

Abstract
Talacotuzumab, a humanized anti-CD123 monoclonal antibody, was evaluated in combination with decitabine in elderly patients with acute myeloid leukemia (AML) not eligible for intensive chemotherapy. A multicenter, phase 2/3 study was initiated to determine the recommended phase 2 dose (RP2D) of talacotuzumab (Part A) followed by an open-label, randomized comparison of talacotuzumab in combination with decitabine versus decitabine alone to assess achievement of complete response (CR) and overall survival (OS) in Part B. Ten patients were enrolled in Part A and 316 in Part B; the results presented here are based on a database lock on January 25, 2018. Part A confirmed the RP2D of talacotuzumab to be 9 mg/kg. In Part B, CR was achieved in 12/80 (15%) patients receiving combination therapy and in 9/82 (11%) patients receiving decitabine alone (odds ratio: 1.4; 95% confidence interval [CI]: 0.6–3.6; p = 0.44). Median (95% CI) OS was 5.36 (4.27–7.95) months for combination therapy versus 7.26 (6.47–8.64) months for decitabine alone (hazard ratio: 1.04; 95% CI: 0.79–1.37; p = 0.78). Combination therapy showed no improvement in efficacy versus decitabine alone, resulting in the Independent Data Monitoring Committee’s recommendation of early termination of enrollment and discontinuation of talacotuzumab treatment.
Subject terms: Acute myeloid leukaemia, Cancer
Introduction
Treatment options for older patients with acute myeloid leukemia (AML) have been limited; although younger patients with AML can generally tolerate induction chemotherapy, potential risks of standard induction outweigh the benefits in many older AML patients [1]. Treatment alternatives for these patients were limited for many years to either supportive care or low-intensity treatments such as low-dose cytarabine or hypomethylating agents (decitabine or azacitidine) [2]. Only modest improvements in overall survival (OS) were achieved with both decitabine and azacitidine compared to low-dose cytarabine, with median OS still <1 year for patients over 65 years of age [2]. Combination strategies based on these agents have been explored to improve outcome while maintaining adequate tolerability for older patients. Venetoclax combined with decitabine or azacitidine has recently gained accelerated approval in the US based on response rates from a nonrandomized study [3].
Leukemic stem and progenitor cells (LSPCs) possess biological properties that render these cells resistant to conventional chemotherapies [4]. Thus, targeted approaches to eliminate LSPCs are currently being investigated to improve prognosis and prevent relapses in patients with AML [5]. Human interleukin-3 receptor alpha chain (IL-3Rα or CD123), the major binding protein for IL-3, is overexpressed on AML blast cells and LSPCs [6]. Upregulated expression of CD123 has been associated with higher blast cells counts at diagnosis and poorer complete response (CR) to standard treatment and survival rates in AML [7]. As a result, increased expression of CD123 on AML-LSPCs provides an opportunity for therapeutic monoclonal antibody (mAb) targeting [8, 9].
Therapeutic targeting of LSPCs, in addition to leukemic blast cells, could potentially enhance the rate and duration of response in AML patients [4]. The dismal prognosis of patients with advanced age not eligible for intensive chemotherapy suggests that this setting may be the best match to demonstrate clinical benefit with talacotuzumab. Classified as a second generation humanized anti-CD123 mAb, talacotuzumab, binds to CD123 and inhibits the downstream IL-3 signaling cascade. Talacotuzumab has been engineered to have a 100-fold higher affinity to Fc receptor CD16 on natural killer cells, resulting in enhanced ability to induce antibody-dependent cellular cytotoxicity (ADCC) against target cells expressing CD123 (such as AML blasts and LSPCs) [9]. Decitabine promotes re-expression of tumor suppressor genes [10] and has been used extensively in patients not candidates for intensive chemotherapy [2]. However, the median OS following decitabine treatment is <8 months for patients ≥65 years of age [11, 12]. The combination of decitabine and talacotuzumab was hypothesized to have complimentary/synergistic effects on leukemic cells due to different underlying mechanisms of action and differences in the leukemic cell populations targeted by these two agents.
The current study was designed to confirm the recommended phase 2 dose (RP2D) of talacotuzumab and to evaluate the CR rate and OS following either talacotuzumab plus decitabine or decitabine alone in patients with previously untreated AML not eligible for intensive induction chemotherapy.
Methods
Patients
In Part A, patients with AML (per WHO 2008 criteria) were eligible if they were treatment naïve or had refractory or relapsed disease. Part B included patients ≥65 years old with previously untreated AML not eligible for standard intensive induction chemotherapy or allogeneic hematopoietic stem cell transplantation. Inclusion and exclusion criteria are described in detail in the supplementary file.
Study design
This study was a two-part, open-label (Part A), and randomized (Part B) phase 2/3 study conducted across 96 sites in 15 countries from August 2015 to January 2018. The screening phase was up to 28 days prior to treatment initiation (Part A) or randomization (Part B). Part A was conducted to assess the safety and confirm the RP2D of talacotuzumab in patients with AML eligible for experimental therapy. Part B was conducted to assess the CR rate and OS in previously untreated patients with AML considered unfit for intense induction chemotherapy or hematopoietic stem cell transplantation.
In Part A, at least six patients were to be enrolled and receive one dose of talacotuzumab monotherapy at 9 mg/kg on Day 1 of every 28-day cycle as a 180-min intravenous (IV) infusion. Dose-limiting toxicities (DLTs) were assessed during the subsequent 14-day DLT evaluation period. Pharmacokinetic (PK) and pharmacodynamic (PD) assessments were conducted during the 14-day talacotuzumab evaluation period. RP2D was established by a study evaluation team (SET). Patients in Part A continued the study (in Part A) and started subsequent cycles of combined study therapy with IV decitabine (20 mg/m2/d) on Days 1–5 followed by 9 mg/kg (or a lower dose if determined by the SET) talacotuzumab on Day 8 and Day 22 of a 28-day cycle.
Part B was initiated after the RP2D of talacotuzumab was confirmed; eligible patients were randomized in a 1:1 ratio using an interactive web response system to receive talacotuzumab + decitabine or decitabine alone. Randomization was stratified by baseline Eastern Cooperative Oncology Group (ECOG) performance status score (0–1 versus 2), type of AML (de novo versus secondary), and cytogenetic risk by European Leukemia Net 2010 [13] cytogenetic characterization (adverse versus others).
