

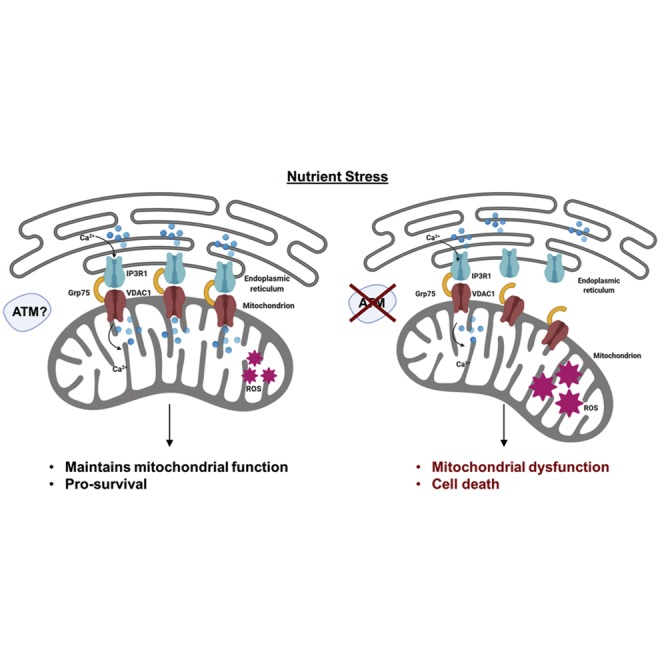
Summary
There is evidence that ATM mutated in ataxia-telangiectasia (A-T) plays a key role in protecting against mitochondrial dysfunction, the mechanism for which remains unresolved. We demonstrate here that ATM-deficient cells are exquisitely sensitive to nutrient deprivation, which can be explained by defective cross talk between the endoplasmic reticulum (ER) and the mitochondrion. Tethering between these two organelles in response to stress was reduced in cells lacking ATM, and consistent with this, Ca2+ release and transfer between ER and mitochondria was reduced dramatically when compared with control cells. The impact of this on mitochondrial function was evident from an increase in oxygen consumption rates and a defect in mitophagy in ATM-deficient cells. Our findings reveal that ER-mitochondrial connectivity through IP3R1-GRP75-VDAC1, to maintain Ca2+ homeostasis, as well as an abnormality in mitochondrial fusion defective in response to nutrient stress, can account for at least part of the mitochondrial dysfunction observed in A-T cells.
Subject areas: Cell Biology, Organizational Aspects of Cell Biology, Functional Aspects of Cell Biology
Graphical Abstract
Highlights
-
•
Hypersensitivity to glucose deprivation in ATM-deficient cells
-
•
Defective ER-mitochondrion cross talk after nutrient stress in these cells
-
•
Markedly reduced Ca2+ transfer between these two organelles in ATM-deficient cells
-
•
Mitochondrial dysfunction in response to nutrient stress in the absence of ATM
Cell Biology; Organizational Aspects of Cell Biology; Functional Aspects of Cell Biology
Introduction
Ataxia-telangiectasia (A-T) is a multisystem, autosomal recessive disorder characterized by immunodeficiency, neurodegeneration, lung disease, radiosensitivity, and cancer predisposition (Lavin, 2008; McKinnon, 2012). A-T is caused by functional mutations in a gene that codes for a phosphoinositide-3-kinase-like kinase, ATM (Savitsky et al., 1995). ATM plays a central role in the response to DNA double-strand breaks (DSBs) by phosphorylating a multitude of substrates involved in cell cycle control, DNA repair, and other cellular processes (Shiloh and Ziv, 2013). In response to DNA DSBs, ATM is activated at a low level initially followed by recruitment to the sites of DSB by the MRE11/Rad50/NBS1(MRN) complex leading to amplification of the activation (Mirzoeva and Petrini, 2003; Uziel et al., 2003). Activation occurs by autophosphorylation at several sites (Bakkenist and Kastan, 2003; Kozlov et al., 2006, 2011), and activation involves conversion of an inactive dimer to an active monomer leading to phosphorylation of all three members of the MRN complex and in turn multiple downstream substrates (Lavin et al., 2015). ATM is also activated by oxidative stress in the absence of DNA damage by a distinct mechanism that involves formation of a disulfide-cross-linked dimer (Guo et al., 2010). Activation also occurs after exposure to mitochondrial electron transport inhibitors (Valentin-Vega et al., 2012), and A-T cells are hypersensitive to H2O2 (Yeo et al., 2017, 2019). These data suggest that ATM is a sensor of reactive oxygen species (ROS) for broader protection of the cell. Although the majority of ATM is present in the nucleus, in most cell types, it is also localized to mitochondria (Valentin-Vega et al., 2012; Blignaut et al., 2019), peroxisomes (Watters et al., 1999; Zhang et al., 2015) and membrane vesicles (Lim et al., 1998). ATM binds to and phosphorylates PEX5, the peroxisome import receptor, which leads to PEX5 mono-ubiquitination and recognition by the autophagy adaptor protein p62 to induce pexophagy (Zhang et al., 2015). The FATC domain at the C-terminal of ATM can interact with a range of membrane mimetics and thereby may act as a membrane anchoring unit (Abd Rahim et al., 2019). The localization of ATM to mitochondria and its activation by mitochondrial dysfunction supports a role in mitochondrial homeostasis. Loss of ATM leads to elevated ROS, increased aberrant mitochondria, higher membrane potential, high cellular respiratory capacity, and decreased mitophagy (Valentin-Vega et al., 2012). Aberrant mitochondrial morphology and respiration has been observed in A-T lymphoblastoid cells (Ambrose et al., 2007), mitochondrial instability is observed in A-T fibroblasts (Eaton et al., 2007), and a role for ATM has been demonstrated in mitochondrial redox signaling (Zhang et al., 2018). Developmental lethality of a Drosophila ATM mutant is rescued by Ronnel (an organophosphate) by inhibiting the function of mitochondrial ATM (Rimkus and Wassarman, 2018). This protein is essential in mitochondrial radiation responses (Wei et al., 2018), and senescence control by the lysosomal-mitochondrial axis is modulated by ATM activity (Kang et al., 2017). In addition, loss of ATM induces mitochondrial dysfunction and compromised mitophagy due to NAD+ insufficiency (Fang et al., 2016) and ATM mediates spermidine-induced mitophagy via PINK1 and PARKIN regulation in human fibroblasts (Qi et al., 2016). All these reports suggest that there is an inherent defect in mitochondrial function in A-T cells, but the nature of that defect remains unknown. We have employed glycolysis inhibition to investigate this and provide evidence that a defect in signaling from the endoplasmic reticulum (ER) to the mitochondrion in A-T cells contributes to the mitochondrial abnormalities.
