

Abstract
Cardiac fibrosis represents an enormous health concern as it is prevalent in nearly every form of cardiovascular disease, the leading cause of death worldwide. Fibrosis is characterized by the activation of fibroblasts into myofibroblasts, a contractile cell type that secretes significant amounts of extracellular matrix components; however, the onset of this condition is also due to persistent inflammation and the cellular responses to a changing mechanical environment. In this review, we provide an overview of the pro-fibrotic, pro-inflammatory, and biomechanical mechanisms that lead to cardiac fibrosis in cardiovascular diseases. We then discuss cadherin-11, an intercellular adhesion protein present on both myofibroblasts and inflammatory cells, as a potential link for all three of the fibrotic mechanisms. Since experimentally blocking cadherin-11 dimerization prevents fibrotic diseases including cardiac fibrosis, understanding how this protein can be targeted for therapeutic use could lead to better treatments for patients with heart disease.
Keywords: Cadherin-11, Cardiac Fibrosis, Myofibroblast, Extracellular Matrix, Inflammation
I. Introduction
The cardiac extracellular matrix (ECM) is the structural scaffold in which cardiomyocytes, fibroblasts, inflammatory cells, and endothelial cells (among others) reside [1]. This fibrillar network is composed of a complex arrangement of structural and signaling proteins that regulate, in part, the biochemical and biomechanical functions of the cardiac tissue. Further, through cell-matrix interactions, the ECM plays a critical role in force transmission such that the contractile force of a single cardiomyocyte is transmitted throughout the entire heart.
Fibrosis, the excessive deposition of ECM components, occurs in response to tissue injury or stress and is a well-recognized cause of morbidity and mortality [2]. In response to cardiac injury (e.g., myocardial infarction) or chronic stress (e.g., hypertension, atherosclerosis, and aortic stenosis), fibrotic remodeling of the ECM provides the necessary structural components and growth factors needed for healthy cardiac tissue to regrow [3]. Prolonged and/or unrestrained fibrosis can lead to subdued regeneration and eventual loss of tissue function through a variety of mechanisms. In the heart, excessive fibrotic remodeling can disrupt excitation-contraction coupling increasing the risk of arrhythmias and increase tissue stiffness leading to systolic, diastolic, and valvular dysfunction [4–8]. However, if insufficient fibrotic remodeling occurs, defective scar tissue may form resulting in ventricular dilation and, in the most adverse instances, tissue rupture [9,10]. Given that alterations in the ECM alter the active and passive mechanical properties of the heart tissue as well as the cellular signaling that occurs within the tissue, a thorough understanding of the biochemical and biomechanical underpinnings of cardiac fibrosis is necessary in order to find cardiovascular disease therapeutics.
In this review, we broadly consider the biochemical and biomechanical mechanisms that lead to cardiac fibrosis in cardiovascular diseases by discussing the cells involved in the fibrotic response. We then focus on the importance of cadherin-11, an intercellular adhesion protein that is required for myofibroblast activation and implicated in inflammatory remodeling of cardiac tissue. We propose that cadherin-11 is a therapeutic target for treating cardiac fibrosis, as it represents a common feature of the biochemical and biomechanical underpinnings of fibrosis.
II. Biochemical Causes of Cardiac Fibrosis
In healthy cardiac tissue, resident fibroblasts maintain the ECM through the synthesis of ECM proteins (e.g., fibrillar collagens I and III, non-fibrillar collagens, elastin, and laminin) as well as their degradation through production of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs) (Figure 1) [11]. The balance of this synthesis and degradation allows for ECM homeostasis. Recent work has shown that resident cardiac fibroblasts are not a homogenous group of cells but rather two fibroblast sub-populations defined by the high (F-SH) or low (F-SL) expression of Sca1 [12]. Both of these resident populations express canonical fibroblast markers, however, F-SH cells have a distinct cell adhesion phenotype while F-SL cells exhibit a secretory phenotype. F-SH fibroblasts seem to preferentially differentiate into activated fibroblasts and myofibroblasts while F-SL fibroblasts preferentially differentiate into the newly described Wif1-expressing fibroblasts that act to inhibit fibrosis through expression of Wnt and TGF-β signaling inhibitors. This suggests that a balance of these resident fibroblast populations may also be needed to maintain a healthy cardiac ECM, though more work is needed to describe the function of these novel cell types.
Figure 1:
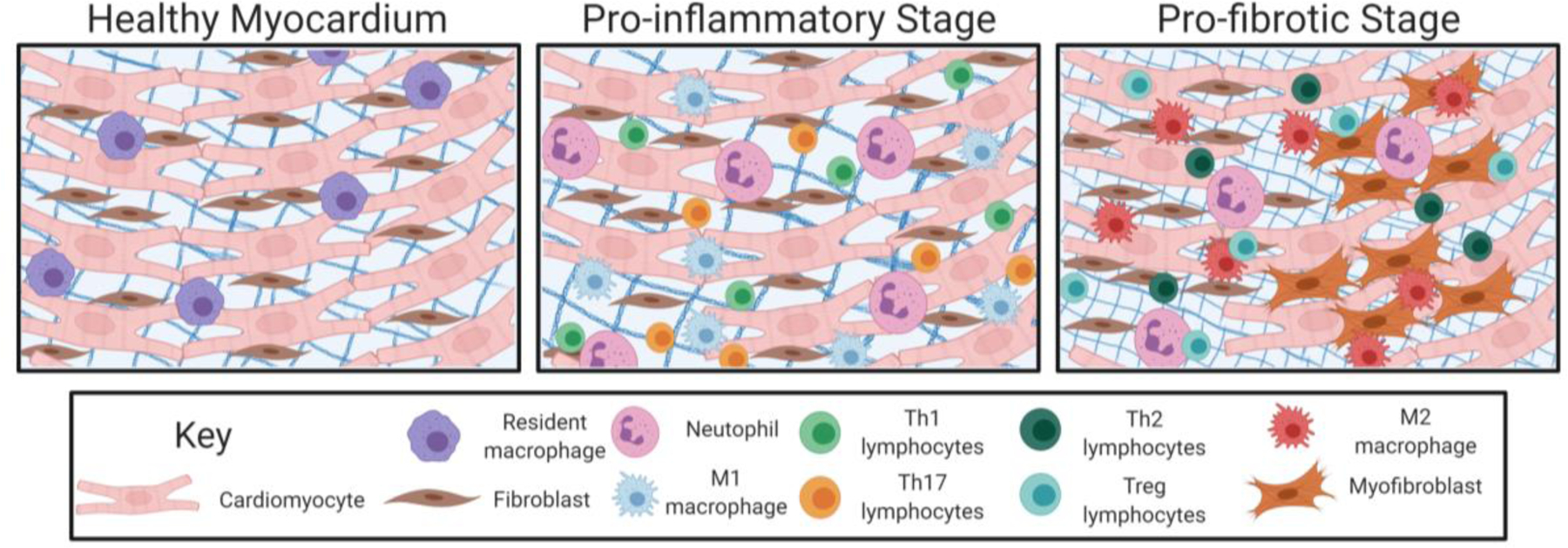
Cell involvement in healthy cardiac tissue and during the cardiac fibrotic response. Left Panel: In the healthy myocardium, healthy cardiomyocytes (red), resident fibroblasts (brown), and resident immune cells (i.e., macrophages; purple) reside. In this state, resident fibroblasts help to maintain a normal ECM through the production of collagens, elastin, and laminin. These resident cells also produce a balance of MMPs and TIMPs to help maintain a normal ECM in which these cells can reside. Center Panel: In the pro-inflammatory stages of cardiac fibrosis, DAMPs secreted by stressed or necrotizing cells lead to neutrophil (pink) invasion. This sets off an inflammatory cascade whereby macrophages are recruited and activated so that these cells take on an M1 pro-inflammatory phenotype (blue). In later stages of inflammation, Th1 (green) and Th17 (orange) lymphocytes are recruited and help to maintain activation of other inflammatory cells. Right Panel: As inflammatory signaling persists, macrophages take on an M2, pro-fibrotic phenotype (red). Th2 (dark green) and Treg (teal) lymphocytes also secrete anti-inflammatory and pro-fibrotic mediators leading to this switch toward increased fibrosis. Neutrophils (pink) also persist at this stage and directly secrete ECM components. Signaling from each these cell types activates pathways leading to fibroblast (brown) proliferation (left) and activation into myofibroblasts (dark orange; right). Myofibroblasts are contractile and secretory and are the predominant cell type secreting extracellular matrix proteins. The activation and accumulation of these cells results in a dense ECM (blue grid behind cells). These cells eventually differentiate into matrifibrocytes (not shown) to maintain a stable fibrotic scar. Created with BioRender.com
Cardiac tissue injury during myocardial infarction [13,14], aortic valve disease [15], hypertension [16], and atherosclerosis [17] results in injured and necrotizing cells releasing damage-associated molecular patterns (DAMPs) as the first stage of the healing process. Leukocytes sense this damage and produce pro-inflammatory cytokines leading to inflammatory cell recruitment to the site of damage [18]. The resultant increase in inflammatory cells leads to clearance of the damaged tissue and the release of pro-fibrotic signals that increase proliferation, recruitment, and activation of fibroblasts into secretory, contractile myofibroblasts. Myofibroblasts are required for the injury response since collagen fibrillogenesis and scar formation are necessary to stabilize the tissue architecture due to the limited regenerative capacity of cardiac tissues [11,19–21]. In healthy repair, clearance of inflammatory cells occurs after the heart’s structure is stabilized leaving behind a stable fibrotic scar and healthy cardiomyocytes. In most tissues, myofibroblasts are also cleared at this point; however, a significant number of quiescent myofibroblasts in the heart are found long after a stable scar has formed (>60 days post myocardial infarction) [22]. Recent lineage tracing studies have shown that these cells are matrifibrocytes, differentiated myofibroblast-lineage cells that express bone-cartilage markers, lose their proliferative ability, and no longer express α-SMA [23]. While these cells were described in the context of post-myocardial infarct fibrosis, matrifibrocytes have also been shown to be similar to fibroblast sub-populations in Ang-II induced cardiac fibrosis [24]. Thus, these cells potentially arise following multiple types of cardiac injury to stabilize fibrotic scar, but more work is needed to understand the role of these cells in this broader context. If clearance of inflammatory cells and differentiation of myofibroblasts into matrifibrocytes does not occur, feedforward signaling begins whereby inflammatory cells and myofibroblasts continue to promote fibrosis through chronically activated proinflammatory and profibrotic signaling pathways.
