

ABSTRACT
Anaplasma phagocytophilum infects neutrophils to cause granulocytic anaplasmosis. It poorly infects mice deficient in acid sphingomyelinase (ASM), a lysosomal enzyme critical for cholesterol efflux, and wild-type mice treated with desipramine that functionally inhibits ASM. Whether inhibition or genetic deletion of ASM is bacteriostatic or bactericidal for A. phagocytophilum and desipramine's ability to lower pathogen burden requires a competent immune system were unknown. Anaplasma phagocytophilum-infected severe combined immunodeficiency disorder (SCID) mice were administered desipramine or PBS, followed by the transfer of blood to naïve wild-type mice. Next, infected wild-type mice were given desipramine or PBS followed by transfer of blood to naïve SCID mice. Finally, wild-type or ASM-deficient mice were infected and blood transferred to naïve SCID mice. The percentage of infected neutrophils was significantly reduced in all desipramine-treated or ASM-deficient mice and in all recipients of blood from these mice. Infection was markedly lower in ASM-deficient and desipramine-treated wild-type mice versus desipramine-treated SCID mice. Yet, infection was never ablated. Thus, ASM activity contributes to optimal A. phagocytophilum infection in vivo, pharmacologic inhibition or genetic deletion of ASM impairs infection in a bacteriostatic and reversible manner and A. phagocytophilum is capable of co-opting ASM-independent lipid sources.
Keywords: Anaplasma phagocytophilum, acid sphingomyelinase, obligate intracellular bacteria, desipramine, Anaplasmataceae, functional inhibitor of acid sphingomyelinase
Pharmacologic inhibition or genetic deletion of acid sphingomyelinase impairs Anaplasma phagocytophilum infection in vivo in a bacteriostatic and reversible manner.
INTRODUCTION
Anaplasma phagocytophilum is a granulocytotropic obligate intracellular bacterium in the family Anaplasmataceae that zoonotically cycles between Ixodes spp. ticks and small animal reservoirs. When transmitted via the tick bloodmeal to humans or other accidental mammalian hosts, including horses, sheep and dogs, A. phagocytophilum causes granulocytic anaplasmosis, an acute non-specific febrile illness that can progress to severe sequelae, such as rhabdomyolysis, acute respiratory distress syndrome, increased susceptibility to secondary infections, shock and death particularly in the elderly and immunocompromised (Bakken and Dumler 2015; Ismail and McBride 2017; Eisen and Eisen 2018). First confirmed as a human disease in 1994 (Chen et al. 1994), granulocytic anaplasmosis is the second most common tick-transmitted infection in the United States with >5700 cases reported to the Centers for Disease Control and Prevention in 2017 (https://www.cdc.gov/anaplasmosis/stats/index.html). Seroprevalence data suggest that its true incidence in certain endemic areas of the United States is underrepresented potentially by as much as an order of magnitude (Bakken et al. 1996, 1998; Belongia et al. 2001; Aguero-Rosenfeld et al. 2002; Leiby et al. 2002; Dahlgren et al. 2011, 2015). The disease is also present in numerous European countries, Scandinavia and eastern Asia (Bakken and Dumler 2015). Anaplasma phagocytophilum invades neutrophils, myeloid progenitors and susceptible immortalized cell lines via receptor-mediated endocytosis to reside in a host cell-derived vacuole (Ismail and McBride 2017).
C57BL/6 mice are useful models for studying granulocytic anaplasmosis (Hodzic et al. 1998). Following syringe inoculation of host cell-free A. phagocytophilum bacteria, neutrophils harboring A. phagocytophilum-occupied vacuoles (ApVs) or A. phagocytophilum DNA can be detected in peripheral blood smears beginning around day 4 with infection peaking between days 8 and 16 and subsequently declining to near or completely undetectable levels by day 28 (Carlyon et al. 2003; Naimi et al. 2018; Cockburn et al. 2019; Green et al. 2020). Male C57BL/6 mice exhibit significantly higher A. phagocytophilum peripheral blood burdens than females, reflecting epidemiological trends for the incidence of granulocytic anaplasmosis in humans (Bakken and Dumler 2006; Dumler et al. 2007; Naimi et al. 2018). In contrast to being cleared from immunocompetent mice, A. phagocytophilum establishes a persistent infection in severe combined immunodeficiency disorder (SCID) mice (Telford et al. 1996; Hodzic 1998).
As an obligate intracellular bacterium having a reduced genome and limited metabolic capacity (Dunning Hotopp et al. 2006), A. phagocytophilum parasitizes host cell nutrients in order to establish a productive infection, a key one of which is cholesterol. The pathogen incorporates the sterol into its peptidoglycan-deficient cell wall, is halted in its intracellular growth and rendered less infectious when it takes up the cholesterol analog methyl-β-cyclodextrin, and more efficiently infects hypercholesterolemic mice versus mice having lower cholesterol levels (Lin and Rikihisa 2003; Xiong, Wang and Rikihisa 2007). Evidence to date indicates that A. phagocytophilum exclusively obtains cholesterol from its host cell by hijacking the Niemann–Pick type C protein 1 (NPC1) pathway that mediates lysosomal efflux of cholesterol exogenously acquired by the low-density lipoprotein (LDL) receptor (Xiong, Lin and Rikihisa 2009; Xiong and Rikihisa 2012). Acid sphingomyelinase (ASM) is a lysosomal enzyme that hydrolyzes sphingomyelin to generate phosphorylcholine and ceramide (Vanier 2013). ASM genetic deficiency or pharmacologic inhibition leads to sphingomyelin accumulation in lysosomes, which blocks LDL-derived cholesterol efflux (Brady et al. 1966; Schuchman and Miranda 1997; Lloyd-Evans et al. 2008; Vanier 2013). Functional inhibitors of acid sphingomyelinase (FIASMAs) are lysosomotropic compounds that indirectly inactivate ASM by promoting detachment of the enzyme from the lysosomal inner membrane, which renders it susceptible to proteolytic degradation (Hurwitz, Ferlinz and Sandhoff 1994; Kolzer, Werth and Sandhoff 2004; Kornhuber et al. 2010). FIASMAs have broad therapeutic potential and many are approved for human use (Kornhuber et al. 2010; Kuzu et al. 2017). Treatment of A. phagocytophilum-infected promyelocytic HL-60 cells or human neutrophils with the FIASMA, desipramine, blocks NPC1-mediated delivery of cholesterol to the ApV and bacteriostatically halts the organism's replication, ApV expansion and production of infectious progeny. Anaplasma phagocytophilum poorly infects ASM-deficient mice, and administration of desipramine to infected wild-type mice reduces the bacterial burden severalfold (Cockburn et al. 2019). Yet, it remains unclear whether pharmacologic inhibition or genetic deletion of ASM exerts a bacteriostatic or bactericidal effect against A. phagocytophilum in vivo, whether the ability of desipramine to reduce pathogen burden in mice requires functional B and T cells and whether A. phagocytophilum potentially co-opts NPC1-independent cholesterol sources.
