

Ancient genomes reveal a complex demographic picture since the post-LGM in northeast Asia and report the presence of Y. pestis.
Abstract
We present genome-wide data from 40 individuals dating to c.16,900 to 550 years ago in northeast Asia. We describe hitherto unknown gene flow and admixture events in the region, revealing a complex population history. While populations east of Lake Baikal remained relatively stable from the Mesolithic to the Bronze Age, those from Yakutia and west of Lake Baikal witnessed major population transformations, from the Late Upper Paleolithic to the Neolithic, and during the Bronze Age, respectively. We further locate the Asian ancestors of Paleo-Inuits, using direct genetic evidence. Last, we report the most northeastern ancient occurrence of the plague-related bacterium, Yersinia pestis. Our findings indicate the highly connected and dynamic nature of northeast Asia populations throughout the Holocene.
INTRODUCTION
Siberia covers an extensive area in northeast Asia, stretching eastward from the Ural Mountains to the Pacific Ocean and northward from the Altai Saian Mountains to the Arctic Ocean. Despite being one of the most sparsely populated regions on Earth (1), this large territory has an intriguing human history as home to multiple ethnic groups and a source area for the peopling of Americas (2).
Anatomically modern humans pioneered the northeastern part of Siberia ~38,000 years ago, in parallel with the dispersal of the parts of megafauna, i.e., mammoth, woolly rhino, and cave lion (3–5). These dispersals took place before the beginning of the Last Glacial Maximum (LGM) ~33,000 years ago. By the time the glaciation came to an end, ~20,000 to 18,000 years ago, both humans and the megafauna in the area had suffered the consequences of the LGM, but new data have contradicted earlier speculation of a marked settlement decline in the area (5–7). Traces of LGM and post-LGM human activity in northeast Asia, as evidenced by the presence of lithic technologies and pre-Neolithic pottery, are found across the east Siberian plateau as well as the Trans-Baikal area, east of Lake Baikal (2, 8–11). Throughout the Holocene, multiple cultural complexes emerged gradually across the northeast Asia, some of which spread eastward through northern Alaska and reached the previously uninhabited Greenland around 4500 years ago (11–13).
Recent genetic studies have revealed complex and dynamic population histories in northeast Asia, shaped by multiple admixture and gene flow events. These studies reported substantial changes in human population genetic structure associated with migrations across the area (5, 14–17). Still, further investigation is required to fully assess the population dynamics of this vast geographical territory across a broad time span. Increased resolution can be achieved by analyzing ancient genetic data from unexplored regions and archaeological sites of northeast Asia. For example, genetic data from sites in the Trans-Baikal area carrying the earliest traces of post-LGM human occupation and from sites in Yakutia associated with the ancestors of the Paleo-Inuit Saqqaq cultural complex can provide new investigative venues. Here, focusing on these regions, we explore the population history of northeast Asia, particularly throughout the Lena, Angara, and Kolyma river basins and the Lake Baikal area. In studying a comprehensive sample of ancient genomes, we infer to what extent mobility, admixture processes, or even pandemics have affected northeast Asia over the Holocene.
RESULTS AND DISCUSSION
Genome-wide ancient DNA data
We produced whole-genome sequence data from 40 ancient individuals spanning from the Late Upper Paleolithic to the Medieval era (table S1) and representing five distinct administrative regions in the Russian Federation encompassing Yakutia, Trans-Baikal, Cis-Baikal, Krasnoyarsk Krai, and Amur Oblast (Fig. 1). Genome sequence data from Yakutia include 10 individuals spanning a time period between c.16,900 and 2490 calibrated years before the present (cal BP) (0.03×–8.9× genome coverage); data from the Trans-Baikal area consist of 8 individuals dated to between c.8515 and 3000 cal BP (0.1×–4.7×); data from the Cis-Baikal area encompass 20 individuals spanning a time period between c.8980 and 550 cal BP (0.1×–14.5×); data from Krasnoyarsk Krai encompass an individual dated to c.4280 to 4085 cal BP (13.6×); and data from Amur Oblast cover an individual dated to c.1345 to 1270 cal BP (0.7×) (fig. S1 and table S1). Fifteen individuals were classified as biologically female, and 25 individuals were found to be male (table S1). All individuals were accredited to either Y macro-haplogroup Q or N and non-African mitochondrial macrohaplogroups of M, N, and R (18). We excluded biologically related individuals from the frequency-based analyses (table S1 and see Supplementary Text).
Fig. 1. Geographical and chronological information concerning the ancient individuals.
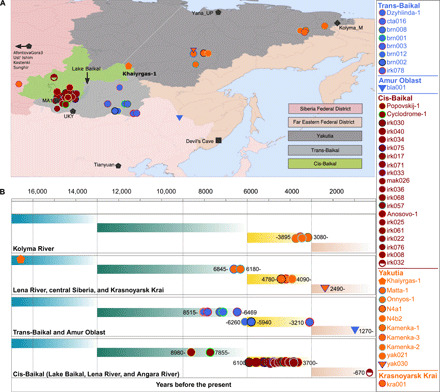
(A) Geographical map showing the locations of the individuals sequenced in this study (orange, blue, and red). Genomes published elsewhere are shown as black (see table S2 for information about all published individuals used in comparative analysis). (B) Timeline showing the ages of the ancient individuals as calibrated years before the present.
The degree of modern DNA contamination was negligible for all data (table S1). Principal components analysis (PCA) using present-day world populations (table S2) displayed a genetic gradient extending from west Eurasia to Central and East Asia (Fig. 2A). Projecting ancient individuals onto the space defined by the first two components indicated that the analyzed ancient northeast Asian individuals harbored genetic affinities to present-day populations of Central and East Asia (Fig. 2A). To infer post-LGM human population dynamics in northeast Asia, we first analyzed the oldest individual in our data and then investigated the population transformations in time until the Medieval era.
Fig. 2. PCA and ADMIXTURE analysis.
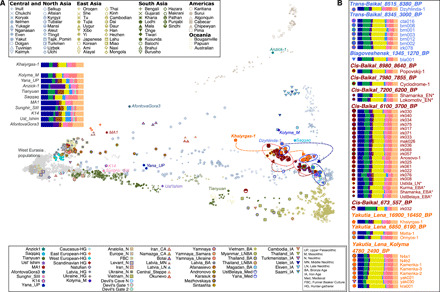
(A) PCA calculated using a set of world populations. Ancient individuals were projected onto the inferred PC space (see table S2 for information about individuals). Arrows indicate the direction of population changes in time. Asterisk denotes being published in (16). (B) A subset of ADMIXTURE result for K = 14 clusters showing the ancestral composition of investigated ancient individuals.
Population dynamics during and after the LGM in northeast Asia
The Khaiyrgas Cave on the middle Lena is one of the earliest locations of post-LGM human occupation in northeast Asia (8, 19, 20). However, origin and legacy of people who settled this part of northeast Asia remain unknown. We sequenced ancient DNA from a deciduous tooth of a c.16,900-year-old subadult female (Khaiyrgas-1) excavated from the upper layer of the Paleolithic horizon of the Khaiyrgas Cave to investigate the post-LGM population dynamics in northeast Asia. This individual is one the first known post-LGM representatives of the settlers of the Central Siberian Plateau. Human groups geographically represented by this individual retracted from the area during the Bølling-Allerød warming period dated to c.15,000 to 13,000 cal BP (8).
Khaiyrgas-1 exhibited genetic affinity toward present-day Selkups, a north Siberian Uralic-speaking population, on the PCA (Fig. 2A). This individual shared more alleles with indigenous populations of Native America, i.e., Chane, Guarani, and Karitiana, but to a lesser degree with Selkups compared with other world populations as measured by outgroup f3-statistics (fig. S2 and table S3). We estimated ancestral clusters in the genome of Khaiyrgas-1 using ADMIXTURE (21) (Fig. 2B and fig. S3). On the basis of K = 14 ancestral components, a genetic component that is maximized in the present-day Nganasan population from northeast Asia (yellow) and another component that is maximized in the present-day Native America populations (purple) were present at high levels in the genome of Khaiyrgas-1 compared to other Upper Paleolithic individuals from Eurasia and Asia (Fig. 2, figs. S3 and S4, and table S4).
According to the PCA, Khaiyrgas-1 marked the first major genetic shift throughout the region upon the end of the LGM. This individual representing a distinct line of ancestry in a maximum likelihood tree fitted using TreeMix (22) (fig. S5) was positioned on the PCA between two distinct lineages from Siberia encompassing Ancient North Siberians (ANS), the first inhabitants of northeast Siberia, represented by a ~38,000-year-old Upper Paleolithic individual from the Yana RHS (Rhinoceros Horn Site) (Yana_UP), and Ancient Paleo-Siberians (AP), the Siberian ancestors of Native Americans, represented by a ~9800-year-old individual from the Kolyma region (Kolyma_M) (5). To test whether there was a local continuity during and after the LGM, i.e., genetic continuity between the ~38,000-year-old Yana_UP (5), the ~16,900-year-old Khaiyrgas-1, and the ~9800-year-old Kolyma_M (5), or whether there was a possible gene flow from distant sources into the region, we performed formal tests of shared genetic drift through f4-statistics for different tree-like topologies. F4-statistics in the form of f4(Yoruba, Popx; Yana_UP, Khaiyrghas-1) where Popx is either the ~24,000-year-old MA1 (23) or the ~16,000-year-old AfontovaGora3 (24) [Ancient North Eurasians (ANEs)] revealed that Khaiyrgas-1 shares more genetic drift with ANE lineage than Yana_UP (ANS), thus implying a possible gene flow from west Eurasia into the region during the LGM (table S5). F4-statistics in the form of f4(Yoruba, Khaiyrghas-1; Popx, Kolyma_M) and f4(Yoruba, Kolyma_M; Popx, Khaiyrghas-1) (figs. S6 and S7 and table S6) revealed that Khaiyrgas-1 and Kolyma_M share more drift with each other compared to Yana_UP and other Upper Paleolithic individuals from Eurasia and Asia including MA1 (23) and AfontovaGora3 (24), the ~45,000-year-old Ust’ Ishim (25), the ~37,000-year-old Kostenki14 (26) and the ~34,000-year-old Sunghir from Russia (27), and the ~40,000-year-old Tianyuan from China (28). The only exceptions were the ~12,000-year-old Anzick-1 from North America (29) and the ~4000-year-old Saqqaq from Greenland (30), both of which shared more drift with Kolyma_M (5) (fig. S8). Recently, a 14,500-year-old Upper Paleolithic individual from the south of Lake Baikal (UKY) (17) was found to be closely related with Kolyma_M (5). These two individuals (UKY and Kolyma_M) have genetic affinity toward Native American populations. In addition, f4(Yoruba, Anzick-1; Khaiyrghas-1, UKY) revealed that Anzick-1 shared more alleles with the UKY compared to the Khaiyrgas-1 (Z = 2.1). To further assess the complex relationship between Khaiyrgas-1, UKY, and Kolyma_M, we calculated f4-statistics in the form of f4(Yoruba, UKY; Khaiyrghas-1, Kolyma_M); unexpectedly, Khaiyrgas-1 and Kolyma_M appeared symmetrically related to the UKY individual (Z = 0.2). Furthermore, f4(Yoruba, Khaiyrghas-1; Kolyma_M, UKY) also suggested that UKY and Kolyma_M were symmetrically related to Khaiyrgas-1 (Z = 0.01). These results, likely due to a lack of power since these tests are based on a relatively low number of single-nucleotide polymorphisms (SNPs) (9366), leave multiple scenarios consistent with the data. Last, we modeled the Kolyma_M as a two-way mixture of Khaiyrgas-1 (80 ± 19%) and Devils_Cave_N (19 ± 19%) using qpAdm (P = 0.28). Our results show the presence of a new post-LGM lineage in northeast Asia, represented by Khaiyrgas-1, which is distinct from the older ANS who settled the region around 38,000 years ago. This lineage contributed directly to the later groups in the region, the AP who settled the area around 9800 years ago.
