNA sequencing without any culture and isolation. By comparing the frequency of genes mapped to the gene catalog, functional differences between samples can be studied. Thus, gene abundance data are affected by high levels of systematic variability, which can greatly reduce statistical power and increase false positives (7). There are many reasons for the systematic variation in metagenome data that can affect the observed abundance of genes and microorganisms. One of the important reasons is the difference in the depth of sequencing so that each sample has a different number of sequencing reads (8). Other reasons for systematic variability include inconsistencies in sampling methods, DNA extraction, variation in the quality of sequencing runs, errors in read mapping and incomplete reference gene catalogs (9). In addition, the systematic variability can be due to differences in the average genome size of microorganisms, species richness and GC-content related to reads, which can affect the observed gene abundance (7,10).
Next-Generation Sequencing (NGS) data are also inherently compositional. Compositional means that the relative abundance of each nucleotide fragments is dependent to the abundance of other fragments. This property is related to the sequencing equipment and underlying methodology, and the resulted sequences are affected by the bias involved in amplification and subsequent nucleotide sampling (11). Hence, the composition is a result of this ambiguity in measurements that are an unclear part of the whole (e.g. metagenomic count data generated by NGS sequencing). The compositional data analysis (CoDA) refers to handling and resolving this bias (12).
Metagenomic count data also faces more severe challenges compared to the other NGS data. One of these challenges is the highly variable number of sequenced reads or sequencing depth in different samples. The second challenge is the very high percentage of zeros in metagenomic count data referred to as zero-inflation (roughly between 50 and 90%) (13). Also, metagenomic data are very high dimensional in comparison with the other NGS data (e.g. in a sample of the gut microbiome gene catalog, there are ∼10 M gene sequences or features (6)). On the other hand, due to the low frequency of DNA sampling, the very rare taxa are not recorded, which is called technical zeros. Also, some taxa may not be captured through their missing population, known as structural zeros. Another challenges are the size of the study and large variance in taxa distributions (over-dispersion) (14).
Normalization processes can identify and eliminate systematic variability and compositional bias, so it is an essential step in data preprocessing and analysis. Many normalization methods have been proposed for high-dimensional count data, but for most of them, their performance has not been evaluated on metagenomic data (7). Various approaches have been proposed yet to address the challenges involving the compositional data. For example, to solve the problem of uneven sequencing depth, two approaches are introduced. First, rescaling the read counts in different samples to achieve a fixed value for their library size, and second, re-sampling reads to achieve the number of fixed reads for all samples (2). Also, many CoDA methods use transformation instead of normalization. These methods map the data to real space using log-ratio transformation, which makes it possible to use conventional statistical methods for further analysis. These methods try to use a reference such as the geometric mean of the subset feature for data transformation (15).
As explained, one of the major challenges in metagenomic compositional data is sparsity or zero-inflation, which becomes more acute for gene-centric count data. Several packages and tools have been developed to handle this problem and improve the accuracy of comparative gene abundance studies. Some packages have been developed specifically to deal with the zeros in the metagenomic data. The metagenomeSeq uses a zero-inflated log-normal model for gene abundance data (16,17). This method assumes that the zero-inflation is sample-specific and depends on the depth of sequencing (18). Ratio approach for identifying differential abundance (RAIDA) used a statistical model that first converts counts into relative frequencies, which are described by a log-normal distribution. RAIDA assumes that most features are not differentially abundant which makes it suitable to analyze metagenomic data at the taxa and gene levels. Also, this tool was developed to comparative analysis of metagenomic data samples in two different conditions, which can be generalized to more than two conditions (19). Also, there are several zero-inflated statistical models for metagenomic data, including zero-inflated negative-binomial and zero-inflated beta regression models (20,21).
However, other packages and tools for CoDA are provided, including ANCOM (22), ZIBSeq (21), CPL (23), DESeq2 (24) and edgeR (25). These methods exploit different statistical methods and try to handle the compositional bias and zero-inflation in the high-throughput sequencing data. It is important to note that most of the packages described above, including DESeq2 and edgeR, were originally developed for RNA-seq data analysis. Some of the most widely used CoDA packages and their properties are organized in Table 1.