METHODS
Cultivation of uninfected and A. phagocytophilum-infected cells
Uninfected and A. phagocytophilum str. NCH-1-infected human promyelocytic HL-60 cells (ATCC CCL-240; American Type Culture Collection [ATCC], Manassas, VA) were cultivated as described (Green et al. 2020).
Mouse studies
All mouse research was conducted under the approval of the Institutional Animal Care and Use Committee at Virginia Commonwealth University (Protocol Number AM10220). C57BL/6 and C57BL/6 SCID mice were purchased from Jackson Laboratories (Bar Harbor, ME). ASM-deficient mice (sphingomyelin phosphodiesterase 1 knockout; Smpd1−/−) on a C57BL/6 genetic background have been described (Horinouchi et al. 1995; Paris et al. 2001; Gupta et al. 2004). Because A. phagocytophilum multiplies intracellularly as microcolonies, a formula for estimating the number of host cell-free bacteria recovered from host cells following mechanical disruption was previously devised by Kim and Rikihisa (2000, 2002). We employed this formula to estimate the number of dense-cored organisms following isolation from infected HL-60 cells by sonication and differential centrifugation as described (Troese and Carlyon 2009; Huang et al. 2010). All mice were intraperitoneally inoculated by syringe injection of a phosphate-buffered saline (PBS) suspension containing 1 × 108 A. phagocytophilum dense-cored organisms. Mice were age-matched and male due to the higher susceptibility of male C57BL/6 mice to A. phagocytophilum infection versus female mice (Naimi et al. 2018). Unless otherwise stated, 10 mice were used per group. On specified days, blood was collected from the tails followed by the addition of heparin (Sigma-Aldrich, St Louis, MO) at 100 U mL−1. The percentage of peripheral blood neutrophils containing ApVs was determined via microscopic examination of triplicate blood smears for a total of at least 100 neutrophils per time point per mouse as described (Naimi et al. 2018). In some instances, A. phagocytophilum-infected mice were intraperitoneally injected for a specified number of days with 10 mg kg−1 desipramine (Millipore Sigma, Burlington, MA), a dosage that was selected because it is within the range approved for administration to humans (Teichgraber et al. 2008; Hayasaka et al. 2015), or PBS as a negative control.
For the experiment in which blood was transferred from infected C57BL/6 SCID mice that had received either desipramine or PBS on days 16 through 39 to naïve C57BL/6 mice, the infected C57BL/6 SCID mice were euthanized on day 40 and blood collected by cardiac puncture. Heparin-treated blood from the four C57BL/6 SCID mice that had been injected with PBS and exhibited the highest bacterial loads or from the four infected C57BL/6 SCID mice that had been treated with desipramine and had the lowest bacterial loads was, respectively, pooled. One hundred microliters of either pooled sample was intraperitoneally injected into each of five C57BL/6 mice and infection was monitored as described above. Mice in this experiment were 5–8 weeks old at the time of inoculation.
For the experiment in which blood was transferred from infected C57BL/6 mice that had received desipramine or PBS on days 7 through 12 to naïve C57BL/6 SCID mice, the infected C57BL/6 mice were euthanized on day 12 and blood collected via cardiac puncture. Heparin-treated blood from all 10 mice from each of the two respective groups was pooled. One hundred microliters of pooled blood from each condition was intraperitoneally injected into each of 10 C57BL/6 SCID mice and infection was monitored as described above. Mice in this experiment were 5–8 weeks old at the time of inoculation.
For the experiment assessing whether ASM deficiency is bacteriostatic or bactericidal against A. phagocytophilum, 16–18-week-old ASM-deficient or age-matched C57BL/6 mice were injected intraperitoneally with A. phagocytophilum bacteria and monitored for infection. Mice were euthanized on day 12 and blood was recovered by cardiac puncture. One hundred microliters of pooled heparin-treated blood from either group was intraperitoneally injected into each of 10 C57BL/6 SCID mice followed by monitoring of infection as described above.
Statistical analyses
Statistical analyses were performed using the Prism 7.0 software package (GraphPad, San Diego, CA). Two-way analysis of variance with Tukey's post-hoc test was used to test for significant differences among the groups. P-values <0.05 were considered statistically significant.
