

Summary
The MRE11-RAD50-NBS1 complex plays a central role in response to DNA double-strand breaks. Here, we identify a patient with bone marrow failure and developmental defects caused by biallelic RAD50 mutations. One of the mutations creates a null allele, whereas the other (RAD50E1035Δ) leads to the loss of a single residue in the heptad repeats within the RAD50 coiled-coil domain. This mutation represents a human RAD50 separation-of-function mutation that impairs DNA repair, DNA replication, and DNA end resection without affecting ATM-dependent DNA damage response. Purified recombinant proteins indicate that RAD50E1035Δ impairs MRE11 nuclease activity. The corresponding mutation in Saccharomyces cerevisiae causes severe thermosensitive defects in both DNA repair and Tel1ATM-dependent signaling. These findings demonstrate that a minor heptad break in the RAD50 coiled coil suffices to impede MRE11 complex functions in human and yeast. Furthermore, these results emphasize the importance of the RAD50 coiled coil to regulate MRE11-dependent DNA end resection in humans.
Keywords: RAD50, coiled-coil, MRE11, MRN, immunodeficiency, nuclease activity, DNA repair, replication
Graphical Abstract
Highlights
-
•
A human RAD50 mutant impairs DNA repair but not ATM-dependent DDR
-
•
A single amino acid deletion disrupts the coiled-coil structure of RAD50
-
•
A Rad50 mutant yeast strain exhibits an extremely severe thermosensitive phenotype
Chansel-Da Cruz et al. provide evidence that a mutation in the human RAD50 coiled coil located away from the MRE11 interaction interface uncouples the signaling function of the MRN complex from its broken DNA end processing activities. This demonstrates the crucial role of the RAD50 coiled coil in the MRE11 complex regulation.
Introduction
DNA double-strand breaks (DSBs) are considered the most toxic form of DNA damage. Although unrepaired DSBs can cause cell death, improperly repaired DSBs represent a source of mutation and translocation that challenges genome stability and can lead to cancer development (Scully et al., 2019). MRE11-RAD50-NBS1 (MRN; Mre11-Rad50-Xrs2 [MRX] in yeast) is a conserved complex that plays a central role in the sensing of and response to DSBs (Lisby et al., 2004; Mirzoeva and Petrini, 2001). MRN is one of the first molecular entities detected at DSBs, where it activates the ataxia telangiectasia mutated (ATM) kinase to trigger DNA damage response (DDR) signaling to induce cell-cycle checkpoints and apoptosis (Lisby et al., 2004; Stracker and Petrini, 2011). The MRN complex also influences DSB repair via the endo- and exonuclease activities of MRE11 and initiates DNA end resection to generate, in concert with other nucleases, single-strand DNA (ssDNA) ends that drive homology-directed repair (HDR) (Scully et al., 2019; Syed and Tainer, 2018).
RAD50 belongs to the highly conserved structural maintenance of chromosome (SMC) family of proteins (de Jager et al., 2004; Hopfner and Tainer, 2003). RAD50 possesses an extended coiled-coil domain that folds back upon itself to form an intramolecular, antiparallel coiled-coil structure of about 500 Å that leads to juxtaposition of the N- and C-terminal Walker A and B domains and generates a functional ATPase that directly interacts with MRE11. The coiled-coil structure, which is conserved across the SMC proteins and all known Rad50 orthologs (de Jager et al., 2004; Paull, 2018; Stracker and Petrini, 2011; Syed and Tainer, 2018), comprises a heptad repeat pattern wherein the first and the fourth residues are hydrophobic to allow the association of antiparallel helices via their hydrophobic faces (de Jager et al., 2001; Truebestein and Leonard, 2016; van Noort et al., 2003). The apex of the RAD50 coiled coil consists of a zinc (Zn)-hook domain by which two MRE11-RAD50 complexes can dimerize to form intra- and intermolecular complexes that have been proposed to bridge DNA ends (Hohl et al., 2015; Hopfner, 2014; Park et al., 2017; Paull, 2018; Stracker and Petrini, 2011; Syed and Tainer, 2018; Tatebe et al., 2020). It has also been recently suggested that MRE11-RAD50 complexes could promote DNA tethering of sister chromatids at stalled forks by facilitating Cohesin loading (Delamarre et al., 2020). Although the flexibility of the RAD50 coiled-coil domain has prevented determination of its whole structure at atomic resolution (Käshammer et al., 2019; Park et al., 2017), several studies suggested that this domain participates in the regulation of MRN functions by propagating information from the Zn-hook domain to the globular domain (Hohl et al., 2011, 2015; Park et al., 2017). Analyses using crystallographic microscopy, cryo-electron microscopy (cryo-EM), and high-throughput single-molecule microscopy suggested that dimers of the ATP-bound MRE11-RAD50 intracomplex (M2R2) mostly adopt a ring conformation that can interact and scan the DNA homoduplex to recognize the DNA end and trigger ATM/Tel1-dependent DDR (Deshpande et al., 2014; Hopfner, 2014; Käshammer et al., 2019; Myler et al., 2017; Tatebe et al., 2020). A model suggested that upon ATP and DNA binding, the MRE11-RAD50 intracomplex undergoes a conformational switch, leading to the interaction of intermolecular coiled-coil domains and resulting in a rod-shaped structure. This structural modification would allow exposure of the catalytic domain of MRE11 to the DNA end and stimulate nuclease activity and DNA end processing (Deshpande et al., 2014; Hopfner, 2014; Käshammer et al., 2019; Lammens et al., 2011; Lim et al., 2011; Liu et al., 2016; Rojowska et al., 2014). Several studies conducted in yeast and bacteria suggested that the RAD50 coiled-coil domain was particularly important during these conformational transitions by propagating spatial information from the Zn hook to the globular domain (Hohl et al., 2011, 2015; Hopfner et al., 2002; Käshammer et al., 2019; Lee et al., 2013; Park et al., 2017). Nonetheless, despite progress in structure determination of all or part of the MRN complex, our knowledge of the RAD50 coiled-coil functional role in the regulation of the multiple MRE11 complex functions remains limited, especially in mammals (Paull, 2018).
Here, we identified an individual, P1, who presented with immunodeficiency and developmental defects and has compound heterozygous mutations in RAD50. One of the mutations creates a null allele, and the other appears to be hypomorphic because of the loss of a single amino acid residue in the coiled-coil domain of RAD50 (denoted as RAD50E1035Δ). The analysis of the human RAD50E1035Δ mutant, as well as its yeast counterpart, provided unprecedented information, emphasizing the fundamental role of the RAD50 coiled-coil domain in modulating MRE11 complex functions. Furthermore, this study reports the first RAD50 separation-of-function mutation that does not affect ATM-dependent DDR but severely impairs DSB repair. These findings demonstrate the crucial role of the RAD50 coiled-coil domain to regulate MRE11-dependent DNA end resection and repair in humans.
Results
Clinical Features of Individual P1
This study was initiated by the analysis of individual P1, who was diagnosed at 7 years of age with bone marrow failure and developmental defects. In the family, the two other children and parents were healthy (Figure 1A). P1’s cell blood count revealed thrombocytopenia (platelet count of 113 G/L) associated with anemia (hemoglobin level of 10 g/dL) and neutropenia (neutrophil count of 1 G/L) (Table S1). Bone marrow smears and biopsies showed reduced cellularity (15%) without myelodysplastic features, consistent with aplastic anemia. Immunophenotyping highlighted a virtual absence of circulating B lymphocytes (CD19+), whereas T lymphocytes (CD3+) that were normal in number exhibited an increased proportion of central memory T cells (CD3+ CCR7− CD45A−) (Table S1). Conversely, the proportions of both naive CD4+ T cells (evaluated by the CD45RA marker) and recent thymus emigrant (characterized by CD45RA+CD31+ markers) were markedly reduced, suggesting impaired T lymphopoiesis (Table S1). Moreover, physical examination of P1 revealed failure to thrive (height of 108.5 cm, <3rd percentile; weight of 17.2 kg, <3rd percentile), skin pigmentation, nail dysplasia, leukoplakia, dental loss, microcephaly (cranial perimeter of 48 cm, <3rd percentile), and dysmorphia. P1 also developed a cataract at 10 years of age that necessitated surgical intervention. Collectively, P1’s clinical features were reminiscent of a defect in DNA repair and/or telomere maintenance (Glousker et al., 2015; Rivera-Munoz et al., 2007). However, telomere restriction fragment (TRF) analysis indicated that telomere length in P1’s peripheral blood cells was not reduced compared with his parents and his healthy sister (Figure S1).
Figure 1.

Biallelic RAD50 Mutations Identified in Individual P1
(A) (Upper panel) Pedigree of P1’s family. (Lower panel) Genetic approach used to identify the disease-causing gene.
(B) Sequencing of cloned RAD50 PCR products in the patient.
(C) Schematic of the domain architecture of human RAD50, showing the position of disease-associated mutations. NBD, nucleotide binding domain.
(D) (Left) Immunoblot of indicated proteins in B-LCL lysates from a healthy donor, P1, and his mother. KU70 was used as loading control. (Right) Relative abundance of indicated proteins normalized to KU70.
(E) (Left) Schematic view of a RAD50 protein. (Right) Schematic view of the MRN complex in its intracomplex conformation13.
(F) Marcoil analysis revealed a break in the heptad repeats near the E1035Δ mutation (blue arrow).
(G) (Upper panels) Representative view of the coiled-coil probability along the full WT and RAD50E1035Δ sequences determined by the Marcoil tool. The red area corresponds to the probable coiled-coil regions. (Lower panels) Zoom of the region encompassing the E1035Δ mutation. The blue arrow indicates a break in the coiled-coil probability.
Individual P1 Carries Biallelic RAD50 Mutations
To determine the molecular etiology of the disease, we performed whole-exome sequencing analysis in P1 by considering coding sequences carrying rare biallelic variants (frequency less than 0.1% in 1,000 genomes, Exome Variant Server (EVS), Single Nucleotide Polymorphism Database (dbSNP), and our in-house database of 8,319 individuals) predicted to be deleterious (Figure 1A). This approach highlighted two mutations in RAD50 that were confirmed by Sanger sequencing and present in a compound heterozygous configuration (Figure 1B). One consisted in a nucleotide insertion, leading to a frameshift and a premature stop codon (c.2165dup; p.Glu723Glyfs∗5) (Figures 1B and 1C), inherited from the asymptomatic mother and present in the healthy sister and brother (Figure S2A). The other mutation was an in-frame deletion of three nucleotides, resulting in the loss of a single glutamic-acid residue at position 1035 (c.3109_3111del; p.Glu1035del) located in the coiled-coil domain of RAD50 (Figures 1B and 1C; Figure S2A). This deletion corresponded to a de novo mutation, because it was absent for the siblings and parents (Figure S2A), whereas microsatellite analysis ascertained for the genetic paternity of P1’s father (Figure S2B). Both RAD50 mutations were absent from the gnomAD database (>120,000 individuals tested). Sequencing of RAD50 cDNA from P1’s blood cells did not detect the c.2165dup; p.Glu723Glyfs∗5 mutation, likely because of nonsense-mediated decay (NMD) (data not shown). Hence, the only RAD50 transcripts present in P1’s cells encoded a RAD50 protein lacking a unique glutamic-acid residue in the coiled-coil domain of RAD50 (p.Glu1035del, hereafter dubbed RAD50E1035Δ) (Figure 1C). RAD50 immunoblots with B lymphoblastoid cell line (B-LCL) lysates from P1 and his mother revealed approximately half of the RAD50 amount compared with B-LCL lysate from a healthy donor (Figure 1D). This result was consistent with the presence of a null allele in both P1 and his mother (i.e., c.2165dup; p.Glu723Glyfs∗5). Moreover, this finding indicated that RAD50E1035Δ was correctly expressed and as stable as its wild-type (WT) counterpart. Congruent with an interdependent stability of the human MRN components (Stewart et al., 1999; Waltes et al., 2009), we noticed a reduced amount of NBS1 and MRE11 in cell lysates from P1 and his mother (Figure 1D). The RAD50E1035Δ mutation was located in the coiled-coil structure composed of heptad amino acid repeats commonly labeled abcdefg, where a and d represent hydrophobic positions (Truebestein and Leonard, 2016) (Figure 1E). The program Marcoil, which calculates coiled-coil probability (Delorenzi and Speed, 2002), indicated that the loss of the residue E1035 led to a break in the heptad repeats (blue arrow in Figure 1F) that was predicted to disrupt the RAD50 coiled-coil structure near the mutation, as inferred by the drop in coiled-coil probability in this region (blue arrow in Figure 1G). Thus, we decided to investigate the functional consequence of this peculiar mutation in P1’s cells.
