

Abstract
Cardiac fibrosis occurs in most cardiac diseases, which reduces cardiac muscle compliance, impairs both systolic and diastolic heart function and, ultimately, leads to heart failure. Long noncoding RNAs (lncRNAs) have recently emerged as important regulators of a variety of biological processes; however, little is known about the expression and function of lncRNAs in cardiac fibrosis. Using unbiased transcriptome profiling in a mouse model of myocardial infarction (MI), we identified a cardiac fibroblast-enriched lncRNA (AK048087) named cardiac fibroblast-associated transcript (Cfast), which is significantly elevated after MI. Silencing Cfast expression by small interfering RNAs (siRNAs) or lentiviral short hairpin RNAs (shRNAs) resulted in suppression of fibrosis-related gene expression and transdifferentiation of myofibroblasts into cardiac fibroblasts. Depletion of Cfast by lentiviral shRNAs in mouse hearts significantly attenuated cardiac fibrosis induced by MI or isoproterenol-infusion. Importantly, inhibition of Cfast ameliorated cardiac function following cardiac injury. RNA pull-down followed by mass spectrometry analyses identified COTL1 (coactosin-like 1) as one of the Cfast interacting proteins. Mechanistically, Cfast competitively inhibits the COTL1 interaction with TRAP1 (transforming growth factor-β receptor-associated protein 1), which enhances TGF-β signaling by augmenting SMAD2/SMAD4 complex formation. Therefore, our study identifies Cfast as a novel cardiac fibroblast-enriched lncRNA that regulates cardiac fibroblast activation in response to pathophysiological stress. Cfast could serve as a potential therapeutic target for the prevention of cardiac fibrosis and cardiac diseases.
Keywords: lncRNA, myocardial infarction, cardiac fibroblast, fibrosis, cardiac function, COTL1, TRAP1, TGF-β
Graphical Abstract
Here, we describe a novel cardiac fibroblast-enriched lncRNA called Cfast, which regulates cardiac fibroblast activation and fibrosis in response to pathophysiological stress in the heart. Cfast could serve as a potential therapeutic target for the prevention of fibrosis in cardiac diseases.
Introduction
Cardiovascular diseases remain a major cause of health loss and cost in the world.1 As a common pathophysiologic companion, cardiac fibrosis occurs in most heart diseases, impacting both systolic and diastolic function.2,3 Cardiac fibroblasts (CFs) exert essential roles in fibrotic responses following acute and chronic stress resulting from myocardial infarction (MI), hypertension, scar formation, and tissue repair.4,5 Under pathological conditions, CFs proliferate and transdifferentiate into myofibroblasts, which are characterized by the expression of α-smooth muscle actin (ACTA2) and are responsible for the production of excessive extracellular matrix (ECM) and the formation of a collagen-rich fibrotic scar.5,6 The progressive interstitial fibrosis and fibrotic scar accumulates throughout the myocardium, resulting in a reduction of cardiac muscle compliance, impairment of cardiac function, and, eventually, heart failure.3, 4, 5 Despite the appreciation of the importance of pathological remodeling in heart diseases, there is still a lack of effective therapies directly targeting CF responses and fibrotic remodeling. In addition, the molecular mechanisms underlying fibrosis-associated heart diseases remain unclear.
It is now widely recognized that the majority of the mammalian genome is actively transcribed to RNA. A small part of the RNAs are protein-coding transcripts, while the majority of these transcribed RNAs constitute non-coding RNA, including a large number of long noncoding RNAs (lncRNAs), which are more than 200-nucleotides in length. Emerging evidence suggests that lncRNAs play important roles in diverse biological processes and diseases.7,8 In particular, recent studies have reported the function of lncRNAs are associated with a variety of cardiovascular disease responses,9,10 including the regulation of atherosclerosis and plaque inflammation,11 modulation of cardiac hypertrophy,12, 13, 14, 15 and cardiomyocyte proliferation and cardiac regeneration.16, 17, 18, 19 We have previously found that long intergenic noncoding RNA (lincRNA)-p21 plays a critical role in cell proliferation and neointimal formation during cardiovascular remodeling.20 More recently, we reported that many lncRNAs are dynamically expressed in ischemic cardiomyopathy (ICM) and these lncRNAs regulate the expression and function of the ECM and cardiac fibrosis during the development of ICM.21 Several lncRNAs, like Wisper, Meg3, and Safe, have been demonstrated as powerful regulators of cardiac fibrosis.22, 23, 24 At the same time, the significance of lncRNAs in regulating cardiac fibrosis and their underlying molecular mechanisms have yet to be fully addressed.
To gain insight into fibrosis-related lncRNAs in the heart, we performed unbiased screening using a mouse model of MI and detected 752 differentially expressed lncRNAs in the infarcted heart. We identified a novel cardiac fibroblast-enriched lncRNA, named cardiac fibroblast-associated transcript (Cfast), and found that it modulates cardiac fibrosis upon acute and chronic cardiac injury. We showed that Cfast participates in the regulation of the transforming growth factor-β (TGF-β) signaling pathway by interacting with COTL1 (coactosin-like 1). Inhibition of Cfast during and/or after cardiac injury effectively prevents pathological fibrotic remodeling and improves heart function in a mouse model of MI. As a result, our study identifies a fibroblast-enriched lncRNA, which may represent a novel regulator and potential therapeutic target for cardiac fibrotic remodeling in cardiac disorders.
Results
Cfast is a cardiac fibroblast-enriched lncRNA upregulated in the heart after MI
To explore the key lncRNAs involved in regulating essential pathophysiological processes of the adult heart, we performed microarray-based transcriptome profiling on mouse hearts 3 days after MI or sham surgery (Figure 1A). Microarray analysis revealed a total of 7,339 differentially expressed transcripts in MI hearts compared to sham controls (fold change [FC] ≥ 2, p < 0.05; Figure 1B). Kyoto Encyclopedia of Genes and Genomes (KEGG) or Gene Ontology (GO) pathway analyses revealed that oxidative phosphorylation, ECM-receptor interaction, cell proliferation, and inflammatory responses were among the top dysregulated genes in MI hearts (Figures S1A and S1B), consistent with prior reports.4,25 Interestingly, 209 lncRNAs are upregulated and 543 lncRNAs are downregulated in MI hearts (FC ≥ 2, p < 0.05; Figure 1C). Among these lncRNA transcripts, we found that 8 lncRNAs are mostly upregulated and 6 lncRNAs mostly downregulated (FC ≥ 2, p < 0.05), whereas the expression level of the AK048087 transcript was increased by 25-fold (Figure 1D). Interestingly, lncRNA AK034241 appears to be the most downregulated lncRNA (Figures 1C and 1D), which will be reported in a separate study.
Figure 1.

Cfast is a cardiac fibroblast-enriched lncRNA upregulated in the heart after MI
(A) Workflow of discovery of lncRNAs in mouse model of MI. (B) Heatmap of differentially expressed lncRNAs in the heart 3 days after MI (n = 5) versus sham (n = 4; microarray FC ≤ 0.5 or ≥ 2, unpaired t test p value ≤ 0.05). (C) Enrichment plot of Cfast and dysregulated lncRNAs in the heart 3 days after MI. The x axis shows p value and the y axis shows lncRNAs fold change versus sham. (D) qRT-PCR of top up- and downregulated lncRNAs in the heart 3 days after MI. AK048087 is highlighted in blue. Graphs show means normalized to sham ± SEM (n = 6 to 8 animals per group). p values were determined by Student’s t test. (E) qRT-PCR of Cfast expression in the border zone (left panel), infarct zone (middle panel), and remote zone (right panel) of infarcted hearts at 3, 14, and 28 days. Graphs show means normalized to sham ± SEM (n = 6 to 8 animals per group). p values were determined by one-way ANOVA. (F) Time course of ECM relative gene expression after MI. Graphs show means normalized to sham ± SEM (n = 6 to 8 animals). p values were determined by one-way ANOVA (Dunnett’s multiple comparisons test). (G) Positive correlation of Cfast expression and fibrosis genes. Pearson’s correlation test (r; 95% confidence interval [CI]). (H) qRT-PCR of Cfast expression in cardiomyocytes and cardio-fibroblasts. Data show mean ± SEM (n ≥ 4). p values are determined by Student’s t test. (I) Coding potential of Cfast. Protein coding mRNAs and known non-coding lncRNAs serve as positive and negative controls, respectively. (J) Cfast is enriched in nucleus of cardio-fibroblasts. Percentage of nuclear (black bar) and cytoplasmic (gray bar) RNA concentrations of Cfast, Gapdh, and β-actin (cytoplasmic markers), and Neat1 and Xist (nuclear markers) measured by qRT-PCR after subcellular fractionation in CFs. Data represent mean ± SEM (n = 3). (K) qRT-PCR of Cfast expression in human heart samples from dilated cardiomyopathy (DCM) patients and controls. Data show mean ± SEM (n = 3). p values were determined by Student’s t test.
