

Summary
Background:
Rare coding mutations underlying deficiencies of antithrombin and proteins C and S contribute to familial venous thromboembolism (VTE). It is uncertain whether rare variants play a role in the etiology of VTE in the general population.
Objectives:
We conducted a deep whole-exome sequencing (WES) study to investigate the associations between rare coding variants and the risk of VTE in two population-based prospective cohorts.
Patients/Methods:
WES was performed in the Longitudinal Investigation of Thromboembolism Etiology (LITE), which combines the ARIC study (316 incident VTE events among 3,159 African Americans (AAs) and 458 incident VTEs among 7,772 European Americans (EAs)) and the CHS study (60 incident VTEs among 1,751 EAs). We performed gene-based tests of rare variants (allele frequency <1%, exome-wide significance p<1.47x10−6) separately in each study and ancestry group, and meta-analyzed the results for the EAs in ARIC and CHS.
Results:
In the meta-analysis of EAs, we identified one gene, PROC, in which the burden of rare, coding variants was significantly associated with increased risk of VTE (HR=5.42 [3.11, 9.42] for carriers versus non-carriers, p=2.27 x 10−9). In ARIC EAs, carriers of the PROC rare variants had on average 0.75 SD lower concentrations of plasma protein C and 0.28 SD higher D-dimer (p<0.05) than non-carriers. Adjustment for low protein C status did not eliminate the association of PROC burden with VTE. In AAs, rare coding PROC variants were not associated with VTE.
Conclusions:
Rare coding variants in PROC contribute to increased VTE risk in EAs in this general population sample.
Keywords: genomics, rare mutations, protein C, venous thrombosis, whole exome sequencing
Introduction
Venous thromboembolism (VTE) has important genetic determinants [1]. For many decades, rare, coding mutations underlying deficiencies of antithrombin, protein C, and protein S have been reported to contribute to familial forms of VTE [2–4]. Recently, large scale candidate-gene and genome-wide association studies (GWAS) have reported associations of common variants (frequency ≥ 1%) at more than a dozen loci with VTE risk [5–8]. However, most of these common variants were associated with only modestly increased risk for VTE (e.g., relative risk estimate ˂ 2.0). As the cost has fallen, whole-exome and whole-genome sequencing approaches have been increasingly applied to common complex conditions in general population samples, and have successfully identified rare variants (minor allele frequency (MAF) < 1%) with large effects, including for type 2 diabetes [9], Alzheimer’s disease [10] and early-onset myocardial infarction [11]. Moreover, a rare variant burden approach has been shown to be more powerful than single-variant analysis when multiple rare variants in a gene region are associated with the disease [10–12]. For example, Cruchaga et al. identified the gene PLD3 that contained at least 3 rare variants (carrier frequency < 1%) contributing to the risk for Alzheimer’s disease [10]. The single-variant analysis did not yield p-values less than 1x10−6, while the gene-based test yielded a p-value of 1x10−11. Similar observation was made for the APOA5 and LDLR genes for early-onset myocardial infarction (about 2-fold increased risk associated with carrier status of rare, nonsynonymous or damaging mutations) [11]. It is unknown whether rare variants, individually or collectively within a gene, contribute to VTE in the general population. We therefore conducted a deep whole-exome sequencing (WES) study of VTE in the prospective Longitudinal Investigation of Thromboembolism Etiology (LITE), to elucidate the association of rare, coding variants with VTE incidence in the general population.
Methods
Study population
LITE comprises 21,680 individuals from two population-based cohorts: the Atherosclerosis Risk in Communities Study (ARIC) (n=15,792) and the Cardiovascular Health Study (CHS) (n=5,888). LITE has identified and validated hospitalized VTE events (leg deep vein thrombosis [DVT] or pulmonary embolus [PE]) in ARIC from 1987–89 through 2015 and in CHS from 1989–1990 through 2001. Details on the study design, methods, and VTE ascertainment in LITE have been reported elsewhere [13]. Nearly all events were confirmed by imaging. The institutional review committees at each study center approved the study, and ARIC and CHS staff obtained informed participant consent.
Exome sequencing, variant calling, and annotation
As part of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium’s exome sequencing effort, ARIC and CHS DNA samples were sequenced and called together at the Baylor College of Medicine Human Genome Sequencing Center. Descriptions of the sequencing protocol, variant calling, and quality control procedures are published elsewhere [14, 15]. In total, there were 2,184,103 single nucleotide variants (SNVs) and 62,510 indels that were polymorphic in the analyzed samples after quality control filtering, including 184,011 that had a minor allele count (MAC) ≥ 40 in the combined sample of EAs from ARIC and CHS. The mean depth of coverage was 92-fold. After taking into account available follow-up information for VTE and sequencing data, our analytic sample included 316 incident VTE events (123 PE with or without DVT and 193 DVT) among 3,159 ARIC African Americans (AAs), 458 incident events (211 PE and 247 DVT) among 7,772 ARIC European Americans (EAs), and 60 incident events (21 PE and 39 DVT) among 1,751 CHS EAs.