Treatment
Patients in both treatment arms received decitabine 20 mg/m2/d on Days 1–5 of each 28-day cycle. Patients in the talacotuzumab + decitabine group were administered 9 mg/kg IV talacotuzumab on Day 8 and Day 22 every 28 days. An Independent Data Monitoring Committee (IDMC) was established to evaluate safety and efficacy data during Part B, including the formal efficacy interim analyses (IA) and make recommendations for study conduct.
An Institutional Review Board and Independent Ethics Committee approved the study protocol and amendments. The study was conducted in accordance with ethical principles of the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. All patients provided written informed consent before participating in the study. This trial was registered at ClinicalTrials.gov (NCT02472145).
Efficacy assessments
The primary efficacy endpoints were CR rate (proportion of patients achieving CR) and OS (time from randomization to death from any cause). Disease responses were evaluated according to ELN response criteria [13] by investigators and a blinded independent data review committee (IRC). Disease status based on independent central review was the primary source for efficacy analyses.
Key secondary efficacy endpoints included: event-free survival (EFS; time from randomization to treatment failure, relapse from CR or CR with incomplete blood count recovery [CRi], or death, whichever occurred first), overall response rate (ORR; CR + CRi), time to response and duration of response for patients who achieved CR or CRi, minimal residual disease (MRD) negativity (defined as <1 blast or leukemic stem cell in 10,000 leukocytes [i.e., MRD level <10−4]). MRD assessments were performed on bone marrow aspirates and/or whole blood collected at various time points in Part B. Bone marrow aspirate specimens were evaluated for MRD status for patients who achieved a response using flow cytometry at a central laboratory. Additional secondary efficacy endpoints and their details are defined in the Supplementary file.
Pharmacokinetic assessments
In Part A, all patients received a single IV infusion of 9 mg/kg talacotuzumab as monotherapy on Day 1. Serum concentrations of talacotuzumab were measured for PK parameters during the 14-day evaluation period. In Part B, blood samples were collected for decitabine and talacotuzumab PK assessments. The time points for sample collection are described in the Supplementary file. Serum concentrations of talacotuzumab and plasma concentrations of decitabine were determined using validated and sensitive methods with a lower limit of quantification of 10 μg/mL for serum talacotuzumab and 1 ng/mL for plasma decitabine concentrations. PK parameters such as Cmax (maximum observed concentration) and Cmin (minimum observed concentration) were evaluated in both Part A and Part B. Methods for biomarkers and PD assessment are included in the Supplementary file.
Immunogenicity
The immunogenicity of talacotuzumab alone (in Part A) and in combination with decitabine (in Part B) was evaluated based on the development of antidrug antibodies (ADAs) for talacotuzumab. Blood samples were collected for immunogenicity evaluation at predose, end of study, and follow-up phase for talacotuzumab.
Safety assessments
Safety evaluations were performed based on treatment-emergent adverse events (TEAEs), laboratory analyte values (clinical hematology and chemistry), vital sign measurements, and periodic electrocardiogram data. AEs were coded using Medical Dictionary for Regulatory Activities Version 20.0 and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, Version 4.03.
Statistical analysis
Sample size determination
Part B of the study was planned to enroll up to 400 patients (approximately 200 for each arm). Both primary endpoints, CR rate and OS, were to be powered for 80% with an overall α allocation of 0.01 and 0.04, respectively. For CR rate, the effect size to be detected was 40% for talacotuzumab + decitabine group versus 15% for decitabine alone group, requiring a total of 160 patients (80 per arm). For OS, the targeted effect size in terms of median OS was 11.4 months for talacotuzumab + decitabine group versus 8.0 months for decitabine alone group, or a hazard ratio (HR) of 0.70, requiring total of 270 deaths.
Interim analysis
For Part B of the study, three IAs were planned. The first IA occurred as planned after ~80 patients (40 per arm) were randomized and had follow up for at least 4 months. Guided by predefined statistical criteria based on CR and CR + CRi rate, the IDMC were to determine if the study should continue enrollment to the full prespecified phase 3 sample size of 400 patients. Subsequently, another two IAs were planned: (1) after 160 patients were randomized and followed for at least 4 months (final analysis for CR rate and the first IA for OS), (2) when 180 deaths had occurred (second IA of OS); final analysis of OS was planned with a total of 270 deaths. O’Brien-Fleming α spending procedure was utilized for the IA and final analysis for OS.
Analysis sets
Intent-to-treat (ITT): all patients randomized into the study, grouped per treatment assigned by randomization, regardless of the actual treatment received. Safety: all randomized patients who received at least one dose of study medication, grouped according to actual treatment received.
Planned analyses
For PK analysis, descriptive statistics were used to summarize serum talacotuzumab and plasma decitabine concentrations at each sampling timepoint. Number and percent of patients achieving CR were presented by treatment arm. Odds ratio (talacotuzumab + decitabine versus decitabine alone) was reported along with the associated 95% CI based on a stratified logistic regression with treatment as the only covariate. The p value was based on a stratified Cochran–Mantel–Haenszel test. Stratification factors are those used in randomization. Kaplan–Meier estimates for OS were presented graphically, and the p value was calculated using the stratified log-rank test. Time to response and duration of response were summarized by treatment arm using the Kaplan–Meier method. The safety results were summarized descriptively.
Results
Patient characteristics and disposition
In Part A of the study, ten patients were enrolled. All the patients were Caucasian, with a median age of 67.5 (range: 51–78) years, and the majority were men (60%) (Table 1). At the time of final clinical cutoff, all patients in Part A had discontinued the study due to death (including one death before starting the combination treatment with decitabine) (Fig. 1).
Table 1.
