

Zinc anodes are a powerful platform for understanding metal deposition and for low-cost electrical energy storage.
Abstract
Scalable approaches for precisely manipulating the growth of crystals are of broad-based science and technological interest. New research interests have reemerged in a subgroup of these phenomena—electrochemical growth of metals in battery anodes. In this Review, the geometry of the building blocks and their mode of assembly are defined as key descriptors to categorize deposition morphologies. To control Zn electrodeposit morphology, we consider fundamental electrokinetic principles and the associated critical issues. It is found that the solid-electrolyte interphase (SEI) formed on Zn has a similarly strong influence as for alkali metals at low current regimes, characterized by a moss-like morphology. Another key conclusion is that the unique crystal structure of Zn, featuring high anisotropy facets resulting from the hexagonal close-packed lattice with a c/a ratio of 1.85, imposes predominant influences on its growth. In our view, precisely regulating the SEI and the crystallographic features of the Zn offers exciting opportunities that will drive transformative progress.
AFFORDABLE ELECTROCHEMICAL STORAGE: PROMISES AND CHALLENGES OF ZINC BATTERIES
The development of affordable energy harvesting and storage technology has in recent times emerged as a grand challenge in lowering humanity’s dependence on fossil fuels. Cost-effective storage of electricity has, at the same time, emerged as a crucial requirement to lower humanity’s carbon footprint by electrifying transportation and by enabling scalable use of renewable-sourced power on the electric grid. After decades of research and commercialization efforts, photovoltaic (PV) technology has, in recent years, advanced rapidly as a high-efficiency and cost-effective option for harvesting renewable electricity from the Sun (1). Advances made in lowering cost and scaling supply of renewable electricity generated from wind are as impressive (2). Figure 1A illustrates one of the two barriers to broad-based use of solar (and wind) as sources of renewable energy. First, the supply of energy from the Sun is intermittent and most concentrated during periods of moderate or low electric power demand. Second, the supply varies seasonally; meaning that variability of the power supply can extend over periods ranging from hours to months. Power interruptions on either of these time scales are disruptive and cost tens to hundreds of billion U.S. dollars in 2002 and 2015, respectively (3, 4). It is straightforward to see that complementary, cost-effective energy storage systems (ESS) are a requirement for “leveling” the supply, shifting the energy from peak to off-peak production periods.
Fig. 1. Assessment of the requirements for affordable EES technologies that are suitable for integration into clean energy generation systems.
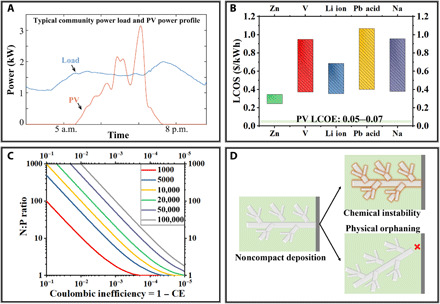
(A) Hourly power profiles for typical power demand and supply from solar-PV. Adapted with permission from (12). (B) Levelized costs of energy (LCOE) production from solar-PV compared with levelized energy of storage (LCOS) costs of representative battery chemistries. Replotted according to (18). Zn-based batteries show the lowest LCOS of ~$0.3/kWh. (C) The plating/stripping Coulombic efficiency (CE) requirements for metal anodes to achieve the cycle life targets. Each curve depicts the correlation between N:P ratio and the Coulombic inefficiency (=1 − CE), assuming 80% capacity retention after a certain number of cycles as specified in the legend. For example, the red curve depicts the relation between N:P ratio and Coulombic inefficiency to meet the goal of a battery cycle life equal to 1000 cycles. N:P ratio is defined as the ratio between the capacities of active materials in the negative electrode(anode) and the positive electrode(cathode), respectively, assuming the materials operate at their theoretical specific capacities. The curves are plotted by solving , where y is N:P ratio, x is Coulombic inefficiency, and T is the cycle life target. (D) Schematic cartoon showing the two key irreversibilities associated with Zn metal electrodes. Noncompact Zn electrodeposits lose electrochemical activity after they chemically react with the electrolyte (chemical instability) or physically detach from the current collector (physical orphaning).
Energy can, of course, be stored via multiple mechanisms, e.g., mechanical, thermal, and electrochemical. Among the various options, electrochemical energy storage (EES) stands out for its potential to achieve high efficiency, modularity, relatively low environmental footprint, and versatility/low reliance on ancillary infrastructure (5, 6). Despite these advantages, the relatively high cost of EES systems limits market penetration, particularly in situations where long-term storage is required to address variability in the energy supply. EES technology based on the intrinsically low-cost element, zinc (Zn), are among the “oldest” batteries; Alessandro Volta’s letter (7) in 1800 described a pile of Cu-Zn couples to generate stable electricity. Primary Zn-MnO2 batteries have been available commercially since the early 20th century (8), but despite a very large body of work, efforts to create rechargeable Zn batteries have been less fruitful. The low cycle life (<100 cycles) (9) typical for these batteries have been attributed to fundamental issues at both electrodes: The metal oxide cathodes exhibit large electrochemical irreversibility above a certain depth of discharge (DOD) (10); the Zn metal anode undergoes a so-called “shape change” process irreversibly depleting the anode metal (11). In recent decades, the cycle life of Zn batteries has been extended by approximately one order of magnitude (i.e., ~1000 cycles). In addition, very recently, fundamental advances in electrolyte, interface, and electrode design suggest that additional substantial increases in reversibility of the Zn electrode may be possible.
To more concretely establish the benefits of these advances in Zn electrode reversibility, we first look at their potential impact on the system (electric power generation and storage) levelized cost (LCOEsystem). On this basis, we will establish basic performance requirements for achieving practically relevant storage. The LCOEsystem is computed using the following formula
| (1) |
The formula takes into account cost (C) and energy (E) terms associated with power generation (in this example, from PV) and storage (12). We may likewise define a levelized cost of storage (LCOS) as a quantitative metric for more formally comparing EES and other storage options, as well as for guiding choices among EES options
| (2) |
Here, I0 is the one-time installment cost, r is the discount rate that relates future value to present value (usually 5 to 8%), CESS,t and EESS,t are the maintenance cost and the energy production in year t after installation of the EES system (13). We note that for the EES systems of interest in this review, the annual maintenance cost CESS is minimal, e.g., 1 to 2% of the initial investment, I0 (14), which means that the CESS,t term in Eq. 2 is generally small and we may approximate it as
| (3) |
Figure 1B reports LCOS values for a number of EES technologies of contemporary interest. These values are evidently 6 to 20 times larger than those typically reported for pumped-storage hydroelectricity and compressed air energy storage (15), explaining the traditional dominance of these ESS options. They are also substantially higher than reported LCOE for renewable electric power generation using solar PV [$0.05 to $0.07/kilowatt-hour (kWh)] or wind ($0.04 to $0.08/kWh) installations (2, 16). At this level of comparison, even Zn batteries, which have the lowest LCOS ranging from $0.245 to $0.345/kWh, will introduce nontrivial additional cost when integrated with solar PV or wind generation technology. Considering that comparable cost of electric power generation from fossil fuels range from $0.03 to $0.15/kWh (17, 18), we predict that meaningful market penetration of PV-EES integrated system will require substantial reduction in the cost of EES, at least to levels comparable with the LCOE of solar-PV technology, i.e., <$0.1/kWh, preferably <$0.05/kWh. An examination of Eq. 3 suggests two approaches for achieving LCOS reductions of this scale: (i) substantially lower I0 and/or (ii) increase EESS. The former is mainly dictated by materials chemistry and the latter by the cycle life of the EES system. That is, a truly complementary EES technology for enabling penetration of renewable power generation on the grid must have at least three traits: inherently low materials cost, scalability for long-term storage, and long cycle life. Secondary Zn batteries easily achieve the first two of these requirements but require a cycle life of 30 to 50 years and one 100% DOD cycle per day, corresponding to 10,000 to 20,000, 100% DOD cycles to be of practical interest for addressing intermittency and variability of the power supply from renewable sources. For example, a Zn-based storage system able to achieve a cycle life of 10,000 is required to achieve a LCOS ≈ $0.1/kWh, and one able to achieve a cycle life above 20,000 is required to achieve a LCOS of <$0.05/kWh. Promising progresses of cathode development toward this goal have been made in the recent years through the rational design of the chemistry and morphology of the active materials, e.g., MnO2 and its derivatives. Cycle lives above a few thousands stable cycles, at practical areal capacities were demonstrated (19, 20). The performance of Zn anodes is one order of magnitude inferior to what has been achieved at the cathodes. While the contemporary Zn electrodes have already demonstrated such cycle life (tens of thousands cycles) at a low DOD of <1% (21), it quickly falls to below 1000 cycles as the battery operates at a greater DOD of 40 to 50% (21, 22).
Multiple big leaps in the design of all components of the electrochemical cell are required to achieve Zn systems that achieve cycle life of 10,000 to 20,000 cycles. Figure 1C, for example, quantitatively assesses the reversibility metrics for the Zn metal anode needed to meet these goals. It shows that with a reasonable N:P ratio below 5:1, a >99.96% plating/stripping efficiency is necessary. We note that this value is widely reported to be <90% in cells without proper control over the plating/stripping reaction, and the reversibility further deteriorates as areal capacity increases. We note further that unlike the liquid-phase reactions that can be treated in a classical electrochemistry framework with quite good accuracy, the metal plating/stripping reaction that controls the reversibility of the Zn battery anode involves the repeated creation and dissolution of a solid, crystalline phase over cycling. This means that non-negligible, oftentimes appreciable, morphology evolution of the electrode is constantly taking place during normal battery operation. This attribute differentiates metal plating/stripping from classical pure liquid-phase electrochemical reactions—For liquid-phase reactions, the reversibility is mainly dictated by the chemistry and the energy surface associated with the kinetics (23); in contrast, the reversibility of a metal plating/stripping redox couple is dominated by the morphology of the electrodeposits through two main mechanisms: chemical instability and physical orphaning (24, 25). The former describes metals’ propensity for undergoing side reactions with other battery components, e.g., an electrolyte; the latter for detaching from the current collector and thereby losing the electrical connection (Fig. 1D). Both of the two mechanisms are fundamentally attributable to the porous, heterogeneous morphology of the Zn electrodeposits formed in battery recharge and ultimately result in permanent materials loss. As a result, the real plating/stripping efficiency achieved experimentally in electrochemical cells is lower (i.e., oftentimes <90%, sometimes <50%) than one would expect. The noncompact nature of the porous electrodeposits also creates volume expansion in the anode chamber. A perhaps more obvious aftermath of heterogeneous Zn growth is—the metallic, electrically conductive Zn deposits, which exhibit a high modulus of 108 GPa (versus 5 and 10 GPa, for Li and Na, respectively), penetrate the separator with ease, and physically bridge the two electrodes—the battery short (22, 26, 27).
Regulating the morphology evolution of Zn electroplating/stripping with microscopic or even atomic precision is therefore, in our view, the key to high-performance Zn metal anodes for applications in affordable battery systems. With the specific aim of discussing the underlying scientific challenges in-depth, the review focuses on electrolyte, interphase, and anode design principles for achieving highly reversible Zn anodes. A more specific goal is to deconvolute the fundamental principles that govern the electrochemical deposition morphology of Zn. On the basis of that framework, we will assess promising approaches for regulating Zn electrodeposition morphology for achieving the exceptional levels of reversibility required to drive practical interest.
THE POLYMORPHISM OF ZINC METAL DEPOSITS
The electrodeposits formed at any metal battery anode upon recharge are crystalline in nature. For example, zinc is a hexagonal close-packed (HCP) crystal. Although the Zn electrodeposits shown in Fig. 2 share the same HCP crystal lattice, they exhibit diverse morphologies as the deposition condition varies. The first challenges to controlling the reversibility of the process are therefore to categorize and to understand this obvious polymorphism. Here, we use two main descriptors of morphology: (i) the geometry of the microstructural building block and (ii) the assembly mode of the building blocks. The former describes the symmetry and dimension of the microstructural building blocks, e.g., one-dimensional (1D) wires with Φ = 1 μm and 2D plates with Φ = 5 μm; whereas the latter depicts the alignment/orientation of the building blocks with respect to the electrode surface in creating the assemblies that form the macroscopic electrodeposition layer, e.g., randomly oriented (Fig. 2, A to C), horizontally oriented (Fig. 2D), and vertically oriented (Fig. 2E) structures. Notably, the dendritic structure in Fig. 2E points outward from the electrode surface into the bulk electrolyte as a manifestation of the chemotaxis-like growth, which will be discussed later. The significance of this categorization becomes clear when one examines the plating/stripping reversibility for each of the morphologies: horizontally aligned plate-like (over 99%) > nonaligned plate-like (80 to 90%) ≈ randomly oriented moss/wire-like (80% to 90%) > vertically oriented dendrites (<50%) (28–30).
Fig. 2. Representative morphologies of Zn metal deposits obtained in electrodeposition or chemical reactions.

Scanning electron microscopy (SEM) images of electrodeposited (A) wire-like, (B) moss-like, (C) randomly oriented plate-like, (D) horizontally aligned plate-like, and (E) dendritic Zn metal. SEM images of chemically synthesized (F) horizontally aligned plate-like and (G) moss-like Zn metal. Adapted with permission from (22, 28, 130, 131, 136).
The low reactivity of Zn suggests that, unlike Li where a considerable portion of the Coulombic inefficiency comes from active material loss via parasitic reactions with electrolyte components, the inefficiency for Zn is more closely associated with loss of electrochemical access to the deposits (24, 25). This can be understood in a straightforward manner by picturing the microscopic ion conduction/electron conduction contact interface—For dendritic structures, the ion-electron contact spans across the surface of the deposit, meaning that dissolution can take place at the root, breaking the entire dendrite off the electrode (Fig. 1D); for aligned plate-like structures, the interface sits between the compact electrodeposition layer and the liquid electrolyte, and dissolution from the bottom is therefore prohibited. The randomly oriented structures situate in between these two extremes.
On this basis, we contend that to achieve Zn electrodes with exceptional reversibility benchmarked in Fig. 1C, horizontally aligned compact Zn electrodeposition in the anode is required. Likewise, a clear understanding of the fundamental origins of transitions from operating regime where compact deposition gives way to less compact/more open Zn electrodeposit morphologies is an important first step in the search for interventions that eliminate these instabilities. We note further that while we focus here on Zn electrodeposition, these goals are not limited to electrochemically deposited Zn: For example, randomly oriented moss-like and horizontally oriented plate-like Zn crystals (Fig. 2, F and G) are, in fact, observed in vapor deposition and liquid-state chemical reactions, respectively; the moss-like morphology is also widely seen in Li metal anodes. We will soon see that this broader perspective provides a promising path for elucidating the governing principles and for leveraging the conclusions for meaningful control of Zn crystallization in general. For this purpose, we, in the next section, discuss a theoretical framework built upon the classical electrochemistry perspective and then, in the subsequent sections, move on to the critical physicochemical processes distinctive to Zn, e.g., the solid-electrolyte interphase (SEI) and the crystal anisotropy.
FLUXES NEAR THE METAL DEPOSITION INTERFACE
Electrochemical deposition of metals requires reduction of metal ions in an electrolyte at an electronically conductive substrate. The progress of the deposition can therefore be quantified in terms of the reaction flux—the amount of metal ions reduced to elemental metal per unit area of the substrate, per unit time. Multiple transport fluxes control the rate of delivery of metal ions to the interface and the rate of dispersal of the formed metal atoms: ion transport in the bulk electrolyte, ion transport near the electrode, and adatom self-diffusion of the deposited metal on the electrode (Fig. 3A). Because electroneutrality must be maintained outside of the thin equilibrium space charge layer, the interplay among these fluxes and the local electric field can have nontrivial effects on the deposition morphology; in certain regimes, the underlying crystal anisotropy of electrodeposits may be preserved, and in others, it may not.
