

Abstract
The CD27-CD70 costimulatory pathway is essential for the full activation of T cells, but some studies show that blocking this pathway exacerbates certain autoimmune disorders. Herein, we report on the impact of CD70/CD27 signaling on disease progression in the NOD mouse model of type 1 diabetes (T1D). Specifically, our data demonstrates that CD70 ablation alters thymocyte selection and increases circulating T cell levels. CD27 signaling was particularly important for the thymic development and peripheral homeostasis of Foxp3+Helios+ regulatory T cells (Tregs), which likely accounts for our finding that CD70-deficient NOD mice develop more aggressive T1D onset. Interestingly, we found that CD27 signaling suppresses the thymic development and effector functions of T1D-protective invariant natural killer T (iNKT) cells. Thus, rather than providing costimulatory signals, the CD27-CD70 axis may represent a coinhibitory pathway for this immunoregulatory T cell population. Moreover, we showed that a CD27 agonist Ab reversed the effects of CD70 ablation, indicating that the phenotypes observed in CD70-deficient mice were likely due to a lack of CD27 signaling. Collectively, our results demonstrate that the CD27-CD70 costimulatory pathway regulates the differentiation program of multiple T cell subsets involved in T1D development, and may be subject to therapeutic targeting.
Keywords: CD70, CD27, regulatory T cells, invariant natural killer T cells, type 1 diabetes, NOD mice
Introduction
In conjunction with antigen recognition by the T cell receptor (TCR), T cell activation is carefully regulated by various receptor/ligand molecules on the surface of responder T cells and antigen presenting cells (APC). This second signal, referred to as costimulation, enables full T cell activation, proliferation, and differentiation. Early T cell priming events require CD28 costimulatory molecule interactions with B7 ligands (1). However, later phases of the immune response rely on differing combinatorial signals drawn from a collection of tumor necrosis factor receptor superfamily receptor (TNFR)/TNF superfamily members (including CD27-CD70, CD30/CD30L, OX40/OX40L, CD137/CD137L, HVEM and GITR/GITRL) which vary considerably in location and timing of expression (2–4). Costimulatory molecule interactions also contribute to thymocyte selection, which is controlled by the intensity with which MHC-peptide complexes on hematopoietic and epithelial cells induce TCR signaling in CD4/CD8 double positive (DP) thymocytes (5). Currently, most data support that costimulatory molecule interactions alter thymic selection by modifying the TCR signaling threshold (5). This is especially evident for immunoregulatory T cell populations such as invariant natural killer T (iNKT) cells and regulatory T cells (Tregs) that seemingly require higher levels of signaling for their development (6, 7).
CD27 is a transmembrane homodimer with subunits of 55 kDa that is expressed on B cells, NK cells and T cells (8–10). CD27 is constitutively expressed at high levels on naïve T cells and increases during early T cell activation (11, 12). Expression is also high on central memory T cells, while late-stage effector and effector memory T cells downregulate CD27 (13). CD27 signaling is tightly controlled by the very limited expression of its ligand CD70 that is constitutively expressed by discrete cellular niches within the thymus and gut, and is transiently expressed by mature dendritic cells (DCs), macrophages, activated NK cells, T cells and B cells (14, 15). CD27-CD70 engagement appears to be important for T cell expansion, survival, and long-term memory formation, which manifests in the increased susceptibility of CD27- and CD70-deficient mice to numerous viruses and cancer (16–24).
Several reports show that CD27-CD70 costimulation is involved in autoimmunity, where signaling through this pathway is usually associated with increased pathology. CD70 is overexpressed on pathogenic CD4+ T cells from patients suffering from rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) as well as MRL/lpr mice that develop a lupus-like disease (25, 26). Furthermore, CD70 blockade leads to the amelioration of pathology in mouse models of arthritis and various inflammatory bowel diseases (27, 28). By contrast, CD27-CD70 interactions play a protective role in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, by impairing Th17 effector cells that are critical for autoimmunity (29). Thus, CD27-CD70 interactions can have diametric effects on autoimmune pathologies, which probably depends on the role of various T cell subsets in the pathogenesis of these different disorders.
In this study, we asked whether the CD27-CD70 pathway contributes to autoimmune type 1 diabetes (T1D) in the NOD mouse model given that autoreactive T cells play a central role in disease development by destroying pancreatic βcells. Disrupting the Cd70 gene accelerated T1D development in NOD mice, indicating CD27-CD70 costimulation elicits immunological tolerance induction responses inhibiting disease onset. We show that CD27-CD70 interactions limit circulating T cell levels and modulate thymocyte selection by controlling the ratio of DP to double negative (DN) thymocytes. Notably, CD70 ablation diametrically skewed the development of iNKT cells and Foxp3+Helios+ Tregs, which is significant because T1D development in NOD mice is partly due to numerical and functional deficiencies in both cell types (30–39). Indeed, NOD mice are protected from T1D by treatment with the iNKT cell agonist α-galactosylceramide (36–38, 40) or adoptively transferred iNKT cells (41, 42) and CD1d-deficient mice that lack iNKT cells develop accelerated T1D (36, 39). Moreover, compared to other mouse strains, NOD Tregs have a reduced capacity to suppress conventional T cells (33, 43, 44), due in part to a hypomorphic IL2 allelic variant and additional susceptibility genes (45). Furthermore, it has been reported that NOD Treg functionality declines with age (33, 34), although later studies have disputed this finding (46, 47). Since CD27-CD70 signaling was found to regulate both immunoregulatory and conventional T lymphocyte subsets involved in β-cell autoimmunity, the aim of the current study was to test the hypothesis that alterations in one or more T cell populations accounts for CD70-mediated modulation of T1D.
Materials and methods
Mice
CD70-deficient NOD/ShiLtDvs mice (Cd70−/−) were generated at the Jackson Laboratory using transcription activator-like effector nucleases (TALENs) designed to disrupt the Cd70 start site. The TALENs were designed using the online software “ZIFIT” (48, 49) and were constructed using the fast ligation-based automatable solid-phase high-throughput assembly system (50). These were designed to target the cleavage domain and a DNA binding domain in exon 1. The left TALEN targets the 18bp sequence: TGTTTATTACTGTGTTTT, while the right TALEN targets the 18bp sequence: TAGCGGACTACTCAGTAA. Following binding and dimerization, the TALEN pair should cause a double-stranded genomic break within the 16 bases of intervening sequence (GCTGTTGGTTTCATTG). The two TALENs were prepared as mRNA and microinjected directly into NOD/ShiLtJ zygotes at a concentration of 50 µg/µl each. A total of 304 embryos were transferred to pseudopregnant females, ultimately yielding 99 offspring. DNA was extracted from tail tips and screened for TALEN-induced mutations by PCR amplification using forward (5’-CAACAACGTGTGCGTCAATC-3’) and reverse (5’-CAGCCCAATACTGAACCTCTAAT-3’) primers. Sanger Sequencing of the PCR products identified a male mutant mouse with a 363 base pair deletion at the start site of Exon 1 of Cd70. This founder was backcrossed to NOD/ShiLtJ females, and the heterozygous progeny intercrossed to generate littermates of all three genotypes. Genotyping was performed by PCR using the primers described above (WT allele = 785bp, mutant allele = 422bp). This strain, NOD/ShiLtJ-Cd70<em1Mvw>/Mvw (Stock no. 24167), was imported from The Jackson Laboratory and maintained in a specific pathogen-free research colony. A previously described NOD stock carrying an inactivated Rag1 gene [NOD.129S7(B6)-Rag1<tm1Mom>/SzJ, designated NOD.Rag1−/−] (51) was used to create NOD.Rag1−/−.Cd70−/− mice (NOD.Cg-Rag1<tm1Mom> Cd70<em1Mvw>/Jpd). Previously described NOD stocks respectively transgenically expressing the TCR from the diabetogenic BDC2.5 CD4+ T cell clone (NOD.Cg-Tg(TcraBDC2.5,TcrbBDC2.5)1Doi/DoiJ) (52) and expressing GFP off the mouse Foxp3 promoter (NOD/ShiLt-Tg(Foxp3-EGFP/cre)1cJbs/J) (53) were intercrossed with Cd70−/− mice to generate NOD.BDC2.5.Foxp3eGFP.Cd70−/− mice. Previously described NOD.FOXP3-GFP-Cre.Rosa26-YFP mice (54) were obtained as a gift from the laboratory of Jeffrey A. Bluestone.
Assessment of diabetes and insulitis
Diabetes was monitored weekly by measuring glycosuria with Bayer Diastix, (Bayer, Diagnostics Division, Elkhart, IN), with disease onset defined by values of ≥3 on two consecutive days. Insulitis was assessed as previously described (55). Briefly, pancreata from indicated mice were fixed in a 10% neutral buffered formalin solution and embed with paraffin using automatic processors (VIP6, Leica) with graded levels of ethanol (70-100%) and xylene, after which samples were stained with Hematoxylin and Eosin. Insulitis scores were assigned according to a previously designated classification ranging from 0 (normal β-cell mass) to 4 (complete destruction) (56).
Islet isolation and analysis of infiltrates
Preparation of islet infiltrating leukocytes was adapted from a previous publication (57). Pancreata from seven-wk-old NOD.Rag1−/− and NOD.Rag1−/−.Cd70−/− splenocyte or T cell recipients were inflated with a Liberase TL solution (0.91 units/mL, Roche Diagnostics) in HBSS via the bile duct. The inflated pancreata were removed and incubated at 37° C for 14 minutes. After digestion, pancreata were washed 3 times with HBSS, resuspended in 5 ml HBSS and poured into 60 × 15 mm glass petri dishes. Islets from every mouse were hand-picked into a 1.5 ml Eppendorf tube under a dissecting microscope. After centrifugation, islets were resuspended in 750 μl of enzyme-free Cell Dissociation Buffer (Invitrogen) to obtain single cell suspensions. Cells were then washed once with HBSS, resuspended in FACS buffer and stained with the indicated fluorochrome-conjugated Abs.
Flow cytometry and antibodies
Ab staining and flow cytometry analysis protocols have been previously described (35). Whole spleen, thymus, and pancreatic lymph nodes (pLNs) from euthanized mice were dispersed into single cell suspensions using glass homogenizers after which erythrocytes were lysed using an ammonium chloride based lysis buffer and cells were counted using a BD Accuri C6 flow cytometer. Cell suspensions were Fc receptor blocked using anti-CD16/CD32 (2.4G2) from BioXCell (West Lebanon, NH). Abs purchased from BD Bioscience and eBioscience were used to stain the following surface molecules: CD8 (clone; 53-6.7), CD4 (RM4-5), TCRβ (H57-597), Ly-6G (RB6-8C5), CD70 (FR70), isotype control rat IgG2b (A95-1), CD45R (RA3-6B2), CD11b (M1/70), CD25 (PC61), CD86 (GL1), CD1d (1B1) CD27 (LG.7F9), pCDA1 (JF05-1C2.4.1) and TIGIT (GIGD7). iNKT cells were identified using PBS57-loaded and control CD1d tetramers supplied by the National Institutes of Health Tetramer Core Facility. iNKT cell cytokine staining was performed as previously described (58). Briefly, splenocytes from mice injected i.v. two hours previously with α-GalCer (4 μg per mouse) or DMSO were stained with anti-IFNγ (XMG1.2) and anti-IL-4 (11B11) after cells were surface stained with CD1d tetramer, anti-CD4, and anti-TCRβ and fixed and permeabilized with solutions from the BD Biosciences Cytofix/Cytoperm kit. Intranuclear staining was performed using the Transcription Factor Staining Buffer set from eBioscience and Abs against Foxp3 (FJK-16s) and Helios (22F6) for Tregs and PLZF (R17-809, BD Biosciences), T-bet (O4-46, BD Biosciences), and RORγt (B2D, eBiosciences) for iNKT cells. Stained cells were washed once with FACS buffer and acquired using an LSR Fortessa or BD Accuri C6 flow cytometer (BD Biosciences). Fluorescence minus one controls were used to identify positive and negative populations (Supplemental Figures 1–2). Propidium iodide or a fixable viability dye from BD Bioscience was used for exclusion of dead cells during analysis. All flow data were analyzed using FlowJo software (Version 10.5.0, Treestar, Palo Alto, CA).
Cell culture
To validate the ablation of CD70 protein expression in Cd70−/− mice, 2 × 106 splenocytes from Cd70−/− or standard NOD mice were seeded in RPMI 1640 (containing 10% FBS, 1% penicillin-streptomycin-neomycin and 1% β-mercaptoethanol) with or without 1 μg/ml lipopolysaccharide (LPS) and cultured at 37oC for 24 h. Thereafter, cells were stained with Abs against CD11c, CD86, CD70 and an isotype control for flow cytometric analysis. A previously described protocol (59) was used to compare the ability of NOD and Cd70−/− CD25+CD4+ Tregs to suppress CD25−CD4+ effector T cell proliferation. Different concentrations of Tregs were cultured with 1μg/ml anti-CD3 (145-2C11), 5 × 104 carboxyfluorescein succinimidyl ester (CFSE)-labeled NOD effector T cells and 1 × 104 CD11c+ DCs purified from Cd70−/− mice. Tregs, effector T cells, and DCs were purified using magnetic beads (Miltenyi Biotec) according to the manufacturer’s directions. Proliferation was measured after 3 days by flow cytometry.