Inflammatory Signaling in Cardiac Fibrosis
Cardiovascular diseases such as heart failure, aortic valve disease, and atherosclerosis are often initiated by acute inflammatory reactions caused by stressed and necrotic cardiomyocytes as a protective response to infection, injuries such as myocardial infarction, or stress conditions such as hypertension [16,25,26]. As mentioned previously, stressed cardiac cells release DAMPs such as heat-shock proteins, degraded ECM molecules, and DNA fragments. These signals are recognized by toll-like receptors (TLRs) 2 and 4 in the damaged cell as well as in surrounding, healthy cells promoting the production of proinflammatory cytokines (i.e., TNF-α, IFN-γ, IL-1β, and IL-6) and fibroblast growth factors through activation of NF-κB and AP-1 signaling cascades [27,28]. The first cells to respond to these signals are neutrophils that are recruited to the injured site to engulf damaged cells and tissue debris. Neutrophils also release proinflammatory cytokines, MMPs, reactive oxygen species, and serine proteases leading to both the remodeling of the ECM and further recruitment of inflammatory cells [29]. While this clearance is initially beneficial for the tissue, the neutrophil-mediated response lacks specificity and can result in further damage to the healthy cardiac tissue.
Macrophages that reside within the myocardium and those that are recruited via TLR signaling are the next cells to arrive at the injury site [30]. These cells initially take on an M1-like phenotype due to activation of IFN-γ and TNF-α signaling pathways activated following cardiac injury and secrete large amounts of pro-inflammatory mediators and MMPs [18]. M1-like macrophages respond to the localized upregulation and secretion of MCP-1/CCL2 through their expression of C-C chemokine receptor 2 (CCR2) and infiltrate the damaged tissue [31]. Once at the injury site, M1-like macrophages promote further increases to the inflammatory response by producing proinflammatory cytokines (e.g., CCL2, IL-1β, TNF-α, IL-23, IL-12) that both recruit other inflammatory cells and promote a proinflammatory phenotype in cardiomyocytes and resident fibroblasts [32]. Macrophages display marked heterogeneity and change their phenotypes based on their microenvironment [30,33,34]. Thus, during the reparative phase of the fibrotic response, macrophages can take on an anti-inflammatory, M2-like phenotype and lead to inflammatory resolution [34]. These cells secrete IL-10, arginase, and TGF-β, cytokines that act to inhibit M1 macrophage polarization and Th1 lymphocyte activity. M2-like macrophages also produce a variety of pro-fibrotic cytokines, MMPs, and TIMPs that will be discussed in more depth in a later section. The complexity of microenvironments can result in the generation of multiple subpopulations of macrophages leading to a complex balance pro- and anti-inflammatory signaling.
During subsequent stages of cardiac inflammation, T lymphocytes are recruited to the injury site and differentiated by IL-12 into Th1 cells, IL-6 and TGF-β into Th17 cells, TGF-β alone into Treg cells, and IL-4, IL-25, and IL-33 into Th2 cells [35]. T lymphocytes can help to sustain and promote (Th1 and Th17 cells) or resolve (Treg and Th2 cells) inflammation [36]. Th1 cells secrete IFN-γ, TNF-α, and IL-2, cytokines that lead to positive feedback to maintain the proinflammatory phenotypes of M1-like macrophages, neutrophils, cardiomyocytes, and fibroblasts. Th17 cells produce the cytokine IL-17A, which is associated with increased and sustained neutrophil activation [37]. These pro-inflammatory T-cells are counteracted as Treg and Th2 cells are recruited. These anti-inflammatory T-cells secrete TGF-β, IL-4, and IL-13, pro-fibrotic cytokines that inhibit Th1 and Th17 cell differentiation and promote polarization of macrophages toward an M2-like phenotype [38]. The recruitment of these anti-inflammatory cells is essential to resolving the inflammatory cascade and preventing further fibrosis. For a more in-depth review of inflammatory signaling in cardiac fibrosis, please refer to Smolgovsky et al [39].
Fibrotic Signaling in Cardiac Fibrosis
In addition to their roles regulating the inflammatory status of injured cardiac tissues, each of these cell types signal to fibroblasts to migrate to the site of injury and transdifferentiate into secretory, contractile cells termed myofibroblasts that become the primary drivers of cardiac fibrosis. Myofibroblasts are defined by their expression of contractile stress fibers, expression of α-smooth muscle actin (α-SMA), and secretion of an abundance of ECM components [20]. These cells, though predominantly from resident, interstitial fibroblasts, originate from a variety of sources including circulating fibrocytes, endothelial cells, epithelial cells, and pericytes [40–42]. Regardless of their origin, the differentiation of cells into myofibroblasts in the injured heart requires several key factors [20]. First, TGF-β signaling through the Smad2/3 signaling cascades promotes α-SMA transcription. Second, expression of cell surface receptors that can promote growth factor signaling cascades leads to increased myofibroblast activation. Next, alterations to the mechanical properties and composition of the ECM mediates differentiation of fibroblasts into myofibroblasts by modulating their responses to mechanical and growth factor signaling. Last, direct mechanical stimulation of myofibroblasts results in the maintenance of the myofibroblast phenotype through RhoA signaling pathways. The first two of these requirements originate from signals from the cell types previously mentioned as they take on a second, pro-fibrotic role.
While initially recruited to the injury site to remove damaged cells and cellular debris, neutrophils directly and indirectly promote fibrosis. As previously mentioned, neutrophils release MMPs, elastase, and cathepsins that initially help to breakdown and reorganize the ECM so it can be replaced [29]. This breakdown can release latent fibroblast growth factors sequestered in the ECM and directly activate fibrotic pathways. Further, neutrophils release large amounts of reactive oxygen species through NADPH oxidase in a process referred to as the respiratory burst [43]. Reactive oxygen species directly aid in myofibroblast differentiation through the Smad2/3, MAPK, JNK, and Src pathways to activate signaling intermediates within proto-myofibroblasts, myofibroblasts who have formed stress fibers but do not yet express α-SMA [44]. All of these signaling cascades induce the transcription of target genes (i.e., α-SMA, fibrillar collagens, and fibronectin) that are necessary for ECM deposition and myofibroblast activation. Recent work from Daseke et al. also suggests that neutrophils alter their signaling pathways during myocardial infarction such that they also secrete fibronectin and fibrinogen at later stages of injury and directly aid in ECM deposition [45]. Neutrophils also play an indirect role in inducing a fibrotic response by further activating the macrophage response and inducing a switch from pro-inflammatory to pro-fibrotic macrophages when they are phagocytized by M1-like macrophages [46].
Macrophages are functionally plastic, thus their role in regulating fibrosis is complex and dependent on the microenvironment where they reside. These cells are the primary source of several MMPs and TIMPs [47]. As previously mentioned, the MMPs and TIMPs that are most directly related to the pro-fibrotic response tend to be produced by M2-like macrophages. The pro-fibrotic MMP-9 is of particular importance in cardiac fibrosis as its inhibition reduces fibrosis in dilated cardiomyopathy and myocardial infarction, seemingly by inhibiting MMP-9’s activation of TGF-β1 [48]. TGF-β is the best characterized pro-fibrotic growth factor in chronic fibrotic diseases [49]. Of the three TGF-β isoforms, TGF-β1 is the most relevant and abundant in cardiac injury [34,50]. TGF-β neutralization prevents cardiac fibrosis and improves diastolic dysfunction in pressure-overloaded rats [51]. Further, TGF-β1 knockout mice, though exhibiting other disease phenotypes, present with markedly reduced collagen deposition [52]. M2-like macrophages produce large amounts of TGF-β that then binds to TGF-β receptors on fibroblasts leading to the potent activation of myofibroblast differentiation and enhanced synthesis of ECM proteins through the Smad family of transcription factors. Further, TGF-β1 induces the expression of protease inhibitors in myofibroblasts and macrophages resulting in the preservation of the remaining ECM. TGF-β1 can also promote collagen synthesis through Smad-independent pathways through its activation of JNK/MAPK protein kinase cascades [53]. M2-like macrophages in later stages of the fibrotic response produce other pro-fibrotic cytokines such as IL-4, IL-13, and TNF-α [54]. IL-4 and IL-13 lead to further activation of myofibroblasts via STAT6, AKT, and MAPK pathways through which they promote collagen production [55]. These cytokines also potently induce several chemokines (e.g., CCL2/MCP-1 and CCL3) that can recruit fibroblasts to the site of injury [56]. TNF-α secretion by macrophages can promote expression of TGF-β1 and recruit more inflammatory cells resulting in an increased and sustained fibrotic response [57].
Much like macrophages, the pro-fibrotic role of T cells in cardiovascular disease is context-dependent. Th2 cells are characterized by the secretion of IL-4, IL-5, and IL-13, pro-fibrotic cytokines that lead to the phenotypic shift of fibroblasts to myofibroblasts [38]. IL-13 also induces TGF-β expression in macrophages and inhibits MMP synthesis by fibroblasts leading to reduced matrix degradation and excessive collagen deposition [58]. The role of Th1 cells in promoting fibrosis seems to be through IFN-γ recruitment of other inflammatory cells, though IFN-γ can also increase expression TNF-α which promotes maturation of pro-fibrotic, M2-like macrophages [59,60]. Th17 cells are essential for the development and progression of cardiac fibrosis [61]. Their expression of IL-17 causes the proliferation of cardiac fibroblasts and, in parallel, induces expression of MMP-1 and MMP-2, both of which cleave fibrillar collagens and aid in turnover of collagens I and III [62]. Treg cells release IL-10, which can have a dual role in the development of fibrosis. IL-10 inhibits collagen synthesis in cardiac fibrosis via a reduction in STAT3 signaling; however, prolonged expression of IL-10 can promote fibrocyte recruitment and M2-like macrophage activation [63,64].
B-cells, while not as well studied in the context of cardiac fibrosis, produce pro-fibrotic cytokines IL-6, CCL2, TNF-α, and TGF-β in addition to B-cell-activating factor (BAFF) [18]. IL-6 is predominantly known for its pro-inflammatory role as it promotes the infiltration, migration, and polarization of macrophages. However, IL-6 also induces the conversion of fibroblasts into myofibroblasts suggesting that this cytokine can directly result in fibrosis [65]. BAFF induces the production of collagens, TIMPs, MMP-9, and α-SMA in fibroblasts [66]. While B-cells seem to play a role in promoting cardiac fibrosis, the extent of their importance in this pathology requires further study.
In addition to the effects of the aforementioned cell types on myofibroblast differentiation, myofibroblasts also secrete cytokines that act in a paracrine manner to augment the fibrotic process. Activated myofibroblasts secrete IL-6, IL-1β, IL-10, TNF-α, CCL2, and TGFβ, all of which directly promote the fibrotic response by promoting myofibroblast proliferation and activation [67]. Further, these cells secrete cytokines ROS and TNF-α that can maintain the inflammatory response and prolong the injury response through the previously discussed mechanisms.
Cellular Senescence in Cardiac Fibrosis
The aforementioned pro-inflammatory and pro-fibrotic mechanisms are complicated when considered in the context of aging and cellular senescence. While not specifically discussed here, the signaling mechanisms related to fibrosis in the aging heart are largely similar to those in cardiovascular diseases though they are complicated with the addition of cellular senescence. For detailed reviews on cardiac fibrosis in aging, please refer to Shimizu and Minamino [68], Lu et al. [69], and Biernacka and Frangogiannis [70].