Variants were annotated using ANNOVAR [16] and dbNSFP v2.0 [17] according to the reference genome GRCh37 and National Center for Biotechnology Information RefSeq. We used SIFT [18], PolyPhen HDIV [19], and PolyPhen HVAR [19] to predict the possible impact of identified variants on the structure and function of corresponding protein (i.e., PROC variants on protein C). Mutation maps for protein C were generated using the tools at https://www.cbioportal.org/mutation_mapper [20, 21].
Statistical analyses
We performed single variant and gene-based association analyses between the WES data and VTE using the seqMeta R package (http://cran.r-project.org/web/packages/seqMeta/). ARIC EAs, CHS EAs, and ARIC AAs were analyzed separately. Fixed effects, inverse-variance weighted meta-analyses were conducted in seqMeta to pool the association results from the EA samples of ARIC and CHS. Results from the AA samples were compared with those of EA meta-analysis to evaluate the replication of EA findings.
Single Variant Testing:
Variants were included in single variant analysis if their MAC was at least 40 (MAC=40 translates into MAF>0.0042 for this meta-analysis of ARIC and CHS EAs). This threshold was determined a priori to reduce the number of statistical tests performed and to reduce the chance for false positive associations caused by extremely rare variants. Within each race group and cohort, we tested for single variant associations with VTE by a Cox proportional hazards model adjusting for age, sex, and race-specific principal components (PCs). An association, as measured by the hazard ratio (HR), was considered to be significant at p<2.72x10−7 given a Bonferroni correction for testing as many as 184,011 single variant sites.
Gene-based testing:
In WES studies of modest sample size, single-variant analyses typically have limited statistical power to identify associations with rare variants. Gene-based approaches aggregate the cumulative effects of individual, rare variants thereby increasing the frequency of the exposure variable and simultaneously reducing the multiple-testing burden [22]. We hypothesized that deleterious rare alleles (MAF <1%) collectively within a gene may contribute to increased risk of VTE. To test our hypothesis, we performed gene-based tests to determine whether VTE cases (compared to non-cases) have an excess of rare alleles (MAF<1%) annotated as stop-gain, stop-loss, splicing, missense, or small insertion or deletion sites (indels). Using the seqMeta package, we conducted a “T1” test where all variants passing annotation filters mentioned above were summed together to generate a gene burden score [23]. The gene-level test was adjusted for age, sex, and race-specific PCs, and required the gene to have a cumulative MAC of at least 40 in the combined ARIC and CHS EA sample set. There was no lower limit on the allele frequency of individual variants that were included in the burden test. An association was considered to be significant at p<1.47x10−6, corresponding to a Bonferroni-corrected significance threshold for the exome-wide test of 16,987 qualifying genes (i.e., MAC≥40).
For genes that were statistically significant in the T1 burden test, we further calculated the HR for VTE associated with the burden of rare alleles using a Cox regression model that analyzed time-to-event data. Burden was analyzed as a binary variable coded 1 if the participant had one or more rare alleles and 0 if they had none. The analyses were performed in R using the coxph function in the Survival package (https://cran.r-project.org/web/packages/survival/) with the burden of the variants being examined as the predictor, adjusting for age, sex, and race-specific PCs.
Sensitivity analyses for gene-based tests:
1) CHS used three strategies to select participants for the CHARGE exome sequencing project: random sampling of the cohort (19%), those having MRI or cognitive data (57%), or those with extreme values for several cardiovascular risk factors including fibrinogen and waist-hip ratio (WHR) (24%) [15]. Since fibrinogen and WHR might be associated with VTE risk, we conducted a sensitivity analysis by additionally adjusting for these two variables in CHS to evaluate the influence of the sampling design on any significant genetic associations for VTE. 2) It has been noticed that an unbalanced case to non-case ratio can lead to inflated type I error rates in association analysis of rare variants [24]. Therefore, we conducted a sensitivity analysis for the significant association observed in the gene-based analysis (i.e., PROC burden) using SAIGE, which can control for an unbalanced case to non-case ratio in single variant tests [25]. To analyze the burden of the PROC rare coding variants in SAIGE, we assigned a burden score of 0 to participants without any of these PROC variants and 1 to those with at least one copy of such variants. The analysis was run in the EAs of ARIC and CHS separately and then meta-analyzed. Covariate adjustment was the same as in the primary analysis.
Analysis of rare variants in PROC and hemostatic factors:
In ARIC, we evaluated the associations between the burden of rare variants in PROC and hemostatic factors relevant to the detected gene (i.e., PROC), such as plasma levels of protein C, factor VIII activity (FVIIIc), activated partial thromboplastin time (aPTT), and D-dimer. These hemostatic factors were measured at baseline in 1987–89 (protein C, FVIIIc, aPTT) or in the 2nd follow-up visit in 1993–95 samples (D-dimer). Details on the lab measurements were provided elsewhere [26–29]. Users of anticoagulants at the time of blood collection were excluded. Samples with extreme values off 3 standard deviations (SDs) from the mean for these hemostatic factors were excluded to reduce the possibility of spurious associations with rare variants. FVIIIc, aPTT, and D-dimer were natural log (ln) transformed to normalize their distributions.