Adoptive transfer studies
NOD.Rag1−/− mice were injected i.v. with 5 × 106 total splenocytes from 7-10-wk-old NOD and Cd70−/− mice. Blood samples were analyzed by flow cytometry weekly until recipient mice were euthanized at 3 weeks after injection. In another study, NOD.Rag1−/− or NOD.Rag1−/−.Cd70−/− mice were adoptively transferred with 5 × 106 total T cells purified from splenocytes of 6-wk-old NOD mice by magnetic bead separation. One group of recipients were monitored for T1D development until 22 weeks post-transfer, while a second cohort was euthanized at 7 weeks post-transfer to analyze Tregs in the spleen, pancreatic lymph nodes (pLNs) and pancreatic islets. In a separate experiment, NOD.Rag1−/− and NOD.Rag1−/−.Cd70−/− mice were i.v. injected with 4.5 × 106 Foxp3-negative T cells that were sorted from NOD.FOXP3-GFP-Cre.Rosa26-YFP donors using a BD FACS Aria III. Recipient mice were euthanized after the development of T1D or ulcerative colitis. Another experiment injected NOD.Rag1−/− mice with 2 × 106 splenocytes from diabetic NOD mice admixed with CD25+CD4+ Tregs purified from Cd70−/− or standard NOD splenocytes. An aliquot of each Treg preparation was stained for Foxp3 before transfer. Treg concentrations were then adjusted so that each NOD.Rag1−/− recipient received 2 × 105 Foxp3+CD25+CD4+ cells.
Bone marrow chimeras
Bone marrow (BM) cells were collected from tibias and femurs of Cd70−/− and NOD mice. T cells were removed using CD4 and CD8 Ab conjugated microbeads (Miltenyi Biotec). Cd70−/− and NOD mice were lethally irradiated (6 Gy 2 h apart) and injected intravenously with 5 × 106 purified BM cells from Cd70−/− and NOD donors in a crisscross design. After 8 weeks, thymi, spleens and pLNs were collected and analyzed by flow cytometry.
Anti-CD27 Ab treatment
Starting at 5-6 weeks of age, female Cd70−/− mice were intraperitoneally (i.p.) injected twice a wk with PBS or 50 μg of anti-CD27 (AT-124) provided by Celldex Therapeutics, Inc. Hapmton, NJ (60). Standard NOD mice were treated with PBS as a control. Mice were treated for 4 wk after which their spleens and thymi were removed for analysis by flow cytometry.
Statistical analyses
Wilcoxon matched-pairs signed rank test, Mann-Whitney U test, or two-way ANOVA were used for comparisons among groups as indicated. Log-rank test was used for the comparison of T1D incidence. All statistical analyses were performed using GraphPad Prism, version 8.0 (GraphPad software, Inc., La Jolla, CA).
Results
Generation of Cd70−/− mice
A pair of TALENs designed to target the Cd70 gene transcriptional start site were delivered by pronuclear injection into NOD zygotes. TALEN off-target mutations were avoided by choosing a unique target sequence that differed from any other site in the genome (61). Ninety-nine offspring were obtained and analyzed of for mutations in the Cd70 gene. Of these, four mice were identified with deletions capable of disrupting CD70 expression, including a male with a 363 bp deletion that was selected as the founder for the CD70 knockout line. This mouse was backcrossed to standard NOD females to verify germline transmission of the mutant allele (Figure 1A) generating the line NOD/ShiLtJ-Cd70<em1Mvw>/Mvw (stock no.24167). Subsequent generations from this breeding scheme produced a Mendelian inheritance pattern of mutant and wild-type alleles, indicating that the mutation was introduced into the embryo at the one cell stage. CD70 protein ablation was confirmed by flow cytometry using LPS-stimulated splenic DCs isolated from homozygous mutant and WT NOD mice (Figure 1B). We also confirmed that CD70 deletion does not affect the overall ability of APCs to mature, as mutant DCs retained their ability to upregulate the T cell co-stimulatory molecule CD86 in response to LPS stimulation (Figure 1C & D).
Figure 1.
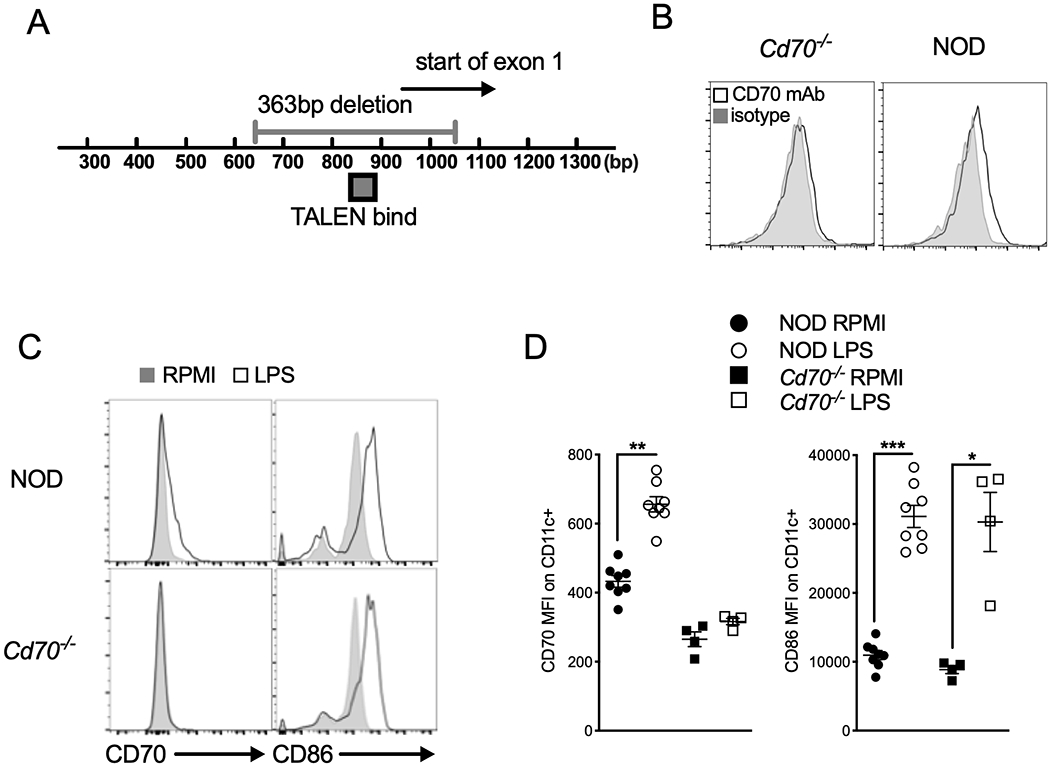
Cd70 gene disruption in NOD mice using transcription activator-like effector nucleases (TALENs). (A) Genomic map showing the TALEN binding site used to create a 363 bp deletion in Exon 1. Numbers below the bar represent the base-pair (bp) location from the Cd70 start site. (B) Absence of CD70 expression confirms its deletion in Cd70−/− mice. Splenocytes from Cd70−/− and standard NOD mice were incubated for 24 h with 1 μg/ml LPS, after which cells were harvested and analyzed for CD70 protein expression by flow cytometry. Representative histograms show CD70 and isotype control staining on CD11c+ DCs. (C & D) Upregulation of CD86 is not affected by Cd70 ablation. Flow cytometric analysis of CD70 and CD86 expression on CD11c+ cells among Cd70−/− and WT NOD splenocytes cultured with complete media alone (RPMI) or LPS for 24 hours. (C) Representative histograms show the expression of CD70 and CD86 on CD11c+ gated cells. (D) Graphs show summarized data for MFI of CD70 and CD86 Ab staining on CD11c+ DCs. Results are shown as mean ± SE from 4–8 individual mice per group. Statistical analysis was carried out by Wilcoxon matched-pairs signed rank test; *P < 0.05, **P < 0.01, ***P < 0.001.
CD70 deficiency accelerates T1D in NOD mice
Male and female NOD mice homozygous and heterozygous for mutant and WT Cd70 alleles were monitored for the development of insulitis and T1D. Cd70−/− mice developed more severe insulitis and accelerated diabetes compared to WT littermates (Figure 2A & B). Diabetes in heterozygous mice was intermediate between WT and Cd70−/− mice, suggesting that disease is partially suppressed by one copy of the WT Cd70 allele. These results show that CD70 elicits immune effects that limit T1D development.
Figure 2.
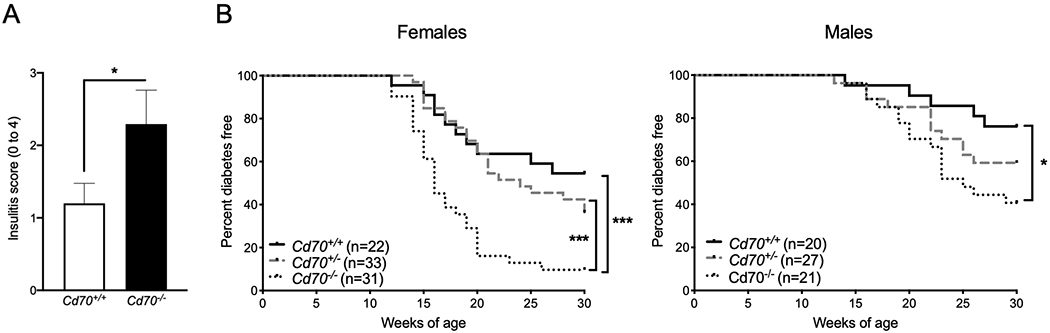
Cd70 ablation exacerbates insulitis and accelerates T1D onset in NOD mice. (A) Mean insulitis scores (0 = no insulitis to 4 = no remaining islet cell mass) in 8- to 10-week-old Cd70−/− and Cd70+/+ female NOD mice. Insulitis severity was significantly greater in Cd70−/− than Cd70+/+ mice according to the Mann-Whitney test. (B) The incidence of T1D in female and male littermates of the indicated genotypes. Survival curves were compared by the Log-rank test. ***P < 0.001, *P < 0.05.
Disrupting CD70 alters thymocyte development and peripheral T cell levels
Splenocytes and thymocytes from 8- to 10-week-old sex-matched Cd70+/+, Cd70+/− and Cd70−/− mice were analyzed by flow cytometry to determine the effect of CD70 ablation on immune cell populations, including subsets that contribute to T1D pathogenesis. No differences were found in the frequency or number of total lymphocytes, DCs, and granulocytes (Table 1). However, both the frequency and number of splenic αβ T lymphocytes were increased in Cd70−/− mice compared to Cd70+/− and Cd70+/+ mice (Figure 3A & B). The ratio of CD4 and CD8 T cells was similar among mice of the different genotypes (data not shown). Cd70−/− mice had a lower frequency of splenic B cells compared to Cd70+/+ and Cd70+/− mice, however, the number of B cells was similar among the different genotypes (Figure 3A & B). CD70 ablation affected thymocyte development by respectively increasing and decreasing the frequency of the DN and DP subsets compared to CD70-intact NOD mice (Figure 3C–E). Numerically, DP and CD4 single positive (SP) thymocytes were lower in CD70-deficient versus -intact mice. These collective findings indicate that CD70 regulates thymic T cell development and peripheral T cell levels.
Table 1.
Flow cytometric analysis of leukocyte populations in spleens of 8- to 10-week-old female (F) and male (M) NOD mice carrying none, one or both copies of Cd70 mutant gene.
| Cd70+/+ F | Cd70+/− F | Cd70−/− F | Cd70+/+ M | Cd70+/− M | Cd70−/− M | |
|---|---|---|---|---|---|---|
| Lymphocytes (x107) | 7.24±0.45 | 6.57±0.57 | 5.64±0.51 | 5.98±0.35 | 5.59±0.24 | 5.85±0.60 |
| Conventional DC (%) | 1.91±0.13 | 1.93±0.12 | 1.94±0.13 | 1.23±0.05 | 1.68±0.11 | 1.57±0.14 |
| Conventional DC (x106) | 0.83±0.08 | 0.93±0.12 | 1.16±0.06 | 0.60±0.04 | 0.77±0.05 | 0.84±0.05 |
| Plasmacytoid DC (%) | 1.47±0.09 | 1.41±0.06 | 1.38±0.15 | 1.06±0.05 | 1.11±0.10 | 0.96±0.02 |
| Plasmacytoid DC (x106) | 0.62±0.05 | 0.66±0.09 | 0.92±0.12 | 0.50±0.03 | 0.49±0.05 | 0.51±0.04 |
| Eosinophils (%) | 7.82±1.11 | 7.90±0.60 | 8.37±1.24 | 5.86±0.28 | 4.99±0.44 | 5.93±0.95 |
| Eosinophils (x106) | 0.12±0.02 | 0.13±0.02 | 0.16±0.03 | 0.08±0.01 | 0.08±0.01 | 0.13±0.02 |
| Macrophages/monocytes (%) | 38.66±1.84 | 36.64±1.82 | 40.85±1.63 | 50.28±2.34 | 49.70±1.14 | 47.82±1.29 |
| Macrophages/monocytes (x106) | 0.61±0.07 | 0.64±0.04 | 0.87±0.15 | 0.68±0.06 | 0.79±0.06 | 1.01±0.05 |
| Neutrophils (%) | 48.77±1.91 | 50.85±2.24 | 45.70±1.36 | 40.42±2.53 | 42.10±0.96 | 42.43±1.37 |
| Neutrophils (x106) | 0.75±0.06 | 0.89±0.09 | 0.95±0.10 | 0.55±0.07 | 0.67±0.05 | 0.89±0.03 |
Single-celled, live lymphocytes were identified according to size and granularity. Dendritic cells (DCs) were identified as CD11c+ cells after gating single live leukocytes. Different DCs subsets were distinguished from each other according to pCDA1 surface expression. Eosinophils, neutrophils, macrophages and monocytes were identified as CD11b+ single live leukocytes with respectively high and low granularity according to side scatter and surface expression of Ly6G. The number of immune cells were determined by calculating the absolute number of each cell type from fluorescent-activated cell analysis profiles and total cell numbers in the spleen. Values represent mean ± SEM. At least 10 individual mice were examined per group. No significant differences (p > 0.05) were detected between the indicated genotypes for any of the immune cell populations according to the Mann-Whitney test.