Results
A-T cells are hypersensitive to nutrient stress
To investigate the source of mitochondrial dysfunction in A-T cells we disrupted the ATM gene in the human bronchial epithelial cell line HBEC3-KT using CRISPR-Cas9 to create a syngeneic cell pair to minimize variability due to genetic and/or biochemical differences. Two ATM-deficient cell lines, B3 and C5, which did not express normal full-length ATM mRNA or ATM protein (Figure 1A), were generated. We selected B3 for additional studies because it was shown to be most susceptible to metabolic stress (see Figure S1A). This cell line also demonstrated increased radiosensitivity, characteristic of the A-T cellular phenotype (Figures S1B–S1D). We have previously shown that primary nasal epithelial cells from patients with A-T are hypersensitive to oxidative stress (Yeo et al., 2017, 2019). However, because exposure to H2O2 and other chemicals causing oxidative stress can cause damage to DNA, we minimized that risk by selecting another non-DNA damage form of stress, glycolysis inhibition, to investigate the response to metabolic stress (Xie et al., 2019; Yin et al., 2002). Cells were exposed to 2-deoxyglucose (2DG) to inhibit glycolysis, which would be expected to lead to a greater reliance on mitochondria for energy production and perhaps expose a greater susceptibility in A-T cells given the range of mitochondrial abnormalities reported for these cells (Fang et al., 2016). However, as 2DG treatment also causes ER stress we initially checked whether this was the case using expression of GRP78, a molecular chaperone that is expressed during ER stress (Kishi et al., 2010). The results and their quantitation are included in Figures S1E and S1F, revealing that both cell types display approximately the same degree of ER stress. These results suggest that the effect we are seeing here on ATM-deficient cells is due to glycolysis inhibition. We initially screened for ATM activation in the presence of 2DG in control HBEC cells and showed that inhibition of glycolysis activated ATM, peaking at 4–6 h and declining over 24 h (Figure 1B). In contrast, the ATM protein was not detected in ATM knockout B3 cells, and consequently no activity was found in these cells. ATM was activated in HBEC cells under these conditions, whereas no or minimal phosphorylation of H2AX, an established marker of DNA DSB, or the ATM downstream effector KAP1 was evident, pointing to the lack of DNA damage (Figure 1B). We verified this using immunofluorescent staining, demonstrating that ATM was activated only in HBEC cells as evidenced by ATMpS1981 staining (Figure 1C). In addition, there was only residual staining with γH2AX pointing to the absence of DNA DSB, again providing evidence that DNA damage is not causing ATM activation (Figure 1D). The data in Figures 1C and 1D show that ATM activation is present in the nucleus, which is similar to that reported for oxidative stress where mitochondrial H2O2 signaling can promote ATM dimerization in the nucleus (Zhang et al., 2018). ATM activation (ATM pS1981) has also been observed in the nucleus in primary mouse neurons after ATP depletion-induced oxidative stress, where there was no evidence of DNA damage (Chow et al., 2019). Previous results have shown that a fraction of ATM is localized to mitochondria and that mitochondrial damage can directly activate ATM kinase in the absence of DNA damage (Valentin-Vega et al., 2012; Blignaut et al., 2019). We thus determined whether ATM was present in mitochondria in this case by depriving cells of glucose rather than using 2DG inhibition. As was observed in Figure 1C glucose deprivation also activated ATM without activating the DNA damage response (Figure S2A), and activation was evident as early as 15 min in glucose-deprived medium (Figure S2B). ATM was present in the vesicular fraction in untreated control cells but increased significantly during glucose deprivation (Figure 1E). Activation of ATM by glycolysis inhibition does not appear to be mediated through ROS because the antioxidant N-acetylcysteine (NAC) did not interfere with activation (Figure S3). To check this further, we employed an iso-indoline nitroxide (CTMIO) also an effective antioxidant (Gueven et al., 2006) and again failed to see a reduction in ATM activation (results not shown). Although these results appear to rule out ROS, it is not clear at this stage what it is that activates ATM in response to glycolysis inhibition.
Figure 1.

Generation of ATM-deficient cells and hypersensitivity to glucose deprivation
(A) ATM-deficient HBEC3-KT cells (B3) were generated using the CRISPR-Cas9 system. A 15-bp deletion was generated in exon 57 of the ATM gene as shown in the sequence alignment and genotyping PCR. Immunoblotting confirmed the absence of the ATM protein and kinase activity in B3 cells following exposure to H2O2. C5 was another ATM-deficient cell line generated and used as a negative control.
(B) Immunoblotting using antibodies against ATM and phospho-ATM S1981 demonstrated that ATM is activated following exposure to 2DG. This was in the absence of DNA damage because neither H2AX nor KAP1 was phosphorylated. α-Tubulin was used as a loading control. No signal for ATM or phospho-ATM was observed in B3.
(C) Immunostaining of cells exposed to 2DG confirms that glucose deprivation is capable of activating ATM. Green, ATM protein; red, phospho-ATM S1981; green, YH2AX as a marker for DNA damage; blue, DAPI. Scale bar, 5 μm. ∗p < 0.01; unpaired two-tailed Student's t test.
(D) Immunostaining of cells exposed to 2DG confirmed activation of ATM in the absence of DNA damage as marked by γH2AX. Scale bar, 5 μm. Quantitation of both ATM pS1981 and γH2AX are provided below. ns, not significant.
(E) Increased association of ATM with mitochondria in response to glucose deprivation. HFF cells were cultured in DMEM with (control) 10 mM glucose and 10 mM glutamine or 0 mM glucose and 0 mM glutamine (stress) for 6 h before whole-cell lysate isolation.
(F) ATM-deficient cells are more sensitive to 2DG. Approximately 60% cell death was observed in B3 cells when compared with ∼10% observed in 2DG-treated HBEC3-KT cells.
(G) Nasal epithelial cells taken from 5 patients with A-T also showed the same sensitivity to 2DG. All data are plotted as a mean ± SD. n = 3.
As glycolysis inhibition activated ATM, we determined whether in its absence A-T cells would be more sensitive to this treatment. The results in Figure 1F show that B3 ATM-deficient cells are significantly more sensitive to glycolysis inhibition (approximately 3-fold) than HBEC controls with approximately 40% of cell death after 48 h when compared with only 10% in controls. A second CRISPR-generated cell line, C5, with no evidence of ATM expression (Figure 1A) also showed increased sensitivity to glycolysis inhibition but not to the same extent as B3 (Figure S1A). As both ATM-deficient cell lines had increased sensitivity to glycolysis inhibition but showed a difference, we investigated this further in primary airway epithelial cells from 5 patients with A-T again revealing an approximately 4-5-fold greater sensitivity at 48 and 72 h after treatment (Figure 1G). To investigate the wider applicability of this sensitivity we also analyzed different A-T cell types for this hypersensitivity to glycolysis inhibition that include induced pluripotent stem cells (iPSCs) and olfactory neural stem (ONS) cells from patients with A-T, shown previously to be more sensitive to infrared (Nayler et al., 2012; Stewart et al., 2013). Again in both cases we observed enhanced sensitivity to glycolysis inhibition (Figure S4). In addition, employment of the ATM kinase inhibitor KU55933 demonstrated that the abrogation of ATM kinase activity was sufficient to sensitize these cells to 2DG-induced cell killing (Figure S5). Furthermore, treatment with antioxidant did not enhance cell killing in control cells treated with 2DG in keeping with the ATM activation data (results not shown). These results demonstrate that ATM-deficient primary cells and cell lines are hypersensitive to inhibition of glycolysis. ATM is activated under these conditions, but this does not appear to be mediated by ROS.
