

Cyanobacterial light-harvesting complex features efficient energy capture, transmission, and regulation to reaction centers.
Abstract
In cyanobacteria and red algae, the structural basis dictating efficient excitation energy transfer from the phycobilisome (PBS) antenna complex to the reaction centers remains unclear. The PBS has several peripheral rods and a central core that binds to the thylakoid membrane, allowing energy coupling with photosystem II (PSII) and PSI. Here, we have combined chemical cross-linking mass spectrometry with homology modeling to propose a tricylindrical cyanobacterial PBS core structure. Our model reveals a side-view crossover configuration of the two basal cylinders, consolidating the essential roles of the anchoring domains composed of the ApcE PB loop and ApcD, which facilitate the energy transfer to PSII and PSI, respectively. The uneven bottom surface of the PBS core contrasts with the flat reducing side of PSII. The extra space between two basal cylinders and PSII provides increased accessibility for regulatory elements, e.g., orange carotenoid protein, which are required for modulating photochemical activity.
INTRODUCTION
In natural photosynthesis, light-harvesting complexes capture and transmit solar energy to the reaction centers (RCs) where photochemistry takes place, leading to long-term energy storage (1, 2). In cyanobacteria and red algae, phycobilisomes (PBSs) harvest the energy in the spectral range between 450 and 650 nm, which is complementary to that of Chl a (chlorophyll a) in the RCs, i.e., photosystem I (PSI) and PSII, and substantially increases the utilization of the solar energy spectrum (3). PBSs, with a molecular mass range of 5 to 20 MDa, are located on the cytoplasmic side (or the stromal side of the red algal chloroplasts) of RCs. PBSs are highly organized assemblies of brightly colored phycobiliproteins and colorless linker polypeptides (4). Each PBS consists of a central cylindrical core made of allophycocyanin (Apc), from which several outwardly oriented rods radiate. Light energy collected by phycobiliproteins in the peripheral rods is transferred to PSI and PSII, with the PBS core Apc acting as a link between phycocyanin and the Chl a in RCs. The detailed structural orientation of the PBS and the RCs, however, remains to be determined, probably because of the structurally weak interactions of PBS and RCs, which could be advantageous for easy regulation of the excitation energy transfer under varying light conditions (5, 6). The PBS core not only serves as a cornerstone for the rods to attach (7, 8) but also acts as an anchoring module that enables efficient energy flow from PBSs to RCs (9–13).
The building blocks of the PBS core are two homologous proteins, α- and β-phycocyanin subunits, which form a heterodimer, αβ, and further self-assemble into disc-like (αβ)3 trimers (4). Several discs of Apc (ApcA/B) stack into a cylinder, varying in numbers in different organisms. The best-known PBS structure is described as hemidiscoidal PBS (7, 14). In this group, there are bicylinder type of PBS in Synechococcus sp. PCC 6301 (15) and pentacylinder type in Mastigocladus laminosus as well (16). The core structure of Synechocystis sp. PCC 6803, however, is tricylindrical as determined in 1986 by electron microscopy (EM) (17). Later, advanced EM methodology yielded a resolution structure at 13 Å using a genetically modified strain in Synechocystis 6803 (CK strain) (7), which contains an intact core complex but no phycocyanin (PC) rods. The EM structure reveals that this PBS core consists of three stacked core cylinders: two on the bottom and one on the top. The two-dimensional (2D) EM map further highlights a twofold rotational symmetry of the triangular core, indicating that the two basal cylinders are arranged in an antiparallel fashion. Although the overall architecture is resolved, a detailed molecular model showing the location of each terminal energy emitter, ApcE, ApcD, and the tomography of the bottom surface of the PBS core that directly interacts with the thylakoid membranes are not known.
Recently, two reports notably advanced the understanding of the PBS structure, specifically, in two red algae Griffithsia pacifica (18) and Porphyridium purpureum (19) by using cryo-EM at resolutions of 3.5 and 2.8 Å, respectively. GpPBS and PpPBS contain numerous protein subunits, linker proteins, and chromophores, revealing in great detail the PBS architecture. GpPBS is morphologically categorized as a block-shaped type (18) in contrast to PpPBS, which is a hemiellipsoidal-shaped PBS, both of which, however, are likely evolutionary derivatives of the core of hemidiscoidal PBS (18). Both GpPBS core and PpPBS core are characterized by two unique features: lack of one trimeric ApcA/B disc at the distal end of ApcD in each basal cylinder (Fig. 1A) and thus a decreased copy number (by comparing to a four-Apc-trimer cylinder) of ApcC (Apc C; formerly LC), a small polypeptide linker protein that stabilizes and caps each end of the core cylinders (20, 21). The top cylinder in both GpPBS core and PpPBS core is featured by two Apc trimers, instead of four. Two Apc discs are stacked back to back (or tail to tail). Parts of the second and third linker domains of ApcE (18, 19), which is usually buried in the core of four-Apc-trimer cylinder (3), are exposed and do not interact with the Apc trimers.
Fig. 1. Model construction of a tricylindrical cyanobacterial PBS core.
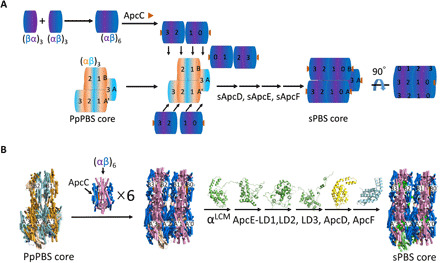
(A) Cartoon illustration of disc extension based on PpPBS core structure [Protein Data Bank (PDB) ID: 6KGX] and cyanobacterial structure. PpApcA (salmon), PpApcB (cyan), ApcA (cyanobacteria, purple), and ApcB (dark blue). (B) Detailed representation of disc extension based on PpPBS core structure and cyanobacterial structure. Disc extension of PpPBS core using ApcA/B hexamer, cartoon (A) or x-ray structure (B) (PDB ID: 4F0U). Other Apc domains, i.e., αLCM (green), ApcE-LD1 (green), ApcE-LD2 (green), ApcE-LD3 (green), ApcD (yellow), and ApcF (light cyan), are constructed similarly. The final core model of sPBS is shown (side view). All structure figures in (B) were prepared using PyMOL.
In this study, we build a 3D structure of a cyanobacterial PBS core (sPBS core of Synechocystis sp. PCC 6803) starting with the available crystal structure of cyanobacterial Apc and the red algal PBS cryo-EM structure in combination with protein structure and function prediction software suites. The generated model represents a cyanobacterial tricylindrical core with two basal cylinders and one top cylinder, each of which contains four Apc trimers (or disc). Other Apc subunits, including ApcD, ApcE, and ApcF, were all modeled to afford the core architecture. We also performed chemical cross-linking using three types of PBSs isolated from Synechocystis 6803, mapped the identified cross-links onto the proposed 3D model, a portrayal of the native sPBS core, and calculated the distances between each pair of the cross-linked residues. The proposed sPBS core was then justified and evaluated by the experimental distance restraints with respect to individual subunits. The described framework reveals a side-view X-shaped configuration of the two basal cylinders, which only allows the protrusion of the PB loops on ApcE and ApcD interacting with the thylakoid membranes. The bottom surface of the PBS that touches the thylakoid membrane/RCs is not perfectly flat, in contrast to a flat surface of the reducing side of PSII. The increased accessibilities to the bottom surface of the PBS core and the reducing side of PSII, however, provide perspectives on the excitation energy regulation and RC photoprotection.