RAD50E1035Δ Mutation Impairs DNA Repair
First, we assessed whether P1’s cells were able to cope with various DNA lesions produced by distinct genotoxic agents. P1’s SV40-transformed fibroblasts, similar to cells from an ATM-deficient patient, exhibited a strong sensitivity to ionizing radiation (IR), suggesting a DNA repair defect (Figure 2A). P1’s cells also had a pronounced sensitivity to the DSB-inducing agent phleomycin (Figure 2B) and to etoposide (Figure 2C), a drug that generates topoisomerase 2-DNA adducts. P1’s cells were also sensitive to the DNA interstrand-crosslink (ICL)-inducing agent mitomycin C (MMC), although to a lesser extent than cells from a Fanconi anemia patient (Figure 2D) and not associated with an impaired MMC-induced FANCD2 ubiquitination (data not shown). Of note, a human HT1080 cell line carrying a nonsense mutation on one allele of the RAD50 gene (RAD50+/−) generated by CRISPR-Cas9 only exhibited modest sensitivity to genotoxics at higher doses, ruling out haploinsufficiency as the cause of the severe DNA repair defect in P1’s cells (Figure S3). Altogether, these results demonstrated that P1’s cells have a general DNA repair defect that is likely the cause of the bone marrow failure and developmental anomalies found in this patient.
Figure 2.

Defective DNA Repair but Normal DNA Damage Sensing and Signaling in P1’s Cells
(A–D) Survival of SV40 fibroblasts after IR (A), phleomycin (B), etoposide (C) and MMC (D) treatment. Results are expressed as the fraction of surviving cells in relation to untreated cells. Each point represents the mean value and standard deviation of three separate determinations. ATM- and Cernunnos-deficient cells were used as sensitive controls. Control-sensitive fibroblasts were from ATM-, Cernunnos-, and Fanconi-deficient patients.
(E) Sensitivity of P1’s cells to phleomycin after WT-RAD50 or empty vector transduction. This experiment was performed three times. Control-sensitive fibroblasts were from a Cernunnos-deficient patient.
(F) Functional complementation of phleomycin sensitivity provided by WT-RAD50 transduced into the patient’s cells compared with the empty vector. The selective growth advantage is scored as the increase in the index of mCherry-positive cells/mCherry-negative cells at various times compared with the initiation of the culture (index = 1). This experiment was performed twice.
(G) Recruitment of GFP-NBS1 to DNA damage upon laser microirradiation in indicated cells. GFP-NLS (nuclear localization signal) is used as negative control. Irradiated zones are located in the red circles. Representative of three independent experiments.
(H) Kinetics of recruitment of GFP-NBS1 and GFP-NLS to DNA damage in the control and the patient’s cells.
(I) Immunofluorescence detection of P-KAP1 in primary fibroblasts before and 1 h after 0.5 Gy irradiation. ATM- and NBS1-deficient cells are used as negative controls. The image is representative of three independent experiments.
(J) Phosphorylated forms of CHK2 and NBS1 detected by western blot analysis of whole-cell lysates from SV40 fibroblasts from a healthy control, an ATM-deficient patient, and P1 untreated or 1 h after 5 or 10 Gy irradiation. Vinculin immunoblot is used as loading control. An asterisk indicates a nonspecific band.
(K) (Left) DNA content and histone H3 phosphorylation (P-H3) in G2 (pink rectangle) were analyzed by fluorescence-activated cell sorting (FACS). (Middle) FACS images of P-H3 in G2 in untreated and irradiated cells. (Right) G2/M checkpoint measured by the percentage of inhibition of entry into mitosis in cells after IR. Results represent the mean and SD from at least three independent experiments. Statistical significances are noted.
To verify that the RAD50 deficiency in P1’s cells was responsible for the DNA repair defect, we transduced the cells with a vector expressing WT-RAD50 (Figure S4). Although the empty vector had no effect, the WT-RAD50-expressing vector complemented the phleomycin sensitivity of P1’s cells (Figure 2E). We confirmed this result using a multicolor competition assay after transduction of P1 and control cells with a lentiviral Ires-mCherry vector containing, or not containing, the WT-RAD50 coding sequence (Smogorzewska et al., 2007). A mix of transduced (mCherry+) and nontransduced (mCherry−) cells was analyzed for selective advantage in culture, as evaluated by the mCherry index of transduced (mCherry+) over nontransduced (mCherry−) cells upon treatment with phleomycin (Figure 2F). Ectopic expression of mCherry/WT-RAD50, but not mCherry alone, gave in P1’s cells a strong selective advantage over nontransduced cells upon treatment with phleomycin, as determined by the 40-fold increase in mCherry/WT-RAD50-expressing cells after 18 days in this culture condition (Figure 2F). Altogether, these results established a causal link between the DNA repair defect and the expression of RAD50E1035Δ in P1’s cells.
RAD50E1035Δ Mutation Does Not Affect DNA Damage Sensing and Signaling
To determine whether the DNA repair defect in P1’s cells could be associated with impaired MRN recruitment at DNA damage sites, we monitored the dynamics of NBS1 at DSBs. We transfected cells with a vector expressing green fluorescent protein (GFP)-NBS1 fusion protein and analyzed by live microscopy GFP-NBS1 behavior following the generation of localized DNA damage induced by a laser microbeam (Zhang et al., 2016). Kinetics and intensity of GFP-NBS1 recruitment at DNA lesions was similar in control and P1’s cells (Figures 2G and 2H), indicating that RAD50E1035Δ did not affect the capacity of the ΜRΝ complex to sense the DNA lesion and accumulate at DNA DSBs.
We next assessed whether the DNA repair defect in P1’s cells was associated with impaired ATM-dependent DDR functions (Stracker and Petrini, 2011). As expected, KAP1 phosphorylation (P-KAP1), analyzed by immunofluorescence, was undetectable in primary fibroblasts from ATM- and NBS1-mutated patients, demonstrating the need for functional ATM and MRN in this process (Figure 2I). To our surprise, IR-induced P-KAP1 was readily detected in primary fibroblasts from P1, suggesting that RAD50E1035Δ did not impair ATM-dependent P-KAP1. Furthermore, western blot analysis demonstrated that P1’s SV40 fibroblasts normally induced phosphorylation of NBS1 and CHK2 following IR, whereas ATM-deficient control cells did not (Figure 2J). Consistent with normal IR-induced CHK2 phosphorylation, P1’s SV40-transformed fibroblasts exhibited a functional G2/M checkpoint, as determined by the reduction of phospho-histone 3 in the G2 phase, a marker of chromosome condensation, following IR (Figure 2K). As expected, ATM-deficient cells used as negative control had a defective G2/M checkpoint (Figure 2K).
Collectively, these results demonstrated that P1’s cells behaved differently from ATM- and NBS1-mutated cells and provided evidence that the human RAD50E1035Δ mutation, despite leading to a DNA repair defect, did not impair recruitment of the MRN complex at DNA damage sites or the subsequent activation of ATM.
RAD50E1035Δ Mutation Impairs DNA End Resection and HDR
MRE11 initiates double-strand DNA end resection via its nuclease activity, a process that culminates in the formation of ssDNA that is used to scan sequence homology and promote HDR (Stracker and Petrini, 2011; Syed and Tainer, 2018). During this process the ssDNA molecules are coated by replication protein A (RPA) and then by RAD51 (Zhao et al., 2019). To gain insight into the DNA repair pathway that might be affected in this patient, we assessed DNA end resection following IR treatment in P1’s cells. In contrast to P-KAP1, which was readily detected in P1’s primary fibroblasts 6 h after IR (Figure 3A), RPA foci (Figure 3B) and RAD51 foci (Figure 3C; Figure S5) (p < 0.0001) were virtually absent from these cells, suggesting impaired DNA end resection at the IR-induced DSB. The impaired RAD51 foci formation upon IR treatment was confirmed in SV40-hTERT P1’s fibroblasts (p < 0.0001) (Figure 3D). Transduction with WT-RAD50, but not with an empty vector, complemented the impaired IR-induced RAD51 foci formation in P1’s cells, a result demonstrating the causal link between compromised DNA end resection and RAD50 deficiency in these cells (Figure 3D). We next tested the HDR pathway by measuring the efficiency of insertion in a chromosomal context of a DNA sequence encoding Clover, a GFP variant, mediated by CRISPR-Cas9-mediated HDR within the first exon of the Lamin-A (LMNA) gene (Figure S6A) (Pinder et al., 2015). Cells were transfected with an mCherry-expressing vector to gate on transfected cells (mCherry+), in combination with the Clover-donor vector with or without CRISPR-Cas9 vector targeting the Lamin-A locus (Pinder et al., 2015). This approach induced a significant increase in Clover-positive cells in control cells transfected with both the donor and the CRISPR-Cas9 vectors (p < 0.0001), asserting efficient HDR in these cells (Figure 3E). In contrast, in the same experimental conditions, we did not detect a significant increase in Clover expression in P1’s cells, suggesting that HDR was impaired in these cells (Figure 3E). Because the HDR pathway is mainly active during the S and G2 phases, when a DNA template generated by DNA replication is available (Jasin and Rothstein, 2013), we analyzed the cell-cycle profile of P1’s cells. As determined by the combined detection of bromodeoxyuridine (BrdU) incorporation and propidium iodide staining, the cell-cycle profiles in control and P1’s cells were similar, ruling out that the defective HDR in P1’s cells was caused by an abnormal cell cycle (Figure S6B).
Figure 3.

P1’s Cells Exhibit a Reduced DNA End Resection and HDR
(A–C) Immunofluorescence detection of P-KAP1 (A), RPA32 (B), and RAD51 (C) in primary fibroblasts from healthy control and P1 before and 6 h after 10 Gy irradiation. The image is representative of three independent experiments.
(D) Quantitative analysis of RAD51 foci before and after IR in SV40-hTERT fibroblasts untransduced or transduced with an empty or WT-RAD50-expressing vector. The number of nuclei scored and statistical significances are noted.
(E) Efficiency of HDR assessed by the Cas9-directed knockin of Clover in the Lamin-A (LMNA) coding sequence. Cells were transfected with the mCherry vector alone (used to control transfection efficiency) or in combination with the repair template sequence containing the Clover coding sequence in conjunction, or not, with the pX330-LMNA1 vector encoding Cas9 and the guide RNAs (gRNAs) targeting the LMNA sequence (denoted as CRISPR-Cas9). Relative HDR efficiency was measured by the percentage of Clover+ cells triple transfected relative to cells transfected without the CRISPR-Cas9-gRNA-expressing vector. Results represents the mean and SD of triplicates from three independent experiments.
We concluded from these experiments that the DNA repair defect observed in P1’s cells was associated with a reduced resection of IR-induced DSBs and impaired HDR efficiency.