To confirm the above results and identify top lncRNA candidates for further study, we performed an independent series of mouse heart infarctions (sham n = 8 versus MI n = 7). We examined the expression, using qRT-PCR, of the top up- and downregulated lncRNAs identified from the above microarray screening in MI hearts and we verified that AK048087 expression increased the most in the hearts 3 days after MI. To better characterize the expression of this lncRNA during cardiac remodeling in response to MI, we carefully examined its expression in the heart 3, 14, and 28 days after MI. In particular, we examined its expression in the MI border zone, infarct zone, and remote zone. Substantially increased expression of lncRNA AK048087 was observed in all zones of the heart 3 days after MI (Figure 1E), which diminished by 14 and 28 days. Cardiac remodeling after MI is associated with the formation of scar and the expression of fibrosis-related genes.22 Therefore, we tested the expression of Col1a1, Col3a1, and Fibronectin (Fn1) and found their expression increased in the MI hearts (Figure 1F). Interestingly, the expression pattern of these fibroblast-related genes is closely correlated with that of lncRNA AK048087 in infarcted hearts (Figure 1G).
Given its increased expression and positive correlation with that of fibrosis-related genes in infarcted hearts, we decided to focus on lncRNA AK048087 and renamed it Cfast. To investigate cell type distribution of Cfast in the heart, we dissociated the cardiomyocytes (CMs) and CFs of adult mouse hearts. qRT-PCR profiling showed that the Cfast transcript is enriched in CFs. As positive controls, we show that Col3a1 is enriched in CFs, while cTnT is enriched in CMs (Figure 1H). The gene encoding Cfast is located on mouse chromosome 4 and appears to produce a single exon transcript (Figure S2A). Cfast is highly conserved in both DNA sequence and its genome locus across multiple species (Figure S2B). In addition to the heart, Cfast expression is also detected in other mouse tissues (Figure S2C). We performed 3′ and 5′ rapid amplification of cDNA end (RACE) assays, using RNAs isolated from adult mouse hearts and we obtained the full-length Cfast transcript of 2,418 nt (Figures S3A and S3B). To test whether Cfast, which contains several putative open reading frames (ORFs), is a noncoding RNA, we employed the Protein Coding Potential Calculator26 to evaluate the protein-coding potential of full-length Cfast transcript. As expected, Cfast has low coding-potential score, similar to that of well-studied lncRNAs Xist, Bvht, and Fendrr. In contrast, protein-coding genes, like Gapdh, Act-B, α-Mhc, and troponin T, all exhibit high coding potential scores (Figure 1I).
Next, we asked whether Cfast has the potential to encode micro-peptides. Using mass spectrometry assays at the detecting sensitivity of 10 pg, we were unable to detect any predicted micro-peptides from adult hearts, suggesting Cfast is unlikely to encode micro-peptides (Figure S3C). Finally, we performed subcellular fractionation assays to determine the subcellular location of Cfast transcripts. qRT-PCR assays revealed that the Cfast transcript is predominantly in the nuclear fraction of cardiac fibroblasts, similar to the previously identified lncRNAs Xist and Neat1 (Figure 1J). To assess the clinical relevance of our findings, we studied the expression of the Cfast human homolog and confirmed its expression as the previous reported human lncRNAs n384785 and n338538.21 Expression of Cfast was significantly increased in patients with dilated cardiomyopathy (DCM) (Figure 1K)
Knockdown of Cfast inhibits myofibroblast differentiation in isolated cardiac fibroblasts
To determine the function of Cfast, we employed a loss-of-function approach to assess the effect of downregulation of Cfast in cardiac fibroblasts isolated from neonatal mouse hearts. Using a lentivirus-based knockdown approach to specifically target Cfast, we consistently achieved more than 60% knockdown of Cfast levels (Figure 2A). qRT-PCR revealed a significant downregulation of the expression of fibrotic related genes Col1a1, Col3a1, fibronectin (Fn1), and elastin (Eln) when Cfast is knocked down (Figure 2B). Similarly, we observed that protein levels of Col1A1, Col1A2, and Col3A1 were reduced in Cfast knockdown cells (Figures 2C and 2D). Next, we performed immunofluorescence staining of α-smooth muscle actin (α-SMA) as a marker of myofibroblast transdifferentiation. We observed less α-SMA positive cells upon Cfast silencing (Figure 2E) and quantification confirmed this observation (Figure 2F). Furthermore, we found that the transcript level of α-Sma was reduced when Cfast is diminished (Figure 2G). In addition, the expression of Vimentin and Periostin, which are molecular markers of fibroblast activation, was reduced (Figure 2G).
Figure 2.

Knockdown of Cfast inhibits myofibroblast differentiation in isolated cardiac fibroblasts
(A) Expression of Cfast after lentivirus-based knockdown in cardiac fibroblasts. Data represent mean ± SEM (n = 6); p values were determined by Student’s t test. (B) Reduced expression of fibrosis genes after Cfast knockdown (Kd) in cardiac fibroblasts. Data represent mean ± SEM (n = 6), p values were determined by Student’s t test. (C) Western blot showing the expression of indicated protein in control and Cfast kd fibroblasts. (D) Quantification of western blot band density. n = 3. p values were determined by Student’s t test. (E) Immunostaining of α-smooth muscle actin (α-SMA) in control and Cfast kd fibroblasts. α-SMA labels activating cardiac fibroblasts (green) and DAPI labels nuclei (blue). (F) Quantification of α-SMA positive cells in control and Cfast kd fibroblasts. p values were determined by Student’s t test. (G) qRT-PCR of the expression of α-Sma, Vimentin, and Periostin genes in control and Cfast kd fibroblasts. p values were determined by Student’s t test. (H and I) Mouse cardiac fibroblasts were transfected with lentivirus-Cfast to overexpress Cfast (Cfast Over) or with lentivirus-GFP (control) for 48 h. Gene expression of Cfast (H) and fibrosis related genes (I), Col1a1, Col3a1, Fn1, Eln, and α-SMA were measured by qRT-PCR. Data represent mean ± SEM (n = 6). p values were determined by Student’s t test.
We applied an independent small interfering RNAs (siRNA) that targets a distinct site of the Cfast gene in isolated mouse CFs and confirmed that the abovementioned fibrosis-related genes, Col1a1, Col3a1, Fn1, Eln, and α-Sma, are all markedly downregulated (Figure S4A). Conversely, we found that overexpression of Cfast by lentivirus (Figure 2H) resulted in significant upregulation in the fibrosis-related genes Col1a1, Col3a1, Fn1, Eln, and α-Sma (Figure 2I) in cardiac fibroblasts. Together, these findings indicate that Cfast regulates the expression fibrotic genes and the differentiation of cardiac fibroblasts.
Inhibition of Cfast reduces scar size and improves cardiac function in response to MI
Given its anti-fibrotic effects in cultured cardiac fibroblasts, we tested the therapeutic potential of Cfast inhibition in mouse hearts in response to MI. The left anterior descending coronary artery was permanently ligated to create MI and a Cfast-depletion lentivirus was directly injected into the myocardium at regions adjacent to the ligation site. Cardiac function and the development of fibrosis was analyzed after 28 days (Figure 3A). We first determined the cell-type specificity and knockdown efficiency 7 days after MI and Cfast-depletion lentivirus injection. Indeed, we found that Cfast expression was significantly downregulated in the isolated adult cardiac fibroblast fraction, but not in the adult cardiomyocyte fraction, indicating that lentivirus knockdown of Cfast is specifically effective in cardiac fibroblasts opposed to cardiomyocytes (Figure 3B). This observation was not due to the isolation process because both cardiomyocyte and fibroblast-specific marker genes were detected and confirmed in the respective isolated cells (Figures S5A and S5B).
Figure 3.