Results
Single variant test results
Single variant testing at a MAC threshold of ≥40 identified only one variant, the well-known Factor V Leiden, that was significantly associated with VTE (EA p=8 x 10−15, MAF=0.03; AA p=0.01, MAF=0.006).
Gene-based test results
Fig. 1 shows the quantile–quantile (QQ) plot for the p-values from the meta-analysis of the T1 gene burden test in ARIC EAs and CHS EAs. There was little evidence of genomic inflation for the gene burden test (λ=1.01 in ARIC EAs and 0.99 in the meta-analysis of ARIC and CHS EAs). Based on the pre-defined statistical threshold (p<1.47x10−6), the T1 burden test revealed one significant gene-level association with VTE in EAs (Table 1): PROC (EA meta-analysis: OR=5.23 [95% CI: 2.80, 9.80] for carriers versus non-carriers, p=2.81 x 10−8). PROC had strong and consistent associations in the EA samples of both ARIC and CHS (ARIC OR=4.13; CHS OR=12.72). This association remained consistent when analyzed with a Cox model (EA meta-analysis: HR=5.42 [3.11, 9.42], p=2.27 x 10−9). The percentage of samples with 1 or more rare coding variants in PROC is 0.6% in the EAs of ARIC and CHS and 1.4% in ARIC AAs. Fig. 2 shows specific mutations in PROC in VTE cases and non-cases in ARIC EAs. When analyses were restricted to only private variants (i.e., MAC=1) in PROC, results for EAs in ARIC and CHS remained significant: OR=9.35 [3.75, 23.31], p=1.61x10−08 from T1 burden test, HR=7.43 [3.67, 15.08], p=2.62x10−08 from Cox regression. Supplementary Table S1 presents the T1 burden test results for the other top 6 genes with subthreshold signals at p<0.0005 (ranked after PROC). In ARIC AAs, the T1 burden test detected no association at PROC (HR=0.80 for carriers versus non-carriers, p=0.71), and one variant (N371D) accounted for nearly half of all rare variants in PROC. When that variant was removed, the association remained nonsignificant (HR=1.01 for carriers versus non-carriers, p=0.91) in AAs. There was little overlap in PROC rare variants found in EAs and AAs (Fig. 3).
Fig.1.
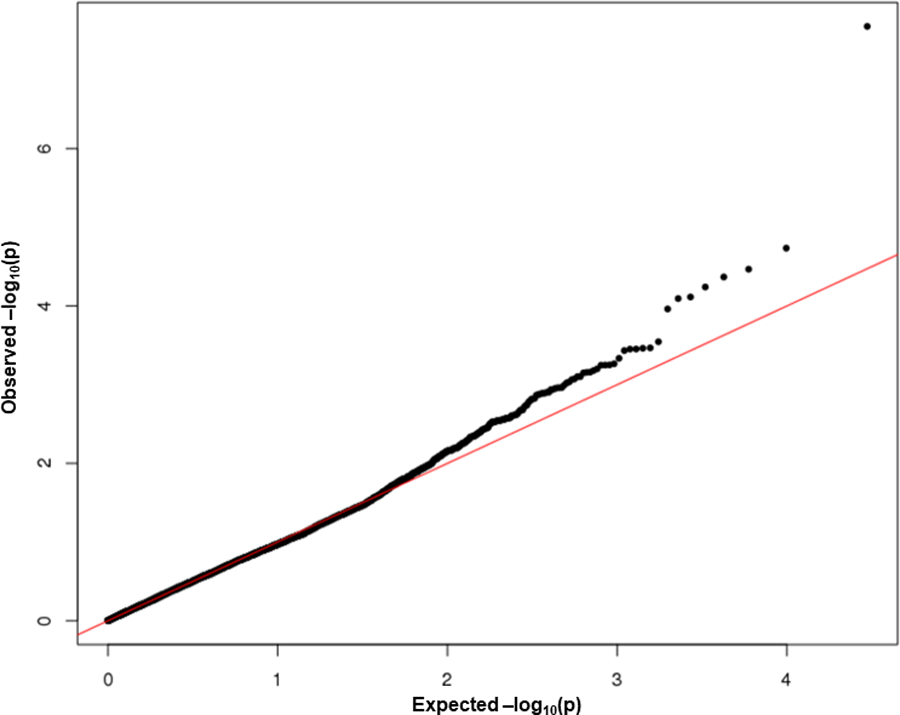
Quantile–quantile (Q-Q) plot for the meta-analysis of gene-based burden test in ARIC EAs and CHS EAs (genomic inflation factor (lambda) = 0.99).
Table 1.