Population transformations across Yakutia and the origins of the Paleo-Inuits
To further investigate population dynamics in northeast Asia between ~13,000 and 2500 years ago, a period of substantial cultural and social changes marked by transition from the Paleolithic to the Neolithic (31), we analyzed chronological genome sequence data from the Lena and Kolyma river regions of Yakutia. These data included a ~6800-year-old individual associated with the Syalakh cultural complex (Matta-1), a ~6200-year-old individual associated with the Belkachi cultural complex (Onnyos-1), and seven individuals dated to c.4780 to 2490 cal BP (Fig. 1 and table S1). The PCA revealed a west-east genetic cline extending from the Late Upper Paleolithic to the Iron Age and the presence of three major genetic changes during this period throughout Lena and Kolyma regions (Fig. 2A). These include a shift ~9800 years ago between the Late Upper Paleolithic and Mesolithic (5), another one ~6800 years ago between the Mesolithic and Early/Middle Neolithic, and ~4700 years ago between Early/Middle Neolithic and Late Neolithic/Iron Age (Fig. 2A).
Matta-1 and Onnyos-1 formed a distinct group (Yakutia_Lena_6850_6190_BP) on the PCA between Khaiyrgas-1, Kolyma_M (5), and a group of succeeding individuals (Yakutia_Lena_Kolyma_4780_2490_BP) (Fig. 2A and fig. S9). The Bronze Age (c.4280 to 4085 BP) individual (kra001) from the Krasnoyarsk Krai (Fig. 1A) grouped with Yakutia_Lena_Kolyma_4780_2490_BP. Yakutia_Lena_6850_6190_BP (table S7 and fig. S10) and Yakutia_Lena_Kolyma_4780_2490_BP (table S8 and fig. S11) exhibited genetic affinities toward present-day northeast Asia populations (32). ADMIXTURE analysis revealed a pattern of increase in northeast Asia–related and a pattern of decrease in Native America–related genetic ancestry from the Late Upper Paleolithic to the Iron Age (Fig. 2B, figs. S4 and S12, and table S4). Consistently, Devil’s Cave individuals representing the East Asian ancestry and the Anzick-1 individual representing the Native American ancestry shared more alleles with the succeeding and preceding groups, respectively, implying the presence of gene flow into the Lena and Kolyma regions from Far Eastern sources during the Neolithic and the Bronze Age (figs. S13 and S14 and table S9).
During the Neolithic, various cultural complexes emerged throughout Yakutia, notably represented by large Neolithic cemeteries in the Angara and Lena river basins. Of these, the Belkachi lithic complex is of particular interest since it has been hypothesized that the people associated with it might be ancestral to the Paleo-Inuit Saqqaq cultural complex, albeit without genetic evidence (12, 13). Here, Matta-1 and Onnyos-1 (Yakutia_Lena_6850_6190_BP) exhibited genetic affinities toward the Paleo-Inuit Saqqaq individual (Fig. 2A), which was confirmed by f4-statistics in which Yakutia_Lena_6850_6190_BP and Saqqaq shared more drift with each other compared to other ancient groups (figs. S15 and S16 and table S10). Consistently, distribution of ancestral components in the genomes of Yakutia_Lena_6850_6190_BP and Saqqaq was similar (Fig. 2B). Genetic affinities between Paleo-Inuit and Chukotko-Kamchatkan speakers have been reported previously (33). Here, outgroup f3-statistics in the form of f3(Yakutia_Lena_6850_6190_BP, PopX, Yoruba) further revealed that Yakutia_Lena_6850_6190_BP shares more alleles with Chukotko-Kamchatkan speakers, i.e., Itelmen and Koryak populations (33) (table S7 and fig. S10). To confirm the ancestry proportions in the genome of the Saqqaq, we used qpAdm (34) estimating the Saqqaq as a mixture of 90 ± 10% Yakutia_Lena_6850_6190_BP and 10 ± 10% west Eurasians (K14) (P = 0.40) (table S11). These results provide the first direct evidence of Belkachi-related genetic ancestry of the Paleo-Inuits.
Origins and interactions of the Baikal populations
The region around Lake Baikal, particularly the Trans-Baikal, serves as another unique area to study post-LGM human population dynamics in northeast Asia since the region carries the earliest evidence of post-LGM human activity indicated by pre-Neolithic pottery (8). The appearance of pre-Neolithic ceramics ~13,000 to 11,000 years ago in this area was associated with possible migration from southeast Asia (35). Genetic studies revealed population transitions during the Holocene west of Lake Baikal (Cis-Baikal) (16), although the Trans-Baikal remained unexplored. Here, to fully understand the dynamics behind the post-LGM peopling of northeast Asia and to address the ancestry and interactions of Baikal populations, we analyzed genome sequence data from 8 Trans-Baikal individuals spanning c.8515 to 3000 cal BP and from 20 Cis-Baikal individuals spanning c.8980 to 560 cal BP (Fig. 1 and table S1).
In the PCA, an ~8515-year-old individual from the Trans-Baikal region, Dzyhlinda-1, appeared in close proximity to the Yakutia_Lena_6850_6190_BP. Dzyhlinda-1 exhibited genetic affinities toward the Saqqaq and Kolyma_M (Fig. 2A) and shared alleles with present-day Itelmen, Chukchi, and Inuits (fig. S17 and table S12). Dzyhlinda-1 shared more drift with Yakutia_Lena_6850_6190_BP compared to other ancient groups (Fig. 3A, figs. S18 and S19, and table S13), which might implicate that people who settled the Trans-Baikal area ~8515 years ago would be ancestral to those who settled the Lena region.
Fig. 3. Key f4-statistics summarizing the population transitions across the Lake Baikal area.

(A) f4(Yoruba, Trans-Baikal_8515_8380_BP; PopX, Yakutia_Lena_6850_6190_BP). (B) f4(Yoruba, Trans-Baikal_8345_3000_BP; PopX, Cis-Baikal_8980_8640_BP). (C) f4(Yoruba, Cis-Baikal_8980_8640_BP; PopX, Trans-Baikal_8345_3000_BP).
The other Trans-Baikal individuals formed a distinct genetic group, suggesting a genetic continuity without major demographic shifts in the region between ~8345 and ~3000 years ago (Fig. 2A). This group (Trans-Baikal_8345_3000_BP) was genetically close to the Neolithic individuals from the Devil’s Cave (5, 36) and exhibited genetic affinities toward present-day Central and East Asian populations (fig. S20 and table S14). In contrast to Dzyhlinda-1, individuals in this group carried comparably more northeast and southeast Asia–related and fewer Native America–related genetic components (Fig. 2B, fig. S4, and table S4) revealed by ADMIXTURE analysis and supported by f4-statistics (table S15). A ~1345-year-old individual from Amur Oblast had genetic affinities toward Trans-Baikal_8345_3000_BP (Fig. 2, fig. S21, and table S16). Thus, the results yield further support of genetic continuity over time and over vast geographical area reaching from Trans-Baikal to Far East, contrasting the pattern of population changes throughout Yakutia.
On the contrary, our data revealed multiple population transformations across the Cis-Baikal area spanning a time transect between ~8980 and ~560 years ago (Fig. 2): An ~8980-year-old Cis-Baikal individual, Popovskij-1, falls within the genetic variation of the Trans-Baikal_8345_3000_BP (Fig. 2). Popovskij-1 shared more drift with the Trans-Baikal_8345_3000_BP compared to the other ancient groups (Fig. 3, B and C, and table S17), suggesting the presence of a uniform gene pool across the area ~8900 years ago. A ~7900-year-old individual (Cyclodrome-1) marked the first genetic shift around the Cis-Baikal region, and this individual together with individuals spanning from ~7200 to ~6200 years ago (Cis-Baikal_7200_6200_BP) formed a distinct genetic group on the PCA (16). Yet, Cis-Baikal individuals spanning 6100 to 3700 years ago formed another group (Cis-Baikal_6100_3700_BP), carrying comparably high amount of west Eurasia–related and low amount of northeast Asia–related components (Fig. 2, fig. S4, and table S4). A Medieval Cis-Baikal individual (c.670 to 550 cal BP) was genetically distinct from the other individuals with increased genetic affinities toward East Asia populations (Fig. 2). Y haplogroup distribution was in agreement with the observed genetic groupings (table S18).
In a recent study (17) analyzing ancient human genomes from the Lake Baikal area, Cis-Baikal individuals spanning approximately 6700 to 3700 BP formed a genetic group that exhibits a genetic profile similar to our Cis-Baikal_6100_3700_BP group. However, two approximately 4300-year-old Cis-Baikal individuals (GLZ001 and GLZ002) formed another group, which we refer to as GLZ. These two individuals had genetic affinities toward Neolithic individuals from the Devil’s Cave (5, 36) similar to our Trans-Baikal_8345_3000_BP group that also has East Asian genetic affinities. To test mutual relations between these individuals and our Trans-Baikal_8345_3000_BP group, we performed f4-statistics in the form of f4(Yoruba, GLZ001; Cis-Baikal_6100_3700_BP, Trans-Baikal_8345_3000_BP) and f4(Yoruba, GLZ002; Cis-Baikal_6100_3700_BP, Trans-Baikal_8345_3000_BP). The tests revealed that these two individuals share more alleles with the Trans-Baikal_8345_3000_BP group. F4-statistics in the form of f4 (Yoruba, Devils_Cave_N; GLZ_individuals, Trans-Baikal_8345_3000_BP) further revealed that GLZ individuals and Trans-Baikal_8345_3000_BP were symmetrically related to the Devil’s Cave Neolithic individual (table S17). These results might implicate Trans-Baikal_8345_3000_BP as one of the source populations for these two individuals.
Our findings indicate a complex pattern of demographic change throughout the Baikal area consistent with (17), which revealed dynamic changes in population structure around Lake Baikal area. Before ~8500 years ago, people inhabited the Trans-Baikal region. They became ancestral to the human groups settled in Lena basin in Yakutia. About 8300 years ago, a major genetic shift happened in the Trans-Baikal area. This new group with East Asian genetic ancestry (Fig. 2B) populated the west and east side of Lake Baikal during the Mesolithic. Although they remained in the Trans-Baikal region until ~3000 years ago, an extensive gene flow from distant sources affected the Cis-Baikal region ~7900 years ago and then ~6100 years ago.