Inhibition of Cfast reduces scar size and improves cardiac function in response to MI
(A) Workflow of testing Cfast function during MI. (B) qRT-PCR of Cfast in isolated cardiomyocytes and fibroblasts 7 days after cardiac-injection of Cfast depletion lentivirus and MI. Data are mean fold change relative to sham ± SEM (n = 3). p values versus sham determined by two-way ANOVA Tukey’s test. (C) M-mode echocardiography of mice 28 days after cardiac-injection of Cfast depletion lentivirus and MI. (D) Measurement of cardiac function of mice by echocardiography 28 days after cardiac injection of Cfast depletion lentivirus and control lentivirus subjected to MI or sham. Graphs show mean values ± SEM. Sham treatment: control n = 3 versus Cfast Kd n = 3. MI treatment: control n = 8, Cfast Kd n = 10. p values were calculated by Student’s t test. (E) qRT-PCR of cardiomyopathy marker genes of mouse hearts 28 days after cardiac-injection of Cfast depletion lentivirus and MI. Bars represent means normalized to control ± SEM (n ≥ 8). p values were determined by Student’s t test. (F) Representative images of series of transverse sections of mouse hearts 28 days after cardiac injection of Cfast depletion lentivirus and MI. Sirius red/fast green collagen staining marks myocardium (green) and scar (red). Scale bar, 2 mm. (G) Quantification of the size of scar. (Control n = 5, Cfast Kd n = 6). p values were calculated by Student’s t test. (H) Immunohistochemistry of left ventricle of mouse hearts 28 days after cardiac injection of Cfast depletion lentivirus and MI. Wheat germ agglutinin (WGA) marks fibrotic area (red) and MF20 labels myocardium (green). (I) qRT-PCR of Cfast and fibrosis genes of mouse hearts 28 days after cardiac-injection of Cfast depletion lentivirus and MI. (control n = 8, Cfast Kd n = 10) p values were calculated by Student’s t test. (J) Positive correlation of Cfast expression and fibrosis genes in mouse hearts 28 days after cardiac injection of Cfast depletion lentivirus and MI. Pearson’s correlation test (r; 95% CI).
28 days after MI and Cfast inhibition, cardiac dimensions, and function were measured using echocardiography. We found that mice with Cfast depletion exhibited improved cardiac function, as evidenced by preserved ejection fraction (EF%), with prevention of cardiac dilation (LVID;d(mm)), when compared to the control group. In the absence of MI injury (sham), Cfast inhibition did not impact cardiac function compared to the control group. (Figures 3C and 3D; Table S1). Furthermore, the heart weight to bodyweight ratio is substantially lower in Cfast-depleted hearts (Figure S6A). Molecular marker analysis demonstrated significant reduction of the expression of cardiomyopathy marker genes such as Nppa, Nppb, and Mhy7 in the heart after Cfast inhibition and MI (Figure 3E). This further supports the view that inhibition of Cfast ameliorates cardiac function after MI. Interestingly, we found decreased cardiomyocyte size in Cfast depleted hearts (Figure S6B).
We next assessed whether Cfast inhibition prevents cardiac fibrosis and pathological remodeling. Histological analyses revealed that Cfast inhibition reduced scar formation in infarcted hearts (Figures 3F and 3G). Interestingly, immunohistochemical analysis showed that more MF20-positive cardiomyocytes exist in the scar regions of Cfast knockdown hearts when compared with that of control hearts (Figure 3H). Consistent with the above observation, we found that the expression levels of fibrosis-related genes, Col1a1, Col3a1, Fn1, Eln, and α-Sma are all markedly reduced in Cfast knockdown hearts after MI (Figure 3I). In support of these results, we found that post-infarction fibrosis is positively correlated with Cfast expression, as well as the fibrosis-associated genes, Col1a1, Col3a1, and α-Sma (Figure 3J); yet, suppression of fibrotic gene expression was not observed in the sham treatment (Figure S7).
Given that Cfast inhibition resulted in the reduction of cardiomyocyte size in the heart (Figures S6A and 6B), while Cfast is predominately expressed in cardiac fibroblasts, we set out to test the hypothesis that cardiac fibroblast-expressed Cfast represses cardiomyocyte hypertrophic growth via a paracrine mechanism. Cultured neonatal mouse cardiomyocytes were treated with conditioned media from cultured cardiac fibroblasts transfected with Control or Cfast knockdown lentivirus (Figure S6C). We found cardiomyocyte size was substantial reduced after treatment with Cfast knockdown conditioned medium (Figure S6D). These results suggest a paracrine mechanism where Cfast silencing in fibroblasts affects cardiomyocyte responses.
Figure 6.

Cfast associates with coactosin-like 1 (COTL1) protein to regulate the TGF-β signaling pathway
(A) Summary of distribution of proteins identified from cardiac fibroblasts using Cfast RNA pull-down coupled mass spectrometry. (B) Western blot of Cotl1 protein after lncRNA Cfast or control pull-down. Whole protein was used as positive control and poly(A) RNA used as negative control. (C) qRT-PCR of Cfast level after RNA immunoprecipitation using anti-COTL1 or control antibodies, in control or Cfast knockdown cardiac fibroblasts. Graph shows means ± SEM (n ≥ 3). p values were determined by two-way ANOVA (Sidak’s test). (D) Co-immunoprecipitation using anti-COTL1 or control IgG antibodies and western blot using anti-TRAP1 antibody, in control or Cfast knockdown cardiac fibroblasts. (E) Cardiac fibroblasts were infected with AD-COTL1 or control and COTL1 expression detected by qRT-PCR. Graph shows means ± SEM (n ≥ 5). p values were determined by Student’s t test. (F) qRT-PCR of the expression of indicated fibrotic genes from cardiac fibroblasts infected with AD-COTL1 or control. Graph shows means ± SEM (n ≥ 5). p values were determined by Student’s t test. (G) Cardiac fibroblasts were infected with AD-COTL1 or control, treated with TGF-β, and COTL1 expression detected by qRT-PCR. Graph shows means ± SEM (n ≥ 5). p values were determined by Student’s t test. (H) qRT-PCR of the expression of indicated fibrotic genes from cardiac fibroblasts infected with AD-COTL1 or control, treated with TGF-β. Graph shows means ± SEM (n ≥ 5). p values were determined by Student’s t test. (I) Gene set enrichment analysis of RNA-seq showed decreased TGF-β signaling in cardiac fibroblasts following Cfast knockdown.
Collectively, our results demonstrate that Cfast plays an important role in pathological cardiac fibrosis and remodeling in response to MI.
Cfast depletion blocks isoproterenol-induced cardiac fibrosis in mouse hearts
The above results, in which inhibition of Cfast in the heart reduced scar formation and affected remodeling of cardiomyocytes in response to MI, prompted us to more specifically investigate the role of Cfast in regulating cardiac fibrosis during remodeling. We decided to employ a chronic cardiac pathological model by daily injection of isoproterenol (ISO, 30 mg/kg) for 21 days in mice—an experimental procedure that has been well-documented to induce cardiac fibrosis and impair cardiac function.27 At day 8, we injected lentivirus to silence Cfast, while a scrambled sequence was used as a control (Figure 4A). We assessed cardiac phenotypes at 21 days and found that ISO injection resulted in a reduction of cardiac function and Cfast-depletion in these hearts significantly improved cardiac function compared to controls (Figure 4B; Table S2). Improved cardiac function in these hearts is supported by a reduction in the expression of cardiomyopathy molecular marker genes, including Nppa, Nppb, and Myh7 (Figure 4C).
Figure 4.

Cfast depletion blocks ISO-induced cardiac fibrosis in mouse hearts
(A) Workflow of testing Cfast function in ISO-induced cardiac fibrosis and heart failure. (B) Measurement of cardiac function by echocardiography 21 days after cardiac injection of Cfast depletion lentivirus and ISO-infusion. Graphs show mean values normalized to the average value of the control group ± SEM (control n = 6, Cfast Kd n = 6); p values were calculated by Student’s t test. (C) qRT-PCR of cardiomyopathy marker genes of mouse hearts 21 days after cardiac injection of Cfast depletion lentivirus and ISO-infusion. Bars represent means normalized to control ± SEM (n = 6). p values were determined by Student’s t test. (D) Representative images of series of transverse sections of mouse hearts 21 days after cardiac injection of Cfast depletion lentivirus and ISO-infusion. Sirius red/fast green collagen staining marks myocardium (green) and scar (red). Scale bars, 1 mm. (E) Quantification of the size of scar. (Control n = 5, Cfast Kd n = 4). p values were calculated by Student’s t test. (F) Representative images of Sirius red/fast green collagen staining. Sections were visualized under polarized light microscopy. Collagen type I is shown in yellow orange. Collagen type III is shown in green. Scale bars, 100 μM. (G) qRT-PCR of Cfast and fibrosis genes of mouse hearts 21 days after cardiac injection of Cfast depletion lentivirus and ISO-infusion. (Control n = 8, Cfast Kd n = 10); p values were calculated by Student’s t test. (H) Positive correlation of Cfast expression and fibrosis genes in mouse hearts 21 days after cardiac injection of Cfast depletion lentivirus and ISO-infusion. Pearson’s correlation test (r; 95% CI).
