

Abstract
MicroRNAs (miRNAs) are important regulators in the process of cardiac hypertrophy and heart failure. Previous studies have shown that miR-199a is upregulated in pressure-overload cardiac hypertrophy and that inhibition of miR-199a attenuates cardiac hypertrophy in vitro. However, the therapeutic role of anti-miR-199a treatment in the cardiac hypertrophy in vivo model is less known. Here, we show an efficient and useful method to treat mouse cardiac hypertrophy and restore cardiac function through injection of adeno-associated virus (AAV)-mediated anti-miR-199a tough decoys (TuDs). RNA-seq transcriptome analysis indicated that genes related to cytoplasmic translation and mitochondrial respiratory chain complex assembly were upregulated in anti-miR-199a-treated recovered hearts. We further validated that PGC-1α is the direct target of miR-199a involved in the therapeutic effect and the regulation of the PGC-1α/ERRα axis and that the downstream pathway of mitochondrial fatty acid oxidation and oxidative phosphorylation constitute the underlying mechanism of the restored mitochondrial structure and function in our anti-miR-199a-treated mice. Our study highlights the important regulatory role of miR-199a in cardiac hypertrophy and the value of the AAV-mediated miRNA delivery system.
Keywords: adeno-associated virus, microRNA-199a, cardiac hypertrophy, heart failure, PGC1-alpha
Graphical Abstract
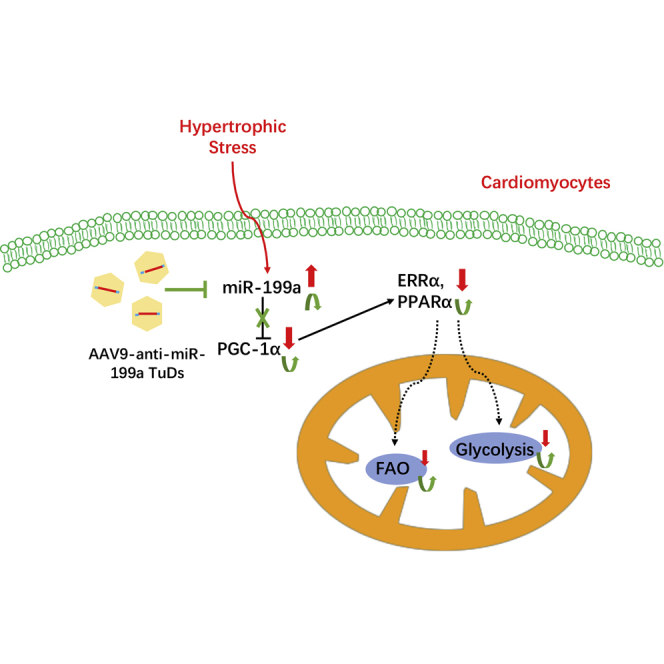
Previous studies showed that miR-199a is upregulated in pressure-overload cardiac hypertrophy, and inhibition of miR-199a attenuated cardiac hypertrophy in vitro. This study showed an efficient and useful way to treat mice cardiac hypertrophy and restored cardiac function through injection of adeno-associated virus-mediated anti-miR-199a tough decoys targeting the PGC-1α/ERR axis.
Introduction
Pathological cardiac hypertrophy is a compensatory cardiac remodeling response characterized by cardiac enlargement and cardiomyocyte hypertrophy in response to neurohormonal stimulation and mechanical forces under pathological conditions. Prolonged exposure to stress stimuli causes cardiac remodeling to become maladaptive decompensation, accompanied by the following processes: cell death, fibrosis, dysregulation of Ca2+-handling proteins, mitochondrial dysfunction, metabolic reprogramming, increase of reactive oxygen species, and altered sarcomere structure.1 Among them, mitochondrial dysfunction and metabolic remodeling can be the direct cause of pathological cardiac hypertrophy.1 Mitochondrial biogenesis, metabolic reprogramming (changes in fatty acid oxidation [FAO] and glycolysis pathway), mitochondrial oxidative stress, and oxidative phosphorylation (OXPHOS) have been shown to associated with pathological cardiac hypertrophy.2, 3, 4, 5, 6
MicroRNAs (miRNAs) are a class of small, noncoding RNAs that regulate gene expression at the post-transcription level. Growing evidence has shown that miRNAs play important roles in the progression of cardiac hypertrophy7, 8, 9, 10 and they can regulate cardiac mitochondrial dysfunction, which results in pathological cardiac hypertrophy and heart failure (HF).2, 3, 4,6,11 The manipulation of these miRNAs can be a therapeutic strategy to treat cardiac hypertrophy. However, common chemically modified miRNAs, such as miRNA agomirs/antagomirs, miRNA mimics, and miRNA sponges, have transient lifespans with short-term effects and require large-dose and repetitive delivery.12 Recent studies demonstrated that adeno-associated virus (AAV)-mediated delivery of miRNA interference sequence in vivo achieved efficient and persistent therapeutic effects on cardiovascular diseases.7,8,13, 14, 15 Compared with chemically modified miRNAs, AAV9 has high tropism for the heart, and miRNA interference sequence expression can be cell specific when we used cardiac-specific promoter, such as cTNT or α-MHC.12,16 In addition, AAV exhibits long-term (25 weeks) effects of miRNA intervention and is safe, without adverse side effects.12,17
Our previous study found that miR-199a was upregulated in isoproterenol (ISO)-induced cardiac hypertrophy.18 miR-199a was identified as an important regulator in primary cardiomyocyte hypertrophy.19 The inhibitory effect of miR-199a on hypertrophic primary cardiomyocytes results in reduced cell size and attenuated hypertrophy.20 We hypothesize that the in vivo inhibition of miR-199a in a cardiac hypertrophy model through AAV delivery can result in amelioration of cardiac hypertrophy and restored cardiac function. Here, we aimed to investigate the role of miR-199a in pathological cardiac hypertrophy through AAV-mediated miRNA delivery and to achieve therapeutic effects on cardiac hypertrophy and heart function.