Associations between burden of rare coding variants in PROC with incident venous thromboembolism (VTE) in ARIC and CHS
| T1 Burden Test in SeqMeta* |
Cox Regression* |
|||||||
|---|---|---|---|---|---|---|---|---|
| Study sample | N cases: non-cases | CMAC (CMAF) in cases | CMAC (CMAF) in non-cases | N SNVs | P-value | OR (95% CI) | HR (95% CI) | P-value |
| ARIC EAs | 458:7314 | 10 (0.01) | 38 (0.003) | 29 | 2.02x10−5 | 4.13 (2.04, 8.36) | 4.26 (2.27, 7.98) | 6.04x10−06 |
| CHS EAs | 60:1691 | 3 (0.025) | 8 (0.002) | 8 | 2.87x10−6 | 12.72 (3.25, 49.80) | 11.44 (3.54, 36.94) | 4.57x10−05 |
| All EAs | 518:9005 | 13 (0.01) | 46 (0.003) | 35 | 2.81x10−8 | 5.23 (2.80, 9.80) | 5.42 (3.11, 9.42) | 2.27x10−09 |
| ARIC AAs | 316:2843 | 3 (0.005)† | 42 (0.007)† | 18 | 0.47 | 0.65 (0.20, 2.11) | 0.80 (0.26, 2.51) | 0.71 |
| ARIC AAs‡ | 315:2823 | 2 (0.003) | 22 (0.004) | 17 | 0.45 | 0.81 (0.19, 3.48) | 1.01 (0.27, 4.38) | 0.91 |
Coding variants= stop-gain, stop-loss, splicing, missense, or small insertion or deletion sites
CMAC= cumulative minor allele count; CMAF= cumulative minor allele frequency; SNV=single nucleotide variants belonging to the coding variants mentioned above; HR=hazard ratio for carriers vs. non-carriers obtained from Cox regression analysis of time to VTE event
Adjusted for age, sex, and principal components capturing ancestry (2 for ARIC EAs, 3 for CHS EAs, 4 for ARIC AAs)
One AA case and 20 non-cases had the N371D variant
After exclusion of carriers of the N371D variant.
Fig. 2.
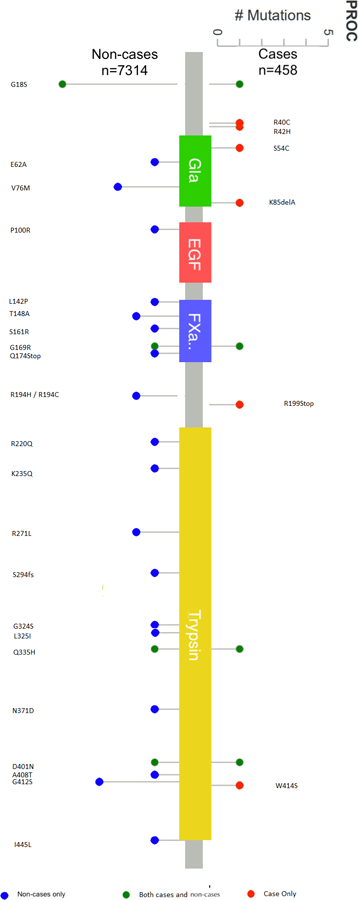
PROC mutations in VTE cases and non-cases of European American individuals in ARIC. Red=cases only, blue=non-cases only, green=both cases and non-cases. The protein domains are as defined by PFAM (http://pfam.xfam.org/), which include a Gla site (vitamin K-dependent carboxylation/gamma-carboxyglutamic domain), an EGF site (epidermal growth factor-like domain), a FXa.. site (coagulation factor Xa inhibitory site), and a Trypsin (a serine protease) site.
Fig. 3.
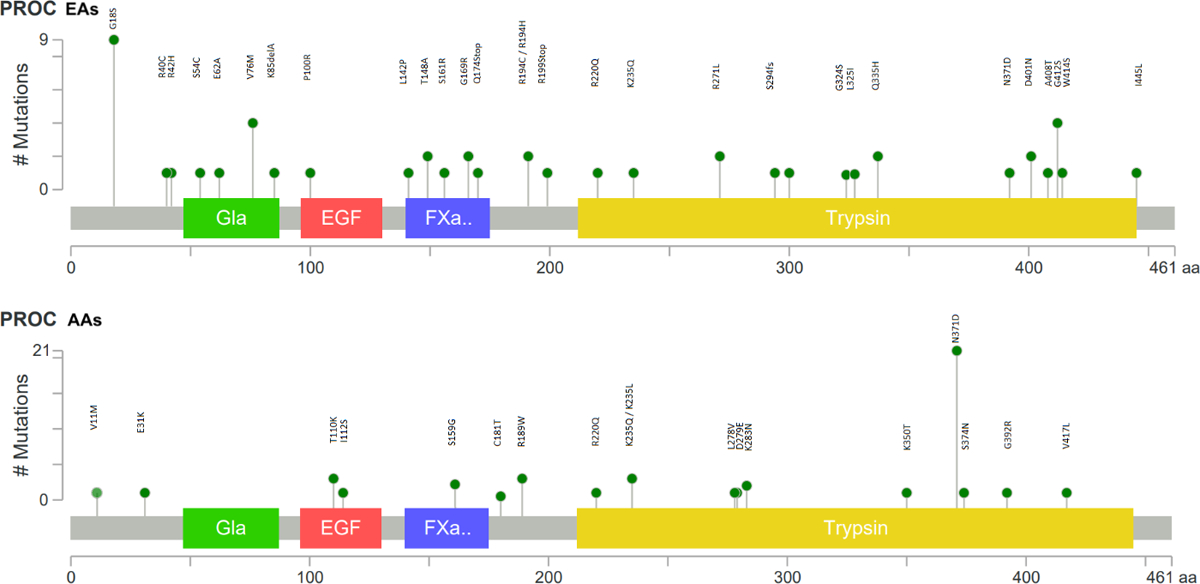
Mutation plots of rare mutations in PROC in ARIC EAs (A) and AAs (B). The protein domains are as defined by PFAM (http://pfam.xfam.org/), which include a Gla site (vitamin K-dependent carboxylation/gamma-carboxyglutamic domain), an EGF site (epidermal growth factor-like domain), a FXa.. site (coagulation factor Xa inhibitory site), and a Trypsin (a serine protease) site.