Table 1.
Some of the most widely used packages and their normalization methods for handling composition bias and zero-inflation in high-throughput sequencing data
| Software Packages/tools | Distribution of taxon | Normalization method | Most specific usage | Advantage | Ref. |
|---|---|---|---|---|---|
| edgeR | Negative Binomial | Trimmed mean of M values (TMM) | RNA-Seq data | suitable for detecting the similarity of expression in RNA-Seq data and over-dispersion. | (25) |
| DESeq2 | Negative Binomial | Relative Log Expression (RLE) | RNA-Seq data | suitable for detecting the similarity of expression in RNA-Seq data and over-dispersion. | (24) |
| ANCOM | Non-parametric | Log-ratio transformations | Metagenomic data (taxa level) | calculates the relation between taxa even in repeated samples to reduce false positives in differentially abundant taxa. | (22) |
| ZIBSeq | Zero-inflated beta | Total sum scaling (TSS) | Metagenomic data (taxa level) | developed to handle sparsity in metagenome data. It is also more efficient for detecting differentially abundant features in multiple conditions. | (21) |
| CPL | Non-parametric | Centered log ratio (CLR) | Metagenomic data (taxa level) | determines the relationship between taxa regardless of sparsity on distribution. | (23) |
| RAIDA | Zero-inflated log-normal | Ratio Approach | Metagenomic data (taxa and gene level) | suitable for sparse data. It is not affected by the amount of difference in total abundance of differentially abundant features (DAFs) in different conditions. Assumes that most features are not differentially abundant and was developed for two conditions. | (19) |
| metagenomeSeq | Zero-inflated log-normal and Gaussian | Cumulative Sum Scaling (CSS) | Metagenomic data (taxa and gene level) | suitable for handling sparsity in very high dimensional data. It is also highly efficient for detecting rare samples in metagenomic data. | (16) |
In addition, a large number of normalization methods have been proposed for compositional data. Some of these methods are provided for RNA-seq data (26), some for operational taxonomic units (OTUs) data generated by amplicon sequencing (27) and some for metagenome data. But comparative studies of these methods show that there is a large dependency between performance and data characteristics. For example, the methods that have better performance for RNA-seq data will not necessarily be suitable for metagenome data (7). Some of these normalization methods are described below. Total sum scaling (TSS) is a standard normalization method for count data that is obtained by dividing the individual counts by the total counts in the sample such that the sum of the normalized values is 1 (28). TSS with a fixed scaling factor may harm OTU counts due to technological sequencing biases. Cumulative sum scaling (CSS) (16) re-scales samples based on the low-frequency (relatively constant and independent) quartiles, and does not eliminate the effect of high-frequency samples. Additionally, CSS as a highly cited log-normal model, has been used at taxa/gene level for metagenomic CoDA in many studies (2,18,29,30). Trimmed mean of M-values (TMM) estimates scale factors between samples for use in statistical analysis to identify differential expression. TMM normalization assumes that most genes are not differentially expressed between samples (31). Another family of methods for normalizing compositional data is based on the log-ratio transformation. The mutual dependence between features in a composition implies that the analysis of individual features is performed to a reference baseline that transforms each sample into a new space and the statistical analysis will be done in this new space. Based on the choice of reference, different log-ratio based methods were developed. The centered log-ratio (CLR), additive log-ratio (ALR) and relative log expression (RLE) transformations use different strategies for selecting the reference (11,32). Table 1 details some of the packages, related normalization methods, properties and their advantages for CoDA.