Figure 3.
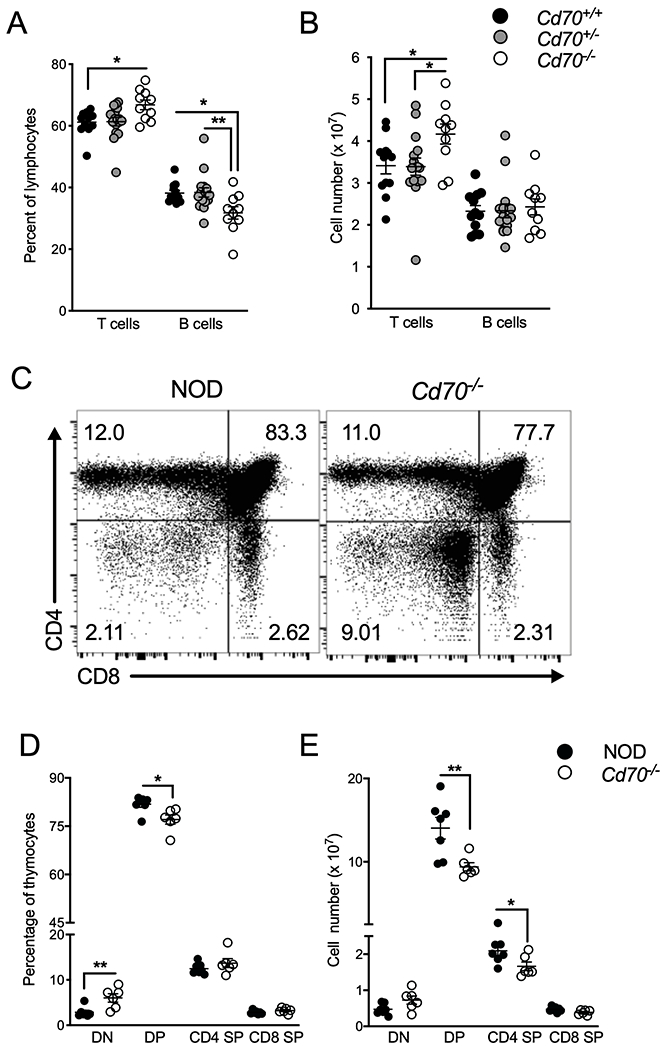
Peripheral T cell levels and thymocyte development are altered by Cd70 deletion. The frequency (A) and number (B) of splenic αβ T cells and B cells from 8- to 10-week-old Cd70+/+, Cd70+/− and Cd70−/− mice. (C) Representative FACS plots showing thymocyte subsets in NOD and Cd70−/− mice. The frequency (D) and number (E) of CD4/CD8 DN, DP and SP thymocytes from 7- to 8- week-old NOD and Cd70−/− mice. Data represent results from four independent experiments analyzed by the Mann-Whitney test; error bars correspond to mean ± SEM. *P < 0.05; **P < 0.01.
CD70 deficiency impairs Helios+ Treg generation
To establish whether CD70 ablation affects CD4+Foxp3+ Tregs that are important for suppressing diabetogenic T cell responses, CD70-deficient and -intact NOD mice were compared for Treg levels. The frequency of Tregs was significantly reduced in thymus, spleen, pLNs, and blood of CD70-deficient mice (Figure 4A). Furthermore, Tregs were numerically reduced in the thymus and spleen of CD70-deficient compared to -intact mice (Figure 4B). Splenic Tregs from Cd70−/− mice expressed less Foxp3 compared to CD70-intact mice (Figure 4C). However, they expressed more of the immune receptor T cell Ig and ITIM domain (TIGIT) in the thymus and spleen and more CD25 in the thymus (Figure 4D & E). To determine whether CD70 is involved in maintaining the stability of Tregs, thymic and splenic Tregs were co-stained with an Ab against the transcription factor Helios, which is expressed very early in Foxp3+Treg development and is required for maintaining the anergic and suppressive phenotype of these cells (62). We found that the Treg reduction in Cd70−/− mice was due to a decrease in Helios+ and not Helios− Tregs, suggesting that Treg stability is affected by CD70 ablation (Figure 4F & G). Next, we assessed if CD70 deletion impacts the development of β-cell islet-specific Tregs, as antigen-specific Tregs play an important role in suppressing diabetogenic immune responses (63). Tregs were analyzed from CD70-deficient and -intact NOD mice transgenically expressing the TCR from the BDC2.5 CD4 T cell clone and also Green Fluorescent Protein off the Foxp3 promoter (NOD.BDC2.5.Foxp3eGFP). BDC2.5 clonotypes make up >80 percent of T cells in NOD.BDC2.5 TCR transgenic mice (64). The frequency of Tregs was significantly reduced in the thymus, spleen and pLN of CD70-deficient than -intact NOD.BDC2.5.Foxp3eGFP mice (Figure 4H). This stock also had reduced numbers of Tregs in the thymus and pLN, but not the spleen (Figure 4I), which is likely because CD70-deficient NOD.BDC2.5.Foxp3eGFP mice had larger spleens than their CD70-sufficient counterparts (data not shown). Interestingly, ablation of CD70 led to a greater reduction in the frequency of Tregs in NOD.BDC2.5.Foxp3eGFP than nontransgenic NOD mice (Figure 4A vs. 4H). These results show that CD70 is important for the development of Tregs, including clonotypes that recognize β-cell antigens, and that CD70 promotes the generation of Helios+ Tregs. The reduced number of Tregs in Cd70−/− mice may underlie why this strain has higher levels of circulating T cells compared to standard NOD mice (Figure 3A & B). Thus, young 3-week-old mice were analyzed to determine whether increased T cell levels develop downstream of or alongside reduced Treg levels in Cd70−/− compared to standard NOD mice. We found that the number of total splenic T cells and Tregs were respectively similar and reduced between Cd70−/− and NOD mice (Figure 4J & K). This result suggests that the higher levels of T cells in Cd70−/− mice are not the consequence of altered thymic development, but rather from impaired peripheral homeostasis, which is probably due to fewer Tregs.
Figure 4.
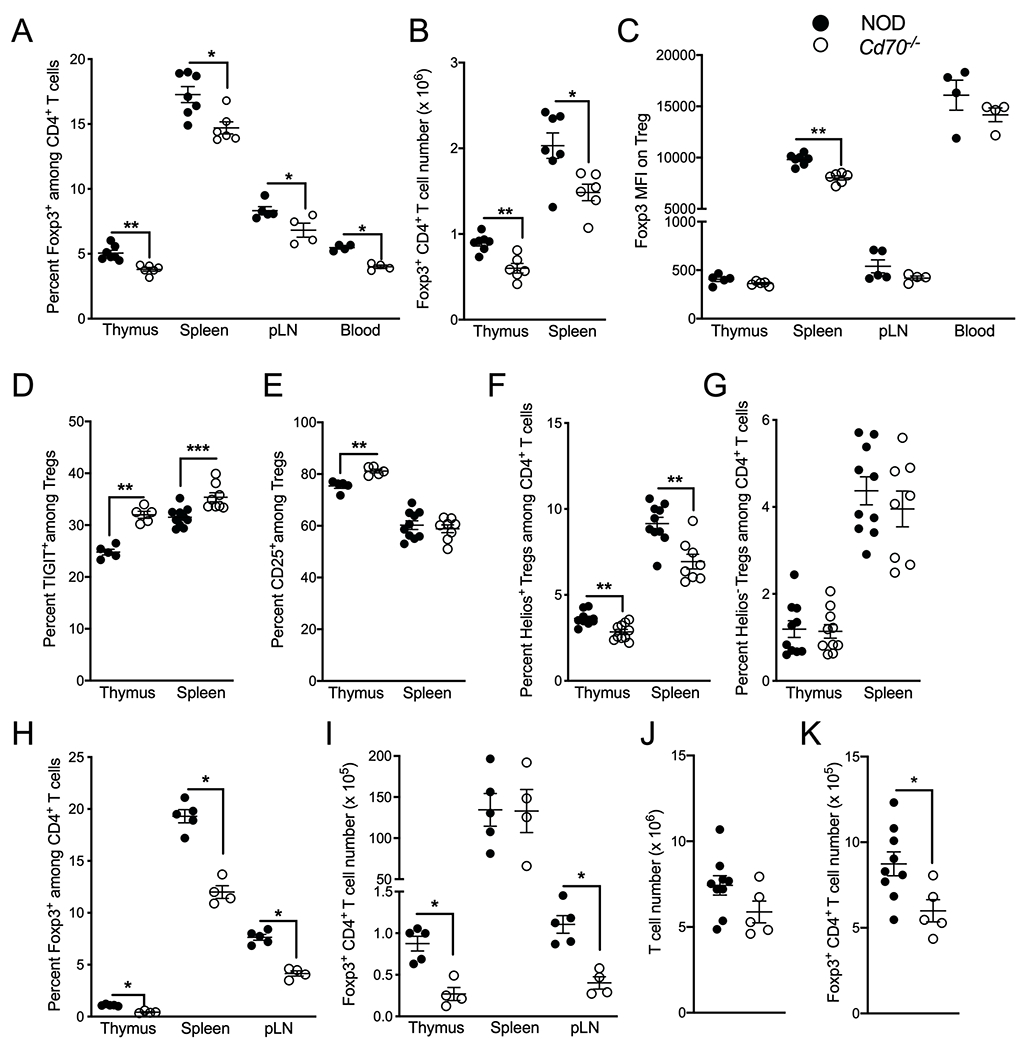
CD70 deficiency impairs Helios+ Treg development. (A) The proportion CD4+ T cells that express Foxp3 among thymocytes, splenocytes, pancreatic lymph nodes (pLNs) and blood of 7- to 10-week-old NOD and Cd70−/− mice. (B) The number of Foxp3+CD4+ Tregs from thymus and spleen of NOD and Cd70−/− mice. (C) The MFI of Foxp3 Ab staining of Tregs among thymocytes, splenocytes, pLN and blood of NOD and Cd70−/− mice. (D & E) Proportion of thymic and splenic Tregs that express TIGIT (D) and CD25 (E). (F & G) Percentage of Helios+ Tregs (F) and Helios− Tregs (G) among thymic and splenic CD4+ T cells of NOD and Cd70−/− mice. (H & I) The proportion CD4+ T cells that express Foxp3 (H) and the number of Foxp3+CD4+ Tregs (I) from thymus, spleen, and pLN of 8-week-old BDC2.5 TCR transgenic NOD and Cd70−/− mice. (J & K) The number of T cells (J) and Foxp3+CD4+ Tregs (K) from spleens of 3-week-old NOD and Cd70−/− mice. Each symbol represents a single mouse. Results are displayed as mean ± SEM and represent cumulative data from at least two independent experiments that were analyzed by the Mann-Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001.
CD70 regulates Treg development through a hematopoietic cell type
Both hematopoietic and non-hematopoietic cell types are required for intrathymic Treg generation (65). To investigate whether one or both of these compartments affects Treg development through CD70 signaling, we created chimeric mice by transferring T cell-depleted NOD or Cd70−/− BM into lethally irradiated NOD or Cd70−/− recipients in a criss-cross experimental design. Analysis of mice 8 weeks after reconstitution found that recipients of Cd70−/− BM had lower frequencies of thymic and splenic Tregs than recipients of WT BM (Figure 5A). However, it was only in Cd70−/− recipients of Cd70−/− BM that the reduction in Tregs was statistically lower than mice that received NOD BM. Tregs were numerically lower in the thymus and spleen of Cd70−/− BM recipients compared to NOD recipients (Figure 5B). Further analysis found that the frequency and number of Helios+ and Helios− Tregs was lower in recipients of Cd70−/− than NOD BM (Figure 5C & D). These results indicate that CD70 signaling supports Treg development, including Helios+ and Helios− subsets, through a hematopoietic rather than non-hematopoietic cell type(s), although non-hematopoietic cells appear to make some contribution.
Figure 5.