RAD50E1035Δ Mutation Prevents Nascent DNA Degradation after Replication Stress
In the absence of genotoxic stress, spontaneous 53BP1 focus formation is an indicator of replicative stress (Pasero and Vindigni, 2017). We noticed an increase in 53BP1 foci in P1’s primary fibroblasts in the absence of exogenous stress (Figure 4A). Automated detection and quantification of 53BP1 foci reported a mean of 7 events/nucleus in P1’s cells (n = 9,894) and 2.7 events/nucleus in control cells (n = 19,632; p < 0.0001) (Figure 4B), with 63.9% of P1’s cells exhibiting 5 or more 53BP1 foci versus 16.3% in control cells (p < 0.0001) (Figure 4C). We then performed a replication-combing assay (RCA), a technique that enables the measurement of fork speed, fork asymmetry, and interorigin distance (Bianco et al., 2012; Schurra and Bensimon, 2009) (Figure 4D). SV40-transformed fibroblasts from P1 consistently exhibited defective DNA replication inferred by a significant reduction of fork speed (p < 0.0001) (Figure 4E). We also noticed a shortening of interorigin distance in P1’s cells (p < 0.01) (Figure 4F), suggesting increased dormant origin firing to compensate for reduced fork speed, as previously described (Anglana et al., 2003; Courbet et al., 2008; Mokrani-Benhelli et al., 2013). P1’s cells also showed an increase in asymmetric replicons (p < 0.001), likely attributable to stochastic replication fork stalling and/or collapse (Figure 4G). Transduction of P1’s cells with a lentiviral vector allowing the expression of WT-RAD50 restored a fork speed comparable to that of the control cells, demonstrating a causal link between replication defects and RAD50 deficiency in P1’s cells (Figure 4H).
Figure 4.

DNA Replication and Resection at Stalled Forks in P1’s Cells
(A) Detection of 53BP1 foci in primary fibroblasts from the patient and a healthy control at a similar passage.
(B) Mean of 53BP1 foci in primary fibroblasts from a healthy control and the patient. The number of nuclei scored and statistical significances are noted.
(C) Percentages of fibroblasts from a control and P1 with the indicated number of 53BP1 foci are indicated. Statistical significances are noted.
(D) (Up) Representative image of combed DNA (yoyo labeling). (Down) Picture representing the detection of newly synthesized DNA by molecular combing (successive pulse labels of IdU and CldU). DNA is detected by counterstaining (blue).
(E) Fork velocity in SV40 fibroblasts from 3 independent experiments in control and P1 are represented.
(F) Interorigin distances in SV40 fibroblasts from control and P1 are represented. The statistical significances are noted.
(G) (Left) Asymmetric replicon with sister forks with a more than 25% length difference in SV40 fibroblasts from control and P1. Plots of the distances covered by right-moving and left-moving sister forks during the IdU and CldU pulses from two independent experiments. (Right) Percentage of replicons with more than 25% asymmetry in SV40-transformed fibroblasts from control and P1. The statistical significance is noted.
(H) Fork velocity of SV40-transformed fibroblasts from control and P1 before or after transduction with a WT-RAD50-expressing vector or empty vector. Results are representative of two independent experiments. The statistical significances are noted (Mann-Whitney statistical test).
(I) (Upper panel) Protocol used to measure DNA resection after replicative stress induction by HU. (Lower panel) Representative tracks of CldU (red) and IdU (green) incorporation in control and patient’s cells with or without HU treatment. DNA is counterstained in blue.
(J) DNA resection at stalled forks is determined by the ratio between CldU and IdU track lengths after 3 h of HU treatment. Results are representative of two independent experiments.
(K) DNA resection at stalled forks after 3 h of HU in control and P1 cells before or after transduction with an empty or WT-RAD50-expressing vector. Results are representative of two independent experiments. The statistical significances are noted (Mann-Whitney statistical test).
Fork restart depends on fork resection initiated by the nuclease activity of MRE11 that degrades newly synthesized DNA strands (Bryant et al., 2009; Pasero and Vindigni, 2017; Trenz et al., 2006). To test whether replicative stress observed in P1’s cells could be accompanied by a defect in DNA resection at arrested forks, after successive pulses of 5-Chloro-2′-deoxyuridine (CldU) and 5-Iodo-2′-deoxyuridine (IdU), we treated cells with a high dose of hydroxyurea (HU) for 3 h to provoke replicative stress and examined DNA resection by molecular combing (Coquel et al., 2019) (Figure 4I). As expected, HU treatment led to nascent DNA degradation in control cells, as revealed by a IdU/CldU ratio < 1 (Figures 4I and 4J). In contrast, IdU track length did not change after HU treatment in P1’s cells (Figures 4I and 4J), suggesting that RAD50E1035Δ mutation led to impaired DNA resection at arrested forks. Transduction of WT-RAD50 in P1’s cells complemented this defect (Figure 4K), supporting the notion that RAD50E1035Δ impaired DNA resection at arrested forks.
Collectively, the sharp reduction of DNA end resection at IR-induced DSBs (Figures 3B–3D; Figure S5) and at arrested forks (Figure 4J) observed in P1’s cells suggested that the RAD50E1035Δ mutation impairs MRE11 nuclease activity.
Impairment of Nuclease Activity of a Recombinant MR(N) Complex Containing RAD50E1035Δ
Because complete loss of MRE11 nuclease activity is incompatible with cellular viability (Buis et al., 2008; Hoa et al., 2015, 2016), we surmised that RAD50E1035Δ could not abolish this process. To directly evaluate the impact of RAD50E1035Δ on MRE11 nuclease activity, we expressed and purified human MRE11 and RAD50 (MR) as a complex from baculovirus-infected Spodoptera frugiperda 9 (Sf9) cells as previously described (Anand et al., 2016) (Figure S7A). For simplicity, we denominated the wild-type MRE11-RAD50 complex as MR WT and the MRE11-RAD50 complex containing RAD50E1035Δ as MR E1035Δ. As depicted in Figure 5A, MR WT and MR E1035Δ were similarly purified, reinforcing our previous results showing that the RAD50E1035Δ mutation did not affect protein stability (Figures 1D and 2J; Figure S4A). Electrophoretic mobility shift assay (EMSA) using a 70-bp probe revealed a slight decrease in the DNA binding capacity of the MR E1035Δ complex compared with its WT counterpart (Figures 5B and 5C). In vitro assay with 50-bp unprotected DNA substrates revealed a consistent decreased exonuclease activity of MR complexes with MR E1035Δ compared with MR WT (Figures 5D and 5E). In particular, the lower products corresponding to the most resected DNA substrates were sharply underrepresented with MR E1035Δ, asserting reduced exonuclease activity (Figures 5D and 5F). CtBP-interacting protein (CtIP) is a cofactor that potentiates the DNA binding activity of the MR complex, especially in its nonphosphorylated form (Anand et al., 2016). We produced recombinant CtIP treated or not treated with lambda phosphatase to generate either nonphosphorylated CtIP or phosphorylated CtIP (pCtIP), respectively (Figure S7B). As previously described (Anand et al., 2016), adding the nonphosphorylated recombinant CtIP led to a strong increase in the DNA binding capacity of the MR complex that, in this condition, was similar with MR E1035Δ and MR WT (Figures S7C and S7D). This result indicated that in the presence of nonphosphorylated CtIP, the RAD50 E1035Δ mutation did not impair the DNA binding capacity of the MR complex. However, in the presence of pCtIP, we observed a slight reduction in DNA binding capacity of the mutated MR E1035Δ complex (Figures S7C and S7D). Next, we added MBP-NBS1 and pCtIP to the complex to assess the endonuclease activity of MRE11, as described previously (Anand et al., 2019). This analysis revealed a subtle reduction of endonuclease activity of the MR E1035Δ complex, evident only at a higher concentration (Figures 5G and 5H). Salt titration experiments performed to examine whether the effect of the mutant may be more apparent after more restrictive conditions confirmed the slightly decreased endonuclease activity of the MR E1035Δ complex and showed that a high salt concentration did not worsen this defect (Figures S7E and S7F). The detection of MRE11 endonuclease activity, which depends on ATP hydrolysis (Anand et al., 2016; Cannavo et al., 2019; Deshpande et al., 2017; Hopfner et al., 2000; Liu et al., 2016; Paull and Gellert, 1999), suggested that recombinant RAD50E1035Δ did not abolish ATP hydrolysis, at least in vitro. Accordingly, in vitro ATPase activity was similar with MR E1035Δ and MR WT (Figures 5I and 5J).
Figure 5.

Impairment of Nuclease Activity of Recombinant MRE11 and RAD50E1035Δ Proteins
(A) Recombinant MRE11-RAD50 (MR) with WT RAD50 (MR WT) or mutated RAD50E1035Δ (MR E1035Δ) used in this study.
(B) Representative DNA binding analysis by electrophoretic mobility shift assay with recombinant MR WT and MR 1035Δ using 70-bp-long double-strand DNA (dsDNA) as a substrate.
(C) Quantitation of experiments shown in (B). Error bars, SEM; n = 3.
(D) Exonuclease activity of MR WT or MR E1035Δ using a 50-bp-long dsDNA substrate.
(E) Plots of signals shown in (D) reveal a reduction of the most resected DNA substrate (higher migration) with MR E1035Δ compared with MR WT.
(F) Quantitation of experiments shown in (C). Error bars, SEM; n = 3.
(G) Representative nuclease assay with MR WT or MR E1035Δ in the presence of MBP-NBS1 and pCtIP on 5′ end-labeled 70-bp dsDNA with all ends blocked with streptavidin.
(H) Quantitation of experiments shown in (G). Error bars, SEM; n = 3.
(I) ATP hydrolysis activity of MR WT or MR E1035Δ with or without MBP-NBS1. The MRN complexes containing the RAD50 K40A (denoted as MR(KA)N) and RAD50 K40R (denoted as MR(KR)N) mutants, defective in ATP binding and hydrolysis (Cannavo et al., 2019; Chen et al., 2005), were used as negative controls.
(J) Quantitation of experiments shown in (I). n = 2.
In summary, our experiments using recombinant proteins indicated that the RAD50E1035Δ-containing MRN complex exhibits defects in MRE11 nuclease activity in vitro, a result congruent with the conclusions drawn from the observations obtained in P1’s RAD50E1035Δ-expressing cells.
Modeling the Human RAD50E1035Δ Mutation in Saccharomyces cerevisiae
We next modeled the human RAD50E1035Δ mutation in S. cerevisiae by producing a yeast strain deleted of the corresponding residue, i.e., the glutamic acid at position 1042 (rad50-E1042Δ) (Figure 6A). In addition, we generated a strain lacking a nearby glutamic acid at position 1044 (rad50-E1044Δ) Figure 6A) and created two other yeast strains carrying missense mutations instead of deletion at position 1042, where the glutamic acid residue was substituted by either an alanine (rad50-E1042A) or a lysine (rad50-E1042K). Western blot and coimmunoprecipitation experiments showed normal expression and interaction with Mre11 of the deletion and missense mutants at both 30°C and 37°C, indicating that these mutations did not impair Mre11 complex formation and stability (Figure 6B). However, unlike the WT strain, rad50-E1042Δ and rad50-E1044Δ deletion mutants exhibited high sensitivity to camptothecin (CPT) (Figure 6C), methyl methane sulfonate (MMS) (Figure 6D), and HU (Figure 6E) at 37°C (but not at 30°C), indicating a strong temperature-sensitive effect of these mutations on DNA repair. In contrast, both rad50-E1042A and rad50-E1042K missense mutants showed WT levels of survival in the presence of CPT, MMS, and HU at both 30°C and 37°C (Figures 6C–6E). These observations provided evidence that the glutamic acid residue at position 1042 (or position 1044) in the coiled-coil domain of Rad50 is not critical for DNA repair. Instead, the presence of any residue at this position is required, presumably to preserve the phasing of the local heptad-repetitive disposition of the coiled-coil region. Consistent with this result, Marcoil analysis predicted that Rad50 deletion mutations caused a heptad break in the coiled-coil domain, whereas this structure was not affected in missense mutants (Figure S8A). Thus, this observation supported the hypothesis that the temperature-dependent DNA repair defect observed in rad50-E1042Δ and rad50-E1044Δ strains resulted from an abnormal coiled-coil conformation.