Results
Inhibition of miR-199a attenuated hypertrophy of neonatal rat ventricular cardiomyocytes (NRVMs)
The expression level of miR-199a in NRVMs was significantly decreased to 13% in the inhibitor group compared with the control group (Figure 1A). Based on the calculated cardiomyocyte size in each group, inhibition of miR-199a by the miR-199a inhibitor significantly reduced the cardiomyocyte size of hypertrophic NRVMs, while overexpression of miR-199a by the miR-199a mimic did not change the size of hypertrophic cardiomyocytes (Figure 1B). The elevated hypertrophic markers ANP and Acta1 were significantly decreased by inhibition of miR-199a, while BNP and β-MHC were not significantly changed after inhibition of miR-199a (Figure 1C).
Figure 1.

Inhibition of miR-199a attenuated hypertrophy of neonatal rat ventricular cardiomyocytes
(A) The expression of miR-199a in NRVMs with or without miR-199a inhibitor treatment (n = 3). (B) Representative confocal images of sarcomeric alpha actinin (SAA, red) and DAPI (nuclei, blue) staining of NRVMs in different groups. NRVM size in each group was calculated by ImageJ. Scale bar, 20 μm. Approximately 200 counted cells in each group. (C) The expression of hypertrophic markers ANP, BNP, β-MHC, and Acta1 in each NRVM group (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
AAV9 technology enabled safe, cardiac-specific, and highly efficient cardiac infection and long-term miR-199a inhibition in vivo
The injection of AAV9-anti-miR-199a tough decoys (TuDs) or AAV9-cTNT-EGFP virus (1 × 1011 viral genomes [Vg] per neonatal mouse) achieved a very high infection efficiency since 8 days of age (Figures 2A and 2B). Calculated by the percentage of GFP that both AAV viruses expressed, the average infection efficiency of AAV9-anti-miR-199a TuDs and AAV9-cTNT-EGFP virus was 30% and 24% at 8 days of age, respectively, and both AAVs sustained high expression until 13 weeks of age (Figure 2C). The high and sustained expression of AAV9-anti-miR-199a TuDs virus achieved stable inhibition of miR-199a in the heart throughout the observation period (Figure 2D). As AAV expression was driven by the cardiac-specific cTNT promoter, the expression was almost absent in other organs, such as the liver, brain, and skeletal muscle of the injected mice (Figure 2E), and miR-199a expression was not inhibited in liver, brain, and skeletal muscle of the injected mice (Figure 2F).
Figure 2.

AAV-mediated miR-199a inhibition in vivo
(A and B) Representative confocal images of wheat germ agglutinin (WGA, red) and DAPI (nuclei, blue) staining of hearts in 8-day-old, 4-week-old, and 13-week-old mice injected with AAV9-anti-miR-199a TuDs or AAV9-EGFP. Green, autofluorescence of cardiomyocytes with AAV9 virus infection. Scale bars, 200 μm; n = 4–5. (C) Calculated relative percentage of GFP-positive cells in the hearts of 8-day-old, 4-week-old, and 13-week-old mice injected with AAV9-anti-miR-199a TuDs or AAV9-EGFP. n = 4–5 different sections in each group. NS represents not significant compared with that in 8-day-old mice. (D) The expression of miR-199a in the hearts of 8-day-old, 4-week-old, and 13-week-old mice injected with AAV9-anti-miR-199a TuDs or AAV9-EGFP (n = 6). (E) Representative confocal images of WGA (red) and DAPI (nuclei, blue) staining of skeletal muscle, liver, and brain tissue in 13-week-old mice injected with AAV9-anti-miR-199a TuDs or AAV9-EGFP. Arrow, green autofluorescence of cardiomyocytes with AAV9 infection. Scale bars, 200 μm; n = 3. (F) The expression of miR-199a in liver, brain, and skeletal muscle of 13-week-old mice with AAV9-anti-miR-199a TuDs or AAV9-EGFP injection (n = 5). NS, not significant. (G) The plasma cytokine levels of IL-1β, IL-6, and IFN-γ in AAV-injected mice and wild-type control mice (n = 6). IFN-γ in each group was lower than 7.8 pg/mL. ∗p < 0.05, ∗∗p < 0.01.
Moreover, AAV was safe in terms of the systemic inflammatory response. The inflammatory cytokines interleukin-1β (IL-1β), IL-6, and interferon-γ (IFN-γ) showed no significant increase in the serum of the injected mice compared with that in wild-type mice (Figure 2G).
Injection of AAV9-anti-miR-199a TuDs virus alleviated cardiac hypertrophy and restored cardiac function in vivo
The injection of AAV9-anti-miR-199a TuDs virus significantly reduced the hypertrophic cardiac size induced by ISO (Figure 3A). The cardiac hypertrophy index, the ratio of heart weight and tibia length, significantly decreased to normal value (Figure 3B). Treatment with AAV9-anti-miR-199a TuDs virus significantly reduced the hypertrophic cardiomyocyte size calculated from the wheat germ agglutinin (WGA) staining (Figure 3C). Furthermore, miR-199a inhibition treatment restored cardiac function. The left ventricular ejection fraction (LVEF) of treated mice increased to 72%, and the left ventricular fractional shortening (LVFS) of treated mice increased to 40%, significantly (Figure 3D; detailed echocardiographic assessment in Table S1). The cardiac hypertrophy markers ANP, BNP, and Acta1 were all dramatically reduced after miR-199a inhibition treatment (Figure 3E). However, the cardiac fibrosis and apoptosis were not significantly changed after anti-miR-199a treatment (Figures S1 and S2). Overall, miR-199a inhibition treatment by AAV9-anti-miR-199a TuDs virus not only attenuated cardiac hypertrophy but also restored heart function of cardiac hypertrophy mice.
Figure 3.

AAV9-anti-miR-199a TuDs virus attenuated cardiac hypertrophy
(A) The representative gross morphology (scale bars, 1 mm) and H&E staining (scale bars, 400 μm) of heart sections in control and cardiac hypertrophy mice injected with AAV9-anti-miR-199a TuDs or AAV9-EGFP. n = 8. (B) The ratio of heart weight (HW, mg) and tibia length (TL, mm) in different mouse groups. n = 8. (C) Representative images of WGA (green) and DAPI (nuclei, blue) staining of hearts in different mouse groups (left). Scale bars, 20 μm. Calculated relative cell areas of cardiomyocytes in different mouse groups (right). n = 8. (D) Representative M-mode echocardiography images in different mouse groups (top). The percentages of left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) in the different mouse groups (bottom). n = 8. (E) The expression of hypertrophic markers ANP, BNP, β-MHC, and Acta1 in different mouse groups. n = 8. (F) The expression of miR-199a in different mouse groups. n = 8 in each group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
As shown by the efficiency of AAV in mice without cardiac hypertrophy (Figure 2D), the elevated expression level of miR-199a in the cardiac hypertrophy model was significantly decreased by AAV9-anti-miR-199a TuDs virus (Figure 3F), which was the basis for the therapeutic effect against cardiac hypertrophy and cardiac dysfunction. However, AAV9-anti-miR-199a TuDs virus treatment had no effect on the expression of miR-199b (Figure S3).