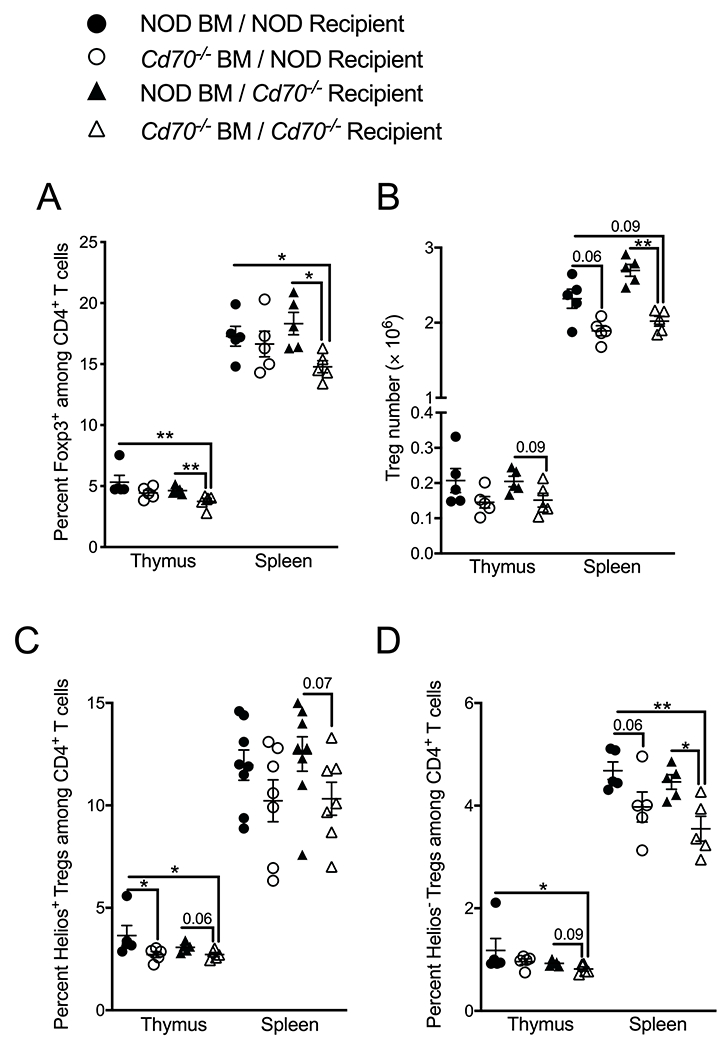
CD70 expression by hematopoietic rather than non-hematopoietic cell types contribute to thymic Treg development. NOD and Cd70−/− mice were lethally irradiated and reconstituted with BM cells from CD70-deficient or -intact mice. Thymus and spleen were analyzed eight weeks after BM reconstitution for the (A) frequency and (B) number of total Tregs as well as the frequency of Helios+ Tregs (C) and Helios− Tregs (D). The results represent two independent experiments with 3 to 5 mice per group. Each symbol represents an independent biological replicate comprised of a single recipient mouse reconstituted with BM from a single donor animal. Data were analyzed by the Mann-Whitney test. *P < 0.05, **P < 0.01. Actual P valves are provided when 0.1>p-value>0.05.
CD70 deficiency does not inherently alter Treg suppressor function
Because Tregs from Cd70−/− mice express less Foxp3 but more TIGIT and CD25 than WT Tregs (Figure 4C–E), we tested whether CD70 and WT Tregs have different effector functions. On a per cell basis, Tregs from NOD or Cd70−/− mice equally suppressed the activation of conventional NOD CD25−CD4+ T cells in an in vitro suppression assay that used DCs from CD70-deficient mice as APCs (Figure 6A & B). To determine whether the suppressor function of Tregs is fundamentally altered when these cells develop in the absence of CD70 signaling, NOD.Rag1−/− mice were injected with either NOD or Cd70−/− Tregs and diabetic NOD splenocytes. T1D developed at a similar rate in both groups demonstrating that the suppressive functions of NOD and Cd70−/− Tregs were comparable (Figure 6C). The frequency and number of splenic Tregs at diabetes onset was similar between groups (Figure 6D & E), indicating that the experiment was not affected by changes in Treg levels after adoptive transfer. Together, these results demonstrate that Tregs isolated from Cd70−/− and NOD mice are functionally equivalent and that thymic development in the absence of CD70 does not inherently alter the capacity of Tregs to suppress conventional T cell responses.
Figure 6.
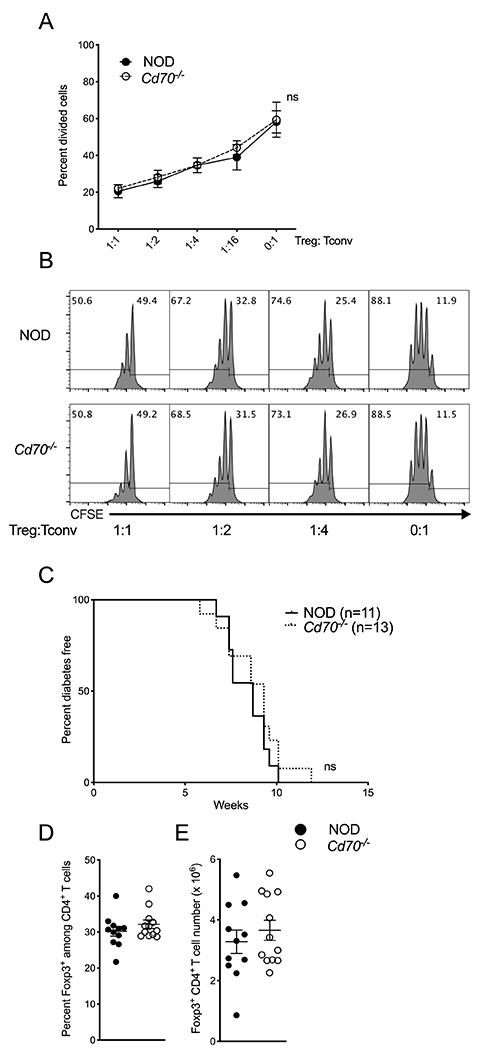
CD70-deficient Tregs possess normal suppressor function. (A & B) An in vitro Treg suppression assay comparing the suppressor activity of NOD and Cd70−/− Tregs. Splenic CD4+CD25+ cells isolated from NOD or Cd70−/− mice were cocultured at the indicated ratios with 5 × 104 carboxyfluorescein succinimidyl ester (CFSE)-labeled NOD CD4+CD25− T cells, 1 × 104 Cd70−/− CD11c+ dendritic cells and 1μg/ml anti-CD3 Ab. (A) The proportion of CD4+CD25− cells which underwent at least one round of cell division was measured after 3 days. Suppression assays were done in technical triplicates. Combined data from three independent experiments are shown and results are displayed as mean ± SEM with each symbol representing the mean of three experiments. (B) Representative flow cytometric profiles of responder cell CSFE dilution in the presence of the indicated sources of Tregs at the indicated ratios. (C) T1D incidence for NOD.Rag1−/− mice co-transferred with 2 × 106 diabetic NOD splenocytes and 2.5 × 105 Foxp3+ Tregs from standard NOD or Cd70−/− mice. (D & E) The frequency (D) and number (E) of splenic Tregs from recipient mice after diabetes development. Results are displayed as mean ± SEM with each symbol representing a single mouse. Statistical differences were analyzed by the Mann-Whitney test. Survival curves were compared by log-rank test. ns, not significant.
Peripheral CD70 expression regulates Treg levels and T1D development
To determine whether accelerated T1D in Cd70−/− mice is associated with the reduced level of Tregs in this strain, we adoptively transferred 5 ×106 splenocytes from CD70-deficient or -intact mice into CD70-sufficient NOD.Rag1−/− mice. As expected, freshly isolated splenocytes from Cd70−/− donors had a lower frequency of Tregs than WT splenocytes (Figure 7A). However, analysis of peripheral blood from recipient mice starting from 6 days post transfer, found that Tregs from Cd70−/− and WT donors had expanded to the same level (Figure 7B). Since this study was no longer useful to investigate an association between Treg levels and T1D susceptibility, all recipient mice were sacrificed at 3 weeks after injection. No differences were found in the frequency or number of splenic Tregs between Cd70−/− and WT recipients (Figure 7C & data not shown). Our finding that Cd70−/− Tregs are capable of increasing to WT levels when transferred in to a CD70-intact environment indicates that the Treg deficit of Cd70−/− mice is due to an absence of CD70 signaling from an extrinsic source rather than from a Treg-intrinsic defect. To establish if CD70 expressed by a non-lymphoid cell population is responsible for controlling circulating Treg levels, NOD.Rag1−/−.Cd70−/− and NOD.Rag1−/− mice were analyzed 7 to 10 weeks after injection with purified T cells from standard NOD mice. Tregs were significantly reduced in spleen, pLN and pancreatic islet infiltrates of NOD.Rag1−/−.Cd70−/− mice compared to NOD.Rag1−/− recipients (Figure 7D). This was due to a decrease in Helios+ rather than Helios− Tregs (Figure 7E). Unlike Tregs, CD4+ and CD8+ T cell levels were similar between treatment groups (Figure 7F & G). In a separate cohort of mice, we found that T1D was accelerated in NOD.Rag1−/−.Cd70−/− versus NOD.Rag1−/− NOD T cell recipients (Figure 7H), which was not affected by CD27 expression on Tregs (Supplemental Figure 3A). These results show that CD70 expressed by a non-lymphoid population of cells sustains circulating Helios+ Treg levels and inhibits T1D development. To address the possibility that differences in T1D progression in Cd70−/− mice and NOD mice were due to differential Treg levels, we asked whether in the absence of Tregs the two strains would develop a similar rate of disease. NOD.Rag1−/−. Cd70−/− and NOD.Rag1−/− mice were adoptively transferred with T cells from NOD.FOXP3-GFP-Cre.Rosa26-YFP mice after flow cytometric depletion of Foxp3+Tregs and cells that had at some point expressed Foxp3. We found that without these cells, Cd70−/− mice did not develop accelerated T1D compared to WT mice (Figure 7I). Indeed, while all NOD.Rag1−/− mice succumbed to T1D by four weeks post transfer, two out of four NOD.Rag1−/−.Cd70−/− recipients remained T1D-free until seven weeks post transfer when they developed ulcerative colitis instead. This result indicates that Tregs are required for a non-lymphoid population of cells to suppress T1D through CD70 signaling.
Figure 7.
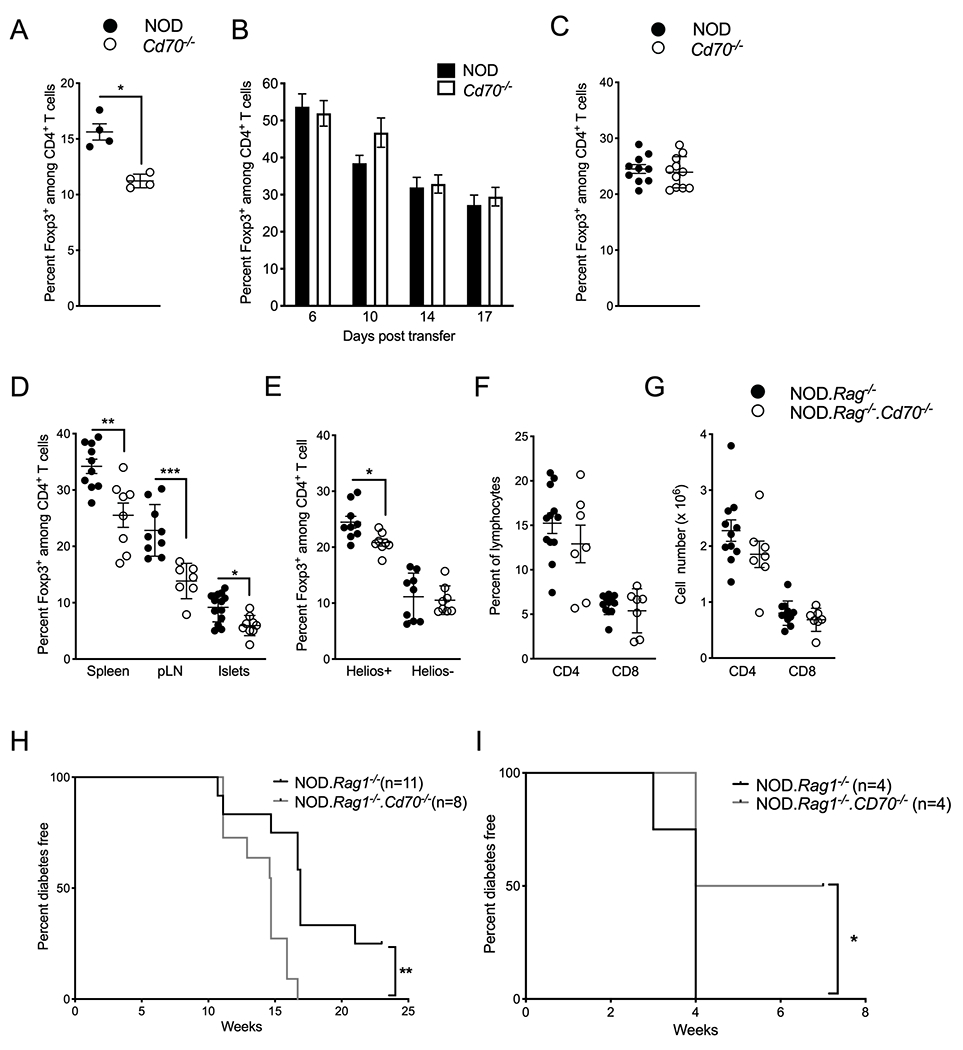
CD70 expression by a non-lymphoid cell population(s) regulates peripheral Treg levels and T1D development. (A-C) Tregs were analyzed in 7- to 10-week-old female NOD.Rag1−/− mice adoptively transferred with 2 × 106 splenocytes from NOD or Cd70−/− donors. (A) Frequency of Foxp3+ cells among CD4+ T cells in NOD or Cd70−/− donor splenocytes before adoptive transfer. (B) Frequency of Foxp3+ cells among peripheral blood CD4+ T cells of recipient mice at the indicated time points. Results are displayed as mean ± SEM for 10 and 11 recipients of NOD and Cd70−/− splenocytes, respectively. (C) Frequency of Foxp3+ cells among splenic CD4+ T cells of recipient mice 3 weeks after adoptive transfer. (D) The proportion Foxp3+ CD4+ T cells in the spleen, pLN, and pancreatic islet infiltrates of NOD.Rag1−/− and NOD. Rag1−/−.Cd70−/− mice 7 to 10 weeks post-adoptive transfer with 5 × 106 NOD T cells. (E) Percentage of Helios+ and Helios− Tregs among splenic CD4+ T cells of NOD.Rag1−/− and NOD. Rag1−/−.Cd70−/− recipient mice. (F) Frequency and (G) number of CD4+ and CD8+ T cells among splenocytes of NOD.Rag1−/− and NOD. Rag1−/−.Cd70−/− recipient mice. (H) Incidence of T1D in NOD.Rag1−/− and NOD.Rag1−/−.Cd70−/− mice injected with 5 × 106 T cells isolated from 6- to 8-wk old female NOD mice. (I) T1D incidence of NOD.Rag1−/− and NOD.Rag1−/−.Cd70−/− mice injected with 5 × 106 T cells isolated from 6- to 8-wk old NOD.FOXP3-GFP-Cre.Rosa26-YFP females after depletion of Foxp3+ cells and cells that had at some point expressed Foxp3. Recipients were analyzed for the development of T1D or until the mice developed ulcerative colitis. Results are displayed as mean ± SEM with each symbol representing a single mouse, unless otherwise indicated. Statistical differences were analyzed by the Mann-Whitney test. Survival curves were compared by Log-rank test. *P < 0.05, **P < 0.01, ***P < 0.001.