For functional analysis of metagenomic data, it is necessary to use a pipeline that considers the compositional nature of metagenomic data. In the present paper, we introduce CAMAMED, a mapping-based software pipeline to perform taxonomic and functional analysis of metagenomic data. Therefore, the proper normalization method is very important for metagenomic data analysis. Considering the properties previously described for this data (e.g. sparsity, high-dimensionality, rarefaction, undersampling and vast differences in sequencing depths, etc.), the metagenomeSeq and CSS as its normalization method is one of the most widely used methods in metagenomic studies (2,17,18,33). However, one of the most important drawbacks reported for metagenomeSeq in different studies is the high false-positive rate for differential abundance analysis when the sample size is small (34,35). CAMAMED uses the metagenomeSeq due to its high capability of handling compositional bias in sparse metagenomic data in taxa, gene, KO, EC number and reaction levels. CAMAMED is implemented using Python3 and Shell programming based on the Linux operating system. It is designed for non-professional users and is relatively easy to execute. Also, for easier use of CAMAMED, two Docker images have been constructed that enables users to run the CAMAMED pipeline without involving in installation details and dependencies. These images are available at www.hub.docker.com.
MATERIALS AND METHODS
Metagenomic dataset
In this study, we used 80 metagenomic shotgun-sequenced fecal samples. This dataset (named originally ‘cohort1’) includes samples from 24 healthy (control), 27 colorectal adenoma and 29 colorectal carcinoma individuals. The samples had been sequenced using the Illumina platform and paired-end sequencing methods (36). Also, to obtain the abundance of genes in the samples, we used a previously-reported gene catalog with 9.88 × 106 non-redundant gene sequences. This catalog, called the integrated gene catalog (IGC), contains the nucleotide and protein gene sequences of the human gut microbiome (6). For more information on the ‘Materials and Methods’, see Supplementary File 1.
Pipeline overview
CAMAMED is an automatic and easy-to-use computational pipeline for taxonomic and functional profiling of metagenomic data. Figure 1 shows the overall workflow of this pipeline.
Figure 1.

Overall workflow for the CAMAMED pipeline.
Pre-processes
CAMAMED can get the input data in FASTQ, FASTA or SRA file formats, in both paired-end and single-end modes. If the sequence format is SRA, it will be converted to FASTQ or FASTA format using the SRA Toolkit v2.8.2 (http://ncbi.github.io/sra-tools). Then, the quality control of the sequence dataset is performed using FastQC v0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). The statistical characteristics of samples are extracted using SeqKit v0.10.1 (37).
Mapping processes
To find the bacterial frequency of the samples at different taxonomic levels, we use MetaPhlAn v2.0. MetaPhlAn (Metagenomic Phylogenetic Analysis) is a computational tool for profiling the composition of microbial communities from metagenomic shotgun sequencing data (38). CAMAMED uses a mapping-based strategy for metagenome data processing. To obtain the best mapping results, an appropriate gene catalog should be provided (see Ref. (6) as an example). If an in-house gene catalog is created, one can use CD-HIT v4.7 to remove potential duplicate sequences from the gene catalog. CD-HIT is a widely used program for clustering and comparing protein or nucleotide sequences (39). At this step, metagenomic read sequences are mapped to the gene catalog using MOSAIK v2.2.3 software. This software uses the hash-based algorithm to map nucleotide sequences very quickly to the nucleotide gene catalog (40).
KEGG annotations
After mapping reads to the gene catalog, one can compute the genes frequencies in each sample. For functional profiling, we use the KEGG database, which includes a comprehensive list of genes and genomes, together with their biochemical annotations (41). To extract the KEGG orthology (KO) groups associated with each of the genes, KEGG Automatic Annotation Server (KAAS, Ver. 2.1) (42) can be used. By using this tool, nucleotide and amino acid sequences can be mapped to KEGG and the associated KOs are retrieved. Alternatively, one may use GhostKOALA Ver. 2.2 (43), which maps amino acid sequences to KEGG. After extracting the KOs associated with each gene, for each KO, we obtain the enzyme commission (EC) numbers and reaction IDs. CAMAMED currently includes the KO-EC-reaction relations associated with KEGG. It is always possible for the user to retrieve the latest update of these data from KEGG, but this might be a time-consuming task. Then, for each sample, CAMAMED extracts the frequency of each gene, KO, EC number and reaction.