Patient demographics and baseline characteristics.
| Characteristics | Part A (All enrolled set) | Part B (ITT analysis set) | ||
|---|---|---|---|---|
| Talacotuzumab alone (n = 10) | Decitabine alone (n = 159) | Talacotuzumab + Decitabine (n = 157) | Total (n = 316) | |
| Age, median (range), years | 67.5 (51–78) | 75.0 (65–89) | 75.0 (65–92) | 75.0 (65–92) |
| Gender, n (%) | ||||
| Men | 6 (60.0) | 91 (57.2) | 80 (51.0) | 171 (54.1) |
| Race, n (%) | ||||
| White (Caucasian) | 10 (100.0) | 133 (84.2) | 141 (89.8) | 274 (87.0) |
| Type of AML, n (%) | ||||
| De novo | 106 (66.7) | 107 (68.2) | 213 (67.4) | |
| Secondary | 53 (33.3) | 50 (31.8) | 103 (32.6) | |
| History of MDS | 24 (45.3) | 21 (42.0) | 45 (43.7) | |
| History of myeloproliferative disorder | 19 (35.8) | 23 (46.0) | 42 (40.8) | |
| Previous leukemogenic exposure | 10 (18.9) | 6 (12.0) | 16 (15.5) | |
| ECOG performance status, n (%) | ||||
| 0 | 29 (18.2) | 30 (19.1) | 59 (18.7) | |
| 1 | 69 (43.4) | 65 (41.4) | 134 (42.4) | |
| 2 | 61 (38.4) | 62 (39.5) | 123 (38.9) | |
| Bone marrow blast—Aspiration, median (range) | 39.50 (15.0; 100.0) | 45.00 (14.0; 100.0) | 42.00 (14.0; 100.0) | |
| Bone marrow blast—Biopsy, median (range) | 38.00 (4.0; 90.0) | 40.00 (0.0; 92.0) | 40.00 (0.0; 92.0) | |
| Classification of risk, n (%) | ||||
| Favorable | 8 (5.0) | 10 (6.4) | 18 (5.7) | |
| Intermediate-I | 61 (38.4) | 51 (32.5) | 112 (35.4) | |
| Intermediate-II | 33 (20.8) | 32 (20.4) | 65 (20.6) | |
| Adverse | 47 (29.6) | 57 (36.3) | 104 (32.9) | |
| Patients with previous cancer-related therapy, n (%) | ||||
| Systemic therapy | 33 (20.8) | 24 (15.3) | 57 (18.0) | |
| Radiotherapy | 1 (0.6) | 5 (3.2) | 6 (1.9) | |
| Cancer-related surgery/procedure | 10 (6.3) | 10 (6.4) | 20 (6.3) | |
AML acute myeloid leukemia, ECOG Eastern Cooperative Oncology Group, ITT intent-to-treat, MDS myelodysplastic syndrome.
Fig. 1. Study design and patient disposition.

aPatients randomized in the decitabine + talacotuzumab arm who received only decitabine and not talacotuzumab were grouped in the decitabine alone arm; bpatients still being treated with decitabine as they continued to derive benefit from the treatment. RP2D recommended phase 2 dose.
In Part B of the study, between October 26, 2015 and July 28, 2017 a total of 316 patients were randomized 1:1 to either talacotuzumab + decitabine (n = 157) or decitabine alone (n = 159). The patient demographics were similar between the treatment groups. The patients had a median age of 75 (range: 65–92) years and were mostly Caucasian (87.0%). The majority of patients presented with de novo AML (67.4%) and with a baseline ECOG score of 1 or 2 (81.3%) (Table 1). The safety analysis set included 312 patients, 147 (93.6%) patients in the talacotuzumab +decitabine group and 165 (103.8%) in the decitabine alone group (Fig. 1). At the final clinical cutoff (which occurred 6 months after the IDMC recommendation to discontinue treatment with talacotuzumab), 262 (82.9%) patients had discontinued treatment, and treatment with decitabine alone was ongoing for 52 (16.5%) patients. Death (n = 65; 24.8%) was the most common reason for treatment discontinuation including 35 patients randomized in the talacotuzumab + decitabine arm (Fig. 1).
The median treatment duration was 3.71 (range: 0.3–24) months in the talacotuzumab + decitabine arm, with 67 (45.6%) patients receiving <3 months of treatment and 4 (2.7%) patients receiving at least 18 months of treatment. The median number of cycles received was 2 (range: 1–17 cycles). In the decitabine alone arm, the median treatment duration was 3.61 (range: 0.0–22.5) months, with 79 (47.9%) patients receiving <3 months of treatment and 3 (1.8%) patients receiving at least 18 months of treatment. The median number of cycles received was 4 (range: 1–22 cycles).
Primary endpoints
As per the IRC assessment based on 162 patients (80 in the talacotuzumab + decitabine group and 82 in the decitabine alone group) from the second IA, CR was achieved in 12 (15.0%) patients receiving talacotuzumab + decitabine versus in 9 (11.0%) patients receiving decitabine alone. The odds ratio for CR rate was 1.4 (95% CI: 0.6, 3.6); p = 0.44. In the ITT population of Part B, CR was achieved in 26 (16.6%) patients receiving talacotuzumab +decitabine versus in 19 (11.9%) patients receiving decitabine alone. The odds ratio for CR rate was 1.5 (95% CI: 0.8, 2.8); p = 0.47. No clinically meaningful or statistically significant improvement in CR was observed with combined talacotuzumab + decitabine treatment, compared with treatment with decitabine alone in either the IRC assessed group or the overall ITT population. In the talacotuzumab +decitabine arm, 4 (10.3%) patients with low baseline CD123 blasts achieved CR; whereas, more patients (8 [20.5%]) with high baseline CD123 blasts achieved CR. In the decitabine alone arm, 4 (10.0%) patients with low baseline CD123 blasts and 5 (12.5%) patients with high baseline CD123 blasts achieved CR.
In the ITT population, death occurred in 101 (64.3%) patients in the talacotuzumab + decitabine group, and 102 (64.2%) patients in the decitabine alone group. The median OS was 5.36 (95% CI: 4.27, 7.95) months for talacotuzumab + decitabine group and 7.26 (95% CI: 6.47, 8.64) months for decitabine alone group, with an estimated HR (talacotuzumab + decitabine versus decitabine alone) of 1.04 (95% CI: 0.79, 1.37; p = 0.78) (Table 2 and Fig. 2). No improvement in the OS was observed with combined talacotuzumab + decitabine treatment as compared with treatment with decitabine alone.