RESULTS
Desipramine temporally reduces the A. phagocytophilum load in a bacteriostatic manner in SCID mice
Administration of desipramine to A. phagocytophilum-infected wild-type C57BL/6 mice on days 7 through 12 post-infection suppresses the peripheral blood bacterial load severalfold (Cockburn et al. 2019). Whether the compound's effect is bacteriostatic or bactericidal and requires B and/or T cell responses to reduce pathogen burden was unclear. As a first step in addressing these knowledge gaps, 20 C57BL/6 SCID mice were inoculated with A. phagocytophilum, and the infection was allowed to proceed unhindered for 15 days (Fig. 1A). Beginning on day 16 and continuing through day 39, 10 of the mice were treated daily with desipramine while the other 10 received PBS. The mice were euthanized on day 40. The bacterial load was determined via light microscopic examination of peripheral blood smears obtained every four days for the presence of neutrophils containing ApVs. On days 20, 24, 28 and 32, the percentage of A. phagocytophilum-infected peripheral blood neutrophils was significantly reduced by as much as 2.8-fold in desipramine-treated mice (Fig. 1B). However, by day 36, the bacterial load had recovered to comparable levels in control mice. To determine the competency of A. phagocytophilum organisms from both treatment groups for establishing productive infections, 100 μL of blood pooled from the four SCID mice that had been administered PBS and exhibited the highest bacterial burdens or from the four desipramine-treated SCID mice that had the lowest pathogen loads was transferred to each of five naïve C57BL/6 wild-type mice per group. There was a 1.4-fold difference in the mean A. phagocytophilum load between the pooled inocula from control and desipramine-treated mice. Infection was permitted to progress for 28 days (Fig. 1A). On days 4, 8, 12 and 16, the percentage of infected neutrophils in wild-type mice that had been inoculated with blood from infected desipramine-treated SCID mice was 1.4-, 1.4-, 1.5- and 2.1-fold lower, respectively, than in wild-type mice that received blood from infected control mice (Fig. 1C). Thus, A. phagocytophilum bacteria from both treatment groups were capable of establishing infection and the initial difference in bacterial load between the inocula was relatively maintained throughout the course. These results indicate that in mice lacking functional B and T cell responses, desipramine is not bactericidal, but rather is bacteriostatic and/or compromises bacterial fitness. Notably, this inhibition is temporal as the A. phagocytophilum peripheral blood burden eventually recovers.
Figure 1.
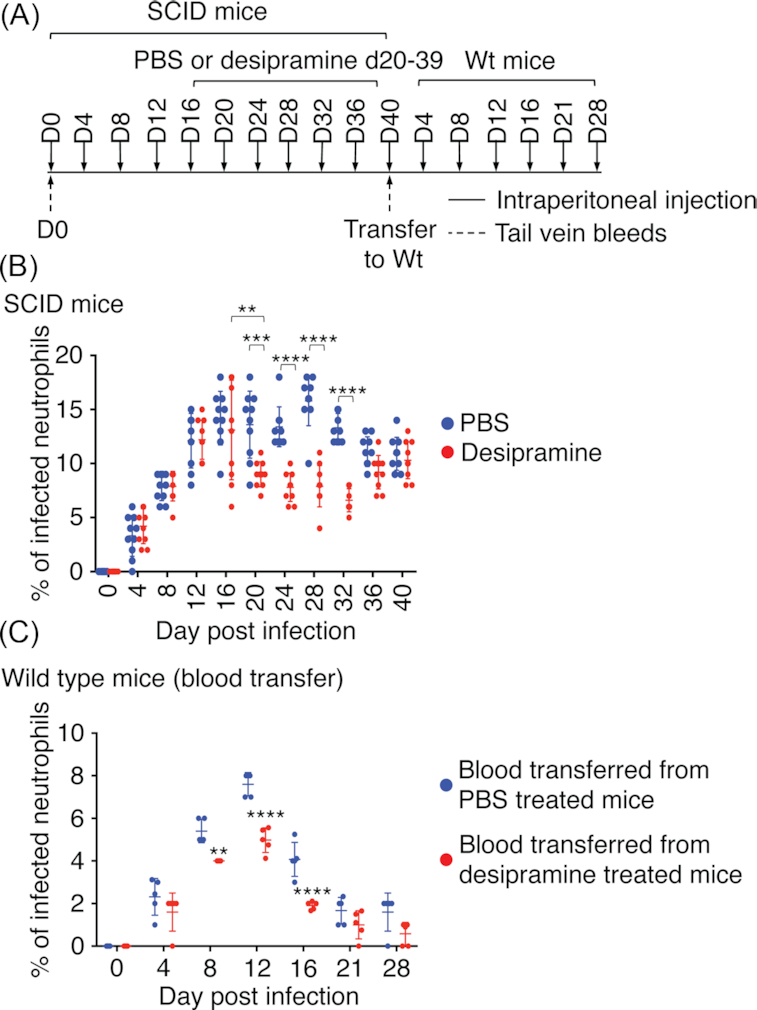
Desipramine temporally reduces the A. phagocytophilum load in SCID mice in a bacteriostatic manner. (A, B) Twenty C57BL/6 SCID mice were intraperitoneally injected with 1 × 108 A. phagocytophilum bacteria. Desipramine or PBS was administered on days 16 through 39 to 10 mice per group. All mice were tail bled on days 0 (pre-challenge), 4, 8, 12, 16, 20, 24, 28, 32 and 36, sacrificed on day 40 and blood collected via cardiac puncture. Peripheral blood smears were microscopically examined for neutrophils containing ApVs. (A, C) Heparin-treated blood from the four C57BL/6 SCID mice in (B) that had been injected with PBS and exhibited the highest bacterial loads or from the four infected C57BL/6 SCID mice in (B) that had been treated with desipramine and had the lowest bacterial loads was, respectively, pooled. One hundred microliters of either pooled sample was intraperitoneally injected into each of five C57BL/6 mice (wild-type, Wt) and infection was monitored by examination of smears from peripheral blood obtained on days 0, 4, 8, 12, 16, 21 and 28 post-injection. Each symbol corresponds to the percentage of A. phagocytophilum-infected neutrophils as determined by examining at least 100 neutrophils per mouse. Data are the mean ± standard deviation (SD) of the percentages determined for 10 C57BL/6 SCID mice per group (B) and 5 C57BL/6 mice per group (C). Error bars indicate SD among the samples per time point. Statistically significant values are indicated. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Desipramine bacteriostatically limits A. phagocytophilum infection in immunocompetent mice
It was next examined whether desipramine administration to wild-type mice is bacteriostatic or bactericidal. Twenty C57BL/6 mice were inoculated with A. phagocytophilum, 10 of which received desipramine and 10 were given PBS on days 7 through 12 (Fig. 2A). Consistent with prior observations in wild-type (Cockburn et al. 2019) and SCID mice (Fig. 1), desipramine significantly reduced infection on days 8 and 12 (Fig. 2B). The peak difference in bacterial load of 4.3-fold was observed on day 12. Blood recovered from all mice per respective group on day 12 was pooled, inoculated into 10 naïve C57BL/6 mice each and infection was monitored for 28 days (Fig. 2A). All mice became infected (Fig. 2C). However, the percentage of infected neutrophils was reduced by 2.1–3.0-fold on days 8, 12, 16, 21 and 28 in mice that received blood from the desipramine-treated cohort. Thus, desipramine had bacteriostatically limited the growth of A. phagocytophilum in the wild-type mice. The mean bacterial burden in recipient SCID mice infected with A. phagocytophilum that had been exposed to desipramine versus PBS was not as reduced as in the respective donor wild-type mice. This observation suggests that A. phagocytophilum infection proceeded more efficiently when no longer hindered by both desipramine and the host response, which enabled the bacterial population to partially recover. Collectively, data from this and the previous experiment confirm that desipramine exerts a bacteriostatic effect on A. phagocytophilum in vivo whether in the absence or presence of a competent immune response and that the infection resumes upon removal of the drug.
Figure 2.
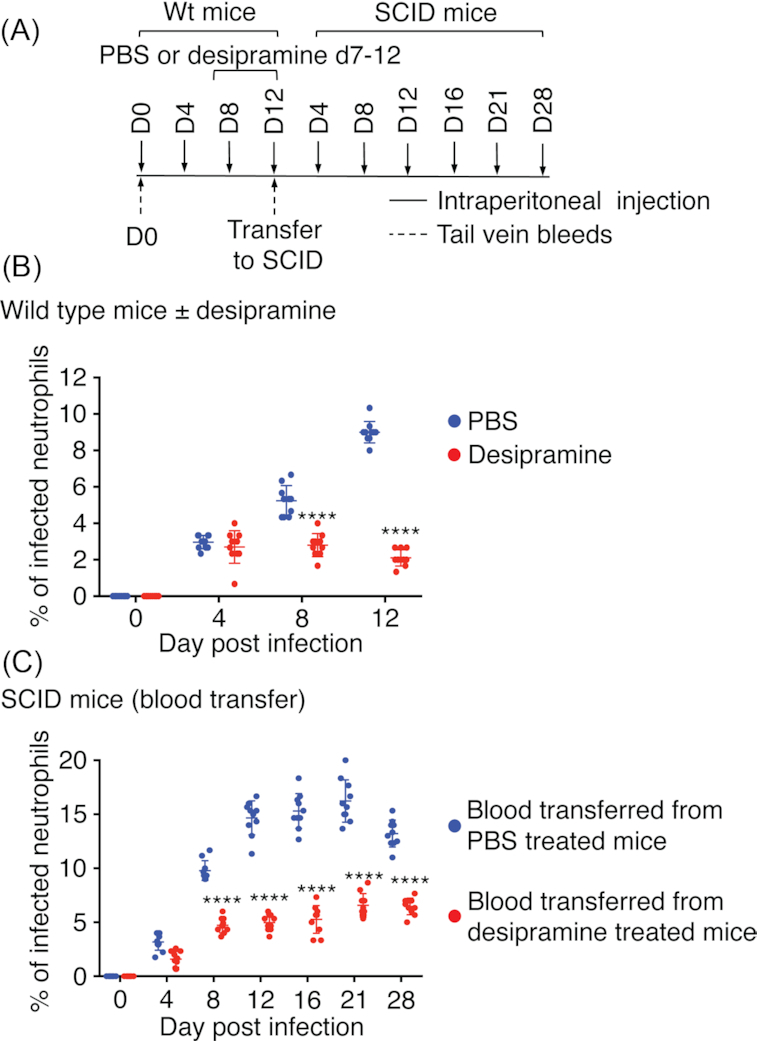
Desipramine bacteriostatically limits A. phagocytophilum infection in immunocompetent mice. (A, B) Twenty C57BL/6 (Wt) mice were intraperitoneally injected with 1 × 108 A. phagocytophilum bacteria followed by the administration of desipramine or PBS on days 7 through 12 to 10 mice per group. Mice were tail bled on days 0 (pre-challenge), 4 and 8. The mice were euthanized and blood collected on day 12 via cardiac puncture. Peripheral blood smears were microscopically examined for neutrophils containing ApVs. (A, C) Heparin-treated blood from all desipramine- or PBS-treated mice was pooled. One hundred microliters of pooled blood was transferred to naïve C57BL/6 SCID mice such that 10 received blood from infected desipramine-treated mice and 10 received blood from infected PBS-treated mice. Infection was monitored by examination of smears from peripheral blood obtained from the SCID mice on days 0, 4, 8, 12, 16, 21 and 28 post-injection. Each symbol corresponds to the percentage of A. phagocytophilum-infected neutrophils as determined by examining at least 100 neutrophils per mouse. Data are the mean ± SD of the percentages determined for 10 mice per group. Error bars indicate SD among the samples per time point. Statistically significant values are indicated. ****P < 0.0001.