Defect in ER-mitochondrial interaction in ATM-deficient cells
The ER and mitochondria are physically linked and signal through the mitochondria-associated membrane (MAM) to control the transmission of stress signals from the ER to the mitochondria assisting in the regulation of energy production, lipid transfer, Ca2+ homeostasis, and apoptosis (Honrath et al., 2017). Thus we hypothesized that the mitochondrial dysfunction in A-T cells might be explained by defective interaction between these two organelles. We initially determined the number of contact sites between ER and mitochondria by transmission electron microscopy (TEM) as a means of measuring interaction between these organelles in response to stress. The results in Figure 2A show an outline of ER-mitochondrial association in HBEC and B3 cells with contact points identified in both untreated and 2DG-treated cells. There were no obvious morphological differences in either mitochondria or ER in both cell types before or after treatment. We next analyzed the number of contact sites between the ER and mitochondria in wild-type (WT) and knockout cells, with and without 2DG treatment. The number of contact sites between mitochondria and ER were not different in untreated control and ATM-deficient cells, but after glycolysis inhibition they increased significantly in control cells but did not change appreciably in B3 cells (Figure 2B). Failure to see an increase in the number of contact sites in response to stress in ATM-deficient cells suggests that there may be a defect in signaling through the MAM to regulate ER-mitochondrial tethering and transfer of Ca2+ from ER stores into the mitochondrial matrix, which could affect mitochondrial function and in the longer term cell viability (Honrath et al., 2017). Accordingly, because the tripartite complex between VDAC1-IP3R1, mediated by GRP75, is critical for Ca2+ transfer into mitochondria, we determined whether this complex was properly integrated into the MAM in ATM-deficient cells after stress. GRP75 staining co-localized with MitoTracker in untreated control HBEC cells and ATM-deficient B3 cells to the same extent (Figure 2C). On exposure to 2DG for 2 h, the intensity of GRP75 increased markedly in control cells but remained approximately the same in ATM-deficient cells (Figure 2C). Quantitation of this revealed a significantly greater increase in control cells (Figure 2D). This might be explained by an increase in GRP75 protein in the control cells as observed recently where overexpression of GRP75 plays a key role in promoting regrowth of injured axons (Lee et al., 2019). However, it is evident that over a time course up to 24 h with glucose deprivation no increase in GPR75 protein occurred in either cell type and this was also the case for the other two members of the complex, VDAC1 and IP3R1, using immunoblotting (Figure 2E). To determine whether the increased intensity of GRP75 staining might be explained by increasing physical links between the ER and mitochondria by mediating the interaction between VDAC1 and IP3R1, we employed the proximity ligation assay (PLA). This assay allows visualization of in situ endogenous protein-protein interactions by the appearance of fluorescent spots because of amplification and subsequent incorporation of fluorescent nucleotides on complementary strands that are conjugated to target antibodies (Yuan et al., 2000). In this case we examined proximity between GRP75 on the mitochondrion with IP3R1 on the ER. The results show that there is an increase in cytoplasmic puncta in control cells after 2DG treatment, demonstrating increased association between GRP75 and IP3R1, which would be expected if the number of contact sites between the mitochondria and ER were increased (Figure 2F). On the other hand, there was only a minimal increase in cytoplasmic puncta in ATM-deficient cells. Quantitation showed a similar degree of interaction between GRP75 and IP3R1 in both types of untreated cells, but a significant increase was only observed in control treated cells after nutrient stress (Figure 2G). Although interaction between IP3R1 and GRP75 was also demonstrated by co-immunoprecipitation (results not shown), it was not possible to detect the increased association demonstrated by PLA, which is a more sensitive approach (Tubbs and Rieusset, 2017; Gomez et al., 2016). Overall these data show that the number of ER-mitochondrial contact sites under basal conditions is approximately the same in control and ATM-deficient cells, as determined by TEM and IP3R1-GRP75-VDAC1 bridge formation. However, the increase in these contact sites that occurs in response to nutrient stress in WT cells was abrogated in ATM-deficient cells.
Figure 2.

Defect in ER-mitochondrial interaction in ATM-deficient cells
(A) Electron microscopy of HBEC3-KT and B3 cells with and without exposure to 2DG. A distance of ≤25 nm between mitochondria and ER is considered as a contact site. Each contact site is highlighted in pink. An increase in the number of contact sites were observed in HBEC3-KT cells exposed to 2DG, whereas the opposite was observed for B3.
(B) Quantitation of the number of contact sites between mitochondria and ER in HBEC3-KT and B3 cells in the presence or absence of 2DG performed in a blind fashion.
(C and D) (C) Co-staining using an antibody against Grp75 (green) and MitoTracker (red) was performed on HBEC3-KT and B3 cells in the presence or absence of 2DG. An increase in the fluorescence intensity was observed in HBEC3-KT cells following exposure to 2DG but not in B3. These data are quantitated in (D). Scale bar, 5 μm.
(E) Immunoblotting for Grp75, IPR1, and VDAC1 in cells following exposure to 2DG showed no differences in protein levels between HBEC3-KT and B3. This suggested a change in Grp75 localization in 2DG-treated HBEC3-KT cells and not an increase in protein levels. α-Tubulin was used as a loading control.
(F and G) (F) Proximity ligation assay (PLA) using antibodies against IP3R1 (ER) and GRP75 (mitochondria) was performed as a means of showing increased interaction between GRP75 and IP3R1. An increase in the number of foci in HBEC3-KT cells following exposure to 2DG was observed. Quantitation of these data is shown in (G). Scale bar, 5 μm. Appropriate negative control with no primary antibody was performed. All data are plotted as a mean ± SD. n = 3, ∗p < 0.01, unpaired two-tailed Student's t test.
Defect in Ca2+ transfer between ER and mitochondria in ATM-deficient cells after 2DG
The ER houses the major Ca2+ store in mammalian cells, and this store is released through IP3R channels on the ER membrane (Phillips and Voeltz, 2016). Signaling cross-talk from the ER to the mitochondria is expected to be more efficient at tight interfaces, and control of Ca2+ concentration within tight limits is of primary relevance to cell physiology (Pallafacchina et al., 2018). Here we provided evidence of reduced interaction between ER and mitochondria in ATM-deficient cells in response to metabolic stress, and consequently, we predicted that there would be a defect in Ca2+ release from the ER and transfer to mitochondria in these cells under these conditions. The results in Figure 3A reveal that Ca2+ is being released to approximately the same extent in both cell types under basal conditions, but on exposure to 2DG, a rapid release from the ER into the cytoplasm occurred for HBEC control cells. On the other hand, whereas an increased release of Ca2+ was observed for ATM-deficient cells, it was significantly reduced compared with controls after 2DG (Figure 3A). To determine whether reduced levels of Ca2+ storage in the ER in B3 might account for this defect, we employed thapsigargin to release total ER Ca2+. The results in Figure S6A showed similar levels of Ca2+ release in both HBEC3-KT and B3. We also employed primary A-T epithelial cells to determine Ca2+ release. As observed with the cell line these cells too showed a defect in rate and extent of Ca2+ release after 2DG treatment (Figure S6B). As both the release of Ca2+ from the ER and its subsequent uptake into mitochondria are coupled to achieve Ca2+ homeostasis, we investigated whether uptake of Ca2+ into mitochondria might also be defective in ATM-deficient cells. The results in Figure 3B show that ATM-deficient cells are also defective in uptake of Ca2+ into mitochondria when compared with control after 2DG treatment, and the difference is of the same order as observed for Ca2+ release from the ER. To understand the significance of this difference in Ca2+ kinetics, we employed 2-aminoethoxydiphenyl-borate (2APB) to inhibit IP3R1 and determine the effect on establishment of the IP3R1-GRP75-VDAC channel and on cell survival (Sun et al., 2011). We initially checked the effect of blocking IP3R1 with 2APB on 2DG-induced formation of the IP3R1-GRP75-VDAC1 channel. The results in Figure 3C show that the increased intensity of the GRP75 signal after 2DG in HBEC control cells after treatment with the IP3R inhibitor was reduced to levels comparable to that for ATM-deficient cells indicating that the signaling complex between the ER and mitochondria was being disrupted. Exposure of control cells to 2APB in the presence of 2DG also led to a marked enhancement in cell killing under conditions that prevented Ca2+ release from the ER (Figure 3D). ATM-deficient cells were also significantly more sensitive to killing by 2DG when additionally treated with 2APB. We also employed the calcimimetic, NPS-R568, which activates the calcium-sensing receptor to increase intracellular Ca2+ (Di Mise et al., 2018), to determine whether this might be protective for ATM-deficient cells. NPS-R568 was more toxic to ATM-deficient cells and enhanced the toxicity of 2DG in both cell types, further supporting a role for specific ER release and mitochondrial uptake of Ca2+ in protecting the cell (Figure 3E). These results demonstrate that the defective linkage between the ER and mitochondria in ATM-deficient cells (both primary cells and a cell line) in response to glycolysis inhibition, is accompanied both by significantly reduced release of Ca2+ from the ER and reduced uptake into mitochondria. It is evident that this affects mitochondrial function and cell survival because inhibition of the IP3R and consequently Ca2+ release and transfer in control cells led to significantly increased killing similar to that seen with ATM-deficient cells treated with 2DG alone.