Fig. 3. Classical electrochemistry framework of Zn metal electrodeposition.
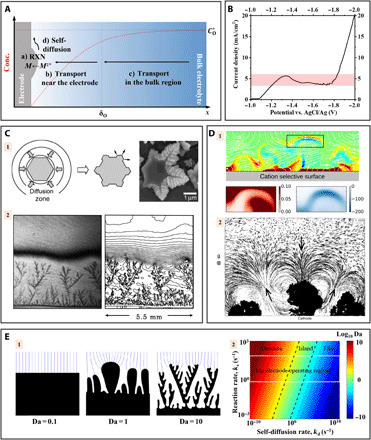
(A) Schematic diagram showing the four fluxes near the electrochemical interface. The red curve shows the concentration (conc.) profile of the metal cations. (B) Current density–overpotential relation of Zn deposition in a 0.05 M ZnSO4 aqueous electrolyte. (C) Mass transport limit induced dendritic growth of Zn electrodeposits and corresponding concentration profile. Adapted with permission from (52, 53). (D) Electroconvection formed near an ion-selective membrane or metal electrodeposits. Adapted with permission from (67, 70). (E) Effects of dimensionless Damköhler numbers on electrodeposition morphology. (E-1) Exchange current density versus diffusion-limited current density, and (E-2) reaction rate versus self-diffusion rate. Adapted with permission from (37, 38).
The electrodeposition reaction flux can be quantified in terms of the faradaic current density J, as described by the Butler-Volmer equation: J = J0[e−αnfη − e(1 − α)nfη]. Here, J0 is the exchange current density, α is the transfer coefficient, n is the number of electrons transferred, f is the inverse of thermal voltage, and η is the overpotential (23). J0 is related to the intrinsic reaction rate: J0 = nFCk0, where F is the Faradaic constant, C is the concentration of the reactant, and k0 is the standard chemical reaction rate constant. It is then possible to connect the observed reaction flux J to the applied overpotential η and the intrinsic chemical kinetics of the reaction k0. In turn, this relation allows one to experimentally interrogate the reaction kinetics by measuring the J and η. For example, Tafel plot (η versus log J) is used to obtain J0 by extrapolating the measured J to the equilibrium state (η ➔ 0) and thereby calculate k0. The experimentally measured k0 is an apparent parameter that reflects the overall reaction process, which, in most cases, is multistep in nature. For example, it includes the desolvation (also called dissociation) of the complex ions in the vicinity of the electrode and the charge transfer on the electrode surface (31). The measured apparent k0 values can be used to infer detailed mechanistic information about the reaction and in favorable cases, allowing the details such as the reaction order, , to be determined from knowledge of the activity ai of cationic species i in the electrolyte. Ready access to this sort of information is considered a hallmark of the Zn electrochemical deposition; it has spawned a rich body of literature on the reaction mechanisms that govern electrodeposition of Zn in multiple types of electrolyte, e.g., sulfate (32, 33), chloride (34, 35), and alkaline baths (31, 36).
A general conclusion is that a smaller exchange current density, i.e., slower electroreduction kinetics, promotes relatively compact electrodeposition. As we will show later in this section, the beneficial effects of low J0 values can be theoretically understood by comparing the interfacial reaction kinetics to the other kinetic variables in the problem through several dimensionless Damköhler numbers: e.g., based on (i) the cation diffusion rate in the electrolyte bulk (37), (ii) the self-diffusion rate of the deposited metal (38), and (iii) the rate at which electrons move in an external circuit. An important discovery is that electrodeposition of Zn can be tuned by adding polymers, such as poly(ethylene glycol) (PEG), e.g., PEG-200, at such low concentrations in liquid electrolytes that the polymer does not have any obvious effect on the ion or adatom transport fluxes. Instead, studies such as the work reported by Banik and Akolkar (39) suggest that the polymer additive directly alters J0. The authors showed, for example, that J0 decreases systematically (from 2.28, 2.22, and 1.30 to 0.39 mA/cm2) as the concentration of PEG-200 increases from 0, 100, and 1000 to 10,000 parts per million in aqueous electrolytes. The authors further observed that the growth of Zn dendrites is inhibited by PEG addition, which is consistent with other studies (40, 41). Analogous studies of Cu electrodeposition show by means of in situ electrochemical quartz crystal microbalance analysis that dynamic adsorption of the polymer additive may instead influence the reaction kinetics by introducing a new rate-limiting step, namely, the ion adsorption kinetics at the electrode (42).
Assuming that the surface concentration of ions at the electrode does not appreciably differ from the bulk concentration, i.e., = 1, the Butler-Volmer equation can be linearized for small η, and a plot of J versus η would be approximately linear and monotonic (see Fig. 3B). Above a certain η, the electrode reaction consumes the metal cations near the electrode surface, C(0, t) will deviate from C* and, if the ion diffusion rate is small, may even fall to zero. Under these conditions, the current density-overpotential relation is time dependent, . As CO(0, t) approaches zero, the electroreduction reaction is mass transport limited—any metal cations that arrives at the electrode surface is reduced instantaneously. In this so-called “diffusion-limited” regime, a diffusion layer of thickness δO is developed near the electrode, within which CO(0, t) substantively deviate from C*, as illustrated by the red curve in Fig. 3A. The maximum diffusion flux, i.e., the diffusion-limited current density Jlim, is an intrinsic property of the electrolyte, which can be estimated to be (43), where DO is the diffusivity of the metal cation and δO is the diffusion layer thickness (44, 45).
In pure solution-phase reactions, the diffusion-limited regime is observed as a plateau in a J versus η plot, similar to what is observed in the red region in Fig. 3B. In contrast, the nature of electrodeposition, in which a solid, conductive phase is created, allows the morphology of the electrode surface to evolve as the deposition proceeds. Since the diffusion layer thickness δO is proportional to t1/2 (23, 46), the electrodeposits can penetrate this diffusion layer and maintain contact with sufficient cations if it grows at a rate faster than δO. This leads to the observation of an overlimiting regime in metal deposition reactions (Fig. 3B) that can be quite different from classical expectations (47). A second consequence of this chemotaxis-like electrodeposit growth is the increase in deposit porosity and the transition in crystal texturing—Horizontally oriented Zn plates observed at a underlimiting current density are tilted up above Jlim, which can be captured by x-ray diffraction (see the section discussing crystallography for details) (28). When a rotating disk electrode (RDE) is applied to artificially limit the diffusion layer thickness δO to a small value of <10 μm, the reorientation from horizontal to vertical is completely suppressed, and the strong (002) texturing of Zn is preserved throughout the deposition.
In relatively dilute electrolytes (CZn2+ < 0.5 M), which have greater diffusion layer thicknesses, a group of “diffusion-limited aggregate” (DLA) patterns form, including dendritic, ramified, densely branched, etc. (Fig. 3C) (48–51). The hexagonal crystal symmetry of Zn is reflected in the dendritic growth [see the scanning electron microscopy (SEM) image on the right of Fig. 3C-1]. López and Choi (52) interpret this as a result of the interaction between the hexagonal shape of the deposit and the “concentric” diffusion gradient (see the scheme on the left of Fig. 3C-1). Growth therefore preferentially occurs at the protrusions. An early in situ interferometry study experimentally monitored the concentration field near the deposition front (Fig. 3C-2) (53). The isocontours exhibit a strong propensity for stacking in the vicinity of the foremost dendrite tips. The physical significance of this observation can be discerned through the relationship, Jdiff = − D∇C, where Jdiff is the diffusional flux. The initiation and the growth of DLA type of metal dendrites can thus be understood to arise inherently unstable solid/liquid interface, where electronically conductive heterogeneities are self-reinforcing; analogies can be found across many branches of physics, e.g., the formation of thermal dendrites in metallurgical processes (54). There is a far richer body of literature discussing these DLA patterns owing to their broad implications in physics and some approaches exist for changing one DLA pattern to another. In the battery context, their formation is simply because of their large propensity for detaching from the electrode upon battery discharge and for penetrating the separator (55). It means that the operation of Zn batteries should veer off such diffusion-limited regime. The cycle life of Zn batteries has been shown to be negatively correlated to the applied current density (56). The instability arising at high current density is reportedly attributable to the initiation of dendritic growth.
Caution, however, should be made against oversimplifications of these transport analyses based on the measured current i because the electrode surface in a metal electrodeposition is not invariant as expected in classical liquid-phase electrochemistry. The straightforward consequence is that does not hold as A0 evolves over time in a nontrivial manner particularly in a diffusion-limited regime where the electrodeposits fundamentally alter the electrode landscape; the relation between experimentally measured nominal current and the scientifically meaningful real current density is no longer explicit. In addition, because of the patterning of the electrode surface by metal deposits, the mass transport field determined by the gradients will, as discussed, deviate from the unidirectional situation for planar electrode, which needs additional treatment particularly when the length scale of the morphological heterogeneities (roughness) is comparable to that of the diffusion boundary layer thickness as pointed out by Bard and Faulkner (23).
Another complication arises from the activation of electroconvective flows in liquid electrolytes at large overpotentials. Such flows introduce a hydrodynamic component that influences both the transport and morphology evolution during metal deposition (Fig. 3D). The general effect is that morphological heterogeneities present at the metal electrode are exacerbated by hydrodynamic flow field (Fig. 3D-2), the details are complicated, however, because the boundary condition on the hydrodynamic flow changes as the metal deposit front progresses into the electrolyte bulk. We therefore consider first the analogous problem of electroconvective flow at an ion-selective membrane (see Fig. 3D-1). The current-voltage response at an ion-selective membrane includes three regimes: underlimiting, limiting, and overlimiting, similar to that of a metal electrode (Fig. 3B). The overlimiting regime can no longer be interpreted as a result of ramifying metal deposits penetrating the diffusion boundary layer. Initial clues to understanding this overlimiting behavior near an ion-selective membrane were provided by Maletzki et al. (57), who found that the noisy overlimiting regime completely disappears after the 0.01 M CuSO4(aq) electrolyte is “immobilized” by agarose gel. This observation suggests that the overlimiting current involves the contribution from convective mass transport, which has been corroborated by multiple studies using microparticles to monitor the hydrodynamic states (58, 59) or using interferometry to visualize the concentration field (60, 61). This voltage-driven convective flow is thought to stem from the electroosmotic slip in a space charge layer developed near the membrane (62–64). Following studies show that the onset of the hydrodynamic instability occurs at , which is ~𝒪(0.1) V (65, 66); this is consistent with experimental observations (43). The initiation and development of such convective flow has been evaluated using “direct numerical simulation (DNS) to solve the Poisson-Nernst-Planck and Navier-Stokes equation in 3D space (Fig. 3D-1) (67). The chaotic vortices as delineated by the flow streamlines disrupt the extended space charge layer and induce charge imbalance outside the extended space charge region, e.g., the ribbon indicated by the rectangle, which has a steep concentration gradient and a large induced electric field. However, note that the size of 3D DNS simulations is constrained to 1/ϵ ≪ 104 (where ϵ is the dimensionless Debye length) due to the computational cost, while experimental systems typically have a 1/ϵ ≫ 104, making the matching between experimental and computational parameters challenging (68).
In comparison with an ion-selective membrane, additional morphological heterogeneities and instabilities are at play on a reacting electrode surface where metal deposition reactions occur. While there have been a few studies of the electroosmotic flow near reacting electrodes (69), a quantitative framework that integrated both the morphological and hydrodynamic instabilities is not yet available (68). The qualitative understanding of a convective flow’s effect on the electrodeposition morphology appears, however, straightforward. It can be understood as one considers the Nernst-Planck equation governing the mass transport in a dilute electrolyte: , where the terms describe the contribution from diffusion, migration, and convection, respectively. As captured by the convection term, the pattern of convective vortices will strongly influence the mass transport flux, and the metal deposition morphology in a diffusion-limited regime therefore exhibits an obvious correlation with the hydrodynamic states of the liquid electrolyte—The metal preferentially grows at localities with normal flow velocities toward the electrode surface (Fig. 3D-2) (70). It is therefore imperative that this type of convective flows, regardless of the specific origins (e.g., buoyancy, charge imbalance, etc.), be effectively suppressed in an operating battery anode. One may naively conclude that this can be fulfilled by adopting battery cycling protocols that ensure the system stays away from the diffusion limit. In reality, the operation of a macroscopic battery system creates local states [e.g., around hotspots of metal deposition (71)] that are far away from the apparent condition. This means that additional intervention is needed to address the issue. Recent reports show that addition of high–molecular weight polymers delays the onset of convective flow in metal deposition and substantially enlarges the diffusion-limited regime (72). The entanglement among the polymer chains above a threshold concentration introduces elasticity to the liquid electrolyte that attenuates convective flows (73). DNS study further shows that the flow velocity is reduced mainly due to the polymers’ strong resistance to extensional motion (74).
It should be pointed out that the initial adatom distribution landscape is not necessarily the ultimate electrodeposition morphology, owing to the contribution from surface mobility of the metal atoms (75). Self-diffusion of the metal is a flux that could alter the deposition landscape. The main driving force of self-diffusion is energy minimization (Fig. 3A). The origin of the energy release is manifold—(i) The possible binding between the metal deposit and a “metal-philic” substrate provides a chemical driving force (76), and (ii) the possible difference between surface tensions acts as a physical driving force. It has been argued in many studies that Mg metal does not form dendrites because of its “vanishing small” surface diffusion barrier Ediff that gives rise to a very large surface diffusivity Ds (77). Jäckle et al. (78) performed an ab initio density function theory (DFT) simulation to calculate the diffusion energy barrier of the metals used as metal anodes. According to the DFT calculation, the Ediff of Zn is comparable with, in some situations smaller than, that of Mg. The authors, however, attributed the observations of Zn dendrites in the literature to the precipitation of a resistive ZnO layer onto the Zn surface. This argument connects to the role played by the SEI that will be discussed in the next section. The effect of self-diffusion can be deconvoluted from the complex electrodeposition process and be critically evaluated using annealing experiments. At an elevated temperature of 378 K, a 10-min annealing will fundamentally change the shape and crystallographic texture of the Zn—Horizontally aligned, (002)-textured Zn crystallites are formed (79). The fast reorientation in annealing is consistent with the high Ds suggested by the DFT study by Jäckle et al. (78). We note that Zn also has a relatively low melting point Tm is 693 K, meaning that its homologous temperature TH is 0.5 at 347 K (74°C). As an empirical rule, above 0.5 Tm, self-diffusion becomes prominent in metals, giving rise to phenomena such as plastic flow and creep (80). While elevated operation temperatures are less practical for aqueous Zn batteries, strategies such as local self-heating induced by high current cycling (81, 82) might be of interest for Zn.
As noted earlier, the Damköhler number is a dimensionless group that allows the absolute values of fluxes and electroreduction reaction rates to be compared. Because mass transport fluxes take many forms at a metal electrode, there are multiple Damköhler numbers. The second Damköhler number, ≈ J0/Jlim, is among the most useful because it quantifies the relative rates of the electrode reaction and diffusion . Here, L is the interelectrode distance, D is the electrolyte diffusivity, and Vm is the molar volume (37). Phase-field analysis shows that Da, II plays a key role in determining the electrodeposition morphology of Zn metal; smooth deposition is favored at small Da, II ≪ 1, while obvious dendritic deposition occurs at Da, II ≫ 1 (Fig. 3E-1) (37). The physical significance is that the deposition is kept within a reaction-limited regime owing to the sluggish chemical reaction kinetics, and the propagation of the dendrite growth front is therefore slowed. Consistent with this finding, the phase-field modeling study by Enrique et al. (83) revealed that the current distribution on the surface of a columnar dendrite is homogenized at a low Da, II ≈ 0.5, while the current concentrates on the dendrite tip (roughly one order of magnitude higher than on the side) at a high Da, II ≈ 50. These simulation results provide a theoretical explanation of the finding by Banik and Akolkar (39) that lowering J0 by adding low–molecular weight polymer to a liquid electrolyte stabilizes Zn deposition. The diffusivity D and conductivity κ of a dilute electrolyte are related through the Nernst-Einstein equation. Da, II can therefore be written as J0/κ, which can explain the widely reported effect of high ionic conductivity electrolyte in stabilizing electrodeposition of metals (84).