Genetic diversity of ancient northeast Asia populations
We further evaluated the degree of genetic diversity of the human groups in ancient northeast Asia. We restricted these analyses to individuals with coverages >4.5× including nine individuals (6.9×–14.5×) from Cis-Baikal_6100_3700_BP, one individual (4.7×) from Trans-Baikal_8345_3000_BP, and three individuals (5.5×–13.6×) from Yakutia_Lena_Kolyma_4780_2490_BP. For comparison, we included two west European individuals, i.e., Loschbour (Mesolithic) and Stuttgart (Neolithic) (37). Heterozygosity estimates revealed that genetic diversity in all groups ranged between the diversity of west European Mesolithic and Neolithic populations (Fig. 4A). Yakutia_Lena_Kolyma_4780_2490_BP had the lowest genetic diversity followed by the Trans-Baikal_8345_3000_BP (Fig. 4A). Cis-Baikal_6100_3700_BP had the highest estimates of heterozygosity with the exception of two individuals dating to ~4400 years ago. Consistently, Cis-Baikal_6100_3700_BP had low levels of short runs of homozygosity (RoH) implying a high ancestral population size compared to the geographically more isolated Trans-Baikal_8345_3000_BP and Yakutia_Lena_Kolyma_4780_2490_BP (Fig. 4B). Long RoH levels were high in Yakutia_Lena_Kolyma_4780_2490_BP and in three ~4400-year-old Cis-Baikal individuals pointing toward more limited mobility in recent times (fig. S22). A multiple sequentially Markovian coalescent (MSMC) analysis revealed that the effective population size of Yakutia_Lena_Kolyma_4780_2490_BP was low ~4700 years ago, consistent with the low genetic diversity observed in this group. Although effective population size estimates of human groups in Cis-Baikal ~6100 to 5600 years ago were comparably high, they decreased ~4400 years ago (Fig. 4C). Similarity between the observed low effective population sizes in Cis-Baikal and Yakutia 4700 to 4400 years ago could mirror global climatic changes, i.e., cooling of the Subboreal Period (38), and might indicate a possible population collapse ~4700 to 4000 years ago.
Fig. 4. Estimating the level of genetic diversity and population size changes in time.
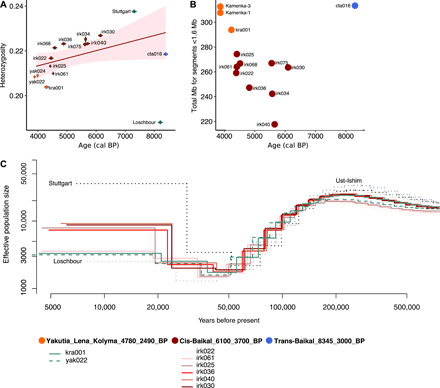
(A) Heterozygosity estimates. (B) Amount of short RoH in the genomes of Lake Baikal and Yakutia individuals. Mb, megabase. (C) Effective population size change in time estimated using MSMC.
Yersinia pestis in ancient northeast Asia
We examined the functional SNPs (table S19 and see Supplementary Text) and the possible presence of a plague-related bacterium that was involved in historical pandemics, Y. pestis, in the genomes of ancient northeast Asian individuals. The earliest evidence of Y. pestis was found in Late Neolithic and Bronze Age Eurasia ~5000 years ago (39). To assess the presence of this bacterium in northeast Asia and its possible effect on population dynamics throughout the region, we analyzed sequence data of all 40 individuals. We identified 9395 Y. pestis–specific sequencing reads (3.66% genome coverage for CO92) in a ~4400-year-old individual from Cis-Baikal, Anosovo-1, and 4176 Y. pestis–specific sequencing reads (1.65% genome coverage for CO92) in a ~3800-year-old individual from Yakutia, Kamenka-2. The Kamenka burial contained three young individuals, all related to each other including a parent-child kin (table S1).
Conclusions
Northeast Asia, particularly the Baikal adjacent area and the entire Russian Far East, presents a complex demographic picture with hitherto unknown genetic shifts since the post-LGM. The Trans-Baikal area displays few genetic turnovers with an extended period of genetic continuity over a period of c.6000 years. This unique demographical pattern throughout the Holocene stands in sharp contrast to the recurrent gene flow events of Cis-Baikal and Yakutia. We document that the human group that was represented by Khaiyrgas-1 must have dispersed to Yakutia after the LGM. This group was genetically distinct from the first inhabitants of the Siberia who settled the area before the LGM. The genetic legacy of this group is visible among human groups in the area ~6000 years later. Our data fit well with Belkachi groups as having key position in the ancestry of Paleo-Inuits who launched the second wave of gene flow into the Americas c.5000 years ago. We also document the presence of the most northeastern occurrence of ancient Y. pestis in the less populated Yakutia region and in the highly connected Cis-Baikal area. The bacterium may well have had consequences in shaping human population dynamics in both regions, visible in the reduction in the effective population size and the genetic diversity levels ~4400 years ago. Consistent with the finding of the same bacterium in the Lake Baikal region during the Bronze Age (17), this finding suggests that a plague pandemic in this part of northeast Asia could be a hypothesis worth exploring with more data. Our results demonstrate a complex demography in northeast Asia from the Late Upper Paleolithic up until the Medieval era in which Siberian populations expanded interacting with each other and with populations from distant geographical areas.
MATERIALS AND METHODS
Ancient DNA extraction and sequencing
DNA extractions from prehistoric bone and teeth and all further laboratory work including library preparation were performed at the ancient DNA laboratory facilities at the Archeological Research Laboratory at Stockholm University. We cleaned the samples from earth and potential contaminants using 1 to 3% NaOCl solution and ddH2O mechanical cleansing. We thereafter subjected the samples to ultraviolet radiation at about 6 J/cm2 at 254 nm to further decontaminate the surfaces. Bone powder or bone fragments were obtained using a Dremel drill. Weight of taken sample ranged from 50 to 400 mg. The samples were digested in 1 M urea, EDTA (0.5 M), and proteinase K (10 mg/ml) at temperatures between 37° and 55°C. A blank negative control was added for every 8 to 10 samples at this step. Upon dissolving, we concentrated the DNA extract using Amicon filters (Millipore) and purified it with silica-based MinElute spin columns (Qiagen) (40–42), eluting the DNA in 110 μl of elution buffer (Qiagen). Double-stranded blunt-end–repaired Illumina libraries were prepared from 20 μl of DNA. Each library was amplified in at least five replicates using 0.5 μl of 10 μM index primer per library. The thermocycling conditions used were as in (42), and the number of cycles was estimated individually for each library using quantitative polymerase chain reaction (qPCR). For some samples, damage-repaired double-stranded libraries (table S1) were prepared with USER enzyme (NEB/BioNordika) to remove deaminated cytosines (43). All libraries were sequenced using Illumina HiSeq X platform at the SciLife sequencing center at Stockholm.
Sequencing data processing
Paired-end sequencing reads were merged using MergeReadsFastQ_cc.py requiring a minimum 11–base pair (bp) overlap, and residual adapter sequences were trimmed (44). Merged reads were mapped to the human reference genome (version hs37d5) using BWA (version 0.7.13) aln algorithm (45) in single end mode (samse) with parameters “-n 0.01 -o 2 -l 16500” (37). PCR duplicates were filtered using a modified version of FilterUniqSAMCons_cc.py (44) to prevent the biased choice of the nucleotides. Reads mapped to the reference genome with more than 10% mismatches and shorter than 35 bp and with mapping quality below 30 were filtered. For each sample, all libraries including the damage-repair and non–damage-repair libraries were merged into a single bam file using SAMtools (46) merge module. These files were used for further variant discovery (C/T-type transitions excluded) to be used in population genetics analysis. We also merged only damage-repair libraries for the samples to be used in diversity analysis.
Evaluation of authenticity
We assessed the authenticity of all sequencing reads based on three different approaches including (i) evaluation of cytosine (C)-to thymine (T)-type transitions at the 5′ ends of all sequencing reads, (ii) mitochondrial DNA (mtDNA)–based estimation of contamination for all samples, and (iii) X chromosome–based estimation of contamination for male samples (table S1). All sequences showed increased frequency of C/T-type transitions at 5′ ends (up to 37% at the last five bases) (table S1). To estimate contamination levels based on mtDNA, we used two different methods: (i) According to the method developed in (47), proportion of contamination per sample ranged between 1.8 and 20.5% (table S1). In this method, private alleles that have a frequency of less than 5% in 311 modern reference mtDNAs with a minimum base quality of 30 and a minimum genomic depth of 10× were used. Point estimate of contamination was calculated on transversion-type variations by summing the consensus and alternative allele counts across all allele counts. (ii) In the second mtDNA-based method developed in (48) that provides Bayesian estimates of contamination, the probability of being authentic per sample was estimated to be between 79 and 99% (table S1). For this method, we first produced consensus mitochondrial sequences using ANGSD (49) with parameters “-doFasta 2 -doCounts 1-minQ 30 -minMapQ 30 -setMinDepth 3” and calculated the probabilities for being authentic per sequence using contamMix library in R. Last, according to X chromosome–based estimation of degree of contamination in male individuals, contamination estimates were between 0.0001 and 0.009 (table S1). This method that was implemented in ANGSD (49) is based on examination of heterozygous positions on the X chromosome. To run this method, we first created a binary count file with the command “angsd -r X:5000000-154900000 -doCounts 1 -setMinDepth 3 -setMaxDepth 100 -i Counts 1 -minMapQ 30 -minQ 30” and then calculated the contamination estimate on transversion-type SNPs by “contamination -d 3 -e 100.” Contamination estimates and two different CIs including sampling all reads from each site (method 1) and sampling one read randomly per site (method 2) are reported in table S1. We included samples in the downstream analysis if the sample did not fail at least two of the applied tests; we thus included all samples for further analysis.
Biological sex determination
Biological sex of the individuals was determined using the Ry method, which was developed in (50). This method relies on dividing the total number of reads that were mapped to the Y chromosome to the total number of reads that were mapped to the X and Y chromosome. Computation was performed on sequencing reads with a minimum read length of 35 bp, with a mapping quality minimum of 30, and that were mapped to the reference genome with a maximum of 10% mismatches. We used ry_compute.py script, which is available at https://github.com/pontussk/ry_compute/blob/master/ry_compute.py.
Biological relatedness estimation
We have used the program lcMLkin to detect related individuals (51). We chose only transversion-type SNPs (n = 1,065,109) from the Estonian Genome Diversity Project (EGDP) (52) to avoid bias due to postmortem damage in our samples. The genotype likelihoods of the selected SNP positions were called using “SNPbam2vcf.py” with a minimum frequency of 0.15 and using the population allele frequencies from the ancient individuals generated in this study. After a first run with all the ancient individuals, we used only the unrelated individuals as founders to estimate the allele frequencies of each pair. We inferred the most probable relationship between the ancient samples analyzed here comparing the obtained values of k0, k1, and k2 with the expected values of these parameters for the different degrees of relationship shown in (53) (see Supplementary Text).
Uniparental markers
To determine Y chromosome haplogroups, variants at haplogroup defining SNPs were identified on the basis of haplogroup definitions from PhyloTreeY (54) (http://phylotree.org/Y/tree/) and nomenclature from the International Society of Genetic Genealogy database (https://isogg.org) (v.11.349). The variants were called using SAMtools (v.1.8) mpileup with “–B” parameter (46), and positions with base and mapping quality of less than 30 were filtered. We also excluded insertions and deletions and sites that displayed multiple alleles from the analysis. Depending on the library preparation mode (damage-repaired or non–damage-repaired libraries), we used either transversions (non–damage-repaired libraries) or both transitions and transversions (damage-repaired libraries) in Y haplogroup assignment (table S18).
Mitochondrial haplogroups of 38 individuals were reported in (18) and those of 2 individuals (N2a-Matta-1 and cta016) were reported in this study (table S1). In brief, using sequence alignment files (BAM files) as input, consensus mitochondrial sequences were produced using ANGSD with parameters -doFasta 2 -doCounts 1-minQ 30 -minMapQ 30 -setMinDepth 3. Using HaploFind (55) and HaploGrep (56), mitochondrial haplogroups were determined. For the final assignment of haplogroups, visual inspection against PhyloTree mtDNA tree (build 17) was performed (table S1) (18) (see Supplementary Text).