RESULTS
Homology modeling and construction of a tricylindrical cyanobacterial PBS core
We adopted the high-resolution PpPBS core structure from red algae P. purpureum [Protein Data Bank (PDB) ID: 6KGX] (19) as a building chassis. Although the overall appearance of PpPBS is a hemiellipsoidal-shaped PBS–type PBS, it essentially retains the classical twofold symmetry with its symmetrical axis oriented perpendicularly to the thylakoid membranes, and both block- and hemiellipsoidal-shaped PBSs are considered to be evolutionally derived from a core of hemidiscoidal PBS core by eliminations of the exterior Apc disc (Apc trimer) of three cylinders. This hypothesis is consistent with a hemidiscoidal model proposed in earlier reports (7). We also noticed that GpPBS core and PpPBS core share high structural homology (18, 19) with root mean square deviation (RMSD) of 0.78 Å. We are interested in building a tricylindrical cyanobacterial hemidiscoidal PBS core with four stacked Apc trimers per cylinder.
The cryo-EM structure of both GpPBS core and PpPBS core revealed several features: Each basal cylinder contains three ApcA/B trimers, namely, discs A1–3 and A′1–3, and the top cylinder contains only two discs with tail-to-tail orientation (three ApcBs in one disc bordering to three ApcBs in another disc), i.e., discs B1 and B2 (see Fig. 1, A and B, left, for simplified and detailed cartoon representation, respectively). Each disc contains three copies of ApcA and ApcB. This structure is in contrast to that of a model in Synechocystis 6803 (7, 11), which indicates that each core cylinder contains four discs, arranged in a face-to-face (three ApcAs in one disc to three ApcAs in another disc), tail-to-tail, and face-to-face fashion. Using homology modeling, we swapped all the PpPBS core Apc discs by using cyanobacterial Apc (sApc) discs. Each cylinder was also extended up to four discs. First of all, a hexameric cyanobacterial Apc (PDB ID: 4F0U) was generated and used as a building unit. This hexamer contains two discs arranged in face-to-face fashion. To extend the top cylinder, a cyanobacterial Apc hexamer was adopted to align with the disc B1. The alignment of the B1 disc results in the replacement of the red algal counterpart and in an extension of one cyanobacterial Apc trimer, i.e., the B0 disc (see Fig. 1, A and B, middle, for simplified and detailed cartoon representation, respectively). Similarly, the top cylinder was extended with the alignment of B2 disc by another hexameric Apc (two discs, face to face), leading to an addition of B3 disc to make a total of four-disc top cylinder. The replacement and extension for the two bottom cylinders were performed on A1 and A′1 discs, giving additional Apc trimers as disc A0 and A′0, respectively. The remaining red algae A2/A3 and A′2/A′3 hexamers were further swapped with cyanobacterial Apc hexametric units, contributing to an sPBS core architecture with four cyanobacterial discs on each of the three cylinders (see Fig. 1, A and B, right, for a simplified and detailed cartoon representation, respectively). The selection of cyanobacterial crystal structure (PDB ID: 4F0U) took into account its high-resolution structure and high protein sequence homology with those of Synechocystis 6803 (22), which was used in our following chemical cross-linking studies. Extension of A0/A′0 not only fulfills the face-to-face geometry in adjacent discs (A1 and A′1, respectively) but also enables the second linker domain, i.e., ApcE-LD2/ApcE-LD2′, being buried inside the cylinder, as the ApcE-LD1/ApcE-LD1′ is concealed within discs A3/A2 and A′3/A′2 in two basal cylinders, respectively (18, 19). During the swapping and elongation of the B1 disc (PpPBS core), a value of less than 1-Å RMSD was always obtained (table S1), indicative of high structural similarities of red algal ApcA/B trimer and the cyanobacterial ApcA/B trimer assembly. The protein sequence identities between the cyanobacteria and red algae strongly support this conclusion (fig. S1). The remaining PpApcA/B trimers in each basal cylinder that contain ApcD and ApcE were also substituted by sApcA/B hexamer with consistent low RMSD, contributing to a complete framework of an sPBS core (Fig. 1B). The last step is to model other PBS core components, such as ApcC, ApcD, ApcE, and ApcF; we either used the available crystal structure (ApcE, PDB ID: 4XXI; ApcD, PDB ID: 4PO5) (23) or predicted their structures by using protein sequence from Synechocystis 6803 on the Zhang Server (24) and then defined their positions by using homology modeling. Their copy numbers were based on the characteristic features of a tricylindrical cyanobacterial PBS (7).
ApcE, also termed as core-membrane linker or LCM (25), is a multifunctional protein containing both phycocyanin binding domain, known as PB domain (or αLCM), and several linker domains that are essential for connecting Apc trimers (discs) and thus for assembly of Apc discs into cylinders. The αLCM is located in discs A2 and A′2 in the red algal structure (18, 19). The ApcE linker domains serve to stitch all the discs and subsequently cylinders together to form the PBS core (7). The large ApcE protein in tricylindrical PBS in Synechocystis 6803 contains four domains, namely, αLCM (PB domain/α domain in LCM), ApcE-LD1, ApcE-LD2, and ApcE-LD3 (fig. S2B), which were treated individually for structure prediction and modeling. For example, when the ApcE-LD3 peptide sequence was submitted to the I-TASSER server (24), the homology-modeling algorithm screened through the database and adopted the PpApcE (PDB ID: 6KGX) and GpApcE (PDB ID: 5Y6P) and another two protein structures (PDB ID: 2KY4, http://dx.doi.org/10.2210/pdb2ky4/pdb; 3OHW and http://dx.doi.org/10.2210/pdb3ohw/pdb) among others as references, giving the top five high-scoring models (Fig. 1B and table S1). These models resemble the overall configuration and helical content that are consistent with protein sequence identity (fig. S2A). Each generated model was then aligned with both PpApcE-LD3 domains in the top cylinders, evaluated in terms of RMSD for all corresponding alpha carbons (table S1). Using the smallest RMSD value as a criterion, we chose sApcE-LD3-M1 as the representative model. We use both an I-TASSER–generated model and the x-ray crystal structure of αLCM (PDB ID: 4XXI) (26) in our modeling, since the former model contains the PB loop that allows model building of the PBS-core-PSII (13), while the latter contains the chromophore structure information.