Figure 3.

Defect in Ca2+ transfer between ER and mitochondria in ATM-deficient cells in response to glucose deprivation
(A) Intracellular calcium was measured using FURA-2 as a calcium indicator. A significant increase in calcium was observed in HBEC3-KT cells treated with 2DG when compared with B3.
(B) Mitochondrial calcium was measured using Rhodamine-2AM. Similar to results observed with FURA-2, a significant increase in calcium was seen in HBEC3-KT cells treated with 2DG when compared with B3.
(C) To determine the effect of an inhibitor of IP3R1, 2APB, on formation of the IP3R1-GRP75-VDAC1 bridge, Grp75 labeling was attenuated in HBEC3-KT cells exposed to 2DG. Scale bar, 5 μm.
(D) Effect of 2DG + 2APB and 2APB alone on cell killing in both HBEC3-KT and B3 cells.
(E) Effect of the calcium mimetic, R568, on cell survival in the presence of 2DG. All data are plotted as mean ± SD. n = 3.
Mitochondrial function is defective in glucose-deprived ATM-deficient cells in response to nutrient stress
Previous data have shown that absence of ATM is associated with elevated levels of ROS (Barzilai and Yamamoto, 2004; Mercer et al., 2010). We also observed elevated basal levels of intracellular ROS in B3 compared with control, and although ROS increased in both B3 and control cells in response to 2DG treatment, the overall amount was significantly higher in B3 cells (Figure 4A). Elevated basal levels of ROS were also evident in A-T ONS stem cells (Figure S7). Elevated ROS has been linked to increased mitochondrial membrane potential and other mitochondrial abnormalities including increased mitochondrial mass in Atm−/- thymocytes and in ATM-deficient primary neurons (Valentin-Vega et al., 2012; Fang et al., 2016). Not unexpectedly we also observed increased mitochondrial membrane potential in glucose-deprived B3 cells compared with control with time (Figure 4B). Use of FCCP treatment to destroy membrane potential demonstrated a similar extent of loss for both cell types at 60 min as expected. We also observed increased mitochondrial mass in ATM-deficient cells compared with control using mtDNA/nDNA ratios (Figure 4C). This is consistent with a possible defect in mitochondrial fusion/fission in A-T cells. A recent report shows that BRCA1 deficiency results in a higher level of MFN1/2 protein expression, accelerating mitochondrial fusion, which can block the separation of damaged mitochondria from healthy ones and negatively affect mitophagy (Chen et al., 2020). In addition, clearing damaged mitochondria is defective in Atm-deficient cells and increasing intracellular NAD+ is associated with improving mitochondrial quality via mitophagy (Valentin-Vega et al., 2012; Fang et al., 2016). We showed here that this was also the case after inhibition of glycolysis for ATM-deficient cells (Figure 4D). The extent of mitophagy was approximately 50% of that seen in control cells after 2DG treatment. We also determined mitophagy by measuring the expression levels of BNIP3, a gene associated with mitophagy (Sunderland et al., 2020). The results confirm the deficiency in ATM-deficient cells showing a significantly reduced level of expression after 2DG treatment in B3 (Figure 4E). To understand the nature of the defect we determined whether the increased mitochondrial mass and reduced mitophagy in Atm-deficient cells might be explained by defective mitochondrial fusion. The results in Figure S8 show that in response to nutrient stress the level of MFN2 protein falls rapidly by 1 h in HBEC control cells but negligibly in B3 ATM-deficient cells as determined by immunofluorescence. Immunoblotting confirmed the rapid degradation of MFN2 in control cells, but on the contrary, MFN2 levels remained unchanged in B3 ATM-deficient cells (Figure 4F). Quantitation appears in Figure 4G. These results are consistent with a shift in the balance to mitochondrial fusion in ATM-deficient cells. There was no evidence for any difference in the mitochondrial fission protein DRP1 in the two cell types (Figure 4F). Degradation of MFN2 occurs in a proteasome and p97 AAA + ATPase-dependent manner, and during the process p97 accumulates on mitochondria (Tanaka et al., 2010). We found that basal levels of p97 associated with mitochondria, determined by co-localization with TOMM20, were approximately the same in the two cell types (Figure 4H). However, after treatment with 2DG the number of cells with perinuclear p97 rings increased markedly in control cells but remained unchanged in ATM-deficient cells. Quantitation of these data appears in Figure 4I. Under these conditions there was no change in the amount of p97 protein (Figure 4J), suggesting that changes in p97 expression levels did not account for the differences seen in Figure 4H. In summary we observed a number of mitochondrial abnormalities in ATM-deficient cells including increased mitochondrial mass and reduced mitophagy, which is consistent with reduced MFN2 degradation in these cells and reduced recruitment of p97.
Figure 4.

Mitochondrial function is defective in glucose-deprived ATM-deficient cells in response to nutrient stress
(A) Mitochondrial ROS was measured using MitoSox, a mitochondrial superoxide indicator. Basal levels of mitochondrial ROS were higher in B3 when compared with HBEC3-KT, and an increase was observed in both cell types exposed to 2DG. Increase in mitochondrial ROS was also observed following exposure to H2O2, which was used as a positive control.
(B) Mitochondrial membrane potential was measured using JC-1. A significant increase in membrane potential was observed in B3 cells exposed to 2DG. Treatment with FCCP was used as a control to decrease membrane potential.
(C) Mitochondrial mass as determined by the ratio of mtDNA to nDNA.
(D) Analysis of mitophagy was performed using MtPhagy Dye (red) co-stained with MitoTracker (green). An increase in mitophagy was observed in HBECs exposed to 2DG but not in B3. Data are plotted as the mean ± SD. A minimum of 50 cells were analyzed. Scale bar, 5 μm.
(E) BNIP expression in HBEC3-KT and B3 cells ± 2DG as a measure of mitophagy.
(F) Immunoblotting for the detection of MFN2 and DRP1. α-Tubulin was used as a loading control.
(G) Quantitation of Mfn2 band intensity.
(H) Recruitment of p97 to mitochondria as determined by co-immunostaining with TOMM20. Scale bar, 5 μm.
(I) Quantitation of perinuclear p97 rings in HBEC and B3 with and without 2DG treatment.
(J) Immunoblotting of p97. VDAC1 was used as a loading control.
(K and L) (K) Mitochondrial function analysis was performed using the Seahorse XF24 extracellular flux analyzer. Results showed a higher respiratory capacity in basal (upper) and 2DG-treated (lower) B3 cells when compared with HBEC cells in (L). All data are plotted as mean ± SD. n = 3, ∗p < 0.01; unpaired two-tailed Student's t test.