iNKT cell differentiation, subsets and effector responses are regulated by CD70
Invariant NKT cells are another immunoregulatory T cell population that modulates T1D development in the NOD mouse. Since T cell costimulatory molecules regulate TCR signaling which directs lineage determining factors, such as Foxp3 for Tregs, we asked whether CD27-CD70 interactions affect the development of iNKT cells. iNKT cells were quantified in Cd70+/+, Cd70+/− and Cd70−/− littermates using a mouse PBS57-loaded CD1d tetramer. Thymic and splenic iNKT cells were proportionally and numerically higher in Cd70−/− than Cd70+/+ mice, while iNKT cell levels in Cd70+/− mice were respectively similar to Cd70+/+ and Cd70−/− mice in the thymus and spleen (Figure 8A & B). Previous reports showed an inverse relationship between CD1d expression on mouse DP thymocytes and the frequency of thymic iNKT cells (66–68). Therefore, we analyzed littermates of all three genotypes for CD1d expression. We observed that CD70 expression was correlated to CD1d expression (Figure 8C), which subsequent reciprocal bone marrow transfer experiments identified is due to CD70 signaling from a non-lymphoid source more than the lymphoid compartment (Figure 8D). Next, we assessed how CD70 ablation affects the development of thymic iNKT cell lineages that segregate into iNKT1, iNKT2 and iNKT17 subsets according to the differential expression of transcription factors T-bet, PLZF, and RORγt (69). The absence of CD70 respectively increased and decreased the frequency of iNKT1 and iNKT2 cells (Figure 8E–G) so that the iNKT1:iNKT2 ratio was ~8:1 in Cd70−/− mice compared to ~3:1 in WT mice. CD70 deficiency had no effect on the frequency of iNKT17 cells, nor did it alter the ratio of CD4+ and CD8−CD4− DN iNKT cells in central and peripheral lymphoid organs (data not shown). To test if CD70 deficiency alters iNKT cell function, splenic iNKT cells from NOD and Cd70−/− mice were measured for IFNγ and IL4 production 2 h after injection with the iNKT cell superagonist α-galactosylceramide (α-GalCer). A higher frequency and number of IFNγ and IL4 positive iNKT cells was detected in Cd70−/− mice (Figure 8H & I). We also found that Cd70−/− iNKT cells produced more cytokines than NOD iNKT cells on a per cell basis, (Supplemental Figure 1E). These results suggest that CD70 signaling decreases the fraction of iNKT cells that become activated after stimulation while also limiting the amount of cytokines they produce. Because Tregs have been reported to negatively regulate iNKT cell development (70), we measured iNKT cells in standard NOD mice after Treg depletion using an anti-CD25 Ab. iNKT cell levels did not change despite a large decrease in Tregs (Supplemental Figure 4), which suggests that reduced Treg levels is not the underlying reason why Cd70−/− mice produce more iNKT cells. In summary, these data indicate that CD70 inhibits iNKT cell development, limits the differentiation of iNKT1 cells in favor of iNKT2 cells, and reduces iNKT cell effector functions after activation by a strong cognate ligand.
Figure 8.
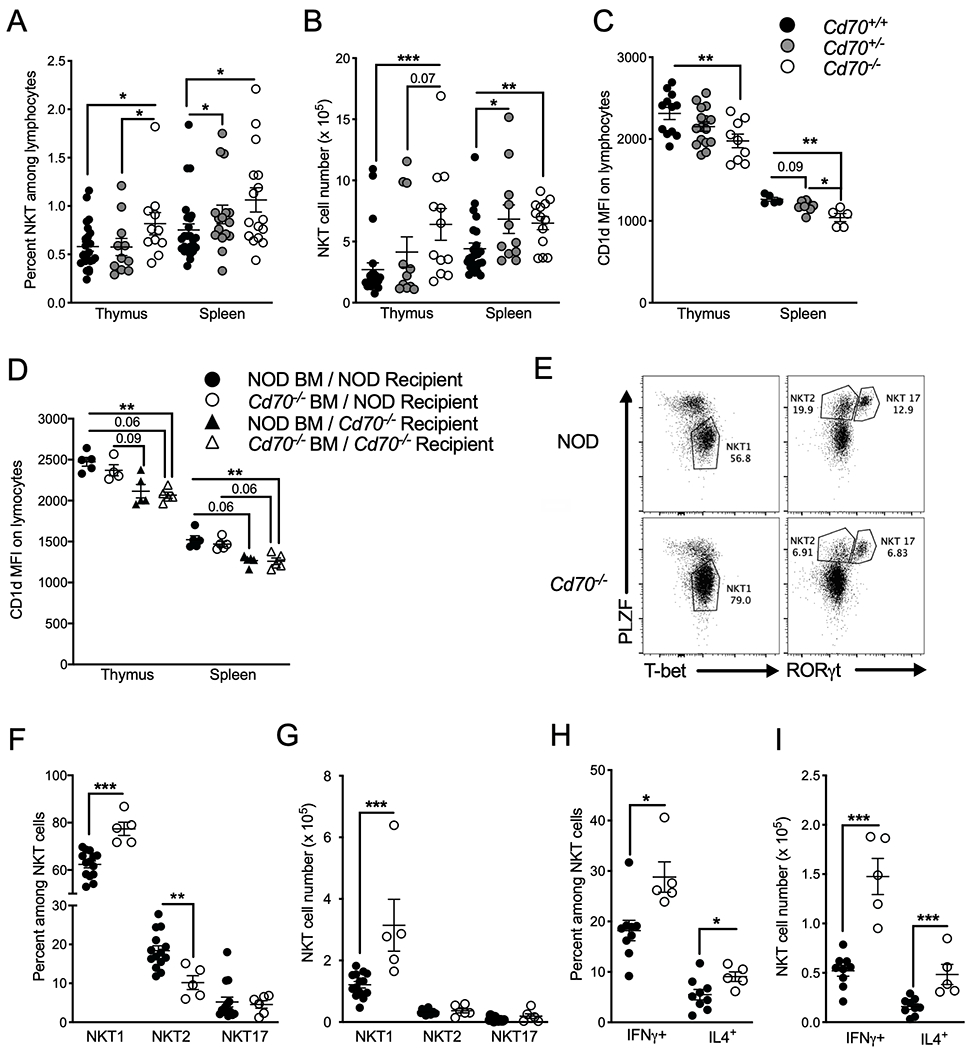
CD70 regulates iNKT cell development and function. The frequency (A) and number (B) of thymic and splenic CD1d tetramer+ TCRβ+ iNKT cells in 7- to 10-wk old female mice of the indicated genotypes. (C) MFI of CD1d Ab staining of thymocytes and splenocytes from 7- to 10-wk old female mice of the indicated genotypes. (D) Thymic and splenic CD1d expression in NOD and Cd70−/− mice 8 weeks after lethal irradiation and reconstitution with BM cells from CD70-deficient or -intact mice. (E) Characterization of thymic iNKT cell subsets by flow cytometric analysis of the key transcription factors PLZF, T-bet, and RORγt. (F) Frequency and (G) number of iNKT1 (T-bet+ PLZFlo), iNKT2 (PLZFhi RORγt−) and iNKT17 (PLZFhi RORγt+) iNKT cells from 7- to 8-week-old female NOD and Cd70−/− thymi. (H & I) Intracellular IFNγ and IL4 staining of iNKT cells resident in spleens of 7- to 8-wk old NOD and Cd70−/− mice injected i.v. 2 h previously with 4 μg per mouse of α-GalCer or DMSO. (H) Frequency and (I) number of IFNγ and IL4 producing iNKT cells after α-GalCer treatment. Data represent results from two independent experiments analyzed by Mann-Whitney test; error bars correspond to mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Actual P values are provided when 0.1>p-value>0.05.
CD27 agonist Ab treatment alters Treg and iNKT cell levels and reduces insulitis
Most of the effects of CD70 have been attributed to intracellular signaling events induced by CD27. However, experimental evidence exists that CD70 ligation can evoke reverse signaling cascades that regulate the expansion and differentiation of immune cells (20, 71). To determine whether the altered iNKT cell and Treg phenotypes in Cd70−/− mice are due to a lack of CD70 or CD27 signaling, we injected Cd70−/− mice for four weeks with a CD27 agonist Ab (clone AT-124). The treatment downregulated CD27 expression on Tregs, CD4+ T cells, and CD8+ T cells, which was probably a response to chronic CD27 stimulation (Supplemental Figure 3B). Ab treatment increased the frequency, but not the number, of thymic Tregs compared to control mice (Figure 9A & B). We observed a similar but more striking effect in the spleen where Ab treatment increased Treg numbers beyond those found in NOD mice. Analysis of Treg subsets found that anti-CD27 treatment respectively increased and did not affect the frequency of Helios+ and Helios− Tregs (Figure 9C & D). In contrast to Tregs, anti-CD27 Ab treatment decreased the number of thymic iNKT cells, while splenic iNKT cell frequency and numbers were reduced below standard NOD mouse levels (Figure 9E & F). We also found that anti-CD27 treatment increased CD1d expression compared to PBS treated Cd70−/− mice, but only in the spleen (Figure 9G). To determine whether the immune responses elicited by anti-CD27 Ab administration would reduce islet inflammation in Cd70−/− mice, pancreata from the different experimental groups were examined for insulitis levels after four weeks of treatment. Insulitis was significantly lower in Cd70−/− mice treated with anti-CD27 compared to PBS (Figure 9H). However, anti-CD27 Ab administration failed to suppress the onset of T1D in either CD70-deficient or -intact mice when injections were continued until 30 weeks of age (not shown). This is likely because AT-124 eventually lost its effect on Tregs that were found at similar levels between anti-CD27 Ab and control treated mice at the end of the study. Overall, these results support that the altered levels of immunoregulatory cell populations and islet inflammation observed in Cd70−/− mice is due to an absence of CD27 intercellular signaling rather than from a lack of reverse signaling from CD70 ligation.
Figure 9.