Table 2.
Primary and secondary efficacy endpoints (intent-to-treat population).
| Per investigators assessment | Per the IRC assessment | |||
|---|---|---|---|---|
| Parameter | Decitabine + Talacotuzumab | Decitabine | Decitabine + Talacotuzumab | Decitabine |
| Primary endpoints | ||||
| Na | 157 | 159 | 80 | 82 |
| Complete response (CR), n (%) | 26 (16.6) | 19 (11.9) | 12 (15.0) | 9 (11.0) |
| Odds ratio (95% CI)b | 1.5 (0.8; 2.8) | 1.4 (0.6; 3.6) | ||
| p valuec | 0.4747 | 0.4403 | ||
| N | 157 | 159 | ||
| Overall survival, median (95% CI), months | 5.36 (4.27; 7.95) | 7.26 (6.47; 8.64) | ||
| Hazard ratio (95% CI)d | 1.04 (0.79; 1.37) | |||
| p valuee | 0.7817 | |||
| Secondary endpoints | ||||
| Na | 157 | 159 | 80 | 82 |
| CR with incomplete blood counts recovery (CRi), n (%) | 16 (10.2) | 13 (8.2) | 9 (11.3) | 8 (9.8) |
| Overall response (CR + CRi), n (%) | 42 (26.8) | 32 (20.1) | 21 (26.3) | 17 (20.7) |
| Odds ratio (95% CI)b | 1.4 (0.9; 2.4) | 1.4 (0.7; 2.8) | ||
| p valuec | 0.4145 | 0.2805 | ||
| MLFS, n (%) | 14 (8.9) | 24 (15.1) | 9 (11.3) | 12 (14.6) |
| Time to initial responsef, median (range), weeks | 15.57 (7.1–60.0) | 9.43 (6.4–46.1) | ||
| Time to best responseg, median (range), weeks | 16.71 (7.4–60.0) | 15.43 (7.1–46.1) | ||
| Duration of best responseh, median (95% CI), weeks | 56.43 (16.00; 56.43) | 23.43 (8.71; 33.71) | ||
| Event-free survival, median (95% CI), months | 4.60 (3.61; 7.20) | 4.24 (3.32; 6.70) | ||
| Hazard ratio (95% CI)d | 0.76 (0.53; 1.09) | |||
| p valuee | 0.1286 | |||
| Na | 80 | 80 | ||
| MRD-Negative (CR + CRi + MLFS), n (%) | 13 (16.3) | 8 (10.0) | ||
| p valuei | 0.3493 | |||
AML acute myeloid leukemia, CI confidence interval, CR complete response, CRi CR with incomplete blood counts recovery, ECOG Eastern Cooperative Oncology Group, IRC Independent Review Committee, MLFS morphologic leukemia-free state, MRD minimal residual disease.
aInclude patients whose data were reviewed by IRC.
bOdds ratio >1 favors talacotuzumab + decitabine; Odds ratio is from a logistic regression with treatment as the only covariate.
cp value is from a CMH chi-square test adjusted for stratification factors.
dHazard ratio is from a stratified proportional hazards model. A hazard ratio <1 favors talacotuzumab + decitabine.
ep value is from a log-rank test stratified by two randomization stratification factors: baseline ECOG performance status (0–1 versus 2) and type of AML (de novo versus secondary).
fTime to initial response is calculated as the time from the randomization date to first documented response for patients who achieved CR or CRi.
gTime to best response is calculated as the time from the randomization date to the first documented date for the best response for patients who achieved CR or CRi.
hDuration of best response is calculated as the number of weeks from documented best response (CR or CRi) for patients who achieved CR or CRi to relapse, death due to relapse, or date of censoring.
ip values are based on Fisher’s Exact test.
Fig. 2. Kaplan–Meier curves for overall survival; ITT analysis set.
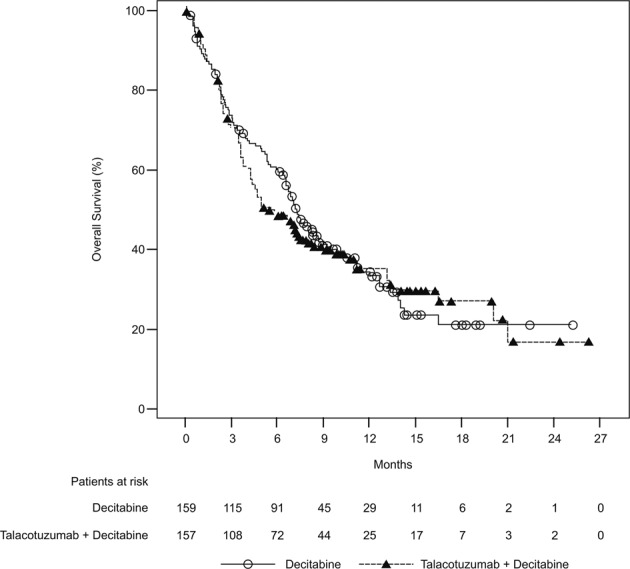
ITT Intent-to-treat.
Furthermore, the median OS in adverse cytogenetic risk subgroup was 4.90 months in the talacotuzumab + decitabine arm and 3.91 months in the decitabine alone arm, with an estimated HR (talacotuzumab + decitabine versus decitabine alone) of 0.78 (95% CI: 0.49, 1.24). The median OS in the nonadverse cytogenetic risk subgroup was 6.90 months in the talacotuzumab + decitabine arm and 8.25 months in the decitabine alone arm with an estimated HR (talacotuzumab + decitabine versus decitabine alone) of 1.18 (95% CI: 0.84, 1.66).