Cellular senescence is the stable state of cell cycle arrest resulting in cellular resistance to growth stimuli-induced proliferation that is associated with age. Senescence can be caused by either telomere attrition during cell division leading to accumulated DNA damage or through various external and internal stress signals including oxidative stress, metabolic stress, and constitutive activation of mitogenic stimuli [68]. These signals trigger activation of signaling pathways, primarily the p53/p21 and p16 signaling pathways, and the expression of other senescence-associated markers such as the pro-inflammatory senescence-associated secretory phenotype and MAPK signaling activation. The p53 and p16 signaling pathways are broadly responsible for cell cycle arrest, DNA damage repair, and apoptosis [71]. Senescent cells produce a variety of pro-inflammatory cytokines (e.g., IL-6, IL-1β, IL-13), chemokines (e.g., CCL8 and CCL13), MMPs/TIMPs, reactive oxygen species, and ECM components (e.g., collagens, fibronectin, laminin) as part of the senescence-associated secretory phenotype [71]. Because these components are involved in both the pro-inflammatory and pro-fibrotic pathways described above, the role of cellular senescence in the progression of cardiac fibrosis is complex.
While we know cellular senescence is broadly associated with cardiac disease and cardiac fibrosis, the relationship does not seem to be simple. Humans with end-stage heart failure have increased p53 levels and markers of apoptosis in the myocardium [72]. Further, p53 expression is also elevated in patients with hypertrophic cardiomyopathy and dilated cardiomyopathy compared to the non-failing heart [73,74]. Elevated p53 in the myocardium has also been associated with the suppression of angiogenesis, tissue hypoxia, and cardiac dysfunction [75]. However, studies in cardiac fibroblasts have shown that increased p16 suppresses age-induced cardiac fibrosis [76]. The reduction of senescence-associated markers ataxia telangiectasia mutated, p53, and p21 enhances fibrosis in the non-infarct area following myocardial infarction [77]. These studies suggest that while cardiomyocyte senescence may result in increased fibrosis through increasing pro-inflammatory and pro-fibrotic signaling in other cell types, cardiac fibroblast senescence may actually be beneficial.
III. Biomechanical Mechanisms of Cardiac Fibrosis
The fibrotic response is regulated at the site of cardiac injury through the complex signaling within and between the previously discussed cell types. These biochemical mechanisms that promote fibrosis are further complicated when considered with the mechanical environment in which the cells reside. The pumping function of the heart requires that cardiac tissue maintain enough stiffness to resist its mechanical load yet be compliant enough to contract [78]. To balance these mechanical needs, fibroblasts must respond to changes in load by increasing production of ECM components, MMPs, and TIMPs as necessary. In cardiac disease, the previously mentioned biochemical signals induce tissue remodeling and ECM degradation. This loss of ECM, as well as pressure overload, results in an increase in diastolic strain [9]. In response to pathological strains, aortic valve interstitial cells and cardiac fibroblasts proliferate and increase expression of MMPs and α-SMA [11,79–81]. The expression of these proteins allows for increased cardiac fibroblast migration into the injured region and contraction of 2D and 3D substrates in vitro that contributes to the fibrotic response [82,83]. Cardiac fibroblasts subjected to increased cyclic strain also increase production of ECM components and TGF-β which, as previously discussed, strongly promotes further pro-fibrotic signaling [84].
In later stages of cardiac fibrosis, fibroblasts experience increased substrate stiffness due to the deposition and cross-linking of ECM components. In 2D culture systems, stress fiber formation in fibroblasts is prevented on very soft (1–3 kPa) substrates [85]. Progressively increasing substrate stiffness promotes myofibroblast differentiation such that α-SMA negative stress fibers form in fibroblasts on stiffer (3 kPa) substrates and very stiff (>20 kPa) substrates are required for α-SMA positive myofibroblasts to completely differentiate [86]. In addition to α-SMA, cardiac myofibroblasts increase expression of TGF-β1 and collagen I on stiff substrates demonstrating that myofibroblasts respond to increased substrate stiffness by exacerbating the fibrotic phenotype [87,88]. Further, myofibroblasts increase internal cellular tension by contracting to match the increased extracellular tension of the ECM [89]. This contraction results in a pulling force on both the ECM and surrounding cells because myofibroblasts are bound to both the ECM, cardiomyocytes, and other myofibroblasts. Thus, myofibroblast contraction can increase the substrate stiffness of nearby cells leading to a progressive and pathological fibrotic phenotype.
Tissue mechanics are sensed via mechanosensitive adhesion proteins that are incorporated into focal adhesions and adherens junctions, the physical tethers that, respectively, link cells to the ECM and to other cells. These mechanosensors include the integrin and cadherin protein families with the specific isoform of each family’s members being associated with specific cellular and disease phenotypes. Integrins are transmembrane glycoproteins consisting of α- and β-subunits that bind to the ECM and recruit other focal adhesion proteins that bind to the actin cytoskeleton, providing a direct mechanical link between stress fibers and ECM proteins [90]. Through focal adhesions, fibroblasts periodically pull on the ECM to sample the stiffness of their microenvironment via actomyosin contractions [91]. On soft matrices, the matrix resistance is not sufficient to deform focal adhesion proteins, thus fibroblasts remain in a quiescent state. Stiff matrices, however, resist this pulling force and allow for mechanical deformation of focal adhesion proteins resulting in activation of stretch-dependent signaling pathways.
The intracellular tails of integrin subunits bind to signaling proteins such that the combination of integrin subunits determines both ECM binding specificity and downstream intracellular signaling [92]. Thus, the precise pathways activated during cardiac fibrosis is a product of which integrin subunits are present. Others have provided detailed reviews on the role of specific integrins in regulating cardiac fibrosis, thus we will focus on the signaling mediators activated downstream of focal adhesions [92–94]. One signaling mediator of particular importance in cardiac fibrosis is focal adhesion kinase (FAK), a tyrosine kinase that colocalizes with integrins through its association with other focal adhesion proteins [80,95]. This protein is activated by cyclic stretch and activates the AKT and MAPK pathways to promote myofibroblast differentiation [80,96]. However, FAK’s role in cardiac fibrosis is complicated as activation of FAK through FGF-2 signaling prevents myofibroblast differentiation and reverses TGF-β induced α-SMA expression [95,97]. Silencing of FAK by siRNA or pharmacologically blocking this protein attenuates fibrosis, collagen content, and MMP-2 activity in pressure overload cardiac hypertrophy, thus FAK can broadly be considered as a pro-fibrotic mediator though this activity is context-dependent [98].
An additional pathway activated downstream of focal adhesions is the RhoA small GTPase activation of SRF/MRTF-A transcription factors. In healthy conditions, G-actin binds to and sequesters MRTF-A in the cytoplasm [99]. RhoA activation promotes actin stress fiber formation and stabilization leading to the release of MRTF-A as G-actin is recruited to form these filaments [100]. Once released, MRTF-A translocates to the nucleus where it pairs with SRF and transcriptionally activates genes needed for myofibroblast differentiation and migration to further the fibrotic response. MRTF-A deficient mice with isoproterenol-induced myocardial stress or myocardial infarction develop reduced fibrosis and a smaller scar signifying the importance of this transcriptional activation for the development of cardiac fibrosis [101].
As previously mentioned, TGF-β is a primary mediator of fibrotic disease and can be sequestered in a latent form in the ECM. TGF-β is non-covalently linked to the ECM through a protein complex recognized by αv integrins referred to as the large latent complex [102–104]. With increasing traction forces as myofibroblasts become contractile in the context of a stiff ECM, a conformational change occurs in the latency associated peptide within the latent complex leading to the release of active TGF-β that promotes myofibroblast differentiation and ECM deposition. Blocking αvβ3 and αvβ5 integrins in cardiac fibroblasts prevents activation of latent TGF-β and myofibroblast differentiation in vitro suggesting this pool of TGF-β is important for the fibrotic response.
In addition to the signaling pathways originating from outside the cell, integrins can participate in inside-out signaling whereby focal adhesion proteins activate integrins by modifying their intracellular domains resulting in strengthened ECM adhesions. Vinculin is a focal adhesion protein that strengthens and stabilizes focal adhesions under high tension [105–107]. Activated vinculin also recruits FAK to further activate fibrotic signaling [105]. Through these focal adhesion mediated pathways, myofibroblasts increase expression of α-SMA positive stress fibers and become more contractile creating a positive feedback loop.
In addition to the cell-ECM mechanosensing pathways, cells also interact with each other through adherens junctions. The mechanosensitive protein mediating cell-cell interactions are cadherins, a family of transmembrane proteins linked to actin stress fibers that bind to cadherins of the same type on adjacent cells [108,109]. When myofibroblasts increase their intracellular forces, adherens junctions allow for the cells to mechanically activate signaling pathways in neighboring cells by physically pulling on homophilic cadherin dimers. Cadherins will be discussed in the next section with a special focus on cadherin-11, so the focus here is the signaling pathways downstream of cadherin activation.
Cadherins are linked to multiple intracellular pathways that promote myofibroblast differentiation and cardiac fibrosis [110–113]. β-catenin is a dual-function protein that both links cadherins to the actin cytoskeleton and acts as a transcription factor when activated along with TGF-β or Wnt signaling pathways to promote a mesenchymal cell phenotype [114]. β-catenin binds to TGF-β and Wnt signaling genes as well as ECM genes in cardiac fibroblasts, thus its activation through adherens junctions can promote an increased fibrotic response in a positive feedback loop [115]. In addition to activation of β-catenin signaling pathways, cadherins act through SRF/MRTF-A to increase expression of α-SMA promoting myofibroblast differentiation and through MAPK to induce expression of IL-6 and MMPs further promoting inflammation [114,116]. Cadherin engagement can also lead to vinculin recruitment to adherens junctions by α-catenin [117–119]. This recruitment stabilizes the intercellular adhesion and increases cell contractility which can further activate adherens junction signaling. For more information on the role of mechanobiology in the development of cardiac fibrosis, please see the review by Herum et al [120].
IV. Cadherin-11 in Fibrosis
As previously mentioned, classical cadherins regulate calcium-dependent, cell-cell interactions at adherens junctions [108,109]. These proteins are most well known for their roles in cellular migration and adhesion in embryonic development as well as providing solid tissues with mechanical and functional integrity in adulthood. Classical cadherins share a common protein structure consisting of five extracellular domains, a single-pass transmembrane domain, and a cytoplasmic tail domain [121]. This family of cadherins is subdivided into two types based on the presence of a histidine-alanine-valine (HAV) tripeptide motif and one, conserved, N-terminal tryptophan (type I) or the lack of an HAV motif and two, conserved, N-terminal tryptophans (type II) [122]. The extracellular domain of type II classical cadherins dimerize in a homophilic manner between neighboring cells (in trans) via the two tryptophans (W2 and W4) in the EC1 domain. Intracellularly, the cytoplasmic tail binds to β-catenin, which in turn binds to α-catenin (Figure 2) [123]. α-catenin interacts directly with the filamentous actin cytoskeleton, which helps to provide tension and stability for maintenance of these intercellular junctions.