Figure 6.

Modeling the Human RAD50E1035Δ Mutation in Saccharomyces cerevisiae
(A) Schematic representation of the Rad50 structure and multiple sequence alignment of the C-terminal coil domain of Rad50 carrying the human E1035Δ mutation. The human E1035 residue and corresponding amino acid in Saccharomyces cerevisiae and Mus musculus are highlighted in green. (Sc, S. cerevisiae; Mm, M. musculus; Hs, H. sapiens).
(B) Coimmunoprecipitation and western blot with Rad50 or Mre11 antisera assessed the Mre11 complex integrity in WT and indicated Rad50 mutants. IP, immunoprecipitation.
(C–E) Sensitivities of WT and the indicated Rad50 mutant to the indicated concentration of CPT (C), MMS (D), and HU (E). Plates were incubated at 30°C and 37°C as indicated.
(F) Telomere lengths of WT and rad50 mutants after 4 days of growth at 30°C and 37°C. Heterozygote diploids were included as the zero generation of growth.
(G) Cell survival of rad50 mutants in Mec1- and Mec1-Sae2-deficient backgrounds upon MMS treatment at 30°C and 37°C.
In S. cerevisiae, telomere maintenance relies on a functional Tel1ATM pathway. Both rad50-E1042Δ and rad50-E1044Δ deletion mutants, but not rad50-E1042A and rad50-E1042K missense mutants, exhibited shorter telomeres at 37°C (and to a lesser extent at 30°C) than in the WT strain, suggesting impaired Tel1 activation in deletion mutants (Figure 6F; Figure S8B). We previously showed that MMS sensitivity of mec1ATRΔ cells is strongly rescued by sae2CtIPΔ, because mec1Δ sae2Δ cells hyperactivate Tel1 kinase signaling and compensate for a lack of the Mec1 kinase (Park et al., 2017; Usui et al., 2001). Thus, to examine Tel1 function, we evaluated the phosphorylation of Rad53CHK2 and the survival upon MMS treatment of Rad50 mutants deficient for both Mec1 and Sae2. Rad50-E1042Δ mec1Δ sae2Δ and rad50-E1044Δ mec1Δ sae2Δ mutant strains exhibited defective survival (Figure 6G) and Rad53 phosphorylation (Figure S8C) upon MMS exposure at 37°C, confirming impaired Tel1 signaling at this temperature. In contrast, rad50-E1042A mec1Δ sae2Δ and rad50-E1042K mec1Δ sae2Δ mutants behaved as WT at both 30°C and 37°C (Figure 6G; Figure S8C).
Altogether, the experiments conducted in yeast demonstrated that the loss of a unique residue breaking the heptad repeats in the coiled-coil domain of Rad50 did not affect Rad50 expression or Mre11 complex formation but severely affected DNA repair and Tel1 checkpoint signaling in a temperature-dependent manner.
Discussion
In this study, we identified biallelic mutations in the RAD50-encoding gene in an individual (P1) exhibiting bone marrow failure, immunodeficiency, microcephaly, and developmental defects. Consistent with the function of RAD50 as a part of the MRN complex, most of P1’s clinical characteristics were reminiscent of, although apparently less severe than, those found in the autosomal-recessive diseases Nijmegen breakage syndrome (NBS) and ataxia-telangiectasia-like disorder (ATLD), caused by biallelic mutations in the genes encoding NBS1 and MRE11, respectively (O’Driscoll, 2012; Stracker and Petrini, 2011). The symptoms observed in these disorders, as well as in P1, most likely originate from the genome instability and DNA damage accumulation that particularly affect proliferating tissues and lead to apoptotic attrition in those contexts (Taylor et al., 2019). P1 also developed a cataract at 10 years of age that necessitated surgical intervention. To our knowledge, cataracts have not been reported in patients with NBS1 and MRE11 deficiencies. However, a mouse model has implicated Nbs1 in terminal differentiation of the lens fiber cells and cataractogenesis (Yang et al., 2006). Thus, the early-onset cataract in P1 suggests that RAD50 might also participate in the development and maintenance of the lens in humans.
To our knowledge, RAD50 hypomorphism has only been reported twice in two individuals who presented with microcephaly, mental retardation, and short stature but no immunodeficiency (Taylor et al., 2019; Waltes et al., 2009). These patients carried biallelic RAD50 mutations that strongly reduced the expression of RAD50 (Ragamin et al., 2020; Waltes et al., 2009). Waltes et al. (2009) and Ragamin et al. (2020) demonstrated that the highly pronounced reduction of functional MRN in the cells from these patients caused DNA repair defects, genome instability, and impaired ATM-dependent DDR similar to that observed in cells from patients with ATM, MRE11, and NBS1 mutations (Carney et al., 1998; Gatei et al., 2011; Shiloh and Lederman, 2017; Stewart et al., 1999; Taylor et al., 2019; Varon et al., 1998; Waltes et al., 2009). In contrast, the present study showed that the cells from P1 exhibited severe defects in DNA replication, DNA repair, and DNA end resection, whereas the ATM-dependent DDR remained intact (Figure 7A). Because one of P1’s RAD50 mutations generated a null allele, we attributed this unprecedented phenotype to the RAD50E1035Δ mutation producing a normally expressed and stable RAD50 protein that lacks a unique glutamic acid, causing a break in the heptad repeats within the coiled-coil domain. Knowing that the complete loss of Rad50 is lethal in mice (Adelman et al., 2009) and having observed that P1’s cells were proficient in ATM-dependent DDR, we deduced that the RAD50E1035Δ mutation is hypomorphic.
Figure 7.

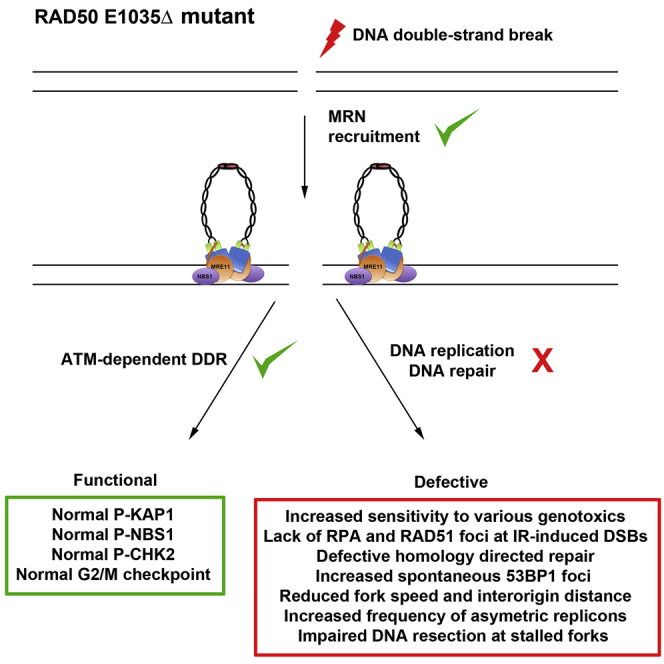
Speculative Model of the Functional and Structural Consequences of RAD50E1035Δ in Human Cells
(A) Experimental findings conducted in P1’s cells suggested that the MRE11-RAD50E1035Δ-NBS1 (MRE1035ΔN) complex is proficient in DNA DSB detection and ATM-dependent DDR activation. In contrast, MRE1035ΔN was associated with DNA replicative stress, impaired resection at stalled forks and the IR-induced DNA end, a DNA repair defect, and defective HDR.
(B) Speculative model proposing that the ring-shaped MRE1035ΔN intracomplex (open) is functional and can sense the DNA end and activate ATM-dependent DDR but that the RAD50E1035Δ mutation provokes a hindrance in the structural transition from the ring toward the rod-shaped (closed) intracomplex conformation necessary to allow MRE11 access to its DNA substrate to promote nuclease activity.
P1’s cells expressing RAD50E1035Δ mirrored the phenotype observed in mouse embryonic fibroblasts (MEFs) expressing the Mre11 nuclease dead mutant (Mre11H129N) (Buis et al., 2008). Indeed, in both Mre11H129N MEFs and P1’s RAD50E1035Δ cells, MRN stability and recruitment to DSB, as well as ATM-dependent DDR, were functional (Buis et al., 2008). In contrast, both Mre11H129N MEFs and P1’s RAD50E1035Δ cells exhibited increased sensitivity to genotoxics, defective IR-induced RPA and RAD51 focus formation, and reduced HDR efficiency (Buis et al., 2008; Myler et al., 2017). This resemblance is consistent with the interpretation that the RAD50E1035Δ mutation affected the MRE11 nuclease activity in vivo. This hypothesis was supported by in vitro assays with purified recombinant proteins that showed impaired MRE11 exonuclease activity when in complex with RAD50E1035Δ, whereas endonuclease activity was only slightly affected. However, because animal model expressing the Mre11 nuclease dead mutant at a homozygous status (Mre11H129N/H129N) and human MRE11−/H129N lymphoblast cell lines are unviable (Buis et al., 2008; Hoa et al., 2015, 2016), we surmise that RAD50E1035Δ does not abolish MRE11 nuclease activity in vivo and/or that an alternative mechanism allows it to cope with defective MRE11-dependent DNA end resection in P1.
Hence, the RAD50E1035Δ mutation represents the first human RAD50 separation-of-function mutation impairing DNA repair, DNA replication, and DNA end resection without affecting MRN recruitment to DSBs and ATM-dependent DDR (Figure 7A). It is unlikely that RAD50E1035Δ affected RAD50-ATP binding and hydrolysis, because (1) this step is required for ATM-dependent DDR (Cannavo and Cejka, 2014; Deshpande et al., 2014; Lee et al., 2013), (2) we observed an in vitro ATP hydrolysis with recombinant RAD50E1035Δ (Figure 5), and (3) in vitro MRE11 endonuclease activity, which depends on ATP hydrolysis (Anand et al., 2016; Cannavo et al., 2019; Deshpande et al., 2017; Hopfner et al., 2000; Liu et al., 2016; Paull and Gellert, 1999), was detected with recombinant RAD50E1035Δ (Figure 5). Instead, our observation that the recombinant MR E1035Δ complex exhibited a reduced DNA binding and impaired exonuclease activity in vitro (Figure 5), combined with the reduction of IR-induced RPA and RAD51 foci and HDR in P1’s cells (Figure 3), supports the notion that RAD50E1035Δ impairs the MRN complex nuclease activity required to promote DNA end resection.
These results led us to propose a unified speculative model in which the human RAD50E1035Δ mutation creates (or loses) a structural constraint in the coiled-coil domain that propagates to the globular domain to hinder the proper conformational transition of the MRN intracomplex from the ring to the rod shape (Figure 7B). This abnormally structured MRN intracomplex would retain its ATP binding and hydrolysis, as well as most of its capacity to stimulate MRE11 endonuclease activity. However, the mutant would corrupt the subsequent conformational change required for MRE11-dependent exonuclease activity and DNA end resection (Shibata et al., 2014). Because it has been suggested that the MRE11 intracomplex in its rod-shaped conformation clamps DNA (Käshammer et al., 2019), the observation of a reduced capacity of recombinant MR E1035Δ proteins to interact with DNA in vitro supports this model. However, one cannot rule out that other functionally important coiled-coil interactions both within the antiparallel coiled coil and between the dimeric coiled-coil assemblies could be affected by RAD50E1035Δ. Alternatively, we cannot exclude that the RAD50E1035Δ mutation could compromise MRN to interact with and/or activate other factors, such as EXO1, DNA2, and EXD2, required for efficient DNA end resection in vivo (Broderick et al., 2016; Cejka et al., 2010; Delamarre et al., 2020; Myler et al., 2017; Pasero and Vindigni, 2017; Paull, 2018; Stracker and Petrini, 2011; Syed and Tainer, 2018; Zhu et al., 2008).