Davidson et al. (38) introduced another Damköhler number , where ke is the electrochemical reaction rate and kd is the surface diffusion rate. As shown in Fig. 3E-2, a large kd value results in a small Da ≪ 1 and predicts planar, film-like metal deposition. In contrast, a small kd value leads to Da ≫ 1 and rough/dendritic electrodeposition. Together, it is generally agreed that a moderately small exchange current (slow reaction kinetics), large diffusion-limited current density (high electrolyte ion diffusivity), and high adatom self-diffusivity are favorable for achieving smooth electrodeposition morphology. We, however, point out that the real scenario in metal deposition is more complicated than what have been assumed in the modeling studies; the main additional degree of complication stems from the presence of a SEI formed in between the metal deposits and the electrolyte. The role played by such SEI layer is treated in the next section.
CHEMISTRY OF THE ELECTROLYTE AND THE SEI
The electrolyte choice influences the Zn deposition morphology via a variety of processes—It determines the anisotropy of the energy landscape of the crystal facets, it influences the interface reaction kinetics, and it affects the rate and stability of mass transport processes. Here, we focus on the interphases formed by electrolytes in Zn batteries. We classify interphases as solid, by-product layers or precipitates that form a physical boundary between an electrolyte and an electrode, e.g., the SEI first proposed by Peled (85) in 1979 to interpret the behavior of alkaline metal in nonaqueous electrolytes. The SEI has received enormous attention because of its hypothetical role in the formation of moss-like Li metal deposition (84, 86, 87). While it is generally agreed that the SEI between the electrically conductive metal and the ionically conductive electrolyte notably complicates the transport phenomena at the interface, why and how it serves to regulate the morphology of metal electrodeposits remains controversial. One school of thought is that electrons access the arriving metal ions by tunneling through the SEI, another is that ions access electrons from the underlying metal via transport through the SEI (88). In either scenario, a SEI that is simultaneously heterogeneous and insulative (in terms of electron and/or ion transport) would results in heterogeneous metal deposition by inducing preferential growth along local high-diffusivity pathways (84). It is therefore important to reduce, even eliminate, the heterogeneity of the SEI layer and to promote the formation of a uniform, thin SEI that allows fast but homogeneous transport. As a point of departure toward this goal, the formation mechanisms of SEI in the electrolytes need to be understood.
Figure 4A compares the typical electrolyte systems for Zn batteries: aqueous, walt-in-salt, molten salt, organic, and polymeric electrolytes. While all these systems exhibit some favorable features, it is clear that the aqueous electrolytes (89) appear as the most desirable option for its high operation current density (≈50 mA/cm2) and low cost (<$1/kg) when paired with cost-effective cathodes such as MnO2 (20, 90). Compared with electrolytes that generate multiple inorganic/organic/polymeric products, aqueous Zn electrolytes have a relatively simple chemistry as delineated in Fig. 4B. The Pourbaix diagram (Fig. 4B-1) and the distribution functions (Fig. 4B-2) of Zn-H2O illustrate the stable species in the electrolyte under certain conditions, e.g., potential pH. To avoid a pure solid conversion of Zn(s)↔ZnO(s) that has slow kinetics, a Zn anode operates either in an acidic regime or in an alkaline regime, where the oxidative Zn2+ is soluble in the liquid phase. However, there is a thermodynamic potential for Zn metal to react with the acidic or alkaline electrolyte and produce H2 gas in these regimes, creating a local pH deviation toward the neutral regime, which favors ZnO as a solid precipitate from electrolyte. Such precipitates can adhere to the surface of Zn metal, creating the interphasial layer.
Fig. 4. Designing electrolytes for highly reversible Zn metal plating/stripping.
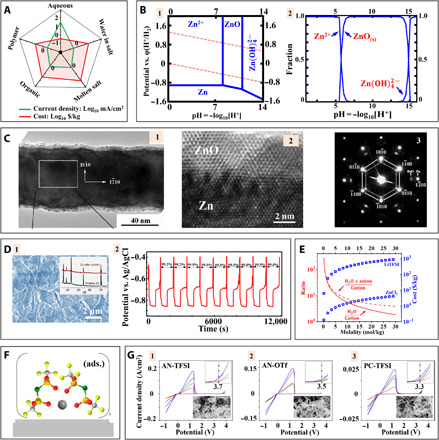
(A) Spider chart comparing electrolytes for Zn batteries. (B) Distribution of chemical species in aqueous Zn2+ electrolytes. (B-1) Pourbaix diagram. The top and bottom dashed lines represent oxygen evolution and hydrogen evolution reactions (HERs), respectively. (B-2) Fractions of different Zn2+-based species. (C) Transmission electron microscopy (TEM) characterization of Zn electrodeposits. Adapted with permission from (199). (C-1 and C-2) TEM images and (C-3) selected area electron diffraction pattern. (D) Water-in-salt electrolyte for Zn metal deposition. Adapted with permission from (107). (D-1) SEM image of Zn after cycling in water-in-salt electrolyte. (D-2) The plating/stripping voltage profile of Zn in water-in-salt electrolyte. (E) The molality-dependent parameters of high-concentration electrolytes. (F) Schematic diagram showing reaction kinetics of adsorbed (ads.) solvated Zn2+ in acetonitrile (AN) on electrode surface. Adapted with permission from (114). (G) Cyclic voltammetry of organic Zn battery electrolytes: (G-1) AN-Zn(TFSI)2, (G-2) AN-Zn triflate (OTf), and (G-3) propylene carbonate (PC)–Zn(TFSI)2. Adapted with permission from (115).
This type of acid-base reactions constitutes the first of the two formation mechanisms of the SEI on Zn. The SEI species formed via this mechanism usually belongs to the category of oxides or hydroxides. Figure 4C reports a transmission electron microscopy (TEM) investigation of the Zn nanowires electrodeposited from ~0.2 M ZnCl2 aqueous solution. As can be observed in Fig. 4C-1, the nanowire is covered with a thin, low-contrast layer (~5 nm), which is confirmed to be ZnO by high-resolution TEM in Fig. 4C-2 and electron diffraction pattern in Fig. 4C-3. The observation of ZnO on the surface of Zn metal anode is supported by other ex situ electron microscopy studies (91, 92) and in situ characterization techniques, e.g., electrochemical-acoustic time-of-flight analysis (93).
SEI prevents the direct contact between Zn metal and electrolyte. It, on the one hand, suppresses the gassing reactions and, on the other, introduces heterogeneity into the transport pathways as discussed. For Li metal, which has a stronger propensity for SEI formation owing to the low electrochemical potential, it is generally thought that the persisting moss-like deposition morphology in multiple electrolytes (24) is caused by heterogeneous SEI (84, 87). We note that the moss-like Li deposition morphology is highly analogous to the moss-like Zn morphology (Fig. 2B), in terms of dimension and geometry. The moss-like Zn deposition from alkaline electrolyte are observed at small overpotential (~20 mV) (94) or low current densities of <0.4 (95). This indicates that SEI is playing a more dominant role at low current density/overpotential than it does at large overpotential or high current densities. We hypothesize that this is because of the ratio between SEI-forming reaction rate and the Zn deposition rate; at a small deposition current, the parasitic reaction can proceed steadily and form the SEI layer, which results in the moss-like deposition morphology. Initial experimental evidence for this hypothesis was provided by a high-resolution TEM study of the Zn electrodeposits formed in alkaline electrolytes at a low overpotential (50 mV) and at a moderate overpotential (150 mV) (92). Existence of ZnO crystallites is consistently detected under these two conditions but in different geometries and quantities. At higher overpotentials, the ZnO forms a uniform, thin layer of a ~𝒪(10 nm) thickness and exhibits an epitaxial growth relation with the Zn metal; at low overpotential, the ZnO has a substantially higher content and a heterogeneous spatial distribution. The Zn metal deposits under these two conditions show morphologies in stark contrast: (002)-textured single-crystalline Zn plates at large overpotential and tortuous, polycrystalline nanoaggregates without specific orientational order. These direct TEM observations of SEI suggest that the excessive formation of heterogeneous ZnO crystallites is playing a crucial role in the deposition at low overpotential/current density. An analogous in-depth TEM study of the Zn electrodeposits formed in ZnSO4-based electrolytes will be of immediate interest in understanding the structure and composition of the SEI in mild-pH environment and its influence on Zn deposition morphology.
This hypothesis is also consistent with the observation of moss-like Li deposition at current density far below the diffusion limit. It is also worth pointing out that TEM studies of moss-like Li deposits reveal that the long dimension is parallel to low-index zone axes, e.g., [111], which allow the exposure of low-energy facets, e.g., (110) (96). Similarly, the Zn structure shown in Fig. 4C also extends along a low-index zone axis, i.e., , allowing the exposure of (002) and (100). The analogy between Li and Zn further suggests that the moss-like Zn electrodeposits are formed in a SEI-dominated regime. The composition and the structure of SEI on Zn and its effect on deposition morphology are still under active investigation. For example, some claim that Zn4SO4(OH)6·4H2O is also detected on electrodeposited Zn metal (97–99), as opposed to the ZnO observed in other studies as mentioned. Advanced characterization techniques, e.g., high-resolution TEM/scanning TEM, could provide important insights into the SEI of Zn metal to resolve these remaining questions about the nature of SEI and the role it plays in electrodeposition, particularly considering that Zn metal samples are not as sensitive to atmosphere or to beam damage as Li samples are (100).
The moss-like electrodeposition morphology is obviously undesirable, because of the low plating/stripping efficiency and the gaseous products generated during the side reactions. To stabilize the aqueous electrolyte against such side reactions, groups of water-in-salt electrolytes are designed, where the salt concentration is unusually high, e.g., >20 m (101, 102). The water-in salt electrolyte can comprise a high solubility Zn salt, e.g., ZnCl2 (103–106), or a high solubility Li salt [e.g., Li bis(trifluoromethanesulfonyl)imide (LiTFSI)] plus a Zn salt of normal concentration of ~1 m (107). In such electrolytes, Zn deposits in a more compact morphology without ZnO as detected by x-ray diffraction (XRD), featuring a high Coulombic efficiency of >99% over a moderate cycle life (>200 hours) at 1 mA/cm2 (Fig. 4D), in contrast to the <50% and the <80% Coulombic efficiencies achieved in alkaline and mildly acidic ZnSO4 electrolytes, respectively (107). The stabilizing mechanism of the water-in-salt electrolytes is illustrated in Fig. 4E—The ratio between the number of water molecules and the number of cations is below the conventional solvation numbers as depicted by the solid red line. When the anions that are also able to solvate the metal cations are taken into calculation, the ratio still remains at a low level, i.e., 3.6 for 1-m Zn(TFSI)2 + 20-m LiTFSI. The change in solvation sheath structure under such conditions is anticipated to alter the SEI chemistry on the Zn electrode (108) and the electrokinetics of the deposition reaction (109). Note also that water-in-salt electrolytes show an enhanced electrochemical stability against oxidation, allowing the reversible operation of high-voltage cathodes in aqueous electrolytes, e.g., Li2Mn2O4 (107) and LiFePO4 (110). These stabilizing effects increase the energy throughput per cycle and guarantee a long cycle life of the full batteries. While the cost of electrolyte is positively correlated to the salt concentration as shown by the blue curves in Fig. 4E, this can be overcome using a low-cost salt or reducing the manufacturing cost of a salt as its production scales up. Of particular interest is that a subset of recent studies reports on a concept of localized high-concentration electrolytes for alkali metal anodes (111–113)—A secondary “diluent” solvent, miscible with the primary solvent but does not dissolve the salt, is added into the electrolyte; the outcome is that the solvation structure is maintained because the ratio between the numbers of the cations and the primary solvent molecules remains constant, while the apparent salt concentration is lowered. Wisdom might be borrowed from this progress in designing highly concentrated electrolytes for Zn.
Seemingly, a more outright approach for overcoming the issue of parasitic reactions in aqueous solution is to use aprotic organic nonaqueous electrolytes. As illustrated in Fig. 4F, computation work suggests that Zn electrodeposition can proceed via a one-step, two-electron transfer mechanism in organic electrolyte, e.g., Zn(TFSI)2 in acetonitrile (AN), which is comparatively faster than the kinetics of Mg, another HCP metal (114). Han et al. (115) reported on a comprehensive experimental study of the electrochemical properties of nonaqueous Zn electrolytes including the combinations between one of the salts: Zn(TFSI)2, Zn(CF3SO3)2, Zn(BF4)2, Zn(PF6)2, and one of the organic solvents: diglyme (G2), AN, propylene carbonate (PC), and N,N′-dimethylformamide (DMF). Among these combinations, AN-Zn(TFSI)2, AN-Zn(CF3SO3)2, and PC-Zn(TFSI)2 are singled out as promising Zn battery electrolytes owing to the high-voltage stability(>3.3 V versus Zn2+/Zn) and the absence (or negligible observation) of undesirable additional redox reactions, as well as the reversible Zn plating/stripping behaviors (see cyclic voltammetry scans in Fig. 4G). An additional finding according to the cyclic voltammetry is that AN-based electrolytes show fastest kinetics as evidenced by the current density. The authors show that AN-based electrolytes have remarkably high ionic conductivities [e.g., using Zn(TFSI)2 salt, the ionic conductivity equals to 11, 6, 2, and 2 mS/cm for AN, DMF, PC, and G2, respectively], which is attributable to the low viscosity and weak coordination between the solvent and the ionic species. This finding is consistent with a few separate studies that report on Zn full batteries using an AN-based electrolyte (116–118). It could also be attributable to the one-step, two-electron transfer mechanism of Zn as predicted by simulation (114), which results in a large reaction constant five orders of magnitudes larger than that of Mg electrodeposition from tetrahydrofuran that includes two steps. Together, the kinetics in the bulk (i.e., diffusivity) and at the interface (i.e., reaction constant) single out AN-based systems as a group of promising electrolytes for Zn batteries.
It is, however, quite unexpected that moss-like Zn electrodeposition persists across all the three electrolytes as shown by the SEM images in the bottom insets of Fig. 4G. Clues to interpreting this observation could be found in a few studies that report the parasitic decomposition reactions of the organic solvent and the S-containing salts (119, 120). DFT calculations show that the coordination with Zn2+ cations destabilizes the TFSI− anion, lifting its reduction potential to 0.37-V Zn2+/Zn. This means that, at least, a portion of the TFSI− anions decompose before the occurrence of Zn metal deposition at 0-V Zn2+/Zn (109).
This exemplifies the second mechanism of SEI formation on Zn—redox reaction. Spectral studies reveal that the decomposition product of TFSI− is complicated—ZnF2, ZnO, sulfide species, N-rich species, and organic functional groups are detected. Calculations further show that the ion diffusion activation energy across these species is comparable to that of typical Li metal SEI formed in Li hexafluorophosphate ethylene carbonate (EC)/dimethyl carbonate (DMC) (49.7 kJ/mol versus 51 kJ/mol). This comparison suggests the possibility that the SEI is imposing a strong regulation on the transport near the electrode, similar to what is claimed for Li metal deposition.
The SEI-forming nature of organic electrolyte systems explains the observed formation of mossy growth at certain low-current regimes. These observations can be compared to a series of recent reports about phosphate-based Zn battery electrolytes, where plate-like Zn electrodeposits are consistently observed at comparable current densities (121–123). This straightforward contrast in Zn deposition morphology confirms that even in aprotic systems, the SEI can play a dominant role in shaping the Zn deposition landscape and, therefore, may need as precise regulation as for Li metal deposition. Understanding the SEI in organic systems with spatial resolution remains a relatively untapped area. Using direct imaging techniques, e.g., TEM, coupled with spectral measurements to probe the structure/composition of SEI, the ion transport mechanism across SEI and its influence on the deposition morphology would constitute a fruitful course of future studies centering on SEI formed in Zn metal deposition.
