

Abstract
Neurodevelopmental disorder with microcephaly, hypotonia and variable brain anomalies (NMIHBA) is an autosomal recessive neurodevelopmental and neurodegenerative disorder characterized by global developmental delay and severe intellectual disability. Microcephaly, progressive cortical atrophy, cerebellar hypoplasia and delayed myelination are neurological hallmarks in affected individuals. NMIHBA is caused by biallelic variants in PRUNE1 encoding prune exopolyphosphatase 1. We provide in-depth clinical description of two affected siblings harboring compound heterozygous variant alleles, c.383G > A (p.Arg128Gln), c.520G > T (p.Gly174*) in PRUNE1. To gain insights into disease biology, we biochemically characterized missense variants within the conserved N-terminal aspartic acid-histidine-histidine (DHH) motif and provide evidence that they result in the destabilization of protein structure and/or loss of exopolyphosphatase activity. Genetic ablation of Prune1 results in midgestational lethality in mice, associated with perturbations to embryonic growth and vascular development. Our findings suggest that NMIHBA results from hypomorphic variant alleles in humans and underscore the potential key role of PRUNE1 exopolyphoshatase activity in neurodevelopment.
Introduction
Prune exopolyphosphatase 1 (PRUNE1) belongs to the DHH (Asp-His-His) superfamily of proteins, which includes phosphoesterases, exopolyphosphatases and pyrophosphatases (1–3). Members of this superfamily are characterized by the DHH motif within the N-terminal domain that contains highly conserved charged residues required for binding metal cofactors and enzymatic function (4). PRUNE1 is ubiquitously expressed across multiple tissues with widespread central nervous system expression during fetal development in humans (5). Previous studies have implicated PRUNE1 in a complex network of interactions regulating cell cycle and motility, with elevated PRUNE1 expression associated with cancer metastasis (6–8).
Karaca et al. reported five individuals from four unrelated families segregating biallelic predicted pathogenic variants in PRUNE1 with a novel neurodevelopmental disorder characterized by microcephaly, hypotonia and variable brain anomalies (NMIHBA, MIM #617481) (9). The authors reported two different homozygous missense variants (p.D30N and p.D106N) in three consanguineous families and compound heterozygous (p.R128Q; p.G174*) variants in a non-consanguineous European ancestry family. Several groups have since identified other disease associated recessive alleles (p.P54T; p.R297W; c.521-2A > G: IVS4-2A > G) clustering throughout the conserved N-terminal domain (8,10,11). The observed incidence of NMIHBA in geographically distant families establishes this disorder as a pan-ethnic neurodevelopmental disorder. In addition to its nondescript developmental delay (DD)/intellectual disability (ID) nervous system hallmarks, NMIHBA patients may manifest more variable clinical features, including craniofacial anomalies (sloped forehead, large prominent ears, prominent eyes and narrow palate), scoliosis, joint contractures, muscle atrophy, hyperreflexia, profound global DD, fronto-temporal atrophy, cortical and cerebellar atrophy, seizures, and spastic quadriplegia.
Although the neurodevelopmental and neurobiological etiology of NMIHBA remains unclear, the severe clinical and developmental aspects of this disorder suggest an important role for PRUNE1 in fundamental cellular functions. Indeed, prior studies have implicated PRUNE1 in cell proliferation and migration, processes mediated in part through interactions with nucleoside diphosphate kinase 1 (NME1), glycogen synthase kinase-3β (GSK-3β) and β-tubulin respectively (4,8,12). Although in silico predictions of likely damaging effects of missense alleles classify them as likely pathogenic, the direction of effect (i.e. loss-of-function versus gain-of-function) and functional consequences of these variants have not been established. The observation of biallelic variants for a recessive disease trait, and the identification of putative loss of function splicing and nonsense variants support the notion of a reduced or loss of PRUNE1 function in NMIHBA pathophysiology. In agreement, patients harboring compound heterozygous mutations p.R128Q and p.G174* (described herein) manifest typical NMIHBA similar to those patients with homozygous missense variants spanning the conserved DHH (p.D30N, p.P54T, p.D106N and p.R128Q) and DHHA2 domains (p.R297W) (8–10). Heterozygous carriers of reported pathogenic missense and truncating variants in PRUNE1 do not appear to have any phenotypic manifestations in the spectrum of NMIHBA, indicating that PRUNE1 haploinsufficiency in heterozygous carrier parents is permissive for neurotypical development. To gain further neurodevelopmental and neurobiological insights into NMIHBA, we investigated the functional effects of PRUNE1 variant alleles and generated a knock-out mouse model of Prune1.