Other ApcE linker domains, i.e., ApcE-LD1 and sApcE-LD2, which are composed similarly of a Pfam00427 (PDB IDs: 2KY4, 2L06, and 3OHW, released by the Northeast Structural Genomics Consortium of America), and the αLCM were performed for homology modeling in a comparable fashion. Five generated top-scoring models for each domain were evaluated individually by structural alignment in terms of calculated RMSD (table S1). We always chose the models with the smallest RMSD value to give the best likelihood of being the most reasonable configuration for αLCM, ApcE-LD1 and ApcE-LD2. ApcE-LD1 and ApcE-LD2 are in the cavity of the basal rod-like protrusions between the A2/A3 (or A′2/A′3) and A0/A1 (or A′0/A′1) ApcA/B discs, respectively, stabilizing two basal cylinders. Two copies of αLCM situate at the A2 and A′2 of the basal cylinders with a large loop (PB loop) from peptides 78 to 143 (fig. S2B), extending toward the thylakoid membrane and anchoring the whole PBS assembly to RCs (2, 7, 27, 28).
PpApcD, an Apc α-like subunit that is required for energy delivery from PBS to PSI (29), was substituted by a resolved structure of ApcD at 1.75 Å from a previous study (PDB ID: 4PO5) (23). The two PpApcD subunits are located at the A3 and A′3 discs in the basal cylinders, respectively, both of which are well aligned with the sApcD and affording RMSD differences that are around 0.912 Å (table S1).
After swapping ApcE and ApcD in the structure, we next performed homology modeling of ApcC, which belongs to the CpcD superfamily and upholds a cylinder in shape (20). The structure was based on an x-ray crystallographic study of a trimeric ApcC from Fischerella sp. PCC 6703 (PDB ID: 1B33) (20). The SPICKER program, an algorithm to discern near-native models from a pool of decoyed protein structures (30), converged the simulation and identified only one large cluster, indicating the high quality of the generated models. The alignment of ApcC onto PpApcC shows small RMSD value (table S1), indicative of a good model. The copy numbers of ApcC in the characterized PpPBS and GpPBS structures, however, are only two per PBS, unexpectedly low, comparing to six per PBS in tricylindrical cyanobacterial PBS core, probably because there is one disc missing on each basal cylinder and two discs (Apc trimers) missing on the top cylinder where the ApcC subunit usually binds (20). It has been accepted that in cyanobacteria, each core cylinder has two ApcC proteins located at both ends (20), totaling six per tricylindrical PBS core. We substituted the existing PpApcC with sApcC in the ApcA/B trimer (note that PpApcA/B trimer has been swapped by ApcA/B trimer). The complex of ApcC/sApcA/B was then regarded as an independent object and aligned onto the end of each core cylinder, resulting in A0, A′0, B3, and B0 discs containing ApcC.
ApcF, an Apc β-like unit, affects state transitions and energy transfer through interactions with αLCM (31). sApcF is modeled at the same trimeric disc where the αLCM is located (A2 and A′2) with replacement of the PpApcF. Thus, ApcF interacts with αLCM (although they are not in an αβ units), sApcE-LD1, and potentially, with the nearby pigment, αLCM phycocyanobilin (18, 19, 31), to allow energy transmission to PSII (32). Only one model was generated from I-TASSER, and its RMSD value is less than 1 Å, indicative of an excellent modeling (table S1). By combining the protein sequence analysis of three ApcFs from PpPBS/GpPBS and Synechocystis 6803, we noticed some sequence variations in detail (fig. S3): an indel (insertion/deletion) around 145 between three structures. In cyanobacteria, this region is composed of five amino acid residues less than GpApcF and one less than PpApcF. This indel is supposedly in a fragment of an α-helix (G-H) in red algal ApcF. For both of the red algal PBS structures (PDB ID: 6KGX or 5Y6P), ApcF has an extension at the tip of the G-H helix hairpin (18, 19). ApcF is considered to be an important factor that fine-tunes the spectroscopic properties of the PCB in αLCM. We hypothesize that combination of an indel and a fragment of an α-helix extension not only affects the spectroscopic properties of αLCM but also helps each basal cylinder to build up its polarity so that only a homogeneous trimer [A1/A′1, (αβ)3] can be recruited with a back-to-back configuration (Fig. 1B). In other words, such structures (indel, and the C-terminal extension of ApcF) could help each basal cylinder to build up its polarity so that only an A1 trimer (αβ)3 can assemble with the trimer A2/A′2 (αLCMβ∙αβApcF∙αβ). In terms of the assembly of disc A2/A′2, we propose that it is the linker domain (i.e., ApcE-LD1) that plays an important role to recruit an ApcF, a β-like unit, to build a (A2/A′2) trimer rather than a regular β unit. Further biochemical and spectroscopic research on this region by using genetically tractable cyanobacteria may shed light on its function.
Chemical cross-linking and strategy
Chemical cross-linking coupled with mass spectrometry (MS) provides information of proximal amino acid residues within proteins and protein complexes. It is particularly useful for protein complexes that are recalcitrant to conventional structural biology studies. Successful observation of two red algal PBS structures at the atomic level using cryo-EM took advantage of a particularly stable PBS species that allows PBS sample preparation for cryo-EM analysis at low-salt conditions. The cyanobacterial PBS, however, is much less stable, at least to the current reports, and the structure at the atomic level remains unclear. Here, we used residue-level chemical cross-linking to provide structural restraints to justify and evaluate a protein complex model that we generated by using bioinformatics methods (Fig. 1B).
In this experiment, we used wild-type PBS of cyanobacteria Synechocystis 6803 (WT-sPBS) and two mutant PBS for cross-linking analysis (Fig. 2 and fig. S4). In WT-sPBS, we identified a total of 33 cross-links, in which 17 inter-subunit cross-links are within the PBS core (Fig. 2A and table S2). We also found numerous loop-links and mono-links that provide limited structural information for subunit-subunit interactions. In contrast, when CK-PBS, a rod-less PBS (33), was cross-linked and submitted to MS interrogation, 35 cross-links were identified (Fig. 2B and table S3), and all 17 cross-links in the WT-sPBS sample were identified in this sample. Eighteen cross-links, however, have been uniquely found in CK-PBS. The differences in identified cross-links between WT-sPBS and CK-PBS are probably due to the shielding effect in WT-sPBS by six outward radiating PC rods that have extensive contacting interfaces and large steric hindrance to limit the accessibility of cross-linkers to core components, indicative of our successful strategy of picking up a genetically modified cell strain. A CpcL-PBS mutant (34) containing only the rods was also used for a negative control. For the CpcL-PBS mutant (Fig. 2C and table S4), we identified 14 cross-links of rod subunits and associated proteins, especially ferredoxin-NADP+ (nicotinamide adenine dinucleotide phosphate) oxidoreductase (FNRL) that has been successfully located in two types of PBSs in our recent research (27). There are 85 primary amine groups in all the subunits in sPBS core, among which 79 are from lysine residues and 6 are N-terminal amine group from six core components (table S5). Because of the redundancy of each PBS core subunit, there are 688 primary amine groups in total that are theoretically available for our cross-linking chemistry (table S5), underlying our identification of the cross-linking network, which allows distinct distance restraints for constructing and validating our sPBS core model (Fig. 3).