Sensitivity analyses:
1) In CHS, additional adjustment for fibrinogen or WHR had little impact on the association between VTE and the burden of rare coding variants in PROC (data not shown). 2) In the sensitivity analysis for the PROC burden test in the EAs of ARIC and CHS using SAIGE, we observed consistent results for the association of VTE with the burden of PROC variants: p=4.70 x 10−4 in ARIC, 0.03 in CHS, and 3.97 x 10−5 in the meta-analysis.
PROC and hemostatic factors
In ARIC EAs, carriers of the PROC rare variants showed significantly lower levels of protein C and higher levels of D-dimer compared to non-carriers, independent of the influence of age, gender, and prevalent VTE status (Table 2). The burden of PROC rare variants was not significantly associated with FVIIIc or aPTT (p>0.05). Notably, the burden of PROC rare variants was also significantly associated with lower protein C in ARIC AAs but with a smaller effect size as compared to that in EAs (AAs: mean/SD=2.78/0.61 in carriers and 3.12/0.61 in non-carriers, p=0.0003 after adjustment for age, sex and prevalent VTE, difference=0.56 SD in contrast to 0.75 SD in EAs).
Table 2.
Association between burden of rare, coding variants of PROC and relevant plasma hemostatic variables in EAs of ARIC
| Trait | Carriers |
Non-carriers |
P-value* | P-value† | ||
|---|---|---|---|---|---|---|
| N | Mean/SD | N | Mean/SD | |||
| Protein C, mg/L | 45 | 2.73/0.65 | 7655 | 3.18/0.58 | <0.0001 | <0.0001 |
| FVIIIc, % ‡ | 47 | 130.0/34.8 | 7693 | 124.3/31.3 | 0.38 | 0.22 |
| aPTT, seconds‡ | 45 | 28.5/2.73 | 7648 | 28.9/2.7 | 0.27 | 0.70 |
| D-dimer, μg/mL‡ | 41 | 0.61/0.85 | 6619 | 0.48/1.42 | 0.004 | 0.005 |
Coding variants= stop-gain, stop-loss, splicing, missense, or small insertion or deletion sites
Mean/SD: in original unit for the trait
T-test comparison on the residual values between carriers and non-carriers after adjusting for age and sex (with appropriate transformation of the trait to normalize its distribution if necessary)
T-test comparison on the residual values between carriers and non-carriers after adjusting for age, sex, and prevalent VTE status (with appropriate transformation of the trait if necessary)
Natural log transformed values were analyzed in the statistical test.
Given the significant association between the burden of rare PROC coding variants and low protein C levels, we conducted an additional analysis to evaluate whether the association of VTE with the burden of rare PROC variants was explained by low protein C levels. We dichotomized protein C levels based on the cutpoint of 2.0 mg/L, consistent with the previously published LITE study [28]. In a Cox regression model that included both the burden of rare PROC coding variants and low protein C status adjusting for the same covariates as in the primary analysis, the PROC burden remained significant when the status of low protein C was adjusted for (Table 3). Moreover, the adjustment for the burden of rare PROC coding variants resulted in more reduction in the strength of the association between low protein C and VTE than vice versa (21.8% vs 5.0%). We noticed that the HR for low protein C with VTE was lower than the previously reported HR in LITE based on about 8 years of follow-up (1.83 in EAs and AAs combined vs 3.36 from the previous report) [28]. This difference was mainly due to the attenuation of the association of low protein C with VTE over time.
Table 3.
Association of incident VTE with PROC burden and low protein C levels in ARIC EAs
| Test Model* | Exposure Variable | HR (95% CI) | P-value |
|---|---|---|---|
| Low protein C† | Low protein C | 1.97 (0.88, 4.41) | 0.10 |
| PROC burden | PROC burden | 4.80 (2.56, 9.00) | 9.79x10−7 |
| Low protein C + PROC burden | Low protein C | 1.54 (0.67, 3.52) | 0.16 |
| PROC burden | 4.56 (2.40, 8.67) | 3.74x10−6 |
Analysis was limited to those who had values for protein C levels and PROC burden score (n=7749; 23 participants were not included due to missing data on protein C)
Adjusted for age, sex, and the 2 principal components related to ancestry for ARIC EAs
Defined as protein C level < 2.0 mg/L,23 account for 0.72% of ARIC EAs.