We undertook comprehensive clinical phenotyping of two siblings from one of the initially reported families (SZ51) with NMIHBA and summarize the phenotypic spectrum across all NMIHBA patients reported to date. Additionally, we provide experimental evidence that disease-associated missense variant alleles that map within the conserved N-terminal domain can result in destabilization of the protein structure and consequent loss of exopolyphosphatase activity. Furthermore, we demonstrate that homozygous null alleles in Prune1−/− mice results in multiple vascular anomalies including abnormal vascular network within the yolk sac and disrupted cephalic vascular plexus culminating in midgestational lethality (E9.5). Although embryonic lethality of Prune1 homozygous null mice precluded the molecular exploration of the neurodevelopmental phenotypes, our study provides a potential biochemical basis to NMIHBA pathophysiology and implicitly an avenue for therapeutic exploration.
Results
Clinical description of NMIHBA patients
Family SZ51 is composed of two affected siblings (Patient 1 and Patient 2), an unaffected female sibling and non-consanguineous parents of European ancestry (Fig. 1A). Familial genomic analyses of these two affected patients and available family members through whole exome sequencing identified, as previously reported, shared compound heterozygous variants in PRUNE1 (9). Both affected siblings inherited a missense c.383G > A; p.Arg128Gln variant from their unaffected father and a nonsense c.520G > T; p.Gly174* variant from their unaffected mother (Supplementary Material, Fig. S1). The unaffected sister is a heterozygous carrier for the paternal missense variant only.
Figure 1.
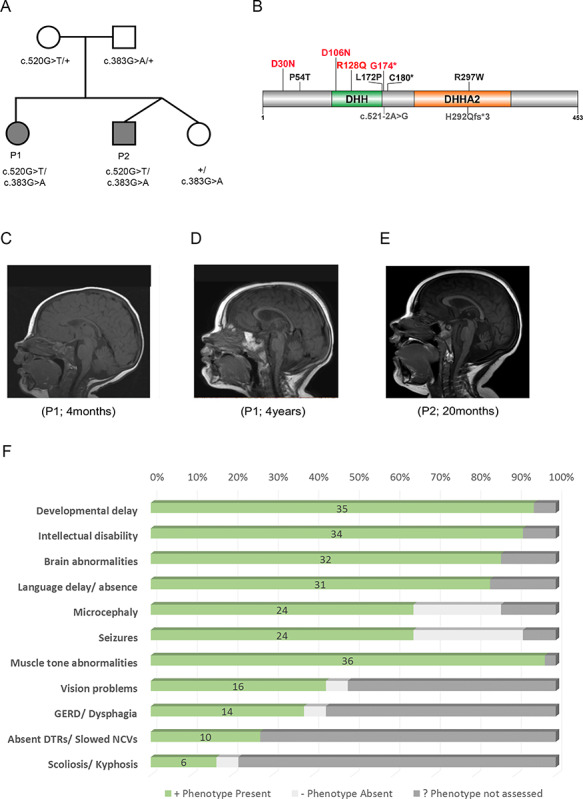
Clinical presentation of NMIHBA patients. (A) Immediate family pedigree and genotypes of patient 1 (P1) and patient 2 (P2). (B) Pathogenic variants in PRUNE1 identified in NMIHBA patients reported to date. The majority of pathogenic variants cluster in the DHH and DHHA2 domains. Variants characterized in this study are highlighted in red. (C−E) Sagittal T1-weighted brain MRI images. (C) Image of patient 1 at 4 months of age showing no significant findings. (D) Image of patient 1 at 4 years of age showing severe cortical and cerebellar atrophy and milder corpus callosum and brainstem atrophy. The craniofacial ratio is decreased. The cerebellar vermis decreased in height from 29.5 to 25.6 mm between the time the two images were obtained. (E) Image obtained for patient 2 at 20 months of age demonstrating cerebellar vermis hypoplasia and mild cortical atrophy. (F) Frequency of clinical manifestations ascertained in reported NMIHBA cases.