Secondary efficacy endpoints
The median EFS was 4.60 (95% CI: 3.61, 7.20) months for talacotuzumab + decitabine versus 4.24 (95% CI: 3.32, 6.70) months for decitabine alone (HR 0.76 [95% CI: 0.53, 1.09]; p = 0.1286). In the talacotuzumab + decitabine group, 9/80 (11.3%) patients achieved CRi and 8/82 (9.8%) patients receiving decitabine alone achieved CRi. As per the IRC assessment, the ORR (CR + CRi) was observed in 21 (26.3%) patients following combined treatment with talacotuzumab +decitabine versus 17 (20.7%) patients in the decitabine alone arm, with an odds ratio of 1.4 (95% CI: 0.7, 2.8); p = 0.2805. Per investigator assessment, the ORR was observed in 42 (26.8%) patients following combined treatment with talacotuzumab + decitabine versus 32 (20.1%) patients in the decitabine alone arm, with an odds ratio of 1.4 (95% CI: 0.9, 2.4); p = 0.41 (Table 2). In the talacotuzumab +decitabine group, morphologic leukemia-free state (MLFS) was observed in 9 (11.3%) patients, partial response (PR) was observed in 1 (1.3%) patient, and stable disease (SD) was observed in 30 (37.5%) patients. In the decitabine alone group, MLFS was reported for 12 (14.6%) patients, and SD was reported for 28 (34.1%) patients. In the talacotuzumab +decitabine group, 6 (15.4%) patients with low baseline CD123 blasts and 3 (7.7%) patients with high baseline CD123 blasts achieved CRi. In the decitabine alone group, 4 (10.0%) patients with low baseline CD123 blasts and 4 (10.0%) patients with high baseline CD123 blasts achieved CR. Response rates based on baseline CD123 blasts are summarized in the supplemantary Table S1.
The median time to initial response was 15.57 weeks in the talacotuzumab + decitabine group versus 9.43 weeks for decitabine alone; while the median time to best response was 16.71 weeks in the talacotuzumab + decitabine group and 15.43 weeks for those receiving decitabine alone. The median duration of best response for the talacotuzumab + decitabine group was longer compared with decitabine alone; 56.43 (95% CI: 16.00, 56.43) weeks and 23.43 (95% CI: 8.71, 33.71) weeks, respectively.
Analysis of MRD was based on 160 patients included in the second IA, and assessed at the time of response (CR, CRi, or MLFS), as confirmed by investigators. MRD negativity occurred in 13 (16.3%) receiving talacotuzumab + decitabine versus 8 (10.0%) patients treated with decitabine alone (p = 0.3493). Overall, there were no clinically meaningful or statistically significant improvements in EFS, ORR, or MRD negativity for the talacotuzumab + decitabine group compared with decitabine alone.
Pharmacokinetics
In Part A, patients who received a single IV infusion of talacotuzumab as monotherapy at 9 mg/kg on Day 1 and who were evaluable for PK analysis had observed mean Cmax (C1D1 30 min) of 135.7 µg/mL and observed mean Cmin (C2D1 predose) of 9.8 µg/mL (Fig. 3a). As the mean Cmin concentration of talacotuzumab following the first IV dose was above the desired target of 9 µg/mL, the predefined criteria for Part B initiation at the RP2D of 9 mg/kg every 14 days was met. In Part B, the observed mean plasma decitabine Cmax in cycle 1 was higher in the talacotuzumab + decitabine group (215.9 ng/mL) than that in the decitabine alone group (164.1 ng/mL). Mean serum talacotuzumab Cmax after cycle 1 (179.6 µg/mL) and cycle 4 (187.6 µg/mL) were comparable (Fig. 3b). The observed mean trough concentration at cycle 2 (14.7 µg/mL) was lower than that at cycle 4 (30.1 µg/mL). In both parts of the study, interindividual variability in talacotuzumab PK was observed.
Fig. 3. Serum concentration of talacotuzumab with/without decitabine.

a Serum concentration of talacotuzumab alone in Part A. b Serum concentration of talacotuzumab + decitabine in Part B. EOT end of trial.
Immunogenicity
Immunogenicity samples were available from 10 patients in Part A and 136 patients in Part B (the talacotuzumab + decitabine group only) treated with talacotuzumab. Among these patients, 1 (10.0%) patient in Part A and 35 (26.0%) patients in Part B developed ADA. Neutralizing antibody (Nab) was evaluated in 33 of 35 ADA positive patients and 21 (63.6%) patients were found to be Nab positive.
Biomarkers and pharmacodynamics
In Part A, treatment with talacotuzumab demonstrated a reduction of basophils, plasmacytoid dendritic cells (pDCs), NK cells, and blasts in the peripheral blood, indicative of a PD response to a single IV infusion, which supported the RP2D of 9 mg/kg. In Part B, rapid reduction of basophils and pDCs were noted equally for both treatment arms. Other efficacy biomarkers (blasts and NK cells) were also reduced similarly between both treatment arms. The frequency of peripheral blood target positive (CD123+) blasts as a percentage of total blasts was reduced in patients treated with talacotuzumab + decitabine compared with decitabine alone.
Safety
In Part A, all ten patients reported at least 1 TEAE; 7 (70.0%) patients reported drug-related TEAEs, and 7 (70.0%) patients with Grade ≥3 TEAEs. Serious TEAEs were reported for 4 (40.0%) patients. One patient died due to a TEAE of a pre-existing condition aggravated with the combination of talacotuzumab + decitabine. Nine patients from Part A were subsequently treated with combination treatment, and all were reported with at least 1 TEAE. Eight (88.9%) patients reported with drug-related TEAEs, which were attributed to decitabine and talacotuzumab. Two (22.2%) patients reported TEAEs leading to treatment discontinuation and 5 (55.6%) patients died due to TEAEs (Table 3).
Table 3.