Data normalization
After extracting the frequencies of genes, these data should be corrected for the compositional bias. Before this step, we remove the genes to which less than five reads in all of the samples have been mapped (44). We then, divide the frequency of each gene to the length of the gene, in order to compute a normalized abundance of the genes with different length to calculate KO, EC number and reaction frequencies. Then, we use the metagenomeSeq package with CSS method for compositional correction and normalization (16). We also use this method to correct compositional bias in species-level in taxonomic data. Now, to calculate the normalized frequencies of KO, EC number, reaction, we use the sum of normalized frequency for their subset genes.
In the following a brief explanation of the CSS method is provided. Suppose raw data is represented as a count matrix M (m, n) where m and n represent the number of features and samples, respectively. The raw data in the count matrix with cij represents the number of times that feature i was observed in sample j. Also, the sum of counts for sample j is calculated as  . A usual method for normalizing feature value is
. A usual method for normalizing feature value is  , which is called total-sum normalization and has its own important drawbacks (improper handling of compositional bias). To avoid these drawbacks, CSS considers the lth quantile of sample j to be
, which is called total-sum normalization and has its own important drawbacks (improper handling of compositional bias). To avoid these drawbacks, CSS considers the lth quantile of sample j to be 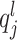 , meaning that sample j has l features with counts less than
, meaning that sample j has l features with counts less than 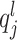 . For
. For 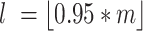 .
. 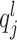 represents the 95th percentile of the count distribution for sample j. Also,
represents the 95th percentile of the count distribution for sample j. Also, 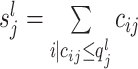 is the sum of feature counts up to lth quantile in sample j (16).
is the sum of feature counts up to lth quantile in sample j (16).
CSS selects 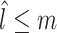 to calculate the normalization scaling factor in each sample and defines normalized counts as
to calculate the normalization scaling factor in each sample and defines normalized counts as 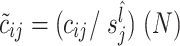 , where N is the normalization constant that is selected equally for all samples. It is recommended to select N as the median of scaling factors
, where N is the normalization constant that is selected equally for all samples. It is recommended to select N as the median of scaling factors 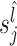 of all samples. The counts for samples with a scaling factor close to N can be considered as reference samples, and the counts for other samples can be calculated relative to the reference samples (16). Selecting an appropriate quantile based on
of all samples. The counts for samples with a scaling factor close to N can be considered as reference samples, and the counts for other samples can be calculated relative to the reference samples (16). Selecting an appropriate quantile based on 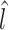 is critical to the normalization of data, and its value is project-specific and is chosen based on data driven methods that use experimental details such as sample preparation and sequencing (16,26). Also, CSS-normalized sample abundance can be well approximated with zero-inflated log-normal model in studies with a large number of samples. Therefore, logarithmic transformation is used on normalized count data. This transformation controls the diversity of features measured in the samples (16).
is critical to the normalization of data, and its value is project-specific and is chosen based on data driven methods that use experimental details such as sample preparation and sequencing (16,26). Also, CSS-normalized sample abundance can be well approximated with zero-inflated log-normal model in studies with a large number of samples. Therefore, logarithmic transformation is used on normalized count data. This transformation controls the diversity of features measured in the samples (16).
Statistical analyses
A possible application of metagenome analysis is when a comparative study is performed, to detect those ‘biomarkers’ which are under-represented in case versus control samples. Using the non-parametric Kruskal-Wallis H test, one can identify the components that are changed significantly in different sample groups. Kruskal–Wallis test is used for comparing the value of a variable in two or more groups. The one-way analysis of variance (ANOVA) test can also be used in this pipeline assuming the data distribution is normal. We then use the Benjamini–Hochberg (BH) method to correct for false discovery rates.
By this stage, we have normalized data for the frequency of taxa, genes, KOs, EC numbers and reactions. We also specify the group labels to which each sample belongs. All the code and tools used in CAMAMED are collected in two Docker images that the user can easily run CAMAMED without having to worry about the installation details.