SNP discovery and datasets
We prepared four different datasets for population genetics and diversity analysis including the following: (i) The Human Origins dataset consists of pseudohaploidized genotypes of ancient individuals sequenced in this study and pseudohaploidized genotypes of published ancient individuals (table S2) for a total of 594,924 SNPs genotyped in 2730 individuals from 203 present-day populations, which is available as curated Human Origins SNP Array dataset from (37, 57). This dataset was used in PCA, model-based clustering analysis, and outgroup f3-statistics. (ii) The 1000 Genomes dataset consists of pseudohaploidized genotypes of ancient individuals sequenced in this study as well as pseudohaploidized genotypes of published ancient individuals (table S2) for a total of 1,938,919 transversion-type SNPs ascertained with a minor allele frequency of 10% in the Yoruba population (n = 108 individuals) from phase 3 of the 1000 Genomes Project (58). This dataset was used in f4-statistics, qpAdm, and admixture graph-fitting analysis. (iii) The EGDP dataset consists of pseudohaploidized genotypes of ancient individuals sequenced in this study for 42,971,058 SNPs genotyped in 483 humans from 148 present-day populations, which is available from (52). This dataset was used for kinship analysis. (iv) The diploid dataset consists of diploid genotypes of 13 ancient individuals sequenced in this study with coverages above 4.5× including Kamenka-1, Kamenka-3, kra001, cta016, irk022, irk025, irk030, irk034, irk036, irk040, irk061, irk068, and irk075 (table S1), as well as 3 published individuals including Stuttgart (37), Loschbour (37), and Ust’ Ishim (25). This dataset was used in diversity analysis including estimation of heterozygosity, RoH, and also effective population size changes in time.
To prepare the diploid dataset, we used bam files that were prepared by merging damage-repaired libraries of Kamenka-1, Kamenka-3, kra001, cta016, irk022, irk025, irk030, irk034, irk036, irk040, irk061, irk068, and irk075, as well as bam files including Stuttgart (37), Loschbour (37), and Ust’ Ishim (25), which were processed with the same parameters. The last two bases of each read were trimmed using TrimBam of bamutils (59). Using Picard (http://broadinstitute.github.io/picard/), “read groups” were added to the files. Using indels of phase 1 of the 1000 Genomes Project (58) as reference, indel realignment was performed using GATK (60) IndelRealignment module. To call the diploid genotypes, the UnifiedGenotyper module of GATK was used with the parameters -stand_call_conf 50.0, -stand_emit_conf 50.0, -mbq 30, -contamination 0.02, and --output_mode EMIT_ALL_SITES. Using vcftools, SNP sites overlapping with 1,938,919 transversion-type SNPs were extracted. To prepare the other datasets, we used SAMtools (46) mpileup, producing pileup of each read for every genomic position of each ancient individual overlapping with 594,924 SNPs from the Human Origins SNP Array dataset (37, 57), overlapping with 42,971,058 SNPs (52), and overlapping with 1,938,919 transversion-type SNPs of African Yoruba individuals (58). Subsequently, a random read with a minimum mapping quality of 30 was selected and encoded as homozygous in the ancient individual. In the Human Origins dataset, C/T-type transitions were encoded as missing data. The 1000 Genomes dataset contained only transversion-type SNPs.
Principal components analysis
PCA was performed on the Human Origins dataset, which was prepared as described above. Principal components were calculated on present-day populations from Asia, Eurasia, Oceania, and America (table S2) using smartpca tool (v.1600) of the EIGENSOFT (61) with numoutlieriter: 0, shrinkmode: YES, and lsqproject: YES options. The result was plotted using the ploteig program of the EIGENSOFT (61).
Model-based clustering
Unsupervised clustering was performed to estimate the ancestry components of ancient and present-day individuals. This analysis was conducted using ADMIXTURE (21) on the Human Origins dataset, which was described above. This dataset was divided into two sub-datasets to encompass (i) modern individuals and (ii) ancient individuals using the “--keep” option in PLINK (v1.90) (62). As described in (63), to prevent influence of missingness in ancient samples and the difference in number of overlapping SNPs across samples on analysis, we first run ADMIXTURE on the modern dataset and inferred the cluster memberships of each ancient individual using the ancestral allele frequencies that were obtained from the modern dataset (63). Before ADMIXTURE analysis, the modern dataset consisting of a total of 2340 individuals from present-day Asia, Eurasia, America, Oceania, and Africa populations was filtered for linkage disequilibrium using the --indep-pairwise option in PLINK (v1.90) with parameters (--indep-pairwise 200 25 0.2) (62), yielding a total of 594,924 SNPs. The ancient dataset was filtered by selecting the same set of SNPs using the “--extract” option in PLINK (v1.90) (62). For every hypothetical cluster number (K) ranging between K = 2 and K = 16, ADMIXTURE was run 30 times with different random seeds on the modern dataset, and cluster memberships of each ancient individual for each K and each run were computed using the ancestral allele frequencies (63). After merging the ancestral clusters of modern and ancient individuals for each K and each run, common signals between different replicate runs were determined using the LargeKGreedy algorithm of CLUMPP (64). Results were visualized using rworldmap, ggplot2, SDMTools, and RColorBrewer packages of GNU R version 3.3.0. Since we run the ADMIXTURE on modern samples and projected the ancient samples on this, we did not perform cross-validation to decide which K explains the observed pattern better; instead, we examined each K and present the results for all K values (fig. S3, table S4, and see Supplementary Text).
qpAdm analysis
To estimate the admixture patterns in ancient Siberian genomes, qpAdm analysis was performed. Using the qpAdm program of ADMIXTOOLS (57) package (https://github.com/DReichLab/AdmixTools) with the “allsnps: YES” option on the Human Origins dataset prepared as described above, we estimated the ancestry proportions in the genomes of Saqqaq (30) and Kolyma_M (5). First, to estimate ancestry proportions in the genome of Saqqaq, the following set of six populations was used as outgroup and provided as “right populations” to the program: MbutiPygmy, Yoruba, Mota, Onge, Ju_Hoan_north, and Papuan. Providing the “left populations” list as Saqqaq (30), K14 (26), and Yakutia_Lena_6850_6190_BP, we modeled the Saqqaq as a two-way admixture of the K14 and Yakutia_Lena_6850_6190_BP (table S11). To model the Kolyma_M as a two-way admixture of Khaiyrgas-1 and Devils_Cave_N (5), another set of six populations including Yoruba, Mota (65), Onge, Ju_Hoan_north, Papuan, and Ust_Ishim was used as in the right populations list as outgroup. We considered the models with a P value of >0.05 (indicating a small deviation of data from the model expectation) as consistent with the tested model.
f3- and f4-statistics
Shared genetic drift between two populations (or individuals) since their divergence from an ancestral population was computed using the outgroup f3-statistics. The qp3pop program of ADMIXTOOLS (57) package was used with the “inbreed: YES” option on the Human Origins dataset described above to calculate f3-statistics for the tree-like topology in the form of f3(Outgroup, Test Population, Modern Population) in which the Yoruba population was used as Outgroup, each of the 40 ancient Siberians was used as Test Population, and 194 modern populations from the Human Origins dataset were used as Modern Population. Significant deviations from zero indicated a deviation from the proposed tree-like topology; thus, more positive values indicated an excess of shared genetic drift between the Test Population and Modern Population. The f4-statistics for the tree-like topology of the shape f4(Outgroup, Test; Group1, Group2) measures the shared genetic drift between Test Population and Group1 and Group2. Assuming no recent interactions between each of these four groups, tree topologies are balanced at zero, and deviation from zero implies a deviation from this proposed tree. Positive values indicate that Test population shares more alleles with Group1, and negative values indicate that Test population shares more alleles with Group2. Using the qpDstat program of ADMIXTOOLS (57) package with the “f4mode:YES” option, f4-statistics were calculated for a set of tree-like topologies.
Admixture graph fitting
A statistical frame implemented in TreeMix (22) was used to examine the genetic relationships between ancient individuals and Khaiyrgas-1. The method relies on building a maximum likelihood tree of the given populations using the covariance matrix of allele frequencies. This method was applied to Khaiyrgas-1 (0.3×), irk034 (14.5×), kra001 (13.6×), Tianyuan (28), Kolyma_M (5), Yana_UP (5), Anzick (29), MA1 (23), and AfontovaGora3 (24). Tree was rooted with the Yoruba population, and analysis was run on the 1000 Genomes dataset described above. To inactivate the correction for low sample size, the program was run with the “-noss” option. Using “-k 500 -se” parameters, SEs were estimated using blocks of 500 SNPs. The analysis was run for m = 0 zero-migration model. The analysis was run for 30 different random seeds, and the graph with the highest likelihood and most common topology was reported (fig. S5).
Heterozygosity estimation
We estimated the heterozygosity levels in the genomes of 13 ancient individuals with coverages above 4.5× (Fig. 4A and table S1) as well as in the genomes of Loschbour (6.2×, European Mesolithic) and Stuttgart (9.2×, European Neolithic) (37). Heterozygosity levels were computed using ANGSD (49) on the alignment files (BAM) of the individuals as “angsd -i ${bamfile} -GL 1 -doGlf 2 -doMajorMinor 1 -sites $sites -doSaf 1 -anc $ref. -minQ 30 -minmapq 30 -noTrans 1 -out ${filebase}” and “realSFS ${filebase}.saf.idx -bootstrap 100 -P 16 > ${filebase}_est_boot.ml.”
Runs of homozygosity
We computed the RoH in the genomes of 13 ancient Siberian individuals with coverages above 4.5× (Fig. 4B and table S1). For this analysis, the diploid dataset as described above was used. First, the dataset was converted to plink file format using vcftools (66) by extracting Yoruba-ascertained transversion SNPs. To compute the RoH levels, plink (62) was used as “plink --file ${filename} --homozyg --homozyg-density 50 --homozyg-gap 100 --homozyg-kb 500 --homozyg-snp 50 --homozyg-window-het 1 --homozyg-window-snp 50 --homozyg-window-threshold 0.05 --out ${filename}.”
Multiple sequentially Markovian coalescent
MSMC was used to infer the changes in effective population size through time. The analysis was restricted to the samples with coverages above 7.5× coverage. Input files were prepared using scripts provided with the release of MSMC (https://github.com/stschiff/msmc-tools), and MSMC was run with the parameters --fixedRecombination and -r 0.88. Effective population sizes were plotted on the basis of a mutation rate of 1.25 × 10−8 and a generation time of 30 years, and the curves were shifted on the basis of approximate ages of the samples.
Analysis for Y. pestis
We used 205 DNA libraries from 40 ancient individuals. Quality of the sequencing libraries was evaluated using fastqc (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were first mapped to a hybrid Yersinia genome (39), and those mapping to Y. pestis CO92 reference genome were extracted. To judge the taxonomical origin of Y. pestis sequences, edit distance distribution, length distribution, deamination patterns, and breadth of coverage information were used. Malt was used to create an alignment database from reference and representative genomes of bacteria, fungi, archaea, and viruses from the National Center for Biotechnology Information RefSeq database (67). DustMasker was used to mask repetitive regions in the reference genomes. Each DNA library was aligned to the reference genome database by using the malt, and reads that were aligned to the Y. pestis reference genome were extracted using the malt-extract tool (https://github.com/rhuebler/HOPS) (68) with the default options. Reads with mapping quality of <30 and <20 nucleotides were filtered. Picard (http://broadinstitute.github.io/picard/) with MarkDuplicates option was used to remove the PCR duplicates. Edit distances were extracted using the NM field from the SAM file and visualized in R (version 3.6.2, R Core Team, 2013). MapDamage (69) was used to calculate the deamination patterns and the deamination rate of Y. pestis and human DNA sequences. To examine the coverage, SAMtools (46) “depth -a” command was used (see Supplementary Text).