Valentin-Vega et al. (2012) have demonstrated that Atm−/− thymocytes and fibroblasts both exhibit increased cellular respiration under basal conditions. The oxygen consumption rate (OCR) was elevated in Atm−/− cells, and Beclin-1 heterozygosity significantly rescued this abnormality. We determined whether this was the case in ATM-deficient B3 cells. The results in Figure 4K show that there is elevated basal respiration and increased respiratory capacity in ATM-deficient cells compared with control, providing evidence for a higher rate of metabolism to support basic cellular function in these cells. Quantitation of these data appears in Figure 4L. Exposure of ATM-deficient cells to 2DG led to marked increases in OCRs per viable cell compared with controls including a further increase in basal respiration and a very significant increase in maximal respiratory capacity in these cells, caused by the sudden surge in energy requirement in an attempt to overcome glycolysis inhibition (Figure 4K). Quantitation of these data appears in Figure 4L. We interpret the higher value for A-T to be overcompensation in response to stress in keeping with the higher basal respiration to maintain basic cellular function. Previous data have also shown that the process of clearing damaged mitochondria is defective in Atm-deficient cells and that increasing intracellular NAD+ is associated with improving mitochondrial quality via mitophagy (Valentin-Vega et al., 2012; Fang et al., 2016). These results demonstrate that glycolysis inhibition exacerbates mitochondrial dysfunction in ATM-deficient cells. The capacity of this form of stress to cause accumulation of ATM on mitochondria supports further an extranuclear role for this protein.
Discussion
Hypersensitivity to oxidative stress supports an extranuclear role for ATM, further substantiated by the description of a separate mechanism of activation for ATM by oxidative stress, in the absence of DNA damage (Guo et al., 2010; Yeo et al., 2017, 2019). We have shown here that ATM-deficient cells are also hypersensitive to 2DG-induced glycolysis inhibition compared with controls without evidence for detectable DNA damage. This hypersensitivity is characteristic of several different ATM-deficient cells. We also observed increased sensitivity to this treatment in primary airway epithelial cells from five patients with A-T and in A-T iPSC and ONS stem cells. ATM was activated under these conditions in the absence of DNA damage and was localized to the nucleus, similar to that reported for menadione treatment of HeLa cells and depletion of ATP in mouse neurons, both of which induce ROS (Zhang et al., 2018; Chow et al., 2019). The involvement of ATM in the response to oxidative stress and its localization to the mitochondrion suggests a role in the function of that organelle (Valentin-Vega et al., 2012; Blignaut et al., 2019). In addition, there is accumulating evidence for mitochondrial abnormalities in A-T cells, and it has been suggested that A-T should be considered, at least in part, as a mitochondrial disease (Valentin-Vega et al., 2012). We also observed mitochondrial abnormalities after 2DG treatment including increased levels of ROS. However, it seems unlikely that the same mechanism of ATM activation occurs because the antioxidant NAC failed to interfere with ATM activation under these conditions but ATM still localized to mitochondria. In addition to NAC, a second antioxidant CTMIO did not affect ATM activation or cell survival induced by glycolysis inhibition (data not shown). These results suggest that although ROS is generated during 2DG treatment, it does not appear to represent the agent activating ATM to protect control cells against cell death. On the other hand, neither loss of ATM kinase activity nor inhibition of ATM kinase in MCL, A-T, and HeLa cell lines mitigated CCCP-induced mitophagy, but stable knockdown of ATM conferred resistance to mitophagy suggesting that ATM protein and not ATM kinase activity is required for mitophagy (Chow et al., 2019; Zhang et al., 2018). At this stage it is unclear as to how metabolic stress achieves this.
We have provided evidence here for a new explanation for the mitochondrial abnormalities in these cells that centers on the interaction between the ER and mitochondria in response to metabolic stress. We showed that GRP75 fails to efficiently assemble the IP3R1-GRP75-VDAC1 complex in response to glycolysis inhibition in ATM-deficient cells. This protein complex localizes to the MAM and has been shown to play a major role in bridging between the ER and mitochondria, acting as an important control point of cell fate in conditions of oxidative and other forms of stress (Szabadkai et al., 2006; Honrath et al., 2017). While stress-induced formation of the complex is deficient in A-T cells, it is evident that constitutive activity of this channel appears to be relatively normal and thus able to support to some extent basal mitochondrial function. This is consistent with Cardenas et al. (2010) who demonstrated that mitochondrial uptake of InsP3R-released Ca2+ is an essential cellular process that is required for efficient mitochondrial respiration and maintenance of normal cell bioenergetics. Our results show that IP3R1-GRP75-VDAC1- facilitated release of Ca2+ from the ER and uptake into mitochondria in A-T cells is defective in response to nutrient stress, suggesting that this is responsible for defective mitochondrial function in these cells. Clearly this is a tightly controlled process because enhanced Ca2+ release from the ER can lead to Ca2+ overload and cell death (Rizzuto et al., 2009; Jones et al., 2010). GRP75 has been shown to mediate both beneficial and harmful effects, which is dependent on both the cell type and pathological context (Honrath et al., 2017). Interaction between GRP75 and IP3R1 has been demonstrated to increase upon axonal injury, contributing to increased ER-mitochondrial tethering, elevated mitochondrial Ca2+, and enhanced ATP generation to promote axonal regrowth (Lee et al., 2019). GRP75 overexpression is also associated with protection against cell death after exposure to cellular stress such as radiation and in astrocytes exposed to oxygen-glucose deprivation (Guo et al., 2012; Voloboueva et al., 2008). Overexpression studies also give rise to both mitochondrial stress response and protection against stress-induced ROS formation and loss of mitochondrial membrane potential in different cell types (Jin et al., 2006; Burbulla et al., 2014). GRP75 overexpression in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia (Xu et al., 2009) and has been shown to suppress apoptosis induced by glucose deprivation in PC12 cells (Yang et al., 2008). On the other hand, glutamate-induced cell death was prevented in GRP75 knockdown cells, but the protective effect was specific for glutamate toxicity as it failed to protect against ER stress (Honrath et al., 2017). Here, in the case of glucose deprivation stress our data suggest that the reduced IP3R1-GRP75-VDAC1 channel activity, resulting in reduced import of Ca2+ into mitochondria, affects negatively both mitophagy and survival in ATM-deficient cells. Reduced survival and mitophagy is also consistent with a reduced number of contact sites between these organelles in response to nutrient stress in A-T cells, reflected in a significantly reduced transfer of Ca2+. We suggest that the flow of Ca2+ after glucose deprivation is an early event designed to protect the cell. Failure to establish sufficient contacts in the MAM between ER and mitochondria resulting in reduced Ca2+ transfer would negatively affect mitophagy and cell survival in the longer term. This is consistent with an essential role for GRP75 in maintaining contact between these organelles and facilitating the propagation of the Ca2+ signal into mitochondria to regulate various mitochondrial functions (Phillips and Voeltz, 2016; Honrath et al., 2018). In addition, previous results have shown that contact sites between ER and mitochondria represent sites of autophagosome formation during starvation-induced autophagy and that autophagosome biogenesis is impaired in cells with defective ER-mitochondrial tethering (Hamasaki et al., 2013; Kishi-Itakura et al., 2014). Thus the defect in apposition between ER and mitochondria in ATM-deficient cells and in turn the defect in Ca2+ transfer in response to stress is likely to contribute to the mitophagy defect in these cells. However, it is more complex than this because mitochondria-ER contacts are critical to mitochondrial fission/fusion and mitophagy, which is consistent with the increased mitochondrial mass that we observed here. We also showed here that MFN2 degradation and p97 recruitment to mitochondria are defective in ATM-deficient cells, which would seem to favor the model that MFN2 binds to both ER and mitochondria acting as a negative regulator to keep them apart to control the non-specific transfer of Ca2+ and avoid apoptosis (Leal et al., 2016). In that study knockdown of MFN2 increased contact between ER and mitochondria and increased Ca2+ transfer. What we show here is that MFN2 is preferentially degraded in control compared with A-T cells (Figures 4F and 4G) and also that the number of ER contact points increases in control in response to 2DG but shows little change in A-T. As expected by Leal et al. this would also lead to a significant difference in Ca2+ transfer between ER and mitochondria in control over A-T, which we see (Figures 3A and 3B). We can account for this by increased contact and increased channel formation between IP3R1 and VDAC1. In short, our data are completely consistent with a model wherein MFN2 functions to minimize contact between ER and mitochondria under normal physiological conditions but on exposure to stress it is degraded to regulate these contacts to ensure controlled Ca2+ transfer. In control cells number of IP3R1-GRP75-VDAC1 contacts would increase and increase the flow of Ca2+, but in ATM-deficient cells they would show little change leading to reduced Ca2+ transfer. We also showed here that p97 recruitment to mitochondria is defective in ATM-deficient cells, which would favor mitochondrial fusion. It is of interest that mutations in MFN2 also result in neuronal dysfunction (Jiang et al., 2018), MFN2 has also been associated with diabetes (Schrepfer and Scorrano, 2016), and this observation could account for the metabolic phenotypes seen in patients with A-T. So in short, our data suggest that the defect in mitophagy in ATM-deficient cells can be explained both by defective communication between ER and mitochondria through IP3R1-GRP75-VDAC1 to supply Ca2+ and by a failure to remove MFN2 cross-links between mitochondria to enable mitophagy to occur. These results agree with previous reports that mitophagy is decreased in A-T cells leading to accumulation of damaged mitochondria (Valentin-Vega et al., 2012; Fang et al., 2016; Chow et al., 2019). We have provided greater insight into the nature of that defect here.