Detailed clinical evaluation of both affected siblings was performed through the NIH Undiagnosed Diseases Program (UDP) at ages 4 years and 20 months, respectively. Both patients presented with profound DD, ID, brain atrophy (Fig. 1C–E), seizures and absent language. Additional features not documented in the initial report include optic atrophy, esotropia, scoliosis, gastrointestinal reflux (GERD), hypotonia and spasticity. Onset of seizures in both siblings occurred at 6 months of age; it was observed initially as infantile spasms that developed into gelastic complex partial myoclonic seizures in Patient 1. EEG showed slow waking background and slow spike waves with progression to multifocal epileptiform discharges. Additional detailed clinical information for these patients can be found in supplementary information (including Supplementary Material, Tables S2 and S3).
Additionally, we performed detailed literature and clinical review of all 35 NMIHBA patients reported to date in order to better understand the phenotypic spectrum of NMIHBA (Table 1,Fig. 1B and F) (8–11,13–15). Consistent features across the majority of patients include severe global DD (94.59%), profound ID (91.89%) with absent language (83.78%) and brain abnormalities (86.48%) characterized primarily by cerebral and cerebellar atrophy, thin corpus callosum and white matter changes. Hypotonia and spasticity are features observed in the majority of patients (97.29%), whereas microcephaly and seizures (64.86%) appear to be more variable than initially reported, as more cases are documented. Visual (43.24%) and gastrointestinal (37.83%) problems are additional clinical features in a sizable number of patients. Interestingly, Alhaddad et al. reported about half of their cohort having absent deep tendon reflexes (DTRs) and/or reduced nerve conduction velocities (NCVs) suggesting peripheral neuropathy as part of the clinical spectrum of NMIHBA (13). Other patients also showed neurogenic findings on electromyography (EMG) studies and spinal motor neuron involvement. However, the majority of patients were not assessed for these clinical phenotypes and the true prevalence of peripheral nervous system involvement in this disorder may still be underappreciated. A few patients including probands described in this report developed scoliosis as well as foot anomalies such as talipes equinovarus or clubfoot, whether these features are secondary to the peripheral neuropathy and spinal muscular atrophy like phenotype or independent skeletal findings remains to be determined. Attempts at establishing a genotype–phenotype correlation for patients with NMIHBA were unsuccessful given that the clinical severity of the phenotype was variable even among individuals harboring the same variant allele (e.g. the recurrent D106N variant). No relationship between severity of the clinical presentation or presence/absence of specific clinical features with the type of mutation or zygosity of variants was evidently observed.
Table 1.
Summary of clinical findings of individuals with biallelic PRUNE1 mutations
| Patient 1 (SZ51-1) | Patient 2 (SZ51-2) | Karaca et al. (2015) | Zollo et al. (2017) | Costain et al. (2017) | Karakaya et al. (2017) | Iacomino et al. (2018) | Alfadhel et al. (2018) | Alhaddad et al. (2018) | Papuc (2019) | Fujii (2019) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at evaluation | 4 yo | 20 mo | 1.5−5.5 yo | 0.3−21.0 yo | 2.2 yo | 3 yo | 9 mo | 12 mo; 30 mo | 0 m−12 yo | 1.2 yo | 12 mo |
| Sex | F | M | 1 F, 2 M | 9 F; 4 M | M | M | M | 2 F | 10 F; 2 M | M | F |
| Ethnicity | European | European | 1 Saudi; 2 Turkish | Omani, Iranian, Italian and Indian | Ojibwe-Cree | Turkish | European (Italian) | Saudi | Lebanese; Turkish; European; North African | Sri Lankan | Japanese |
| Variant Zygosity | Comp Het | Comp Het | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous | Comp Het |