Histological examination using Sirius Red and Fast Green staining to assess fibrosis indicates that Cfast-depletion markedly prevented ISO-induced cardiac pathological fibrosis (Figure 4D), which was confirmed by quantification (Figure 4E). The above observation was further confirmed by the analysis of collagen content with polarized light, where we found a dramatic reduction of collagen in Cfast-inhibited hearts (Figure 4F). Finally, we found the expression of cardiac fibrosis-associated genes, Col1a1, Col3a1, Eln, and α-Sma, was repressed by Cfast depletion (Figure 4G). In line with the role of Cfast in the regulation of cardiac fibrosis, we found that ISO-induced fibrosis is correlated with the expression of Cfast and cardiac fibrosis-related genes Col1a1 and α-Sma (Figure 4H). Furthermore, the heart weight to body weight ratio is substantially lower in Cfast-depleted hearts (Figure S8A). Consistent with the view that Cfast inhibition prevents cardiac hypertrophic growth, we found reduced cardiomyocyte size in Cfast-depleted hearts (Figure S8B). Together, these results establish an important role of Cfast in the development of cardiac fibrosis, cardiac remodeling, and function.
Cfast is an important regulator of the fibrosis program
To better understand how loss of Cfast affects gene expression in cardiac fibroblasts, we performed RNA sequencing (RNA-seq) using Cfast knockdown CFs. We identified 3,951 differentially expressed genes (log2 FC > 0.5; p < 0.05) in Cfast-knockdown compared to control CFs, of which 1,880 genes were upregulated and 2,071 downregulated (Figure 5A). GO term enrichment analysis revealed that downregulated genes primarily related to “cell differentiation, cell migration, extracellular matrix organization, response to TGF-β, and cell proliferation,” are among the most enriched terms (Figure 5B). Similarly, KEGG pathway analysis indicated that ECM-receptor interaction and focal adhesion, which are linked to fibrosis, are among the most affected pathways in Cfast-silenced CFs (Figure 5C). We subsequently examined genes related to ECM organization in detail.
Figure 5.

Cfast is an important regulator of fibrosis program
(A) Heatmap of differentially expressed gene in Cfast knockdown and control cardiac fibroblasts. (fold change, > 2; adjusted p < 0.05). (B) Gene Ontology (GO) analysis of downregulated genes. (C) KEGG pathway analysis of downregulated genes. (D) Enrichment plots showing downregulation of genes related to extracellular matrix organization, including ECM proteoglycans, collagen formation, and non-integrin membrane-ECM interactions in Cfast knockdown cardiac fibroblasts. (E) Heatmaps of differentially expressed genes related to matrix, cell proliferation, cell migration, and cell differentiation in Cfast knockdown cardiac fibroblasts.
Using gene set enrichment (GSE) analysis, we found that “extracellular matrix organization,” including “ECM proteoglycans, collagen formation, and non-integrin membrane ECM interactions” were dramatically downregulated in Cfast-knockdown CFs (Figure 5D). In addition to the downregulation of ECM genes (e.g., Col4a3 and Col4a4) closely-associated with fibrosis in Cfast-knockdown CFs, we observed that genes related to cell proliferation (e.g., Mmp2 and Sox4), cell migration (e.g., Tgfbr1 and Tgfbr3), and cell differentiation (e.g., Akap6 and Cap2) are also downregulated in Cfast-knockdown CFs (Figure 5E). This decreased expression of fibrosis-related genes in Cfast-knockdown CFs is consistent with the reduced cardiac fibrosis observed in MI- and ISO-stimulated hearts in vivo. Together, these results support the view that Cfast plays an important role in fibrotic gene expression and cardiac fibrosis processes during cardiac remodeling in response to pathophysiological stress.
Cfast is associated with COTL1 protein to regulate the TGF-β signaling pathway
To elucidate the molecular mechanism by which lncRNA Cfast modulates cardiac fibrosis, we attempted to identify Cfast-interacting proteins. We performed RNA pull-down assays by labeling full-length Cfast RNA with biotin and mixing it with cellular lysates of cardiac fibroblasts. Putative Cfast binding proteins were then identified by mass spectrometry. Using the criteria of unique sequence coverage and fold enrichment, we identified five proteins that potentially interact with Cfast (Figure 6A). These include ABCF1 (ATP-binding cassette, sub-family F [GCN20], member 1), AIMP2 (aminoacyl tRNA synthetase complex-interacting multifunctional protein 2), S100A11 (S100 calcium binding protein A11), COTL1, and STAU1 (staufen double-stranded RNA binding protein 1).
Previous studies showed that ABCF1 functions as a regulator of immune responses,28,29 AIMP2 plays roles in cancer development30 and Parkinson’s disease,31,32 S100A11 is required for efficient plasma membrane repair,33 and STAU1 promotes cell survival.34,35 Therefore, none of these proteins appear functionally relevant with the cardiac fibrotic phenotype. Interestingly, COTL1 was reported to be involved in TGF-β signaling36 and inhibit neuronal migration during mouse corticogenesis,37 suggesting that COTL1 could be a Cfast binding protein to regulate cardiac fibrosis. To confirm the mass spectrometry data, we performed a Cfast RNA pull-down assay coupled with western blotting to detect COTL1 after the lncRNA Cfast pull-down. The binding of COTL1 was readily detected in the Cfast pull-down sample but not in the control (Figure 6B). The association of COTL1 with Cfast was independently confirmed by RNA immunoprecipitation (RIP). Using qRT-PCR, we found that Cfast transcripts were associated with the COTL1 immunoprecipitants, but not that of control IgG samples (Figure 6C). The Cfast-COTL1 interaction is specific, since knockdown of Cfast abolished COTL1-associated Cfast RNA (Figure 6C).
A previous study using a yeast two-hybrid screen identified that 5-lipoxygenase (5LO) interacts with COTL1, and that 5LO also interacts with TRAP1,38 which is a SMAD4 chaperone involved in TGF-β signaling by facilitating the interaction of SMAD4 with SMAD2.39 It is tempting to speculate that COTL1 could associate with TRAP1 to mediate the downstream signaling. Therefore, the identification of the interaction between Cfast and COTL1 prompted us to test the hypothesis that lncRNA Cfast and its interaction with COTL1 will competitively abrogate the interaction between COTL1 and TRAP1, leading to an interaction between TRAP1 and SMAD4; thus, increasing formation of the SMAD2/4 complex. As a result, the TGF-β/SMAD-mediated fibrotic signal would be enhanced. If this hypothesis is correct, we would predict that Cfast will enhance TGF-β signaling and increase fibrosis. Conversely, silencing or knockdown Cfast could diminish TGF-β signaling and fibrosis.
To test this hypothesis, we first performed co-immunoprecipitation assays and demonstrated the interaction between COTL1 and TRAP1 (Figure 6D). Interestingly, the interaction between COTL1 and TRAP1 proteins is substantially enhanced when Cfast is silenced, indicating that Cfast and TRAP1 indeed compete for their association with COLTL1 (Figure 6D). Next, we examined whether COTL1 is sufficient to modulate the activity of the TGF-β signaling and fibrosis program in cardiac fibroblasts. Using an adenovirus-mediated overexpression of COTL1 in cardiac fibroblasts (Figure 6E), we found that COTL1 overexpression inhibited the expression of ECM-related genes, including Col1a1, Col3a1, and Fn1 (Figure 6F). Importantly, overexpression of COTL1 in cardiac fibroblasts (Figure 6G), markedly reduced TGF-β induced expression of these ECM related genes (Figure 6H). Furthermore, COTL1 is able to repress the expression of TGF-β activated Postn gene, which is a well-known fibroblast activation marker gene (Figure 6H).
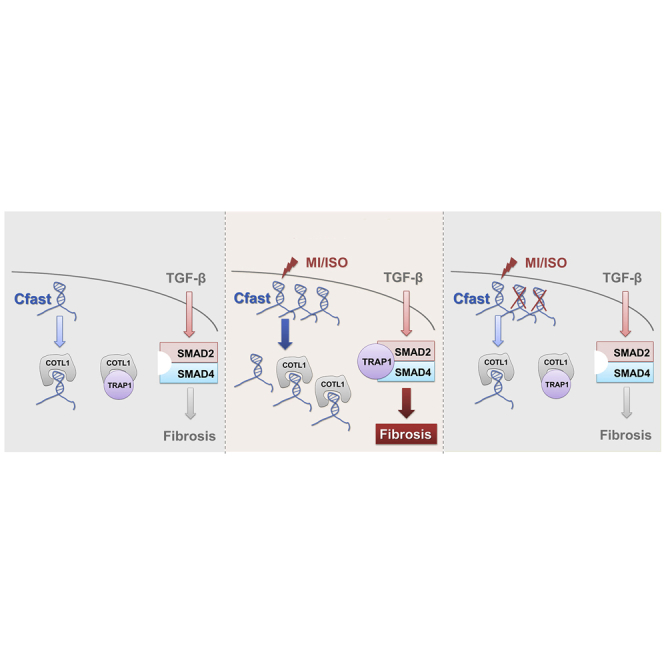
An earlier study showed COTL1 inhibits TGF-β signaling in breast cancer via non-canonical signaling involving interleukin-24 (IL-24)/p53 apoptosis effector related to PMP-22 (PERP). We asked whether Cfast knockdown affects non-canonical TGF-β signaling by altering the levels of IL-24 and PERP. However, Cfast depletion mediated suppression of cardiac fibrosis without affecting the expression levels of IL-24 and PERP in both in vivo models of MI and ISO treated hearts, as well as in cardiac fibroblasts in vitro (Figures S9A–S9C). Conversely, GSE analysis with RNA-seq data of Cfast knockdown in cardiac fibroblasts demonstrated that the TGF-β signaling pathway is downregulated, further supporting the view that Cfast regulates the canonical TGF-β signaling pathway in cardiac fibroblasts (Figure 6I). Together, these results indicate that cardiac fibroblast-enriched lncRNA Cfast participates in the regulation of ECM gene expression and fibrosis process by modulating the TGF-β signaling pathway (Figure 7).