Our analysis of the rad50-E1042Δ yeast strain modeling the human RAD50E1035Δ mutation pinpointed a phenotype strikingly different from human cells. The rad50-E1042Δ strain (and rad50-E1044Δ) behaved as WT at 30°C, whereas it was almost as severe as the rad50Δ strain for both DNA repair and Tel1ATM activation at 37°C. To our knowledge, this extremely severe thermosensitive phenotype in rad50-E1042Δ (and rad50-E1044Δ) is unprecedented in Rad50 mutants. Furthermore, the observation that yeast strains carrying missense mutations at position E1042 (rad50-E1042A and rad50-E1042K) behaved as WT at both 30°C and 37°C proved that the deletion of a single amino acid at this position is deleterious, not the nature of the residue. From these yeast experiments, we propose a model in which the rad50-E1042Δ (and rad50-E1044Δ) mutations, by breaking the heptad repeats, increase the flexibility of the coiled coil at 37°C, but not at 30°C. This temperature-dependent change would destabilize the coiled-coil interface-dependent dimerization, the conformation of the globular domain, the Zn-hook-mediated dimerization, or a combination of these modifications that would result in the extremely severe phenotype at 37°C. The phenotype of the rad50-E1042Δ strain at 37°C was similar to the one of the yeast mutant lacking the Zn hook, in which MR intra- and intercomplex formations are abolished (Hopfner et al., 2002; Stracker and Petrini, 2011). Moreover, it has been recently reported that the Zn-hook-proximal coiled coil participated in the stabilization of the intracomplex Mre11 complex assembly (Park et al., 2017). One can therefore hypothesize that the temperature sensitivity of the rad50-E1042Δ mutant results from a defective MRN intracomplex assembly at 37°C (Hohl et al., 2011; Wiltzius et al., 2005). Interestingly, other yeast strains carrying mutations affecting the Rad50 Zn-hook and/or coiled-coil domains exhibited thermosensitive phenotypes (Hohl et al., 2011, 2015). However, the molecular cause of the temperature-dependent phenotype in these Rad50 mutants remains unclear, and future studies are warranted to research the structural and functional consequences of these mutants.
In summary, our study provided evidence that a single amino acid deletion that breaks the heptad repeats in the coiled-coil domain of yeast and human RAD50 compromised MRE11 functions. Furthermore, our demonstration that human RAD50E1035Δ is a separation-of-function mutation that impairs DNA repair, DNA replication, and DNA end resection without affecting ATM-dependent DDR supports the idea that the integrity of the RAD50 coiled-coil domain is essential to enable switch of the MR intracomplex toward functional rod-shaped conformation promoting DNA clamping and DNA end resection (Hopfner, 2014; Käshammer et al., 2019; Park et al., 2017; Rojowska et al., 2014). Further studies using cryo-EM, atomic force, and high-throughput single-molecule microscopy are warranted to better characterized the structural hindrance caused by RAD50E1035Δ and decipher the rules delineating communication among the hook, the coiled-coil, and the globular domains of the MR complex in mammals. Lastly, the development of a Rad50E1035Δ mouse model should provide crucial information on the functional consequence of compromised Mre11-dependent DNA end resection in vivo.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 goat F[Ab′]2 | Invitrogen | A31556 |
| Anti-53BP1 | Novus Biological | NB100-304; RRID: AB_10003037 |
| Anti-vinculin | Santa Cruz | SC73614; RRID: AB_1131294 |
| Anti-MRE11 | Novus | NB100-473; RRID: AB_10001780 |
| Anti-RAD50 | Genetex | GTX 70228; RRID: AB_372854 |
| Anti-NBS1 | Novus | NB100-143E1; RRID: AB_350080 |
| Anti-CHK2 | Santa Cruz | SC17748; RRID: AB_627260 |
| Goat anti-rabbit Ig IRDye 800CW Infrared Dye | Li-Cor | 926-32211; RRID: AB_621843 |
| Anti-Phospho-KAP1 | Bethyl | S824 IHC 00073; RRID: AB_577234 |
| Anti-RAD51 | Millipore Calbiochem | PC130; RRID: AB_2238184 |
| Anti-RPA32 | Bethyl | A300-244A; RRID: AB_185548 |
| Anti-BrdU | Abcam | clone BU1/75-ICR, Ab6326; RRID: AB_305426 |
| Cy5 goat anti rat Ig | Abcam | AB6565; RRID: AB_955063 |
| Cy3.5 goat anti mouse Ig | Abcam | AB6946; RRID: AB_955045 |
| Anti-ssDNA antibodies | Merck Millipore | MAB3034; RRID: AB_94645 |
| Anti-P-H3 | Millipore | 06-570; RRID: AB_310177 |
| Anti-KU70 | Thermo Scientific Pierce | MA5-13110; RRID: AB_10976973 |
| Anti-Phospho-NBS1 | Cell Signaling | 3001S; RRID: AB_10829154 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Triton X-100 | Sigma | 101958312 |
| 4’-6-diamidino-2-phenylindole dihydrochloride | Thermo Scientific | 62248 |
| Phleomycin | InvivoGen | 11006-33-0 |
| MMC | Sigma | M0503 |
| Etoposide | Sigma | E1383 |
| IdU | Sigma | I-7125 |
| CldU | Sigma | C-6891 |
| Fluorsave | Calbiochem | 345789 |
| Bradford | Bio-Rad | 500-0006 |
| Protease Inhibitor | Roche | 4693159001 |
| Phosphatase inhibitor #1 | Sigma | P0044-5ML |
| Phosphatase inhibitor #2 | Sigma | P5726-5ML |
| PBS | GIBCO | 70011-36 |
| PFA | Sigma | 1002491639 |
| Glycine | Euromedex | 26-1286405C |
| BSA | Sigma | A2153-100G |
| Trizol | Ambion | 15596026 |
| Tris | Invitrogen | 15504-020 |
| EDTA | Invitrogen | 15575-038 |
| Blocking buffer (TBS) | Li-Cor | 927-60001 |
| Tween-20 | MP Biochemicals | 103168 |
| Ethanol | VWR | 20821.330 |
| IP | Sigma | P4864 |
| Protéinase K | Invitrogen | P100005393 |
| β-Agarase | New England Biolabs | M0392S |
| BrdU | Becton Dickinson | 550891 |
| Critical Commercial Assays | ||
| jetPRIME | PolyPlus Transfection | 114-07 |
| FluorSave | Calbiochem | 345789 |
| SureSelect Human All Exon Kit | Agilent Technologies | V5 |
| SuperScript First-Strand Synthesis Kit | Invitrogen | 18064-014 |
| QuikChange site-directed mutagenesis kit | Agilent Technologies | 200521 |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda 9 (Sf9) cells | Cejka lab | N/A |
| HT1080 | Revy Lab | N/A |
| HT1080 RAD50+/− | This paper | N/A |
| HT1080 RAD50-/del669-672 | This paper | N/A |
| Primary fibroblast from healthy donor | This paper | N/A |
| Primary fibroblast from patient P1 | This paper | N/A |
| SV40-transformed hTERT-immortalized fibroblast from healthy donor | This paper | N/A |
| SV40-transformed hTERT-immortalized fibroblast from patient P1 | This paper | N/A |
| SV40-transformed hTERT-immortalized fibroblast from healthy donor transduced with pLVX-IRES-mCherry | This paper | N/A |
| SV40-transformed hTERT-immortalized fibroblast from patient P1 transduced with pLVX-IRES-mCherry | This paper | N/A |
| SV40-transformed hTERT-immortalized fibroblast from healthy donor transduced with pLVX-wtRAD50-IRES-mCherry | This paper | N/A |
| SV40-transformed hTERT-immortalized fibroblast from patient P1 transduced with pLVX- wtRAD50-IRES-mCherry | This paper | N/A |
| All S.cerevisiae strains used in this study are listed in Table S2 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| All S.cerevisiae strains used in this study are listed in Table S2 | This paper | N/A |
| Oligonucleotides | ||
| codon-optimized oligonucleotides for recombinant protein: RAD50_ΔE1035_F (5′- AACGAGGAGCT GAAGGAGGTGGAGGAGCGAAAGCAGCACCTG-3′ |
This paper | N/A |
| Recombinant DNA | ||
| PGK-H2BmCherry | Addgene | #21217 |
| pX330-LMNA-gRNA1 | Addgene | #122507 |
| pCR2.1 Clover-LMNA donor | Addgene | #122508 |
| pLVX-EF1alpha-IRES-mCherry | Takara Bio | 631987 |
| pLVX-EF1a-wtRAD50-IRES-mCherry | This paper | N/A |
| pX330-hRAD50-gRNA | This paper | N/A |
| pEGFPN1-NBS1 | Lukas lab | N/A |
| pEGFP-NLS | Zhang et al., 2016 | N/A |
| pLV-hTERT-IRES-hygro | Addgene | #85140 |
| Software and Algorithms | ||
| FlowJo | FlowJo | V10.1 |
| GraphPad Prism version 8 | GraphPad | V6.0 |
| FiberStudio® | Genomic Vision | V2.0 |
| Marcoil program | Delorenzi and Speed, 2002 | https://waggawagga.motorprotein.de/ |
| ImageJ | ImageJ | https://imageJimagej.net/Welcome |
| Other | ||
| FiberVision® scanner | Genomic Vision | SCN-002 |
| FiberComb® Molecular Combing System | Genomic Vision | MCS-001 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Patrick Revy (patrick.revy@inserm.fr).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
All used software is listed in the Key Resources Table. This study did not generate any unique datasets or new code.
Experimental Model and Subject Details
Human fibroblasts used in this study were obtained from skin biopsies from pediatric healthy donors (3 years of age) and patients. When indicated transformed (with large T antigen from SV40T) and immortalized (after transduction with a hTERT-expressing vector, Addgene #85140) fibroblasts were used. All cell lines were from male individuals and were checked for mycoplasma contamination. Informed and written consent was obtained from donors and patients. The study and protocols comply with the 1975 Declaration of Helsinki as well as with the local legislation and ethical guidelines from the Comité de Protection des Personnes de l’Ile de France II and the French advisory committee on data processing in medical research.
Saccharomyces cerevisiae strains used in this paper are listed in Table S2. Yeast strains were grown in liquid cultures and on plates in YPD media supplemented with 100 mg/L adenine. Further specifications are mentioned within the Method Details section.
Method Details
Study approval
Informed and written consent was obtained from donors and patients. The study and protocols comply with the 1975 Declaration of Helsinki as well as with the local legislation and ethical guidelines from the Comité de Protection des Personnes de l’Ile de France II and the French advisory committee on data processing in medical research.
Cells
Control fibroblasts were obtained from skin biopsies from pediatric healthy donors (3 years of age). When indicated transformed (with large T antigen from SV40T) and immortalized (after transduction with a hTERT-expressing vector, Addgene #85140) fibroblasts were used. All cell lines were checked for mycoplasma contamination.