Comparison with known variants
We checked the PROC rare coding variants identified in this study against those in the Human Gene Mutation Database (HMGD, http://www.hgmd.cf.ac.uk/ac/index.php), and found that 18 of the 29 variants identified in EAs and 6 of the 18 variants identified in AAs were present and reported to cause protein C deficiency (Supplementary Table S2). To determine if our results were driven by these known variants, we ran an additional Cox burden analysis after exclusion of carriers of these variants. We found that the association between the burden of PROC rare coding variants and VTE remained consistent (ARIC EAs: HR = 5.20 [1.67, 16.20], p = 0.0045, CMAC: 3 in 451 cases and 10 in 7,286 non-cases).
Discussion
These data, from two population-based cohorts, demonstrate that rare coding variants in PROC are present at an appreciable frequency in the general population and that their presence is associated with a more than 5 fold increased risk for VTE. Exclusion of known PROC mutations in HMGD did not eliminate the association, indicating the effect of the newly identified PROC variants on VTE risk in these general population samples. The association of PROC rare coding variants with plasma D-dimer, a marker of fibrin turnover and an important risk marker for VTE [29], corroborates the influence of these rare coding variants on VTE risk and increases our understanding of why D-dimer is such a strong VTE risk factor.
Protein C deficiency due to rare mutations in PROC is well known as a strong VTE risk factor in families [30–33], where heterozygous presence of mutations that are uncommon in the general population raise the risk about 10-fold [34]. Common risk-elevating variants of PROC have not been identified in published GWASs in general population samples [7, 8]. Protein C, encoded by PROC, is an important anticoagulant; low plasma levels of protein C are associated with a marked increase in risk of VTE [28, 32, 35], and activated Protein C resistance, often caused by Factor V Leiden, also is a risk factor for VTE [36]. In our data, carriers of the rare, coding variants in PROC had protein C levels that were on average 0.75 SDs lower than those of non-carriers (Table 2), and explained an important part of the association between low protein C and VTE risk. Our data show that individuals in the general population harbor PROC mutations that are associated with low protein C levels and an increased risk of VTE. Therefore, deep sequencing of PROC gene among VTE cases, at least in EAs, may be warranted, especially when the cause of VTE cannot be determined by standard clinical tests.
We failed to observe any association of VTE with the burden of PROC rare coding variants in AAs. While power was likely lower in AAs due to a smaller number of VTEs, the estimated relative risk in AAs was also near 1. We noticed that many of the PROC variants in EAs were predicted to be probably or possibly damaging, while only one variant in AAs was predicted to be damaging (Supplementary Table S2), which might contribute to the different associations of PROC rare variants with VTE risk between EAs and AAs. Interestingly, we also observed that, in both EAs and AAs, the burden of PROC rare coding variants was strongly associated with lower protein C levels, but the status of low protein C was only modestly associated with increased risk of VTE (HR=1.97 [0.88, 4.41] and 1.63 [0.61, 4.39] in EAs and AAs, respectively, p for interaction by race>0.05). Therefore, the association between the PROC burden and VTE risk in EAs but not in AAs suggests that the effect of PROC rare coding variants on VTE risk in EAs was largely mediated through mechanisms other than low protein C levels. The nonsynonymous variants likely influence the structure and function of protein C, and it is possible that the rare coding variants observed in the AAs may have less impact on these aspects of protein C than those in EAs. This speculation is supported by the mutual adjustment analysis in which the adjustment for low protein C only had a minor influence on the association between the burden of PROC rare, coding variants and VTE risk in EAs. These findings indicate that future functional studies are needed to delineate the underlying mechanisms by which these PROC rare coding variants increase the risk of VTE in EAs.
A prior targeted sequencing study identified 3 rare coding variants in sporadic, unprovoked VTE patients that resulted in abnormal protein C levels; no rare coding variants were found in the controls [37]. Of note, only one variant (2:128179014:G:A) was present in our sample (n=4 EA non-cases), which demonstrates that rare variants can be specific to study population. Our results extend those findings by showing that a burden of rare, coding variants in PROC contributes to VTE risk in the general population, and the effect of rare variants in aggregate can be captured by a deep exome-wide sequencing approach. More recently, de Haan et al reported a target sequencing study of 734 hemostasis genes in 899 European DVT patients and 599 controls, and failed to identify an association between burden of rare variants and DVT risk.[38] The discrepant results between that study and ours may be explained by differences in patient characteristics (i.e., de Haan et al focused on DVT while our study included both DVT and PE) and sequencing approach (i.e., depth and coverage of sequencing).