RESULTS
Test case: colorectal adenoma and carcinoma
To test the applicability of CAMAMED, we used 80 metagenomic shotgun-sequenced fecal samples obtained from 24 healthy (control), 27 colorectal adenoma and 29 colorectal carcinoma individuals (36). The results of the mapping analysis can depend on the reference gene catalog used. We used IGC, a previously-reported gene catalog of the human gut (6), to evaluate CAMAMED.
By applying CAMAMED for mapping the reads to the gene catalog, we found 3 354 281 genes to which at least one read was mapped. Table 2 shows the results of the Kruskal–Wallis test for levels of the species, gene, KO, EC number and reaction (for a significance level of P-value ≤ 0.01).
Table 2.
The results of the Kruskal–Wallis test for significance level P-value ≤ 0.01 at species, gene, KO group, EC number and reaction levels
| Test Level | Total number of entities | Number of significant entities (P-value ≤ 0.01) | Percentage of significantly changed entities | Percentage of significantly-changed entities after P-value adjustment |
|---|---|---|---|---|
| Species | 374 | 10 | 2.67 | 0.53 |
| Gene | 3 354 281 | 64 402 | 1.92 | 0.44 |
| KO group | 16 482 | 491 | 2.98 | 0.25 |
| EC number | 3377 | 86 | 2.55 | 0.21 |
| Reaction | 3183 | 78 | 2.45 | 0.19 |
The results in Table 2 show that the ratio of significantly changed genes (that is, 1.92%) is not different from what is expected by chance (as p was assumed to be significant at the level of 0.01). This observation suggests that functional analysis cannot be ideally performed at the level of genes, in contrast to what has been previously proposed (45). In contrast, the ratio of significantly changed species is 2.67%, which explains why taxonomic profiling of microbiome data is widely used in the literature. However, one should note that 2.67% of 374 species means only 10 species, which might not provide us with enough number of features to be used in a successful classification of samples to control, adenoma and carcinoma groups.
In case of other functional features, with the same level of significance (P ≤ 0.01) a greater ratio of KO groups, EC numbers and reactions might be detected as significant (2.98, 2.55 and 2.45%, respectively). Also, the last column of Table 2 shows the percentage of significantly changed entities after P-value adjustment, using the BH method associated with the fourth column. Therefore, we recommend the metagenome functional analysis to be performed at these levels, rather than the gene level (30). CAMAMED is currently the only available pipeline for this kind of analysis. Note that CAMAMED is an integrated pipeline of more than ten well-known bioinformatics tools that were previously used in other studies. To ensure the correctness of our implementation, we also applied CAMAMED on another datasets (30), and observed that all the results are reproducible.
DISCUSSION
Handling metagenomic data is a time-consuming and elaborate task. A number of pipelines are currently available for facilitating metagenomic analysis. Different aspects of the available metagenome analysis pipelines were compared in Table 3. These tools use assembly or mapping-based methods to process sequences. The level of annotation (taxon, gene, etc.) reported to the user is a vital aspect when tools are compared. The level of annotation in these tools is commonly taxon or gene, while CAMAMED annotates the samples at five levels, including taxon, gene, KO, EC number and reaction. This exclusive feature enables the user to analyze the samples at the functional level, which is reported to be more robust compared to taxonomic or genomic changes (30,46).
Table 3.