Yeast
Yeast strains were generated by integration of the rad50-coiled coil mutants at their native chromosomal locus in a diploid WT strain. rad50 mutant spores were obtained by tetrad dissection and verified by PCR genotyping and sequencing using genomic DNA. Details of yeast strains and plasmid constructions are available upon request. All strains used in this study are listed in Table S2. Yeast strains were grown in liquid cultures and on plates in YPD media supplemented with 100 mg/L adenine. 5-fold serial cell dilutions (250,000 cells per spot to 80 cells per spot) were spotted on plates without or with S-phase clastogens and plates were incubated for 3 days (30°C) or 2 days (37°C). Mre11 complex co-immunoprecipitation were done as described previously (Hohl et al., 2011). Telomere lengths were assessed following 40 generation of growth using PstI-digested genomic DNA by Southern blot using a telomere specific probe as previously described (Hohl et al., 2011).
Homology directed repair assay
Cells were plated in 6 well plates and transfected in triplicate with jetPRIME (Polyplus transfection) according to the manufacturer’s instructions. 1 μg of mCherry vector (PGK-H2BmCherry, Addgene #21217), 4 μg of CRISPR/cas9 vector (pX330-LMNA-gRNA1, Addgene #122507) and 1 μg of donor vector (pCR2.1 Clover-LMNA donor, Addgene #122508) were used. The percent of clover-positive cells among the transfected cells (inferred by mCherry staining) was analyzed by flow cytometry using a LSR-Fortessa FACS (BD Biosciences) and results were ploted using the FlowJo software. Statistical significance was determined using the Student’s t test.
Automated count of 53BP1 foci
Early passaged primary fibroblasts were cultured on coverslips for 24h. Cells were washed with PBS, fixed with 4% paraformaldehyde for 15 minutes and incubated for 20 minutes with PBS/0.1 mol/L glycine. Cells were then permeabilized in 0.5% Triton X-100 in PBS for 15 minutes. After each step, coverslips were rinsed 3 times with PBS. Thereafter, cells were incubated for 30 minutes with PBS-BSA 1% and labeled (45 minutes) with primary antibody (Anti-53BP1; Novus Biological; NB100-304). Cells were washed with PBS/BSA 1% solution and incubated (30 minutes) with secondary antibody (Alexa Fluor 488 goat F[Ab′]2; Molecular Probes). Slides were stained for 5 minutes with 0.1 μg/mL 4′-6-diamidino-2-phenylindole dihydrochloride and mounted in FluorSave (Calbiochem). Slides were analyzed by using the FiberVision® scanner (Genomic Vision). Nucleus and foci were automatically detected and visualized in the FiberStudio® (Genomic Vision) software. Statistical significance was determined using the Student’s t test.
Whole exome sequencing
Exome capture was performed using the SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA). Agilent SureSelect Human All Exon (54 Mb, Clinical research Exome) libraries were prepared from 3 μg of genomic DNA sheared with an Ultrasonicator (Covaris, Woburn, MA) as recommended by the manufacturer. Barcoded exome libraries were pooled and sequenced using a HiSeq2500 (Illumina, San Diego, CA) generating 130 × 130 paired-end reads. After demultiplexing, sequences were mapped on the human genome reference (NCBI build37/hg19 version) with BWA. The mean depth of coverage obtained from the exome library was 138X with > 99% of the targeted exonic bases covered by at least 15 independent reads and > 97% by at least 30 independent sequencing reads (> 99% at 15 × and > 97% at 30 × ). Variant calling was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard Tools. Single nucleotide variants were called with GATK Unified Genotyper, whereas indel calls were made with the GATK IndelGenotyper_v2. All variants with a read coverage ≤ 2 × and a Phred-scaled quality of ≤ 20 were filtered out. All the variants were annotated and filtered using Polyweb, an in-house developed annotation software.
Constructs and cDNA analysis
Total RNA from patient and control fibroblasts or B-LCL was extracted using TRIzol reagent (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions. Reverse transcription was performed using a SuperScript First-Strand Synthesis Kit (Invitrogen). Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. The initiation codon is codon 1. RAD50 ORF was PCR-amplified from cDNA and cloned into a pLVX-EF1α-mCherry-lentiviral vector (Invitrogen) for complementation experiments.
Coiled-coil probability
Marcoil program (https://waggawagga.motorprotein.de/) (Delorenzi and Speed, 2002; Simm et al., 2015) was used to predict coiled-coil probability.
Complementation assay
For complementation of phleomycin sensitivity, patients’ SV40-transformed fibroblasts were transduced with a wtRAD50-IRES–mCherry expressing lentivirus vector (pLVX-EF1alpha-IRES-mCherry Vector, Takara Bio) or empty mCherry-expressing vector. Cells were cell sorted thanks to mCherry expression to obtain 100% of transduced cells. Sensitivity assays were then conducted as described above.
Selective advantage assay
Selective advantage was performed through multicolor competition assay as previously described (Smogorzewska et al., 2007; Zhang et al., 2016). Fibroblasts were transduced with a RAD50-IRES–mCherry expressing lentivirus vector (pLVX-EF1alpha-IRES-mCherry Vector, Takara Bio) or mCherry-expressing vector and treated or not with 100 ng/ml phleomycin for up to 18 days. The number of mCherry-positive cells was scored at various time points to assess for relative selective advantage of transduced (mCherry+) over untransduced (mCherry-) cells.
Western blotting
Cells were lysed for 20 min on ice in lysis buffer containing 50 mM Tris (pH 8.0), 2 mM EDTA, 1% Triton X-100, 1% phosphatase inhibitor cocktails (Sigma), and protease inhibitors (Roche). After centrifugation, the supernatant was harvested and protein concentration was quantified with the Bradford assay. After SDS–PAGE, proteins were transferred to PVDF Immobilon-P membrane (Millipore). Then, the membrane was incubated for 1h in Odyssey blocking buffer (TBS), followed by incubation with the appropriate antibody, then washed and incubated with goat anti-rabbit secondary antibody (Li-Cor IRDye 800CW Infrared Dye, 1:10,000 dilution). The presence of proteins was detected by infrared fluorescence according to the manufacturer’s protocol (Odyssey CLx Imaging System). The blot was then incubated with anti-vinculin (Santa Cruz, SC73614; 1:1,000) as loading control. The antibodies used for WB analysis were anti-MRE11 (Novus 12D7, NB100-473; 1:500), anti-RAD50 (Genetex 13B3, GTX 70228; 1:500), anti-NBS1 (Novus, NB100-143E1; 1:500), anti-CHK2 (Santa Cruz, SC17748; 1:1,000).
Immunofluorescence
For P-KAP1, RPA32 and RAD51 detection, cells were cultured on coverslips 24h before experiments and treated with 10 Gy of IR (137Cs) with a 6 hr incubation post IR. Cells were pre-permeabilized with PBS/0.5% Triton X-100 on ice for 5 minutes and fixed with 2% paraformaldehyde for 15 minutes at RT under agitation. Cells were then permeabilized in PBS/0.5% Triton X-100 for 5 minutes under agitation. Thereafter, cells were incubated for 30 minutes with PBS/BSA 2%/Tween 0.05% with agitation and labeled 1h in humid chamber at 37°C with primary antibodies (anti-P-KAP1, Bethyl, S824 IHC 00073, 1/200; anti-RAD51, Millipore Calbiochem, PC130, 1/200; anti-RPA32, Bethyl, A300-244A, 1/200). Cells were then washed 10 minutes 3 times with PBS/Tween 0.05% solution under agitation and incubated 45 minutes in humid chamber at 37°C with secondary antibody (Alexa Fluor 488 goat F[Ab′]2; Molecular Probes, 1/1000). Slides were washed 10 minutes 3 times under agitation with PBS/Tween 0.05%, stained for 5 minutes with 0.1 μg/mL 4′-6-diamidino-2-phenylindole dihydrochloride and mounted in FluorSave (Calbiochem). Slides were analyzed by using epifluorescence microscopy (Axioplan; Zeiss, Oberkochen, Germany). Images were processed for quantification with ImageJ software.
G2/M checkpoint analysis
3.105 immortalized SV40-transformed fibroblasts were harvested 1 hr after 5 Gy X-irradiation, washed with PBS, fixed with cold 70% ethanol, and kept at –20°C for 16 hr. After washing, the cells were resuspended in PBS 0.25% with Triton X-100 and incubated on ice for 15 min. Cells were washed in PBS and incubated with 1 mg of anti-phospho-H3 in PBS-1% BSA for 3 hr. Anti-P-H3 was detected with FITC-conjugated F(Ab’)2 goat anti-rabbit IgG antibody. The cells were washed and resuspended in PBS containing 25 mg/ml PI. Fluorescence was measured by using a Becton Dickinson FACScan flowcytometer.
DNA repair analysis
Cells were plated at 25,000 per well in 48 wells plates and imaged for 7 days using the Incucyte Live-Cell Analysis System upon exposure to radiation (X-ray) or increased doses of phleomycin (invivoGen, 11006-33-0), MMC (Sigma, M0503) or etoposide (Sigma, E1383).
DNA replication analysis
Cells were sequentially pulse labeled with 25 μM CldU (Sigma, C-6891) and 25 μM IdU (Sigma, I-7125) for 30 min (or 15 min when indicated) as previously described by Lebofsky and Bensimon (2005). Cells were harvested and embedded in low-melting agarose plugs in which DNA was subjected to deproteinization by Proteinase K treatment. Agarose was then removed by digestion with Agarase and the high molecular DNA yielded was used for combing as previously described by Michalet et al. (1997) by using the FiberComb Molecular Combing System (Genomic Vision). CldU and IdU was respectively stained with rat anti-BrdU antibody (clone BU1/75-ICR, Ab6326) and mouse anti-BrdU (BD Biosciences 347580), followed by staining with Cy5 goat anti rat Ig (AB6565) and Cy3.5 goat anti mouse Ig (AB6946). DNA fibers were counterstained with anti-ssDNA antibodies (Merck Millipore MAB3034) to distinguish fork pausing/stalling from fiber breakage. DNA fibers were visualized using the FiberVision scanner (Genomic Vision). Data analysis was performed as described by Rimmelé et al. (2010).
Stalled fork resection
Cells were sequentially pulse labeled with 25 μM CldU (Sigma, C-6891) and 25 μM IdU (Sigma, I-7125) for 15min and treated with HU (4mM) for 3h. Forks were then detected as described in the “DNA replication analysis” section. The ratio between CldU and IdU track lengths was calculated and statistical significance was determined using the Student’s t test.
Laser microirradiation
NBS1-EGFP plasmids were provided by C. Lukas (University of Copenhagen, Copenhagen, Denmark). SV40-transformed fibroblasts were transfected with NBS1-GFP or NLS-GFP constructs using Jet Prime reagent (Polyplus transfection). 24 h after transfection, cells were presensitized with BrdU (BD) for another 28 h. Microirradiation was performed using a 405-nm UV laser focused through a 63 × objective lens in a confocal microscope system (TCS sp8; Leica Biosystems). Laser settings (100% laser power for 10% of laser duration) were used to generate DNA damage specifically at the laser path (dot) in a BrdU-dependent manner. The cells were analyzed in time course mode with a 0.329 s interval between pictures for up to 215 pictures. The interval between pictures was then shifted to 30 s for another 15 pictures. Images were exported as LIF files and analyzed using ImageJ.