Investigating the functional SNPs
We inspected the allele distribution in SNP positions associated with a selection of biological traits using SAMtools mpileup (v1.3) (46). The traits that we investigated include the status of acetylator phenotype (NAT2), diet, and a set of phenotypic traits, i.e., risk of hypertension, cancers, and drug metabolism. We further evaluated the blood group variation among ancient individuals (see Supplementary Text).
Supplementary Material
Acknowledgments
We thank A. Munters for processing the data, V. Sobrado for helping with experiments, E. Altınışık for helpful discussions, S. Fedoseeva and A. Alekseev for providing archaeological material, and E.Ko. for providing archaeological material as well as helpful discussions about the prehistory of northeast Asia. Funding: This work has been funded by the Knut and Alice Wallenberg Foundation (to A.G.). Computations were performed at UPPMAX resources under the projects uppstore2018029 and SNIC-2018-8-43. Author contributions: A.G., J.S., and M.J. initiated the study. A.G., J.S., M.J., M.S., M.K., N.K., and G.M.K. designed the study. G.M.K. led the computational part of the study. M.K. and N.K. produced the data. D.S., G.I., D.Ki., K.P., D.V., P.M., A.K., A.T., E.I., E.Ko., and A.S. excavated and sampled the archaeological material and led the archaeological part of the study with J.S. and A.G. L.D. and E.Ki. led the microbiome analysis. G.M.K., D.Ko., N.B., H.M.D., R.R.-V., E.Ki., M.K., and T.G. analyzed the data. G.M.K., A.G., N.K., and J.S. wrote the manuscript with contributions from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Genome sequence data are available at the European Nucleotide Archive (ENA) with accession number PRJEB39378.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabc4587/DC1
REFERENCES AND NOTES
- 1.J. F. Richards, The Unending Frontier: An Environmental History of the Early Modern World (University of California Press, ed. 1, 2003). [Google Scholar]
- 2.Goebel T., Waters M. R., O’Rourke D. H., The late Pleistocene dispersal of modern humans in the Americas. Science 319, 1497–1502 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Fiedel S. J., Kuzmin Y. V., Radiocarbon date frequency as an index of intensity of Paleolithic occupation of Siberia: Did humans react predictably to climate oscillations? Radiocarbon. 49, 741–756 (2007). [Google Scholar]
- 4.Pitulko V. V., Nikolsky P. A., Girya E. Y., Basilyan A. E., Tumskoy V. E., Koulakov S. A., Astakhov S. N., Pavlova E. Y., Anisimov M. A., The Yana RHS site: Humans in the Arctic before the last glacial maximum. Science 303, 52–56 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Sikora M., Pitulko V. V., Sousa V. C., Allentoft M. E., Vinner L., Rasmussen S., Margaryan A., de Barros Damgaard P., de la Fuente C., Renaud G., Yang M. A., Fu Q., Dupanloup I., Giampoudakis K., Nogués-Bravo D., Rahbek C., Kroonen G., Peyrot M., McColl H., Vasilyev S. V., Veselovskaya E., Gerasimova M., Pavlova E. Y., Chasnyk V. G., Nikolskiy P. A., Gromov A. V., Khartanovich V. I., Moiseyev V., Grebenyuk P. S., Fedorchenko A. Y., Lebedintsev A. I., Slobodin S. B., Malyarchuk B. A., Martiniano R., Meldgaard M., Arppe L., Palo J. U., Sundell T., Mannermaa K., Putkonen M., Alexandersen V., Primeau C., Baimukhanov N., Malhi R. S., Sjögren K.-G., Kristiansen K., Wessman A., Sajantila A., Lahr M. M., Durbin R., Nielsen R., Meltzer D. J., Excoffier L., Willerslev E., The population history of northeastern Siberia since the Pleistocene. Nature 570, 182–188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henry A., Bezrukova E. V., Teten’kin A. V., Kuz’min M. I., New data on vegetation and climate reconstruction in the Baikal-Patom Highland (Eastern Siberia) in the last glacial maximum and early holocene. Dokl. Earth Sci. 478, 241–244 (2018). [Google Scholar]
- 7.G. Medvedev, in The Paleolithic of Siberia: New Discoveries and Interpretations, A. P. Derevianko, D. B. Shimkin, W. R. Powers, Eds. (Institute of Archaeology and Ethnography, Siberian Division, Russian Academy of Sciences/University of Illinois Press, 1998), pp. 122–132. [Google Scholar]
- 8.A. D. Stepanov, A. S. Kirillin, S. A. Vorob’ev, E. N. Solov’eva, N. N. Efimov, in Ancient cultures of North-Eastern Asia. Astroarchaeology. Paleoinformatics (Nauka, 2003), pp. 98–113. [Google Scholar]
- 9.Derevianko A. P., Kuzmin Y. V., Burr G. S., Jull A. J. T., Kim J. C., AMS 14C age of the earliest pottery from the Russian Far East: 1996–2002 results. Nucl. Instrum. Methods Phys. Res., Sect. B 223–224, 735–739 (2004). [Google Scholar]
- 10.Kuzmin Y. V., Orlova L. A., The neolithization of Siberia and the Russian Far East: Radiocarbon evidence. Antiquity 74, 356–364 (2000). [Google Scholar]
- 11.Y. A. Mochanov, S. A. Fedoseeva, in American Beginnings. The Prehistory and Palaeoecology of Beringia, F. H. West, Ed. (The University of Chicago Press, 1996), pp. 215–218. [Google Scholar]
- 12.Alekseyev A. N., Dyakonov V. M., Radiocarbon chronology of neolithic and bronze age cultures in Yakutia. Archaeol. Ethnol. Anthropol. Eurasia 37, 26–40 (2009). [Google Scholar]
- 13.Powers W. R., Jordan R. H., Human biogeography and climate change in Siberia and Arctic North America in the fourth and fifth millennia BP. Philos. Trans. Royal Soc. A 330, 665–670 (1990). [Google Scholar]
- 14.Wong E. H. M., Khrunin A., Nichols L., Pushkarev D., Khokhrin D., Verbenko D., Evgrafov O., Knowles J., Novembre J., Limborska S., Valouev A., Reconstructing genetic history of Siberian and Northeastern European populations. Genome Res. 27, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pugach I., Matveev R., Spitsyn V., Makarov S., Novgorodov I., Osakovsky V., Stoneking M., Pakendorf B., The complex admixture history and recent southern origins of Siberian populations. Mol. Biol. Evol. 33, 1777–1795 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Barros Damgaard P., Martiniano R., Kamm J., Moreno-Mayar J. V., Kroonen G., Peyrot M., Barjamovic G., Rasmussen S., Zacho C., Baimukhanov N., Zaibert V., Merz V., Biddanda A., Merz I., Loman V., Evdokimov V., Usmanova E., Hemphill B., Seguin-Orlando A., Yediay F. E., Ullah I., Sjögren K.-G., Iversen K. H., Choin J., de la Fuente C., Ilardo M., Schroeder H., Moiseyev V., Gromov A., Polyakov A., Omura S., Senyurt S. Y., Ahmad H., McKenzie C., Margaryan A., Hameed A., Samad A., Gul N., Khokhar M. H., Goriunova O. I., Bazaliiskii V. I., Novembre J., Weber A. W., Orlando L., Allentoft M. E., Nielsen R., Kristiansen K., Sikora M., Outram A. K., Durbin R., Willerslev E., The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science 360, eaar7711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu H., Spyrou M. A., Karapetian M., Shnaider S., Radzevičiūtė R., Nägele K., Neumann G. U., Penske S., Zech J., Lucas M., LeRoux P., Roberts P., Pavlenok G., Buzhilova A., Posth C., Jeong C., Krause J., Paleolithic to Bronze Age Siberians reveal connections with first Americans and across Eurasia. Cell 181, 1232–1245.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Kılınç G. M., Kashuba N., Yaka R., Sümer A. P., Yüncü E., Shergin D., Ivanov G. L., Kichigin D., Pestereva K., Volkov D., Mandryka P., Kharinskii A., Tishkin A., Ineshin E., Kovychev E., Stepanov A., Alekseev A., Fedoseeva S. A., Somel M., Jakobsson M., Krzewińska M., Storå J., Götherström A., Investigating Holocene human population history in North Asia using ancient mitogenomes. Sci. Rep. 8, 8969 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitulko V. V., Nikolskiy P. A., The extinction of the woolly mammoth and the archaeological record in Northeastern Asia. World Archaeol. 44, 21–42 (2012). [Google Scholar]
- 20.Kuzmin Y. V., Kosintsev P. A., Stepanov A. D., Boeskorov G. G., Cruz R. J., Chronology and faunal remains of the Khayrgas cave (Eastern Siberia, Russia). Radiocarbon 59, 575–582 (2017). [Google Scholar]
- 21.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickrell J. K., Pritchard J. K., Inference of population splits and mixtures from genome-wide allele frequency data. PLOS Genet. 8, e1002967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raghavan M., Skoglund P., Graf K. E., Metspalu M., Albrechtsen A., Moltke I., Rasmussen S., Stafford T. W. Jr., Orlando L., Metspalu E., Karmin M., Tambets K., Rootsi S., Mägi R., Campos P. F., Balanovska E., Balanovsky O., Khusnutdinova E., Litvinov S., Osipova L. P., Fedorova S. A., Voevoda M. I., DeGiorgio M., Sicheritz-Ponten T., Brunak S., Demeshchenko S., Kivisild T., Villems R., Nielsen R., Jakobsson M., Willerslev E., Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505, 87–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Q., Posth C., Hajdinjak M., Petr M., Mallick S., Fernandes D., Furtwängler A., Haak W., Meyer M., Mittnik A., Nickel B., Peltzer A., Rohland N., Slon V., Talamo S., Lazaridis I., Lipson M., Mathieson I., Schiffels S., Skoglund P., Derevianko A. P., Drozdov N., Slavinsky V., Tsybankov A., Cremonesi R. G., Mallegni F., Gély B., Vacca E., González Morales M. R., Straus L. G., Neugebauer-Maresch C., Teschler-Nicola M., Constantin S., Moldovan O. T., Benazzi S., Peresani M., Coppola D., Lari M., Ricci S., Ronchitelli A., Valentin F., Thevenet C., Wehrberger K., Grigorescu D., Rougier H., Crevecoeur I., Flas D., Semal P., Mannino M. A., Cupillard C., Bocherens H., Conard N. J., Harvati K., Moiseyev V., Drucker D. G., Svoboda J., Richards M. P., Caramelli D., Pinhasi R., Kelso J., Patterson N., Krause J., Pääbo S., Reich D., The genetic history of Ice Age Europe. Nature 534, 200–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Q., Li H., Moorjani P., Jay F., Slepchenko S. M., Bondarev A. A., Johnson P. L. F., Aximu-Petri A., Prüfer K., de Filippo C., Meyer M., Zwyns N., Salazar-García D. C., Kuzmin Y. V., Keates S. G., Kosintsev P. A., Razhev D. I., Richards M. P., Peristov N. V., Lachmann M., Douka K., Higham T. F. G., Slatkin M., Hublin J.