Figure 2:
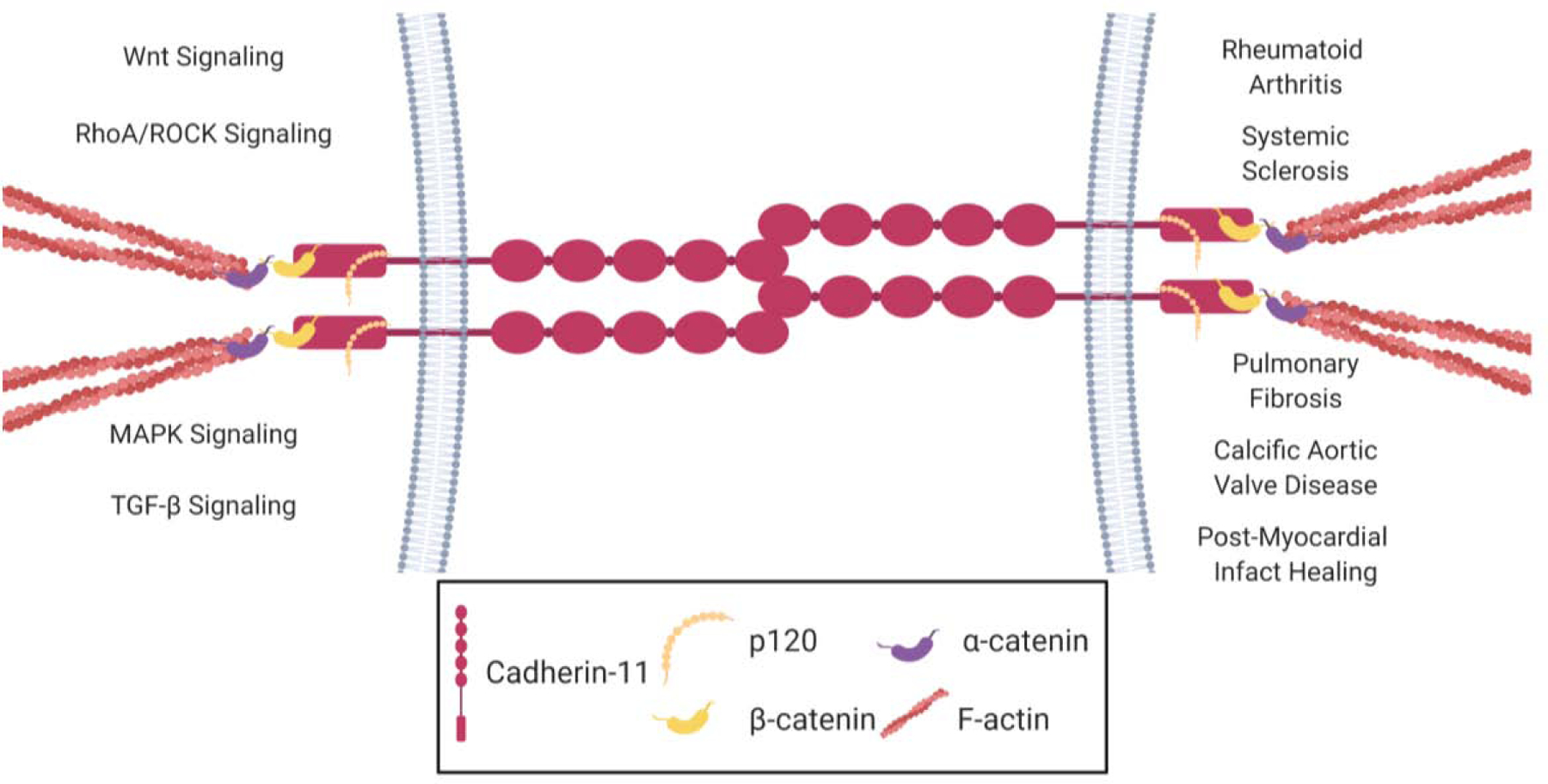
Cadherin-11’s role in signaling pathways and disease. Cadherin-11 forms homophilic dimers between neighboring cells. These intercellular linkages aid in transmitting mechanical signals between cells to their respective cytoskeletons by binding to p120 catenin and β-catenin. β-catenin binds to α-catenin which is linked to the actin cytoskeleton. Through these physical tethers, cadherin-11 transmits mechanical signals to molecular signaling pathways in the cells (left) that are associated with fibrotic diseases (right). Created with BioRender.com
Cadherin expression is differentially regulated during development and disease leading to strengthened or weakened cell-cell adhesion and differential activation of cadherin-associated signaling pathways [124]. The primary cadherin expressed in quiescent fibroblasts in the heart is cadherin-2 (CDH2 or N-Cadherin) whose expression is associated with increased β-catenin stability and low expression of α-SMA in aortic valve interstitial cells and stromal cells in ischemic heart injury [125,126]. During myofibroblast differentiation, the predominant cadherin expressed switches from cadherin-2 to cadherin-11, which can withstand two-fold higher forces than N-cadherin allowing for stronger contractile forces and transmission of higher intracellular tension [127–130].
Cadherin-11 (CDH11 or OB-Cadherin) is a type II classical cadherin that was first identified in osteoblasts but has since been noted in other cells of mesenchymal origin [131]. While this protein was originally studied in the context of tissue development and morphogenesis, cadherin-11 has more recently been recognized as a driver of the myofibroblast phenotype and regulator of fibroblast inflammation [127,129,132–134]. These findings have uncovered this protein’s regulatory role in fibroblast mediated diseases such as pulmonary fibrosis [130], rheumatoid arthritis [112], systemic sclerosis [135], metastatic cancer [136], and kidney fibrosis [137] as well as fibrotic diseases of the cardiovascular system, calcific aortic valve disease [128,138,139], and myocardial infarction [93] (Figure 2).
Cadherin-11 is upregulated during myofibroblast differentiation through TGF-β1 signaling concurrently with a decrease in expression of cadherin-2 [127]. This cadherin switch promotes myofibroblast invasion [140]. Further, cadherin-11 expression has been observed on TGF-β producing macrophages [113]. Since cadherin-11 is involved in cell migration, its expression in macrophages raises the possibility that this protein both recruits inflammatory cells to the injury site and maintains these cells in close proximity to promote the profibrotic niche. Cadherin-11 is also the only cadherin known to participate in focal adhesions where it promotes cell-substrate adhesion [141]. In addition to its mechanical role in strengthening adherens junctions and focal adhesions, cadherin-11 also activates intracellular signaling pathways to influence cellular behavior. Cadherin-11 engagement activates SRF through the RhoA pathway leading to increased expression of α-SMA and cadherin-11 and also promotes differentiation of smooth muscle cells by upregulating expression of TGF-β1 [142]. Likewise, overexpression of cadherin-11 in fibroblasts increases expression of Wnt and β-catenin and extracellular matrix components [143,144]. Thus, cadherin-11 expression and engagement promotes differentiation of myofibroblasts and activates downstream pathways that further serve to strengthen and stabilize cadherin-11 containing adherens junctions. Additionally, cadherin-11 engagement promotes IL-6, MCP-1, and MMP-1 expression in synovial fibroblasts which could further promote inflammatory invasion in fibrotic diseases.[133,145] Altogether, cadherin-11 expression promotes the differentiation of myofibroblasts and modulates both the mechanical and inflammatory pathways associated with fibrotic diseases.
Cadherin-11 expression is highly expressed in the endothelial (VECs) and interstitial cells (VICs) of diseased heart valves and in the non-cardiomyocyte cells of the infarcted heart [93,128,138,139]. In the diseased aortic valve, interstitial cells take on a myofibroblast phenotype and mediate the structural remodeling that leads to aortic stenosis [20,146–148]. In these cells, cadherin-11 is upregulated downstream of TGF-β through both the Smad2/3 and MAPK signaling pathways and this upregulation occurs both during development then again in aortic valve pathogenesis [149]. Further, cadherin-11 expression regulates dystrophic calcific nodule formation in aortic valves, a process that follows an increase in myofibroblast contractility [128]. Our group has more recently shown that cadherin-11 knockout mice have reduced aortic valve calcification and decreased expression of myofibroblast and inflammatory markers including IL-6 [134]. Further, cadherin-11 was shown to regulate VIC contractility through its association with focal adhesions. Other groups have supported cadherin-11’s role in promoting myofibroblast differentiation and subsequent calcification in valve disease [138,139,150]. Overexpressing cadherin-11 in adult mouse aortic valves leads to increased ECM components, numbers of focal adhesions, and valve calcification [150]. Further, cadherin-11 overexpression in VICs increases cell migration, adhesion, and stress fiber bundles suggesting that expression of this protein promotes the myofibroblast phenotype. Our group has also shown that these markers of aortic valve disease can be prevented through blocking cadherin-11 with a monoclonal antibody (SYN0012) in the Notch1+/− mouse model of this disease [151]. Along with cadherin-11’s known role in strengthening adherens junctions, these findings suggest that cadherin-11 broadly regulates the fibrotic response in calcific aortic valve disease.
While less is known about cadherin-11’s role in the myocardium, it is expressed in cardiac fibroblasts [152]. This expression and the cadherin-11’s regulation of a broad range of fibrotic diseases led our group to recently study the role of this protein in the fibrotic response to myocardial infarction [93]. Following myocardial infarction, cadherin-11 expression increased nearly 10-fold in non-cardiomyocyte cells of the heart and was particularly high among infiltrating M2-like macrophages. This increase following myocardial infarction is transient as cadherin-11 expression increases by day-3 post-infarct, stays elevated through 2-weeks post-infarct, but returns back to baseline expression when scar formation is stable 4-weeks post-infarct [23]. Importantly, outcomes following myocardial injury were improved when wild-type mice given cadherin-11 null bone marrow. Further, treating wild-type mice with SYN0012 also improved cardiac function following myocardial infarction by limiting transcription and secretion of IL-6. In angiotensin II-induced fibrosis, cadherin-11 expression seems to remain elevated in matrifibrocyte-like, fibrosis-associated cell populations suggesting that this protein could play a prolonged role in stress-associated cardiac fibrosis [24]. Cadherin-11 has also been described as senescence-responsive whereby its expression is suppressed in senescent endothelial cells and this suppression is greater when senescent cells are under shear stress [153]. p53 is also suppressed downstream of cadherin-11 in a STAT3-dependent manner [154]. This senescence responsiveness and downregulation of p53 downstream of cadherin-11 suggests that a reciprocal relationship exists between cadherin-11 and senescence-associated signaling pathways. Thus, cadherin-11 expression may prevent the beneficial effects of cardiac fibroblast senescence. Through these findings and the findings in the context of aortic valve disease, cadherin-11’s role in regulating cardiac fibrosis through the biochemical and biomechanical signaling pathways that drive this pathology are becoming clear.