Summary of available software pipelines for taxonomic and/or functional analysis of metagenomic data
| Tools | Sequence processing | Taxonomic profiling | Functional profiling | Annotation level | Ref. |
|---|---|---|---|---|---|
| MetaCRAM | Mapping (Bowtie2) | Kraken | Taxon, gene | (5) | |
| SUPER-FOCUS | Mapping (RAPSearch2) | FOCUS | GenBank database | Taxon, gene | (47) |
| MOCAT2 | Assembly (SOAPdenovo) and gene prediction (MetaGeneMark) | eggNOG database | eggNOG database and others | Taxon, gene | (4) |
| MetaStorm | Assembly (IDBA-UD) and gene prediction (PRODIGAL) | SILVA and GREENGENES databases | CARD and CAZy databases | Taxon | (48) |
| CAMAMED | Mapping (MOSAIK) | MetaPhlAn2 | KAAS and GhostKOALA web services and KEGG database | Taxon, gene, KO, EC number and reaction | Present work |
In recent years, in most of the metagenomic analyses, taxonomic profiles have been used as markers in case-control groups (49). In the functional analysis of metagenomes, on the other hand, case-control differences are studied at the gene level (50). Using the CAMAMED pipeline, not only one can easily analyze metagenome data at taxonomical (taxon) and functional (gene) level, but also it is possible to go further by analyzing the potential functional differences at other functional levels, that is, KO, EC number and reaction.
Methods for analyzing microbiome data sometimes assume, although implicitly, that sequencing data can be used equivalently in place of environmental data. However, microbiome sequencing data is often compositional and may not represent the original distribution of the samples in the environment (51). Due to the sparse nature of metagenomic data, different methods have been proposed for compositional bias correction. To this end, the CSS method is one of the most popular and powerful methods to handle this challenge (17,52). To demonstrate the composition-awareness of CSS and hence CAMAMED, Norouzi-Beirami et al. used two independent gut metagenome datasets on colorectal cancer (30). In this study, taxa and gene abundance data were extracted and compositional bias removal was performed independently on the data using the CSS method. The feature set extracted as a colorectal cancer marker from the first dataset has been accurately evaluated on the second dataset. However, without applying the CSS, the results from these datasets were not consistent (30). Furthermore, CAMAMED considers the correction for zero-inflation and compositional bias in the metagenomic data, which is, to the best of our knowledge, largely neglected in other pipelines.
The CAMAMED pipeline performs all the steps involved in the analysis of metagenomic data in a (semi-)automatic and step-by-step manner. Most of the tools used in this pipeline do not need to be installed separately by the user, which liberates the nonprofessional user from being engaged in potential software installation obstacles. It is necessary to emphasize that CAMAMED is a mapping-based pipeline for analyzing metagenomic data at the taxonomic and functional level. After running CAMAMED on metagenomic samples, normalized datasets are extracted at five levels of taxon, gene, KO, EC number and reaction. Such output can be then exploited by the user for additional machine learning and statistical studies.
Also, preparing the Docker images for CAMAMED make it possible that the user can employ it without involving in installation details and dependencies. These images also make using CAMAMED easier and increase the life of the software.
DATA AVAILABILITY
The software manual and the software package are available from https://github.com/mhnb/camamed and the Docker images with titled camamed_pipeline and camamed_pipeline_db at www.hub.docker.com.
Supplementary Material
Contributor Information
Mohammad H Norouzi-Beirami, Laboratory of Complex Biological systems and Bioinformatics (CBB), Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran 1417614335, Iran.
Sayed-Amir Marashi, Department of Biotechnology, College of Science, University of Tehran, Tehran 1417614411, Iran.
Ali M Banaei-Moghaddam, Laboratory of Genomics and Epigenomics (LGE), Department of Biochemistry, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran 1417614335, Iran.