-J., Reich D., Kelso J., Viola T. B., Pääbo S., Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seguin-Orlando A., Korneliussen T. S., Sikora M., Malaspinas A.-S., Manica A., Moltke I., Albrechtsen A., Ko A., Margaryan A., Moiseyev V., Goebel T., Westaway M., Lambert D., Khartanovich V., Wall J. D., Nigst P. R., Foley R. A., Lahr M. M., Nielsen R., Orlando L., Willerslev E., Genomic structure in Europeans dating back at least 36,200 years. Science 346, 1113–1118 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Sikora M., Seguin-Orlando A., Sousa V. C., Albrechtsen A., Korneliussen T., Ko A., Rasmussen S., Dupanloup I., Nigst P. R., Bosch M. D., Renaud G., Allentoft M. E., Margaryan A., Vasilyev S. V., Veselovskaya E. V., Borutskaya S. B., Deviese T., Comeskey D., Higham T., Manica A., Foley R., Meltzer D. J., Nielsen R., Excoffier L., Lahr M. M., Orlando L., Willerslev E., Ancient genomes show social and reproductive behavior of early Upper Paleolithic foragers. Science 358, 659–662 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Fu Q., Meyer M., Gao X., Stenzel U., Burbano H. A., Kelso J., Pääbo S., DNA analysis of an early modern human from Tianyuan cave, China. Proc. Natl. Acad. Sci. U.S.A. 110, 2223–2227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasmussen M., Anzick S. L., Waters M. R., Skoglund P., DeGiorgio M., Stafford T. W. Jr., Rasmussen S., Moltke I., Albrechtsen A., Doyle S. M., Poznik G. D., Gudmundsdottir V., Yadav R., Malaspinas A.-S., White S. S. V, Allentoft M. E., Cornejo O. E., Tambets K., Eriksson A., Heintzman P. D., Karmin M., Korneliussen T. S., Meltzer D. J., Pierre T. L., Stenderup J., Saag L., Warmuth V. M., Lopes M. C., Malhi R. S., Brunak S., Sicheritz-Ponten T., Barnes I., Collins M., Orlando L., Balloux F., Manica A., Gupta R., Metspalu M., Bustamante C. D., Jakobsson M., Nielsen R., Willerslev E., The genome of a late Pleistocene human from a Clovis burial site in western Montana. Nature 506, 225–229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen M., Li Y., Lindgreen S., Pedersen J. S., Albrechtsen A., Moltke I., Metspalu M., Metspalu E., Kivisild T., Gupta R., Bertalan M., Nielsen K., Gilbert M. T. P., Wang Y., Raghavan M., Campos P. F., Kamp H. M., Wilson A. S., Gledhill A., Tridico S., Bunce M., Lorenzen E. D., Binladen J., Guo X., Zhao J., Zhang X., Zhang H., Li Z., Chen M., Orlando L., Kristiansen K., Bak M., Tommerup N., Bendixen C., Pierre T. L., Grønnow B., Meldgaard M., Andreasen C., Fedorova S. A., Osipova L. P., Higham T. F. G., Ramsey C. B., Hansen T. V. O., Nielsen F. C., Crawford M. H., Brunak S., Sicheritz-Pontén T., Villems R., Nielsen R., Krogh A., Wang J., Willerslev E., Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463, 757–762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuzmin Y. V., Orlova L. A., Radiocarbon chronology of the Siberian Paleolithic. J. World Prehist. 12, 1–53 (1998). [Google Scholar]
- 32.Friesen T. M., On the naming of Arctic Archaeological Traditions: The case for Paleo-Inuit. ARCTIC 68, iii–iv (2015). [Google Scholar]
- 33.Flegontov P., Altınışık N. E., Changmai P., Rohland N., Mallick S., Adamski N., Bolnick D. A., Broomandkhoshbacht N., Candilio F., Culleton B. J., Flegontova O., Friesen T. M., Jeong C., Harper T. K., Keating D., Kennett D. J., Kim A. M., Lamnidis T. C., Lawson A. M., Olalde I., Oppenheimer J., Potter B. A., Raff J., Sattler R. A., Skoglund P., Stewardson K., Vajda E. J., Vasilyev S., Veselovskaya E., Hayes M. G., O’Rourke D. H., Krause J., Pinhasi R., Reich D., Schiffels S., Palaeo-Eskimo genetic ancestry and the peopling of Chukotka and North America. Nature 570, 236–240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haak W., Lazaridis I., Patterson N., Rohland N., Mallick S., Llamas B., Brandt G., Nordenfelt S., Harney E., Stewardson K., Fu Q., Mittnik A., Bánffy E., Economou C., Francken M., Friederich S., Pena R. G., Hallgren F., Khartanovich V., Khokhlov A., Kunst M., Kuznetsov P., Meller H., Mochalov O., Moiseyev V., Nicklisch N., Pichler S. L., Risch R., Guerra M. A. R., Roth C., Szécsényi-Nagy A., Wahl J., Meyer M., Krause J., Brown D., Anthony D., Cooper A., Alt K. W., Reich D., Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vetrov V. M., Ineshin E. M., The most ancient ceramics of Baikal Siberia in the context of the traditions of ceramics of east Asia. Vestnik Sankt-Peterburgskogo Universiteta, Istoriya 64, 453–473 (2019). [Google Scholar]
- 36.Siska V., Jones E. R., Jeon S., Bhak Y., Kim H.-M., Cho Y. S., Kim H., Lee K., Veselovskaya E., Balueva T., Gallego-Llorente M., Hofreiter M., Bradley D. G., Eriksson A., Pinhasi R., Bhak J., Manica A., Genome-wide data from two early Neolithic East Asian individuals dating to 7700 years ago. Sci. Adv. 3, e1601877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazaridis I., Patterson N., Mittnik A., Renaud G., Mallick S., Kirsanow K., Sudmant P. H., Schraiber J. G., Castellano S., Lipson M., Berger B., Economou C., Bollongino R., Fu Q., Bos K. I., Nordenfelt S., Li H., de Filippo C., Prüfer K., Sawyer S., Posth C., Haak W., Hallgren F., Fornander E., Rohland N., Delsate D., Francken M., Guinet J.-M., Wahl J., Ayodo G., Babiker H. A., Bailliet G., Balanovska E., Balanovsky O., Barrantes R., Bedoya G., Ben-Ami H., Bene J., Berrada F., Bravi C. M., Brisighelli F., Busby G. B. J., Cali F., Churnosov M., Cole D. E. C., Corach D., Damba L., van Driem G., Dryomov S., Dugoujon J.-M., Fedorova S. A., Romero I. G., Gubina M., Hammer M., Henn B. M., Hervig T., Hodoglugil U., Jha A. R., Karachanak-Yankova S., Khusainova R., Khusnutdinova E., Kittles R., Kivisild T., Klitz W., Kučinskas V., Kushniarevich A., Laredj L., Litvinov S., Loukidis T., Mahley R. W., Melegh B., Metspalu E., Molina J., Mountain J., Näkkäläjärvi K., Nesheva D., Nyambo T., Osipova L., Parik J., Platonov F., Posukh O., Romano V., Rothhammer F., Rudan I., Ruizbakiev R., Sahakyan H., Sajantila A., Salas A., Starikovskaya E. B., Tarekegn A., Toncheva D., Turdikulova S., Uktveryte I., Utevska O., Vasquez R., Villena M., Voevoda M., Winkler C. A., Yepiskoposyan L., Zalloua P., Zemunik T., Cooper A., Capelli C., Thomas M. G., Ruiz-Linares A., Tishkoff S. A., Singh L., Thangaraj K., Villems R., Comas D., Sukernik R., Metspalu M., Meyer M., Eichler E. E., Burger J., Slatkin M., Pääbo S., Kelso J., Reich D., Krause J., Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.P. Ya. Groisman, T. A. Blyakharchuk, A. V. Chernokulsky, M. M. Arzhanov, L. B. Marchesini, E. G. Bogdanova, I. I. Borzenkova, O. N. Bulygina, A. A. Karpenko, L. V. Karpenko, R. W. Knight, V. Ch. Khon, G. N. Korovin, A. V. Meshcherskaya, I. I. Mokhov, E. I. Parfenova, V. N. Razuvaev, N. A. Speranskaya, N. M. Tchebakova, N. N. Vygodskaya, in Regional Environmental Changes in Siberia and Their Global Consequences, P. Ya. Groisman, G. Gutman, Eds. (Springer Netherlands, 2013), pp. 57–109. [Google Scholar]
- 39.Rasmussen S., Allentoft M. E., Nielsen K., Orlando L., Sikora M., Sjögren K.-G., Pedersen A. G., Schubert M., Van Dam A., Kapel C. M. O., Nielsen H. B., Brunak S., Avetisyan P., Epimakhov A., Khalyapin M. V., Gnuni A., Kriiska A., Lasak I., Metspalu M., Moiseyev V., Gromov A., Pokutta D., Saag L., Varul L., Yepiskoposyan L., Sicheritz-Pontén T., Foley R. A., Lahr M. M., Nielsen R., Kristiansen K., Willerslev E., Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago. Cell 163, 571–582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang D. Y., Eng B., Waye J. S., Dudar J. C., Saunders S. R., Improved DNA extraction from ancient bones using silica-based spin columns. Am. J. Phys. Anthropol. 105, 539–543 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Malmström H., Svensson E. M., Gilbert M. T. P., Willerslev E., Götherström A., Holmlund G., More on contamination: The use of asymmetric molecular behavior to identify authentic ancient human DNA. Mol. Biol. Evol. 24, 998–1004 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Günther T., Valdiosera C., Malmström H., Ureña I., Rodriguez-Varela R., Sverrisdóttir Ó. O., Daskalaki E. A., Skoglund P., Naidoo T., Svensson E. M., Bermúdez de Castro J. M., Carbonell E., Dunn M., Storå J., Iriarte E., Arsuaga J. L., Carretero J.-M., Götherström A., Jakobsson M., Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc. Natl. Acad. Sci. U.S.A. 112, 11917–11922 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.A. W. Briggs, P. Heyn, Preparation of Next-Generation Sequencing Libraries from Damaged DNA, in Ancient DNA: Methods and Protocols, B. Shapiro, M. Hofreiter, Eds. (Humana Press, 2012), pp. 143–154. [DOI] [PubMed] [Google Scholar]
- 44.M. Kircher, in Methods in Molecular Biology (Clifton, N.J.) (Humana Press Inc., 2012), vol. 840, pp. 197–228. [DOI] [PubMed] [Google Scholar]