Genetic deficiency in ASM severely reduces the A. phagocytophilum burden, but does not ablate infection
FIASMAs do not induce complete degradation of ASM in vitro or in vivo, leaving residual basal ASM activity (Kornhuber et al. 2005; Becker et al. 2010; Kornhuber et al. 2010). Accordingly, the hypothesis that such residual ASM activity accounts for the ability of A. phagocytophilum to survive in desipramine-treated mice, albeit at reduced fitness, was evaluated. C57BL/6 mice genetically deficient for ASM or wild-type controls (10 mice each) were infected with A. phagocytophilum followed by transfer of pooled blood obtained from all mice per group on day 12 to cohorts of 10 C57BL/6 SCID mice (Fig. 3A). The percentage of infected peripheral blood neutrophils was monitored through day 12 for the ASM-deficient and wild-type mice and through day 28 for the recipient SCID mice. Consistent with our prior report (Cockburn et al. 2019), A. phagocytophilum poorly infected ASM-deficient mice, exhibiting a 6.1-fold reduction in infected neutrophils relative to that for wild-type mice on day 12 (Fig. 3B). Following transfer of blood from both groups to naïve SCID mice, the difference in bacterial load was maintained through day 4, as there were 6.6-fold fewer neutrophils containing ApVs in SCID mice that had been inoculated with blood from infected ASM-deficient mice than in those that had been injected with blood from wild-type infected mice (Fig. 3C). However, the difference was reduced to 2.3-fold by day 8 and to 1.8- to 1.5-fold on days 12, 16, 21 and 28. Thus, once the bottleneck imposed on infection by both the lack of ASM activity and a competent immune system was released, the A. phagocytophilum population rapidly recovered. Moreover, whereas the ability of A. phagocytophilum to infect neutrophils in mice genetically deficient in ASM is pronouncedly compromised, the impairment occurs at the level of bacterial fitness rather than being bactericidal.
Figure 3.
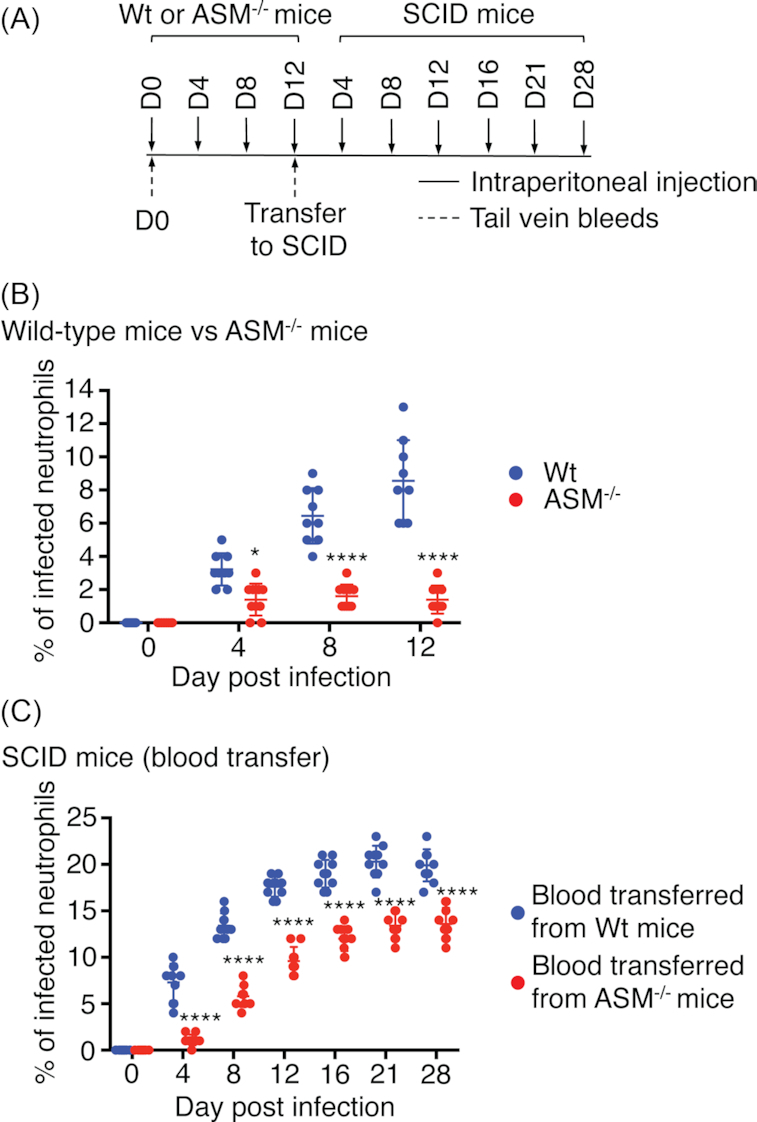
Genetic deficiency in ASM severely reduces the A. phagocytophilum burden, but does not ablate infection. (A, B) Ten C57BL/6 (Wt) or 10 ASM-deficient (ASM−/−) mice were intraperitoneally inoculated with 1 × 108 A. phagocytophilum organisms. Mice were tail bled on days 0 (pre-challenge), 4 and 8. The mice were euthanized and blood collected on day 12 via cardiac puncture. Peripheral blood smears were microscopically examined for neutrophils containing ApVs. (A, C) Heparin-treated blood from all Wt or ASM−/− mice was pooled. One hundred microliters of pooled blood was transferred to naïve C57BL/6 SCID mice such that 10 received blood from infected Wt mice and 10 received blood from infected ASM−/− mice. Infection was monitored by examination of smears from peripheral blood obtained from the SCID mice on days 0, 4, 8, 12, 16, 21 and 28. Each symbol corresponds to the percentage of A. phagocytophilum-infected neutrophils as determined by examining at least 100 neutrophils per mouse. Data are the mean ± SD of the percentages determined for 10 mice per group. Error bars indicate SD among the samples per time point. Statistically significant values are indicated. *P < 0.05; ****P < 0.0001.