RNA-seq revealed the possible underlying protective mechanism of anti-miR-199a treatment in cardiac hypertrophy
To better understand the molecular mechanisms underlying anti-miR-199a-mediated effects against cardiac hypertrophy, we performed next-generation RNA sequencing (RNA-seq) in AAV9-anti-miR-199a TuDs-treated and AAV9-EGFP control heart samples. Compared to the control samples, a total of 2,429 genes were significantly upregulated, and 2,619 genes were significantly downregulated (Figure 4A). Gene Ontology (GO) enrichment analysis revealed that genes related to cytoplasmic translation, mitochondrial respiratory chain complex assembly, and ribonucleoprotein complex biogenesis were enriched among anti-miR-199a-upregulated genes (Figure 4B). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that the ribosome pathway and OXPHOS pathway are the main pathways involved (Figure 4C). Further PCR validation showed that representative ribosome genes Rps27a, Mrps21, and Mrpl14 all increased significantly after anti-miR-199a treatment, indicating active translation in the anti-miR-199a-treated group (Figure 4D). Representative genes involved in OXPHOS, Ndufa6, Sdhb, Uqcrh, Uqcrq, Cox4i1, and Atp5l, all increased significantly after anti-miR-199a treatment in the cardiac hypertrophy model (Figure 4E), suggesting that OXPHOS and the related mitochondrial metabolism pathways constitute a possible underlying mechanism of the anti-miR-199a protective effect.
Figure 4.

RNA sequencing results of anti-miR-199a treatment in cardiac hypertrophy
(A) Heatmap of all genes with significantly altered expression between the cardiac hypertrophy group and the anti-miR-199a-treated group. Red and blue colors indicate upregulated or downregulated genes. n = 3 per group. (B) Gene Ontology (GO) enrichment analysis of all significantly upregulated genes. The y axis shows the top 10 enriched pathways. The x axis is the −log10 of the adjusted p value. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of all significantly upregulated genes. The y axis shows the top enriched pathways. The x axis is the −log10 of the adjusted p value. (D) The expression of representative ribosome genes in the cardiac hypertrophy group and the anti-miR-199a-treated group. n = 8. (E) The expression of representative genes involved in oxidative phosphorylation in the cardiac hypertrophy group and the anti-miR-199a-treated group. n = 8 per group. ∗p < 0.05, ∗∗p < 0.01.
Target prediction and validation of miR-199a in cardiac hypertrophy in vitro and in vivo
Through the target prediction results in TargetScan v7.2 (http://www.targetscan.org/) and TarBase v7.0 (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index) and the reported validated miR-199a targets in heart diseases,4,21, 22, 23 we chose six potential targets related to mitochondrial metabolism or cardiac hypertrophy for further study (Table 1). Among these six potential targets, Ppargc1a (PGC-1α) was the only target that was derepressed after inhibition of miR-199a both in vitro and in vivo (Figures 5A and 5B). The nucleotide position 1518–1525 of the 3′UTR of the Ppargc1a gene was the predicted binding site of miR-199a, and it was conserved across different species (Figure 5C). Furthermore, a dual luciferase reporter assay showed that the miR-199a mimic significantly inhibited the luciferase activity of the Luc-PGC-1α-3′UTR reporter plasmid, while the luciferase activity remained unchanged in the mutant plasmid or miRNA mimic control group (Figure 5D), which suggests that PGC-1α is a direct target of miR-199a.
Table 1.
Predicted targets of miR-199a in cardiac hypertrophy
| Predicted target gene | Representative transcript in Mus musculus | Gene name |
Site in untranslated regions (UTR) (Lower row is the sequence of mmu-miR-199a) |
Reported pathway in cardiac hypertrophy and heart failure | |
|---|---|---|---|---|---|
| Ppargc1a | NM_008904.2 | peroxisome proliferative activated receptor, gamma, coactivator 1 alpha | Position 1518-1525 of Ppargc1a 3' UTR | mitochondrial biogenesis28, cardiac metabolic remodeling25 | |
| Dyrk1a | NM_001113389.1 | dual specificity tyrosine-phosphorylation-regulated kinase 1A | Position 1547-1554 of Dyrk1a 3' UTR | the Calcineurin-NFAT Pathway41 | |
| Sirt1 | NM_001159589.2 | sirtuin 1 | Position 451-457 of Sirt1 3' UTR | the ERR transcriptional pathway42 | |
| Hif1a | NM_001313919.1 | hypoxia inducible factor 1 subunit alpha | Position 237-243 of Hif1a 3' UTR | HIF1α-PPARγ glycolytic and lipid anabolic pathways43 | |
| Ppard | NM_011145.3 | peroxisome proliferator activated receptor delta | Position 902-908 of Ppard 3' UTR | cardiac glucose utilization and metabolic regulatory44 | |
| Gsk3b | NM_001347232.1 | glycogen synthase kinase 3 beta | Position 61-67 of Gsk3b 3' UTR | mToR signaling and inhibit autophagy pathway45 | |
| Position 3781-3787 of Gsk3b 3' UTR | |||||
Figure 5.

Validation of PGC-1α as a target of miR-199a in cardiac hypertrophy
(A) The expression of six predicted targets of miR-199a in hypertrophic NRVMs with and without miR-199a inhibitor treatment (n = 3). (B) The expression of six predicted targets of miR-199a in cardiac hypertrophy mice and the anti-miR-199a-treated mice (n = 8). (C) Schematic representation of genomic localization of miR-199a binding site in the 3′UTR of Ppargc1a gene and the conserved seed sequences in different species. (D) Dual luciferase activity assay of H293T cells co-transfected miR-199a mimic or mimic control with Luc-PGC-1α-3′UTR reporter plasmid or Luc-PGC-1α-Mut-3′UTR plasmid (n = 4). ∗p < 0.05, ∗∗p < 0.01.