Figure 7.

A working model summarizing the function of Cfast in TGF-β signaling and fibrotic gene expression
Under normal physiological conditions, Cfast transcript associates with COTL1 protein, leaving excessive COTL1 protein to bind to TRAP1. Increased expression of lncRNA Cfast under stress conditions results in the association of Cfast and COTL1, releasing the COTL1 partner protein TRAP1, which in turn binds to SMAD2/SMAD4 complex to enhance the TGF-β signaling and fibrosis. Cfast knockdown allows the association between COTL1 and TRAP1, reducing the formation of the TRAP1/SMAD2/SMAD4 complex, therefore the TGF-β signaling and fibrotic gene expression.
Discussion
Here, we report the identification and characterization of the cardiac fibroblast-enriched lncRNA Cfast and show this lncRNA is an important regulator of cardiac fibrosis. We found that Cfast depletion results in a decrease in fibrotic gene expression both in vitro and in vivo; and most importantly, inhibition of Cfast in the heart protects it from pathological fibrotic remodeling and improves cardiac function upon pathological stress. Our results indicate that Cfast competitively interacts with COTL1 to prevent the binding of COTL1 from TRAP1, leading to the formation of the TRAP1/SMAD2/SMAD4 complex. As a result, TGF-β signaling is enhanced and fibrotic gene expression increased. Inhibiting Cfast expression eventually leads to the repression of this signaling cascade and the reduction of cardiac fibrosis (Figures 7A and 7B). Interestingly, overexpression of COTL1 in CFs markedly reduced the fibrotic response with or without TGF-β treatment, placing COTL1 as a lncRNA binding protein with a protective role in cardiac fibrosis. Therefore, our studies identify Cfast as a novel lncRNA and assign its function in cardiac fibroblasts and fibrosis. In addition to its expression in the heart, high Cfast expression is also detected in the brain, kidney, liver, and lung. It will be important to determine its function in these organs.
Acute and chronic cardiac injuries and/or stress often result in decrease of cardiac function and cardiomyopathy. Although the damage to the myocardium (cardiomyocytes) is well-established, recent reports indicate that cardiac fibroblasts transdifferentiate into myofibroblasts in response to injuries, which results in the deposition of large amounts of extracellular matrix proteins and the formation of interstitial fibrosis or cardiac scars the development of cardiac fibrosis.4, 5, 6 Several recent studies reported that cardiac fibroblast expressed lncRNAs, including Wisper,22 Meg3,23 and Safe,24 are involved in the regulation of cardiac fibrosis. Mechanistically, it is suggested that Wisper interacts with and modulates the function of TIAR (TIA1-related protein) to control collagen cross-linking and the stabilization of the matrix. On the other hand, Meg3 interacts with transcription factor P53 to control the expression of MMP-2 gene, and, therefore, fibrosis. Our previous studies also linked the expression and function of lncRNAs to atherosclerosis and cardiac fibrosis in mice and humans.20,21
We have identified Cfast as a novel cardiac fibroblast-enriched lncRNA, and we demonstrated that Cfast modulates pathological cardiac fibrosis in two independent models of cardiac remodeling and heart failure. Our investigation shows a distinct molecular mechanism by which lncRNA Cfast competitively binds to its partner protein, COTL1, resulting in disassociation of this protein with a chaperone protein TRAP1, which is important for an essential signaling cascade (i.e., TGF-β) related to fibrotic gene expression and fibrosis (Figure 7). Our study, together with prior reports, reveals that fibroblast-enriched lncRNAs play primary roles in cardiac fibrotic remodeling and cardiac function. These fibroblast specific lncRNAs may have therapeutic potential for fibrosis-related cardiomyopathy.
Although we have offered a molecular explanation by which Cfast and COTL1 interact to ultimately modulate the TGF-β signaling pathway in the cardiac fibrotic response, this may not be the exclusive molecular mechanism. Many lncRNAs could exert their roles via sponging microRNAs (miRNAs) and influencing miRNA-targeted mRNAs as competing endogenous RNAs (ceRNAs).14,16,17,19 However, we have not found promising candidate Cfast sponging miRNAs that are directly relevant to cardiac fibrosis, based on bioinformatics analysis. In our study, we focused on Cfast binding protein-related molecular mechanisms. Given the complicated regulation of the ceRNA network in pathophysiological processes, future studies will likely expand upon the molecular mechanisms by which Cfast contributes to cardiac fibrosis.
In addition to the observation that silencing Cfast reduces cardiac fibrosis, we also found that depletion of Cfast prevents cardiomyocyte hypertrophy, as evidenced by the blunted fetal gene (Nppa, Nppb, Myh7) reactivation and by the reduction of heart weight verse body weight ratios in both MI- and ISO-induced cardiac injury models, which likely contribute to the improvement of heart function under stress conditions. Intriguingly, the endogenous expression and experimental inhibition of Cfast is primarily limited to cardiac fibroblasts, not cardiomyocytes, raising the question of how cardiac fibroblast-enriched Cfast affects the function of cardiomyocytes under stress conditions. We speculate that though the functional improvement by Cfast depletion is primarily attributed to the prevention of cardiac fibrotic remodeling, there is crosstalk between fibroblasts and cardiomyocytes, which consequently ameliorates hypertrophic remodeling and improves heart function. Our observation does not stand alone—previous studies also document that the fibroblast-enriched lncRNAs, Wisper,22 and Meg3,23 exhibit indirect benefits for cardiomyocyte hypertrophy after MI or TAC, respectively, and this may result from paracrine effects.23
In addition to COTL1, we also detected four other proteins by mass spectrometry after Cfast pulldown; namely, ABCF1, AIMP2, S100A11, and STAU1. There is no evidence that indicates these candidates have direct impact on cardiac fibroblasts and fibrotic remodeling. These proteins are involved in immune responses, cancer development, Parkinson’s disease, plasma membrane repair, and cell survival.28, 29, 30, 31, 32, 33, 34, 35 However, whether these candidates are indeed involved in cardiac fibrosis and heart diseases were not examined in the present study. Therefore, we cannot rule out the possibility that their binding with Cfast may also impact the behavior of cardiac fibroblasts and cardiac remodeling in response to injury. Clearly, their function associated with Cfast warrants further investigation.
In summary, we have identified and characterized lncRNA Cfast as a novel regulator of cardiac fibroblasts and fibrotic remodeling in the heart in response to cardiac injuries. Cfast exerts its function via binding to COTL1 and, consequently, affects TRAP1/SMAD-mediated fibrotic signaling cascades. Our study indicates that Cfast may represent a potential target in antifibrotic RNA therapy in heart diseases.
Materials and methods
Study approvals
The use of animals in this study conformed to the Public Health Service Guide for Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee (IACUC) of Boston Children’s Hospital and by the IACUC of Zhejiang University. Briefly, mice were maintained on 12 h light:dark cycles and were fed with germ-free diets and fresh sterilized water. For post-operative analgesia, mice were injected with 0.1 mg/kg of Buprenorphine intraperitoneally. Repeated subcutaneous injections of 0.1 mg/kg of Buprenorphine were made every 8 h for 3 days. The human study protocol with using heart tissue from DCM patients and control patients was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University. Patients provided written informed consent.
Microarray-based lncRNA profiling
For microarray analysis, RNAs were prepared from the hearts of mice 3 days after MI surgery (n = 5 hearts) or sham surgery (n = 4 hearts). These RNA samples were subjected to global lncRNA profiling using the lncRNA Microarray SurePrint G3 Gene Mouse GE 8∗60K Kit (Agilent Technologies). Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile-normalization and subsequent data processing were performed with the GeneSpring GX v12.1 software package (Agilent Technologies). Differentially expressed lncRNAs were identified through filtering on FC (≥2) and p value (p < 0.05).
qRT-PCR
Total RNA was isolated using TRIzol reagent (Life Technologies) from heart tissue or cell samples and was incubated with DNase I (Life Technologies) to remove residual genomic DNA. For detecting gene expression with qRT-PCR, 1.0 μg total RNA samples were reverse transcribed to cDNA using M-MLV reverse transcriptase and random hexamers (Life Technologies) according to the manufacturer’s instruction. Primers were designed to span introns. All the qPCR analyses, cDNA prepared without reverse transcriptase was served as a negative control. QPCR Signal was detected by the VII7 Real-time PCR System with SYBR green qPCR Master Mix (Vazyme Biotech). Data were normalized by 18S signal. Primers used for qRT-PCR in this study is listed in Table S3.