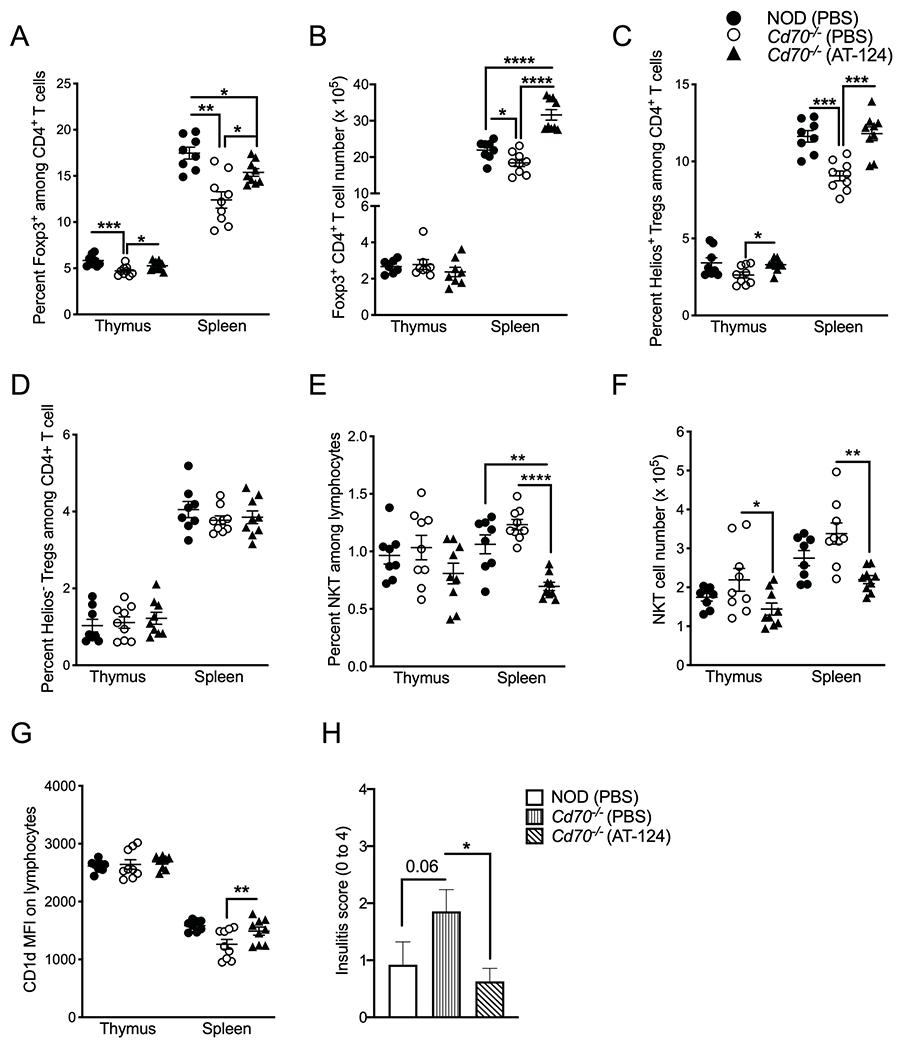
Administration of CD27 agonist Ab inversely regulates Treg and iNKT cell levels and reduces insulitis. Starting at five weeks of age, NOD and Cd70−/− mice were i.p. injected with 50 μg CD27 agonist Ab (AT-124) or PBS twice a wk. After four weeks, thymi and spleens were analyzed for Tregs, iNKT cells and CD1d expression. (A) The frequency and (B) number of thymic and splenic Foxp3+ CD4+ T cells. The frequency of thymic and splenic (C) Helios+ Tregs (D) and Helios− Tregs. (E) The frequency and (F) number of thymic and splenic CD1d tetramer+ iNKT cells. (G) MFI of CD1d Ab staining on thymocytes and splenocytes. (H) Mean insulitis scores at the end of 4 weeks of treatment in the indicated mice (n ≥ 8). Statistical differences were analyzed by the Mann-Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Actual P valves are provided when 0.1>p-value>0.05.
Discussion
Previous studies which examined the contribution of the CD27-CD70 costimulation axis to autoimmunity have found that this pathway often plays Janus-like opposing roles in different disorders (25, 27–29, 72). This is likely because individual CD27-expressing cell types, such as T cells, make distinct contributions to these various diseases. The current study is the first, to our knowledge, to describe the contribution of the CD27-CD70 axis for T1D. This disease develops primarily from defects in T cell tolerance mechanisms. These defects include the aberrant apoptosis-mediated deletion of autoreactive T cells (73, 74) and the impaired ability of immunoregulatory populations, such as Tregs and iNKT cells, to limit the development and/or activation of pathogenic T lymphocytes (30–34). While CD27 signaling modulates both conventional and immunoregulatory T cells, our finding that CD70 ablation exacerbates T1D indicates that the combined effects of CD27-CD70 costimulation favors tolerogenic over pathogenic immune responses in this disease.
We focused on how CD70 regulates T lymphocyte subsets because we found that the T cell compartment was significantly altered in CD70-deficient NOD mice. Our observation that CD70 ablation reduced the number of DP thymocytes is consistent with a previous report that CD27 mAb blockade in 129Sv mixed background mice impedes pre-T cell expansion and differentiation, which resulted in a specific decrease in the absolute number of DP thymocytes without affecting the size of the DN compartment (75). In contrast, studies using B6 mice lacking either CD27 or CD70 found normal steady-state immune compartments including thymocyte subsets (22, 76). This discrepancy might be due to the genetic background of the mice employed as several negative selection-associated and pro-survival genes are reported to be improperly regulated during negative selection in the NOD mouse (77–80). The altered thymic selection in CD70-deficient NOD mice did not underlie why peripheral T cell levels are increased in this strain. Rather, this phenomenon is probably because CD70 ablation impairs the development of Tregs that usually control peripheral T cell expansion. These higher circulating T cell levels may contribute to why T1D onset is more aggressive in CD70-deficient NOD mice.
Although several studies have shown that the CD27-CD70 pathway is involved in the development and/or the induction of Tregs (21, 76, 81, 82), no evidence has been reported that the reduced levels of Tregs in CD27- and CD70-deficient mice renders these stocks more susceptible to spontaneous autoimmunity. This is likely because knockout mice were generated on the B6 genetic background, which is a non-autoimmune prone strain. In contrast, the NOD mouse that spontaneously develops autoimmunity provides a useful model to address this question, particularly as NOD Tregs are already impaired due to a hypomorphic Il2 allelic variant (83) and additional susceptibility genes (46). Overall, Treg levels were reduced by ~20% in CD70-deficient NOD mice compared to controls, which is similar to the reduction in CD27- and CD70-deficient B6 mice. An important mechanism through which CD27-CD70 signaling is reported to increase Treg levels is by augmenting IL2 production by Foxp3-negative CD4+ T cells (82). Thus, it is somewhat surprising that overall Treg levels were not more reduced on the NOD background that produces less IL2 than B6 mice (83, 84). Our study shows that CD27 costimulation supports the generation of β-cell-specific Tregs that exert their suppressive function within the Islets of Langerhans, which is vital for controlling pancreatic inflammation (63). BDC2.5 TCR Tregs were more dependent on CD27 costimulation compared to polyclonal Tregs in standard NOD mice. This could be due to the affinity with which the BDC2.5 TCR binds self-Ag MHC complexes during thymic selection, as Croquet et al have shown that CD27 costimulation is more important for the development of high than low affinity Tregs (76).
CD70 expression in the thymus is mostly restricted to Aire+ medullary thymic epithelial cells (mTECs) and a small number of DCs (76). Our reciprocal BM transfers found that CD70 on hematopoietic cell types rather than mTECs is more important for promoting Treg development in NOD mice. This differs from a previous report showing that mTECs and DCs jointly contribute to Treg development in B6 background mice (76). The discrepancy may be due to variation in positive and negative selection between B6 and NOD mice, including that NOD mice have comparatively high numbers of DP thymocytes competing for limited selection niches, including mTECs (85). We found that CD27-CD70 costimulation is respectively supportive and dispensable for the generation of Helios+ and Helios− Tregs. Although the presence or absence of Helios expression does not unambiguously distinguish Tregs of thymic and peripheral origin (62, 86, 87), there is general consensus that immediate precursors of thymic-derived Tregs have high expression of Helios, and that this molecule identifies more thymic-derived Tregs than any other single biomarker (87, 88). Therefore, one explanation of our findings is that CD27-CD70 costimulation enhances the selection of thymic-derived Tregs. Another possibility is that CD70 signaling improves Treg stability after thymic selection as Helios expression is associated with enhanced Treg differentiation and survival (89, 90). Our contrary finding that CD70 deficiency reduces both Helios− and Helios+ Tregs in BM chimeras is likely an artifact of the BM reconstitution procedure as thymic selection of Tregs in lethally irradiated bone marrow recipients probably differs from normal neonatal Treg selection in unmanipulated mice.
We found no difference on a per cell basis in the suppressive function of Tregs from CD70-intact and -deficient mice, even though Tregs from the latter strain respectively expressed less Foxp3 and more TIGIT, which are both markers of Treg effector function (91, 92). In our in vivo suppression assay, it is possible that Tregs from CD70-deficient mice were initially impaired but regained their suppressor functions when transferred into a CD70-intact environment. However, CD70 most likely regulates T1D development by affecting the number rather than the function of Tregs as a previous report has shown that CD27-deficient Tregs that are incapable of responding to CD70 signaling perform normally in suppression assays (76).
Our finding that Tregs from CD70-intact mice become reduced when transferred into CD70-deficient recipients demonstrates that CD27-CD70 interactions in the periphery are at least as important as in the thymus for regulating circulating Treg levels. Our data resembles a previous study showing that CD28/B7 interactions regulate both thymic development and peripheral homeostasis of Tregs (93). There may be some overlap between how each pathway supports Treg levels; both CD27 and CD28 signaling promote Treg survival and self-renewal by stimulating IL2 production by conventional T cells (82, 93). However, while CD28 is critical for maintaining high levels of CD25 on Tregs (93), CD27 appears to be dispensable for this purpose, and even reduced CD25 expression on thymic Tregs. We have not yet determined which host cells use CD70 to sustain peripheral Treg levels, but even the limited number of CD70-constitutively-expressing cells within the thymus and Peyers patches may be sufficient given that systemic Treg frequencies increase in mice transplanted with CD70-containing tumor fragments (82). Another source may be subsets of DCs and lymphocytes that transiently express CD70, perhaps as a consequence of autoimmune inflammation.
A surprising result was that the loss of CD27 signaling increased iNKT cell development and cytokine production. This contrasts with other costimulatory molecules expressed by iNKT cells (CD28, CD40L, ICOS, OX40, 4-1BB), which have been found to support iNKT cell development and/or effector functions (94–99). We ruled out that Treg and iNKT cell levels are reciprocally regulated (Supplemental Figure 4). Thus, the most plausible explanation for our findings is that loss of CD70 alters how iNKT cells are selected in the thymus. Although iNKT cells differ from conventional αβ T cells because they are positively selected by strong rather than weak interactions with TCR-ligand complexes, studies using mice transgenically overexpressing CD28, B7, or CD1d show that intense TCR signals promote negative selection of thymic iNKT cells (67, 95). It is possible that the loss of CD70 signaling increases iNKT development by allowing a greater proportion of high-affinity iNKT cells to escape negative selection, either through lack of CD27 engagement on iNKT cells or by reducing the expression of CD1d on DP thymocytes. The iNKT1 cell subset was most affected, which is the only population that arises from Stage 3 of iNKT cell development. Our observations that CD27-CD70 costimulation limits α-GalCer-induced production of IFNγ and IL4 contrasts with other well-known iNKT cell costimulatory pathways that augment TCR-induced cytokine production, including CD28/B7, ICOS/ICOS-L and CD40/CD40L (94, 97, 100). Instead, they resemble a previous report that blocking the PD-1/PD-L1 coinhibitory pathway increases α-GalCer-induced production of IFNγ and IL4 (101), which suggests that the CD27-CD70 axis could be a coinhibitory pathway for iNKT cells. Another possibility is that CD27-CD70 costimulation facilitates the deletion of highly reactive iNKT cells in the thymus, leaving less responsive iNKT cells to accumulate in the periphery. In either event, it appears that one function of the CD27-CD70 pathway is to attenuate peripheral iNKT cell effector responses, possibly to limit inflammation caused by the pathogenic activation of these cells.
The ability of the AT-124 CD27 agonist Ab to respectively increase and decrease Helios+ Treg and iNKT cell levels in vivo demonstrates that CD70 signaling modulates these immunoregulatory subsets through interacting with CD27 rather than through reverse signaling cascades. Interestingly, the effect of this Ab was much more pronounced for iNKT cells than Tregs, even though the latter express more CD27. Although short-term AT-124 treatment was able to reduce islet inflammation, the inability of long-term Ab administration to reduce T1D development or support increased Treg levels may have been due to the development of neutralizing Abs against AT-124 or the observed downregulation of CD27 expression on T cells.
In conclusion, we established that the overall effect of CD27-CD70 costimulation limits T1D in the NOD mouse, most likely by supporting the development and homeostasis of Helios+ Tregs through primarily a hematopoietic cell type(s). In addition, the CD27-CD70 pathway was found to be distinct from other T cell costimulatory pathways for how it plays an inhibitory role in the development and effector functions of iNKT cells. Finally, we showed that a CD27 agonist Ab reversed the effects of CD70 ablation in NOD mice, suggesting that there may be potential to alter the course of T1D by therapeutically targeting CD27.
Supplementary Material
Key points :
CD27-CD70 costimulation delays type 1 diabetes onset in NOD mice
Disease protection is through supporting the development of Helios+ Tregs
CD27 agonist Ab therapy raises Treg levels and reduces βcell autoimmunity
Acknowledgments:
The National Institutes of Health Tetramer Core Facility provided the CD1d tetramers under the contract HHSN272201300006C. MVW and BEL were funded in part by The Jackson Laboratory. The authors are grateful for the assistance of Dr. Yi-Guang Chen for critically reading the manuscript and providing constructive advice. We thank Dr. Michael Clare-Salzler for providing us with NOD.BDC2.5.Foxp3eGFP mice and Celldex Therapeutics for donating the AT-124 Ab.