Anti-miR-199a treatment attenuated cardiac hypertrophy via PGC-1α and the mitochondrial metabolic pathway
Through transmission electron microscopy (TEM), the mitochondrial ultrastructure was found to be disrupted in hypertrophic cardiomyocytes, while anti-miR-199a treatment attenuated mitochondrial network disorganization and maintained their linear array (Figure 6A).
Figure 6.

The disrupted mitochondrial ultrastructure and the altered genes related to mitochondrial metabolism are recovered by anti-miR-199a treatment
(A) Representative transmission electron microscopy (TEM) images in control, cardiac hypertrophy mice, and the anti-miR-199a-treated mice. Scale bars, 500 nm. Arrowhead, normal mitochondrial ultrastructure; arrow, disrupted mitochondrial ultrastructure (n = 3). (B) The expression of representative genes in glycolysis in each mouse group (n = 8). (C) The expression of representative genes in fatty acid oxidation (FAO) in each mouse group (n = 8). (D) The expression of important transcription factors involved in the mitochondrial metabolism in each mouse group (n = 8). (E) Representative western blot results and the relative protein expression of ERRα and PGC-1α in each mouse group (n = 6). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To explore the altered genes related to mitochondrial metabolism, we detected the expression levels of genes involved in glycolysis and FAO and important transcription factors involved in mitochondrial metabolism. Glucose transporter 1 (Glut1) and glucose transporter 4 (Glut4) are important uniporter proteins that facilitate the transport of glucose across cell membranes. Hexokinase 1 (Hk1) is the rate-limiting enzyme that catalyzes glucose into glucose-6-phosphate (G-6-P). Glut1, Glut4, and Hk1 levels all returned to normal expression levels upon miR-199a inhibition, indicating restored glycolysis in treated mice (Figure 6B). In the FAO pathway, Cd36, or fatty acid translocase, imports fatty acids inside cells. Carnitine palmitoyl transferase Ib (Cpt1b), acyl-coenzyme A oxidase 1 (Acox1), acyl-coenzyme A dehydrogenase medium chain (Acadm), and acyl-CoA dehydrogenase very long chain (Acadvl) are all important fatty acid beta-oxidation enzymes. The expression levels of Acox1 and Cpt1b were restored after anti-miR-199a treatment, and Acadm and Acadvl even increased to approximately twice the normal expression levels, suggesting activated FAO (Figure 6C).
Crucial transcription factors in mitochondrial metabolism, ERRα, PPARα, and PGC-1α, were restored upon anti-miR-199a treatment (Figure 6D). The corresponding protein levels of ERRα and PGC-1α were consistent with the mRNA changes induced by miR-199a inhibition (Figure 6E).
Discussion
In the present study, we successfully ameliorated mouse cardiac hypertrophy and cardiac dysfunction by injection of AAV9-anti-miR-199a TuDs. RNA-seq indicated that OXPHOS and the related mitochondrial metabolic pathway constituted the possible mechanism underlying this therapeutic phenotype. Furthermore, we confirmed that PGC-1α was the direct target of miR-199a involved in this pathway. Our results revealed that the regulation of the PGC-1α/ERRα axis and the downstream pathway of mitochondrial FAO and OXPHOS is the underlying mechanism of the restored mitochondrial structure and function in our anti-miR-199a-treated mice.
The PGC-1/ERR axis has been considered a candidate therapeutic target for pathological cardiac hypertrophy and heart failure for decades.24 Growing evidence supports that the PGC-1/ERR axis plays a key role in myocardial mitochondrial metabolism and pathological cardiac hypertrophy. The cardiac-specific knockout (KO) of PGC-1α in mice induces suppressed glucose oxidation and FAO, decreased OXPHOS, and reduction in mitochondrial density, which leads to dilating cardiomyopathy and heart failure.25 The lack of PGC-1α also accelerated transverse aortic constriction (TAC)-induced heart failure with decreased FAO and OXPHOS.26 However, the cardiac-specific overexpression of PGC-1α in mice resulted in mitochondrial ultrastructural abnormalities and dilating cardiomyopathy, which was reversible after cessation of PGC-1α overexpression.27,28 Interestingly, PGC-1α acts in a developmental stage-dependent manner: the induction of PGC-1α in the neonatal heart leads to dramatic mitochondrial biogenesis, while the induction of PGC-1α in adult mice causes mitochondrial structural derangements and cardiomyopathy.28 These findings explain why maintaining PGC-1α expression through a transgenic mouse model failed to attenuate pressure-overload cardiac hypertrophy and improve cardiac function29,30. In our study, the derepression of PGC-1α by miR-199a inhibition was modest, with only 1.3-fold change compared with that in control mice (Figure 6D). In addition, PGC-1α may not be the only target of miR-199a involved in mitochondrial recovery and cardiac relief. The results of increased glucose oxidation, FAO and OXPHOS, were in line with the activation of the PGC-1/ERR axis.
Previous studies show that PGC-1α directly coactivates ERRα and that ERRα is required for PGC-1α function in mitochondrial metabolism.31,32 ERRα is a key regulator in energy substrate oxidation, OXPHOS, and ATP synthesis. The ERRα KO mice developed heart failure after TAC, with reduced mitochondrial metabolism and ATP synthesis.33 In contrast, overexpression of ERRα activates PPARα and upregulates most genes involved in mitochondrial energy-producing pathways in cardiomyocytes.34 Our study shows that the expression of both PGC-1α and ERRα increased in anti-miR-199a-treated hearts and that the expression of the downstream mitochondrial metabolism pathway was upregulated, which indicates that the PGC-1/ERR axis is the possible underlying protective mechanism of anti-miR-199a treatment.