We have shown that ER-mitochondrial signaling in response to nutrient stress is defective in ATM-deficient and primary epithelial cells from cells of patients with A-T and that this form of stress activates ATM likely by a mechanism that appears to be different to that described for oxidative stress (Zhang et al., 2018). Loss of ATM under these conditions is linked to mitochondrial dysfunction and dramatically reduced cell survival. A major challenge is to understand the role of ATM in protecting mitochondrial function by identifying the substrate(s) phosphorylated by ATM in response to this form of stress. A relevant candidate in the present context is the BH-3-only protein c-bid, which has been shown to be phosphorylated by ATM (Kamer et al., 2005; Maryanovich et al., 2012). However, we failed to see phosphorylation of this protein after glycolysis inhibition. In addition, we failed to see phosphorylation of MFN2 using a pS/TQ antibody after 2DG treatment (results not shown). Our results provide evidence that the kinase activity of ATM is important in protecting against nutrient stress-induced cell death because cells are sensitized after inhibition of that activity, but on the other hand, a recent report provides evidence that ATM, but not its kinase activity, is associated with induction of mitophagy through ATM-Parkin interaction in cancer cells (Sarkar et al., 2020). Regardless of the exact role of ATM in protecting mitochondrial function and controlling mitophagy, protection against ROS accumulation and oxidative stress is a significant feature of the A-T phenotype. Overexpression of catalase targeted to mitochondria alleviates A-T-related pathology in Atm−/− mice, including reduced propensity to develop thymic lymphoma, improved bone marrow hematopoiesis and macrophage differentiation in vitro, and partial rescue of memory T cell developmental defects (D'souza et al., 2013). In addition, allelic loss of the autophagic regulatory gene, Beclin-1, rescued mitochondrial dysfunction in Atm−/− lymphocytes and delayed tumorigenesis in Atm−/− mice (Valentin-Vega et al., 2012). In this context a recent report describes the activation of the ATM/CHK2/Beclin 1 pathway in response to glucose deprivation and hypoxia to positively regulate autophagy, maintaining cell and tissue homeostasis by suppressing ROS, clearing damaged mitochondria, and preventing apoptosis (Guo et al., 2020).
In summary, we have provided evidence for defective interaction between the ER and mitochondria due to inefficient assembly of the IP3R1-GRP75-VDAC1 complex in response to glycolysis inhibition in ATM-deficient cells leading to a defect in Ca2+ homeostasis and mitochondrial dysfunction. The significance of this to the A-T neurodegenerative phenotype is evident from results showing that the loss of regulation of IP3R1, enriched in Purkinje cells, compromises their function and survival and is a key protein in the pathogenesis of several spinocerebellar ataxias (Shimobayashi and Kapfhammer, 2018). In addition, Atm deficiency in conjunction with chronic DNA damage from Polb inactivation leads to reduced IP3R1 expression in Purkinje cells and severe cerebellar ataxia (Kim et al., 2020). Extending our studies to cerebellar organoids from patients with A-T will provide important insight into ER-mitochondrial signaling in neuronal cells and its role in maintaining Ca2+ homeostasis and mitochondrial function. At present, there is no cure for A-T, although a number of therapeutic approaches are being or have been employed. Targeting ROS production directly, enhancing antioxidant capacity, or increasing redox signaling have been suggested as possible approaches (Zhang et al., 2018), but it is unclear whether these will be effective. A more effective approach may be to focus directly on enhancing mitochondrial function.
Limitations of the study
In this study, we reveal that ATM-deficient cells are hypersensitive to nutrient stress and that ER-mitochondrial connectivity through IP3R1-GRP75-VDAC1 maintains Ca2+ homeostasis, to protect mitochondrial function in the presence of ATM. The exact role of ATM and the substrates involved in this signaling require further studies. In addition, the identification of compounds that protect against this signaling defect is a priority because that may have potential for therapy in this disorder.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Martin Lavin (m.lavin@uq.edu.au).
Materials availability
All cell lines and reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
All data are included in the published article and the Supplemental Information, and any additional information will be available from the lead contact upon request.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We wish to acknowledge Prof. Peter D. Sly and Dr. Emmanuelle Fantino for providing us the cell line HBEC3-KT for the work performed in this paper. We would also like to acknowledge the role of Dr. Peter Scarbrough (Duke University) in the conception of the idea to investigate nutrient stress in A-T cells and Prof. Michael Murphy (Cambridge) for his advice on mitochondrial function. This work was supported by funding from the Australian A-T Foundation, BrAshA-T. This work was also supported by grants and a fellowships from the Children's Hospital Foundation, Australia (RM2018002270 to A.J.Y), National Health and Medical Research Council of Australia (grants APP1140064 and APP1150083 and fellowship APP1156489 to R.G.P.), and the Australian Research Council (Center of Excellence in Convergent Bio-Nano Science and Technology CE140100036 to R.G.P). The authors are grateful to Charles Fergsuon for electron microscopy. We acknowledge NIH grant R01CA157216 awarded to Prof. Michael B. Kastan's laboratory that supported Dr. Adam D. Brown's initial work that led to the observations about glucose deprivation and ATM induction. The authors also acknowledge the use of the Microscopy Australia Research Facility at the Center for Microscopy and Microanalysis at The University of Queensland. The authors would like to acknowledge (thank) the expertise of the QIMR Berghofer Flow Cytometry Facility with help and guidance in flow cytometry data acquisition and analysis.
Author contribution
A.J.Y., M.F.L., D.C., and M.B.K. contributed to the development of the concept and overall design. R.P. and E.W. designed specific experiments and analyzed the data. A.J.Y., C.L.K., M.G., D.Z., R.S., S.W., R.G.P., and A.J.B. carried out the experimental work. A.J.Y. and M.F.L. wrote the initial draft, and all authors contributed to writing and also approved the manuscript.
Declaration of interests
None of the authors have a conflict of interest. This study was not funded by any commercial entity and has not attracted any intellectual property protection or patents.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101972.