Summary of treatment-emergent adverse events (safety population).
| Part A (all enrolled set) | Part B (safety analysis set) | ||
|---|---|---|---|
| n (%) | Talacotuzumaba (n = 10) | Decitabine + Talacotuzumab (n = 147) | Decitabine alone (n = 165) |
| Any TEAEs | 10 (100) | 147 (100) | 164 (99.4) |
| TEAEs | 7 (70) | 128 (87.1) | 124 (75.2) |
| Grade ≥3 TEAEs | 7 (70) | 145 (98.6) | 157 (95.2) |
| Serious TEAEs | 4 (40) | 126 (85.7) | 120 (72.7) |
| Grade ≥3 serious TEAEs | 4 (40) | 124 (84.4) | 115 (69.7) |
| TEAEs leading to treatment discontinuation | 0 | 40 (27.2) | 26 (15.8) |
| Decitabine | – | 33 (22.4) | 26 (15.8) |
| Talacotuzumab | 0 | 32 (21.8) | 2 (1.2) |
| TEAEs leading to death | 1 (10) | 49 (33.3) | 46 (27.9) |
| Serious TEAEs leading to hospitalization | 3 (30) | 112 (76.2) | 99 (60.0) |
| Most common TEAEs (>20% of patients) | |||
| Dyspnea | 5 (50) | 25 (17.0) | 24 (14.5) |
| Pyrexia | 4 (40) | 60 (40.8) | 48 (29.1) |
| Anemia | 3 (30) | 80 (54.4) | 80 (48.5) |
| Asthenia | 2 (20) | 27 (18.4) | 25 (15.2) |
| Chills | 2 (20) | 31 (21.1) | 7 (4.2) |
| Peripheral edema | 2 (20) | 46 (31.3) | 25 (15.2) |
| Nausea | 2 (20) | 38 (25.9) | 33 (20.0) |
| Thrombocytopenia | – | 81 (55.1) | 87 (52.7) |
| Neutropenia | 1 (10) | 66 (44.9) | 61 (37.0) |
| Febrile neutropenia | 1 (10) | 59 (40.1) | 50 (30.3) |
| Hypokalemia | – | 53 (36.1) | 41 (24.8) |
| Diarrhea | 1 (10) | 50 (34.0) | 44 (26.7) |
| Constipation | 1 (10) | 47 (32.0) | 51 (30.9) |
| Pneumonia | 1 (10) | 39 (26.5) | 37 (22.4) |
| Fatigue | – | 31 (21.1) | 31 (18.8) |
| Most common Grade ≥3 TEAEs (>20% of patients) | |||
| Anemia | 3 (30) | 75 (51.0) | 71 (43.0) |
| Dyspnea | 2 (20) | 9 (6.1) | 5 (3.0) |
| Thrombocytopenia | – | 73 (49.7) | 83 (50.3) |
| Neutropenia | 1 (10) | 65 (44.2) | 60 (36.4) |
| Febrile neutropenia | 1 (10) | 59 (40.1) | 50 (30.3) |
| Pneumonia | 1 (10) | 36 (24.5) | 33 (20.0) |
| Infusion-related TEAEs | – | 55 (37.4) | 0 |
| Grade 3 | – | 20 (13.6) | 0 |
| Grade 4 | – | 2 (1.4) | 0 |
| Grade 5 | – | 1 (0.7) | 0 |
TEAE treatment-emergent adverse event.
aPatients on talacotuzumab alone, prior to initiating combination treatment.
In Part B, all 147 (100%) patients in the talacotuzumab +decitabine group and 164 (99.4%) patients in the decitabine alone group reported at least 1 TEAE. The most common TEAEs leading to death (the talacotuzumab + decitabine group versus the decitabine alone group) included sepsis (4.8% versus 0.6%), multiple organ dysfunction syndrome (5.4% versus 5.5%), pneumonia (3.4% versus 5.5%), septic shock (3.4% versus 0.6%), and sudden death (0.7% versus 3.0%). In the talacotuzumab + decitabine group, 55 (37.4%) patients reported infusion-related reactions (IRR)-associated TEAEs, of whom 22 (15.0%) patients had Grade ≥3 TEAEs and 1 (0.7%) patient had Grade 5 TEAEs. The commonly reported IRR-associated TEAEs included chills (16.3%), pyrexia (5.4%), and hypoxia (4.8%). In the decitabine alone group, none of the patients reported an infusion-related TEAE. Blood and lymphatic systems disorders were observed in 82.3% patients in the combination arm and 80.0% in the decitabine alone arm of which most common TEAEs (the talacotuzumab + decitabine group versus decitabine alone group) were anemia (51.0% versus 43.0%), thrombocytopenia (49.7% versus 50.3%), neutropenia (44.2% versus 36.4%), and febrile neutropenia (40.1% versus 30.3%).
Discussion
Results from Part A confirmed the RP2D for talacotuzumab as 9 mg/kg. The results of Part B suggested that addition of talacotuzumab to decitabine monotherapy regimen was not superior to decitabine monotherapy in older patients with AML. No clinically meaningful or statistically significant difference in CR, OS, and other secondary efficacy endpoints were observed in the combination arm as compared with the decitabine alone arm. Higher incidence of IRR in the decitabine + talacotuzumab arm as compared with the decitabine alone arm were reported. The lack of efficacy and a concerning safety profile resulted in an unfavorable risk/benefit profile that did not support the continuation of treatment with talacotuzumab. These results are similar to the finding of a recent study of talacotuzumab conducted in elderly high-risk AML or myelodysplastic syndrome patients [14]. In the current study, the median OS for decitabine (7.26 months) was comparable to the decitabine outcomes in a phase 3 study (7.7 months) conducted in elderly patients with newly diagnosed AML [12]. Furthermore, the median OS following talacotuzumab + decitabine (5.36 months) was lower than that observed in patients (aged ≥65 years) receiving azacitidine (10.4 months). However, a direct comparison between the two studies cannot be drawn because, unlike the current study, AZA-AML-001 included nearly 15% patients eligible for intensive chemotherapy [15].
The incidence of AML increases progressively with age; advanced age, comorbidities, compromised organ functions contribute to poor prognosis in older patients with AML [16]. Intensive and aggressive chemotherapy may improve the efficacy outcomes in these patients, but toxicity and adverse events limit their use in elderly patients unfit for intensive therapy [17]. Until recently, the recommended standard of care for these patients is low-intensity therapies such as decitabine, azacitidine, and low-dose cytarabine, but the prognosis is suboptimal [12, 15, 18]. Studies evaluating new treatments or combinations for older patients with AML are underway, including the emerging data of venetoclax in combination with decitabine or azacitidine that can potentially become standard care for these patients. In the current study, patient population comprised elderly patients (median age: 75 years [range: 65–92 years]), with more than 50% patients having de novo AML, nearly 40% patients with ECOG score of 2, about one-third with adverse-risk cytogenetics, and 42% median baseline blasts in bone marrow. Baseline disease characteristics of patient enrolled in this current study were similar to those who participated in the multicenter phase 3 decitabine study. Furthermore, for the decitabine monotherapy arm, the median OS (7.26 [95% CI: 6.47; 8.64] months) was consistent with the median OS for decitabine reported in the decitabine phase 2 study (7.7 [95% CI: 5.7; 11.6] months) [11] and the decitabine phase 3 study (7.7 [95% CI: 6.2; 9.2] months) [12].