DISCUSSION
This study establishes that pharmacologic inhibition of ASM in immunocompetent and immunodeficient mice as well as genetic deficiency in the enzyme bacteriostatically limits A. phagocytophilum infection. The ability of A. phagocytophilum to initially infect mice was not impaired under any condition evaluated, suggesting that the low pathogen burden results from inhibition of one or more stages of the bacterial infection cycle as has been demonstrated for FIASMA-treated A. phagocytophilum-infected mammalian cells in vitro (Cockburn et al. 2019). These findings are important when considered in the context of desipramine's inhibitory effect on NPC1-dependent delivery of exogenously acquired cholesterol from lysosomes to the ApV (Xiong and Rikihisa 2012; Cockburn et al. 2019) because it indicates that the bacterium also potentially obtains cholesterol from ASM-independent/NPC1-independent/FIASMA-insensitive mechanisms that permit its survival.
Prior reports of interfaces between the ApV and multiple membrane trafficking pathways offer support for NPC1-independent routes facilitating A. phagocytophilum cholesterol acquisition that, in turn, could allow for bacterial survival in the absence of functional ASM. Membrane contact sites are conduits of bidirectional sterol exchange between endosomes/lysosomes and other organelles, including the endoplasmic reticulum, which, in addition to receiving exogenous cholesterol in an NPC1-dependent manner, is a site of endogenous cholesterol synthesis (Meng et al. 2020). The documented membrane contact sites that form between the ApV and endoplasmic reticulum (Truchan et al. 2016a) could enable flux of endogenously synthesized cholesterol to the ApV in desipramine-treated or ASM-deficient cells. The effector, Anaplasma translocated substrate-1 (Ats-1), promotes ApV–autophagosome fusion, which releases inner membrane-enveloped autophagic bodies, another source of endogenous cholesterol (Meng et al. 2020), into the ApV lumen (Niu et al. 2012). Additionally, it was recently discovered that lysosomal integral membrane protein 2 functions independent of the NPC1 pathway to export cholesterol, but at a reduced rate (Heybrock et al. 2019). Whether A. phagocytophilum targets this cholesterol transport route is unknown.
Incorporation of other host cell lipids into the A. phagocytophilum cell wall could complement cholesterol uptake or, in the face of ASM functional inhibition or genetic deficiency, compensate for a lack thereof. Indeed, the ApV intercepts sphingolipid trans-Golgi-derived vesicles that are delivered into the ApV lumen to label the membranes of intravacuolar A. phagocytophilum organisms. Ultra-performance liquid chromatography–electrospray ionization–tandem mass spectrometry performed on A. phagocytophilum bacteria that had naturally egressed from infected myeloid cells confirmed that they were heavily enriched in numerous sphingomyelin and ceramide species (Truchan et al. 2016b). Analogously, 3,3-dioctadecylindocarbocyanine-prelabeled host cell membranes are unidirectionally trafficked into the bacterial membranes of Ehrlichia chaffeensis, another Anaplasmataceae member that parasitizes cholesterol and other host lipids (Lin et al. 2020). The numerous intraluminal membrane-rich vesicles delivered into the ApV are opportune sources of ready-to-use esterified cholesterol, sphingomyelin and ceramide for A. phagocytophilum.
Anaplasma phagocytophilum population growth in the peripheral blood of infected FIASMA-treated or ASM-deficient mice resumes following transfer to naïve untreated mice that express ASM. The reduction in the bacterial load of desipramine-treated and ASM-deficient mice could be due to an actual halt in bacterial replication that would most likely stem from the drug's inhibition of A. phagocytophilum cholesterol parasitism. However, it cannot be ruled out that either condition could impair bacterial fitness such that a portion of the population is killed by the host immune response. Support for this possibility comes from the observation that the percentage of infected neutrophils is markedly lower in ASM-deficient mice and desipramine-treated wild-type mice compared with desipramine-treated SCID mice. Even in this scenario, however, a sub-population survives. Such a sub-population, which could be better adapted at utilizing any of the aforementioned ASM/NPC1-independent lipid sources, is enriched for in desipramine-treated SCID mice, as the bacterial load fully recovers to comparable levels observed in PBS-treated SCID mice by days 38–40. Precedent for selective pressure promoting the emergence of sub-populations of A. phagocytophilum variants comes from the prior report that, although the bacterium primarily uses sialyl Lewis x-capped P-selectin glycoprotein ligand 1 as a receptor for entry, bacteria capable of infecting cells independent of this ligand be enriched for by cultivation in cell lines that are unable to construct sialyl Lewis x (Sarkar, Reneer and Carlyon 2007; Reneer et al. 2008).
In closing, this study demonstrates that ASM activity is critical for optimal A. phagocytophilum infection in vivo and that its functional inhibition or genetic deficiency reversibly impairs, but does not eliminate infection. It also suggests that A. phagocytophilum intracellular survival and replication benefits from ASM/NPC1-dependent and -independent lipid trafficking pathways.
ACKNOWLEDGMENTS
We thank Dr Rebecca Martin of Virginia Commonwealth University for donating C57BL/6 mice for some of these studies and Curtis Read for helpful discussions.