| Variants | c.383G > A; p.Arg128Gln |c.520G > T; p.Gly174* | c.383G > A; p.Arg128Gln |c.520G > T; p.Gly174* | c.88G > A; p.Asp30Asn, c.316G > A; p.Asp106Asn | c.88G > A; p.Asp30Asn, c.160C > A; p.Pro54Thr, c.316G > A; p.Asp106Asn, c.889C > T; p.Arg297Trp | c.521-2A > G | c.874_875insA; p.H292Qfs*3 | c.316G > A; p.Asp106Asn | c.383G > A; p.Arg128Gln | c.316G > A; p.Asp106Asn, c.515 T > C; p.Leu172Pro, g.150984457-151016662del; delExons 2-8 | c.316G > A; p.Asp106Asn | c.316G > A; p.Asp106Asn |c.540 T > A; p.Cys180* |
| Consanguinity | No | No | Yes | Yes | No | Yes | No | Yes | Yes | Yes | No |
| DD | +; profound | +; profound | +; profound (3/3) | +; (13/13) | +; profound | +; regression | + | +; profound (2/2) | + (10/12); 2 NA | + | + |
| ID | + | + | +; (3/3) | +; (13/13) | + | + | NA | +; (1/2) | + (11/12); 1 NA | +; severe | + |
| Brain abnormalities | + | + | +; (3/3) | +; (10/13) | + | + | + | +; (2/2) | + (10/12); 2 NA | + | + |
| Microcephaly | + | + | +; (3/3) | +; (13/13) | − | +; secondary | − | +; (2/2) | + (2/12); 5 Normal; 5 NA | + | − |
| Language delay | +; absent language | +; absent language | +; (3/3) | +; (13/13) | +; absent language | NA | NA | +; (1/2) | + (10/12); 2 NA | + | NA |
| Scoliosis/Kyphosis | + | + | NA | NA | NA | + | NA | − | +; (3/12) | NA | NA |
| Vision problems | +; optic atrophy, esotropia | +; congenital mild intermittent esotropia | NA | +; (5/13) | +; cortical blindness | +; optic atrophy | NA | +, bilateral rudimentary iris strands, congenital cataracts (1/2) | +; cortical blindness (2/12); saccadic eye movements (2/12); nystagmus (2/12) | NA | − |
| Muscle tone abnormalities | +; axial hypotonia, distal limb hypertonia/spasticity | +; increased limb tone, brisk tendon reflexes | +; central hypotonia, spastic quadriparesis, hyperreflexia (2/3) | +; (13/13) | +; neonatal generalized hypotonia, infantile spasms | +; neonatal generalized hypotonia, lower extremity spasticity and clonus | +; profound/severe global hypotonia, diffusely reduced tendon reflexes | +; central/axial hypotonia, spasticity, hyperreflexia (2/2) | +; hypotonia (5/12), spasticity (12/12), absent DTRs (5/12) | +; hypotonia, spastic tetraparesis | +; hypotonia, spastic quadriparesis, hyperreflexia |
| GERD/Dysphagia | +; severe GERD | + | NA | NA | NA | NA | NA | +; (1/2) | +; Dysphagia (11/12) | NA | NA |
| Seizures | + | + | NA | +; (6/13) | + | + | + | − | +; (11/12) | + | + |
| Seizure type and onset | Onset at 6mo; Gelastic, complex partial myoclonic seizures, infantile spasms | Onset at 6 mo; Infantile spasms | NA | NA | NA | Onset at 2 mo; focal motor seizures and flexor spasms | Onset at 6 mo; upper/lower limbs epileptic spasms | − | Onset 1 mo-8 mo; Clonic/myoclonic seizures (4/12) | Onset at. 6 mo; epileptic spasms | Onset at. 8 mo; epileptic spasms |
| Abnormal EEG findings | Background slowing, infrequent temporal spike waves | Modified hypsarrythmia, multifocal epileptiform discharges | NA | NA | Hypsarrhythmia | NA | Slowed background, multifocal epileptic abnormalities | Normal EEG (2/2) | Focal spasms (2/12); Slow multifocal spikes (2/12) | Focal tonic, myoclonic seizures | Hypsarrythmia |
| Abnormal MRI findings | Moderate/severe progressive global brain atrophy; cerebral and cerebellar atrophy | Mildly prominent lateral ventricles and sulci, thinner splenium and corpus callosum | Brain atrophy (3/3); Cortical atrophy, cerebral and cerebellar atrophy, thin corpus callosum, delayed myelination | Delayed myelination (5/13), wide spread white matter hypodensity or abnormalities (4/13), Cerebral/cerebellar atrophy (3/13), thin corpus callosum (2/13) | Cortical atrophy, small cerebellum; bilateral cerebral white matter loss with thinning of corpus callosum | Cerebral and cerebellar atrophy with delayed myelination, inferior vermis hypoplasia | Diffuse cortical atrophy, severe thinning of white matter, signal changes in periventricular white matter and pons, thin corpus callosum | Abnormal corpus callosum and mild cerebral atrophy (2/2); delayed mielination (1/2); prominent CSF spaces (1/2) | Cerebral atrophy (7/12), Cerebellar atrophy (6/12); Hyperintense brain lesions (4/12) | Immature cortex differentiation, punctate cerebellar hemorrhages, discrete signs of hypoxia | Diffuse cerebral and cerebellar atrophy, thin corpus callosum. Midbrain and pontine tegmentum atrophy. White matter damage, nut not delayed myelination |