In the current study, nearly all patients in both treatment arms experienced at least one TEAE. The percentage of patient with serious TEAEs, TEAEs leading to treatment discontinuation, serious TEAEs leading to hospitalization were higher in the talacotuzumab + decitabine arm as compared with the decitabine alone arm. All patients in either arm experienced myelosuppression, however, the proportion was higher in the talacotuzumab + decitabine arm. IRR (pyrexia, peripheral edema, and chills) were substantially higher in the talacotuzumab + decitabine arm as compared with the decitabine alone arm. The number of deaths in the talacotuzumab + decitabine arm was lower as compared with the decitabine alone arm. Insignificant improvement in efficacy outcomes, high risk of myelosuppression, IRR leading to treatment discontinuations and deaths resulted in an unfavorable risk/benefit profile, and contributed to the early discontinuation of talacotuzumab treatment. For mAb-based treatments, development of ADA plays a major role in loss of therapeutic response and treatment failure [19]. In the current study, nearly two-thirds of the patients treated with talacotuzumab + decitabine developed Nab, and this could explain in part, the lack of a striking antileukemic activity of talacotuzumab.
A comparison of complete and overall response between the two treatment arms showed that numerically higher values for CR, ORR, and MRD negativity were observed for the talacotuzumab + decitabine arm versus the decitabine alone arm. Further, reductions of basophils, pDCs, NK cells, blasts/leukocytes, and CD123+ blasts in the peripheral blood were observed suggesting a PD response to talacotuzumab. Rapid reduction of basophils and pDCs for both treatment arms suggests that decitabine may have a cytotoxic effect on talacotuzumab cellular PD biomarkers. Treatment with talacotuzumab reduced the CD123 targets, based on post-dose changes in NK cells, and reduced the CD123+ blasts as a percentage of total blasts. However, lacked optimal safety requirement leading to unacceptable benefit/risk profile. Taken together, these observations suggest that mAbs targeting CD123 with improved tolerability and lesser propensity to develop ADAs could be developed. Furthermore, development of mAbs using additional bioengineering modifications to enhance desirable properties such as ADCC would potentially exhibit improved antileukemic efficacy with an acceptable safety profile and could be valuable in the therapeutic management of AML.
mAbs are known to elicit their responses either via induction of cellular apoptosis or via ADCC. Targeted Fc engineering either by glycosylation or by mutagenesis is known to increase the molecular affinity toward the target cells such as CD16 on the NK cells which further accentuate the NK-mediated ADCC [20]. Talacotuzumab has been engineered toward human IgG (humanized) and is known to have increased affinity to the Fc receptor CD16 on NK cells through which it has effectively induced ADCC of AML blasts in a phase 1 study [21]. In the current study, more patients with high baseline CD123+ blasts responded to the combination treatment versus the low CD123+ blasts. Whereas the response rate in the low and high CD123 blast groups were comparable in the decitabine alone arm. This finding suggests that talacotuzumab exerts its ADCC effects via the CD123. However, in spite of demonstrated ADCC activity of talacotuzumab, the combination treatment could not achieve significant improvement in the current study. One possible explanation to the suboptimal response of the talacotuzumab + decitabine group would be the antagonistic effect of decitabine on ADCC effectors such as NK cells. However, further studies to evaluate this effect are warranted. Kinetics of CD16 and CD14-positive subsets of monocyte were not evaluated in the current study. However, to better understand the mechanisms of primary resistance to mAbs of this class and to identify subgroups of patients who could benefit with this approach of treatment, in depth assessment of immune cells and their immunological effector mechanisms should be included in future studies with such immunotherapeutic agents to better understand mechanisms of primary resistance and to identify subgroups of patients who are best candidates for such approaches.
Conclusion
In summary, the results of the current study demonstrated an unfavorable risk/benefit ratio for the talacotuzumab and decitabine combination for the treatment of AML in the older population not eligible for intensive chemotherapy. However, the results may indicate that CD123 expressions are elevated in AML and can serve as a viable target for the AML treatment paradigm, since targeting of LSPCs is being considered to be a potential strategy to improve the long-term survival of AML patients.
Supplementary information
Acknowledgements
The authors thank all the participants and the investigators involved in this study. The authors thank Pierre Fenaux, Roland Walter, Diane Finkelstein (members of the IDMC committee), Jeff Lancent and David Steensma (Independent Review Committee) for their contributions. The authors also thank Shweta Pitre, MPharm, ISMPP CMPP™ (SIRO Clinpharm Pvt. Ltd, Thane, India) for writing assistance and Harry Ma, PhD (Janssen Global Services, LLC) for additional editorial support for the development of this manuscript.
Funding
The study was sponsored by Janssen Research & Development, LLC.
Author contributions
Authors: JC, MH, UP, GJR, AHW, IS, LX, and JSL participated in the conception and design of the work being described in this publication. Authors: C-EB, SD, JC, HD, VD, SG, FH, SK, PM, YO, GO, MAO, CP, UP, DP, GJR, HJS, MS, AHW, AW, SPY, IS, LX, and JSL participated in the acquisition/collection of data. Authors: JC, SD, HD, SG, MH, FH, SK, PM, GO, CP, UP, GJR, MS, AHW, IS, LX, and JSL participated in the analysis and/or interpretation of data. All authors critically reviewed the manuscript for important intellectual content. All authors approved the final version of this manuscript for submission, had full access to all the data in this study, and take final responsibility for the decision to submit it for publication.