Fig. 2. Identified cross-links in different types of sPBS.
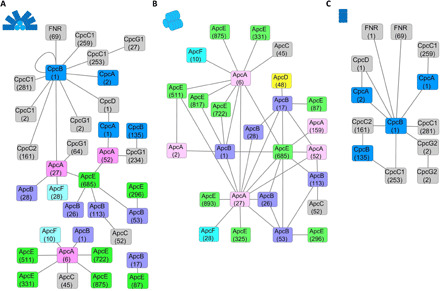
The subunit cross-linking networks, prepared by Cytoscape (41), are shown for (A) WT-sPBS and two mutants including (B) CK-PBS and (C) CpcL-PBS. Cartoon representation of each PBS type is shown in the inset of each panel (top left), where WT-sPBS consists of six rods and a tricylindrical core. CK-PBS contains only a tricylindrical core, and CpcL-PBS is a single rod. The numbers represent the location of the cross-linked residues in each subunit (octagon). ApcA (light pink), ApcB (light purple), ApcC (gray), ApcD (yellow), ApcE (green), and ApcF (cyan). CpcA/B (blue), all other linker proteins (gray)
Fig. 3. sPBS core structure evaluated by observed cross-links.
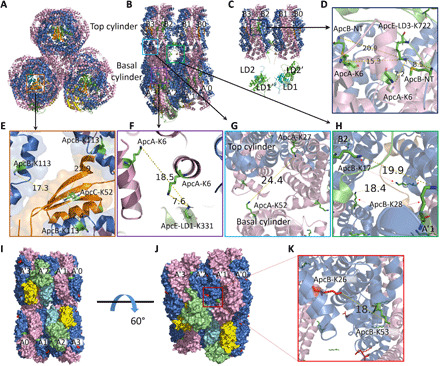
(A) Side view (cylinder perpendicular to the screen) and (B) side view (cylinder parallel to the screen) and (C) another view of (B) with two bottom cylinders removed, showing first two linker domains of ApcE, i.e., LD1/LD1′ and LD2/LD2′, respectively. (D) ApcE-LD3-K722 is cross-linked to ApcA-K6 and ApcB-NT. (E) ApcC-K52 is surrounded by three ApcB-K113. (F) ApcE-LD1-K331 to ApcA-K6 in basal cylinder. (G) Inter-cylindrical cross-link, ApcA-K27–ApcA-K52. (H) Inter-cylindrical cross-link, ApcB-K17–ApcB-K28. (I to K) Inter-hexameric cross-link, ApcB-K26–ApcB-K53. ApcA (light pink), ApcB (blue), ApcC (orange), ApcD (yellow), ApcE (green), and ApcF (cyan).
Model evaluation by chemical cross-links
BS3-H12/D12 is one of the most widely used cross-linkers, having a spacer length of 11.4 Å. The use of an isotopically encoded cross-linker such as this increases the confidence and accuracy of cross-linked peptide identification, with advantages for studying large protein complexes compared to non–isotope encoded cross-linkers. Each end of the cross-linker can form a covalent bond with a primary amine (lysine side chain or protein N terminus). If two such functional groups are within reach of ~30 Å, the cross-linker tends to react with either/both and to yield a mono-link or a cross-link, which can be identified by liquid chromatography–tandem MS (LC-MS/MS), revealing structural information of those two functional groups (35).
In the cross-linked WT-PBS and CK-PBS sample, we successfully identified a total of 35 PBS core subunit interactions [representative MS and product-ion (MS/MS) spectra of cross-linked peptides are shown in figs. S5 and S6]. We then mapped them individually to evaluate the PBS core model, with several focusing regions: first, interfaces between the three linker domains from ApcE and ApcA/B hexamers; second, interfaces of small linker protein, ApcC, and end discs of each cylinder; and last, but not least, relative orientation/twisting mode of three cylinders in our model. For a better visualization, the core structure of sPBS is shown in the side view or bottom view, respectively (Fig. 3, A, B, C, and I). ApcE-LD3-K722, located in the ApcE-LD3/LD3′ domain, which is tucked in the top cylinder, was found to be cross-linked to ApcA-K6 and N-terminal amine of ApcB (ApcB-NT) (Fig. 3D and table S3, #29 and #17, respectively). The measured distances of ApcE-LD3-K722 to ApcA-K6 from two αβ units are 7.2 and 15.3 Å, respectively, and the measured distances of ApcE-LD3-K722 to two ApcB-NTs are 8.5 and 20.9 Å, respectively, consistent with the cross-linking chemistry (11.4-Å arm span). ApcE-LD1-K331, located in one basal cylinder, was found to be cross-linked to K6 from two ApcA subunits at the distances of 7.6 and 18.5 Å (Fig. 3, B and F; table S2, #24; and table S3, #27). This cross-link was actually found in both WT-PBS and CK-PBS. ApcC is a small linker protein, belonging to CpcD linker family. Figure 3 (A and E) shows the cross-links between ApcC-K52 and ApcB-K113 (table S3, #32). That ApcA/B trimer has C3 symmetry does not mean that ApcC can randomly adopt any one of three. It seems that each linker domain of ApcE that faces the ApcC side interacts with ApcC and determines the orientation of ApcC (Fig. 4A).
Fig. 4. The sPBS core structure reveals an acute X-shape of the two basal cylinders.
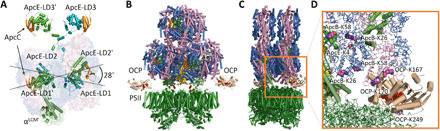
(A) Crossover (side view) of the two basal cylinders, front relative to another (back). Dashed-dotted lines show the general trend of the two sets of linker domains, adopting an acute 28° angle relative to each other. ApcE/ApcE′ (green/teal). ApcC (orange). (B) PBS core sitting on the top of PSII (fully stretched), showing two cavities between uneven PBS basal cylinders and PSII (dark green) that allow orange carotenoid proteins (OCPs) (wheat with red pigment) to interact at the putative geometry. (C) Another view of PBS core (fully stretched) sitting on one PSII dimer. It is the tilting of one end of each basal cylinder that allows access of molecules, such as OCP. (D) Amino acid residues (purple sphere) that are involved in cross-linking to PBS core subunits.