Cloning, expression and purification of recombinant proteins
Recombinant MR wild-type, MBP-NBS1-his and phosphorylated CtIP were expressed in insect Spodoptera frugiperda 9 (Sf9) cells and purified by affinity and ion exchange chromatography as described previously (Anand et al., 2016, 2018, 2019). The RAD50E1035Δ mutant was prepared by mutating the respective codon-optimized pFastBac plasmid by QuikChange site-directed mutagenesis kit following manufacturer’s instructions (Agilent Technology) and using codon-optimized oligonucleotides RAD50_ΔE1035_F (5′- AACGAGGAGCTGAAGGAGGTGGAGGAGCGAAAGCAGCACCTG-3′) and RAD50_ΔE1035_R (5′- CAGGTGCTGCTTTCGCTCCTCCACCTCCTTCAGCTCCTCGTT-3′) Bacmids, primary and secondary baculoviruses were prepared using standard procedures according to manufacturer’s instructions (Bac-to-Bac, Life Technologies). MRE11-RAD50(E1035Δ) heterodimer was purified as described for MR wild-type (Anand et al., 2018). Where indicated, 1 μg of pCtIP was dephosphorylated in 20 μL reactions with 200 units of λ phosphatase (λ-PP, New England Biolabs) in 1x PMP buffer (New England Biolabs) supplemented with 1 mM magnesium chloride for 15 min at 30°C. For “mock” control, λ-PP was excluded from the reaction.
Preparation of oligonucleotide-based substrate
All oligonucleotides were purified by polyacrylamide gel electrophoresis and purchased from Eurogentec. The labeling of oligonucleotides at the 5′ end was carried out by T4 polynucleotide kinase (New England Biolabs) and [γ-32P] ATP (Perkin Elmer). To prepare quadruple blocked 70-bp long DNA substrate, PC210 and PC211 oligonucleotides were used, as described previously (Cannavo and Cejka, 2014). To prepare 50-bp long dsDNA substrate, X12-3 was labeled at 5′ end and annealed with X12-4C (Cejka and Kowalczykowski, 2010).
Nuclease assay
Endonuclease assays (15 μL volume) were performed in nuclease buffer containing 25 mM Tris-HCl pH 7.5, 5 mM magnesium acetate, 1 mM manganese acetate, 1 mM dithiothreitol (DTT), 1 mM ATP, 0.25 mg/mL BSA (New England Biolabs), 1 mM phosphoenolpyruvate (Sigma), 80 U/mL pyruvate kinase (Sigma), and 1 nM oligonucleotide-based DNA substrate (in molecules). The reactions were supplemented with 15 nM streptavidin (Sigma), and incubated for 5 min at room temperature to block the biotinylated ends of the DNA substrates. The recombinant proteins were then added to the reactions on ice and samples were incubated at 37°C for 30 min. Reactions were stopped by adding 0.5 μL ethylenediaminetetraacetic (0.5 M EDTA) and 1 μL Proteinase K (19 mg/mL, Roche), and incubated at 50°C for 30 min. Finally, 16.5 μL loading buffer (5% formamide, 20 mM EDTA, bromophenol blue) was added to all samples and the products were separated on 15% polyacrylamide denaturing urea gels (19:1 acrylamide-bisacrylamide, Bio-Rad), as described (Pinto et al., 2018). The gels were fixed in fixing solution (40% methanol, 10% acetic acid, 5% glycerol) for 30 min at room temperature and dried. The dried gels were exposed to storage phosphor screen (GE Healthcare) and scanned by a Typhoon Phosphor Imager (FLA 9500, GE Healthcare). For salt titration experiment, NaCl was added as indicated before starting the incubation at 37°C. Exonuclease assays with 50-bp long dsDNA substrate were carried out similarly except the reaction buffer does not contained streptavidin and incubation time was extended to 1 h.
ATPase assay
The ATPase assays were performed in 25 mM Tris-HCl (pH 7.5), 5 mM magnesium acetate, 1 mM DTT, 0.1 mg/ml BSA (New England Biolabs), 80 mM NaCl, 8% glycerol, 50 μM ATP, 1 nM of [γ-32P] adenosine 5ꞌ-triphosphate (Perkin Elmer) and 1 nM (in molecules) of the pUC19 plasmid linearized with EcoRI restriction endonuclease (NEB). Recombinant proteins were added on ice and samples were incubated at 37°C for 2 hours. Reactions were stopped with 1.1 μL of 0.5 M EDTA and separated using TLC plates (Merk) and 0.3 M LiCl and 0.3 M formic acid as mobile phase. Dried plates were exposed to storage phosphor screens (GE Healthcare) and scanned by a Typhoon 9500 phosphorimager (GE Healthcare).
Electrophoretic mobility shift assays
The reactions (15 μL volume) were performed in binding buffer containing 25 mM Tris-HCl pH 7.5, 5 mM magnesium acetate, 2 mM manganese acetate, 1 mM DTT, 1 mM ATP, 0.25 mg/ml BSA (New England Biolabs) and 1 nM DNA substrate (in molecules). The reactions were incubated on ice for 15 min. Loading dye (50% glycerol, bromophenol blue) was added and the products were separated by 0.6% agarose gel electrophoresis in Tris-Acetate-EDTA (TAE) buffer. The electrophoresis was carried out in the cold room at 4°C. The gels were squeeze for 15 min and dried on DE81 chromatography paper (Whatman, UK). The dried gels were then exposed to Storage Phosphor screens (GE Healthcare) and scanned by Typhoon Phosphor Imager (FLA 9500, GE Healthcare). When indicated, phosphorylated pCtIP was pretreated with λ phosphatase (λ-PP, New England Biolabs) as described above.
Quantification and Statistical Analysis
Statistical analyses were performed on Prism (GraphPad Software). Groups were analyzed by Student t test or Mann-Whitney test as indicated in figure legends and text, and the difference was considered statistically significant for p < 0.05. All of the statistical details of experiments can be found in the figure legends.
Acknowledgments
We thank the patient and his family for their generous assistance with samples and information, which made this research possible. We acknowledge M. Garfa Traore and the imaging facility of the Imagine Institute for help with microlaser irradiation, the Bioinformatics Department of the Imagine Institute for help with whole-exome sequencing, N. Lambert (CEDI, AP-HP, Necker Hospital, Paris, France) for microsatellite analysis, and A. Fernandes from the Centre de Ressource Biologique of the Imagine Institute, who generated B-LCL. We thank Dr. B. Lopez for the RAD50 construct used as a matrix and technical advice for RAD51 and the RPA immunofluorescence study. We thank Dr. G. Dellaire for the HDR assay components (Pinder et al., 2015). Dr. P. Pasero is acknowledged for his advice with RCAs. P.R. thanks Dr. A. Decottignies, Dr. P. Kannouche, and Dr. E. Brunet for critical reading of the manuscript. Work performed in the GDIS lab was supported by institutional grants from INSERM, Ligue Nationale contre le Cancer (Equipe Labellisée “LIGUE 2020”), INCa (PLBIO19-027 and INCA_13766), and GIS-Institut des Maladies Rares and state funding from the Agence Nationale de la Recherche under the Investissements d’Avenir program (ANR-10-IAHU-01). This work was supported by GM56888 and MSK Cancer Center core grant P30 CA008748 (to J.H.J.P.) and the Swiss National Science Foundation (31003A_17544) and European Research Council grant 681-630 (to P.C.). M.M. is supported by the Ligue Nationale contre le Cancer (EL2028.LNCC/MaM) and the French National Cancer Institute (PLBIO2017-167). M.C.D.-C. benefited from scholarships from the Association Nationale de la Recherche et de la Technologie (ANRT) and La Ligue contre le Cancer. P.R. is a scientist from Centre National de la Recherche Scientifique (CNRS).
Author Contributions
M.C.D.-C. carried out most of the experimental work on human cells, assisted by L.K. and P.R. T.I. identified the affected patient and assisted with related clinical and laboratory studies. I.C. and P.C. produced the recombinant MRN complex and performed in vitro experiments. M.H. and J.H.J.P. conceived and performed experiments on yeast strains. P.R. conceived the project and wrote the manuscript, with editing contributions from J.-P.d.V., I.C., M.H., M.M., P.C., and J.H.J.P.
Declaration of Interests
The authors declare no competing interests.
Published: December 29, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108559.
Supplemental Information
References
- Adelman C.A., De S., Petrini J.H. Rad50 is dispensable for the maintenance and viability of postmitotic tissues. Mol. Cell. Biol. 2009;29:483–492. doi: 10.1128/MCB.01525-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R., Ranjha L., Cannavo E., Cejka P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell. 2016;64:940–950. doi: 10.1016/j.molcel.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Anand R., Pinto C., Cejka P. Methods to Study DNA End Resection I: Recombinant Protein Purification. Methods Enzymol. 2018;600:25–66. doi: 10.1016/bs.mie.2017.11.008. [DOI] [PubMed] [Google Scholar]
- Anand R., Jasrotia A., Bundschuh D., Howard S.M., Ranjha L., Stucki M., Cejka P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019;38:e101005. doi: 10.15252/embj.2018101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglana M., Apiou F., Bensimon A., Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell. 2003;114:385–394. doi: 10.1016/s0092-8674(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Bianco J.N., Poli J., Saksouk J., Bacal J., Silva M.J., Yoshida K., Lin Y.L., Tourrière H., Lengronne A., Pasero P. Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods. 2012;57:149–157. doi: 10.1016/j.ymeth.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Broderick R., Nieminuszczy J., Baddock H.T., Deshpande R., Gileadi O., Paull T.T., McHugh P.J., Niedzwiedz W. EXD2 promotes homologous recombination by facilitating DNA end resection. Nat. Cell Biol. 2016;18:271–280. doi: 10.1038/ncb3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H.E., Petermann E., Schultz N., Jemth A.S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buis J., Wu Y., Deng Y., Leddon J., Westfield G., Eckersdorff M., Sekiguchi J.M., Chang S., Ferguson D.O. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E., Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Cannavo E., Reginato G., Cejka P. Stepwise 5′ DNA end-specific resection of DNA breaks by the Mre11-Rad50-Xrs2 and Sae2 nuclease ensemble. Proc. Natl. Acad. Sci. USA. 2019;116:5505–5513. doi: 10.1073/pnas.1820157116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney J.P., Maser R.S., Olivares H., Davis E.M., Le Beau M., Yates J.R., 3rd, Hays L., Morgan W.F., Petrini J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Cejka P., Kowalczykowski S.C. The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J. Biol. Chem. 2010;285:8290–8301. doi: 10.1074/jbc.M109.083196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J.L., Kowalczykowski S.C. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–116. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Trujillo K.M., Van Komen S., Roh D.H., Krejci L., Lewis L.K., Resnick M.A., Sung P., Tomkinson A.E. Effect of amino acid substitutions in the rad50 ATP binding domain on DNA double strand break repair in yeast. J. Biol. Chem. 2005;280:2620–2627. doi: 10.1074/jbc.M410192200. [DOI] [PubMed] [Google Scholar]
- Coquel F., Neumayer C., Lin Y.L., Pasero P. SAMHD1 and the innate immune response to cytosolic DNA during DNA replication. Curr. Opin. Immunol. 2019;56:24–30. doi: 10.1016/j.coi.2018.09.017. [DOI] [PubMed] [Google Scholar]
- Courbet S., Gay S., Arnoult N., Wronka G., Anglana M., Brison O., Debatisse M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature. 2008;455:557–560. doi: 10.1038/nature07233. [DOI] [PubMed] [Google Scholar]
- de Jager M., van Noort J., van Gent D.C., Dekker C., Kanaar R., Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- de Jager M., Trujillo K.M., Sung P., Hopfner K.P., Carney J.P., Tainer J.A., Connelly J.C., Leach D.R., Kanaar R., Wyman C. Differential arrangements of conserved building blocks among homologs of the Rad50/Mre11 DNA repair protein complex. J. Mol. Biol. 2004;339:937–949. doi: 10.1016/j.jmb.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Delamarre A., Barthe A., de la Roche Saint-André C., Luciano P., Forey R., Padioleau I., Skrzypczak M., Ginalski K., Géli V., Pasero P. MRX Increases Chromatin Accessibility at Stalled Replication Forks to Promote Nascent DNA Resection and Cohesin Loading. Mol. Cell. 2020;77:395–410. doi: 10.1016/j.molcel.2019.10.029. [DOI] [PubMed] [Google Scholar]