Interestingly, PGC-1α can be activated by exercise and promote cardiac mitochondrial biogenesis.27,35 PGC-1α is also involved in physiological cardiac hypertrophy in miR-199a sponge transgenic mice.23 There are two possible explanations of why miR-199 knockdown plays a different role under the physiological and pathological conditions. First, the method we used to inhibit miR-199a is different from that in the study by Li et al.23 Li et al. used sponge RNA to inhibit the expression of miR-199a, which also inhibited miR-199b expression. However, we used TuDs structure with two miR-199a binding sties and no inhibition effect on miR-199b. Second, the regulation of PGC-1α itself is sophisticated in a developmental stage-dependent manner.28 The AAV9 technology we used only drives the expression of anti-miR-199a TuDs postnatally, while the genetic knockin of miR-199a sponge works in the whole fetal development period. However, the underlying mechanism of miR-199a-PCG-1α should be further investigated through the study of different cardiomyocyte gene expression and mitochondrial metabolomics.
el Azzouzi et al.4 also proved that the inhibition of miR-199a and miR-214 using the miRNA antagomir improved cardiac function and restored mitochondrial FAO by targeting PPARδ. Our results are partly in line with their study. The expression of glycolysis genes, FAO genes, and important transcription factors involved in mitochondrial metabolism returned to a normal level compared with that in the cardiac hypertrophy model. However, PPARδ was not significantly changed in cardiac hypertrophy after anti-miR-199a treatment. The main contribution to their recovered phenotype is the inhibition of miR-214 by the antagomir.4 In addition, the AAV9 delivery system achieved cardiac-specific, highly efficient, and long-term inhibition, which is superior to the miRNA antagomir. This may explain why our results of anti-miR-199a treatment for cardiac hypertrophy are more profound. These advantages also make AAV a popular delivery method of miRNA either in mechanistic studies or therapeutic effects on cardiovascular diseases.3,7,8,13, 14, 15
In pressure-overload cardiac hypertrophy, mitochondrial metabolism usually shifts from FAO to glucose utilization. However, metabolic alterations also depend on the animal models, experimental settings, stage and severity of cardiac hypertrophy and dysfunction, and different pathological stimuli.36 Decreased glycolysis also can be detected in abdominal aortic constriction-induced cardiac hypertrophy mice37 and ISO-induced cardiac hypertrophy rats.38 The cardiac insulin resistance in different cardiac hypertrophy models may explain the different glycolysis alterations in our model and the previous ones.
There are several limitations in the present study. First, except for PGC-1α, there may be other contributing targets involved in the regulation of the PGC-1/ERR axis and the related mitochondrial metabolic pathway. A more comprehensive, high-throughput target study may discover new targets. Second, we did not record the dynamic change in anti-miR-199a-treated NRVMs by live cell imaging or Seahorse metabolism analysis, which generate more reliable information on mitochondrial metabolism. Third, the cardiac hypertrophy model induced by ISO injection is mainly generated through neurohormonal stress. Other cardiac hypertrophy models, such as TAC, should be studied. These topics warrant further investigation.
In conclusion, the current study reveals the important regulatory role of miR-199a in cardiac hypertrophy and highlights the efficiency and usefulness of an AAV-mediated miRNA delivery system. Further experiments regarding the mitochondrial metabolism pathway are still required to elucidate the specific mechanism and gain more molecular and functional insight.
Materials and methods
Cardiac hypertrophy model and echocardiographic measurement
Thirty-two C57BL/6 adult male mice (25–30 g) were used for the cardiac hypertrophy model. The establishment of a cardiac hypertrophy model has been previously described in detail.39 In brief, mice were treated with isoproterenol (ISO, Sigma-Aldrich, St. Louis, MO, USA) to induce cardiac hypertrophy. ISO was dissolved in sterile saline and was injected intraperitoneally (60 mg/kg/day) once daily for 14 consecutive days.
Cardiac function was measured by a Vivid 7 ultrasound system (GE Healthcare, Horten, Norway) with a 13 MHz i13L cardiac transducer. Two-dimensional M-mode tracings were used to measure left ventricular internal dimension in diastole (LVIDd), left ventricular internal dimension in systole (LVIDs), and the percentages of LVEF and LVFS. All mice were sacrificed 3 weeks after the final injection of ISO. The Institutional Animal Care and Use Committee of Sichuan University approved all the above experiments.
AAV9-anti-miR-199a TuDs cloning, virus packaging, and systemic delivery
We generated AAV9-U6-anti-miR-199a TuDs-cTNT-EGFP (AAV9-anti-miR-199a TuDs) from AAV9-U6-gRNA-cTNT-EGFP. AAV9-cTNT-EGFP (AAV9-EGFP) was used as a control (AAV9-U6-gRNA-cTNT-EGFP and AAV9-cTNT-EGFP plasmids were a gift from Yuxuan, Guo in William Pu’s lab40). Anti-miR-199a TuDs contain two single-stranded miR-199a-5p binding sites flanked by double-stranded stems that generate a stable hairpin structure (Figure S4). The design of anti-miR-199a TuDs was based on a previous report.17 The anti-miR-199a TuDs RNA was cloned into the AAV9-U6-gRNA-cTNT-EGFP plasmid using AgeI and RsrII restriction enzymes. The design of anti-miR-199a TuDs and the schematic diagram of the AAV9-anti-miR-199a TuDs plasmid are shown in detail in Figure S4.
The AAV9 virus was produced using a previous protocol.40 In brief, the AAV-ITR plasmid (AAV9-anti-miR-199a TuDs or AAV9-cTNT-EGFP) and the AAV9-Rep-Cap and pHelper (pAd-deltaF6, Penn Vector Core, Philadelphia, PA, USA) plasmids were co-transfected into H293T cells. Three days later, cells were collected and lysed. AAV9 was purified and concentrated by gradient ultracentrifugation. The titer of AAV viral particles was quantified by real-time PCR. The viral preparations had titers between 1 × 1013 and 1 × 1014 Vg/mL. The AAV9-anti-miR-199a TuDs or AAV9-cTNT-EGFP virus was injected into 1-day-old (P1) pups of C57BL/6 mice subcutaneously at 1 × 1011 Vg per mouse.
Histological analysis, immunostaining, and microscopy imaging
Freshly dissected heart tissues were fixed in 4% paraformaldehyde overnight and then embedded in paraffin. The myocardial sections (5 μm) were stained with hematoxylin and eosin (H&E) for heart morphology, Masson’s trichrome for cardiac fibrosis, or terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL, DeadEnd Fluorometric TUNEL System, Promega, Madison, WI, USA) for the detection of apoptosis. Images were viewed and captured by a ZEISS Axio Imager 2 microscope (Jena, Germany).