Intralinks, the linked peptides that are from the same protein (subunit), are usually considered as less useful for studying protein-protein interactions. However, if the subunit(s) is redundant, then the nominal intralinks could be authentic cross-links between two redundant subunits, providing 3D structural information of those subunits. A cross-link was found with high confidence (fig. S6 and table S6, #2) between ApcA-K27 and ApcA-K52. We noticed that there are spatial conflicts of this cross-link if it is mapped onto the structure of ApcA. It is also unlikely that this cross-link is from an ApcA/B trimer (disc) or an ApcA/B hexamer or a single cylinder. After a global searching on our PBS core model, we determined that this cross-link can only be successfully mapped onto two ApcA subunits that are from the top cylinder B3/B0 (AcpA-K27) and the basal cylinders A′3/A0 (ApcA-K52), respectively (Fig. 3G), but not other discs, indicating asynchronous staggering/twisting of top cylinder relative to basal cylinder. Another example of a falsely annotated intralink from the software is ApcB-K17–ApcB-K28, which links two ApcB subunits from B2 trimer (top cylinder) and A′1 cylinder, respectively (Fig. 3H and table S6, #3), since spatial conflicts exist in a single ApcB subunit. We consider that these cross-links testified the orientation/twisting of the top cylinder relative to two basal cylinders in our model. In addition, ApcB-K26–ApcB-K53 was found to be cross-linked not in a single subunit; instead, it is between two discs (face to face), A′2/A′1, which are oriented in a back-to-back manner (Fig. 3K and table S6, #1). Overall, the multiple chances of forming structurally relevant cross-links in those cases testify to the reliability of the constructed sPBS core model.
One cross-link showing extra-long cross-linked distances is associated with ApcE-K87 in the middle of a flexible loop on sApcE, i.e., ApcE-K87–ApcB-K17 in both of WT-PBS and CK-PBS (Fig. 2, A and B; table S2, #18; and table S3, #22). The PB loop, consisting of 65 amino acids with primarily basic residues, has been proposed as an anchoring arm attached to the thylakoid membrane (fig. S2B), possibly through electrostatic interaction (2, 36). Consequently, the PB loop can be highly flexible in the absence of the membrane, resulting in the cross-links that cannot form if the PB-loop is anchored on the membrane/RCs. In the cryo-EM structure, the PB loop is not observed (18, 19), possibly owing to the flexible nature of this domain. It should be noted that without the binding partners of the PB-loop, i.e., the membrane/RCs, the structural prediction of this region using any program seems uncertain (13). The cross-linking chemistry and cryo-EM structure may capture and reflect different protein conformers in terms of the loop orientation. To understand its precise location and functionality, various mutants under different chemical environments are needed for adequate cross-linking restraints, which will facilitate the downstream computational simulation. Overall, selection of CK-PBS represented a successful strategy to pinpoint the subunit interaction network. The cross-linked distances supported very well the PBS core structure proposed by computational prediction.
DISCUSSION
The PBS core in Synechocystis 6803 represents a classical tricylindrical core with C2 symmetry (7), which contains various Apc units (copy numbers), i.e., ApcA (32), ApcB (34), ApcD (2), ApcE (2), ApcF (2), and ApcC (6) (table S5). ApcD, ApcE, ApcF, and ApcC are arranged in mirror positions in two basal cylinders. The symmetrical feature of PBS correlates well with that of a dimeric PSII, the most active form in cyanobacteria, red algae, and higher plants. Captured energy is funneled from the PBS rod to the PBS core unidirectionally and then reaches the terminal energy emitters, ApcE and ApcD (37), where excitation energy is lastly transmitted to Chl a in PSII and PSI for photochemical reactions (12). The interface between the PBS core structure and RCs is of great interest in understanding the energy transmission mechanism and dynamic process under varying environmental conditions. Traditionally, two basal cylinders have been described as antiparallel and evenly covering the thylakoid membrane surface/RCs. Previous research techniques have never been able to elucidate the detailed tomography of two basal cylinders relative to the thylakoid membrane surface. Our proposed tricylindrical PBS core model, however, reveals that the two basal cylinders do not adopt a perfect antiparallel configuration, but rather, the two basal cylinders are arranged in an acute X-shape at an angle of ~28° relative to each other (Fig. 4A). We start with our previously proposed PBS-PSII model (13) [described in figure 4 of (13)] constrained by the structural proteomic data (2) and further propose that each basal cylinder is tilted 14° relative to the thylakoid membrane (Fig. 4, B and C). Specifically, ApcD, from the discs A3/A′3, and αLCM, from the discs A2/A′2, comprise a protrusion that preferentially touches the surface of thylakoid membrane. The distal disc A0/A′0, however, tilts away from the thylakoid membrane surface, like an open mouth (Fig. 4, A and C). This orientation leaves an open space between the thylakoid membranes and discs A1/A′1 and A0/A′0 (Fig. 4, B and C). The extra discs on both the top cylinder and two basal cylinders may reflect the functional connection difference between rods and PBS core in the block-shaped PBS (G. pacifica), hemiellipsoidal-shaped PBS (P. purpureum), and the hemidiscoidal PBS (Synechocystis 6803). It seems that cyanobacterial tricylindrical model has increased surface area for attachment of (six) rods, in contrast to that of Griffithsia and Porphyridium, both of which have 14 rods attached to a smaller PBS core directly or indirectly, in a much more compact manner. It seems, although, that six rods on the PBS core in cyanobacteria may have more flexibility relative to each other. This may partially explain why a high-resolution cryo-EM structure is not yet available in cyanobacteria due to the increased rod flexibility.
A previous study indicated that ApcE-K87 tends to interact with CP47-K227 from PSII (2) and could possibly be a critical structural basis dictating excitation energy migration from PBS to the RCs, specifically PSII. With regard to PSI, a chemical cross-link between the N-terminal of PSI and PBS core has also been reported (2), namely, PsaA-K30–ApcB-K17. Pigment-protein complexes involved in light-energy capture and chemical conversion are not evenly distributed in the thylakoid membranes (5, 6, 38). In some of thylakoid membrane regions, PSII is enriched, whereas in other regions, PSI is aggregated in patches that allow biochemical isolation and characterization (5). The reducing side of PSII and PSI has their own characteristics: Unlike the flat surface at the PSII reducing side, PsaC, PsaD, and PsaE of the PSI reducing side comprise a protrusion collectively protecting the iron sulfur centers.
The acute X-shaped configuration at the bottom of the PBS core provides structural accommodations for other regulatory factors on either the PBS core or the reducing side of PSII. Orange carotenoid protein (OCP), for example, is a critical photoprotective protein that dissipates excess excitation energy under stress conditions and disarms harmful reactive oxidative species arising from photosynthetic machinery. It is commonly held that the active form of OCP (OCPR) binds to core subunits in cyanobacterial PBS as the primary energy quenching sites (39). ApcE has been proposed to be a promising target for OCPR (40), given the unique shift of its bilin-binding pocket compared to that of the surrounding ApcA/B subunits, which are all conserved Cys81 bilin sites. The tilted angle of each basal cylinder provides sufficient room for the 34-kDa OCP to move in and bind (Fig. 4B) and, perhaps, to accommodate the fluorescence recovery protein (39), a 14-kDa protein that can accelerate the detachment of OCP from its binding sites followed by subsequent deactivation, and other essential regulators in the photoprotection process. In our model, the extra space between two face-to-face trimers A1/A0 (or A′1/A′0) and the PSII reducing side can accommodate the entrance of OCP without any spatial conflicts (Fig. 4D).