CRYSTALLOGRAPHY OF ZINC METAL ELECTRODEPOSITS
The crystallinity of the metal deposits appears as an oftentimes overlooked aspect in the conventional discussion, but it turns out to be central in understanding the deposition morphologies of Zn as we will see soon in this section. This is, in part, because of the extraordinarily high anisotropy associated with Zn’s crystal structure. As shown in Fig. 5A, Zn metal adopts a HCP lattice (i.e., space group: P63/mmc) that is less symmetric than the cubic lattice. This can be easily understood by considering the fact that there is only one c axis perpendicular to the (002) close-packed plane in HCP, while there are by symmetry multiple such axes perpendicular to the {111} closed-packed planes in a face-centered cubic crystal. Note that the Zn crystal shows an additional elongation along the c axis because of the hybridization between s and p valence bands (124). That is, although the close-packed nature within a basal plane is maintained, the interplanar distance along c is greater than expected for an ideal HCP crystal (as characterized by the c/a ratio of Zn ≈ 1.85 > 1.63), further elevating Zn metal’s crystallographic anisotropy. These qualitative statements can be quantitatively validated by evaluating the surface energy anisotropy factor αγ of each crystal as tabulated in Fig. 5B (125, 126). While the weighted surface energy of Zn is comparable to other metals, the anisotropy αγ of Zn is, by far, the greatest. Specifically, it is two times larger than Mg, an ideal HCP crystal, and approximately four times greater than that of cubic metals.
Fig. 5. Crystallographic characteristics of zinc.
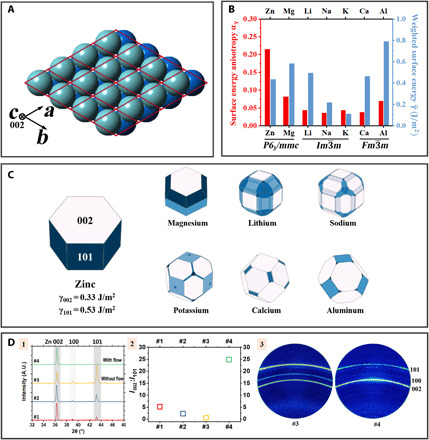
(A) Crystal model of HCP Zn metal. The red lattice denotes the primitive unit cells. The front, light blue layer and the back, dark blue layer are two close-packed layers that stack periodically (…ABABAB…) along the [002] direction (also called the c axis). (B) Weighted surface energy and surface energy anisotropy αγ of representative anode metals., where γhkl is the surface energy of (hkl), and is the area fraction of the (hkl) family in the Wulff shape. . αγ can be viewed as a normalized coefficient of variation of surface energy. A greater αγ value implies a larger anisotropy in the surface energies of the crystal facets exposed in its Wulff shape. In an extreme case of a perfectly isotropic crystal, αγ = 0. Plotted according to data reported in (125). (C) Wulff shapes for metals of contemporary interest as battery anode materials. They depict the shape of the crystals at thermodynamic equilibrium. Adapted with permission using the database reported in (125). (D) XRD analysis of the crystallographic texturing of Zn metal electrodeposits. (D-1) θ−2θ XRD line scans of Zn electrodeposits. (D-2) Peak intensity ratio I002:I101 of the scans shown in Fig. 5D-1. (D-3) 2D XRD of samples #3 and #4 shown in Fig. 5D-1. Adapted with permission from (28). A.U., arbitrary units.
The consequence of this large anisotropy can be visualized by plotting the Wulff shape for each crystal (Fig. 5C). Wulff shape predicts the equilibrium faceted morphology of a crystal by minimizing the overall surface energy (127, 128). While it is apparent that a crystal tends to expose the lowest-energy facet as much as possible, the crystal symmetry imposes nontrivial constraints—For example, the lowest-energy (002) basal planes of Zn are parallel to each other, meaning that the crystal needs to be bounded by vertical (101) facets on the sides. The calculated Wulff construction of Zn is in good accord with the experimentally observed morphology of vapor grown (129) and chemically grown (130) Zn nanocrystals. This also explains the numerous observations of the 2D, plate-like Zn building blocks reported in Fig. 2 (regardless of the alignment with respect to the electrode surface); they preferentially expose the close-packed (002) basal plane owing to an surface energy optimization.
The interpretation of wire-like Zn growth (131) based on the Wulff shape of Zn in Fig. 5C appeases as less successful. Note that these Wulff shapes are built, assuming that the crystal is exposed to vacuum. In a solution, the interphase that adheres to certain facets (in the form of either a loose adsorbent layer or a solid by-product layer) can alter the energy landscape and thereby dominate the stable shape of crystal grown from that solution (132, 133). For example, when the deposit surface is cover by a thin (~5 nm) ZnO interphase, the Zn crystal grows preferentially along the [002] direction, forming 1D wire-like structures (134). Analogous uniaxial growth along the [002] direction is also reported in vapor deposition of Zn under NH3 gas, where a Zn3N2 interphase possibly formed by reaction with the NH3 gas can produce a structure-directing effect (135). This growth mode suggests that the six side facets are stabilized by the interphase and exhibit lower surface energy than the (002) basal plane. These observations are consistent with the argument made in previous section that the interphase formed on the surface of Zn will impose a strong effect on the deposition process, via the alterations of mass transport pathway and/or of crystal surface energy. In addition, of particular interest here is that the moss-like Zn morphology has been observed in vapor deposition (136). This seemingly contradicts with our conclusion that the SEI formed in liquid electrolytes engenders moss-like growth because one may think that such interphase is absent on vapor-deposited Zn metals; the authors, however, interestingly report the existence of “a thin ZnO film on the Zn nanowire surface,” evidenced by the characteristic photoluminescence bands of ZnO. All these observations point toward the conclusion that these wire-like Zn deposition morphologies are fundamentally correlated to the interphasial chemistry.
Briefly, theories based on the equilibrium crystal anisotropy and surface energy are able to provide a fairly good explanation for the geometry of the building blocks. They are, however, unable to explain another critical feature of Zn electrodeposit morphology—the alignment of the building blocks with respect to the electrode surface as shown in Fig. 2 (C to E)—“texturing.”
The crystallographic texture of metal electrodeposits is determined by two main factors: the substrate and the overpotential (87, 92, 137). The influence of substrate can be most fruitfully interrogated using the concept of heteroepitaxy, which describes a group of crystal growths in which the epilayer forms a semi-/coherent interface with the substrate and therefore shows locked orientation relation with the substrate (138). There is a set of well-defined criteria for this heteroepitaxy to occur: (i) the lattice misfit between the two crystals δ is smaller than 15%, and (ii) the substrate is textured with favorable facet(s) being exposed (139). As these criteria are satisfied, the newly nucleated metal spontaneously aligns with the substrate and forms the semi-/coherent interface to minimize surface energy. This heteroepitaxy provides an effective manipulator for regulating the electrodeposition morphology of metals. We showed recently that horizontally aligned graphene layer, which has a δ = 7% with Zn, can effectively promote the formation of horizontally aligned, (002)-textured Zn plate-like electrodeposits that claim plating/stripping efficiencies of >99.6% over thousands of cycles (22, 76). In the early exploration of a group of powder-based Zn electrodes used in alkaline batteries (140), it has been suggested that the addition of some metal oxides (e.g., PbO or SnO) introduces a similar epitaxial effect (141). The metal oxides are reduced to their elemental forms and act as a heterogeneous epitaxial substrate for the Zn electrodeposition. Analogous concept of substrate-induced heteroepitaxy has also been reported for Li metal (142). Even without an epitaxial substrate, metal electrodeposits were found to exhibit some degree of texturing behaviors depending on overpotential as reported in early studies (143, 144). This phenomenon was rationalized in later works by considering work of formation of a 2D nucleus , where NA is the Avogadro number, μ − μ0 is the overpotential, and A and B are constants determined by the works for separating an atoms from a crystal (145, 146). It is argued that when the overpotential is large, the Ahkl term can be neglected and Bhkl dominates Whkl; when the overpotential is small, the Whkl will instead depend much on Ahkl. On the basis of calculation, for HCP crystal, W002 < W101 at low overpotential, and W002 > W101 at large overpotential. This predicts that Zn electrodeposits are (002) textured at small overpotential but (101) textured at large overpotential, which is consistent with experimental observation (Fig. 5D) (28).
However, note that the texture formed at the nucleation stage is not necessarily preserved throughout the entire growth of the crystal, because electrodeposition conditions can deviate from the thermodynamic equilibrium. This means that additional regulation on the kinetic aspects is needed to achieve smooth Zn deposition morphology as we mentioned in the discussions centering on mass transport. For example, a critical thickness exists for heteroepitaxy, beyond which the epicrystal can lose the strong correlation with the substrate (147). In battery electrodes, this transition can be initiated by mass depletion developed near the electrode surface as the deposition proceeds. To lift this “correlation length”–type limitation of regulating nucleation occurring at the bottom interface between the substrate and the electrodeposit, regulation needs to be enforced at the front deposition interface between the electrodeposit and the electrolyte as already detailed in Fig. 3. An artificial convective flow that constrains the diffusion layer thickness can preserve the (002) texturing in prolonged deposition (Fig. 5D) (28). This flow-assisted configuration has been adopted in scaled-up alkaline Zn batteries (~100 Wh) and demonstrated the capability of enhancing the plating/stripping efficiency of Zn metal at elevated current densities (30, 148).
In summary, through the discussions of Figs. 3 to 5, an overarching theoretical framework is established to understand the polymorphism of deposited Zn crystals shown in Fig. 2; this framework serves this purpose reasonably well—The factor(s) dominating each of these morphologies is unraveled. Specifically, the SEI and the crystal anisotropy are the central factors to consider in battery-relevant scenarios away from the diffusion limit. On this basis, we outline in next section (Fig. 6) the most active and fruitful lines of future studies aimed at precisely regulating the Zn deposition morphology in fulfilling the reversibility benchmarks specified in Fig. 1.
Fig. 6. Approaches for regulating Zn electrodeposition morphology at the anode.
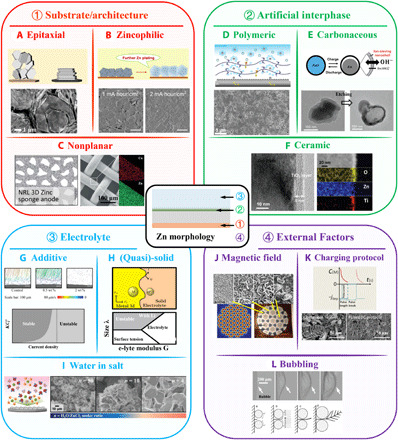
(A to C) Red: Design of electrode substrate/architecture. Adapted with permission from (21, 22, 149, 157). (D to F) Green: Design of artificial SEI (ASEI) . Adapted with permission from (166, 169, 171). (G to I) Blue: Design of electrolyte. Adapted with permission from (72, 84, 103, 179, 185). (J to L) Purple: Design of external factors. Adapted with permission from (176, 187, 196, 200).
APPROACHES FOR REGULATING MORPHOLOGY OF METAL ELECTRODEPOSITS
The cartoon at the center of Fig. 6 provides an overarching picture of the scenarios involved Zn electrodeposition as defined. In the previous sections, the discussions are aimed at decoupling the fundamental physicochemical processes that govern the electrochemical growth of Zn metals in batteries. On the basis of these analyses, we now move on to consider the strategies to proactively intervene such processes and achieve desirable Zn deposition morphologies in batteries. As summarized in Fig. 6, the regulation of Zn deposition can be implemented via (1) substrate/architecture design, (2) artificial interphase design, (3) electrolyte design, and/or (4) manipulation of the external factors. Each of the blocks in Fig. 6 outlines one major category of the state-of-the-art attempts to control the Zn deposition morphology.
We first consider the onset of electrodeposition, namely, nucleation. It occurs at the interface between the electrode surface and the electrolyte. This interface shows dominant effect on the initial stage of the electrodeposition. The first group of strategies approaches the problem from redesigning the electrode—its surface chemistry, geometry, or both (see the red block). The Zn nuclei as crystals have a strong propensity for forming coherent interface with a textured, low lattice misfit graphene layer as an epitaxial substrate (Fig. 6A) (22). The initial heteroepitaxy (i) locks the horizontal alignment and (ii) reduces the nucleation energy barrier, which promotes a homogeneous nucleation landscape. The latter can also be fulfilled by covering the electrode surface with a “zincophilic” coating layer, which shows a good wettability with Zn. The metalphilicity also provides a thermodynamic driving force for smoothening self-diffusion processes as previously discussed. Metal-organic framework materials were recently shown to be capable of facilitating uniform Zn nucleation via similar mechanisms (Fig. 6B) (149–151). The control over the initial nucleation stage by these strategies will show a “guiding” effect on the following growth via homoepitaxy. This suggests that the strategies based on metal-substrate interaction would be particularly effective in the Zn metal anodes compared with Li metal anodes because unfavorable SEI formed on Li surface reportedly blocks the homoepitaxy process (152).
In addition to these approaches that tune the interfacial physiochemical properties, another group of strategies focuses on designing the geometry of the electrode (Fig. 6C). As opposed to the conventional Zn foil used in most of basic battery research, a group of powder-based electrodes in alkaline Zn batteries is made of particulate Zn metal and functional additive salts including fluorides (e.g., KF and NaF), oxides (e.g., Bi2O3 and PbO), and hydroxides [e.g., Ca(OH)2] as the starting materials (140). These functional salts serve a broad range of purposes, such as suppressing H2 evolution, reducing shape changes of the electrode, and trapping zincate species in the anode. The main challenge faced by this group of powder-based Zn electrodes is the gradual collapse of the porous network originally established by the particulate materials. Such electrode shape change over battery cycling reduces the ion and electron transport inside the electrode and results in capacity fading. To address the issue of shape change in a powder-based Zn electrode, Parker et al. (21, 153) proposed to fabricate monolithic Zn sponge that ensures persistent wiring throughout the nonplanar, porous architecture (Fig. 6C, left). The most prominent feature of these 3D sponge Zn electrodes is the high areal capacities of Zn that are deposited in recharge, i.e., >40 mA·hour/cm2, without incurring battery failure either by shape change or internal short. This performance is suggestive of its immediate practical interest as an alternative to Li-ion systems. The authors attributed the stability of Zn deposition to the maximized electrified interface that homogenizes the current distribution and to the confinement of Zn species in the interior of the architecture that retards the shape change (154). An alternative approach toward nonplanar metal anode is to use a nonplanar substrate made of conductive materials that remains inactive throughout battery operation, e.g., Cu skeleton (Fig. 6C, right) (97, 155–157) or interconnected carbon fibers (24, 158–160). This offers additional degree of freedom in tuning the deposition by designing the heterointerface between the host and the metal deposits; for example, Zn and Cu can form solid solution and intermetallic compounds (e.g., CuZn5) (157). As mentioned earlier, it is generally believed that this type of chemical affinity at the interface can reduce the nucleation energy barrier and homogenize the nucleation landscape, which, in part, determines the subsequent growth process; consistent observations are also made in Li electrodepositions (161–163). We speculate that nonplanar architectures with rationally designed surface chemistry could serve as a robust path toward highly reversible, high areal capacity battery, while the usage of an inactive “host” will inevitably introduce additional mass, volume, and materials cost as evaluated in reference (87). Considerable room of optimization exists in designing and fabricating nonplanar architectures that are specifically targeted at battery anode applications. For example, for a Cu skeleton, questions such as what the optimal pore geometry, pore size, and pore volume fraction are for Zn metal deposition remain underexplored (164, 165).