Contributor Information
Waheeda A Naimi, Department of Microbiology and Immunology, Virginia Commonwealth University (VCU) Medical Center, VCU School of Medicine, Richmond, VA, 23398 USA.
Jacob J Gumpf, Department of Microbiology and Immunology, Virginia Commonwealth University (VCU) Medical Center, VCU School of Medicine, Richmond, VA, 23398 USA.
Chelsea L Cockburn, Department of Microbiology and Immunology, Virginia Commonwealth University (VCU) Medical Center, VCU School of Medicine, Richmond, VA, 23398 USA.
Sarah Camus, Department of Pharmacology and Toxicology, Virginia Commonwealth University (VCU) Medical Center, VCU School of Medicine, Richmond, VA, 23298 USA.
Charles E Chalfant, Department of Cell Biology, Microbiology, and Molecular Biology, University of South Florida, Tampa, FL,33620 USA; Department of Biochemistry and Molecular Biology, Virginia Commonwealth University (VCU), Richmond, VA 23298, USA; The Moffitt Cancer Center, Tampa, FL 33620, USA; Research Service, James A. Haley Veterans' Hospital, Tampa, FL 33612, USA.
Pin-Lan Li, Department of Pharmacology and Toxicology, Virginia Commonwealth University (VCU) Medical Center, VCU School of Medicine, Richmond, VA, 23298 USA.
Jason A Carlyon, Department of Microbiology and Immunology, Virginia Commonwealth University (VCU) Medical Center, VCU School of Medicine, Richmond, VA, 23398 USA.
FUNDING
This work was supported by the National Institutes of Health—National Institute of Allergy and Infectious Diseases grants R01 AI072683 (to JAC) and R01 AI139072 (to JAC and CEC), the National Institutes of Health—National Heart, Lung, and Blood Institute grant HL057244 (to PL) and research grants from the Veterans’ Affairs [VA Merit Review, IBX001792 and Senior Research Career Scientist Award, IK6BX004603 (to CEC)]. The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
Conflict of Interest
None declared.
REFERENCES
- Aguero-Rosenfeld ME, Donnarumma L, Zentmaier L et al. Seroprevalence of antibodies that react with Anaplasma phagocytophila, the agent of human granulocytic ehrlichiosis, in different populations in Westchester County, New York. J Clin Microbiol. 2002;40:2612–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken JS, Dumler JS. Clinical diagnosis and treatment of human granulocytotropic anaplasmosis. Ann N Y Acad Sci. 2006;1078:236–47. [DOI] [PubMed] [Google Scholar]
- Bakken JS, Dumler JS. Human granulocytic anaplasmosis. Infect Dis Clin North Am. 2015;29:341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken JS, Goellner P, Van Etten M et al. Seroprevalence of human granulocytic ehrlichiosis among permanent residents of northwestern Wisconsin. Clin Infect Dis. 1998;27:1491–6. [DOI] [PubMed] [Google Scholar]
- Bakken JS, Krueth J, Wilson-Nordskog C et al. Clinical and laboratory characteristics of human granulocytic ehrlichiosis. JAMA. 1996;275:199–205. [PubMed] [Google Scholar]
- Becker KA, Riethmuller J, Luth A et al. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol. 2010;42:716–24. [DOI] [PubMed] [Google Scholar]
- Belongia EA, Gale CM, Reed KD et al. Population-based incidence of human granulocytic ehrlichiosis in northwestern Wisconsin, 1997–1999. J Infect Dis. 2001;184:1470–4. [DOI] [PubMed] [Google Scholar]
- Brady RO, Kanfer JN, Mock MB et al. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann–Pick disease. Proc Natl Acad Sci USA. 1966;55:366–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyon JA, Akkoyunlu M, Xia L et al. Murine neutrophils require alpha1,3-fucosylation but not PSGL-1 for productive infection with Anaplasma phagocytophilum. Blood. 2003;102:3387–95. [DOI] [PubMed] [Google Scholar]
- Chen SM, Dumler JS, Bakken JS et al. Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol. 1994;32:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn CL, Green RS, Damle SR et al. Functional inhibition of acid sphingomyelinase disrupts infection by intracellular bacterial pathogens. Life Sci Alliance. 2019;2:e201800292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren FS, Heitman KN, Drexler NA et al. Human granulocytic anaplasmosis in the United States from 2008 to 2012: a summary of national surveillance data. Am J Trop Med Hyg. 2015;93:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren FS, Mandel EJ, Krebs JW et al. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000–2007. Am J Trop Med Hyg. 2011;85:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler JS, Barat NC, Barat CE et al. Human granulocytic anaplasmosis and macrophage activation. Clin Infect Dis. 2007;45:199–204. [DOI] [PubMed] [Google Scholar]
- Dunning Hotopp JC, Lin M, Madupu R et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006;2:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen RJ, Eisen L. The blacklegged tick, Ixodes scapularis: an increasing public health concern. Trends Parasitol. 2018;34:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RS, Naimi WA, Oliver LD Jr et al. Binding of host cell surface protein disulfide isomerase by Anaplasma phagocytophilum Asp14 enables pathogen infection. mBio. 2020;11:e03141–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Natarajan R, Payne SG et al. Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J Biol Chem. 2004;279:5821–8. [DOI] [PubMed] [Google Scholar]
- Hayasaka Y, Purgato M, Magni LR et al. Dose equivalents of antidepressants: evidence-based recommendations from randomized controlled trials. J Affect Disord. 2015;180:179–84. [DOI] [PubMed] [Google Scholar]
- Heybrock S, Kanerva K, Meng Y et al. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat Commun. 2019;10:3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodzic E, Ijdo JW, Feng S et al. Granulocytic ehrlichiosis in the laboratory mouse. J Infect Dis. 1998;177:737–45. [DOI] [PubMed] [Google Scholar]