In our model, plausible excitation energy transfer routes can be predicted in PBS core and PBS-core-PSII supercomplex as well. In the red algal structure, there are 48 bilins in the core structure (18, 19). In our structure, four extra trimers were modeled onto the red algal PBS core (i.e., B0, B4, A0, and A′0; Fig. 1A), giving a total of 72 bilins in a trycylindrical PBS core (Fig. 5A). The model shows that if the four extra trimers are removed, then the excitation-energy transfer distances between each bilin are comparable to those of red algae. The staggered trimer orientation between two cylinders was retained, similar to that of the red algal structure. In each basal cylinder, we noticed that each trimer adopts a specific angle twist (clockwise) relative to the previous trimer A3/A′3 (Fig. 5, E and F), a feature that has not been reported previously (18, 19). The top cylinder has a different twisting pattern than that of basal cylinders. Each twist angle in the basal cylinder may seem small (11° of A′2 versus A′3, 18° of A′1 versus A′2, and 11° of A′0 versus A′1); however, the twists accumulate to afford a close orientation of the B0 chromophore (B0-α) to that of A′0 (A′0-α), rather than to the α chromophore on another basal cylinder A3 (Fig. 5H). This is due to the discontinuous twisting of four trimer discs (11° of B2 versus B3, −10° of B1 versus B2, and 11° of B0 versus B1). In other words, the A0 disc has a cumulative twisting angle of 40° relative to A3 versus 12° of B0 to B3. The smallest distance between B0-α and A′0-α is estimated to be 34.4 Å, in contrast to that of B0-α and A3, which is 43.5 Å (Fig. 5H). Both distances can allow energy transfer to occur; the former, however, may be more efficient. ApcF in A2/A′2 and the linker domains of ApcE could contribute to these phenomena. Why the top cylinder has a different twisting pattern than the basal cylinders remains unclear. The distance between bilins A′2-βApcF and A′2-αLCM is 20.6 Å (Fig. 5G), the shortest distance between bilin pairs in the core, consistent with the value in red algal PBS (18, 19). The distance between bilin A′2-αLCM and A′3-ApcD is 31.0 Å. The extra discs and the enlarged twisting discs on two ends of each cylinder comprise the major structural differences between red algal PBS core and cyanobacterial PBS core; thus, the functional differences are waiting for future research.
Fig. 5. PBS bilins and energy transfer pathways.
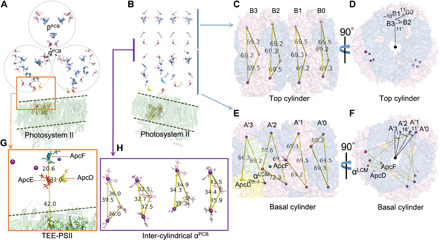
(A) Overview of PBS core bilins in a side view (cylinder perpendicular to the screen) sitting on a PSII dimer (green cartoon). αβPCB are colored as purple and blue, respectively. (B) Side view of top cylinder (parallel to the screen). (C and D) Top cylinder showing disc/bilins twisting in a clockwise-counterclockwise-clockwise fashion, cylinder perpendicular to the screen in (D). (E and F) Cylinder disc/bilins twisting in basal cylinder, cylinder perpendicular to the screen in (F). (G) Overview of terminal energy emitter (TEE) excitation energy transfer pathway in PBS-core-PSII complex in a side view. Bilins are labeled. (H) Orientation of inter-cylindrical bilins.
For our previously reported PBS-core-PSII complex, the energy transfer routes can also be predicted from our new model of bilins in PBS. The shortest distance between the bilin of αLCM and a Chl a in CP47 is 42 Å. Note that the PB loop in ApcE has not been resolved at the present time, and the real distance remains a subject for future research. The distance of bilin of ApcD and Chl a in CP47 is in a comparable range to that of the αLCM bilin. It appears that, on one side of the PBS core, energy transfers from bilins on B0 to A′0 (B3 to A0) and then horizontally transfers to ApcE where it feeds into the Chl a in CP47. Given that the PBS core and PSII share the same C2 symmetry axis, there is another energy transfer route along B3 to A0 and then to A2-αLCM and ends in Chl a in CP47, the mirror component in another PSII monomer.
In summary, we constructed the first detailed PBS core structure of cyanobacteria Synechocystis PCC6803 to reveal an acute X-shape of the two basal cylinders, rather than a perfect antiparallel orientation. Our study provides a structural basis for energy migration and its related regulation associated with the transient association between PBS and RCs. The proximate location of the pigment proteins in the functional complex is essential to promote efficient energy transfer and regulation.
MATERIALS AND METHODS
Cyanobacteria culture and PBS purification
The CK-PBS and CpcL-PBS mutant strains were gifts from G. Ajlani (33, 34). Cyanobacterial strains were grown in BG-11 medium at 30°C with 50 μmol photo m−2. The growth medium was supplemented with 20 mM TES {2-[(2-hydroxy-1,1-bis(hydroxymethyl) ethyl) amino] ethanesulfonic acid, N-[tris(hydroxymethyl)methyl]-2 aminoethanesulfonic acid}–KOH (pH 7.5). The harvested cell cultures were resuspended in 0.8 M K-phosphate buffer and incubated with protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA) and deoxyribonuclease (Sigma-Aldrich, St. Louis, MO). Cell lysates were obtained through three rounds of a French press at 4°C and pH 7.5. Triton X-100 (2%; Sigma-Aldrich, St. Louis, MO) was then added, followed by 30-min incubation at room temperature. The blue supernatant was loaded onto a sucrose gradient for overnight ultracentrifugation (370,000g). The purified PBS was then ready for cross-linking.
Chemical cross-linking and proteolytic digestion
CK-PBS, dissolved in 0.4 M K-phosphate buffer at 0.1 μM, and the isotopic-coded BS3 cross-linker mixture (BS3-H12/D12, Creative Molecule Inc.) were incubated together for 10 min in the dark at 25°C, where the cross-linker was in 10-, 50-, 100-fold excess with respect to the PBS-CK. Tris (1 M) was added to give a final concentration of 50 mM to stop the cross-linking chemistry. For quenching and desalting purposes, Zeba spin columns (Thermo Fisher Scientific, Waltham, MA) were used following the manufacturer’s protocol. The cross-linked CK-PBS were then desalted and purified by acetone precipitation to prepare them for enzymatic digestion. The protein pellets were dissolved in 20 μl of 8 M urea for 30 min at room temperature for denaturation, followed by 30-min incubation with 2.5 mM TCEP [tris(2-carboxyethyl)phosphine] at 37°C. Iodoacetamide was then added to the reaction sample at a final concentration of 5 mM for 30 min at 25°C in dark. The first-step digestion was achieved by Lys-C (0.05 μg/μl) for 2-hour incubation at 37°C. Dilution to 1 M urea with tris buffer (100 mM) was needed for the subsequent trypsin digestion, which required overnight incubation at 37°C with a trypsin:protein ratio of 1:25. The digested mixture was quenched by 0.1% formic acid the next day.