- 45.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Green R. E., Malaspinas A.-S., Krause J., Briggs A. W., Johnson P. L. F., Uhler C., Meyer M., Good J. M., Maricic T., Stenzel U., Prüfer K., Siebauer M., Burbano H. A., Ronan M., Rothberg J. M., Egholm M., Rudan P., Brajković D., Kućan Ž., Gušić I., Wikström M., Laakkonen L., Kelso J., Slatkin M., Pääbo S., A complete neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134, 416–426 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu Q., Mittnik A., Johnson P. L. F., Bos K., Lari M., Bollongino R., Sun C., Giemsch L., Schmitz R., Burger J., Ronchitelli A. M., Martini F., Cremonesi R. G., Svoboda J., Bauer P., Caramelli D., Castellano S., Reich D., Pääbo S., Krause J., A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 23, 553–559 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics. 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skoglund P., Storå J., Götherström A., Jakobsson M., Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 40, 4477–4482 (2013). [Google Scholar]
- 51.Lipatov M., Sanjeev K., Patro R., Veeramah K. R., Maximum likelihood estimation of biological relatedness from low coverage sequencing data. bioRxiv , 023374 (2015). [Google Scholar]
- 52.Pagani L., Lawson D. J., Jagoda E., Mörseburg A., Eriksson A., Mitt M., Clemente F., Hudjashov G., DeGiorgio M., Saag L., Wall J. D., Cardona A., Mägi R., Sayres M. A. W., Kaewert S., Inchley C., Scheib C. L., Järve M., Karmin M., Jacobs G. S., Antao T., Iliescu F. M., Kushniarevich A., Ayub Q., Tyler-Smith C., Xue Y., Yunusbayev B., Tambets K., Mallick C. B., Saag L., Pocheshkhova E., Andriadze G., Muller C., Westaway M. C., Lambert D. M., Zoraqi G., Turdikulova S., Dalimova D., Sabitov Z., Sultana G. N. N., Lachance J., Tishkoff S., Momynaliev K., Isakova J., Damba L. D., Gubina M., Nymadawa P., Evseeva I., Atramentova L., Utevska O., Ricaut F.-X., Brucato N., Sudoyo H., Letellier T., Cox M. P., Barashkov N. A., Skaro V., Mulahasanovic L., Primorac D., Sahakyan H., Mormina M., Eichstaedt C. A., Lichman D. V., Abdullah S., Chaubey G., Wee J. T. S., Mihailov E., Karunas A., Litvinov S., Khusainova R., Ekomasova N., Akhmetova V., Khidiyatova I., Marjanović D., Yepiskoposyan L., Behar D. M., Balanovska E., Metspalu A., Derenko M., Malyarchuk B., Voevoda M., Fedorova S. A., Osipova L. P., Lahr M. M., Gerbault P., Leavesley M., Migliano A. B., Petraglia M., Balanovsky O., Khusnutdinova E. K., Metspalu E., Thomas M. G., Manica A., Nielsen R., Villems R., Willerslev E., Kivisild T., Metspalu M., Genomic analyses inform on migration events during the peopling of Eurasia. Nature 538, 238–242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weir B. S., Anderson A. D., Hepler A. B., Genetic relatedness analysis: Modern data and new challenges. Nat. Rev. Genet. 7, 771–780 (2006). [DOI] [PubMed] [Google Scholar]
- 54.van Oven M., PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic. Sci. Int. Genet. Suppl. Ser. 5, e392–e394 (2015). [Google Scholar]
- 55.Vianello D., Sevini F., Castellani G., Lomartire L., Capri M., Franceschi C., HAPLOFIND: A new method for high-throughput mtDNA haplogroup assignment. Hum. Mutat. 34, 1189–1194 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Kloss-Brandstätter A., Pacher D., Schönherr S., Weissensteiner H., Binna R., Specht G., Kronenberg F., HaploGrep: A fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum. Mutat. 32, 25–32 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D., Ancient admixture in human history. Genetics 192, 1065–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.1000 Genomes Project Consortium, Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., Abecasis G. R., A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jun G., Wing M. K., Abecasis G. R., Kang H. M., An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res. 25, 918–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., Philippakis A. A., del Angel G., Rivas M. A., Hanna M., McKenna A., Fennell T. J., Kernytsky A. M., Sivachenko A. Y., Cibulskis K., Gabriel S. B., Altshuler D., Daly M. J., A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patterson N., Price A. L., Reich D., Population structure and eigenanalysis. PLOS Genet. 2, e190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sikora M., Carpenter M. L., Moreno-Estrada A., Henn B. M., Underhill P. A., Sánchez-Quinto F., Zara I., Pitzalis M., Sidore C., Busonero F., Maschio A., Angius A., Jones C., Mendoza-Revilla J., Nekhrizov G., Dimitrova D., Theodossiev N., Harkins T. T., Keller A., Maixner F., Zink A., Abecasis G., Sanna S., Cucca F., Bustamante C. D., Population genomic analysis of ancient and modern genomes yields new insights into the genetic ancestry of the Tyrolean Iceman and the genetic structure of Europe. PLOS Genet. 10, e1004353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jakobsson M., Rosenberg N. A., CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Llorente M. G., Jones E. R., Eriksson A., Siska V., Arthur K. W., Arthur J. W., Curtis M. C., Stock J. T., Coltorti M., Pieruccini P., Stretton S., Brock F., Higham T., Park Y., Hofreiter M., Bradley D. G., Bhak J., Pinhasi R., Manica A., Ancient Ethiopian genome reveals extensive Eurasian admixture in Eastern Africa. Science 350, 820–822 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., Chetvernin V., Choi J., Cox E., Ermolaeva O., Farrell C. M., Goldfarb T., Gupta T., Haft D., Hatcher E., Hlavina W., Joardar V. S., Kodali V. K., Li W., Maglott D., Masterson P., McGarvey K. M., Murphy M. R., O’Neill K., Pujar S., Rangwala S. H., Rausch D., Riddick L. D., Schoch C., Shkeda A., Storz S. S., Sun H., Thibaud-Nissen F., Tolstoy I., Tully R. E., Vatsan A. R., Wallin C., Webb D., Wu W., Landrum M. J., Kimchi A., Tatusova T., DiCuccio M., Kitts P., Murphy T. D., Pruitt K. D., Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hübler R., Key F. M., Warinner C., Bos K. I., Krause J., Herbig A., HOPS: Automated detection and authentication of pathogen DNA in archaeological remains. Genome Biol. 20, 280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jónsson H., Ginolhac A., Schubert M., Johnson P. L. F., Orlando L., mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mats V. D., Perepelova T. I., A new perspective on evolution of the Baikal Rift. Geosci. Front. 2, 349–365 (2011). [Google Scholar]
- 71.R. S. Losey, M. A. Katzenberg, T. Nomokonova, Middle holocene fishing and hunting in the Baikal Region of Siberia, in The Oxford Handbook of the Archaeology of Diet, J. Lee-Thorp, M. A. Katzenberg, Eds. (Oxford Univ. Press, 2015). [Google Scholar]
- 72.Berdnikova N., Archaeological «Breaks»: Interpretive possibilities (Baikalian Siberia). Archaeol. Ethnol. Anthropol. 1, 178–202 (2012). [Google Scholar]
- 73.M. V. Konstantinov, in Archaeological Quest (North Asia), V. E. Medvedev, Ed. (Nauka, 1980), pp. 16–24. [Google Scholar]
- 74.Bazaliiskii V. I., Burial complexes of the epoch of late mesolithic-neolithic of Baikal Siberia: Burial rites, absolute age. Reports of the Laboratory of Ancient Technologies. Irkutsk National Research Technical University 9, 43–101 (2012). [Google Scholar]
- 75.Konstantinov M. V., Konstantinov A. V., Historical heritage of archaeological discoveries and epochs. Small Enciclopedia of Transbaikalia: Archaoeology. Part 1, 12–40 (2011). [Google Scholar]
- 76.Losey R. J., Waters-Rist A. L., Nomokonova T., Kharinskii A. A., A second mortuary hiatus on Lake Baikal in Siberia and arrival of small-scale pastoralism. Sci. Rep. 7, 2319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mochanov Y. A., Bland R. L., The most ancient paleolithic of the diring and the problem of a nontropical origin for humanity. Arctic Anthropol. 30, 22–53 (1993). [Google Scholar]
- 78.Turkin G. V., Kharinskii A. V., The Shamanka II cemetery: To the question of cultural affinity of the Neolithic-Bronze Age burial complexes in southern Baikal. The Bulletin of Irkutsk State University, 124–158 (2004). [Google Scholar]
- 79.Weber A. W., White D., Bazaliiskii V. I., Goriunova O. I., Savel’ev N. A., Katzenberg M. A., Hunter–gatherer foraging ranges, migrations, and travel in the middle Holocene Baikal region of Siberia: Insights from carbon and nitrogen stable isotope signatures. J. Anthropol. Archaeol. 30, 523–548 (2011). [Google Scholar]
- 80.Weber A. W., Link D. W., Katzenberg M. A., Hunter-gatherer culture change and continuity in the Middle Holocene of the Cis-Baikal, Siberia. J. Anthropol. Archaeol. 21, 230–299 (2002). [Google Scholar]
- 81.A. P. Okladnikov, Neolithic and Bronze Ages of Cis-Baikal. Part I & II (Academy of Sciences USSR, 1950).
- 82.A. P. Okladnikov, in Ancient Population of Siberia and Its Cultures, M. Levin, L. Potapov, Eds. (University of Chicago Press, 1964), pp. 13–98. [Google Scholar]
- 83.V. I. Bazaliiskii, E. V. Menshagin, V. V. Golbert, V. N. Prokopijev, in Problems of Archaeology and Ethnography of Siberia (Irkutsk State University, 1982), pp. 68–69. [Google Scholar]
- 84.P. P. Khoroshikh, in Siberian Archaeological Miscellany, A. P. Okladnikov, Ed. (Nauka, 1966), pp. 84–93. [Google Scholar]
- 85.Ovchinnikov M. P., Materials for the study of the ancient objects in the surroundings of Irkutsk. Proceedings of the Imperial Russian Geographical Society XXXV, 62–76 (1904). [Google Scholar]
- 86.Aksenov M. P., Survey at Lena river. Archaeological Discoveries of 1972. Nauka, 196–197 (1973). [Google Scholar]
- 87.D. L. Shergin, in Ancient cultures of Mongolia and Baikal Siberia: Material from International Scientific Conference (Buryat State University, 2010), pp. 56–60. [Google Scholar]
- 88.V. M. Vetrov, N. E. Berdnikova, A. I. Zhuravljov, A. V. Frolov, in Paleoethnology of Siberia (Irkutsk, 1990), pp. 137–139. [Google Scholar]
- 89.D. E. Kichigin, Y. A. Emel’janova, A. M. Korostelev, in Ancient Cultures of Mongolia, Baikal Siberia and Northern China (2017).