RACE
The full-length cDNA sequence of Cfast was obtained from mouse hearts RNA by applying RACE according to SMARTer RACE 5′/3′ Kit User Manual (TaKaRa). Nested 5′ and 3′ RACE products were obtained using GXL Taq polymerase with GC Buffer (Takara). The primers used for RACE, nested PCR, and PCR are presented in Table S4. Gel products were extracted with a Gel Extraction kit (Vazyme Biotech), cloned into pMD18-T vectors and analyzed by Sanger sequencing.
RNA fractionation
Nuclear and cytoplasmic RNA fractions of cardiac fibroblasts were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) according to the manufacturer’s instructions with addition of RNase inhibitor. The extracted nuclear and cytoplasmic RNA fractions were used to detect Cfast content in subcellular localization according to standard qRT-PCR protocol. The expression of glyceraldehyde phosphate dehydrogenase (Gapdh) and β-actin was used as cytoplasmic controls, whereas the expression of Xist and Neat1 was used as a nuclear control.
Protein coding potential
The protein coding potential of lncRNA candidates was analyzed by using the Coding Potential Calculator (www.cpc.cbi.pku.edu.cn). We used the NCBI ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) to identify the potential peptides and then performed mass spectrometry with these total protein lysates of cardiac fibroblasts to detect whether presence of the predicted peptides.
MI and intra-cardiac injection of lentivirus
MI was performed on 8-week-old male c57BL/6 mice (Jackson Laboratory or Shanghai SLAC Laboratory Animal) by ligation of the left anterior descending (LAD) coronary artery.40,41 For surgery, mice were anesthetized with isoflurane (3% isoflurane for induction, 2% isoflurane for maintenance). After the chest was shaved and cleaned with 75% alcohol, a suture was placed around the front upper incisors and pulled taut so that the neck was slightly extended. For oral intubation, the tongue was retracted and held with forceps, and a 20G catheter was inserted into the trachea. The catheter was then attached to the mouse ventilator via a Y-shaped connector. Ventilation was performed with a tidal volume of 225 μL for a 25 g mouse and a respiratory rate of 130 breaths per minute. 100% oxygen was provided to the inflow of the ventilator. Then the chest was opened through a left parasternal incision, and the heart exposed at the left 3rd–4th intercostal space. Chest retractor was applied to facilitate the view. The pericardium was opened, and the ligation was performed on the LAD coronary artery using 8–0 silk sutures (Ethicon). The lungs were slightly overinflated to assist in removal of air in the pleural cavity. Dissected intercostal space and chest skin were closed using a 6–0 silk suture (Ethicon).
For intra-cardiac injection after MI,41 mice were randomly subjected to intra-cardiac injection of lentivirus- short hairpin RNAs (shRNA)-Cfast, or lentivirus-control (4–5 × 107 viral genome particles per mouse heart), respectively after MI. Immediately after the ligation of LAD coronary artery, lentivirus-shRNA-Cfast, or lentivirus-control in a total volume of 40 μL were injected into ventricle muscular wall but not ventricular cavity using insulin syringe with needle (31 G). The lentiviruses were evenly injected into five sites around the infarcted area (anterior wall, lateral wall, and apex area). Immediately after injection of lentivirus, anesthesia (isoflurane) was stopped to increase survival. Then the chest was closed, and the animal was changed to a prone position until recovery of spontaneous breathing.
ISO administration and intra-cardiac injection of lentivirus
8-week-old male C3H mice (Beijing Vital River Laboratory Animal Technologies) were administered with ISO hydrochloride (Sigma) to induce cardiac fibrotic remodeling. ISO was dissolved in sterile saline and was injected intraperitoneally (30 mg/kg/day) once daily for 21 consecutive days.27
At day 8 of ISO administration, mice were randomly subjected to intra-cardiac injection of lentivirus-shRNA-Cfast, or lentivirus-control (4–5 × 107 viral genome particles per mouse heart). Mice were orally intubated and maintained with a rodent ventilator for artificial respiration with isoflurane. The chest was opened through a left parasternal incision, and the heart was exposed at the left 3rd–4th intercostal space. Chest retractor was applied to facilitate the view. The pericardium was removed and a total volume of 40 μL of lentivirus-shRNA-Cfast or lentivirus-control was intramuscularly injected into anterior left ventricle using insulin syringe with a 31G needle. The lentivirus was evenly injected into five sites around the area of mid-apex of ventricle. The chest was closed, and the animal was changed to a prone position until recovery of spontaneous breathing.
Measurement of cardiac function by echocardiography
Echocardiographic measurements were performed on mice using a Visual Sonics Vevo 2100 Imaging System (Visual Sonics, Toronto, ON, Canada) with a 18–38 MHz transducer (model MS-400).27,40,41 Mice were anesthetized with isoflurane (2.0% isoflurane for induction and 0.5% for maintenance). Heart rate and left ventricular (LV) dimensions, including diastolic and systolic wall thicknesses, LV end-diastolic, and end-systolic chamber dimensions, were measured from 2-D short-axis under M-mode tracings at the level of the papillary muscle. LV mass and functional parameters such as percentage of fractional shortening (FS%) and LV volume were calculated using the above primary measurements and accompanying software.
Isolation and culture of mouse cardiac fibroblasts
Neonatal mouse cardiac fibroblasts were isolated by enzymatic disassociation according to the manual of neonatal heart dissociation Kit (Miltenyi Biotec, Germany). Briefly, the dissected hearts were transferred into a 10 cm dish with PBS and were trimmed off vessels and connective tissue from ventricles. Then the hearts were dissociated using enzyme mix and gentle MACS C tube for 45 min (gentleMACS Program mr_neoheart_01). After termination of the program, the cells were washed and resuspended in culture medium using MACS SmartStrainer (70 μm). Finally, the collected cells were resuspended and differentially plated for 25 min in DMEM medium containing 10% fetal bovine serum (FBS) and penicillin-streptomycin (100 U/mL). After removing the floating cardiomyocytes, cardiac fibroblasts, readily attached to the bottom, were cultured till passages 2–3 for further assays. After 24 h in serum-free medium, fibroblast cultures were treated with variety as indicated.
Adult mouse cardiac fibroblasts and cardiomyocytes were isolated using a previously reported procedure.42 Briefly, following perfusion and digestion of the heart with collagenase II (Worthington Biochemical, Lakewood, NJ, USA), dissociated cells were sedimented by gravity. The supernatant, enriched in cardiac fibroblasts, was collected and centrifuged for 5 min at 1,000 rpm. The bottom layer, rich in adult cardiomyocytes, also was collected. The adult cardiac fibroblasts and cardiomyocytes then were treated with Trizol for RNA extraction and qRT-PCR analyses.
Lentivirus construction and infection of lentivirus
Lentiviral expression vectors were used to generate shRNA expression system. The targeting sites of shRNA for lncRNA Cfast were designed using the siRNA Wizard (https://www.invivogen.com/sirnawizard/design.php). The sequences for shRNA were listed in Table S5. The lentiviral expression vector, namely pGreenPuro shRNA Cloning and Expression Lentivector, was from System Biosciences. For infection, cells plated at density of 1 × 105 /cm2, were infected with indicated lentiviruses expressing of shRNA targeting Cfast or scramble shRNA as control for 24 h at infection of 50 MOI, and then refreshed with fresh medium. Transfection efficacy was evaluated after 72 h of infection via qRT-PCR or western blot assessments of indicated gene expression levels.
siRNA synthesis and transfection
siRNAs targeting Cfast were designed and synthesized by GenePharma (China; sequences listed in Table S5). Targeting siRNAs and scramble siRNA control were transfected into cells at 50 nM of final concentration using Lipofectamine RNAiMAX Reagent (Thermo Scientific).
Western blot analysis
Cultured cells were harvested, homogenized and incubated in Cell Extraction Buffer (Invitrogen) with protease inhibitors cocktail (Sigma) and 1 mM phenylmethylsulfonyl fluoride (PMSF) on ice for 15 min. The lysates were centrifuged at 13,000 × g for 10 min at 4°C. After preparation, samples were mixed with Laemmli buffer containing 5% β-mercaptoethanol and were evenly loaded onto SDS-PAGE gels. The separated proteins on gel were then transferred to polyvinylidene fluoride (PVDF) membranes (BioRad). After blocking in 5% BSA, membranes were incubated overnight at 4°C with primary antibody and then washed three times with TBST buffer before incubation for 1 h with HRP-conjugated secondary antibody at room temperature. Protein bands were visualized by using ECL Reagents (Invitrogen) with the Bio-Rad ChemiDoc imaging system. All the antibody information is listed in Table S6.