- Delorenzi M., Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- Deshpande R.A., Williams G.J., Limbo O., Williams R.S., Kuhnlein J., Lee J.H., Classen S., Guenther G., Russell P., Tainer J.A., Paull T.T. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014;33:482–500. doi: 10.1002/embj.201386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande R.A., Lee J.H., Paull T.T. Rad50 ATPase activity is regulated by DNA ends and requires coordination of both active sites. Nucleic Acids Res. 2017;45:5255–5268. doi: 10.1093/nar/gkx173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M., Jakob B., Chen P., Kijas A.W., Becherel O.J., Gueven N., Birrell G., Lee J.H., Paull T.T., Lerenthal Y. ATM protein-dependent phosphorylation of Rad50 protein regulates DNA repair and cell cycle control. J. Biol. Chem. 2011;286:31542–31556. doi: 10.1074/jbc.M111.258152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glousker G., Touzot F., Revy P., Tzfati Y., Savage S.A. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br. J. Haematol. 2015;170:457–471. doi: 10.1111/bjh.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa N.N., Akagawa R., Yamasaki T., Hirota K., Sasa K., Natsume T., Kobayashi J., Sakuma T., Yamamoto T., Komatsu K. Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells. 2015;20:1059–1076. doi: 10.1111/gtc.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa N.N., Shimizu T., Zhou Z.W., Wang Z.Q., Deshpande R.A., Paull T.T., Akter S., Tsuda M., Furuta R., Tsutsui K. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell. 2016;64:580–592. doi: 10.1016/j.molcel.2016.10.011. [DOI] [PubMed] [Google Scholar]
- Hohl M., Kwon Y., Galván S.M., Xue X., Tous C., Aguilera A., Sung P., Petrini J.H. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat. Struct. Mol. Biol. 2011;18:1124–1131. doi: 10.1038/nsmb.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M., Kochańczyk T., Tous C., Aguilera A., Krężel A., Petrini J.H. Interdependence of the rad50 hook and globular domain functions. Mol. Cell. 2015;57:479–491. doi: 10.1016/j.molcel.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner K.P. ATP puts the brake on DNA double-strand break repair: a new study shows that ATP switches the Mre11-Rad50-Nbs1 repair factor between signaling and processing of DNA ends. BioEssays. 2014;36:1170–1178. doi: 10.1002/bies.201400102. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P., Tainer J.A. Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr. Opin. Struct. Biol. 2003;13:249–255. doi: 10.1016/s0959-440x(03)00037-x. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P., Karcher A., Shin D., Fairley C., Tainer J.A., Carney J.P. Mre11 and Rad50 from Pyrococcus furiosus: cloning and biochemical characterization reveal an evolutionarily conserved multiprotein machine. J. Bacteriol. 2000;182:6036–6041. doi: 10.1128/jb.182.21.6036-6041.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner K.P., Craig L., Moncalian G., Zinkel R.A., Usui T., Owen B.A., Karcher A., Henderson B., Bodmer J.L., McMurray C.T. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Jasin M., Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käshammer L., Saathoff J.-H., Lammens K., Gut F., Bartho J., Alt A., Kessler B., Hopfner K.-P. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell. 2019;76:382–394. doi: 10.1016/j.molcel.2019.07.035. [DOI] [PubMed] [Google Scholar]
- Lammens K., Bemeleit D.J., Möckel C., Clausing E., Schele A., Hartung S., Schiller C.B., Lucas M., Angermüller C., Söding J. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell. 2011;145:54–66. doi: 10.1016/j.cell.2011.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R., Bensimon A. DNA replication origin plasticity and perturbed fork progression in human inverted repeats. Mol. Cell. Biol. 2005;25:6789–6797. doi: 10.1128/MCB.25.15.6789-6797.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Mand M.R., Deshpande R.A., Kinoshita E., Yang S.H., Wyman C., Paull T.T. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the Mre11/Rad50/Nbs1 (MRN) complex. J. Biol. Chem. 2013;288:12840–12851. doi: 10.1074/jbc.M113.460378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H.S., Kim J.S., Park Y.B., Gwon G.H., Cho Y. Crystal structure of the Mre11-Rad50-ATPγS complex: understanding the interplay between Mre11 and Rad50. Genes Dev. 2011;25:1091–1104. doi: 10.1101/gad.2037811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M., Barlow J.H., Burgess R.C., Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Liu Y., Sung S., Kim Y., Li F., Gwon G., Jo A., Kim A.K., Kim T., Song O.K., Lee S.E., Cho Y. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J. 2016;35:743–758. doi: 10.15252/embj.201592462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalet X., Ekong R., Fougerousse F., Rousseaux S., Schurra C., Hornigold N., van Slegtenhorst M., Wolfe J., Povey S., Beckmann J.S., Bensimon A. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 1997;277:1518–1523. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]
- Mirzoeva O.K., Petrini J.H. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol. 2001;21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrani-Benhelli H., Gaillard L., Biasutto P., Le Guen T., Touzot F., Vasquez N., Komatsu J., Conseiller E., Pïcard C., Gluckman E. Primary microcephaly, impaired DNA replication, and genomic instability caused by compound heterozygous ATR mutations. Hum. Mutat. 2013;34:374–384. doi: 10.1002/humu.22245. [DOI] [PubMed] [Google Scholar]
- Myler L.R., Gallardo I.F., Soniat M.M., Deshpande R.A., Gonzalez X.B., Kim Y., Paull T.T., Finkelstein I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell. 2017;67:891–898. doi: 10.1016/j.molcel.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M. Diseases associated with defective responses to DNA damage. Cold Spring Harb. Perspect. Biol. 2012;4:a012773. doi: 10.1101/cshperspect.a012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.B., Hohl M., Padjasek M., Jeong E., Jin K.S., Krężel A., Petrini J.H., Cho Y. Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol. 2017;24:248–257. doi: 10.1038/nsmb.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P., Vindigni A. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu. Rev. Genet. 2017;51:477–499. doi: 10.1146/annurev-genet-120116-024745. [DOI] [PubMed] [Google Scholar]
- Paull T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell. 2018;71:419–427. doi: 10.1016/j.molcel.2018.06.033. [DOI] [PubMed] [Google Scholar]
- Paull T.T., Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinder J., Salsman J., Dellaire G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015;43:9379–9392. doi: 10.1093/nar/gkv993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C., Anand R., Cejka P. Methods to Study DNA End Resection II: Biochemical Reconstitution Assays. Methods Enzymol. 2018;600:67–106. doi: 10.1016/bs.mie.2017.11.009. [DOI] [PubMed] [Google Scholar]
- Ragamin A., Yigit G., Bousset K., Beleggia F., Verheijen F.W., de Wit M.Y., Strom T.M., Dörk T., Wollnik B., Mancini G.M.S. Human RAD50 deficiency: Confirmation of a distinctive phenotype. Am. J. Med. Genet. A. 2020;182:1378–1386. doi: 10.1002/ajmg.a.61570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmelé P., Komatsu J., Hupé P., Roulin C., Barillot E., Dutreix M., Conseiller E., Bensimon A., Moreau-Gachelin F., Guillouf C. Spi-1/PU.1 oncogene accelerates DNA replication fork elongation and promotes genetic instability in the absence of DNA breakage. Cancer Res. 2010;70:6757–6766. doi: 10.1158/0008-5472.CAN-09-4691. [DOI] [PubMed] [Google Scholar]
- Rivera-Munoz P., Malivert L., Derdouch S., Azerrad C., Abramowski V., Revy P., Villartay J.P. DNA repair and the immune system: From V(D)J recombination to aging lymphocytes. Eur. J. Immunol. 2007;37(Suppl 1):S71–S82. doi: 10.1002/eji.200737396. [DOI] [PubMed] [Google Scholar]
- Rojowska A., Lammens K., Seifert F.U., Direnberger C., Feldmann H., Hopfner K.P. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. EMBO J. 2014;33:2847–2859. doi: 10.15252/embj.201488889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurra C., Bensimon A. Combing genomic DNA for structural and functional studies. Methods Mol. Biol. 2009;464:71–90. doi: 10.1007/978-1-60327-461-6_5. [DOI] [PubMed] [Google Scholar]
- Scully R., Panday A., Elango R., Willis N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019;20:698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A., Moiani D., Arvai A.S., Perry J., Harding S.M., Genois M.M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y., Lederman H.M. Ataxia-telangiectasia (A-T): An emerging dimension of premature ageing. Ageing Res. Rev. 2017;33:76–88. doi: 10.1016/j.arr.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Simm D., Hatje K., Kollmar M. Waggawagga: comparative visualization of coiled-coil predictions and detection of stable single α-helices (SAH domains) Bioinformatics. 2015;31:767–769. doi: 10.1093/bioinformatics/btu700. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R., 3rd, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D’Andrea A.D., Elledge S.J. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G.S., Maser R.S., Stankovic T., Bressan D.A., Kaplan M.I., Jaspers N.G., Raams A., Byrd P.J., Petrini J.H., Taylor A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- Stracker T.H., Petrini J.H. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A., Tainer J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018;87:263–294. doi: 10.1146/annurev-biochem-062917-012415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebe H., Lim C.T., Konno H., Shiozaki K., Shinohara A., Uchihashi T., Furukohri A. Rad50 zinc hook functions as a constitutive dimerization module interchangeable with SMC hinge. Nat. Commun. 2020;11:370. doi: 10.1038/s41467-019-14025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M.R., Rothblum-Oviatt C., Ellis N.A., Hickson I.D., Meyer S., Crawford T.O., Smogorzewska A., Pietrucha B., Weemaes C., Stewart G.S. Chromosome instability syndromes. Nat. Rev. Dis. Primers. 2019;5:64. doi: 10.1038/s41572-019-0113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenz K., Smith E., Smith S., Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truebestein L., Leonard T.A. Coiled-coils: The long and short of it. BioEssays. 2016;38:903–916. doi: 10.1002/bies.201600062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T., Ogawa H., Petrini J.H. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol. Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- van Noort J., van Der Heijden T., de Jager M., Wyman C., Kanaar R., Dekker C. The coiled-coil of the human Rad50 DNA repair protein contains specific segments of increased flexibility. Proc. Natl. Acad. Sci. USA. 2003;100:7581–7586. doi: 10.1073/pnas.1330706100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R., Vissinga C., Platzer M., Cerosaletti K.M., Chrzanowska K.H., Saar K., Beckmann G., Seemanová E., Cooper P.R., Nowak N.J. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- Waltes R., Kalb R., Gatei M., Kijas A.W., Stumm M., Sobeck A., Wieland B., Varon R., Lerenthal Y., Lavin M.F. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009;84:605–616. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltzius J.J., Hohl M., Fleming J.C., Petrini J.H. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat. Struct. Mol. Biol. 2005;12:403–407. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- Yang Y.G., Frappart P.O., Frappart L., Wang Z.Q., Tong W.M. A novel function of DNA repair molecule Nbs1 in terminal differentiation of the lens fibre cells and cataractogenesis. DNA Repair (Amst.) 2006;5:885–893. doi: 10.1016/j.dnarep.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Zhang S., Pondarre C., Pennarun G., Labussiere-Wallet H., Vera G., France B., Chansel M., Rouvet I., Revy P., Lopez B. A nonsense mutation in the DNA repair factor Hebo causes mild bone marrow failure and microcephaly. J. Exp. Med. 2016;213:1011–1028. doi: 10.1084/jem.20151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Wiese C., Kwon Y., Hromas R., Sung P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019;88:221–245. doi: 10.1146/annurev-biochem-013118-111058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All used software is listed in the Key Resources Table. This study did not generate any unique datasets or new code.