- 90.A. G. Novikov, O. I. Gorjunova, in Ancient Cultures of Mongolia and Baikal Siberia. Part 2 (Tuvan State University, 2014), pp. 81–85. [Google Scholar]
- 91.N. S. Sosnovkaja, A. V. Kharniskii, in Dulovskij Readings 1997, Section of Archaeology and Ethnography: Materials of Seminar and Announcement (Irkutsk, 1997), pp. 70–73. [Google Scholar]
- 92.A. P. Okladnikov, I. I. Kirillov, South-East Transbaikalia in the Stone Age and Early Bronze Age (Nauka, 1980). [Google Scholar]
- 93.Bazaliiskii V. I., Savelyev V. A., The Wolf of Baikal: The “Lokomotiv” early Neolithic cemetery in Siberia (Russia). Antiquity. 77, 20–30 (2003). [Google Scholar]
- 94.A. V. Kharinskii, E. V. Kovychev, N. N. Kradin, S. G. Bocharov, A. G. Sitdikov, in Dialogue of Urban and Steppe Cultures in the Eurasian Range. Historical Geography of Golden Horde: Materials of the Seventh International Conference, Dedicated to the Memory of Fjodorov-Davydov G.A. (Stratum Plus, 2016), pp. 65–68. [Google Scholar]
- 95.Dyatchina N. G., The description of bone and horn articles found at Priargun Sites of Late Neolith-Bronze Age. Gumanitarnyj vektor 30, 47–59 (2012). [Google Scholar]
- 96.Y. A. Mochanov, S. A. Fedoseeva, Sketches of Prehistory of Yakutia. Stone Age: In 2 Parts. Part 2—Yakutsk (2013), pp. 489–489. [Google Scholar]
- 97.Alekseyev A. N., Zhirkov E. K., Stepanov A. D., Sharaborin A. K., Alekseeva L. L., Burial of an Ymyiakhtakh Warrior in Kyordyughen, Yakutia. Archaeol. Ethnol. Anthropol. Eurasia 26, 45–52 (2006). [Google Scholar]
- 98.Stepanov A. D., Kuzmin Y. V., Hodgins G. W. L., Jall A. J. T., Kyordyugen site, Ymyiakhtakh culture, Yakutia: An interpretation of burial rite. Archaeol. Ethnol. Anthropol. Eurasia N40, 51–61 (2012). [Google Scholar]
- 99.Chikisheva T. A., Zubova A. V., Rahimova N. N., Volkov P. V., Pozdnyakov D. V., Anthropological research of the Neolithic burial on the monument of Pomazkino III (Middle Kolyma river). Theory and Practice of Archaeological Investigations 20, 112–137 (2017). [Google Scholar]
- 100.Kashin V. A., Neolithic burial of children at Middle Kolyma river. Archaeol. Ethnol. Anthropol. Eurasia 2, 78–81 (2001). [Google Scholar]
- 101.Shpakova E. G., Anthropological characteristics of the burial of a child from Late Neolithic Age from the site of Kamenka II. Archaeol. Ethnol. Anthropol. Eurasia 2, 140–153 (2001). [Google Scholar]
- 102.Stepanov A. D., Early Iron Age Dyupsya burial, Central Yakutia. Archaeol. Ethnol. Anthropol. Eurasia 38, 32–32 (2010). [Google Scholar]
- 103.Stepanov A. D., Kuzmin Y. V., Jall E. J. T., New data on the chronology of the Early Iron Age of Yakutia. The Bulletin of Irkutsk State University. Series “Geoarchaeology. Ethnology. Anthropology” , 106–112 (2014). [Google Scholar]
- 104.N. P. Sentekjaeva, in Archeology (Novosibirsk State University, 2015), pp. 11–17. [Google Scholar]
- 105.N. Kashuba, thesis, Uppsala University, Disciplinary Domain of Humanities and Social Sciences, Faculty of Arts, Department of Archaeology and Ancient History, Archaeology (2017). [Google Scholar]
- 106.Bronk Ramsey C., Bayesian analysis of radiocarbon dates. Radiocarbon 51, 337–360 (2009). [Google Scholar]
- 107.Reimer P. J., Bard E., Bayliss A., Beck J. W., Blackwell P. G., Ramsey C. B., Buck C. E., Cheng H., Edwards R. L., Friedrich M., Grootes P. M., Guilderson T. P., Haflidason H., Hajdas I., Hatté C., Heaton T. J., Hoffmann D. L., Hogg A. G., Hughen K. A., Kaiser K. F., Kromer B., Manning S. W., Niu M., Reimer R. W., Richards D. A., Scott E. M., Southon J. R., Staff R. A., Turney C. S. M., van der Plicht J., IntCal13 and Marine13 radiocarbon age calibration curves 0-50,000 years cal BP. Radiocarbon 55, 1869–1887 (2013). [Google Scholar]
- 108.Chain P. S. G., Carniel E., Larimer F. W., Lamerdin J., Stoutland P. O., Regala W. M., Georgescu A. M., Vergez L. M., Land M. L., Motin V. L., Brubaker R. R., Fowler J., Hinnebusch J., Marceau M., Medigue C., Simonet M., Chenal-Francisque V., Souza B., Dacheux D., Elliott J. M., Derbise A., Hauser L. J., Garcia E., Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U.S.A. 101, 13826–13831 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Key F. M., Posth C., Krause J., Herbig A., Bos K. I., Mining metagenomic data sets for ancient DNA: Recommended protocols for authentication. Trends Genet. 33, 508–520 (2017). [DOI] [PubMed] [Google Scholar]
- 110.M. A. Jobling, Human Evolutionary Genetics (Garland Science, ed. 2, 2014). [Google Scholar]
- 111.Hein D. W., Molecular genetics and function of NAT1 and NAT2: Role in aromatic amine metabolism and carcinogenesis. Mutat. Res. 506–507, 65–77 (2002). [DOI] [PubMed] [Google Scholar]
- 112.Sabbagh A., Darlu P., Crouau-Roy B., Poloni E. S., Arylamine N-acetyltransferase 2 (NAT2) genetic diversity and traditional subsistence: A worldwide population survey. PLOS ONE 6, e18507 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Patin E., Barreiro L. B., Sabeti P. C., Austerlitz F., Luca F., Sajantila A., Behar D. M., Semino O., Sakuntabhai A., Guiso N., Gicquel B., McElreavey K., Harding R. M., Heyer E., Quintana-Murci L., Deciphering the ancient and complex evolutionary history of human arylamine N-acetyltransferase genes. Am. J. Hum. Genet. 78, 423–436 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Peng Y., Shi H., Qi X.-b., Xiao C.-j., Zhong H., Ma R.-l. Z., Su B., The ADH1B Arg47His polymorphism in East Asian populations and expansion of rice domestication in history. BMC Evol. Biol. 10, 15 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Magalon H., Patin E., Austerlitz F., Hegay T., Aldashev A., Quintana-Murci L., Heyer E., Population genetic diversity of the NAT2 gene supports a role of acetylation in human adaptation to farming in Central Asia. Eur. J. Hum. Genet. 16, 243–251 (2008). [DOI] [PubMed] [Google Scholar]
- 116.Kuznetsov I. B., McDuffie M., Moslehi R., A web server for inferring the human N-acetyltransferase-2 (NAT2) enzymatic phenotype from NAT2 genotype. Bioinformatics 25, 1185–1186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.He Y. J., Shapero M. H., Mcleod H. L., Novel tagging SNP rs1495741 and 2-SNPs (rs1041983 and rs1801280) yield a high prediction of the NAT2 genotype in HapMap samples. Pharmacogenet. Genomics 22, 322–324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Selinski S., Blaszkewicz M., Lehmann M.-L., Ovsiannikov D., Moormann O., Guballa C., Kress A., Truss M. C., Gerullis H., Otto T., Barski D., Niegisch G., Albers P., Frees S., Brenner W., Thüroff J. W., Angeli-Greaves M., Seidel T., Roth G., Dietrich H., Ebbinghaus R., Prager H. M., Bolt H. M., Falkenstein M., Zimmermann A., Klein T., Reckwitz T., Roemer H. C., Löhlein D., Weistenhöfer W., Schöps W., Rizvi S. A. H., Aslam M., Bánfi G., Romics I., Steffens M., Ekici A. B., Winterpacht A., Ickstadt K., Schwender H., Hengstler J. G., Golka K., Genotyping NAT2 with only two SNPs (rs1041983 and rs1801280) outperforms the tagging SNP rs1495741 and is equivalent to the conventional 7-SNP NAT2 genotype. Pharmacogenet. Genomics 21, 673–678 (2011). [DOI] [PubMed] [Google Scholar]
- 119.Enattah N. S., Sahi T., Savilahti E., Terwilliger J. D., Peltonen L., Järvelä I., Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 30, 233–237 (2002). [DOI] [PubMed] [Google Scholar]
- 120.Ye K., Gu Z., Recent advances in understanding the role of nutrition in human genome evolution. Adv Nutr. 2, 486–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ingram C. J. E., Elamin M. F., Mulcare C. A., Weale M. E., Tarekegn A., Raga T. O., Bekele E., Elamin F. M., Thomas M. G., Bradman N., Swallow D. M., A novel polymorphism associated with lactose tolerance in Africa: Multiple causes for lactase persistence? Hum. Genet. 120, 779–788 (2007). [DOI] [PubMed] [Google Scholar]
- 122.Jeunemaitre X., Soubrier F., Kotelevtsev Y. V., Lifton R. P., Williams C. S., Charru A., Hunt S. C., Hopkins P. N., Williams R. R., Lalouel J.-M., Corvol P., Molecular basis of human hypertension: Role of angiotensinogen. Cell 71, 169–180 (1992). [DOI] [PubMed] [Google Scholar]
- 123.Zeron-Medina J., Wang X., Repapi E., Campbell M. R., Su D., Castro-Giner F., Davies B., Peterse E. F. P., Sacilotto N., Walker G. J., Terzian T., Tomlinson I. P., Box N. F., Meinshausen N., De Val S., Bell D. A., Bond G. L., A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell 155, 410–422 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bangsi D., Zhou J., Sun Y., Patel N. P., Darga L. L., Heilbrun L. K., Powell I. J., Severson R. K., Everson R. B., Impact of a genetic variant in CYP3A4 on risk and clinical presentation of prostate cancer among white and African-American men. Urol. Oncol. 24, 21–27 (2006). [DOI] [PubMed] [Google Scholar]
- 125.Mohamed M. E., Schladt D. P., Guan W., Wu B., van Setten J., Keating B. J., Iklé D., Remmel R. P., Dorr C. R., Mannon R. B., Matas A. J., Israni A. K., Oetting W. S., Jacobson P. A.; DeKAF Genomics and GEN03 Investigators , Tacrolimus troughs and genetic determinants of metabolism in kidney transplant recipients: A comparison of four ancestry groups. Am. J. Transplant. 19, 2795–2804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fondevila M., Phillips C., Santos C., Freire Aradas A., Vallone P. M., Butler J. M., Lareu M. V., Carracedo Á., Revision of the SNPforID 34-plex forensic ancestry test: Assay enhancements, standard reference sample genotypes and extended population studies. Forensic Sci. Int. Genet. 7, 63–74 (2013). [DOI] [PubMed] [Google Scholar]
- 127.Yoshiura K.-i., Kinoshita A., Ishida T., Ninokata A., Ishikawa T., Kaname T., Bannai M., Tokunaga K., Sonoda S., Komaki R., Ihara M., Saenko V. A., Alipov G. K., Sekine I., Komatsu K., Takahashi H., Nakashima M., Sosonkina N., Mapendano C. K., Ghadami M., Nomura M., Liang D.-S., Miwa N., Kim D.-K., Garidkhuu A., Natsume N., Ohta T., Tomita H., Kaneko A., Kikuchi M., Russomando G., Hirayama K., Ishibashi M., Takahashi A., Saitou N., Murray J. C., Saito S., Nakamura Y., Niikawa N., A SNP in the ABCC11 gene is the determinant of human earwax type. Nat. Genet. 38, 324–330 (2006). [DOI] [PubMed] [Google Scholar]
- 128.J. H. Mielke, L. W. Konigsberg, J. H. Relethford, Human Biological Variation (Oxford University Press, 2006). [Google Scholar]
- 129.Günther T., Malmström H., Svensson E. M., Omrak A., Sánchez-Quinto F., Kılınç G. M., Krzewińska M., Eriksson G., Fraser M., Edlund H., Munters A. R., Coutinho A., Simões L. G., Vicente M., Sjölander A., Sellevold B. J., Jørgensen R., Claes P., Shriver M. D., Valdiosera C., Netea M. G., Apel J., Lidén K., Skar B., Storå J., Götherström A., Jakobsson M., Population genomics of Mesolithic Scandinavia: Investigating early postglacial migration routes and high-latitude adaptation. PLOS Biol. 16, e2003703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Melzer D., Perry J. R. B., Hernandez D., Corsi A.-M., Stevens K., Rafferty I., Lauretani F., Murray A., Gibbs J. R., Paolisso G., Rafiq S., Simon-Sanchez J., Lango H., Scholz S., Weedon M. N., Arepalli S., Rice N., Washecka N., Hurst A., Britton A., Henley W., van de Leemput J., Li R., Newman A. B., Tranah G., Harris T., Panicker V., Dayan C., Bennett A., McCarthy M. I., Ruokonen A., Jarvelin M.-R., Guralnik J., Bandinelli S., Frayling T. M., Singleton A., Ferrucci L., A genome-wide association study identifies protein quantitative trait loci (pQTLs). PLOS Genet. 4, e1000072 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabc4587/DC1