As the metal covers the electrode surface, the front interface/phase between the metal and the electrolyte instead plays a pivotal role directing the growth mode. The second set of solutions aims at controllably creating an artificial SEI (ASEI) that exhibits desirable properties (see the green block). This ASEI can be of a variety of chemistries—polymers [Fig. 6D; e.g., polyamide (166), polypyrrole (167), and ionomers (168))], carbons [Fig. 6E; e.g., amorphous carbon nano shell (169) and graphene oxide (170))], and ceramics [Fig. 6F; e.g., TiO2 (171), CaCO3 (172), ZrO2 (173), aluminosilicate (174)), etc.]. The common nature of these coatings is that they regulate the mass transport at the front electrodeposition interface. In the first place, they block the transport of H2O and prevent the direct contact between the electrode surface and the aqueous electrolyte, which is the main origin of undesirable parasitic reactions including hydrogen evolution, salt precipitation, etc. (175). Second, it is thought that the ASEI interphase, which should be uniform in terms of composition and thickness, can homogenize the transport of Zn2+ cations toward the electrode surface. In comparison, the cation transport flux could be dominated by a heterogeneous SEI formed without proper control before or during the electrodeposition (84). Last, we also note that implementing this ASEI strategy at the nanoscale opens up unique opportunities. Wu and coauthors (169) reported a type of nanostructured Zn electrode composed of ZnO nanoparticles coated by electrically conductive carbon nanoshell (Fig. 6E). The main uniqueness is that the active soluble Zn2+ species are confined within the carbon nanoshell. Upon electrodeposition, the locally confined Zn2+ species at the nanoscale are reduced inside the shell. The process does not involve the long-range mass transport near the electrode as described in Fig. 3. This feature therefore substantively differentiates the scenario with the one described by classical theories. The original mass transport limit in the liquid electrolyte might be lifted since no such long-range diffusion process is activated in this scenario of nanoconfinement. These observations suggest that ample opportunities, both to explore fundamental science questions and to solve engineering problems, exist at the nanoscale.
Beyond the interphase is the electrolyte, which influences the electrodeposition via physical (mass transport, electroconvection, etc.) and chemical mechanisms (reaction kinetics, SEI formation, etc.) as already discussed (Fig. 4). Most of the effort devoted to the modification of aqueous electrolytes fall into three subcategories as illustrated in the blue block. Electrolyte additive is perhaps the most extensively explored direction through the recent decades as evidenced by the large volume of works published on it. The additives can work chemically—e.g., trace amount of Pb2+ additive codeposits with Zn, promotes the growth of (002) basal plane, and suppresses the dendrite initiation (176–178) —or physically—e.g., 0.5 to 2 weight % (wt %) of PEG additive (molecular weight, 8 × 106 g/mol) reduces the average flow velocity, eliminates regions of high local velocity, and thereby extends the overpotential range where the electrodeposition remains stable (Fig. 6G, top) (72). A general, physically based framework for understanding the effects of additive on electrodeposition morphology is established by Haataja et al. (179, 180). It is shown that small quantities of molecular additive species will preferentially adsorb onto and accumulate near surface protrusions and thereby stabilize the electrodeposition in a regime below a critical flux J* (Fig. 6G, bottom). In reality, the additives can undergo additional, chemistry-specific interactions with the electrodeposits. Despite the obvious diversity of the additives, we note that most of their work mechanism can be understood when one refers to the previous sections of the present work.
Figure 6H reports the second subcategory, quasi-solid electrolytes—mostly gel polymer electrolytes (GPE)—for Zn batteries. The main advantage of a quasi-solid GPE over an all-solid-state electrolyte is that the fast electrode kinetics of Zn in aqueous electrolyte is preserved, both at the interface(i.e., wetting) and in the bulk (i.e., ionic conductivity). In comparison with conventional aqueous electrolyte, the rationale for using GPE is manifold: (i) It stabilizes water molecules by abundant hydrogen bonds that can form on the polymer matrix; (ii) it expands the temperature window of the electrolyte; and (iii) it reportedly suppresses dendritic growth of Zn. A recent Review provides a comprehensive overview of the existing literature about the materials designs of the quasi-solid electrolytes for Zn (181). An aspect that does not receive as much attention is the mechanoelectrochemical interaction associated with the creation of the solid metal phase at the interface between the electrode and the (quasi-)solid-state electrolyte (Fig. 6H, top). This interaction can be captured by a modified Butler-Volmer equation , where βm is the mechanical cathodic symmetry factor, σh, surf is the hydrostatic pressure on the surface, and Ω is the partial molar volume (182, 183). Phase-field modeling result shows that even in a ZnSO4(aq) liquid electrolyte, nontrivial compressive stress develops at the deposition interface and initiates mossy growth of Zn (183). In viscoelastic liquids and polymers, hydrostatic stresses at the interface stabilize the deposition; a critical shear modulus of the electrolyte exists , where vc, vm, and va,m are the partial molar volumes of the cation, the metal, and the mobile anion in the separator), above which dendritic growth is suppressed (66, 184). Stability analysis shows that unstable transport can be mechanically stopped when the deposit size is smaller than a critical value , meaning that the stable regime expands as GS becomes larger (Fig. 6H, bottom). Recently, a previously unexplored regime was assessed in recent studies by Ahmad and Viswanathan (185, 186); in this regime, vc is smaller than vm, which is usually the case for inorganic solid electrolyte. Under these conditions, hydrostatic stresses destabilize the interface, and stable deposition is achieved at low electrolyte modulus (i.e., GS/GM < 0.7, where GM is the shear modulus of the metal). These modeling and theoretical analyses provide roadmaps in designing (quasi-)solid-state electrolytes for Zn that stabilize the electrodeposition process. The third subcategory of electrolyte innovation centers on the concept of water-in-salt electrolytes (Fig. 6I), which has been discussed in detail in previous section. As is shown in Fig. 6I, the Zn deposition undergoes a transition from a moss-like porous morphology to a more compact morphology as the #H2O/#ZnCl2 ratio decreases (103).
We then would like to draw the readers’ attention to a group of external factors (see the purple block) that can, under some conditions, dominate the electrodeposition morphology but are sometimes overlooked. Although batteries are usually closed electrochemical cells, multiple influences can still be exerted in a dynamic manner, e.g., by imposing a magnetic field (Fig. 6J) or by adopting a pulsed charging protocol (Fig. 6K). The influence of a magnetic field on deposition landscape can be decisive; for example, the deposition is precisely patterned by the magnetic field under mass transport–limited conditions (Fig. 6J) (187). In the generic context of electrodeposition, it is believed that a magnetic field augments the mass transport by a magnetohydrodynamic effect—A convection in the liquid electrolyte is induced because of the Lorentz force, FL = j × B (188). This concept has been recently explored in rechargeable Li metal anodes (189, 190). Of particular interest for Zn is that the crystallographic orientation can be controlled under a high magnetic field (12 T). Taniguchi and co-workers (191) observed that the Zn deposits are strongly (002) textured with a magnetic field perpendicular to the substrate. The authors argue that this is a result of the large anisotropic magnetic susceptibility of HCP Zn. Upon the application of an external field, a magnetization energy is generated: , where μ0 is the vacuum permeability, N is the diamagnetic coefficient, χ is the magnetic susceptibility, and H is the external magnetic field. Magnetic susceptibility χ is a second rank tensor: Mi = χijHj, which has different values along c and the basal directions as dictated by the HCP lattice symmetry. Specifically for Zn, along the c axis, χc = −1.33×10−5, and along the a and b axes, χa, b= −1.81×10−5. Aligning the c axis parallel to the magnetic field therefore minimizes the magnetization energy. We note that the deposition was performed at current densities close to the diffusion limit, meaning that the magnetohydrodynamic convection induced by the high magnetic field may also play a role in determining the texture, as shown in the RDE study as discussed previously (28). Future studies could focus on further exploring the Zn plating/stripping efficiency achieved under a high magnetic field and the possible effect of such magnetic field on the overall operation of a battery.
Charging protocol offers another opportunity to circumvent the diffusion limit–induced dendritic growth (Fig. 6K). The mechanism is quite straightforward as illustrated in the top panel of Fig. 6K: A relaxation period is inserted to allow the reestablishment of the Zn2+ species near the electrode, which prevents ion depletions and the initiation of electroconvective flows. Therefore, the dendritic growth of Zn is not observed when using a pulsed charging protocol (Fig. 6K, bottom). A few studies report on the crystallographic orientation of Zn deposits obtained with pulsed protocol (192, 193). The correlation is, however, not straightforward, unlike what has been shown for Cu deposition—Strong (111), (100), and (101) textures can be achieved respectively by optimization of the pulse parameter (194). In light of the greater crystal anisotropy of Zn than Cu, we speculate that similar quality of texturing can be realized in Zn systems, which warrants further explorations.
To close, we draw the readers’ attention to a phenomenon that can induce large deposition heterogeneity—gassing (bubbling). Hydrogen evolution reaction (HER) generates bubbles at the electrode surface, creating large volume change in closed electrochemical cells. The presence of bubbles on the electrode surface notably influences the deposition morphology; for example, these H2 bubbles template the growth of Zn as visualized by in situ x-ray phase-contrast imaging (195). This effect of bubble templating is exacerbated when trace amount of Cu2+ ions is present, accelerating the rate of HER (Fig. 6L, top) (176). A study by Hsu et al. (196), however, reports different observations, where no such bubble-templating phenomenon is observed; instead, the dendritic Zn preferentially grows among the bubbles formed on the electrode surface (Fig. 6L, bottom). The authors attribute this phenomenon to the locally concentrated electric and mass transport fields created by the bubbles. The differences across these reports could stem from the specific deposition conditions (e.g., Zn2+ concentration). Nonetheless, large local heterogeneity is in either way introduced into the deposition morphology near the bubbles. It highlights the importance of suppressing the gas-generating side reactions in achieving smooth, compact deposition morphology.
CONCLUSIONS AND OUTLOOK
Electrodeposition of metals at battery anodes is, by its very nature, a complex process involving multiple physical and chemical factors, which play different roles in different regimes, as shown in Figs. 2 to 6. Among these factors, we have contended that the SEI and the crystal anisotropy are the two critical but, oftentimes, overlooked aspects of Zn electrodeposition under battery-relevant conditions away from mass transport limit. The transformative progresses may require out-of-the-box approaches conceptualized taking together these fundamental traits of Zn and knowledge borrowed from related fields, e.g., heteroepitaxy or magnetic field-induced alignment of Zn crystals.
In the course of implementing control over electrochemical growth of Zn, cautions need to be made against oversimplifications in the characterization, classification, and interpretation of deposition morphologies—The observation would hardly be scientifically meaningful unless the deposition conditions are chosen with reference to certain intrinsic properties of the system and estimates (at least qualitative) are thereby made to understand which regime the deposition system is in. That is, it is meaningless to draw conclusions about a factor’s influence in a regime governed by another factor. For example, mass transport is a fundamental limit that exists for any electrolyte, above which the system enters a regime where ramified, dendritic electrodeposition dominates. In claiming a system to be “dendrite-free,” it is necessary that one first compares the current density with the diffusion limit. The abuse of terminology may obscure that the fundamental aspects associated the deposition morphology and generate discrepancy across the literature—e.g., there obviously lacks a clear criterion in judging whether a morphology is “dendritic” or “nondendritic” in some contemporary publications. We therefore suggest that a more quantitative, scientific framework be used in describing the morphology. The discussions following Fig. 2 in our view provide a point of departure; the geometry of the building blocks (plate, wire, etc.) and the assembly of the building blocks (horizontally aligned, vertically aligned, random, etc.) could serve as the key descriptors of metal deposition morphologies that ensure a fair, scientifically meaningful comparison across the literature.
This complex nature of electrodeposition, in turn, creates a platform to study the physiochemical processes and their interplay, particularly because these processes are sometimes adequately “internalized” and reflected in the morphology of the deposited, solid metallic phase. For this reason, in-depth characterizations of electrodeposition morphology offer a path toward tracing the dynamics of the physiochemical processes. Among the characterization opportunities, atomic-scale investigations of the composition/structure of the SEI layer and how it regulates the growth of the Zn electrodeposits would generate transformative new insights and be of immediate interest to the broader community of metal anodes including reactive alkali metals. Considering that Zn metal and Li metal share highly analogous deposition morphologies in certain regimes, it could be a quite feasible but as fruitful research direction that one uses Zn as a model system to understand the generic role the SEI plays. This allows Li sample’s notorious problem of sensitivity to be circumvented. Separately, a recent study on Li deposition based on high-resolution TEM shows that the initial nucleation and growth stages of metal deposits could involve the formation of glassy metallic phases (197). It raises a question—“does the deposition of other non-alkali metals adopt a similar mode?” Briefly, nonsensitive Zn provides a platform for understanding mechanisms involved in metal electrodeposition using advanced characterization techniques (92).
Note also that in contrast to the large volume of work focused on the deposition process, the stripping process of metal deposits receives much less attention despite its at least equally important role in determining the ultimate reversibility. As demonstrated by Song et al. (198), in situ techniques (e.g., x-ray phase-contrast imaging) can be used to monitor the electrochemical dissolution of the Zn deposits in various shapes. This could be a research direction that not only is of practical value in improving the electrode reversibility but also opens up some new room for fundamental science research—How these nanostructured metal deposits dissolve upon the anodization. The conclusion from these studies will complement the existing body of knowledge on the deposition process.
As a final remark, the low-cost, environmental-friendly nature of Zn-based batteries, which are the two major advantages over other alternative battery chemistries, should be preserved in any of the proposed strategies. High costs are oftentimes incurred when additional materials in relatively large quantities are used. In these cases, the gain in prolonging cycle life should at least offset the incurred additional costs to make sure it is economically meaningful. Adherence to this simple guiding principle will limit the volume of ultimately unfruitful efforts even in basic battery researches and will help to define the most commercially relevant pathways toward affordable Zn batteries.
Acknowledgments
J.Z. thanked J. Yin, T. Tang, G. Li, R. Luo, S. Jin, and X. Ren for valuable discussions. Funding: This work was supported as part of the Center for Mesoscale Transport Properties, an Energy Frontier Research Center supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award #DE-SC0012673. Author contributions: J.Z. wrote the first draft of the manuscript. L.A.A. and J.Z. conceived the framework of this work and revised and finalized the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Schill W.-P., Residual load, renewable surplus generation and storage requirements in Germany. Energy Policy 73, 65–79 (2014). [Google Scholar]
- 2.T. J. Stehly, P. C. Beiter, 2018 Cost of Wind Energy Review (National Renewable Energy Laboratory, 2020). [Google Scholar]
- 3.J. Eto, The National Cost of Power Interruptions to Electricity Consumers—Revised Update (Lawrence Berkeley National Laboratory, 2017). [Google Scholar]
- 4.K. H. LaCommare, J. H. Eto, Understanding the Cost of Power Interruptions to US Electricity Consumers (Lawrence Berkeley National Laboratory, 2004). [Google Scholar]
- 5.Tong Y., Liang J., Liu H. K., Dou S. X., Energy storage in Oceania. Energy Storage Mater. 20, 176–187 (2019). [Google Scholar]
- 6.Yang Z., Zhang J., Kintner-Meyer M. C. W., Lu X., Choi D., Lemmon J. P., Liu J., Electrochemical energy storage for green grid. Chem. Rev. 111, 3577–3613 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Volta A., XVII. On the electricity excited by the mere contact of conducting substances of different kinds. In a letter from Mr. Alexander Volta, FRS Professor of Natural Philosophy in the University of Pavia, to the Rt. Hon. Sir Joseph Banks, Bart. KBPR S. Philos. Trans. R. Soc. Lond. 90, 403–431 (1800). [Google Scholar]
- 8.T. A. Edison, Primary Battery (Google Patents, 1904).