Some limitations of this study warrant consideration. First, due to a relatively limited sample size, our study had modest statistical power to identify functional rare variants that have weak influences on VTE risk. Second, not all nonsynonymous variants will have an equal impact on protein function. Future studies in animal models may provide insights on the direct effects of these specific mutations on thrombosis. Third, we included all available samples from the cohorts in order to obtain a complete picture of the spectrum of mutations in the general population as well as to maximize precision in the analysis of the longitudinal data. However, an unbalanced case to non-case ratio can lead to inflated type I error rates in association analysis of rare variants [24]. To help alleviate this, we excluded genes with cumulative MAC < 40 in the gene-based analysis and observed a robust genome inflation factor and Q-Q plot. Furthermore, in the sensitivity analysis using SAIGE to control for an unbalanced case to non-case ratio [25], we observed consistent results for the association of VTE with the burden of PROC variants (p= 3.97 x 10−5 in the meta-analysis of ARIC and CHS EAs). Notably, in the analysis of extremely unbalanced case non-case data, SAIGE was observed to be somewhat conservative, corresponding to slightly less stringent empirical α levels than the conventionally used threshold for statistical significance [25]. Altogether, the genome inflation factor, Q-Q plot, and sensitivity analysis with SAIGE suggest that our PROC finding was unlikely to be a false positive finding due to the unbalanced case to non-case ratio.
In conclusion, using a deep whole-exome sequencing approach, we observed that incident VTE cases from two community-based EA cohorts had an excess of rare coding variants in PROC compared to non-cases. The burden of rare PROC coding variants was also associated with lower plasma levels of protein C and higher D-dimer and explained an important part of the association between low protein C and VTE risk. Data from our study suggests that large-scale and deep whole-exome or whole-genome sequencing efforts will detect genetic influences from rare variants among VTE patients ascertained from the general population.
Supplementary Material
Table S1. Top T1 burden test results with subthreshold signals at p<0.0005 in the EAs of our study.
Table S2. PROC rare coding variants identified in the EAs (Data sheet 1) and AAs (Data sheet 2) of our study, their presence in the Human Gene Mutation Database (HGMD) and Clinvar database, and possible impact on the structure and function of protein C predicted by SIFT, PolyPhen HDIV, and PolyPhen HVAR.
Essentials.
The role of rare coding variants in venous thromboembolism in the general population is unclear.
We conducted a whole-exome sequencing study for venous thromboembolism in population-based cohorts.
Rare coding variants in PROC aggregately associated with the risk of venous thromboembolism.
Carriers of the PROC rare variants had lower plasma protein C and higher D-dimer than non-carriers.
Acknowledgements
The National Heart, Lung, and Blood Institute (NHLBI) provided support for LITE via R01 HL059367. This work was carried out in part using computing resources at the University of Minnesota Supercomputing Institute.
Funding support for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by the NIH through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419). Data for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by Eric Boerwinkle on behalf of the Atherosclerosis Risk in Communities (ARIC) Study and Bruce Psaty, principal investigator for the Cardiovascular Health Study (CHS). Sequencing was carried out at the Baylor Genome Center (U54 HG003273).
The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the NHLBI, National Institutes of Health, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HHSN268201700005I). The authors thank the staff and participants of the ARIC study for their important contributions.
The Cardiovascular Health Study (CHS) research was supported by contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086 and grants U01HL080295, HL087652, HL105756, and U01HL130114 from the NHLBI, with additional contribution from National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629 from the National Institutes on Aging (NIA). A full list of CHS principal investigators and institutions can be found at CHS-NHLBI.org.
The authors wish to acknowledge the support of the NHLBI and the contributions of the research institutions, study investigators, field staff and study participants in creating this resource for biomedical research. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interests
None.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Souto JC, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria JM, et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: the GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am J Hum Genet 2000; 67: 1452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353: 1167–73. [DOI] [PubMed] [Google Scholar]
- 3.Simmonds RE, Ireland H, Lane DA, Zoller B, Garcia de Frutos P, Dahlback B. Clarification of the risk for venous thrombosis associated with hereditary protein S deficiency by investigation of a large kindred with a characterized gene defect. Ann Intern Med 1998; 128: 8–14. [DOI] [PubMed] [Google Scholar]
- 4.Egeberg O Inherited Antithrombin Deficiency Causing Thrombophilia. Thromb Diath Haemorrh 1965; 13: 516–30. [PubMed] [Google Scholar]
- 5.Bezemer ID, Bare LA, Doggen CJ, Arellano AR, Tong C, Rowland CM, et al. Gene variants associated with deep vein thrombosis. Jama 2008; 299: 1306–14. [DOI] [PubMed] [Google Scholar]