LC-MS/MS
The peptide mixture was loaded onto a C18 trapping column (180 μm × 2 cm) and C18 Symmetry (5 μm, 100 Å; Waters, MA) for desalting with phase A (water with 0.1% formic acid) of Dionex Ultimate high-performance liquid chromatography (Thermo Fisher Scientific, Waltham, MA). Peptide samples were then eluted and separated on a reversed-phase C18 column (100 μm × 15 cm; C18 Symmetry, 5 μm, 100 Å; Waters, MA) with a 105-min gradient: increasing phase B (80% acetonitrile, 20% water, and 0.1% formic acid) from 2 to 40% for the first 90 min, from 40 to 95% for the next 10 min, and equilibrated at phase A for another 5 min. The flow was controlled at 250 nl/min and sprayed through a Nanospray Flex source coupled with a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Waltham, MA). The mass spectrometer was operated at the following settings: 1.8-kV spray voltage; positive-ion mode; MS1 acquisition at 70,000 resolving power at m/z (mass/charge ratio) 200; Automatic Gain Control (AGC) target 3 × 106; MS/MS acquisition at 17,500 resolving power at m/z 200; AGC target: 1 × 105; maximal injection time of 100 ms. Data-dependent acquisition and with “mass tag” were used. In the latter method, for charge states of 2 to 7, the delta mass differences were set as m/z ±6.03762, ±4.02508, ±3.01881, ±2.41505, ±2.01254, and ±1.72503, respectively. Singly charged species were excluded, and each charge state was acquired independently.
Cross-linked peptide identification
MS and MS/MS data were imported into pLink software for identification. Searching parameters were as follows: Enzyme was trypsin (up to three missed cleavages) with 20 parts per million (ppm) of precursor tolerance and 60 ppm of fragment tolerance. Variable modifications were as follows: oxidation of M and deamidation of N, Q, and N terminus. The minimum number of peptide length is 6 with peptide mass of 600 Da, and the maximum number is 60 with peptide mass of 6000 Da. The false discovery rate was equal to or smaller than 5% at spectral level with a 10-ppm filter tolerance. Isotopic pairs were examined manually in raw files to confirm cross-link identification. The theoretical product-ion mass list was calculated in Protein Prospector. Manual validation of the fragments was further performed as a comparison to the pLink assignment.
The cross-linking dataset was also submitted to isotopically-coded cleavable crosslinking analysis software suite (ICC-CLASS) for cross-linking identification. In the DXMSMSMatch program, the settings were as follows: Cross-linker was DSS (disuccinimidyl suberate) with DX of 12.07532. The DX mass tolerance was 0.013 Da, and the retention time tolerance was 60 s. The filtered DX mass tolerance was 5 ppm, and the filter DX time window was 60 s. The digestion sites were K and R, including cross-link sites. Missed digest sites was up to 4. Assigned cross-link sites are K- and N-terminal primary amines. The precursor tolerance was 5 ppm, and the fragment tolerance was 30 ppm. Manual validation of the fragments was carried out as the same way mentioned above.
Homology modeling and structural construction
Homology simulation was performed on the I-TASSER server. Detailed information and adopted template are described in the homology modeling section.
Supplementary Material
Acknowledgments
We thank G. Ajlani for the CK and ∆AB Synechocystis mutants and helpful discussions. We thank H. Zhang for technical help with the LC-MS/MS and X. (Roger) Liu for technical help with table/figure preparation in SM and helpful discussions and Michael L. Gross for suggestions and edits of the manuscript. Funding: This research was funded by the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Office of Basic Energy Sciences (grant DE-SC0001035). Support was also provided by the DOE, Office of Basic Energy Sciences, Photosynthetic Systems Program (grant DE-FG02-07ER15902 to R.E.B. and H.L. and grant DE-FG02-99ER20350 to H.B.P.). Partial support was also provided by the National Institute of General Medical Sciences of the NIH (grant 2P41GM103422 to Michael L. Gross). Author contributions: H.L. and R.E.B. designed, performed, and supervised the research. M.M.Z. analyzed the data and drafted a preliminary version of the manuscript and prepared figs. S4 and S5. D.A.W. and M.C. analyzed the data. H.L. and D.A.W. prepared tables S2 to S4. M.C. prepared fig. S6. H.B.P., R.E.B., and H.L. wrote and edited the manuscript. H.L. prepared all the other figures and tables. All authors discussed and reviewed the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. MS raw and processed data and PBS core structure PDB (.pse) file were deposited in PRIDE with accession number: PXD017873; project: http://central.proteomexchange.org/cgi/GetDataset?ID=PXD017873; reviewer account details: username: reviewer20380@ebi.ac.uk; password: DDOeVduf. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eaba5743/DC1
REFERENCES AND NOTES
- 1.Mullineaux C. W., Electron transport and light-harvesting switches in cyanobacteria. Front. Plant Sci. 5, 7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu H., Zhang H., Niedzwiedzki D. M., Prado M., He G., Gross M. L., Blankenship R. E., Phycobilisomes supply excitations to both photosystems in a megacomplex in cyanobacteria. Science 342, 1104–1107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adir N., Bar-Zvi S., Harris D., The amazing phycobilisome. Biochim. Biophys. Acta Bioenerg. 1861, 148047 (2020). [DOI] [PubMed] [Google Scholar]
- 4.de Marsac N. T., Cohen-bazire G., Molecular composition of cyanobacterial phycobilisomes. Proc. Natl. Acad. Sci. U.S.A. 74, 1635–1639 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacGregor-Chatwin C., Sener M., Barnett S. F. H., Hitchcock A., Barnhart-Dailey M. C., Maghlaoui K., Barber J., Timlin J. A., Schulten K., Hunter C. N., Lateral segregation of photosystem I in cyanobacterial thylakoids. Plant Cell 29, 1119–1136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho M.-Y., Niedzwiedzki D. M., Gregor-Chatwin C. M., Gerstenecker G., Hunter C. N., Blankenship R. E., Bryant D. A., Extensive remodeling of the photosynthetic apparatus alters energy transfer among photosynthetic complexes when cyanobacteria acclimate to far-red light. Biochim. Biophys. Acta Bioenerg. 1861, 148064 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Arteni A. A., Ajlani G., Boekema E. J., Structural organisation of phycobilisomes from Synechocystis sp. strain PCC6803 and their interaction with the membrane. Biochim. Biophys. Acta Bioenerg. 1787, 272–279 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Glazer A. N., Light guides. Directional energy transfer in a photosynthetic antenna. J. Biol. Chem. 264, 1–4 (1989). [PubMed] [Google Scholar]
- 9.Bald D., Kruip J., Rögner M., Supramolecular architecture of cyanobacterial thylakoid membranes: How is the phycobilisome connected with the photosystems? Photosynth. Res. 49, 103–118 (1996). [DOI] [PubMed] [Google Scholar]