| NCV/EMG findings | NA | NA | NA | NA | NA | Neurogenic findings | Neurogenic findings | NA | Slowed NCVs (5/12) | NA | NA |
| Measurements at evaluation | OFC: 41.5 cm (−5.3 s.d.); Weight: 15.1 kg (24th %ile); Length: 94 cm (−2.0 s.d.) | OFC: 41.0 cm (−5.3 s.d.); Weight: 12.8 kg (70th %ile); Length: 86 cm (77th %ile) | NA | NA | OFC: 49.5 cm (50th %ile); Weight: 16.5 kg (95th %ile); Length: 90 cm (90th %ile) | OFC:48 cm (−2 s.d.) | NA | OFC: −2.1 and −2.3 s.d.; Weight: −2.0 and −2.5 s.d.; Length: −2.1 and − 2.5 s.d. | OFC: −2.96 to +2 s.d.; Weight: −1.08 to +2.63 s.d. | OFC < 3rd %ile | OFC: 43.5 cm (−0.82 s.d.); Weight: 8.4 kg (−0.31 s.d.); Length: 73.8 cm (−0.26 s.d.) |
| Dysmorphic features | NA | NA | +; (2/3) | +; Phlagiocephaly (13/13) | +; tall forehead, bitempral narrowing, low set ears, flat nasal bridge, narrow high arched palate | +; Brachycephaly, large ears | +; Epicanthus, frontal plagiocephaly, brachydactyly, detached and hypoplastic nipples | +; (1/2) Plagiocephaly, epicanthal folds, hypertelorism, flat nasal bridge, abnormal dentition w/widely spaced teeth, micrognathia, low-set ears, hirsutism | +; (1/12): Micrognathia | Retrognathia | Phlagiocephaly |
| Other findings | Exaggerated startle and aversion to various sensory stimuli; Left hip dysplasia | Exaggerated startle; bilateral Babinski signs and sustained ankle clonus | NA | NA | Bilateral talipes equinovarus. Bilateral absent median nerve response. Central hypoventilation and sleep apnea. Bulbar palsy with absent swallow, gastrostomy | Regression. Knee contractures, hip dislocation; respiratory insufficiency (8 mo). Tracheostomy, nasogastric tube dependent | Distal joint arthrogryposis; respiratory distress. Babinksi sign observed. Bilatral talipes equinovarus, bell shaped thorax. Spinal motor neuron involvement | NA | Slow NCVs (5/12); Arthrogryposis/contractures (2/12); Hypertrophic cardiomyopathy (2/12); Pectus excavatum (1/12); Clubfoot (1/12); Death (8/12) | Bilateral deviation of 3rd toe over 4th toe | MR spectroscopy showed decreased concentration of N-acetyl aspartate and choline, and increased concentration of myoinositol |
| Previous Testing | Normal mitochondral respiratory chain activity testing | NA | NA | NA | Normal CMA, Beckwith–Wiedemann, Prader–Willi, SMARD1, DM1, and CCHS testing. Normal metabolic and biochemical testing | NA | Metabolic testing normal. Targeted molecular testing for SMN1, EXOSC3, EXOSC8 | Normal biochemical, metabolic, urine organic acids, and CMA testing (1/2) | Normal metabolic testing (6/12) | Normal CMA testing | NA |
MR: magnetic resonance
Affected subjects have been reported to originate from countries around the globe (Supplementary Material, Fig. S2). As expected, when homozygous alleles were identified, there was often a history of consanguinity. Of note, most homozygous subjects have missense variant alleles suggesting that NMIHBA results in the majority of cases from hypomorphic alleles. The likely loss of function null allele, c.G520T; p.G174* as a compound heterozygote with the missense c.383G > A; p.Arg128Gln variant in patients 1 and 2 is suggestive of partial function in these individuals. More recently, a homozygous splicing variant (11) and microdeletions spanning exon 2-8 in PRUNE1 (13) that possibly result in complete loss of PRUNE1 function have been reported. These patients have similar clinical presentations as those with homozygous missense variants. The neurotypical phenotype of heterozygous loss of function carrier parents with presumed haploinsufficiency at the PRUNE1 locus and the report of either homozygous or compound heterozygous null alleles suggests that NMIHBA results from partial or complete loss of PRUNE1 function.