- Horinouchi K, Erlich S, Perl DP et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann–Pick disease. Nat Genet. 1995;10:288–93. [DOI] [PubMed] [Google Scholar]
- Huang B, Troese MJ, Howe D et al. Anaplasma phagocytophilum APH_0032 is expressed late during infection and localizes to the pathogen-occupied vacuolar membrane. Microb Pathog. 2010;49:273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler. 1994;375:447–50. [DOI] [PubMed] [Google Scholar]
- Ismail N, McBride JW. Tick-borne emerging infections: ehrlichiosis and anaplasmosis. Clin Lab Med. 2017;37:317–40. [DOI] [PubMed] [Google Scholar]
- Kim HY, Rikihisa Y. Expression of interleukin-1beta, tumor necrosis factor alpha, and interleukin-6 in human peripheral blood leukocytes exposed to human granulocytic ehrlichiosis agent or recombinant major surface protein P44. Infect Immun. 2000;68:3394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Rikihisa Y. Roles of p38 mitogen-activated protein kinase, NF-kappaB, and protein kinase C in proinflammatory cytokine mRNA expression by human peripheral blood leukocytes, monocytes, and neutrophils in response to Anaplasma phagocytophila. Infect Immun. 2002;70:4132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolzer M, Werth N, Sandhoff K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004;559:96–8. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Medlin A, Bleich S et al. High activity of acid sphingomyelinase in major depression. J Neural Transm (Vienna). 2005;112:1583–90. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Tripal P, Reichel M et al. Functional inhibitors of acid sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. 2010;26:9–20. [DOI] [PubMed] [Google Scholar]
- Kuzu OF, Gowda R, Noory MA et al. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br J Cancer. 2017;117:513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiby DA, Chung AP, Cable RG et al. Relationship between tick bites and the seroprevalence of Babesia microti and Anaplasma phagocytophila (previously Ehrlichia sp.) in blood donors. Transfusion. 2002;42:1585–91. [DOI] [PubMed] [Google Scholar]
- Lin M, Grandinetti G, Hartnell LM et al. Host membrane lipids are trafficked to membranes of intravacuolar bacterium Ehrlichia chaffeensis. Proc Natl Acad Sci USA. 2020;117:8032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Rikihisa Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect Immun. 2003;71:5324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X et al. Niemann–Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–55. [DOI] [PubMed] [Google Scholar]
- Meng Y, Heybrock S, Neculai D et al. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020;30:452–66. [DOI] [PubMed] [Google Scholar]
- Naimi WA, Green RS, Cockburn CL et al. Differential susceptibility of male versus female laboratory mice to Anaplasma phagocytophilum infection. Trop Med Infect Dis. 2018;3:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Xiong Q, Yamamoto A et al. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci USA. 2012;109:20800–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris F, Grassme H, Cremesti A et al. Natural ceramide reverses Fas resistance of acid sphingomyelinase(-/-) hepatocytes. J Biol Chem. 2001;276:8297–305. [DOI] [PubMed] [Google Scholar]
- Reneer DV, Troese MJ, Huang B et al. Anaplasma phagocytophilum PSGL-1-independent infection does not require Syk and leads to less efficient AnkA delivery. Cell Microbiol. 2008;10:1827–38. [DOI] [PubMed] [Google Scholar]
- Sarkar M, Reneer DV, Carlyon JA. Sialyl-Lewis x-independent infection of human myeloid cells by Anaplasma phagocytophilum strains HZ and HGE1. Infect Immun. 2007;75:5720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchman EH, Miranda SR. Niemann–Pick disease: mutation update, genotype/phenotype correlations, and prospects for genetic testing. Genet Test. 1997;1:13–9. [DOI] [PubMed] [Google Scholar]
- Teichgraber V, Ulrich M, Endlich N et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14:382–91. [DOI] [PubMed] [Google Scholar]
- Telford SR III, Dawson JE, Katavolos P et al. Perpetuation of the agent of human granulocytic ehrlichiosis in a deer tick-rodent cycle. Proc Natl Acad Sci USA. 1996;93:6209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troese MJ, Carlyon JA. Anaplasma phagocytophilum dense-cored organisms mediate cellular adherence through recognition of human P-selectin glycoprotein ligand 1. Infect Immun. 2009;77:4018–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truchan HK, Cockburn CL, Hebert KS et al. The pathogen-occupied vacuoles of Anaplasma phagocytophilum and Anaplasma marginale interact with the endoplasmic reticulum. Front Cell Infect Microbiol. 2016a;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truchan HK, VieBrock L, Cockburn CL et al. Anaplasma phagocytophilum Rab10-dependent parasitism of the trans-Golgi network is critical for completion of the infection cycle. Cell Microbiol. 2016b;18:260–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT. Niemann–Pick diseases. Handb Clin Neurol. 2013;113:1717–21. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Lin M, Rikihisa Y. Cholesterol-dependent Anaplasma phagocytophilum exploits the low-density lipoprotein uptake pathway. PLoS Pathog. 2009;5:e1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Rikihisa Y. Subversion of NPC1 pathway of cholesterol transport by Anaplasma phagocytophilum. Cell Microbiol. 2012;14:560–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Wang X, Rikihisa Y. High-cholesterol diet facilitates Anaplasma phagocytophilum infection and up-regulates macrophage inflammatory protein-2 and CXCR2 expression in apolipoprotein E-deficient mice. J Infect Dis. 2007;195:1497–503. [DOI] [PubMed] [Google Scholar]