For immunostaining, heart tissues were embedded in tissue freezing medium (O.C.T. Compound, VWR, Torrance, CA, USA). Heart sections (5–6 μm) were incubated with primary antibodies at 4°C overnight and secondary antibodies for 2 h. The primary antibody was used as follows: anti-sarcomeric alpha actinin antibody (SAA, 1:200, ab9465, Abcam, Cambridge, MA, USA), WGA (Alexa Fluor 647 Conjugate, 1:400, W32466, Invitrogen). DAPI (62247, Invitrogen) was used to stain the nuclei. Immunostaining images were captured by an Olympus FV1000 confocal microscope (Tokyo, Japan). ImageJ software was used for all image analyses.
TEM imaging
The isolated mouse hearts were immersed in stationary liquid (pH 7.3) containing 3% glutaraldehyde. Tissue samples were then fixed in 2% osmic acid (OsO4, Sigma-Aldrich) in phosphate-buffered solution. After dehydration with a concentration gradient of acetone solutions, tissues were then embedded in Epon (Epon 812, Sigma-Aldrich). Ultrathin sections (50 nm) were cut by microtome, mounted onto formvar-coated slot grids, and stained with uranyl acetate (Zhongjingkeyi, Beijing, China) and lead citrate (Zhongjingkeyi, Beijing, China) at room temperature. Images were captured by a JEM-1400PLUS electron microscope (Tokyo, Japan).
Primary culture of NRVMs, hypertrophic model, and miR-199a treatment
NRVMs were prepared according to the instructions of the neonatal cardiomyocyte isolation kit (nc-6031, Cellutron, Baltimore, MD, USA). Primary cardiomyocytes were isolated by enzymatic disassociation of P1 neonatal rat hearts. Cells were plated on SureCoat solution (sc-9035, Cellutron) coated plates in NS medium (m-8031, with serum, Cellutron). After plating overnight, the cells were changed to NW medium (m-8032, serum-free, Cellutron).
Then, 50 nM (final concentration) miR-199a mimic or 100 nM (final concentration) miR-199a inhibitor and control oligonucleotide (Ribobio, Guangzhou, China) were transfected into cardiomyocytes using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA, USA). Twenty-four hours later, the cells were treated with 20 μM hypertrophic agent, phenylephrine (PE, Sigma-Aldrich). Cells were harvested 24 h after PE treatment for RNA isolation or 48 h after PE treatment for immunostaining.
RNA-seq, GO and KEGG pathway analyses, and miRNA database analysis
Three biological replicates of RNA samples from each group were prepared for RNA-seq. RNA-seq experiments were performed by Novogene (Beijing, China). In brief, RNA sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina), and 150 bp paired-end reads were generated. For the data analysis, raw data in fastq format were first processed through in-house Perl scripts. Clean data were obtained by removing reads containing adapters, reads containing poly-N, and low-quality reads from raw data. Mus musculus GRCm38 (mm10) was the reference genome used, and gene model annotation files were downloaded from the genome website directly.
The index of the reference genome was built using Hisat2 v2.0.5, and paired-end clean reads were aligned to the reference genome using Hisat2 v2.0.5. Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed using the DESeq2 R package (1.16.1). The p values were adjusted using the Benjamini and Hochberg method. GO enrichment and KEGG pathway analyses were implemented using the clusterProfiler R package. The RNA-seq data have been deposited in the GenBank Database (GenBank: GSE155545).
To predict the mRNA targets of miR-199a, we used TargetScan v7.2 (http://www.targetscan.org/) and TarBase v7.0 (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index).
Enzyme-linked immunosorbent assay (ELISA)
Same volumes of plasma samples from mice in each group were used to measure the levels of the inflammatory cytokines IL-1β, IL-6, and tumor necrosis factor alpha (TNF-α) using the corresponding ELISA kits (CME0015, CME0006, CME0004, 4A Biotech Co., Beijing, China) according to the manufacturers’ instructions.
RNA extraction and real-time PCR
Hearts were frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted from cardiac samples using a Total RNA Kit II (Omega, Norcross, GA, USA). RNA (0.5 μg) was reverse transcribed into cDNA using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). For miRNA detection, RNA was reverse transcribed into cDNA using the miRNA qRT-PCR Starter Kit (Ribobio, Guangzhou, China). Real-time PCR was performed with a CFX96 Real-Time System using SYBR Green (Bio-Rad, Hercules, CA, USA).
The reverse transcription (RT) and PCR primers for miR-199a, miR-199b, and the U6 control were from the primer set (Ribobio, Guangzhou, China) according to the accession number in the miRBase database (http://www.mirbase.org). The primer sequences for other mRNAs are listed in Table S2.
Western blot
Heart tissues were lysed in a RIPA lysis buffer system (Beyotime, Shanghai, China) with phenylmethanesulfonylfluoride (PMSF, Beyotime). Total protein concentrations were normalized using a BCA Protein Assay Kit (23227, Thermo Scientific, Rockford, IL, USA). Lysates were subsequently separated by 10% SDS/PAGE gels, transferred to PVDF membranes (IPVH00010, Millipore, Darmstadt, Germany), and blocked in 5% nonfat milk in TBST for 1 h. Then, membranes were incubated with the following antibodies overnight at 4°C: anti-PGC1 alpha antibody (ab54481, Abcam), anti-ERR alpha antibody (ab76228, Abcam), and anti-GAPDH loading control (ab9485, Abcam). Proteins were visualized by ECL Plus western blot detection reagents (Beyotime, Shanghai, China).
Luciferase reporter assay
A total of 50 ng of wild-type Luc-PGC-1α-3′UTR construct, mutant Luc-PGC-1α-Mut-3′UTR construct, 10 nM miR-199a mimic, or negative control was transfected into H293T cells by Lipofectamine 3000 (Invitrogen, Carlsbad, CA). Forty-eight hours after transfection, luciferase activities were measured using a dual luciferase reporter assay system from triplicate transfected wells.
Statistical analysis
Data are shown as the mean ± SD. The significance of intergroup differences was tested using Student’s t test or the Mann-Whitney test as indicated in SPSS v.25.0 software (IBM, Armonk, NY, USA). p < 0.05 was considered statistically significant.