- 9.B. Chalamala, Emerging Energy Storage Technologies Safety and Standards (Sandia National Laboratory, 2018). [Google Scholar]
- 10.Yadav G. G., Wei X., Huang J., Gallaway J. W., Turney D. E., Nyce M., Secor J., Banerjee S., A conversion-based highly energy dense Cu2+ intercalated Bi-birnessite/Zn alkaline battery. J. Mater. Chem. A 5, 15845–15854 (2017). [Google Scholar]
- 11.Karpinski A. P., Makovetski B., Russell S. J., Serenyi J. R., Williams D. C., Silver–zinc: Status of technology and applications. J. Power Sources 80, 53–60 (1999). [Google Scholar]
- 12.Lai C. S., McCulloch M. D., Levelized cost of electricity for solar photovoltaic and electrical energy storage. Appl. Energy 190, 191–203 (2017). [Google Scholar]
- 13.C. Kost, J. N. Mayer, J. Thomsen, N. Hartmann, C. Senkpiel, S. Philipps, S. Nold, S. Lude, N. Saad, T. Schlegl, Levelized Cost of Electricity Renewable Energy Technologies (Fraunhofer Institute for Solar Energy Systems ISE, 2013), vol. 144. [Google Scholar]
- 14.Zakeri B., Syri S., Electrical energy storage systems: A comparative life cycle cost analysis. Renew. Sust. Energ. Rev. 42, 569–596 (2015). [Google Scholar]
- 15.Jülch V., Comparison of electricity storage options using levelized cost of storage (LCOS) method. Appl. Energy 183, 1594–1606 (2016). [Google Scholar]
- 16.U. S. E. I. Administration, A Levelized Cost and Levelized Avoided Cost of New Generation Resources in the Annual Energy Outlook (U.S. Energy Information Administration, 2019).
- 17.Levelized Cost of Energy and Levelized Cost of Storage 2019 (2019); www.lazard.com/perspective/lcoe2019/.
- 18.Lazard’s Levelized Cost of Storage Analysis, Version 1.0 (2015); www.lazard.com/media/2391/lazards-levelized-cost-of-storage-analysis-10.pdf.
- 19.Yadav G. G., Gallaway J. W., Turney D. E., Nyce M., Huang J., Wei X., Banerjee S., Regenerable Cu-intercalated MnO2 layered cathode for highly cyclable energy dense batteries. Nat. Commun. 8, 14424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao D., Zhou W., Ye C., Zhang Q., Chen Y., Gu L., Davey K., Qiao S.-Z., An electrolytic Zn–MnO2 battery for high-voltage and scalable energy storage. Angew. Chem. Int. Ed. 58, 7823–7828 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Parker J. F., Chervin C. N., Pala I. R., Machler M., Burz M. F., Long J. W., Rolison D. R., Rechargeable nickel–3D zinc batteries: An energy-dense, safer alternative to lithium-ion. Science 356, 415–418 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Zheng J., Zhao Q., Tang T., Yin J., Quilty C. D., Renderos G. D., Liu X., Deng Y., Wang L., Bock D. C., Jaye C., Zhang D., Takeuchi E. S., Takeuchi K. J., Marschilok A. C., Archer L. A., Reversible epitaxial electrodeposition of metals in battery anodes. Science 366, 645–648 (2019). [DOI] [PubMed] [Google Scholar]
- 23.A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications (John Wiley and Sons, 2002). [Google Scholar]
- 24.Zheng J., Tang T., Zhao Q., Liu X., Deng Y., Archer L. A., Physical orphaning versus chemical instability: Is dendritic electrodeposition of Li fatal? ACS Energy Lett. 4, 1349–1355 (2019). [Google Scholar]
- 25.Fang C., Li J., Zhang M., Zhang Y., Yang F., Lee J. Z., Lee M.-H., Alvarado J., Schroeder M. A., Yang Y., Lu B., Williams N., Ceja M., Yang L., Cai M., Gu J., Xu K., Wang X., Meng Y. S., Quantifying inactive lithium in lithium metal batteries. Nature 572, 511–515 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Higashi S., Lee S. W., Lee J. S., Takechi K., Cui Y., Avoiding short circuits from zinc metal dendrites in anode by backside-plating configuration. Nat. Commun. 7, 11801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu K., Liu Y., Lin D., Pei A., Cui Y., Materials for lithium-ion battery safety. Sci. Adv. 4, eaas9820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng J., Yin J., Zhang D., Li G., Bock D. C., Tang T., Zhao Q., Liu X., Warren A., Deng Y., Jin S., Marschilok A. C., Takeuchi E. S., Takeuchi K. J., Rahn C. D., Archer L. A., Spontaneous and field-induced crystallographic reorientation of metal electrodeposits at battery anodes. Sci. Adv. 6, eabb1122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei X., Desai D., Yadav G. G., Turney D. E., Couzis A., Banerjee S., Impact of anode substrates on electrodeposited zinc over cycling in zinc-anode rechargeable alkaline batteries. Electrochim. Acta 212, 603–613 (2016). [Google Scholar]
- 30.Desai D., Wei X., Steingart D. A., Banerjee S., Electrodeposition of preferentially oriented zinc for flow-assisted alkaline batteries. J. Power Sources 256, 145–152 (2014). [Google Scholar]
- 31.Hendrikx J., van der Putten A., Visscher W., Barendrecht E., The electrodeposition and dissolution of zinc and amalgamated zinc in alkaline solutions. Electrochim. Acta 29, 81–89 (1984). [Google Scholar]
- 32.Beshore A. C., Flori B. J., Schade G., O’Keefe T. J., Nucleation and growth of zinc electrodeposited from acidic zinc solutions. J. Appl. Electrochem. 17, 765–772 (1987). [Google Scholar]
- 33.Epelboin I., Ksouri M., Wiart R., On a model for the electrocrystallization of zinc involving an autocatalytic step. J. Electrochem. Soc. 122, 1206–1214 (1975). [Google Scholar]
- 34.Kim J. T., Jorné J., The kinetics and mass transfer of zinc electrode in acidic zinc-chloride solution. J. Electrochem. Soc. 127, 8–15 (1980). [Google Scholar]
- 35.Trejo G., Meas Y., Ozil P., Chainet E., Nguyen B., Nucleation and growth of zinc from chloride concentrated solutions. J. Electrochem. Soc. 145, 4090–4097 (1998). [Google Scholar]
- 36.Dirkse T. P., The behavior of the zinc electrode in alkaline solutions: II. Reaction orders at the equilibrium potential. J. Electrochem. Soc. 126, 541–543 (1979). [Google Scholar]
- 37.Cogswell D. A., Quantitative phase-field modeling of dendritic electrodeposition. Phys. Rev. E 92, 011301 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Davidson R., Verma A., Santos D., Hao F., Fincher C., Xiang S., Van Buskirk J., Xie K., Pharr M., Mukherjee P. P., Banerjee S., Formation of magnesium dendrites during electrodeposition. ACS Energy Lett. 4, 375–376 (2018). [Google Scholar]
- 39.Banik S. J., Akolkar R., Suppressing dendrite growth during zinc electrodeposition by PEG-200 additive. J. Electrochem. Soc. 160, D519–D523 (2013). [Google Scholar]
- 40.Lyu L., Gao Y., Wang Y., Xiao L., Lu J., Zhuang L., Improving the cycling performance of silver-zinc battery by introducing PEG-200 as electrolyte additive. Chem. Phys. Lett. 723, 102–110 (2019). [Google Scholar]
- 41.Gu W., Liu C., Tang J., Liu R., Yang H., Hu J., Improving zinc electrodeposition in ammoniacal electrolytes with the saturated dissolved methyltrioctylammonium chloride. Hydrometallurgy 175, 43–51 (2018). [Google Scholar]
- 42.Kelly J. J., West A. C., Copper deposition in the presence of polyethylene glycol: I. Quartz crystal microbalance study. J. Electrochem. Soc. 145, 3472–3476 (1998). [Google Scholar]
- 43.Wei S., Cheng Z., Nath P., Tikekar M. D., Li G., Archer L. A., Stabilizing electrochemical interfaces in viscoelastic liquid electrolytes. Sci. Adv. 4, eaao6243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerra E., Bestetti M., Physicochemical properties of ZnSO4−H2SO4−H2O electrolytes of relevance to zinc electrowinning. J. Chem. Eng. Data 51, 1491–1497 (2006). [Google Scholar]
- 45.Awakura Y., Doi T., Majima H., Determination of the diffusion coefficients of CuSO4, ZnSO4, and NiSO4 in aqueous solution. Metall. Trans. B 19, 5–12 (1988). [Google Scholar]
- 46.O’Brien R. N., Michalik W., Laser interferometric galvanostatic studies in the Zn/ZnSO4/Zn system with gelled electrolyte short time electrolysis. Can. J. Chem. 61, 2316–2320 (1983). [Google Scholar]
- 47.Bai P., Li J., Brushett F. R., Bazant M. Z., Transition of lithium growth mechanisms in liquid electrolytes. Energy Environ. Sci. 9, 3221–3229 (2016). [Google Scholar]
- 48.Elezgaray J., Léger C., Argoul F., Dense branching morphology in electrodeposition experiments: Characterization and mean-field modeling. Phys. Rev. Lett. 84, 3129 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Argoul F., Arneodo A., Grasseau G., Swinney H. L., Self-similarity of diffusion-limited aggregates and electrodeposition clusters. Phys. Rev. Lett. 61, 2558 (1988). [DOI] [PubMed] [Google Scholar]
- 50.Matsushita M., Sano M., Hayakawa Y., Honjo H., Sawada Y., Fractal structures of zinc metal leaves grown by electrodeposition. Phys. Rev. Lett. 53, 286 (1984). [Google Scholar]
- 51.Sawada Y., Dougherty A., Gollub J. P., Dendritic and fractal patterns in electrolytic metal deposits. Phys. Rev. Lett. 56, 1260 (1986). [DOI] [PubMed] [Google Scholar]
- 52.López C. M., Choi K.-S., Electrochemical synthesis of dendritic zinc films composed of systematically varying motif crystals. Langmuir 22, 10625–10629 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Léger C., Elezgaray J., Argoul F., Experimental demonstration of diffusion-limited dynamics in electrodeposition. Phys. Rev. Lett. 78, 5010 (1997). [Google Scholar]
- 54.Langer J. S., Instabilities and pattern formation in crystal growth. Rev. Mod. Phys. 52, 1 (1980). [Google Scholar]
- 55.Yufit V., Tariq F., Eastwood D. S., Biton M., Wu B., Lee P. D., Brandon N. P., Operando visualization and multi-scale tomography studies of dendrite formation and dissolution in zinc batteries. Joule 3, 485–502 (2019). [Google Scholar]
- 56.Yang Q., Liang G., Guo Y., Liu Z., Yan B., Wang D., Huang Z., Li X., Fan J., Zhi C., Do zinc dendrites exist in neutral zinc batteries: A developed electrohealing strategy to in situ rescue in-service batteries. Adv. Mater. 31, 1903778 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Maletzki F., Rösler H. W., Staude E., Ion transfer across electrodialysis membranes in the overlimiting current range: Stationary voltage current characteristics and current noise power spectra under different conditions of free convection. J. Membr. Sci. 71, 105–116 (1992). [Google Scholar]
- 58.Rubinstein S. M., Manukyan G., Staicu A., Rubinstein I., Zaltzman B., Lammertink R. G. H., Mugele F., Wessling M., Direct observation of a nonequilibrium electro-osmotic instability. Phys. Rev. Lett. 101, 236101 (2008). [DOI] [PubMed] [Google Scholar]
- 59.de Valença J. C., Wagterveld R. M., Lammertink R. G. H., Tsai P. A., Dynamics of microvortices induced by ion concentration polarization. Phys. Rev. E 92, 031003 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Vasil’eva V. I., Shaposhnik V. A., Grigorchuk O. V., Petrunya I. P., The membrane–solution interface under high-performance current regimes of electrodialysis by means of laser interferometry. Desalination 192, 408–414 (2006). [Google Scholar]
- 61.Nikonenko V. V., Vasil’eva V. I., Akberova E. M., Uzdenova A. M., Urtenov M. K., Kovalenko A. V., Pismenskaya N. P., Mareev S. A., Pourcelly G., Competition between diffusion and electroconvection at an ion-selective surface in intensive current regimes. Adv. Colloid Interf. Sci. 235, 233–246 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Dukhin S. S., Electrokinetic phenomena of the second kind and their applications. Adv. Colloid Interf. Sci. 35, 173–196 (1991). [Google Scholar]
- 63.Rubinstein I., Mechanism for an electrodiffusional instability in concentration polarization. J. Chem. Soc. Faraday Trans. II 77, 1595–1609 (1981). [Google Scholar]
- 64.Rubinstein I., Zaltzman B., Equilibrium electroconvective instability. Phys. Rev. Lett. 114, 114502 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Kakiuchi T., Electrochemical instability of the liquid∣ liquid interface in the presence of ionic surfactant adsorption. J. Electroanal. Chem. 536, 63–69 (2002). [Google Scholar]
- 66.Tikekar M. D., Archer L. A., Koch D. L., Stabilizing electrodeposition in elastic solid electrolytes containing immobilized anions. Sci. Adv. 2, e1600320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Druzgalski C., Mani A., Statistical analysis of electroconvection near an ion-selective membrane in the highly chaotic regime. Phys. Rev. Fluids 1, 073601 (2016). [Google Scholar]
- 68.Mani A., Wang K. M., Electroconvection near electrochemical interfaces: Experiments, modeling, and computation. Annu. Rev. Fluid Mech. 52, 509–529 (2020). [Google Scholar]
- 69.Davidson S. M., Lammertink R. G. H., Mani A., Predictive model for convective flows induced by surface reactivity contrast. Phys. Rev. fluids 3, 053701 (2018). [Google Scholar]
- 70.Huth J. M., Swinney H. L., McCormick W. D., Kuhn A., Argoul F., Role of convection in thin-layer electrodeposition. Phys. Rev. E 51, 3444 (1995). [DOI] [PubMed] [Google Scholar]