V. Targeting Cadherin-11 in Disease Models
As we have discussed, fibrotic diseases originate as a consequence of chronic inflammation and biomechanical changes leading to prolonged activation of myofibroblasts, yet most available therapies have focused on targeting molecules with central roles in innate and/or adaptive immunity. While these therapies prevent chronic fibrosis with some efficacy, the clinical success of immunomodulatory therapies in cardiac disease is still limited as prolonged immune suppression leads to severe side effects and they fail to address the concurrent mechanical signals that aid in the initiation and progression of fibrotic disease [155]. Given that cadherin-11 is a known regulator of mechanical signaling pathways whose expression increases in fibrotic diseases and given this increased expression is associated with altered inflammatory pathways, this protein seemingly serves as a link between the biomechanical, pro-fibrotic, pro-inflammatory, and senescence signaling pathways. Therefore, cadherin-11 may serve as an ideal therapeutic target in fibrotic disease. This idea has gained support through experiments targeting cadherin-11 with monoclonal antibodies blocking cadherin-11 homotypic bond formation. Treatment with this antibody prevents disease phenotypes in a variety of fibrotic diseases. Specifically, reducing cadherin-11 activity either through genetic knockout or with an anti-cadherin-11 antibody is effective in reducing symptoms in animal models of systemic sclerosis [135], rheumatoid arthritis [112], pulmonary fibrosis [130], calcific aortic valve disease [128,138,139], and improving outcomes following myocardial infarction [93]. These findings were so compelling that the humanized form of this antibody underwent clinical trials for rheumatoid arthritis [156]. These trials were ended, however, due to the drug not providing an increased response compared to TNF-α inhibitors, the current standard of care. Further, the progressive nature of cardiac fibrosis makes administration of an anti-cadherin-11 antibody as a preventative measure difficult as it would require patients to receive monthly injections for an unspecified period of time. Nevertheless, preclinical data suggests that targeting cadherin-11 could be beneficial for diseases that are accompanied by cardiac fibrosis.
As a type II classical cadherin, the method of targeting cadherin-11 that seems effective for preventing fibrotic disease is to block homophilic bond formation via the EC1 domains of these proteins (Figure 3A). As previously mentioned, this binding event requires two tryptophans (W2 and W4) found within a hydrophobic pocket of this domain (Figure 3B and 3C), thus competitive inhibition of homophilic dimerization likely occurs through blocking the availability of these residues. SYN0012, the CDH11-blocking monoclonal antibody, acts through this mechanism as does celecoxib, a cyclooxygenase-2 (COX2) inhibitor and nonsteroidal anti-inflammatory drug used to treat pain and inflammation, and dimethyl celecoxib, an analog of celecoxib that does not inhibit COX2 [112,157]. Celecoxib and dimethyl celecoxib both have a benzene sulfonamide backbone that interact with tryptophan 4 and histidine 25 within this hydrophobic pocket through intermolecular hydrogen bonds [158]. The binding of small molecules to these residues prevents the dimerization of CDH11. The finding that these drugs binds to CDH11 seems to have clinical relevance as well since celecoxib is used to treat rheumatoid arthritis, though its translatability into cardiovascular diseases seems to be more complicated. Our group recently showed that celecoxib promotes aortic stenosis compared to ibuprofen and naproxen controls in human patients [159]. Further, celecoxib induces a myofibroblast phenotype in aortic valve interstitial cells in vitro and promotes dystrophic calcification; however, dimethyl celecoxib prevented the onset of these fibrotic events. Thus, dimethyl celecoxib may be better suited for cardiovascular fibrosis though further research is needed on this topic. Vaidya et al. extended upon these findings by showing that celecoxib and dimethyl celecoxib can both induce osteogenic aortic valve calcification in ex vivo porcine aortic valve leaflets in a glucocorticoid-dependent manner [160]. These findings suggest that any cadherin-11 binding small molecules may be effective at treating cardiac fibrosis, however, these molecules should be evaluated for off target risks in the presence of glucocorticoids prior to approval.
Figure 3:
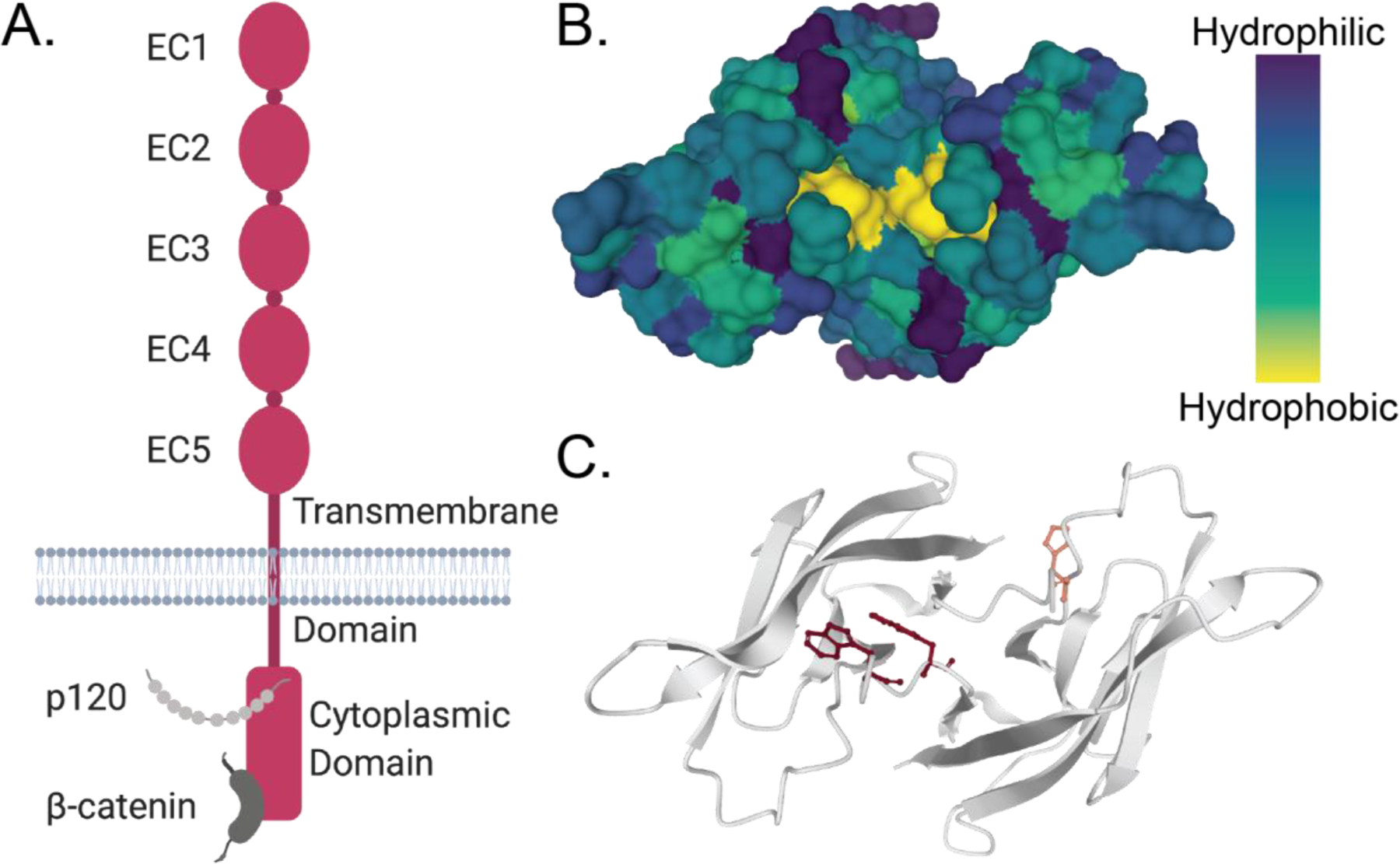
Targeting cadherin-11 to prevent homodimer formation. A. Cadherin-11 is composed of five extracellular (EC) domains, a transmembrane domain, and a cytoplasmic domain. EC1 and EC2 allow cadherin-11 to dimerize while the rest of the protein transmits physical forces for cellular signaling. B. The EC1 domains of a cadherin-11 dimer contain a hydrophobic pocket that be targeted by small molecules to inhibit dimer formation. C. The amino acids that, when blocked, effectively inhibit cadherin-11 dimerization are part of this hydrophobic pocket. These residues should be considered when targeting cadherin-11 with drugs to prevent cadherin-11 mediated disease. Image created using Mol*.[161]
VI. Conclusions
A healthy heart requires maintenance of a healthy ECM to transmit force throughout the tissue and regulate biochemical and biomechanical signaling pathways. Most cardiovascular diseases are accompanied by cardiac fibrosis where ECM accumulation helps to stabilize the tissue while remodeling and/or scar formation occurs; however, the limited regenerative capacity of cardiac tissue often results in excessive ECM deposition leading to systolic dysfunction, diastolic dysfunction, and arrhythmias. Cardiac fibrosis results from a complex interplay of pro-inflammatory, pro-fibrotic, and biomechanical signaling; however, current therapies do not target each of these components of the fibrotic response. Cadherin-11 is a component of focal adhesions and adherens junctions in myofibroblasts and regulates fibroblast inflammation. Preventing homodimerization of this protein effectively prevents fibrotic diseases, including calcific aortic valve disease, and leads to better outcomes following myocardial infarction. While more research is needed to uncover its importance in other types of cardiac fibrosis, cadherin-11 may provide a therapeutic target to effectively treat cardiac fibrosis as this protein acts a link between inflammatory, fibrotic, and biomechanical causes of fibrosis.
Highlights:
Fibrotic diseases are caused by persistent inflammatory, fibrotic, and mechanical signaling.
While myofibroblasts are the effector cells that secrete extracellular matrix components, inflammatory cells are necessary for initiating and sustaining fibrotic remodeling.
Biomechanical signaling is a primary factor generating cardiac fibrosis.
Cadherin-11 engagement increases pro-inflammatory and pro-fibrotic mediators of fibrosis and results in extracellular matrix deposition.
Blocking cadherin-11 dimerization holds potential as a therapeutic target for cardiac fibrosis.
Acknowledgements
This work is supported by the National Institutes of Health [grant numbers: GM007569, HL135790, and HL154596]. Deposited into PMC after 12 months.
Abbreviations
- ECM
Extracellular matrix
- CDH11
cadherin-11
- CDH2
cadherin-2
- MMP
matrix metalloproteinase
- TIMP
tissue inhibitors of metalloproteinase
- F-SH
fibroblast Sca1-high
- F-SL
fibroblast Sca1-low
- DAMP
damage-associated molecular pattern
- TLR
toll-like receptor
- TNF-α
tissue necrosis factor α
- IFN-γ
interferon γ
- IL
interleukin
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- AP-1
activator protein 1
- MCP-1/CCL2
monocyte chemoattractant protein-1/chemokine ligand 2
- CCL8
chemokine lingand 8
- CCL13
chemokine lingand 13
- CCR2
chemokine receptor 2
- TGF-β
transforming growth factor β
- α-SMA
α smooth muscle actin
- Smad2/3
Mothers against decapentaplegic homolog 2/3
- RhoA
Ras homolog family member A
- MAPK
mitogen-activated protein kinase
- JNK
c-Jun N-terminal kinase
- Src
proto-oncogene tyrosine-protein kinase Src
- STAT
signal transducer and activator of transcription
- Akt
protein kinase B alpha
- BAFF
B-cell activating factor
- ROS
reactive oxygen species
- FAK
focal adhesion kinase
- FGF-2
basic fibroblast growth factor
- SRF
serum response factor
- MRTF-A
myocardin-related transcription factor A
- VEC
valve endothelial cells
- VIC
valve interstitial cells
- COX2
cyclooxygenase 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing interests.