Acknowledgments
The authors are grateful to Yuxuan Guo, Donghui Zhang, and other members of the William Pu lab of Boston Children’s Hospital for their theoretical and technical help; to Xiaoyun Hu, Zhan-Peng Huang, and other members of the Da-Zhi Wang lab of Boston Children’s Hospital for their theoretical and technical help; and for the support from the China Scholarship Council (CSC) during the visit (H.Y.) to the Boston Children’s Hospital, Harvard Medical School. This work was funded by the National Natural Science Foundation of China (81671702), the China Postdoctoral Science Foundation (2019M653422), and the Post-Doctor Research Project, West China Hospital, Sichuan University (2018HXBH035).
Author contributions
H.Y., H.W., X.Z., J.H., Y. Li, D.-Z.W., F.Y., and Y. Luo designed and performed experiments. H.Y., Y. Li, K.Z., and Y.H. analyzed and interpreted the data. H.Y. analyzed the RNA-seq data and wrote the manuscript. F.Y. and Y. Luo revised the manuscript. H.Y., F.Y., and Y. Luo supplied funding and supervised the work. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.11.007.
Supplemental information
References
- 1.Nakamura M., Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 2.Liu B.L., Cheng M., Hu S., Wang S., Wang L., Tu X., Huang C.X., Jiang H., Wu G. Overexpression of miR-142-3p improves mitochondrial function in cardiac hypertrophy. Biomed. Pharmacother. 2018;108:1347–1356. doi: 10.1016/j.biopha.2018.09.146. [DOI] [PubMed] [Google Scholar]
- 3.Qiu Y., Cheng R., Liang C., Yao Y., Zhang W., Zhang J., Zhang M., Li B., Xu C., Zhang R. MicroRNA-20b Promotes Cardiac Hypertrophy by the Inhibition of Mitofusin 2-Mediated Inter-organelle Ca2+ Cross-Talk. Mol. Ther. Nucleic Acids. 2020;19:1343–1356. doi: 10.1016/j.omtn.2020.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.el Azzouzi H., Leptidis S., Dirkx E., Hoeks J., van Bree B., Brand K., McClellan E.A., Poels E., Sluimer J.C., van den Hoogenhof M.M. The hypoxia-inducible microRNA cluster miR-199a∼214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab. 2013;18:341–354. doi: 10.1016/j.cmet.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Dai D.F., Johnson S.C., Villarin J.J., Chin M.T., Nieves-Cintrón M., Chen T., Marcinek D.J., Dorn G.W., 2nd, Kang Y.J., Prolla T.A. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T., Zhai M., Xu S., Ponnusamy M., Huang Y., Liu C.Y., Wang M., Shan C., Shan P.P., Gao X.Q. NFATc3-dependent expression of miR-153-3p promotes mitochondrial fragmentation in cardiac hypertrophy by impairing mitofusin-1 expression. Theranostics. 2020;10:553–566. doi: 10.7150/thno.37181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh J.G., Watanabe S., Lee A., Gorski P.A., Lee P., Jeong D., Liang L., Liang Y., Baccarini A., Sahoo S. miR-146a Suppresses SUMO1 Expression and Induces Cardiac Dysfunction in Maladaptive Hypertrophy. Circ. Res. 2018;123:673–685. doi: 10.1161/CIRCRESAHA.118.312751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nie X., Fan J., Li H., Yin Z., Zhao Y., Dai B., Dong N., Chen C., Wang D.W. miR-217 Promotes Cardiac Hypertrophy and Dysfunction by Targeting PTEN. Mol. Ther. Nucleic Acids. 2018;12:254–266. doi: 10.1016/j.omtn.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian C., Hu G., Gao L., Hackfort B.T., Zucker I.H. Extracellular vesicular MicroRNA-27a∗ contributes to cardiac hypertrophy in chronic heart failure. J. Mol. Cell. Cardiol. 2020;143:120–131. doi: 10.1016/j.yjmcc.2020.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu Q., Li A., Chen X., Qin Y., Sun X., Li Y., Yue E., Wang C., Ding X., Yan Y. Overexpression of miR-135b attenuates pathological cardiac hypertrophy by targeting CACNA1C. Int. J. Cardiol. 2018;269:235–241. doi: 10.1016/j.ijcard.2018.07.016. [DOI] [PubMed] [Google Scholar]
- 11.Zaglia T., Ceriotti P., Campo A., Borile G., Armani A., Carullo P., Prando V., Coppini R., Vida V., Stølen T.O. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA. 2017;114:E9006–E9015. doi: 10.1073/pnas.1708772114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie J., Burt D.R., Gao G. Adeno-associated virus-mediated microRNA delivery and therapeutics. Semin. Liver Dis. 2015;35:81–88. doi: 10.1055/s-0034-1397352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H., Fan J., Zhao Y., Zhang X., Dai B., Zhan J., Yin Z., Nie X., Fu X.D., Chen C., Wang D.W. Nuclear miR-320 Mediates Diabetes-Induced Cardiac Dysfunction by Activating Transcription of Fatty Acid Metabolic Genes to Cause Lipotoxicity in the Heart. Circ. Res. 2019;125:1106–1120. doi: 10.1161/CIRCRESAHA.119.314898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao F., Kataoka M., Liu N., Liang T., Huang Z.P., Gu F., Ding J., Liu J., Zhang F., Ma Q. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat. Commun. 2019;10:1802. doi: 10.1038/s41467-019-09530-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou Z., Qin X., Hu Y., Zhang X., Li G., Wu J., Li J., Sha J., Chen J., Xia J. Longterm Exercise-Derived Exosomal miR-342-5p: A Novel Exerkine for Cardioprotection. Circ. Res. 2019;124:1386–1400. doi: 10.1161/CIRCRESAHA.118.314635. [DOI] [PubMed] [Google Scholar]
- 16.Zacchigna S., Zentilin L., Giacca M. Adeno-associated virus vectors as therapeutic and investigational tools in the cardiovascular system. Circ. Res. 2014;114:1827–1846. doi: 10.1161/CIRCRESAHA.114.302331. [DOI] [PubMed] [Google Scholar]
- 17.Xie J., Ameres S.L., Friedline R., Hung J.H., Zhang Y., Xie Q., Zhong L., Su Q., He R., Li M. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods. 2012;9:403–409. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan H., Li Y., Wang C., Zhang Y., Liu C., Zhou K., Hua Y. Contrary microRNA Expression Pattern Between Fetal and Adult Cardiac Remodeling: Therapeutic Value for Heart Failure. Cardiovasc. Toxicol. 2017;17:267–276. doi: 10.1007/s12012-016-9381-z. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H., Li S., Zhou Q., Sun Q., Shen S., Zhou Y., Bei Y., Li X. Qiliqiangxin Attenuates Phenylephrine-Induced Cardiac Hypertrophy through Downregulation of MiR-199a-5p. Cell. Physiol. Biochem. 2016;38:1743–1751. doi: 10.1159/000443113. [DOI] [PubMed] [Google Scholar]