- 71.Zhu Y., Xie J., Pei A., Liu B., Wu Y., Lin D., Li J., Wang H., Chen H., Xu J., Yang A., Wu C.-L., Wang H., Chen W., Cui Y., Fast lithium growth and short circuit induced by localized-temperature hotspots in lithium batteries. Nat. Commun. 10, 2067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang D., Warren A. J., Li G., Cheng Z., Han X., Zhao Q., Liu X., Deng Y., Archer L. A., Electrodeposition of zinc in aqueous electrolytes containing high molecular weight polymers. Macromolecules 53, 2694–2701 (2020). [Google Scholar]
- 73.Tikekar M. D., Li G., Archer L. A., Koch D. L., Electroconvection and morphological instabilities in potentiostatic electrodeposition across liquid electrolytes with polymer additives. J. Electrochem. Soc. 165, A3697–A3713 (2018). [Google Scholar]
- 74.Li G., Archer L. A., Koch D. L., Electroconvection in a viscoelastic electrolyte. Phys. Rev. Lett. 122, 124501 (2019). [DOI] [PubMed] [Google Scholar]
- 75.M. D. Tikekar, “The effect of ion transport and electrolyte rheology on morphological instabilities in electrodeposition,” thesis, Cornell University (2017). [Google Scholar]
- 76.Foroozan T., Yurkiv V., Sharifi-Asl S., Rojaee R., Mashayek F., Shahbazian-Yassar R., Non-dendritic Zn electrodeposition enabled by zincophilic graphene substrates. ACS Appl. Mater. Interfaces 11, 44077–44089 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Muldoon J., Bucur C. B., Oliver A. G., Sugimoto T., Matsui M., Kim H. S., Allred G. D., Zajicek J., Kotani Y., Electrolyte roadblocks to a magnesium rechargeable battery. Energy Environ. Sci. 5, 5941–5950 (2012). [Google Scholar]
- 78.Jäckle M., Helmbrecht K., Smits M., Stottmeister D., Groß A., Self-diffusion barriers: Possible descriptors for dendrite growth in batteries? Energy Environ. Sci. 11, 3400–3407 (2018). [Google Scholar]
- 79.Devan R. S., Lin J.-H., Huang Y.-J., Yang C.-C., Wu S. Y., Liou Y., Ma Y.-R., Two-dimensional single-crystalline Zn hexagonal nanoplates: Size-controllable synthesis and x-ray diffraction study. Nanoscale 3, 4339–4345 (2011). [DOI] [PubMed] [Google Scholar]
- 80.Tegart W. J. M., Sherby O. D., Activation energies for high temperature creep of polycrstalline zinc. Philos. Mag. 3, 1287–1296 (1958). [Google Scholar]
- 81.Li L., Wang Y., Chen Z., Hundekar P., Wang B., Shi J., Shi Y., Narayanan S., Koratkar N., Self-heating–induced healing of lithium dendrites. Science 359, 1513–1516 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Hundekar P., Basu S., Fan X., Li L., Yoshimura A., Gupta T., Sarbada V., Lakhnot A., Jain R., Narayanan S., Shi Y., Wang C., Koratkar N., In situ healing of dendrites in a potassium metal battery. Proc. Natl. Acad. Sci. U.S.A. 117, 5588–5594 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Enrique R. A., DeWitt S., Thornton K., Morphological stability during electrodeposition. MRS Commun. 7, 658–663 (2017). [Google Scholar]
- 84.Tikekar M. D., Choudhury S., Tu Z., Archer L. A., Design principles for electrolytes and interfaces for stable lithium-metal batteries. Nat. Energy 1, 16114 (2016). [Google Scholar]
- 85.Peled E., The electrochemical behavior of alkali and alkaline earth metals in nonaqueous battery systems—The solid electrolyte interphase model. J. Electrochem. Soc. 126, 2047–2051 (1979). [Google Scholar]
- 86.Peled E., Menkin S., SEI: Past, present and future. J. Electrochem. Soc. 164, A1703–A1719 (2017). [Google Scholar]
- 87.Zheng J., Kim M. S., Tu Z., Choudhury S., Tang T., Archer L. A., Regulating electrodeposition morphology of lithium: Towards commercially relevant secondary Li metal batteries. Chem. Soc. Rev. 49, 2701–2750 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Liu Z., Qi Y., Lin Y. X., Chen L., Lu P., Chen L. Q., Interfacial study on solid electrolyte interphase at Li metal anode: Implication for Li dendrite growth. J. Electrochem. Soc. 163, A592–A598 (2016). [Google Scholar]
- 89.Chao D., Zhou W., Xie F., Ye C., Li H., Jaroniec M., Qiao S.-Z., Roadmap for advanced aqueous batteries: From design of materials to applications. Sci. Adv. 6, eaba4098 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chao D., Ye C., Xie F., Zhou W., Zhang Q., Gu Q., Davey K., Gu L., Qiao S.-Z., Atomic engineering catalyzed MnO2 electrolysis kinetics for a hybrid aqueous battery with high power and energy density. Adv. Mater. 32, 2001894 (2020). [DOI] [PubMed] [Google Scholar]
- 91.Liu T., Wang S., Shi Z.-l., Ma G.-B., Wang M., Peng R.-W., Hao X.-P., Ming N.-b., Long-range ordering effect in electrodeposition of zinc and zinc oxide. Phys. Rev. E 75, 051606 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Desai D., Turney D. E., Anantharaman B., Steingart D. A., Banerjee S., Morphological evolution of nanocluster aggregates and single crystals in alkaline zinc electrodeposition. J. Phys. Chem. C 118, 8656–8666 (2014). [Google Scholar]
- 93.Bhadra S., Hsieh A. G., Wang M. J., Hertzberg B. J., Steingart D. A., Anode characterization in zinc-manganese dioxide AA alkaline batteries using electrochemical-acoustic time-of-flight analysis. J. Electrochem. Soc. 163, A1050–A1056 (2016). [Google Scholar]
- 94.Otani T., Nagata M., Fukunaka Y., Homma T., Morphological evolution of mossy structures during the electrodeposition of zinc from an alkaline zincate solution. Electrochim. Acta 206, 366–373 (2016). [Google Scholar]
- 95.Ito Y., Wei X., Desai D., Steingart D., Banerjee S., An indicator of zinc morphology transition in flowing alkaline electrolyte. J. Power Sources 211, 119–128 (2012). [Google Scholar]
- 96.Li Y., Li Y., Pei A., Yan K., Sun Y., Wu C.-L., Joubert L.-M., Chin R., Koh A. L., Yu Y., Perrino J., Butz B., Chu S., Cui Y., Atomic structure of sensitive battery materials and interfaces revealed by cryo–electron microscopy. Science 358, 506–510 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Li C., Shi X., Liang S., Ma X., Han M., Wu X., Zhou J., Spatially homogeneous copper foam as surface dendrite-free host for zinc metal anode. Chem. Eng. J. 379, 122248 (2020). [Google Scholar]
- 98.Qin T., Xu Z., Wang Z., Peng S., He D., 2.5 V salt-in-water supercapacitors based on alkali type double salt/carbon composite anode. J. Mater. Chem. A 7, 26011–26019 (2019). [Google Scholar]
- 99.Dong L., Ma X., Li Y., Zhao L., Liu W., Cheng J., Xu C., Li B., Yang Q.-H., Kang F., Extremely safe, high-rate and ultralong-life zinc-ion hybrid supercapacitors. Energy Storage Mater. 13, 96–102 (2018). [Google Scholar]
- 100.Zachman M. J., Hachtel J. A., Idrobo J. C., Chi M., Emerging electron microscopy techniques for probing functional interfaces in energy materials. Angew. Chem. Int. Ed. Eng. 59, 1384–1396 (2020). [DOI] [PubMed] [Google Scholar]
- 101.Suo L., Borodin O., Gao T., Olguin M., Ho J., Fan X., Luo C., Wang C., Xu K., “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 350, 938–943 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Borodin O., Self J., Persson K. A., Wang C., Xu K., Uncharted waters: Super-concentrated electrolytes. Joule 4, 69–100 (2020). [Google Scholar]
- 103.Chen C.-Y., Matsumoto K., Kubota K., Hagiwara R., Xu Q., A room-temperature molten hydrate electrolyte for rechargeable zinc–air batteries. Adv. Energy Mater. 9, 1900196 (2019). [Google Scholar]
- 104.Zhang C., Holoubek J., Wu X., Daniyar A., Zhu L., Chen C., Leonard D. P., Rodríguez-Pérez I. A., Jiang J.-X., Fang C., Ji X., A ZnCl2 water-in-salt electrolyte for a reversible Zn metal anode. Chem. Commun. 54, 14097–14099 (2018). [DOI] [PubMed] [Google Scholar]
- 105.Wu X., Xu Y., Zhang C., Leonard D. P., Markir A., Lu J., Ji X., Reverse dual-ion battery via a ZnCl2 water-in-salt electrolyte. J. Am. Chem. Soc. 141, 6338–6344 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Zhang L., Rodríguez-Pérez I. A., Jiang H., Zhang C., Leonard D. P., Guo Q., Wang W., Han S., Wang L., Ji X., ZnCl2 "water-in-salt" electrolyte transforms the performance of vanadium oxide as a Zn battery cathode. Adv. Funct. Mater. 29, 1902653 (2019). [Google Scholar]
- 107.Wang F., Borodin O., Gao T., Fan X., Sun W., Han F., Faraone A., Dura J. A., Xu K., Wang C., Highly reversible zinc metal anode for aqueous batteries. Nat. Mater. 17, 543 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Li M., Wang C., Chen Z., Xu K., Lu J., New concepts in electrolytes. Chem. Rev. 120, 6783–6819 (2020). [DOI] [PubMed] [Google Scholar]
- 109.Qiu H., Du X., Zhao J., Wang Y., Ju J., Chen Z., Hu Z., Yan D., Zhou X., Cui G., Zinc anode-compatible in-situ solid electrolyte interphase via cation solvation modulation. Nat. Commun. 10, 5374 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhong X., Wang F., Ding Y., Duan L., Shi F., Wang C., Water-in-salt electrolyte Zn/LiFePO4 batteries. J. Electroanal. Chem. 867, 114193 (2020). [Google Scholar]
- 111.Zheng J., Chen S., Zhao W., Song J., Engelhard M. H., Zhang J.-G., Extremely stable sodium metal batteries enabled by localized high-concentration electrolytes. ACS Energy Lett. 3, 315–321 (2018). [Google Scholar]
- 112.Chen S., Zheng J., Mei D., Han K. S., Engelhard M. H., Zhao W., Xu W., Liu J., Zhang J.-G., High-voltage lithium-metal batteries enabled by localized high-concentration electrolytes. Adv. Mater. 30, 1706102 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Zheng Y., Soto F. A., Ponce V., Seminario J. M., Cao X., Zhang J.-G., Balbuena P. B., Localized high concentration electrolyte behavior near a lithium–metal anode surface. J. Mater. Chem. A 7, 25047–25055 (2019). [Google Scholar]
- 114.Ta K., See K. A., Gewirth A. A., Elucidating Zn and Mg electrodeposition mechanisms in nonaqueous electrolytes for next-generation metal batteries. J. Phys. Chem. C 122, 13790–13796 (2018). [Google Scholar]
- 115.Han S.-D., Rajput N. N., Qu X., Pan B., He M., Ferrandon M. S., Liao C., Persson K. A., Burrell A. K., Origin of electrochemical, structural, and transport properties in nonaqueous zinc electrolytes. ACS Appl. Mater. Interfaces 8, 3021–3031 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Zhang N., Dong Y., Wang Y., Wang Y., Li J., Xu J., Liu Y., Jiao L., Cheng F., Ultrafast rechargeable zinc battery based on high-voltage graphite cathode and stable nonaqueous electrolyte. ACS Appl. Mater. Interfaces 11, 32978–32986 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Pan C., Nuzzo R. G., Gewirth A. A., ZnAlx Co2–xO4 spinels as cathode materials for non-aqueous Zn batteries with an open circuit voltage of≤ 2 V. Chem. Mater. 29, 9351–9359 (2017). [Google Scholar]
- 118.Pan C., Zhang R., Nuzzo R. G., Gewirth A. A., ZnNixMnxCo2−2xO4 spinel as a high-voltage and high-capacity cathode material for nonaqueous Zn-Ion batteries. Adv. Energy Mater. 8, 1800589 (2018). [Google Scholar]
- 119.Guerfi A., Trottier J., Boyano I., De Meatza I., Blazquez J. A., Brewer S., Ryder K. S., Vijh A., Zaghib K., High cycling stability of zinc-anode/conducting polymer rechargeable battery with non-aqueous electrolyte. J. Power Sources 248, 1099–1104 (2014). [Google Scholar]
- 120.Park M. J., Manthiram A., Unveiling the charge storage mechanism in non-aqueous and aqueous Zn/Na3V2 (PO4)2F3 batteries. ACS Appl. Energy Mater. 3, 5015–5023 (2020). [Google Scholar]
- 121.Naveed A., Yang H., Yang J., Nuli Y., Wang J., Highly reversible and rechargeable safe Zn batteries based on a triethyl phosphate electrolyte. Angew. Chem. Int. Ed. Eng. 131, 2786–2790 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Naveed A., Yang H., Shao Y., Yang J., Yanna N., Liu J., Shi S., Zhang L., Ye A., He B., Wang J., A highly reversible zn anode with intrinsically safe organic electrolyte for long-cycle-life batteries. Adv. Mater. 31, 1900668 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Dong Y., Di S., Zhang F., Bian X., Wang Y., Xu J., Wang L., Cheng F., Zhang N., Nonaqueous electrolyte with dual-cations for high-voltage and long-life zinc batteries. J. Mater. Chem. A 8, 3252–3261 (2020). [Google Scholar]
- 124.Häussermann U., Simak S. I., Origin of the c/a variation in hexagonal close-packed divalent metals. Phys. Rev. B 64, 245114 (2001). [Google Scholar]
- 125.Tran R., Xu Z., Radhakrishnan B., Winston D., Sun W., Persson K. A., Ong S. P., Surface energies of elemental crystals. Sci. Data 3, 160080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miller W. A., Carpenter G. J. C., Chadwick G. A., Anisotropy of interfacial free energy of some hexagonal close-packed metals. Philos. Mag. 19, 305–319 (1969). [Google Scholar]
- 127.T. L. Einstein, Equilibrium shape of crystals, in Handbook of Crystal Growth (Elsevier, 2015), pp. 215–264. [Google Scholar]
- 128.Searcy A. W., The equilibrium shapes of crystals and of cavities in crystals. J. Solid State Chem. 48, 93–99 (1983). [Google Scholar]
- 129.X. Ma, A. A. Lall, G. W. Mulholland, M. R. Zachariah, Evaporation anisotropy of free nanocrystals. 115, 16941–16946 (2011).