VII. References
- [1].Frantz C, Stewart KM, Weaver VM, The extracellular matrix at a glance, J. Cell Sci (2010). 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Murtha LA, Schuliga MJ, Mabotuwana NS, Hardy SA, Waters DW, Burgess JK, Knight DA, Boyle AJ, The processes and mechanisms of cardiac and pulmonary fibrosis, Front. Physiol (2017). 10.3389/fphys.2017.00777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dixon IMC, Wigle JT, Cunnington RH, Rattan SG, Cardiac Fibrosis and Heart Failure—Cause or Effect?, Card. Fibros. Hear. Fail. Cause or Eff (n.d.) 1–4. 10.1007/978-3-319-17437-2_1. [DOI] [Google Scholar]
- [4].Ouzounian M, Lee DS, Liu PP, Diastolic heart failure: Mechanisms and controversies, Nat. Clin. Pract. Cardiovasc. Med (2008). 10.1038/ncpcardio1245. [DOI] [PubMed] [Google Scholar]
- [5].Burlew BS, Weber KT, Cardiac fibrosis as a cause of diastolic dysfunction, Herz (2002). 10.1007/s00059-002-2354-y. [DOI] [PubMed] [Google Scholar]
- [6].Harvey PA, Leinwand LA, Cellular mechanisms of cardiomyopathy, J. Cell Biol (2011). 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rosker C, Salvarani N, Schmutz S, Grand T, Rohr S, Abolishing myofibroblast arrhythmogeneicity by pharmacological ablation of α-smooth muscle actin containing stress fibers, Circ. Res (2011). 10.1161/CIRCRESAHA.111.244798. [DOI] [PubMed] [Google Scholar]
- [8].Vasquez C, Benamer N, Morley GE, The cardiac fibroblast: Functional and electrophysiological considerations in healthy and diseased hearts, J. Cardiovasc. Pharmacol (2011). 10.1097/FJC.0b013e31820cda19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Banerjee I, Yekkala K, Borg TK, Baudino TA, Dynamic interactions between myocytes, fibroblasts, and extracellular matrix, in: Ann. N. Y. Acad. Sci, 2006. 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- [10].Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML, Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation, Circ. Res (2013). 10.1161/CIRCRESAHA.111.300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Davis J, Molkentin JD, Myofibroblasts: Trust your heart and let fate decide, J. Mol. Cell. Cardiol (2014). 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JWK, Nordon RE, Harvey RP, Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury, Elife (2019). 10.7554/eLife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Turner NA, Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs), J. Mol. Cell. Cardiol (2016). 10.1016/j.yjmcc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- [14].Christia P, Frangogiannis NG, Targeting inflammatory pathways in myocardial infarction, Eur. J. Clin. Invest (2013). 10.1111/eci.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].García-Rodríguez C, Parra-Izquierdo I, Castaños-Mollor I, López J, San Román JA, Crespo MS, Toll-like receptors, inflammation, and calcific aortic valve disease, Front. Physiol (2018). 10.3389/fphys.2018.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM, Dual Activation of TRIF and MyD88 Adaptor Proteins by Angiotensin II Evokes Opposing Effects on Pressure, Cardiac Hypertrophy, and Inflammatory Gene Expression, Hypertension (2015). 10.1161/HYPERTENSIONAHA.115.06011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roh JS, Sohn DH, Damage-associated molecular patterns in inflammatory diseases, Immune Netw (2018). 10.4110/in.2018.18.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Van Linthout S, Miteva K, Tschöpe C, Crosstalk between fibroblasts and inflammatory cells, Cardiovasc. Res (2014). 10.1093/cvr/cvu062. [DOI] [PubMed] [Google Scholar]
- [19].Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC, Cardiac fibrosis: The fibroblast awakens, Circ. Res (2016). 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G, The myofibroblast: One function, multiple origins, Am. J. Pathol (2007). 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Krenning G, Zeisberg EM, Kalluri R, The origin of fibroblasts and mechanism of cardiac fibrosis, J. Cell. Physiol (2010). 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Willems IEMG, Havenith MG, De Mey JGR, Daemen MJAP, The α-smooth muscle actin-positive cells in healing human myocardial scars, Am. J. Pathol (1994). [PMC free article] [PubMed] [Google Scholar]
- [23].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart, J. Clin. Invest (2018). 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, Rosenthal NA, Pinto AR, High-Resolution Transcriptomic Profiling of the Heart during Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy, Circulation. (2020). 10.1161/CIRCULATIONAHA.119.045115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin L, Knowlton AA, Innate immunity and cardiomyocytes in ischemic heart disease, Life Sci (2014). 10.1016/j.lfs.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mann DL, The emerging role of innate immunity in the heart and vascular system: For whom the cell tolls, Circ. Res (2011). 10.1161/CIRCRESAHA.110.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aoyagi T, Matsui T, The Cardiomyocyte as a Source of Cytokines in Cardiac Injury, J. Cell Sci. Ther (2012). 10.4172/2157-7013.s5-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Frangogiannis NG, The immune system and cardiac repair, Pharmacol. Res (2008). 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ma Y, Yabluchanskiy A, Lindsey ML, Neutrophil roles in left ventricular remodeling following myocardial infarction, Fibrogenes. Tissue Repair. (2013). 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wynn TA, Barron L, Macrophages: Master regulators of inflammation and fibrosis, Semin. Liver Dis (2010). 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG, CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts, Circ. Res (2005). 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- [32].O’Rourke SA, Dunne A, Monaghan MG, The Role of Macrophages in the Infarcted Myocardium: Orchestrators of ECM Remodeling, Front. Cardiovasc. Med (2019). 10.3389/fcvm.2019.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gordon S, Taylor PR, Monocyte and macrophage heterogeneity, Nat. Rev. Immunol (2005). 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- [34].Kong P, Christia P, Frangogiannis NG, The pathogenesis of cardiac fibrosis, Cell. Mol. Life Sci (2014). 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wynn TA, Ramalingam TR, Mechanisms of fibrosis: Therapeutic translation for fibrotic disease, Nat. Med (2012). 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D, Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation, J. Leukoc. Biol (2008). 10.1189/jlb.1107782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Laan M, Cui ZH, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh BE, Lindén A, Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways., J. Immunol (1999). [PubMed] [Google Scholar]
- [38].Wynn TA, Fibrotic disease and the TH1/TH2 paradigm, Nat. Rev. Immunol (2004). 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smolgovsky S, Ibeh U, Tamayo TP, Alcaide P, Adding insult to injury - Inflammation at the heart of cardiac fibrosis, Cell. Signal (2020). 10.1016/j.cellsig.2020.109828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pichler M, Rainer PP, Schauer S, Hoefler G, Cardiac fibrosis in human transplanted hearts is mainly driven by cells of intracardiac origin, J. Am. Coll. Cardiol (2012). 10.1016/j.jacc.2011.11.036. [DOI] [PubMed] [Google Scholar]
- [41].Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM, Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis, J. Clin. Invest (2014). 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fu X, Liu Q, Li C, Li Y, Wang L, Cardiac Fibrosis and Cardiac Fibroblast Lineage-Tracing: Recent Advances, Front. Physiol (2020). 10.3389/fphys.2020.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ciz M, Denev P, Kratchanova M, Vasicek O, Ambrozova G, Lojek A, Flavonoids inhibit the respiratory burst of neutrophils in mammals, Oxid. Med. Cell. Longev (2012). 10.1155/2012/181295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sampson N, Berger P, Zenzmaier C, Redox signaling as a therapeutic target to inhibit myofibroblast activation in degenerative fibrotic disease, Biomed Res. Int (2014). 10.1155/2014/131737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Daseke MJ, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML, Neutrophil proteome shifts over the myocardial infarction time continuum, Basic Res. Cardiol (2019). 10.1007/s00395-019-0746-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lefkowitz DL, Lefkowitz SS, Macrophage-neutrophil interaction: A paradigm for chronic inflammation revisited, Immunol. Cell Biol (2001). 10.1046/j.1440-1711.2001.01020.x. [DOI] [PubMed] [Google Scholar]
- [47].Murray PJ, Wynn TA, Protective and pathogenic functions of macrophage subsets, Nat. Rev. Immunol (2011). 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang M, Pan X, Zou Q, Xia Y, Chen J, Hao Q, Wang H, Sun D, Notch3 Ameliorates Cardiac Fibrosis After Myocardial Infarction by Inhibiting the TGF-β1/Smad3 Pathway, Cardiovasc. Toxicol (2016). 10.1007/s12012-015-9341-z. [DOI] [PubMed] [Google Scholar]
- [49].Biernacka A, Dobaczewski M, Frangogiannis NG, TGF-β signaling in fibrosis, Growth Factors. (2011). 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Schiller M, Javelaud D, Mauviel A, TGF-β-induced SMAD signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing, J. Dermatol. Sci (2004). 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- [51].Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T, Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats, Circulation. (2002). 10.1161/01.CIR.0000020689.12472.E0. [DOI] [PubMed] [Google Scholar]
- [52].Crowe MJ, Doetschman T, Greenhalgh DG, Delayed wound healing in immunodeficient TGF-β1 knockout mice, J. Invest. Dermatol (2000). 10.1046/j.1523-1747.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- [53].Hulsmans M, Sam F, Nahrendorf M, Monocyte and macrophage contributions to cardiac remodeling, J. Mol. Cell. Cardiol (2016). 10.1016/j.yjmcc.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mantovani A, Sica A, Locati M, Macrophage polarization comes of age, Immunity. (2005). 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- [55].Meznarich J, Malchodi L, Helterline D, Ramsey SA, Bertko K, Plummer T, Plawman A, Gold E, Stempien-Otero A, Urokinase Plasminogen Activator Induces Pro-Fibrotic/M2 Phenotype in Murine Cardiac Macrophages, PLoS One. (2013). 10.1371/journal.pone.0057837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sica A, Mantovani A, Macrophage plasticity and polarization: In vivo veritas, J. Clin. Invest (2012). 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sullivan DE, Ferris M, Nguyen H, Abboud E, Brody AR, TNF-α induces TGF-β1 expression in lung fibroblasts at the transcriptional level via AP-1 activation, J. Cell. Mol. Med (2009). 10.1111/j.1582-4934.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A, IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis, Nat. Med (2006). 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- [59].Nathan CF, Prendergast TJ, Wiebe ME, Stanley ER, Platzer E, Remold HG, Welte K, Rubin BY, Murray HW, Activation of human macrophages. Comparison of other cytokines with interferon-gamma., J. Exp. Med 160 (1984) 600–605. 10.1084/jem.160.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Müller DN, Interferon-γ signaling inhibition ameliorates angiotensin ii-induced cardiac damage, Hypertension. (2012). 