Supplemental information
References
- Abd Rahim M.S., Cherniavskyi Y.K., Tieleman D.P., Dames S.A. NMR- and MD simulation-based structural characterization of the membrane-associating FATC domain of ataxia telangiectasia mutated. J. Biol. Chem. 2019;294:7098–7112. doi: 10.1074/jbc.RA119.007653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose M., Goldstine J.V., Gatti R.A. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum. Mol. Genet. 2007;16:2154–2164. doi: 10.1093/hmg/ddm166. [DOI] [PubMed] [Google Scholar]
- Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Barzilai A., Yamamoto K. DNA damage responses to oxidative stress. DNA Repair (Amst) 2004;3:1109–1115. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Blignaut M., Loos B., Botchway S.W., Parker A.W., Huisamen B. Ataxia-Telangiectasia Mutated is located in cardiac mitochondria and impacts oxidative phosphorylation. Sci. Rep. 2019;9:4782. doi: 10.1038/s41598-019-41108-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla L.F., Fitzgerald J.C., Stegen K., Westermeier J., Thost A.K., Kato H., Mokranjac D., Sauerwald J., Martins L.M., Woitalla D. Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis. 2014;5:e1180. doi: 10.1038/cddis.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas C., Miller R.A., Smith I., Bui T., Molgo J., Muller M., Vais H., Cheung K.H., Yang J., Parker I. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Lei J.H., Bao J., Wang H., Hao W., Li L., Peng C., Masuda T., Miao K., Xu J. BRCA1 deficiency impairs mitophagy and promotes inflammasome activation and mammary tumor metastasis. Adv. Sci. (Weinh) 2020;7:1903616. doi: 10.1002/advs.201903616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow H.M., Cheng A., Song X., Swerdel M.R., Hart R.P., Herrup K. ATM is activated by ATP depletion and modulates mitochondrial function through NRF1. J. Cell Biol. 2019;218:909–928. doi: 10.1083/jcb.201806197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'souza A.D., Parish I.A., Krause D.S., Kaech S.M., Shadel G.S. Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol. Ther. 2013;21:42–48. doi: 10.1038/mt.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mise A., Ranieri M., Centrone M., Venneri M., Tamma G., Valenti D., Valenti G. Activation of the calcium-sensing receptor corrects the impaired mitochondrial energy status observed in renal polycystin-1 knockdown cells modeling autosomal dominant polycystic kidney disease. Front. Mol. Biosci. 2018;5:77. doi: 10.3389/fmolb.2018.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton J.S., Lin Z.P., Sartorelli A.C., Bonawitz N.D., Shadel G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest. 2007;117:2723–2734. doi: 10.1172/JCI31604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang E.F., Kassahun H., Croteau D.L., Scheibye-Knudsen M., Marosi K., Lu H., Shamanna R.A., Kalyanasundaram S., Bollineni R.C., Wilson M.A. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016;24:566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez L., Thiebaut P.A., Paillard M., Ducreux S., Abrial M., Da Silva C.C., Durand A., Alam M.R., Van Coppenolle F., Sheu S.S., Ovize M. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3 beta during reperfusion injury. Cell Death Differ. 2016;23:313–322. doi: 10.1038/cdd.2015.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueven N., Luff J., Peng C., Hosokawa K., Bottle S.E., Lavin M.F. Dramatic extension of tumor latency and correction of neurobehavioral phenotype in Atm-mutant mice with a nitroxide antioxidant. Free Radic. Biol. Med. 2006;41:992–1000. doi: 10.1016/j.freeradbiomed.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Guo Q.Q., Wang S.S., Zhang S.S., Xu H.D., Li X.M., Guan Y., Yi F., Zhou T.T., Jiang B., Bai N. ATM-CHK2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. Embo J. 2020:e103111. doi: 10.15252/embj.2019103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Yang L., Li H., Xie Z., Liu W., Zuo J. Glucose-regulated protein 75 overexpression attenuates ionizing radiation-mediated injury in PC12 cells by inducing the expression of topoisomerase IIalpha. Mol. Med. Rep. 2012;6:1423–1427. doi: 10.3892/mmr.2012.1070. [DOI] [PubMed] [Google Scholar]
- Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Honrath B., Culmsee C., Dolga A.M. One protein, different cell fate: the differential outcome of depleting GRP75 during oxidative stress in neurons. Cell Death Dis. 2018;9:32. doi: 10.1038/s41419-017-0148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honrath B., Metz I., Bendridi N., Rieusset J., Culmsee C., Dolga A.M. Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017;3:17076. doi: 10.1038/cddiscovery.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S., Nandy P., Wang W., Ma X., Hsia J., Wang C., Wang Z., Niu M., Siedlak S.L., Torres S. Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol. Neurodegener. 2018;13:5. doi: 10.1186/s13024-018-0238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J., Hulette C., Wang Y., Zhang T., Pan C., Wadhwa R., Zhang J. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol. Cell. Proteomics. 2006;5:1193–1204. doi: 10.1074/mcp.M500382-MCP200. [DOI] [PubMed] [Google Scholar]
- Jones D.P., Lemasters J.J., Han D., Boelsterli U.A., Kaplowitz N. Mechanisms of pathogenesis in drug hepatotoxicity putting the stress on mitochondria. Mol. Interv. 2010;10:98–111. doi: 10.1124/mi.10.2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer I., Sarig R., Zaltsman Y., Niv H., Oberkovitz G., Regev L., Haimovich G., Lerenthal Y., Marcellus R.C., Gross A. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell. 2005;122:593–603. doi: 10.1016/j.cell.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Kang H.T., Park J.T., Choi K., Kim Y., Choi H.J.C., Jung C.W., Lee Y.S., Park S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017;13:616–623. doi: 10.1038/nchembio.2342. [DOI] [PubMed] [Google Scholar]
- Kim J., Kim K., Mo J.S., Lee Y. Atm deficiency in the DNA polymerase beta null cerebellum results in cerebellar ataxia and Itpr1 reduction associated with alteration of cytosine methylation. Nucleic Acids Res. 2020;48:3678–3691. doi: 10.1093/nar/gkaa140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi-Itakura C., Koyama-Honda I., Itakura E., Mizushima N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014;127:4089–4102. doi: 10.1242/jcs.156034. [DOI] [PubMed] [Google Scholar]
- Kishi S., Shimoke K., Nakatani Y., Shimada T., Okumura N., Nagai K., Shin-Ya K., Ikeuchi T. Nerve growth factor attenuates 2-deoxy-D-glucose-triggered endoplasmic reticulum stress-mediated apoptosis via enhanced expression of GRP78. Neurosci. Res. 2010;66:14–21. doi: 10.1016/j.neures.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Kozlov S.V., Graham M.E., Jakob B., Tobias F., Kijas A.W., Tanuji M., Chen P., Robinson P.J., Taucher-Scholz G., Suzuki K. Autophosphorylation and ATM activation: additional sites add to the complexity. J. Biol. Chem. 2011;286:9107–9119. doi: 10.1074/jbc.M110.204065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov S.V., Graham M.E., Peng C., Chen P., Robinson P.J., Lavin M.F. Involvement of novel autophosphorylation sites in ATM activation. Embo J. 2006;25:3504–3514. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin M.F. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- Lavin M.F., Kozlov S., Gatei M., Kijas A.W. ATM-dependent phosphorylation of all three members of the MRN complex: from sensor to adaptor. Biomolecules. 2015;5:2877–2902. doi: 10.3390/biom5042877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal N.S., Schreiner B., Pinho C.M., Filadi R., Wiehager B., Karlstrom H., Pizzo P., Ankarcrona M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J. Cell Mol. Med. 2016;20:1686–1695. doi: 10.1111/jcmm.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Wang W., Hwang J., Namgung U., Min K.T. Increased ER-mitochondria tethering promotes axon regeneration. Proc. Natl. Acad. Sci. U S A. 2019;116:16074–16079. doi: 10.1073/pnas.1818830116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D.S., Kirsch D.G., Canman C.E., Ahn J.H., Ziv Y., Newman L.S., Darnell R.B., Shiloh Y., Kastan M.B. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc. Natl. Acad. Sci. U S A. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanovich M., Oberkovitz G., Niv H., Vorobiyov L., Zaltsman Y., Brenner O., Lapidot T., Jung S., Gross A. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 2012;14:535–U185. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- Mckinnon P.J. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- Mercer J.R., Cheng K.K., Figg N., Gorenne I., Mahmoudi M., Griffin J., Vidal-Puig A., Logan A., Murphy M.P., Bennett M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010;107:1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva O.K., Petrini J.H. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 2003;1:207–218. [PubMed] [Google Scholar]