Kaveh Kavousi, Laboratory of Complex Biological systems and Bioinformatics (CBB), Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran 1417614335, Iran.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
No funders.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sudarikov K., Tyakht A., Alexeev D.. Methods for the metagenomic data visualization and analysis. Curr. Issues Mol. Biol. 2017; 24:24–37. [DOI] [PubMed] [Google Scholar]
- 2. Dhariwal A., Chong J., Habib S., King I.L., Agellon L.B., Xia J.. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017; 45:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lawley T.D., Walker A.W.. Intestinal colonization resistance. Immunology. 2013; 138:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kultima J.R., Coelho L.P., Forslund K., Huerta-cepas J., Li S.S., Driessen M., Voigt A.Y., Zeller G., Sunagawa S.. MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics. 2016; 32:2520–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim M., Zhang X., Ligo J.G., Farnoud F., Veeravalli V. V. MetaCRAM: an integrated pipeline for metagenomic taxonomy identification and compression. BMC Bioinformatics. 2016; 17:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li J., Jia H., Cai X., Zhong H., Feng Q., Sunagawa S., Arumugam M., Kultima J.R., Prifti E., Nielsen T. et al.. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014; 32:834–841. [DOI] [PubMed] [Google Scholar]
- 7. Pereira M.B., Wallroth M., Jonsson V., Kristiansson E.. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genomics. 2018; 19:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mcmurdie P.J., Holmes S.. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014; 10:e1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morgan J.L., Darling A.E., Eisen J.A.. Metagenomic sequencing of an in vitro-simulated microbial community. PLoS Comput. Biol. 2010; 5:e10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Manor O., Borenstein E.. MUSiCC: a marker genes based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 2015; 16:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quinn T.P., Erb I., Gloor G., Notredame C., Richardson M.F., Crowley T.M.. A field guide for the compositional analysis of any-omics data. Gigascience. 2019; 8:giz107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quinn T.P. Visualizing balances of compositional data: a new alternative to balance dendrograms. f1000 Res. 2018; 7:1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu L., Paterson A.D., Turpin W., Xu W.. Assessment and Selection of Competing Models for Zero-Inflated Microbiome Data. PLoS One. 2015; 10:e0129606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H. Microbiome, metagenomics, and high-dimensional compositional data analysis. Annu. Rev. Stat. Its Appl. 2015; 2:73–94. [Google Scholar]
- 15. Quinn T.P., Erb I., Richardson M.F., Crowley T.M.. Understanding sequencing data as compositions: an outlook and review. Bioinformatics. 2018; 34:2870–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paulson J.N., Stine O.C., Bravo H.C., Pop M.. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods. 2013; 10:1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu X., Shyu C.-R., Bromberg Y., Gao J., Gong Y., Korkin K., Yoo I., Zheng H.J.. IEEE Computer Society. IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2017. 2017; MO, USA. [Google Scholar]
- 18. Jonsson Viktor, Osterlund T., Nerman O., Kristiansson E.. Modelling of zero-inflation improves inference of metagenomic gene count data. Stat. Methods Med. Res. 2019; 28:3712–3728. [DOI] [PubMed] [Google Scholar]
- 19. Sohn M.B., Du R., An L.. A robust approach for identifying differentially abundant features in metagenomic samples. Bioinformatics. 2015; 31:2269–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang R., Wagner B.D., Harris J.K.. Zero-inflated negative binomial mixed model: an application to two microbial organisms important in oesophagitis. Epidemiol. Infect. 2016; 144:2447–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peng X., Li G., Liu Z.. Zero-Inflated beta regression for differential abundance analysis with metagenomics data. J. Comput. Biol. 2016; 23:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mandal S., Van Treuren W., White R.A., Eggesb M., Knight R., Peddada S.D.. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Heal. Dis. 2015; 26:27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee C., Lee S., Park T.. Statistical methods for metagenomics data analysis. Int. J. Data Min. Bioinforma. 2017; 19:366–385. [Google Scholar]
- 24. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson M.D., Mccarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marot G., Castel D., Estelle J., Guernec G., Jagla B., Servant N., Jouneau L., Laloe D., Gall C. Le, Schae B.. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2012; 14:671–683. [DOI] [PubMed] [Google Scholar]