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- [61].Feng W, Li W, Liu W, Wang F, Li Y, Yan W, IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure, Exp. Mol. Pathol (2009). 10.1016/j.yexmp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- [62].Xie Y, Li M, Wang X, Zhang X, Peng T, Yang Y, Zou Y, Ge J, Chen H, Chen R, In Vivo Delivery of Adenoviral Vector Containing Interleukin-17 Receptor A Reduces Cardiac Remodeling and Improves Myocardial Function in Viral Myocarditis Leading to Dilated Cardiomyopathy, PLoS One. (2013). 10.1371/journal.pone.0072158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R, IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR, Circ. Res (2009). 10.1161/CIRCRESAHA.108.188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sun L, Louie MC, Vannella KM, Wilke CA, Levine AM, Moore BB, Shanley TP, New concepts of IL-10-induced lung fibrosis: Fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis, Am. J. Physiol. - Lung Cell. Mol. Physiol (2011). 10.1152/ajplung.00122.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL, Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats, Hypertension. (2010). 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].François A, Chatelus E, Wachsmann D, Sibilia J, Bahram S, Alsaleh G, Gottenberg JE, B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis, Arthritis Res. Ther (2013). 10.1186/ar4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bagalad B, Mohan Kumar K, Puneeth H, Myofibroblasts: Master of disguise, J. Oral Maxillofac. Pathol (2017). 10.4103/jomfp.JOMFP_146_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shimizu I, Minamino T, Cellular senescence in cardiac diseases, J. Cardiol (2019). 10.1016/j.jjcc.2019.05.002. [DOI] [PubMed] [Google Scholar]
- [69].Lu L, Guo J, Hua Y, Huang K, Magaye R, Cornell J, Kelly DJ, Reid C, Liew D, Zhou Y, Chen A, Xiao W, Fu Q, Wang BH, Cardiac fibrosis in the ageing heart: Contributors and mechanisms, Clin. Exp. Pharmacol. Physiol (2017). 10.1111/1440-1681.12753. [DOI] [PubMed] [Google Scholar]
- [70].Biernacka A, Frangogiannis NG, Aging and cardiac fibrosis, Aging Dis (2011). [PMC free article] [PubMed] [Google Scholar]
- [71].Cianflone E, Torella M, Biamonte F, De Angelis A, Urbanek K, Costanzo FS, Rota M, Ellison-Hughes GM, Torella D, Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease, Cells. (2020). 10.3390/cells9061558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Song H, Conte JV, Foster AH, McLaughlin JS, Wei C, Increased p53 protein expression in human failing myocardium, J. Hear. Lung Transplant. 18 (1999) 744–749. 10.1016/S1053-2498(98)00039-4. [DOI] [PubMed] [Google Scholar]
- [73].Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM, Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies, Circulation. (2010). 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Birks EJ, Latif N, Enesa K, Folkvang T, Luong LA, Sarathchandra P, Khan M, Ovaa H, Terracciano CM, Barton PJR, Yacoub MH, Evans PC, Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy, Cardiovasc. Res (2008). 10.1093/cvr/cvn083. [DOI] [PubMed] [Google Scholar]
- [75].Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I, p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload, Nature. (2007). 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- [76].Sawaki D, Czibik G, Pini M, Ternacle J, Suffee N, Mercedes R, Marcelin G, Surenaud M, Marcos E, Gual P, Clément K, Hue S, Adnot S, Hatem SN, Tsuchimochi I, Yoshimitsu T, Hénégar C, Derumeaux G, Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by osteopontin production, Circulation. (2018). 10.1161/CIRCULATIONAHA.117.031358. [DOI] [PubMed] [Google Scholar]
- [77].Jia L, Zhang W, Ma Y, Chen B, Liu Y, Piao C, Wang Y, Yang M, Liu T, Zhang J, Li T, Nie S, Du J, Haplodeficiency of ataxia telangiectasia mutated accelerates heart failure after myocardial infarction, J. Am. Heart Assoc (2017). 10.1161/JAHA.117.006349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].van Putten S, Shafieyan Y, Hinz B, Mechanical control of cardiac myofibroblasts, J. Mol. Cell. Cardiol (2016). 10.1016/j.yjmcc.2015.11.025. [DOI] [PubMed] [Google Scholar]
- [79].Balachandran K, Sucosky P, Jo H, Yoganathan AP, Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner, Am. J. Pathol (2010). 10.2353/ajpath.2010.090631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Dalla Costa AP, Clemente CFMZ, Carvalho HF, Carvalheira JB, Nadruz W, Franchini KG, FAK mediates the activation of cardiac fibroblasts induced by mechanical stress through regulation of the mTOR complex, Cardiovasc. Res (2010). 10.1093/cvr/cvp416. [DOI] [PubMed] [Google Scholar]
- [81].Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, Mechanical regulation of cardiac fibroblast profibrotic phenotypes, Mol. Biol. Cell. (2017). 10.1091/mbc.E17-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Driesen RB, Nagaraju CK, Abi-Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV, Reversible and irreversible differentiation of cardiac fibroblasts, Cardiovasc. Res (2014). 10.1093/cvr/cvt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wang X, Chen T, Zhang S, Schaerer J, Qian Z, Huh S, Metaxas D, Axel L, Meshless deformable models for 3D cardiac motion and strain analysis from tagged MRI, Magn. Reson. Imaging. (2015). 10.1016/j.mri.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lee AA, Delhaas T, McCulloch AD, Villarreal FJ, Differential responses of adult cardiac fibroblasts to in vitro biaxial strain patterns, J. Mol. Cell. Cardiol (1999). 10.1006/jmcc.1999.1017. [DOI] [PubMed] [Google Scholar]
- [85].Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA, Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion, Cell Motil. Cytoskeleton (2005). 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- [86].Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B, Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers, J. Cell Biol (2006). 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yip CYY, Chen JH, Zhao R, Simmons CA, Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix, Arterioscler. Thromb. Vasc. Biol (2009). 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- [88].Galie PA, Westfall MV, Stegemann JP, Reduced serum content and increased matrix stiffness promote the cardiac myofibroblast transition in 3D collagen matrices, Cardiovasc. Pathol (2011). 10.1016/j.carpath.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Petersen A, Joly P, Bergmann C, Korus G, Duda GN, The impact of substrate stiffness and mechanical loading on fibroblast-induced scaffold remodeling, Tissue Eng. - Part A. (2012). 10.1089/ten.tea.2011.0514. [DOI] [PubMed] [Google Scholar]
- [90].Baker EL, Zaman MH, The biomechanical integrin, J. Biomech (2010). 10.1016/j.jbiomech.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Geiger B, Spatz JP, Bershadsky AD, Environmental sensing through focal adhesions, Nat. Rev. Mol. Cell Biol (2009). 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- [92].Manso AM, Kang SM, Ross RS, Integrins, focal adhesions, and cardiac fibroblasts, in: J. Investig. Med, 2009. 10.2310/JIM.0b013e3181c5e61f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Schroer AK, Bersi MR, Clark CR, Zhang Q, Sanders LH, Hatzopoulos AK, Force TL, Majka SM, Lal H, Merryman WD, Cadherin-11 blockade reduces inflammation-driven fibrotic remodeling and improves outcomes after myocardial infarction, JCI Insight (2019). 10.1172/jci.insight.131545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chen C, Li R, Ross RS, Manso AM, Integrins and integrin-related proteins in cardiac fibrosis, J. Mol. Cell. Cardiol (2016). 10.1016/j.yjmcc.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK, Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK, FAK-dependent regulation of myofibroblast differentiation, FASEB J (2006). 10.1096/fj.05-4838fje. [DOI] [PubMed] [Google Scholar]
- [96].Zebda N, Dubrovskyi O, Birukov KG, Focal adhesion kinase regulation of mechanotransduction and its impact on endothelial cell functions, Microvasc. Res (2012). 10.1016/j.mvr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Schroer AK, Ryzhova LM, Merryman WD, Network modeling approach to predict myofibroblast differentiation, Cell. Mol. Bioeng (2014). 10.1007/s12195-014-0344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Clemente CFMZ, Tornatore TF, Theizen TH, Deckmann AC, Pereira TC, Lopes-Cendes I, Souza JRM, Franchini KG, Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice, Circ. Res (2007). 10.1161/CIRCRESAHA.107.160978. [DOI] [PubMed] [Google Scholar]
- [99].Miralles F, Posern G, Zaromytidou AI, Treisman R, Actin dynamics control SRF activity by regulation of its coactivator MAL, Cell. (2003). 10.1016/S0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- [100].Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, Treisman R, Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts, Genes Dev (2014). 10.1101/gad.239327.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN, Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction, Circ. Res (2010). 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hinz B, It has to be the αv: Myofibroblast integrins activate latent TGF-β1, Nat. Med (2013). 10.1038/nm.3421. [DOI] [PubMed] [Google Scholar]
- [103].Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, Caldarone CA, Hinz B, Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction, Cardiovasc. Res (2014). 10.1093/cvr/cvu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Wipff PJ, Hinz B, Integrins and the activation of latent transforming growth factor β1 - An intimate relationship, Eur. J. Cell Biol (2008). 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- [105].Carisey A, Tsang R, Greiner AM, Nijenhuis N, Heath N, Nazgiewicz A, Kemkemer R, Derby B, Spatz J, Ballestrem C, Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner, Curr. Biol (2013). 10.1016/j.cub.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA, Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics, Nature. (2010). 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C, Vinculin controls focal adhesion formation by direct interactions with talin and actin, J. Cell Biol (2007). 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Takeichi M, Cadherins: A molecular family important in selective cell-cell adhesion, Annu. Rev. Biochem (1990). 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- [109].Goodwin M, Yap AS, Classical cadherin adhesion molecules: Coordinating cell adhesion, signaling and the cytoskeleton, J. Mol. Histol (2004). 10.1007/s10735-004-1833-2. [DOI] [PubMed] [Google Scholar]
- [110].Suyama K, Shapiro I, Guttman M, Hazan RB, A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor, Cancer Cell. (2002). 10.1016/S1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- [111].Tran NL, Adams DG, Vaillancourt RR, Heimark RL, Signal transduction from N-cadherin increases Bcl-2. Regulation of the phosphatidylinositol 3-kinase/Akt pathway by homophilic adhesion and actin cytoskeletal organization, J. Biol. Chem (2002). 10.1074/jbc.M200300200. [DOI] [PubMed] [Google Scholar]
- [112].Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GFM, Chisaka O, Takeichi M, Brenner MB, Cadherin-11 in synovial lining formation and pathology in arthritis, Science (80-. ). (2007). 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- [113].Lodyga M, Cambridge E, Karvonen HM, Pakshir P, Wu B, Boo S, Kiebalo M, Kaarteenaho R, Glogauer M, Kapoor M, Ask K, Hinz B, Cadherin-11–mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-, Sci. Signal (2019). 10.1126/scisignal.aao3469. [DOI] [PubMed] [Google Scholar]