Histology and immunostaining
Mouse hearts were dissected out, rinsed, and arrested in diastole buffer (4.7 nM KCl and 0.1% 2,3-Butanedione monoxime (BDM) in PBS). Hearts were then fixed in 4% paraformaldehyde (pH 7.4) overnight. After dehydration through a series of ethanol baths, samples were embedded in paraffin wax according to standard laboratory procedures. Sections of 10 μm in thickness were further fixed with prewarmed Bouins’ solution at 55 °C for 1 h and stained with Fast Green and Sirius Red.40,41 The stained sections were used for routine histological examination by light microscopy and polarized light microscopy.
To determine the infarct size, we cut through the embedded paraffin blocks from apex to base. The first 10 sections of every 100 sections were used to stain with Fast Green and Sirius Red. Infarct size was calculated according to the formula: [length of coronal infarct perimeter (epicardial + endocardial) / total LV coronal perimeter (epicardial + endocardial)] × 100.40,41,43
Immunofluorescence staining was performed on 4% paraformaldehyde-fixed and paraffin-embedded heart sections.40,41 After deparaffinization, rehydration, and heat-induced epitope retrieval, sections were incubated with mouse monoclonal anti-myosin heavy chain specific antibody MF20 (5 μg/mL, Developmental Studies Hybridoma Bank) for labeling cardiomyocyte and goat anti-mouse Alexa Fluor 488 secondary antibody (1:400, Invitrogen). The sections were also counterstained with wheat germ agglutinin (WGM; Alexa Fluor 594 conjugate WGA; 1:400, Invitrogen) for labeling fibrotic scar.
Immunofluorescence staining was performed on cultured mouse cardiac fibroblasts for detecting fibroblasts behavior. At the end of the culture, cells were fixed with 3.7% PFA and then permeabilized with 0.5% Triton/PBS. Then the cells were blocked in 5% goat serum and then incubated with indicated antibodies. To identify myofibroblast transdifferentiation, we used mouse monoclonal anti α-SMA-fluorescein isothiocyanate (FITC) antibody (1:400, Sigma Cat# F3777). To detect fibroblast proliferation, we used rabbit anti phospho-histone H3 (pH3, 1:400, Millipore, cat # 06-570) primary antibody and goat anti-rabbit Alexa Fluor 594 secondary antibody (1:400, Invitrogen). Quantitative data were obtained by measuring co-localization of DAPI (nuclear staining) with α-SMA or pH3 on the fibroblasts. All the antibody information is listed in Table S6.
RNA-seq and genome-wide transcriptome analysis
RNA samples from the cardiac fibroblasts after knockdown of Cfast and control were prepared for RNA-seq (three biological replicates for each group). RNA-seq experiments were performed by Novogene (Beijing, China) according to standard procedure.41 Briefly, total RNA was isolated from cultured cells using TRIzol (Invitrogen). mRNA was then purified from total RNA using poly-T oligo-attached magnetic beads. A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform and 150 bp paired-end reads were generated. For the data analysis, raw data (raw reads) in fastq format were first processed through in-house Perl scripts. Clean data (clean reads) were obtained by removing reads containing adapters, reads containing ploy-N and low-quality reads from raw data. Reference genome and gene model annotation files were downloaded from genome website directly. Index of the reference genome was built using STAR and paired-end clean reads were aligned to the reference genome using STAR (v2.5.1b). STAR uses the method of Maximal Mappable Prefix (MMP). HTSeq v0.6.0 was used to count the read numbers mapped to each gene. Analysis of differential expression was performed using the edgeR R package (3.12.1). The p values were adjusted using the Benjamini and Hochberg method. GO and KEGG pathway analyses were implemented using the clusterProfiler R package. The hierarchical clustering heatmap was generated with the ggplot library.
Cfast RNA pull-down
Biotinylated Cfast sense and control RNA (poly[A]) were in vitro transcribed using the T7 RNA polymerase (Invitrogen MEGAscript T7 Transcription Kit) and Biotin RNA Labeling Mix (Thermo Scientific, Pierce RNA 3′ End Desthiobiotinylation Kit) and then purified according to the manufacturers’ instructions. Biotinylated RNA was incubated with magnetic bead (Thermo Scientific, Pierce Magnetic RNA-Protein Pull-Down Kit), and then incubated with cardiac fibroblast lysate for 4 h. The samples were separated via SDS-PAGE and identified using mass spectrometry and retrieved in mouse proteomic library.
RIP and protein co-immunoprecipitation
RIP experiments were performed with DynabeadsTM magnetic beads (Thermo Scientific, Dynabeads Protein G) according to the manufacturer’s instructions. Briefly, we constructed a COTL1 and FLAG fusion protein in adenovirus and overexpressed the COTL1-FLAG in 3T3 cell line. Thereafter, ∼2 × 108 3T3 cells were lysed thoroughly on ice in NP40 Cell Lysis Buffer (Thermo Scientific) supplemented with protease inhibitor cocktail and RNase inhibitor. 10 μg of FLAG antibodies (Table S6) or isotype control immunoglobulin G (IgG) were respectively incubated with magnetic beads at room temperature for 30 min for each immunoprecipitation. Lysate proteins were incubated with indicated antibody-beads complex overnight at 4°C with rotating. Co-immunoprecipitated RNAs were extracted, reverse-transcribed to cDNA, and subjected to qPCR examination. Co-immunoprecipitated proteins were analysis by western blot.
Statistical analysis
Unless otherwise stated, results were presented as mean value ± standard error of the mean (SEM). GraphPad Software (version 7) was used for statistical analysis. Statistical significance between two columns was assessed by two-tailed unpaired Student’s t test; for more than two columns, one-way ANOVA (Dunnett’s multiple comparisons test) analysis was used. Two-way ANOVA (Sidak’s test) was used to evaluate statistical significance between two or more groups. Correlation analysis was performed with Pearson (r or r2 values; 95% confidence interval [CI]) or Spearman (r; 95% CI) test. A value of p <0.05 was considered statistically significant.
Data Availability
RNA-seq data (GEO: GSE161389) and microarray-based transcriptome profiling data (GEO: GSE161427) are available in the Gene Expression Omnibus.
Acknowledgments
The authors thank the Core Facilities of Institute of Translational Medicine, Zhejiang University School of Medicine, and the Laboratory Animal Center of Zhejiang University. This work is supported by National Key R&D Program of China (2017YFA0103700); Zhejiang Provincial NSF project (LZ20H020001 to J.C.); National Natural Science Foundation of China (numbers 81470382, 81670257, and 81970227 to J.C., 81873463 to Z.-P.H., and 82000244 to F.G.,); China Postdoctoral Science Foundation (2020M671751 to F.G.); and Guangdong Science and Technology Department (2018A050506026 to Z.-P.H.). Work in the Wang lab is supported by NIH grant HL125925.
Author contributions
J.C. and D.-Z.W. conceived of and designed the study and wrote and revised the manuscript. F.Z. and X.F. designed the experiments, analyzed the data, and drafted the manuscript. M.K., N.L., Y.W., F.G., T.L., X.D., J.P., and X.H. analyzed and interpreted the data. W.Z., H.Y., D.B.C., X.H., Z.-P.H., and J.W. refined the data analysis and reviewed the manuscript.
Declaration of interest
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.11.013.
Contributor Information
Da-Zhi Wang, Email: dwang@enders.tch.harvard.edu.
Jinghai Chen, Email: jinghaichen@zju.edu.cn.