This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH) (Grant AI130656 to J.P.D.), the American Diabetes Association (Grant 1-14-BS-051 to J.P.D.), the National Institute of Diabetes and Digestive and Kidney Diseases, NIH (Grants DK106191 and AI42288 to T.M.B. and DK46266 and DK95735 to D.V.S), the NIH Office of the Director (OD020351 to D.V.S.), and the Juvenile Diabetes Research Foundation Australia (Grant 2018-568 to D.V.S.). The National Institutes of Health Tetramer Core Facility provided the CD1d tetramers under Contract HHSN272201300006C. M.V.W. and B.E.L. were funded in part by The Jackson Laboratory.
2. Abbreviations used in this paper:
- α-GalCer
α-galactosylceramide
- BM
Bone marrow
- Cd70−/−
CD70-deficient NOD mice
- CFSE
carboxyfluorescein succinimidyl ester
- DCs
dendritic cells
- DN
double negative
- DP
double positive
- EAE
experimental autoimmune encephalomyelitis
- F
female
- iNKT cell
invariant natural killer T cell
- i.p.
intraperitoneal
- i.v.
intravenous
- LPS
lipopolysaccharide
- M
male
- MFI
median fluorescent intensity
- mTECs
medullary thymic epithelial cells
- pLNs
pancreatic lymph nodes
- pTreg
peripherally-induced Tregs
- RA
rheumatoid arthritis
- SLE
systemic lupus erythematosus
- SP
single positive
- TALENs
transcription activator-like effector nucleases
- TCR
T cell receptor
- T1D
type 1 diabetes
- TIGIT
T cell Ig and ITIM domain
- TNFR
tumor necrosis factor receptor superfamily receptor
- Tregs
regulatory T cells, tTreg, thymic-derived Tregs
- WT
wild-type
References
- 1.Chen L, and Flies DB. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13: 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]; [Published erratum appears in 2013. Nat. Rev. Immunol 13: 542.] [Google Scholar]
- 2.Croft M 2009. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 9: 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertram EM, Lau P, and Watts TH. 2002. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol 168: 3777–3785. [DOI] [PubMed] [Google Scholar]
- 4.Dolfi DV, Boesteanu AC, Petrovas C, Xia D, Butz EA, and Katsikis PD. 2008. Late signals from CD27 prevent Fas-dependent apoptosis of primary CD8+ T cells. J Immunol 180: 2912–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurd N, and Robey EA. 2016. T-cell selection in the thymus: a spatial and temporal perspective. Immunol Rev 271: 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, and Hogquist KA. 2011. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med 208: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh CS, Lee HM, and Lio CW. 2012. Selection of regulatory T cells in the thymus. Nat Rev Immunol 12: 157–167. [DOI] [PubMed] [Google Scholar]
- 8.Tortorella C, Schulze-Koops H, Thomas R, Splawski JB, Davis LS, Picker LJ, and Lipsky PE. 1995. Expression of CD45RB and CD27 identifies subsets of CD4+ memory T cells with different capacities to induce B cell differentiation. J Immunol 155: 149–162. [PubMed] [Google Scholar]
- 9.Kashii Y, Giorda R, Herberman RB, Whiteside TL, and Vujanovic NL. 1999. Constitutive expression and role of the TNF family ligands in apoptotic killing of tumor cells by human NK cells. J Immunol 163: 5358–5366. [PubMed] [Google Scholar]
- 10.Tesselaar K, Xiao Y, Arens R, van Schijndel GM, Schuurhuis DH, Mebius RE, Borst J, and van Lier RA. 2003. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J Immunol 170: 33–40. [DOI] [PubMed] [Google Scholar]
- 11.de Jong R, Loenen WA, Brouwer M, van Emmerik L, de Vries EF, Borst J, and van Lier RA. 1991. Regulation of expression of CD27, a T cell-specific member of a novel family of membrane receptors. J Immunol 146: 2488–2494. [PubMed] [Google Scholar]
- 12.Borst J, Sluyser C, De Vries E, Klein H, Melief CJ, and Van Lier RA. 1989. Alternative molecular form of human T cell-specific antigen CD27 expressed upon T cell activation. Eur J Immunol 19: 357–364. [DOI] [PubMed] [Google Scholar]
- 13.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, and Ahmed R. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 4: 225–234. [DOI] [PubMed] [Google Scholar]
- 14.Watts TH 2005. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol 23: 23–68. [DOI] [PubMed] [Google Scholar]
- 15.Lens SM, Tesselaar K, van Oers MH, and van Lier RA. 1998. Control of lymphocyte function through CD27-CD70 interactions. Semin Immunol 10: 491–499. [DOI] [PubMed] [Google Scholar]
- 16.Peperzak V, Veraar EA, Keller AM, Xiao Y, and Borst J. 2010. The Pim kinase pathway contributes to survival signaling in primed CD8+ T cells upon CD27 costimulation. J Immunol 185: 6670–6678. [DOI] [PubMed] [Google Scholar]
- 17.van Oosterwijk MF, Juwana H, Arens R, Tesselaar K, van Oers MH, Eldering E, and van Lier RA. 2007. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int Immunol 19: 713–718. [DOI] [PubMed] [Google Scholar]
- 18.Feau S, Garcia Z, Arens R, Yagita H, Borst J, and Schoenberger SP. 2012. The CD4⁺ T-cell help signal is transmitted from APC to CD8⁺ T-cells via CD27-CD70 interactions. Nat Commun 3: 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao Y, Peperzak V, Keller AM, and Borst J. 2008. CD27 instructs CD4+ T cells to provide help for the memory CD8+ T cell response after protein immunization. J Immunol 181: 1071–1082. [DOI] [PubMed] [Google Scholar]
- 20.García P, De Heredia AB, Bellón T, Carpio E, Llano M, Caparrós E, Aparicio P, and López-Botet M. 2004. Signalling via CD70, a member of the TNF family, regulates T cell functions. J Leukoc Biol 76: 263–270. [DOI] [PubMed] [Google Scholar]
- 21.Allam A, Swiecki M, Vermi W, Ashwell JD, and Colonna M. 2014. Dual function of CD70 in viral infection: modulator of early cytokine responses and activator of adaptive responses. J Immunol 193: 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]; [published erratum appears in 2015. J. Immunol 194: 2033]. [DOI] [PubMed] [Google Scholar]
- 22.Munitic I, Kuka M, Allam A, Scoville JP, and Ashwell JD. 2013. CD70 deficiency impairs effector CD8 T cell generation and viral clearance but is dispensable for the recall response to lymphocytic choriomeningitis virus. J Immunol 190: 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couderc B, Zitvogel L, Douin-Echinard V, Djennane L, Tahara H, Favre G, Lotze MT, and Robbins PD. 1998. Enhancement of antitumor immunity by expression of CD70 (CD27 ligand) or CD154 (CD40 ligand) costimulatory molecules in tumor cells. Cancer Gene Ther 5: 163–175. [PubMed] [Google Scholar]
- 24.He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, Pilsmaker CD, Round SM, Tutt A, Glennie MJ, Marsh H, and Keler T. 2013. Agonist anti-human CD27 monoclonal Ab induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol 191: 4174–4183. [DOI] [PubMed] [Google Scholar]
- 25.Han BK, White AM, Dao KH, Karp DR, Wakeland EK, and Davis LS. 2005. Increased prevalence of activated CD70+CD4+ T cells in the periphery of patients with systemic lupus erythematosus. Lupus 14: 598–606. [DOI] [PubMed] [Google Scholar]
- 26.Park JK, Han BK, Park JA, Woo YJ, Kim SY, Lee EY, Lee EB, Chalan P, Boots AM, and Song YW. 2014. CD70-expressing CD4 T cells produce IFN-γ and IL-17 in rheumatoid arthritis. Rheumatology (Oxford) 53: 1896–1900. [DOI] [PubMed] [Google Scholar]
- 27.Oflazoglu E, Boursalian TE, Zeng W, Edwards AC, Duniho S, McEarchern JA, Law CL, Gerber HP, and Grewal IS. 2009. Blocking of CD27-CD70 pathway by anti-CD70 Ab ameliorates joint disease in murine collagen-induced arthritis. J Immunol 183: 3770–3777. [DOI] [PubMed] [Google Scholar]
- 28.Manocha M, Rietdijk S, Svend R, Laouar A, Liao G, Bhan A, Borst J, Terhorst C, and Manjunath N. 2009. Blocking CD27-CD70 costimulatory pathway suppresses experimental colitis. J Immunol 183: 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]; [Published erratum appears in 2009. J. Immunol 183: 4135.] [Google Scholar]
- 29.Coquet JM, Middendorp S, van der Horst G, Kind J, Veraar EA, Xiao Y, Jacobs H, and Borst J. 2013. The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity 38: 53–65. [DOI] [PubMed] [Google Scholar]
- 30.Novak J, Griseri T, Beaudoin L, and Lehuen A. 2007. Regulation of type 1 diabetes by NKT cells. Int Rev Immunol 26: 49–72. [DOI] [PubMed] [Google Scholar]
- 31.James CR, Buckle I, Muscate F, Otsuka M, Nakao M, Oon J. S. h., Steptoe RJ, Thomas R, and Hamilton-Williams EE. 2016. Reduced interleukin-2 responsiveness impairs the ability of Treg cells to compete for IL-2 in nonobese diabetic mice. Immunol Cell Biol 94: 509–519. [DOI] [PubMed] [Google Scholar]
- 32.D’Alise AM, Ergun A, Hill JA, Mathis D, and Benoist C. 2011. A cluster of coregulated genes determines TGF-beta-induced regulatory T-cell (Treg) dysfunction in NOD mice. Proc Natl Acad Sci U S A 108: 8737–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.You S, Belghith M, Cobbold S, Alyanakian MA, Gouarin C, Barriot S, Garcia C, Waldmann H, Bach JF, and Chatenoud L. 2005. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes 54: 1415–1422. [DOI] [PubMed] [Google Scholar]
- 34.Gregori S, Giarratana N, Smiroldo S, and Adorini L. 2003. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol 171: 4040–4047. [DOI] [PubMed] [Google Scholar]
- 35.Driver JP, Scheuplein F, Chen YG, Grier AE, Wilson SB, and Serreze DV. 2010. Invariant natural killer T-cell control of type 1 diabetes: a dendritic cell genetic decision of a silver bullet or Russian roulette. Diabetes 59: 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang B, Geng Y-B, and Wang C-R. 2001. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. Journal of Experimental Medicine 194: 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, Miura T, Haba T, Scherer DC, and Wei J. 2001. The natural killer T-cell ligand α-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nature medicine 7: 1052. [DOI] [PubMed] [Google Scholar]
- 38.Sharif S, Arreaza GA, Zucker P, Mi Q-S, Sondhi J, Naidenko OV, Kronenberg M, Koezuka Y, Delovitch TL, and Gombert J-M. 2001. Activation of natural killer T cells by α-galactosylceramide treatment prevents the onset and recurrence of autoimmune type 1 diabetes. Nature medicine 7: 1057. [DOI] [PubMed] [Google Scholar]
- 39.Shi FD, Flodstrom M, Balasa B, Kim SH, Van Gunst K, Strominger JL, Wilson SB, and Sarvetnick N. 2001. Germ line deletion of the CD1 locus exacerbates diabetes in the NOD mouse. Proc Natl Acad Sci U S A 98: 6777–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naumov YN, Bahjat KS, Gausling R, Abraham R, Exley MA, Koezuka Y, Balk SB, Strominger JL, Clare-Salzer M, and Wilson SB. 2001. Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc Natl Acad Sci U S A 98: 13838–13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammond KJ, Poulton LD, Palmisano LJ, Silveira PA, Godfrey DI, and Baxter AG. 1998. alpha/beta-T cell receptor (TCR)+CD4-CD8- (NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)-4 and/or IL-10. J Exp Med 187: 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehuen A, Lantz O, Beaudoin L, Laloux V, Carnaud C, Bendelac A, Bach JF, and Monteiro RC. 1998. Overexpression of natural killer T cells protects Valpha14-Jalpha281 transgenic nonobese diabetic mice against diabetes. J Exp Med 188: 1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tritt M, Sgouroudis E, d’Hennezel E, Albanese A, and Piccirillo CA. 2008. Functional waning of naturally occurring CD4+ regulatory T-cells contributes to the onset of autoimmune diabetes. Diabetes 57: 113–123. [DOI] [PubMed] [Google Scholar]
- 44.Gregg RK, Jain R, Schoenleber SJ, Divekar R, Bell JJ, Lee HH, Yu P, and Zaghouani H. 2004. A sudden decline in active membrane-bound TGF-beta impairs both T regulatory cell function and protection against autoimmune diabetes. J Immunol 173: 7308–7316. [DOI] [PubMed] [Google Scholar]
- 45.Yamanouchi J, Puertas MC, Verdaguer J, Lyons PA, Rainbow DB, Chamberlain G, Hunter KM, Peterson LB, Wicker LS, and Santamaria P. 2010. Idd9.1 locus controls the suppressive activity of Foxp3+CD4+CD25+ regulatory T-cells. Diabetes 59: 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mellanby RJ, Thomas D, Phillips JM, and Cooke A. 2007. Diabetes in non-obese diabetic mice is not associated with quantitative changes in CD4+ CD25+ Foxp3+ regulatory T cells. Immunology 121: 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, and Mathis D. 2008. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci U S A 105: 19857–19862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, and Dobbs D. 2010. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res 38: W462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sander JD, Zaback P, Joung JK, Voytas DF, and Dobbs D. 2007. Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res 35: W599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, and Joung JK. 2012. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shultz LD, Lang PA, Christianson SW, Gott B, Lyons B, Umeda S, Leiter E, Hesselton R, Wagar EJ, Leif JH, Kollet O, Lapidot T, and Greiner DL. 2000. NOD/LtSz-Rag1null mice: an immunodeficient and radioresistant model for engraftment of human hematolymphoid cells, HIV infection, and adoptive transfer of NOD mouse diabetogenic T cells. J Immunol 164: 2496–2507. [DOI] [PubMed] [Google Scholar]
- 52.Katz JD, Wang B, Haskins K, Benoist C, and Mathis D. 1993. Following a diabetogenic T cell from genesis through pathogenesis. Cell 74: 1089–1100. [DOI] [PubMed] [Google Scholar]
- 53.Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, and Bluestone JA. 2008. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med 205: 1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, Nakayama M, Rosenthal W, and Bluestone JA. 2009. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 10: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takaki T, Marron MP, Mathews CE, Guttmann ST, Bottino R, Trucco M, DiLorenzo TP, and Serreze DV. 2006. HLA-A*0201-restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J Immunol 176: 3257–3265. [DOI] [PubMed] [Google Scholar]
- 56.Driver JP, Chen YG, Zhang W, Asrat S, and Serreze DV. 2011. Unmasking genes in a type 1 diabetes-resistant mouse strain that enhances pathogenic CD8 T-cell responses. Diabetes 60: 1354–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forsberg MH, Ciecko AE, Bednar KJ, Itoh A, Kachapati K, Ridgway WM, and Chen YG. 2017. CD137 Plays Both Pathogenic and Protective Roles in Type 1 Diabetes Development in NOD Mice. J Immunol 198: 3857–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Driver JP, Lamont DJ, Gysemans C, Mathieu C, and Serreze DV. 2011. Calcium insufficiency accelerates type 1 diabetes in vitamin D receptor-deficient nonobese diabetic (NOD) mice. Endocrinology 152: 4620–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheuplein F, Rissiek B, Driver JP, Chen YG, Koch-Nolte F, and Serreze DV. 2010. A recombinant heavy chain Ab approach blocks ART2 mediated deletion of an iNKT cell population that upon activation inhibits autoimmune diabetes. J Autoimmun 34: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glennie MJ, Tutt AL, and Al-Shamkhani A inventors University of Southampton, assignee, Human immune therapies using a CD27 agonist in combination with another immune agonist to treat cancer. United States patent 9926374; Publication No. 20160215056 2015. December 10.