- 20.Song X.W., Li Q., Lin L., Wang X.C., Li D.F., Wang G.K., Ren A.J., Wang Y.R., Qin Y.W., Yuan W.J., Jing Q. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J. Cell. Physiol. 2010;225:437–443. doi: 10.1002/jcp.22217. [DOI] [PubMed] [Google Scholar]
- 21.Rane S., He M., Sayed D., Vashistha H., Malhotra A., Sadoshima J., Vatner D.E., Vatner S.F., Abdellatif M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z., Song Y., Liu L., Hou N., An X., Zhan D., Li Y., Zhou L., Li P., Yu L. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017;24:1205–1213. doi: 10.1038/cdd.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z., Liu L., Hou N., Song Y., An X., Zhang Y., Yang X., Wang J. miR-199-sponge transgenic mice develop physiological cardiac hypertrophy. Cardiovasc. Res. 2016;110:258–267. doi: 10.1093/cvr/cvw052. [DOI] [PubMed] [Google Scholar]
- 24.Schilling J., Kelly D.P. The PGC-1 cascade as a therapeutic target for heart failure. J. Mol. Cell. Cardiol. 2011;51:578–583. doi: 10.1016/j.yjmcc.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kärkkäinen O., Tuomainen T., Mutikainen M., Lehtonen M., Ruas J.L., Hanhineva K., Tavi P. Heart specific PGC-1α deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc. Res. 2019;115:107–118. doi: 10.1093/cvr/cvy155. [DOI] [PubMed] [Google Scholar]
- 26.Arany Z., Novikov M., Chin S., Ma Y., Rosenzweig A., Spiegelman B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehman J.J., Barger P.M., Kovacs A., Saffitz J.E., Medeiros D.M., Kelly D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russell L.K., Mansfield C.M., Lehman J.J., Kovacs A., Courtois M., Saffitz J.E., Medeiros D.M., Valencik M.L., McDonald J.A., Kelly D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 29.Pereira R.O., Wende A.R., Crum A., Hunter D., Olsen C.D., Rawlings T., Riehle C., Ward W.F., Abel E.D. Maintaining PGC-1α expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. FASEB J. 2014;28:3691–3702. doi: 10.1096/fj.14-253823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karamanlidis G., Garcia-Menendez L., Kolwicz S.C., Jr., Lee C.F., Tian R. Promoting PGC-1α-driven mitochondrial biogenesis is detrimental in pressure-overloaded mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2014;307:H1307–H1316. doi: 10.1152/ajpheart.00280.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huss J.M., Kopp R.P., Kelly D.P. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J. Biol. Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 32.Rangwala S.M., Li X., Lindsley L., Wang X., Shaughnessy S., Daniels T.G., Szustakowski J., Nirmala N.R., Wu Z., Stevenson S.C. Estrogen-related receptor alpha is essential for the expression of antioxidant protection genes and mitochondrial function. Biochem. Biophys. Res. Commun. 2007;357:231–236. doi: 10.1016/j.bbrc.2007.03.126. [DOI] [PubMed] [Google Scholar]
- 33.Huss J.M., Imahashi K., Dufour C.R., Weinheimer C.J., Courtois M., Kovacs A., Giguère V., Murphy E., Kelly D.P. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Huss J.M., Torra I.P., Staels B., Giguère V., Kelly D.P. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safdar A., Little J.P., Stokl A.J., Hettinga B.P., Akhtar M., Tarnopolsky M.A. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 2011;286:10605–10617. doi: 10.1074/jbc.M110.211466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Tran D.H., Wang Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019;8:e012673. doi: 10.1161/JAHA.119.012673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L., Jaswal J.S., Ussher J.R., Sankaralingam S., Wagg C., Zaugg M., Lopaschuk G.D. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ. Heart Fail. 2013;6:1039–1048. doi: 10.1161/CIRCHEARTFAILURE.112.000228. [DOI] [PubMed] [Google Scholar]
- 38.Heather L.C., Catchpole A.F., Stuckey D.J., Cole M.A., Carr C.A., Clarke K. Isoproterenol induces in vivo functional and metabolic abnormalities: similar to those found in the infarcted rat heart. J. Physiol. Pharmacol. 2009;60:31–39. [PubMed] [Google Scholar]
- 39.Huang Z.P., Chen J., Seok H.Y., Zhang Z., Kataoka M., Hu X., Wang D.Z. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 2013;112:1234–1243. doi: 10.1161/CIRCRESAHA.112.300682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Y., VanDusen N.J., Zhang L., Gu W., Sethi I., Guatimosim S., Ma Q., Jardin B.D., Ai Y., Zhang D. Analysis of Cardiac Myocyte Maturation Using CASAAV, a Platform for Rapid Dissection of Cardiac Myocyte Gene Function In Vivo. Circ. Res. 2017;120:1874–1888. doi: 10.1161/CIRCRESAHA.116.310283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Costa Martins P.A., Salic K., Gladka M.M., Armand A.S., Leptidis S., el Azzouzi H., Hansen A., Coenen-de Roo C.J., Bierhuizen M.F., van der Nagel R. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 2010;12:1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- 42.Oka S., Alcendor R., Zhai P., Park J.Y., Shao D., Cho J., Yamamoto T., Tian B., Sadoshima J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011;14:598–611. doi: 10.1016/j.cmet.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krishnan J., Suter M., Windak R., Krebs T., Felley A., Montessuit C., Tokarska-Schlattner M., Aasum E., Bogdanova A., Perriard E. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Burkart E.M., Sambandam N., Han X., Gross R.W., Courtois M., Gierasch C.M., Shoghi K., Welch M.J., Kelly D.P. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhai P., Sciarretta S., Galeotti J., Volpe M., Sadoshima J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011;109:502–511. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.