Supplemental information
References
- 1.Roth G.A., Johnson C., Abajobir A., Abd-Allah F., Abera S.F., Abyu G., Ahmed M., Aksut B., Alam T., Alam K. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017;70:1–25. doi: 10.1016/j.jacc.2017.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frangogiannis N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019;65:70–99. doi: 10.1016/j.mam.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Berk B.C., Fujiwara K., Lehoux S. ECM remodeling in hypertensive heart disease. J. Clin. Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabhu S.D., Frangogiannis N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gourdie R.G., Dimmeler S., Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 2016;15:620–638. doi: 10.1038/nrd.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tallquist M.D., Molkentin J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017;14:484–491. doi: 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Batista P.J., Chang H.Y. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uchida S., Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015;116:737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 10.Viereck J., Thum T. Long Noncoding RNAs in Pathological Cardiac Remodeling. Circ. Res. 2017;120:262–264. doi: 10.1161/CIRCRESAHA.116.310174. [DOI] [PubMed] [Google Scholar]
- 11.Cremer S., Michalik K.M., Fischer A., Pfisterer L., Jaé N., Winter C., Boon R.A., Muhly-Reinholz M., John D., Uchida S. Hematopoietic Deficiency of the Long Noncoding RNA MALAT1 Promotes Atherosclerosis and Plaque Inflammation. Circulation. 2019;139:1320–1334. doi: 10.1161/CIRCULATIONAHA.117.029015. [DOI] [PubMed] [Google Scholar]
- 12.Han P., Li W., Lin C.H., Yang J., Shang C., Nuernberg S.T., Jin K.K., Xu W., Lin C.Y., Lin C.J. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viereck J., Kumarswamy R., Foinquinos A., Xiao K., Avramopoulos P., Kunz M., Dittrich M., Maetzig T., Zimmer K., Remke J. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016;8:326ra22. doi: 10.1126/scitranslmed.aaf1475. [DOI] [PubMed] [Google Scholar]
- 14.Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z., Zhang X.J., Ji Y.X., Zhang P., Deng K.Q., Gong J., Ren S., Wang X., Chen I., Wang H. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016;22:1131–1139. doi: 10.1038/nm.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai B., Ma W., Ding F., Zhang L., Huang Q., Wang X., Hua B., Xu J., Li J., Bi C. The Long Noncoding RNA CAREL Controls Cardiac Regeneration. J. Am. Coll. Cardiol. 2018;72:534–550. doi: 10.1016/j.jacc.2018.04.085. [DOI] [PubMed] [Google Scholar]
- 17.Chen G., Li H., Li X., Li B., Zhong L., Huang S., Zheng H., Li M., Jin G., Liao W. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J. Mol. Cell. Cardiol. 2018;122:152–164. doi: 10.1016/j.yjmcc.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Ponnusamy M., Liu F., Zhang Y.H., Li R.B., Zhai M., Liu F., Zhou L.Y., Liu C.Y., Yan K.W., Dong Y.H. Long Noncoding RNA CPR (Cardiomyocyte Proliferation Regulator) Regulates Cardiomyocyte Proliferation and Cardiac Repair. Circulation. 2019;139:2668–2684. doi: 10.1161/CIRCULATIONAHA.118.035832. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Chen X., Shen D., Ge D., Chen J., Pei J., Li Y., Yue Z., Feng J., Chu M., Nie Y. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell. Cardiol. 2019;127:105–114. doi: 10.1016/j.yjmcc.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Wu G., Cai J., Han Y., Chen J., Huang Z.P., Chen C., Cai Y., Huang H., Yang Y., Liu Y. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Z.P., Ding Y., Chen J., Wu G., Kataoka M., Hu Y., Yang J.H., Liu J., Drakos S.G., Selzman C.H. Long non-coding RNAs link extracellular matrix gene expression to ischemic cardiomyopathy. Cardiovasc. Res. 2016;112:543–554. doi: 10.1093/cvr/cvw201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Micheletti R., Plaisance I., Abraham B.J., Sarre A., Ting C.C., Alexanian M., Maric D., Maison D., Nemir M., Young R.A. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 2017;9:eaai9118. doi: 10.1126/scitranslmed.aai9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piccoli M.T., Gupta S.K., Viereck J., Foinquinos A., Samolovac S., Kramer F.L., Garg A., Remke J., Zimmer K., Batkai S., Thum T. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017;121:575–583. doi: 10.1161/CIRCRESAHA.117.310624. [DOI] [PubMed] [Google Scholar]
- 24.Hao K., Lei W., Wu H., Wu J., Yang Z., Yan S., Lu X.A., Li J., Xia X., Han X. LncRNA-Safe contributes to cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial infarction. Theranostics. 2019;9:7282–7297. doi: 10.7150/thno.33920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu X., Khalil H., Kanisicak O., Boyer J.G., Vagnozzi R.J., Maliken B.D., Sargent M.A., Prasad V., Valiente-Alandi I., Blaxall B.C., Molkentin J.D. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 2018;128:2127–2143. doi: 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Z.P., Chen J., Seok H.Y., Zhang Z., Kataoka M., Hu X., Wang D.Z. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 2013;112:1234–1243. doi: 10.1161/CIRCRESAHA.112.300682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arora H., Wilcox S.M., Johnson L.A., Munro L., Eyford B.A., Pfeifer C.G., Welch I., Jefferies W.A. The ATP-Binding Cassette Gene ABCF1 Functions as an E2 Ubiquitin-Conjugating Enzyme Controlling Macrophage Polarization to Dampen Lethal Septic Shock. Immunity. 2019;50:418–431. doi: 10.1016/j.immuni.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 29.Wilcox S.M., Arora H., Munro L., Xin J., Fenninger F., Johnson L.A., Pfeifer C.G., Choi K.B., Hou J., Hoodless P.A., Jefferies W.A. The role of the innate immune response regulatory gene ABCF1 in mammalian embryogenesis and development. PLoS ONE. 2017;12:e0175918. doi: 10.1371/journal.pone.0175918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim S., Cho H.Y., Kim D.G., Roh Y., Son S.Y., Mushtaq A.U., Kim M., Bhattarai D., Sivaraman A., Lee Y. Targeting the interaction of AIMP2-DX2 with HSP70 suppresses cancer development. Nat. Chem. Biol. 2020;16:31–41. doi: 10.1038/s41589-019-0415-2. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y., Karuppagounder S.S., Shin J.H., Lee Y.I., Ko H.S., Swing D., Jiang H., Kang S.U., Lee B.D., Kang H.C. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 2013;16:1392–1400. doi: 10.1038/nn.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yun S.P., Kim H., Ham S., Kwon S.H., Lee G.H., Shin J.H., Lee S.H., Ko H.S., Lee Y. VPS35 regulates parkin substrate AIMP2 toxicity by facilitating lysosomal clearance of AIMP2. Cell Death Dis. 2017;8:e2741. doi: 10.1038/cddis.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaiswal J.K., Lauritzen S.P., Scheffer L., Sakaguchi M., Bunkenborg J., Simon S.M., Kallunki T., Jäättelä M., Nylandsted J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014;5:3795. doi: 10.1038/ncomms4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong C., Maquat L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeong K., Ryu I., Park J., Hwang H.J., Ha H., Park Y., Oh S.T., Kim Y.K. Staufen1 and UPF1 exert opposite actions on the replacement of the nuclear cap-binding complex by eIF4E at the 5′ end of mRNAs. Nucleic Acids Res. 2019;47:9313–9328. doi: 10.1093/nar/gkz643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia L., Xiao X., Liu W.L., Song Y., Liu T.J.J., Li Y.J., Zacksenhaus E., Hao X.J., Ben-David Y. Coactosin-like protein CLP/Cotl1 suppresses breast cancer growth through activation of IL-24/PERP and inhibition of non-canonical TGFβ signaling. Oncogene. 2018;37:323–331. doi: 10.1038/onc.2017.342. [DOI] [PubMed] [Google Scholar]
- 37.Li G., Yin Y., Chen J., Fan Y., Ma J., Huang Y., Chen C., Dai P., Chen S., Zhao S. Coactosin-like protein 1 inhibits neuronal migration during mouse corticogenesis. J. Vet. Sci. 2018;19:21–26. doi: 10.4142/jvs.2018.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Provost P., Samuelsson B., Rådmark O. Interaction of 5-lipoxygenase with cellular proteins. Proc. Natl. Acad. Sci. USA. 1999;96:1881–1885. doi: 10.1073/pnas.96.5.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wurthner J.U., Frank D.B., Felici A., Green H.M., Cao Z., Schneider M.D., McNally J.G., Lechleider R.J., Roberts A.B. Transforming growth factor-beta receptor-associated protein 1 is a Smad4 chaperone. J. Biol. Chem. 2001;276:19495–19502. doi: 10.1074/jbc.M006473200. [DOI] [PubMed] [Google Scholar]
- 40.Chen J., Huang Z.P., Seok H.Y., Ding J., Kataoka M., Zhang Z., Hu X., Wang G., Lin Z., Wang S. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 2013;112:1557–1566. doi: 10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao F., Kataoka M., Liu N., Liang T., Huang Z.P., Gu F., Ding J., Liu J., Zhang F., Ma Q. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat. Commun. 2019;10:1802. doi: 10.1038/s41467-019-09530-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J., Shearer G.C., Chen Q., Healy C.L., Beyer A.J., Nareddy V.B., Gerdes A.M., Harris W.S., O’Connell T.D., Wang D. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation. 2011;123:584–593. doi: 10.1161/CIRCULATIONAHA.110.971853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfeffer J.M., Pfeffer M.A., Fletcher P.J., Braunwald E. Progressive ventricular remodeling in rat with myocardial infarction. Am. J. Physiol. 1991;260:H1406–H1414. doi: 10.1152/ajpheart.1991.260.5.H1406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data (GEO: GSE161389) and microarray-based transcriptome profiling data (GEO: GSE161427) are available in the Gene Expression Omnibus.