Compliance with ethical standards
Conflict of interest
GJR extended consulting or advisory services to Agios, Amphivena, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Janssen, MEI Pharma, Roche, Shire, Amgen, Astex Pharmaceuticals, Celator, Celgene, Cellectis, CTI, Genoptix, Juno Therapeutics, MedImmune, Novartis, Onconova Therapeutics, Pfizer, Sunesis Pharmaceuticals, received research funding from Abbvie (Inst), Agios (Inst), Astex Pharmaceuticals (Inst), Celgene (Inst), CTI (Inst), Karyopharm Therapeutics (Inst), MedImmune (Inst), MEI Pharma (Inst), Moffitt (Inst), Novartis (Inst), Onconova Therapeutics (Inst) Pfizer (Inst), Sunesis Pharmaceuticals (Inst), Tensha Therapeutics (Inst) and received travel, accommodations and other expenses from AstraZeneca, Shire, Astellas Pharma, Celator, Incyte, Roche, Amphivena, MEI Pharma, Astex Pharmaceuticals, Janssen, Juno Therapeutics. C-EB received support from Amgen, Astellas, Biosafe, Celgene, Chugai, Jazz Pharmaceuticals, Gentium, Gilead, Janssen, Keocyt, Macopharma, Mallinckrodt Pharmaceuticals, MSD, Mundipharma, OrpheliPharm, Pfizer, Pierre Fabre, Sandoz, Sanofi, Spectrum, Takeda, Teva, Therakos, Vifor pharma. MS received research funding from Amgen and Roche. MS received either travel reimbursements or consultant fees from Amgen, Celgene, Pfizer, and Seattle Genetics. UP has received honoraria from Celgene Corporation. AW received honoraria from Celgene. HD provided consultancy services to Agios, Amgen, Astex Pharmaceuticals, Celator, Celgene, Novartis, Roche, Seattle Genetics, Sunesis, and Tolero, and received research funding from Boehringer Ingelheim, Celgene, Novartis, Bristol-Myers Squibb, and Arog Pharmaceuticals research. JC received support from Ariad, Bristol-Myers Squibb, Novartis, and Teva and consultancy support from Ariad, Bristol-Myers Squibb, Novartis, and Pfizer. DAP is a member of advisory boards for Celgene and Pfizer. C.T.J. is an equity holder in Leuchemix Inc GO provided consultancy services to Novartis, Pfizer, Bristol-Myers Squibb, Johnson & Johnson, Sunesis, Celgene, Karypharm, and Amgen, and received research support from Novartis, Johnson & Johnson, Celgene, Immunogene, and Becton Dickinson. AHW provided consultancy services to Novartis, Celgene, Servier, Abbvie, Roche, Amgen, and CTI, and received research funding from Abbvie, Novartis, Celgene, Servier, Ariad, and Amgen. LX, IS, FH, and JSL are employees of Janssen Research and development and may hold company stocks. PM, YO, CP, HJS, VD, SD, SPY, MAO, SK, SG, and MH declared no conflict of interest, financial or otherwise.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41375-020-0773-5) contains supplementary material, which is available to authorized users.
References
- 1.Kantarjian H, Ravandi F, O’Brien S, Cortes J, Faderl S, Garcia-Manero G, et al. Intensive chemotherapy does not benefit most older patients (age 70 years or older) with acute myeloid leukemia. Blood. 2010;116:4422–9. doi: 10.1182/blood-2010-03-276485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129:1627–35. doi: 10.1182/blood-2016-10-696039. [DOI] [PubMed] [Google Scholar]
- 5.Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic stem cells: identification and clinical application. Int J Hematol. 2017;105:549–57. doi: 10.1007/s12185-017-2221-5. [DOI] [PubMed] [Google Scholar]
- 6.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–84. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 7.Graf M, Hecht K, Reif S, Pelka-Fleischer R, Pfister K, Schmetzer H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. Eur J Haematol. 2004;72:89–106. doi: 10.1046/j.0902-4441.2003.00184.x. [DOI] [PubMed] [Google Scholar]
- 8.Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Xie LH, Biondo M, Busfield SJ, Arruda A, Yang X, Vairo G, et al. CD123 target validation and preclinical evaluation of ADCC activity of anti-CD123 antibody CSL362 in combination with NKs from AML patients in remission. Blood cancer J. 2017;7:e567. doi: 10.1038/bcj.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen MY, Liao WS, Lu Z, Bornmann WG, Hennessey V, Washington MN, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer. 2011;117:4424–38. doi: 10.1002/cncr.26073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28:556–61. doi: 10.1200/JCO.2009.23.9178. [DOI] [PubMed] [Google Scholar]
- 12.Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30:2670–7. doi: 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 14.Kubasch AS, Schulze F, Giagounidis A, Götze KS, Krönke J, Sockel K, et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia (2019). 10.1038/s41375-019-0645-z. [DOI] [PMC free article] [PubMed]
- 15.Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126:291–9. doi: 10.1182/blood-2015-01-621664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klepin HD, Balducci L. Acute myelogenous leukemia in older adults. Oncologist. 2009;14:222–32. doi: 10.1634/theoncologist.2008-0224. [DOI] [PubMed] [Google Scholar]
- 17.He PF, Zhou JD, Yao DM, Ma JC, Wen XM, Zhang ZH, et al. Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: a systematic review and meta-analysis. Oncotarget. 2017;8:41498–507. doi: 10.18632/oncotarget.17241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnett AK, Milligan D, Prentice AG, Goldstone AH, McMullin MF, Hills RK, et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007;109:1114–24. doi: 10.1002/cncr.22496. [DOI] [PubMed] [Google Scholar]
- 19.Lazar-Molnar E, Delgado JC. Implications of monoclonal antibody therapeutics use for clinical laboratory testing. Clin Chem. 2019;65:393–405. doi: 10.1373/clinchem.2016.266973. [DOI] [PubMed] [Google Scholar]
- 20.Romain G, Senyukov V, Rey-Villamizar N, Merouane A, Kelton W, Liadi I, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood. 2014;124:3241–9. doi: 10.1182/blood-2014-04-569061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith BD, Roboz GJ, Walter RB, Altman JK, Ferguson A, Curcio TJ, et al. First-in Man, Phase 1 Study of CSL362 (Anti-IL3Rα / Anti-CD123 Monoclonal Antibody) in patients with CD123+ acute myeloid leukemia (AML) in CR at high risk for early relapse. Blood. 2014;124:120–120. doi: 10.1182/blood.V124.21.120.120. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.