- 61.Koo T, Lee J, and Kim JS. 2015. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol Cells 38: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Himmel ME, MacDonald KG, Garcia RV, Steiner TS, and Levings MK. 2013. Helios+ and Helios− cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol 190: 2001–2008. [DOI] [PubMed] [Google Scholar]
- 63.Tarbell KV, Yamazaki S, and Steinman RM. 2006. The interactions of dendritic cells with antigen-specific, regulatory T cells that suppress autoimmunity. Semin Immunol 18: 93–102. [DOI] [PubMed] [Google Scholar]
- 64.You S, Chen C, Lee WH, Wu CH, Judkowski V, Pinilla C, Wilson DB, and Liu CP. 2003. Detection and characterization of T cells specific for BDC2.5 T cell-stimulating peptides. J Immunol 170: 4011–4020. [DOI] [PubMed] [Google Scholar]
- 65.Lee HM, Bautista JL, and Hsieh CS. 2011. Thymic and peripheral differentiation of regulatory T cells. Adv Immunol 112: 25–71. [DOI] [PubMed] [Google Scholar]
- 66.Tsaih SW, Presa M, Khaja S, Ciecko AE, Serreze DV, and Chen YG. 2015. A locus on mouse chromosome 13 inversely regulates CD1d expression and the development of invariant natural killer T-cells. Genes Immun 16: 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chun T, Page MJ, Gapin L, Matsuda JL, Xu H, Nguyen H, Kang HS, Stanic AK, Joyce S, Koltun WA, Chorney MJ, Kronenberg M, and Wang CR. 2003. CD1d-expressing dendritic cells but not thymic epithelial cells can mediate negative selection of NKT cells. J Exp Med 197: 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu H, Chun T, Colmone A, Nguyen H, and Wang CR. 2003. Expression of CD1d under the control of a MHC class Ia promoter skews the development of NKT cells, but not CD8+ T cells. J Immunol 171: 4105–4112. [DOI] [PubMed] [Google Scholar]
- 69.Constantinides MG, and Bendelac A. 2013. Transcriptional regulation of the NKT cell lineage. Curr Opin Immunol 25: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.La Cava A, Van Kaer L, and Fu-Dong-Shi. 2006. CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol 27: 322–327. [DOI] [PubMed] [Google Scholar]
- 71.Arens R, Nolte MA, Tesselaar K, Heemskerk B, Reedquist KA, van Lier RA, and van Oers MH. 2004. Signaling through CD70 regulates B cell activation and IgG production. J Immunol 173: 3901–3908. [DOI] [PubMed] [Google Scholar]
- 72.Sawalha AH, and Jeffries M. 2007. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupus-prone mice. Eur J Immunol 37: 1407–1413. [DOI] [PubMed] [Google Scholar]
- 73.Arreaza G, Salojin K, Yang W, Zhang J, Gill B, Mi QS, Gao JX, Meagher C, Cameron M, and Delovitch TL. 2003. Deficient activation and resistance to activation-induced apoptosis of CD8+ T cells is associated with defective peripheral tolerance in nonobese diabetic mice. Clin Immunol 107: 103–115. [DOI] [PubMed] [Google Scholar]
- 74.Decallonne B, van Etten E, Giulietti A, Casteels K, Overbergh L, Bouillon R, and Mathieu C. 2003. Defect in activation-induced cell death in non-obese diabetic (NOD) T lymphocytes. J Autoimmun 20: 219–226. [DOI] [PubMed] [Google Scholar]
- 75.Gravestein LA, van Ewijk W, Ossendorp F, and Borst J. 1996. CD27 cooperates with the pre-T cell receptor in the regulation of murine T cell development. J Exp Med 184: 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coquet JM, Ribot JC, Bąbała N, Middendorp S, van der Horst G, Xiao Y, Neves JF, Fonseca-Pereira D, Jacobs H, Pennington DJ, Silva-Santos B, and Borst J. 2013. Epithelial and dendritic cells in the thymic medulla promote CD4+Foxp3+ regulatory T cell development via the CD27-CD70 pathway. J Exp Med 210: 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choisy-Rossi CM, Holl TM, Pierce MA, Chapman HD, and Serreze DV. 2004. Enhanced pathogenicity of diabetogenic T cells escaping a non-MHC gene-controlled near death experience. J Immunol 173: 3791–3800. [DOI] [PubMed] [Google Scholar]
- 78.Liston A, Lesage S, Gray DH, O’Reilly LA, Strasser A, Fahrer AM, Boyd RL, Wilson J, Baxter AG, Gallo EM, Crabtree GR, Peng K, Wilson SR, and Goodnow CC. 2004. Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity 21: 817–830. [DOI] [PubMed] [Google Scholar]
- 79.Liston A, Hardy K, Pittelkow Y, Wilson SR, Makaroff LE, Fahrer AM, and Goodnow CC. 2007. Impairment of organ-specific T cell negative selection by diabetes susceptibility genes: genomic analysis by mRNA profiling. Genome Biol 8: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, and Mathis D. 2005. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity 22: 385–396. [DOI] [PubMed] [Google Scholar]
- 81.Pen JJ, De Keersmaecker B, Maenhout SK, Van Nuffel AM, Heirman C, Corthals J, Escors D, Bonehill A, Thielemans K, Breckpot K, and Aerts JL. 2013. Modulation of regulatory T cell function by monocyte-derived dendritic cells matured through electroporation with mRNA encoding CD40 ligand, constitutively active TLR4, and CD70. J Immunol 191: 1976–1983. [DOI] [PubMed] [Google Scholar]
- 82.Claus C, Riether C, Schürch C, Matter MS, Hilmenyuk T, and Ochsenbein AF. 2012. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res 72: 3664–3676. [DOI] [PubMed] [Google Scholar]
- 83.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, Chen SL, Rosa R, Cumiskey AM, Serreze DV, Gregory S, Rogers J, Lyons PA, Healy B, Smink LJ, Todd JA, Peterson LB, Wicker LS, and Santamaria P. 2007. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet 39: 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sgouroudis E, Albanese A, and Piccirillo CA. 2008. Impact of protective IL-2 allelic variants on CD4+ Foxp3+ regulatory T cell function in situ and resistance to autoimmune diabetes in NOD mice. J Immunol 181: 6283–6292. [DOI] [PubMed] [Google Scholar]
- 85.Mingueneau M, Jiang W, Feuerer M, Mathis D, and Benoist C. 2012. Thymic negative selection is functional in NOD mice. J Exp Med 209: 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gottschalk RA, Corse E, and Allison JP. 2012. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol 188: 976–980. [DOI] [PubMed] [Google Scholar]
- 87.Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, and Ignatowicz L. 2015. Differences in Expression Level of Helios and Neuropilin-1 Do Not Distinguish Thymus-Derived from Extrathymically-Induced CD4+Foxp3+ Regulatory T Cells. PLoS One 10: e0141161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Singh K, Hjort M, Thorvaldson L, and Sandler S. 2015. Concomitant analysis of Helios and Neuropilin-1 as a marker to detect thymic derived regulatory T cells in naïve mice. Sci Rep 5: 7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, and Thornton AM. 2016. Helios Controls a Limited Subset of Regulatory T Cell Functions. J Immunol 196: 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, and Kim HJ. 2016. Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci U S A 113: 6248–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, Sharpe AH, Quintana FJ, Mathis D, Benoist C, Hafler DA, and Kuchroo VK. 2014. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40: 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rudensky AY 2011. Regulatory T cells and Foxp3. Immunol Rev 241: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, and Bluestone JA. 2003. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol 171: 3348–3352. [DOI] [PubMed] [Google Scholar]
- 94.Chung Y, Nurieva R, Esashi E, Wang YH, Zhou D, Gapin L, and Dong C. 2008. A critical role of costimulation during intrathymic development of invariant NK T cells. J Immunol 180: 2276–2283. [DOI] [PubMed] [Google Scholar]
- 95.Williams JA, Lumsden JM, Yu X, Feigenbaum L, Zhang J, Steinberg SM, and Hodes RJ. 2008. Regulation of thymic NKT cell development by the B7-CD28 costimulatory pathway. J Immunol 181: 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Akbari O, Stock P, Meyer EH, Freeman GJ, Sharpe AH, Umetsu DT, and DeKruyff RH. 2008. ICOS/ICOSL interaction is required for CD4+ invariant NKT cell function and homeostatic survival. J Immunol 180: 5448–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hayakawa Y, Takeda K, Yagita H, Van Kaer L, Saiki I, and Okumura K. 2001. Differential regulation of Th1 and Th2 functions of NKT cells by CD28 and CD40 costimulatory pathways. J Immunol 166: 6012–6018. [DOI] [PubMed] [Google Scholar]
- 98.Diana J, Griseri T, Lagaye S, Beaudoin L, Autrusseau E, Gautron AS, Tomkiewicz C, Herbelin A, Barouki R, von Herrath M, Dalod M, and Lehuen A. 2009. NKT cell-plasmacytoid dendritic cell cooperation via OX40 controls viral infection in a tissue-specific manner. Immunity 30: 289–299. [DOI] [PubMed] [Google Scholar]
- 99.Vinay DS, Choi BK, Bae JS, Kim WY, Gebhardt BM, and Kwon BS. 2004. CD137-deficient mice have reduced NK/NKT cell numbers and function, are resistant to lipopolysaccharide-induced shock syndromes, and have lower IL-4 responses. J Immunol 173: 4218–4229. [DOI] [PubMed] [Google Scholar]
- 100.Kaneda H, Takeda K, Ota T, Kaduka Y, Akiba H, Ikarashi Y, Wakasugi H, Kronenberg M, Kinoshita K, Yagita H, and Okumura K. 2005. ICOS costimulates invariant NKT cell activation. Biochem Biophys Res Commun 327: 201–207. [DOI] [PubMed] [Google Scholar]
- 101.Wang J, Cheng L, Wondimu Z, Swain M, Santamaria P, and Yang Y. 2009. Cutting edge: CD28 engagement releases antigen-activated invariant NKT cells from the inhibitory effects of PD-1. J Immunol 182: 6644–6647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.