- Nayler S., Gatei M., Kozlov S., Gatti R., Mar J.C., Wells C.A., Lavin M., Wolvetang E. Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl. Med. 2012;1:523–535. doi: 10.5966/sctm.2012-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallafacchina G., Zanin S., Rizzuto R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. 2018;F1000Res:7. doi: 10.12688/f1000research.15723.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M.J., Voeltz G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016;17:69–82. doi: 10.1038/nrm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Qiu Q., Gu X., Tian Y., Zhang Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 2016;6:24700. doi: 10.1038/srep24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimkus S.A., Wassarman D.A. A pharmacological screen for compounds that rescue the developmental lethality of a Drosophila ATM mutant. PLoS One. 2018;13:e0190821. doi: 10.1371/journal.pone.0190821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R., Marchi S., Bonora M., Aguiari P., Bononi A., De Stefani D., Giorgi C., Leo S., Rimessi A., Siviero R. Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta. 2009;1787:1342–1351. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A., Stellrecht C.M., Vangapandu H.V., Ayres M., Kaipparettu B.A., Park J.H., Balakrishnan K., Burks J.K., Pandita T.K., Hittelman W.N. Ataxia telangiectasia mutated interacts with Parkin and induces mitophagy independent of kinase activity. Evidence from mantle cell lymphoma. Haematologica. 2020 doi: 10.3324/haematol.2019.234385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Schrepfer E., Scorrano L. Mitofusins, from mitochondria to metabolism. Mol. Cell. 2016;61:683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Shiloh Y., Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- Shimobayashi E., Kapfhammer J.P. Calcium signaling, PKC gamma, IP3R1 and CAR8 link spinocerebellar ataxias and Purkinje cell dendritic development. Curr. Neuropharmacol. 2018;16:151–159. doi: 10.2174/1570159X15666170529104000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R., Kozlov S., Matigian N., Wali G., Gatei M., Sutharsan R., Bellette B., Wraith-Kijas A., Cochrane J., Coulthard M. A patient-derived olfactory stem cell disease model for ataxia-telangiectasia. Hum. Mol. Genet. 2013;22:2495–2509. doi: 10.1093/hmg/ddt101. [DOI] [PubMed] [Google Scholar]
- Sun X.M., Lu J.H., Qiu Y.H., Liu Z., Wang X.Q., Peng Y.P. Interleukin-6 reduces NMDA-induced Ca2+ overload via prevention of Ca2+ release from intracellular store. Int. J. Neurosci. 2011;121:423–429. doi: 10.3109/00207454.2011.556280. [DOI] [PubMed] [Google Scholar]
- Sunderland P., Augustyniak J., Lenart J., Buzanska L., Carlessi L., Delia D., Sikora E. ATM-deficient neural precursors develop senescence phenotype with disturbances in autophagy. Mech. Ageing Dev. 2020;190:111296. doi: 10.1016/j.mad.2020.111296. [DOI] [PubMed] [Google Scholar]
- Szabadkai G., Bianchi K., Varnai P., De Stefani D., Wieckowski M.R., Cavagna D., Nagy A.I., Balla T., Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A., Cleland M.M., Xu S., Narendra D.P., Suen D.F., Karbowski M., Youle R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs E., Rieusset J. Metabolic signaling functions of ER-mitochondria contact sites: role in metabolic diseases. J. Mol. Endocrinol. 2017;58:R87–R106. doi: 10.1530/JME-16-0189. [DOI] [PubMed] [Google Scholar]
- Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega Y.A., Maclean K.H., Tait-Mulder J., Milasta S., Steeves M., Dorsey F.C., Cleveland J.L., Green D.R., Kastan M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119:1490–1500. doi: 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva L.A., Duan M., Ouyang Y., Emery J.F., Stoy C., Giffard R.G. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab. 2008;28:1009–1016. doi: 10.1038/sj.jcbfm.9600600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters D., Kedar P., Spring K., Bjorkman J., Chen P., Gatei M., Birrell G., Garrone B., Srinivasa P., Crane D.I., Lavin M.F. Localization of a portion of extranuclear ATM to peroxisomes. J. Biol. Chem. 1999;274:34277–34282. doi: 10.1074/jbc.274.48.34277. [DOI] [PubMed] [Google Scholar]
- Wei Y., Chen L., Xu H., Xie C., Zhou Y., Zhou F. Mitochondrial dysfunctions regulated radioresistance through mitochondria-to-nucleus retrograde signaling pathway of NF-kappaB/PI3K/AKT2/mTOR. Radiat. Res. 2018;190:204–215. doi: 10.1667/RR15021.1. [DOI] [PubMed] [Google Scholar]
- Xie G.H., Dai H.J., Liu F., Zhang Y.P., Zhu L., Nie J.J., Wu J.H. A dual role of ATM in ischemic preconditioning and ischemic injury. Cell Mol. Neurobiol. 2019;40:785–799. doi: 10.1007/s10571-019-00773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Voloboueva L.A., Ouyang Y., Emery J.F., Giffard R.G. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J. Cereb. Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Liu X., Hao J., Yang Y., Zhao M., Zuo J., Liu W. Glucose-regulated protein 75 suppresses apoptosis induced by glucose deprivation in PC12 cells through inhibition of Bax conformational change. Acta Biochim. Biophys. Sin (Shanghai) 2008;40:339–348. doi: 10.1111/j.1745-7270.2008.00409.x. [DOI] [PubMed] [Google Scholar]
- Yeo A.J., Fantino E., Czovek D., Wainwright C.E., Sly P.D., Lavin M.F. Loss of ATM in airway epithelial cells is associated with susceptibility to oxidative stress. Am. J. Respir. Crit. Care Med. 2017;196:391–393. doi: 10.1164/rccm.201611-2210LE. [DOI] [PubMed] [Google Scholar]
- Yeo A.J., Henningham A., Fantino E., Galbraith S., Krause L., Wainwright C.E., Sly P.D., Lavin M.F. Increased susceptibility of airway epithelial cells from ataxia-telangiectasia to S. pneumoniae infection due to oxidative damage and impaired innate immunity. Sci. Rep. 2019;9:2627. doi: 10.1038/s41598-019-38901-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin K.J., Chen S.D., Lee J.M., Xu J., Hsu C.Y. ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke. 2002;33:2471–2477. doi: 10.1161/01.str.0000030316.79601.03. [DOI] [PubMed] [Google Scholar]
- Yuan L., Liu J.G., Zhao J., Brundell E., Daneholt B., Hoog C. The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol. Cell. 2000;5:73–83. doi: 10.1016/s1097-2765(00)80404-9. [DOI] [PubMed] [Google Scholar]
- Zhang J., Tripathi D.N., Jing J., Alexander A., Kim J., Powell R.T., Dere R., Tait-Mulder J., Lee J.H., Paull T.T. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Lee J.H., Paull T.T., Gehrke S., D'alessandro A., Dou Q., Gladyshev V.N., Schroeder E.A., Steyl S.K., Christian B.E., Shadel G.S. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci. Signal. 2018;11:eaaq0702. doi: 10.1126/scisignal.aaq0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the published article and the Supplemental Information, and any additional information will be available from the lead contact upon request.