- 27. Weiss S., Xu Z.Z., Peddada S., Amir A., Bittinger K., Gonzalez A., Lozupone C., Zaneveld J.R., Vázquez-baeza Y., Birmingham A. et al.. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017; 5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paulson J.N., Stine C., Bravo H.C., Pop M.. Robust methods for differential abundance analysis in marker gene surveys. Nat. Methods. 2013; 10:1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mcknight D.T., Huerlimann R., Bower D.S., Schwarzkopf L., Alford R.A., Zenger K.R.. Methods for normalizing microbiome data: an ecological perspective. Methods Ecol. Evol. 2019; 10:389–400. [Google Scholar]
- 30. Norouzi-Beirami M.H., Marashi S., Banaei-Moghaddam A.M., Kavousi K.. Beyond taxonomic analysis of microbiomes: a functional approach for revisiting microbiome changes in colorectal cancer. Front. Microbiol. 2020; 10:3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson M.D., Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quinn T.P., Richardson M.F., Lovell D., Crowley T.M.. propr: an R-package for identifying proportionally abundant features using compositional data analysis. Sci. Rep. 2017; 7:16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim J., Kim M.S., Koh A.Y., Xie Y., Zhan X.. FMAP: functional mapping and analysis pipeline for metagenomics and metatranscriptomics studies. BMC Bioinformatics. 2016; 17:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo D., Ziebell S., An L.. An informative approach on differential abundance analysis for time-course metagenomic sequencing data. Bioinformatics. 2017; 33:1286–1292. [DOI] [PubMed] [Google Scholar]
- 35. Ma Y., Luo Y., Jiang H.. A novel normalization and differential abundance test framework for microbiome data. Bioinformatics. 2020; 36:3959–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thomas A.M., Manghi P., Asnicar F., Pasolli E., Armanini F., Zolfo M., Beghini F., Manara S., Karcher N., Pozzi C. et al.. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019; 25:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen W., Le S., Li Y., Hu F.. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016; 11:e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Truong D.T., Franzosa E.A., Tickle T.L., Scholz M., Weingart G., Pasolli E., Tett A., Huttenhower C., Segata N.. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 2015; 12:902–903. [DOI] [PubMed] [Google Scholar]
- 39. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 40. Lee W.P., Stromberg M.P., Ward A., Stewart C., Garrison E.P., Marth G.T.. MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One. 2014; 9:e90581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanehisa M., Goto S.. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M.. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007; 35:182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanehisa M., Sato Y., Morishima K.. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016; 428:726–731. [DOI] [PubMed] [Google Scholar]
- 44. Best M.G., Sol N., Kooi I., Tannous B.A., Wesseling P.. RNA-seq of tumor-educated platelets enables article RNA-seq of tumor-educated platelets enables. Cancer Cell. 2015; 28:666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu J., Feng Q., Wong S.H., Zhang D., Liang Q., Qin Y., Tang L., Zhao H., Stenvang J., Li Y. et al.. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut Microbes. 2015; 66:70–78. [DOI] [PubMed] [Google Scholar]
- 46. Tian L., Wang X., Wu A., Waldor M.K., Weinstock G.M., Fan Y., Friedman J., Weiss S.T., Liu Y., Dahlin A.. Deciphering functional redundancy in the human microbiome. Nat. Commun. 2020; 11:6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Silva G.G.Z., Green K.T., Dutilh B.E., Edwards R.A.. SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics. 2016; 32:354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arango-argoty G., Singh G., Heath L.S., Pruden A., Xiao W., Zhang L.. MetaStorm: a public resource for customizable metagenomics annotation. PLoS One. 2016; 11:e0162442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feng Q., Liang S., Jia H., Stadlmayr A., Tang L., Lan Z., Zhang D., Xia H., Xu X., Jie Z. et al.. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015; 6:6528. [DOI] [PubMed] [Google Scholar]
- 50. Yu J., Feng Q., Wong S.H., Zhang D., Yi Liang Q., Qin Y., Tang L., Zhao H., Stenvang J., Li Y. et al.. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017; 66:70–78. [DOI] [PubMed] [Google Scholar]
- 51. Gloor G.B., Macklaim J.M., Pawlowsky-glahn V., Egozcue J.J.. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 2017; 15:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kumar M.S., Slud E. V, Okrah K., Hicks S.C., Hannenhalli S.. Analysis and correction of compositional bias in sparse sequencing count data. BMC Genomics. 2018; 19:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The software manual and the software package are available from https://github.com/mhnb/camamed and the Docker images with titled camamed_pipeline and camamed_pipeline_db at www.hub.docker.com.