- 10.MacColl R., Cyanobacterial phycobilisomes. J. Struct. Biol. 124, 311–334 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Zlenko D. V., Krasilnikov P. M., Stadnichuk I. N., Structural modeling of the phycobilisome core and its association with the photosystems. Photosynth. Res. 130, 347–356 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Mullineaux C. W., Phycobilisome-reaction centre interaction in cyanobacteria. Photosynth. Res. 95, 175–182 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Liu H., Blankenship R. E., On the interface of light-harvesting antenna complexes and reaction centers in oxygenic photosynthesis. Biochim. Biophys. Acta Bioenerg. 1860, 148079 (2019). [DOI] [PubMed] [Google Scholar]
- 14.W. A. Sidler, in The Molecular Biology of Cyanobacteria (Springer, 1994), pp. 139–216. [Google Scholar]
- 15.Yamanaka G., Glazer A. N., Williams R. C., Molecular architecture of a light-harvesting antenna. Comparison of wild type and mutant Synechococcus 6301 phycobilisomes. J. Biol. Chem. 255, 11104–11110 (1980). [PubMed] [Google Scholar]
- 16.Glauser M., Bryant D. A., Frank G., Wehrli E., Rusconi S. S., Sidler W., Zuber H., Phycobilisome structure in the cyanobacteria Mastigocladus laminosus and Anabaena sp. PCC 7120. Eur. J. Biochem. 205, 907–915 (1992). [DOI] [PubMed] [Google Scholar]
- 17.Elmorjani K., Thomas J.-C., Sebban P., Phycobilisomes of wild type and pigment mutants of the cyanobacterium Synechocystis PCC 6803. Arch. Microbiol. 146, 186–191 (1986). [Google Scholar]
- 18.Zhang J., Ma J., Liu D., Qin S., Sun S., Zhao J., Sui S.-F., Structure of phycobilisome from the red alga Griffithsia pacifica. Nature 551, 57–63 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Ma J., You X., Sun S., Wang X., Qin S., Sui S.-F., Structural basis of energy transfer in Porphyridium purpureum phycobilisome. Nature 579, 146–151 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Reuter W., Wiegand G., Huber R., Than M. E., Structural analysis at 2.2 Å of orthorhombic crystals presents the asymmetry of the allophycocyanin–linker complex, AP LC7.8, from phycobilisomes of Mastigocladus laminosus. Proc. Natl. Acad. Sci. U.S.A. 96, 1363–1368 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dagnino-Leone J., Figueroa M., Mella C., Vorphal M. A., Kerff F., Vásquez A. J., Bunster M., Martínez-Oyanedel J., Structural models of the different trimers present in the core of phycobilisomes from Gracilaria chilensis based on crystal structures and sequences. PLOS ONE 12, e0177540 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marx A., Adir N., Allophycocyanin and phycocyanin crystal structures reveal facets of phycobilisome assembly. Biochim. Biophys. Acta 1827, 311–318 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Peng P. P., Dong L. L., Sun Y. F., Zeng X. L., Ding W. L., Scheer H., Yang X., Zhao K. H., The structure of allophycocyanin B from Synechocystis PCC 6803 reveals the structural basis for the extreme redshift of the terminal emitter in phycobilisomes. Acta Crystallogr. D Biol. Crystallogr. 70, 2558–2569 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy A., Kucukural A., Zhang Y., I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houmard J., Capuano V., Colombano M. V., Coursin T., Tandeau de Marsac N., Molecular characterization of the terminal energy acceptor of cyanobacterial phycobilisomes. Proc. Natl. Acad. Sci. U.S.A. 87, 2152–2156 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang K., Ding W.-L., Höppner A., Zhao C., Zhang L., Hontani Y., Kennis J. T. M., Gärtner W., Scheer H., Zhou M., Zhao K.-H., The terminal phycobilisome emitter, LCM: A light-harvesting pigment with a phytochrome chromophore. Proc. Natl. Acad. Sci. U.S.A. 112, 15880–15885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H., Weisz D. A., Zhang M. M., Cheng M., Zhang B., Zhang H., Gerstenecker G. S., Pakrasi H. B., Gross M. L., Blankenship R. E., Phycobilisomes harbor FNRL in Cyanobacteria. mBio 10, e00669-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang L., Liu X., Li Y., Liu C.-C., Yang F., Zhao J., Sui S.-F., Structural organization of an intact phycobilisome and its association with photosystem II. Cell Res. 25, 726–737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong C., Tang A., Zhao J., Mullineaux C. W., Shen G., Bryant D. A., ApcD is necessary for efficient energy transfer from phycobilisomes to photosystem I and helps to prevent photoinhibition in the cyanobacterium Synechococcus sp. PCC 7002. Biochim. Biophys. Acta Bioenerg. 1787, 1122–1128 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y., Skolnick J., SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865–871 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Calzadilla P. I., Muzzopappa F., Sétif P., Kirilovsky D., Different roles for ApcD and ApcF in Synechococcus elongatus and Synechocystis sp. PCC 6803 phycobilisomes. Biochim. Biophys. Acta Bioenerg. 1860, 488–498 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Gindt Y. M., Zhou J., Bryant D. A., Sauer K., Spectroscopic studies of phycobilisome subcore preparations lacking key core chromophores: Assignment of excited state energies to the Lcm, β18 and αAP-B chromophores. Biochim. Biophys. Acta Bioenerg. 1186, 153–162 (1994). [DOI] [PubMed] [Google Scholar]
- 33.Thomas J. C., Ughy B., Lagoutte B., Ajlani G., A second isoform of the ferredoxin:NADP oxidoreductase generated by an in-frame initiation of translation. Proc. Natl. Acad. Sci. U.S.A. 103, 18368–18373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ajlani G., Vernotte C., Dimagno L., Haselkorn R., Phycobilisome core mutants of Synechocystis PCC 6803. BBA-Bioenergetics 1231, 189–196 (1995). [Google Scholar]
- 35.Sinz A., The advancement of chemical cross-linking and mass spectrometry for structural proteomics: From single proteins to protein interaction networks. Expert Rev. Proteomics 11, 733–743 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Zlenko D. V., Krasilnikov P. M., Stadnichuk I. N., Role of inter-domain cavity in the attachment of the orange carotenoid protein to the phycobilisome core and to the fluorescence recovery protein. J. Biomol. Struct. Dyn. 34, 486–496 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Bryant D. A., Guglielmi G., de Marsac N. T., Castets A.-M., Cohen-Bazire G., The structure of cyanobacterial phycobilisomes: A model. Arch. Microbiol. 123, 113–127 (1979). [Google Scholar]
- 38.Folea I. M., Zhang P., Aro E. M., Boekema E. J., Domain organization of photosystem II in membranes of the cyanobacterium Synechocystis PCC6803 investigated by electron microscopy. FEBS Lett. 582, 1749–1754 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Kirilovsky D., Kerfeld C. A., Cyanobacterial photoprotection by the orange carotenoid protein. Nature Plants 2, 16180 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Stadnichuk I. N., Yanyushin M. F., Maksimov E. G., Lukashev E. P., Zharmukhamedov S. K., Elanskaya I. V., Paschenko V. Z., Site of non-photochemical quenching of the phycobilisome by orange carotenoid protein in the cyanobacterium Synechocystis sp. PCC 6803. Biochim. Biophys. Acta Bioenerg. 1817, 1436–1445 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eaba5743/DC1