- [114].Heuberger J, Birchmeier W, Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling., Cold Spring Harb. Perspect. Biol (2010). 10.1101/cshperspect.a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Xiang FL, Fang M, Yutzey KE, Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice, Nat. Commun (2017). 10.1038/s41467-017-00840-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yap AS, Kovacs EM, Direct cadherin-activated cell signaling: A view from the plasma membrane, J. Cell Biol (2003). 10.1083/jcb.200208156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J, Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling, J. Cell Biol (2012). 10.1083/jcb.201108120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, De Rooij J, Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner, J. Cell Biol (2010). 10.1083/jcb.201001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Yao M, Qiu W, Liu R, Efremov AK, Cong P, Seddiki R, Payre M, Lim CT, Ladoux B, Mège RM, Yan J, Force-dependent conformational switch of α -catenin controls vinculin binding, Nat. Commun (2014). 10.1038/ncomms5525. [DOI] [PubMed] [Google Scholar]
- [120].Herum K, Lunde I, McCulloch A, Christensen G, The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart, J. Clin. Med (2017). 10.3390/jcm6050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L, C-cadherin ectodomain structure and implications for cell adhesion mechanisms, Science (80-. ) (2002). 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- [122].Halbleib JM, Nelson WJ, Cadherins in development: Cell adhesion, sorting, and tissue morphogenesis, Genes Dev (2006). 10.1101/gad.1486806. [DOI] [PubMed] [Google Scholar]
- [123].Horikawa K, Radice G, Takeichi M, Chisaka O, Adhesive subdivisions intrinsic to the epithelial somites, Dev. Biol (1999). 10.1006/dbio.1999.9463. [DOI] [PubMed] [Google Scholar]
- [124].Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR, Cadherin switching, J. Cell Sci (2008). 10.1242/jcs.000455. [DOI] [PubMed] [Google Scholar]
- [125].Xu S, Liu AC, Kim H, Gotlieb AI, Cell density regulates in vitro activation of heart valve interstitial cells, Cardiovasc. Pathol (2012). 10.1016/j.carpath.2011.01.004. [DOI] [PubMed] [Google Scholar]
- [126].Yan W, Lin C, Guo Y, Chen Y, Du Y, Lau WB, Xia Y, Zhang F, Su R, Gao E, Wang Y, Li C, Liu R, Ma XL, Tao L, N-cadherin overexpression mobilizes the protective effects of mesenchymal stromal cells against ischemic heart injury through a β-catenin-dependent manner, Circ. Res (2020). 10.1161/CIRCRESAHA.119.315806. [DOI] [PubMed] [Google Scholar]
- [127].Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister JJ, Myofibroblast development is characterized by specific cell-cell adherens junctions, Mol. Biol. Cell. (2004). 10.1091/mbc.E04-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Hutcheson JD, Chen J, Sewell-Loftin MK, Ryzhova LM, Fisher CI, Su YR, David Merryman W, Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts, Arterioscler. Thromb. Vasc. Biol (2013). 10.1161/ATVBAHA.112.300278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Pittet P, Lee K, Kulik AJ, Meister JJ, Hinz B, Fibrogenic fibroblasts increase intercellular adhesion strength by reinforcing individual OB-cadherin bonds, J. Cell Sci (2008). 10.1242/jcs.024877. [DOI] [PubMed] [Google Scholar]
- [130].Schneider DJ, Wu M, Le TT, Cho S, Brenner MB, Blackburn MR, Agarwal SK, Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-β production and epithelial to mesenchymal transition, FASEB J (2012). 10.1096/fj.11-186098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Okazaki M, Takeshita S, Kawai S, Kikuno R, Tsujimura A, Kudo A, Amann E, Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts, J. Biol. Chem (1994). [PubMed] [Google Scholar]
- [132].Alimperti S, Andreadis ST, CDH2 and CDH11 act as regulators of stem cell fate decisions, Stem Cell Res (2015). 10.1016/j.scr.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM, Brenner MB, Cadherin-11 regulates fibroblast inflammation, Proc. Natl. Acad. Sci. U. S. A (2011). 10.1073/pnas.1019437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Bowler MA, Bersi MR, Ryzhova LM, Jerrell RJ, Parekh A, Merryman WD, Cadherin-11 as a regulator of valve myofibroblast mechanobiology, Am. J. Physiol. - Hear. Circ. Physiol (2018). 10.1152/ajpheart.00277.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wu M, Pedroza M, Lafyatis R, George AT, Mayes MD, Assassi S, Tan FK, Brenner MB, Agarwal SK, Identification of cadherin 11 as a mediator of dermal fibrosis and possible role in systemic sclerosis, Arthritis Rheumatol (2014). 10.1002/art.38275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, Yu-Lee LY, Zhang S, Yeh ET, Hu MCT, Logothetis CJ, Lin SH, Cadherin-11 promotes the metastasis of prostate cancer cells to bone, Mol. Cancer Res (2008). 10.1158/1541-7786.MCR-08-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Craciun FL, Bijol V, Ajay AK, Rao P, Kumar RK, Hutchinson J, Hofmann O, Joshi N, Luyendyk JP, Kusebauch U, Moss CL, Srivastava A, Himmelfarb J, Waikar SS, Moritz RL, Vaidya VS, RNA sequencing identifies novel translational biomarkers of kidney fibrosis, J. Am. Soc. Nephrol (2016). 10.1681/ASN.2015020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wang H, Leinwand LA, Anseth KS, Roles of transforming growth factor-β1 and OB-cadherin in porcine cardiac valve myofibroblast differentiation, FASEB J (2014). 10.1096/fj.14-254623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zhou J, Bowen C, Lu G, Knapp C, Recknagel A, Norris RA, Butcher JT, Cadherin-11 expression patterns in heart valves associate with key functions during embryonic cushion formation, valve maturation and calcification, Cells Tissues Organs (2014). 10.1159/000356762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kiener HP, Niederreiter B, Lee DM, Jimenez-Boj E, Smolen JS, Brenner MB, Cadherin 11 promotes invasive behavior of fibroblast-like synoviocytes, Arthritis Rheum (2009). 10.1002/art.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Langhe RP, Gudzenko T, Bachmann M, Becker SF, Gonnermann C, Winter C, Abbruzzese G, Alfandari D, Kratzer MC, Franz CM, Kashef J, Cadherin-11 localizes to focal adhesions and promotes cell-substrate adhesion, Nat. Commun (2016). 10.1038/ncomms10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Alimperti S, You H, George T, Agarwal SK, Andreadis ST, Cadherin-11 regulates both mesenchymal stem cell differentiation into smooth muscle cells and the development of contractile function in vivo, J. Cell Sci (2014). 10.1242/jcs.134833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Row S, Liu Y, Alimperti S, Agarwal SK, Andreadis ST, Cadherin-11 is a novel regulator of extracellular matrix synthesis and tissue mechanics, J. Cell Sci (2016). 10.1242/jcs.183772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Kim NH, Choi SH, Lee TR, Lee CH, Lee AY, Cadherin 11, a miR-675 target, induces N-Cadherin expression and epithelial-Mesenchymal transition in Melasma, J. Invest. Dermatol (2014). 10.1038/jid.2014.257. [DOI] [PubMed] [Google Scholar]
- [145].Noss EH, Chang SK, Watts GFM, Brenner MB, Modulation of matrix metalloproteinase production by rheumatoid arthritis synovial fibroblasts after cadherin 11 engagement, Arthritis Rheum (2011). 10.1002/art.30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Gu X, Masters KS, Role of the MAPK/ERK pathway in valvular interstitial cell calcification, Am. J. Physiol. - Hear. Circ. Physiol (2009). 10.1152/ajpheart.00099.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS, Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway, Proc. Natl. Acad. Sci. U. S. A (2013). 10.1073/pnas.1306369110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA, Valvular myofibroblast activation by transforming growth factor-β: Implications for pathological extracellular matrix remodeling in heart valve disease, Circ. Res (2004). 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- [149].Bowen CJ, Zhou J, Sung DC, Butcher JT, Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation, Dev. Biol (2015). 10.1016/j.ydbio.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Sung DC, Bowen CJ, Vaidya KA, Zhou J, Chapurin N, Recknagel A, Zhou B, Chen J, Kotlikoff M, Butcher JT, Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves, Arterioscler. Thromb. Vasc. Biol (2016). 10.1161/ATVBAHA.116.307812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Clark CR, Bowler MA, Snider JC, David Merryman W, Targeting Cadherin-11 Prevents Notch1-Mediated Calcific Aortic Valve Disease, Circulation. (2017). 10.1161/CIRCULATIONAHA.117.027771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Thompson SA, Blazeski A, Copeland CR, Cohen DM, Chen CS, Reich DM, Tung L, Acute slowing of cardiac conduction in response to myofibroblast coupling to cardiomyocytes through N-cadherin, J. Mol. Cell. Cardiol (2014). 10.1016/j.yjmcc.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Mun GI, Boo YC, Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells, Am. J. Physiol. - Hear. Circ. Physiol (2010). 10.1152/ajpheart.00835.2009. [DOI] [PubMed] [Google Scholar]
- [154].Geletu M, Mohan R, Arulanandam R, Berger-Becvar A, Nabi IR, Gunning PT, Raptis L, Reciprocal regulation of the Cadherin-11/Stat3 axis by caveolin-1 in mouse fibroblasts and lung carcinoma cells, Biochim. Biophys. Acta - Mol. Cell Res 1865 (2018) 794–802. 10.1016/j.bbamcr.2018.02.004. [DOI] [PubMed] [Google Scholar]
- [155].Frangogiannis NG, The inflammatory response in myocardial injury, repair, and remodelling, Nat. Rev. Cardiol (2014). 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Finch R, Sostelly A, Sue-Ling K, Blaeuer A, Duchateau-Nguyen G, Ukarma L, Petry C, Ravva P, Villiger P, Junker U, OP0224 RESULTS OF A PHASE 2 STUDY OF RG6125, AN ANTI-CADHERIN-11 MONOCLONAL ANTIBODY, IN RHEUMATOID ARTHRITIS PATIENTS WITH AN INADEQUATE RESPONSE TO ANTI-TNFALPHA THERAPY, Ann. Rheum. Dis 78 (2019) 189 LP – 189. 10.1136/annrheumdis-2019-eular.3028. [DOI] [Google Scholar]
- [157].Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, Uren A, Brown ML, Byers SW, Predicting new indications for approved drugs using a proteochemometric method, J. Med. Chem (2012). 10.1021/jm300576q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Assefnia S, Dakshanamurthy S, Auvil JMG, Hampel C, Anastasiadis PZ, Kallakury B, Uren A, Foley DW, Brown ML, Shapiro L, Brenner M, Haigh D, Byers SW, Cadherin-11 in poor prognosis malignancies and rheumatoid arthritis: Common target, common therapies, Oncotarget (2014). 10.18632/oncotarget.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Bowler MA, Raddatz MA, Johnson CL, Lindman BR, Merryman WD, Celecoxib Is Associated With Dystrophic Calcification and Aortic Valve Stenosis, JACC Basic to Transl. Sci (2019). 10.1016/j.jacbts.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Vaidya KA, Donnelly MP, Gee TW, Ibrahim Aibo MA, Byers S, Butcher JT, Induction of aortic valve calcification by celecoxib and its COX-2 independent derivatives is glucocorticoid-dependent, Cardiovasc. Pathol (2020). 10.1016/j.carpath.2019.107194. [DOI] [PubMed] [Google Scholar]
- [161].Sehnal D, Rose AS, Kǒ J, Mol *: Towards a common library and tools for web molecular graphics, (2018). 10.2312/molva.20181103. [DOI] [Google Scholar]