- 6.Smith NL, Hindorff LA, Heckbert SR, Lemaitre RN, Marciante KD, Rice K, et al. Association of genetic variations with nonfatal venous thrombosis in postmenopausal women. Jama 2007; 297: 489–98. 10.1001/jama.297.5.489. [DOI] [PubMed] [Google Scholar]
- 7.Germain M, Chasman DI, de Haan H, Tang W, Lindstrom S, Weng LC, et al. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet 2015; 96: 532–42. 10.1016/j.ajhg.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinds DA, Buil A, Ziemek D, Martinez-Perez A, Malik R, Folkersen L, et al. Genome-wide association analysis of self-reported events in 6135 individuals and 252 827 controls identifies 8 loci associated with thrombosis. Hum Mol Genet 2016; 25: 1867–74. 10.1093/hmg/ddw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet 2014; 46: 294–8. [DOI] [PubMed] [Google Scholar]
- 10.Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014; 505: 550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015; 518: 102–6. 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–41. 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cushman M, Tsai AW, White RH, Heckbert SR, Rosamond WD, Enright P, et al. Deep vein thrombosis and pulmonary embolism in two cohorts: the longitudinal investigation of thromboembolism etiology. Am J Med 2004; 117: 19–25. 10.1016/j.amjmed.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Yu B, Pulit SL, Hwang SJ, Brody JA, Amin N, Auer PL, et al. Rare Exome Sequence Variants in CLCN6 Reduce Blood Pressure Levels and Hypertension Risk. Circ Cardiovasc Genet 2016; 9: 64–70. 10.1161/CIRCGENETICS.115.001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schick UM, Auer PL, Bis JC, Lin H, Wei P, Pankratz N, et al. Association of exome sequences with plasma C-reactive protein levels in >9000 participants. Hum Mol Genet 2015; 24: 559–71. 10.1093/hmg/ddu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 2013; 34: E2393–402. 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc 2016; 11: 1–9. 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–9. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–4. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 2014; 95: 5–23. 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 2008; 83: 311–21. 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma C, Blackwell T, Boehnke M, Scott LJ, GoT2D investigators. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet Epidemiol 2013; 37: 539–50. 10.1002/gepi.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 2018; 50: 1335–41. 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Tracy RP, Aleksic N, et al. Coagulation factors, inflammation markers, and venous thromboembolism: the longitudinal investigation of thromboembolism etiology (LITE). Am J Med 2002; 113: 636–42. [DOI] [PubMed] [Google Scholar]
- 27.Zakai NA, Ohira T, White R, Folsom AR, Cushman M. Activated partial thromboplastin time and risk of future venous thromboembolism. Am J Med 2008; 121: 231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folsom AR, Aleksic N, Wang L, Cushman M, Wu KK, White RH. Protein C, antithrombin, and venous thromboembolism incidence: a prospective population-based study. Arterioscler Thromb Vasc Biol 2002; 22: 1018–22. [DOI] [PubMed] [Google Scholar]
- 29.Folsom AR, Alonso A, George KM, Roetker NS, Tang W, Cushman M. Prospective study of plasma D-dimer and incident venous thromboembolism: The Atherosclerosis Risk in Communities (ARIC) Study. Thromb Res 2015; 136: 781–5. 10.1016/j.thromres.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grundy CB, Chisholm M, Kakkar VV, Cooper DN. A novel homozygous missense mutation in the protein C (PROC) gene causing recurrent venous thrombosis. Hum Genet 1992; 89: 683–4. [DOI] [PubMed] [Google Scholar]
- 31.Grundy CB, Melissari E, Lindo V, Scully MF, Kakkar VV, Cooper DN. Late-onset homozygous protein C deficiency. Lancet 1991; 338: 575–6. [DOI] [PubMed] [Google Scholar]
- 32.Grundy CB, Schulman S, Krawczak M, Kobosko J, Kakkar VV, Cooper DN. Protein C deficiency and thromboembolism: recurrent mutation at Arg 306 in the protein C gene. Hum Genet 1992; 88: 586–8. [DOI] [PubMed] [Google Scholar]
- 33.Lind B, van Solinge WW, Schwartz M, Thorsen S. Splice site mutation in the human protein C gene associated with venous thrombosis: demonstration of exon skipping by ectopic transcript analysis. Blood 1993; 82: 2423–32. [PubMed] [Google Scholar]
- 34.Allaart CF, Poort SR, Rosendaal FR, Reitsma PH, Bertina RM, Briet E. Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet 1993; 341: 134–8. [DOI] [PubMed] [Google Scholar]
- 35.Tuddenham EGD, Cooper DN. The molecular genetics of haemostasis and its inherited disorders Oxford; New York: Oxford University Press, 1994. [Google Scholar]
- 36.Dahlback B Inherited thrombophilia: resistance to activated protein C as a pathogenic factor of venous thromboembolism. Blood 1995; 85: 607–14. [PubMed] [Google Scholar]
- 37.Wu C, Dwivedi DJ, Pepler L, Lysov Z, Waye J, Julian J, et al. Targeted gene sequencing identifies variants in the protein C and endothelial protein C receptor genes in patients with unprovoked venous thromboembolism. Arterioscler Thromb Vasc Biol 2013; 33: 2674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Haan HG, van Hylckama Vlieg A, Lotta LA, Gorski MM, Bucciarelli P, Martinelli I, et al. Targeted sequencing to identify novel genetic risk factors for deep vein thrombosis: a study of 734 genes. J Thromb Haemost 2018; 16: 2432–41. 10.1111/jth.14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Top T1 burden test results with subthreshold signals at p<0.0005 in the EAs of our study.
Table S2. PROC rare coding variants identified in the EAs (Data sheet 1) and AAs (Data sheet 2) of our study, their presence in the Human Gene Mutation Database (HGMD) and Clinvar database, and possible impact on the structure and function of protein C predicted by SIFT, PolyPhen HDIV, and PolyPhen HVAR.