- 130.Cho S., Kim H.-J., Lee K.-H., A method for covering a substrate with highly-oriented single crystalline hexagonal zinc structures under ambient pressure and room temperature. Chem. Commun. , 6053–6055 (2009). [DOI] [PubMed] [Google Scholar]
- 131.Yang J.-M., Hsieh Y.-T., Chu-Tien T.-T., Sun I.-W., Electrodeposition of distinct one-dimensional Zn biaxial microbelt from the zinc chloride-1-ethyl-3-methylidazolium chloride ionic liquid. J. Electrochem. Soc. 158, D235–D239 (2011). [Google Scholar]
- 132.Abbott A. P., Barron J. C., Frisch G., Ryder K. S., Silva A. F., The effect of additives on zinc electrodeposition from deep eutectic solvents. Electrochim. Acta 56, 5272–5279 (2011). [Google Scholar]
- 133.Xu L., Guo Y., Liao Q., Zhang J., Xu D., Morphological control of ZnO nanostructures by electrodeposition. J. Phys. Chem. B 109, 13519–13522 (2005). [DOI] [PubMed] [Google Scholar]
- 134.Pradhan D., Sindhwani S., Leung K. T., Template-free electrochemical growth of single-crystalline zinc nanowires at an anomalously low temperature. J. Phys. Chem. C 113, 15788–15791 (2009). [Google Scholar]
- 135.Khan W. S., Cao C., Zhong J., Liu Y., Iqbal M. A., Synthesis of metallic Zn microprisms, their growth mechanism and PL properties. Mater. Lett. 64, 2273–2276 (2010). [Google Scholar]
- 136.Chen Y. J., Chi B., Zhang H. Z., Chen H., Chen Y., Controlled growth of zinc nanowires. Mater. Lett. 61, 144–147 (2007). [Google Scholar]
- 137.Ohmori Y., Nakai K., Ohtsubo H., Yagi T., Matsumoto T., Crystallographic analysis of electrodeposited zinc crystals on Fe substrate. ISIJ Int. 33, 1196–1201 (1993). [Google Scholar]
- 138.Zheng J. K., Xu X., Luo R., Zeng X., Chen B., Degradation of precipitation hardening in 7075 alloy subject to thermal exposure: A Cs-corrected STEM study. J. Alloys Compd. 741, 656–660 (2018). [Google Scholar]
- 139.M. A. Herman, W. Richter, H. Sitter, Epitaxy: Physical Principles and Technical Implementation (Springer Science & Business Media, 2013), vol. 62. [Google Scholar]
- 140.Turney D. E., Gallaway J. W., Yadav G. G., Ramirez R., Nyce M., Banerjee S., Chen-Wiegart Y.-c. K., Wang J., D’Ambrose M. J., Kolhekar S., Huang J., Wei X., Rechargeable zinc alkaline anodes for long-cycle energy storage. Chem. Mater. 29, 4819–4832 (2017). [Google Scholar]
- 141.McBreen J., Gannon E., The electrochemistry of metal oxide additives in pasted zinc electrodes. Electrochim. Acta 26, 1439–1446 (1981). [Google Scholar]
- 142.Li N., Zhang K., Xie K., Wei W., Gao Y., Bai M., Gao Y., Hou Q., Shen C., Xia Z., Wei B., Reduced-graphene-oxide-guided directional growth of planar lithium layers. Adv. Mater. 32, 1907079 (2020). [DOI] [PubMed] [Google Scholar]
- 143.Bozorth R. M., The orientations of crystals in electrodeposited metals. Phys. Rev. 26, 390–400 (1925). [Google Scholar]
- 144.Glocker R., Kaupp E., Über die Faserstruktur elektrolytischer Metallniederschläge. Z. Phys. 24, 121–139 (1924). [Google Scholar]
- 145.Pangarov N. A., Preferred orientations in electro-deposited metals. J. Electroanal. Chem. 9, 70–85 (1965). [Google Scholar]
- 146.Pangarov N. A., The crystal orientation of electrodeposited metals. Electrochim. Acta 7, 139–146 (1962). [Google Scholar]
- 147.Eaglesham D. J., Gossmann H. J., Cerullo M., Limiting thickness h epi for epitaxial growth and room-temperature Si growth on Si (100). Phys. Rev. Lett. 65, 1227–1230 (1990). [DOI] [PubMed] [Google Scholar]
- 148.Ito Y., Nyce M., Plivelich R., Klein M., Steingart D., Banerjee S., Zinc morphology in zinc–nickel flow assisted batteries and impact on performance. J. Power Sources 196, 2340–2345 (2011). [Google Scholar]
- 149.Wang Z., Huang J., Guo Z., Dong X., Liu Y., Wang Y., Xia Y., A metal-organic framework host for highly reversible dendrite-free zinc metal anodes. Joule 3, 1289–1300 (2019). [Google Scholar]
- 150.Yuksel R., Buyukcakir O., Seong W. K., Ruoff R. S., Metal-organic framework integrated anodes for aqueous zinc-ion batteries. Adv. Energy Mater. 10, 1904215 (2020). [Google Scholar]
- 151.Liu M., Yang L., Liu H., Amine A., Zhao Q., Song Y., Yang J., Wang K., Pan F., Artificial solid-electrolyte interface facilitating dendrite-free zinc metal anodes via nanowetting effect. ACS Appl. Mater. Interfaces 11, 32046–32051 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Xin X., Ito K., Dutta A., Kubo Y., Dendrite-free epitaxial growth of lithium metal during charging in Li–O2 batteries. Angew. Chem. Int. Ed. 57, 13206–13210 (2018). [DOI] [PubMed] [Google Scholar]
- 153.Ko J. S., Geltmacher A. B., Hopkins B. J., Rolison D. R., Long J. W., Parker J. F., Robust 3D Zn sponges enable high-power, energy-dense alkaline batteries. ACS Appl. Energy Mater. 2, 212–216 (2018). [Google Scholar]
- 154.Parker J. F., Chervin C. N., Nelson E. S., Rolison D. R., Long J. W., Wiring zinc in three dimensions re-writes battery performance—Dendrite-free cycling. Energy Environ. Sci. 7, 1117–1124 (2014). [Google Scholar]
- 155.Kang Z., Wu C., Dong L., Liu W., Mou J., Zhang J., Chang Z., Jiang B., Wang G., Kang F., Wu C., 3D porous copper skeleton supported zinc anode toward high capacity and long cycle life zinc ion batteries. ACS Sustain. Chem. Eng. 7, 3364–3371 (2019). [Google Scholar]
- 156.Shi X., Xu G., Liang S., Li C., Guo S., Xie X., Ma X., Zhou J., Homogeneous deposition of zinc on three-dimensional porous copper foam as a superior zinc metal anode. ACS Sustain. Chem. Eng. 7, 17737–17746 (2019). [Google Scholar]
- 157.Zhang Q., Luan J., Fu L., Wu S., Tang Y., Ji X., Wang H., The three-dimensional dendrite-free zinc anode on a copper mesh with a zinc-oriented polyacrylamide electrolyte additive. Angew. Chem. Int. Ed. 58, 15841–15847 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Dong W., Shi J.-L., Wang T.-S., Yin Y.-X., Wang C.-R., Guo Y.-G., 3D zinc@ carbon fiber composite framework anode for aqueous Zn–MnO2 batteries. RSC Adv. 8, 19157–19163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang L.-P., Li N.-W., Wang T.-S., Yin Y.-X., Guo Y.-G., Wang C.-R., Conductive graphite fiber as a stable host for zinc metal anodes. Electrochim. Acta 244, 172–177 (2017). [Google Scholar]
- 160.Zuo T. T., Wu X. W., Yang C. P., Yin Y. X., Ye H., Li N. W., Guo Y. G., Graphitized carbon fibers as multifunctional 3D current collectors for high areal capacity Li anodes. Adv. Mater. 29, 1700389 (2017). [DOI] [PubMed] [Google Scholar]
- 161.Yan K., Lu Z., Lee H.-W., Xiong F., Hsu P.-C., Li Y., Zhao J., Chu S., Cui Y., Selective deposition and stable encapsulation of lithium through heterogeneous seeded growth. Nat. Energy 1, 16010 (2016). [Google Scholar]
- 162.Liu L., Yin Y. X., Li J. Y., Wang S. H., Guo Y. G., Wan L. J., Uniform lithium nucleation/growth induced by lightweight nitrogen-doped graphitic carbon foams for high-performance lithium metal anodes. Adv. Mater. 30, 1706216 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Chen X., Chen X.-R., Hou T.-Z., Li B.-Q., Cheng X.-B., Zhang R., Zhang Q., Lithiophilicity chemistry of heteroatom-doped carbon to guide uniform lithium nucleation in lithium metal anodes. Sci. Adv. 5, eaau7728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang S. H., Yin Y. X., Zuo T. T., Dong W., Li J. Y., Shi J. L., Zhang C. H., Li N. W., Li C. J., Guo Y. G., Stable Li metal anodes via regulating lithium plating/stripping in vertically aligned microchannels. Adv. Mater. 29, 1703729 (2017). [DOI] [PubMed] [Google Scholar]
- 165.Chen K.-H., Sanchez A. J., Kazyak E., Davis A. L., Dasgupta N. P., Synergistic effect of 3D current collectors and ALD surface modification for high Coulombic efficiency lithium metal anodes. Adv. Energy Mater. 9, 1802534 (2019). [Google Scholar]
- 166.Zhao Z., Zhao J., Hu Z., Li J., Li J., Zhang Y., Wang C., Cui G., Long-life and deeply rechargeable aqueous Zn anodes enabled by a multifunctional brightener-inspired interphase. Energy Environ. Sci. 12, 1938–1949 (2019). [Google Scholar]
- 167.Vatsalarani J., Geetha S., Trivedi D. C., Warrier P. C., Stabilization of zinc electrodes with a conducting polymer. J. Power Sources 158, 1484–1489 (2006). [Google Scholar]
- 168.Zhu J., Zhou Y., Effects of ionomer films on secondary alkaline zinc electrodes. J. Power Sources 73, 266–270 (1998). [Google Scholar]
- 169.Wu Y., Zhang Y., Ma Y., Howe J. D., Yang H., Chen P., Aluri S., Liu N., Ion-sieving carbon nanoshells for deeply rechargeable Zn-based aqueous batteries. Adv. Energy Mater. 8, 1802470 (2018). [Google Scholar]
- 170.Zhou Z., Zhang Y., Chen P., Wu Y., Yang H., Ding H., Zhang Y., Wang Z., Du X., Liu N., Graphene oxide-modified zinc anode for rechargeable aqueous batteries. Chem. Eng. Sci. 194, 142–147 (2019). [Google Scholar]
- 171.Zhao K., Wang C., Yu Y., Yan M., Wei Q., He P., Dong Y., Zhang Z., Wang X., Mai L., Ultrathin surface coating enables stabilized zinc metal anode. Adv. Mater. Interfaces 5, 1800848 (2018). [Google Scholar]
- 172.Kang L., Cui M., Jiang F., Gao Y., Luo H., Liu J., Liang W., Zhi C., Nanoporous CaCO3 coatings enabled uniform Zn stripping/plating for long-life Zinc rechargeable aqueous batteries. Adv. Energy Mater. 8, 1801090 (2018). [Google Scholar]
- 173.Liang P., Yi J., Liu X., Wu K., Wang Z., Cui J., Liu Y., Wang Y., Xia Y., Zhang J., Highly reversible Zn anode enabled by controllable formation of nucleation sites for Zn-based batteries. Adv. Funct. Mater. 30, 1908528 (2020). [Google Scholar]
- 174.Deng C., Xie X., Han J., Tang Y., Gao J., Liu C., Shi X., Zhou J., Liang S., A sieve-functional and uniform-porous kaolin layer toward stable zinc metal anode. Adv. Funct. Mater. 2000599 (2020). [Google Scholar]
- 175.Jo Y. N., Kang S. H., Prasanna K., Eom S. W., Lee C. W., Shield effect of polyaniline between zinc active material and aqueous electrolyte in zinc-air batteries. Appl. Surf. Sci. 422, 406–412 (2017). [Google Scholar]
- 176.Chang G., Liu S., Fu Y., Hao X., Jin W., Ji X., Hu J., Inhibition role of trace metal ion additives on zinc dendrites during plating and striping processes. Adv. Mater. Interfaces 6, 1901358 (2019). [Google Scholar]
- 177.Otani T., Fukunaka Y., Homma T., Effect of lead and tin additives on surface morphology evolution of electrodeposited zinc. Electrochim. Acta 242, 364–372 (2017). [Google Scholar]
- 178.Sun B., Zou X.-W., Jin Z.-Z., Morphological evolution in the electrodeposition of the Pb-Sn binary system. Phys. Rev. E 69, 067202 (2004). [DOI] [PubMed] [Google Scholar]
- 179.Haataja M., Srolovitz D. J., Morphological instability and additive-induced stabilization in electrodeposition. Phys. Rev. Lett. 89, 215509 (2002). [DOI] [PubMed] [Google Scholar]
- 180.Haataja M., Srolovitz D. J., Bocarsly A. B., Morphological stability during electrodeposition II. Additive effects. J. Electrochem. Soc. 150, C708–C716 (2003). [Google Scholar]
- 181.Wu K., Huang J., Yi J., Liu X., Liu Y., Wang Y., Zhang J., Xia Y., Recent advances in polymer electrolytes for zinc ion batteries: Mechanisms, properties, and perspectives. Adv. Energy Mater. 10, 1903977 (2020). [Google Scholar]
- 182.Wu B., Lu W., A consistently coupled multiscale mechanical–electrochemical battery model with particle interaction and its validation. J. Mech. Phys. Solids 125, 89–111 (2019). [Google Scholar]
- 183.Yurkiv V., Foroozan T., Ramasubramanian A., Ragone M., Shahbazian-Yassar R., Mashayek F., Understanding Zn electrodeposits morphology in secondary batteries using phase-field model. J. Electrochem. Soc. 167, 060503 (2020). [Google Scholar]
- 184.Monroe C., Newman J., The impact of elastic deformation on deposition kinetics at lithium/polymer interfaces. J. Electrochem. Soc. 152, A396–A404 (2005). [Google Scholar]
- 185.Ahmad Z., Viswanathan V., Stability of electrodeposition at solid-solid interfaces and implications for metal anodes. Phys. Rev. Lett. 119, 056003 (2017). [DOI] [PubMed] [Google Scholar]
- 186.Ahmad Z., Viswanathan V., Role of anisotropy in determining stability of electrodeposition at solid-solid interfaces. Phys. Rev. Mater. 1, 055403 (2017). [DOI] [PubMed] [Google Scholar]
- 187.Dunne P., Coey J. M. D., Patterning metallic electrodeposits with magnet arrays. Phys. Rev. B 85, 224411 (2012). [Google Scholar]
- 188.Fahidy T. Z., Magnetoelectrolysis. J. Appl. Electrochem. 13, 553–563 (1983). [Google Scholar]
- 189.Wang A., Deng Q., Deng L., Guan X., Luo J., Eliminating tip dendrite growth by Lorentz force for stable lithium metal anodes. Adv. Funct. Mater. , 1902630 (2019). [Google Scholar]
- 190.Shen K., Wang Z., Bi X., Ying Y., Zhang D., Jin C., Hou G., Cao H., Wu L., Zheng G., Tang Y., Tao X., Lu J., Magnetic field–suppressed lithium dendrite growth for stable lithium-metal batteries. Adv. Energy Mater. , 1900260 (2019). [Google Scholar]
- 191.Taniguchi T., Sassa K., Yamada T., Asai S., Control of crystal orientation in zinc electrodeposits by imposition of a high magnetic field. Mater. Trans. JIM 41, 981–984 (2000). [Google Scholar]
- 192.Chandrasekar M. S., Shanmugasigamani, Malathy P., Synergetic effects of pulse constraints and additives in electrodeposition of nanocrystalline zinc: Corrosion, structural and textural characterization. Mater. Chem. Phys. 124, 516–528 (2010). [Google Scholar]
- 193.Saber K., Koch C. C., Fedkiw P. S., Pulse current electrodeposition of nanocrystalline zinc. Mater. Sci. Eng. A 341, 174–181 (2003). [Google Scholar]
- 194.Pavithra C. L. P., Sarada B. V., Rajulapati K. V., Ramakrishna M., Gundakaram R. C., Rao T. N., Sundararajan G., Controllable crystallographic texture in copper foils exhibiting enhanced mechanical and electrical properties by pulse reverse electrodeposition. Cryst. Growth Des. 15, 4448–4458 (2015). [Google Scholar]
- 195.Tsai W. L., Hsu P. C., Hwu Y., Chen C. H., Chang L. W., Je J. H., Lin H. M., Groso A., Margaritondo G., Electrochemistry: Building on bubbles in metal electrodeposition. Nature 417, 139 (2002). [DOI] [PubMed] [Google Scholar]
- 196.Hsu P.-C., Seol S.-K., Lo T.-N., Liu C.-J., Wang C.-L., Lin C.-S., Hwu Y., Chen C. H., Chang L.-W., Je J. H., Hydrogen bubbles and the growth morphology of ramified zinc by electrodeposition. J. Electrochem. Soc. 155, D400–D407 (2008). [Google Scholar]
- 197.Wang X., Pawar G., Li Y., Ren X., Zhang M., Lu B., Banerjee A., Liu P., Dufek E. J., Zhang J.-G., Xiao J., Liu J., Meng Y. S., Liaw B., Glassy Li metal anode for high-performance rechargeable Li batteries. Nat. Mater. 19, 1339–1345 (2020). [DOI] [PubMed] [Google Scholar]
- 198.Song Y., Hu J., Tang J., Gu W., He L., Ji X., Real-time x-ray imaging reveals interfacial growth, suppression, and dissolution of zinc dendrites dependent on anions of ionic liquid additives for rechargeable battery applications. ACS Appl. Mater. Interfaces 8, 32031–32040 (2016). [DOI] [PubMed] [Google Scholar]
- 199.Wang J.-G., Tian M.-L., Kumar N., Mallouk T. E., Controllable template synthesis of superconducting Zn nanowires with different microstructures by electrochemical deposition. Nano Lett. 5, 1247–1253 (2005). [DOI] [PubMed] [Google Scholar]
- 200.Garcia G., Ventosa E., Schuhmann W., Complete prevention of dendrite formation in Zn metal anodes by means of pulsed charging protocols. ACS Appl. Mater. Interfaces 9, 18691–18698 (2017). [DOI] [PubMed] [Google Scholar]