Comparative modeling of PRUNE1 mutations
Similar to other members of the DHH superfamily, PRUNE1 contains a common N-terminal DHH domain resulting from four highly conserved motifs, with five invariant aspartates and two conserved histidine residues, one of which is replaced by Arginine (R128). One of the conserved aspartates is thought to act as nucleophile forming a charge-relay system with the conserved histidine, whereas the other aspartates bind divalent cations required for the catalytic activity (4). Interestingly, most of the pathogenic variants map to the highly conserved N-terminal DHH domain. In order to understand how NMIHBA-causing PRUNE1 variants drive the disease phenotype, we chose to characterize four variants (p.D30N, p.D106N, p.R128Q and p.G174*) within the conserved N-terminal domain for which we had patient-derived fibroblasts and/or comprehensive clinical information.
Importance of the D30, D106 and R128 residues to PRUNE1 function is revealed by considering the crystal structure of a homologous enzyme, Saccharomyces cerevisiae cytosolic exopolyphosphatase (PPX1) (PDB ID: 2QB7) (16). PPX1 is 25% identical and 45% similar to human PRUNE1 and has an equivalent enzymatic function (Supplementary Material, Fig. S3A) (4,17). In this structure, a series of divalent metal ions (M1 and M2) and bound phosphate ions (PT, PE1 and PE2) delineate the active site and define residues necessary for binding the polyphosphate substrate as well as the catalytic metal ions. Strikingly, all three of the missense mutations under investigation map to the same region of the PPX1 active site: PPX1 residues D41 and D127 (equivalent to PRUNE1 D30 and D106) coordinate a divalent metal ion, while PPX1 residue H149 (equivalent to PRUNE1 residue R128) forms a charge–charge interaction with the adjacent bound phosphate (Fig. 2A).
Figure 2.
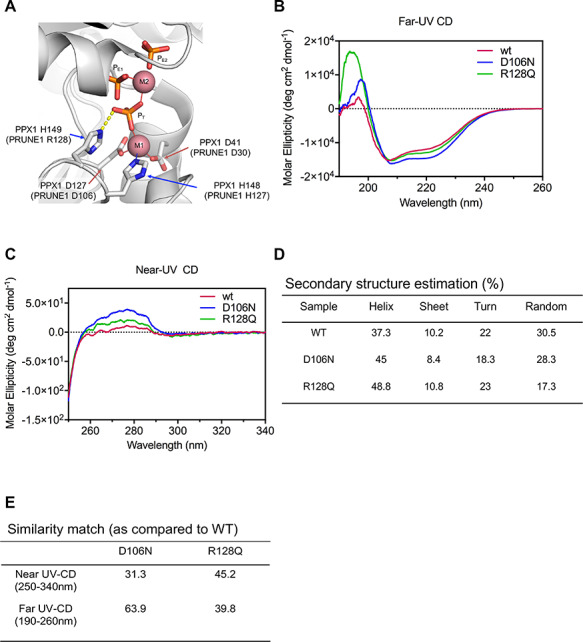
Missense mutations perturb secondary and tertiary structure of PRUNE1. (A) Homology modeling (based on 1.6 Å resolved S.c. PPX1 structure, 2QB7) demonstrated D30, D106 and R128 residues fall within a metal-ion and phosphate binding interface representing the active-site for phosphate hydrolysis. Disruption of these charged residues within the active-site impair metal coordination (D30N and D106N) and substrate binding R128Q. Three bound phosphate moieties PT, PE1 and PE2 shown in ball-and-stick, and the catalytic metal ions (M1 and M2) as pink spheres. (B) Far-UV CD spectra reveal secondary structure differences between wild-type and missense (D106N and R128Q) mutants. (C) Near-UV CD spectra signify change in the tertiary conformation of missense (D106N and R128Q) mutants as compared with the wild-type. (D) Spectral deconvolution of far-UV CD spectra revealed differences in alpha-helical content between wild-type and D106N or R128Q mutants. (E) Similarity match scores (based on quantitative assessment of similarity of far-UV and near-UV spectra) reveal low similarity of the missense mutants as compared with wild-type.
Substitution of Asn for Asp (as in D30N and D106N) will eliminate that amino acid’s ability to coordinate the divalent metal ion, thereby abolishing catalytic activity and destabilizing the local protein structure that was ordered by interactions with that metal ion. Similarly, substitution of Arg 128 by Gln eliminates the positive charge on that residue, likely preventing the mutated residue from interacting with the negatively charged phosphate group of a bound substrate.
To further verify these structural predictions, we performed circular dichroism (CD) on mammalian cell (Expi293) derived recombinant muteins harboring the D106N and R128Q variants. Whereas D106N and R128Q recombinant proteins expressed and migrated similarly to wild-type, rapid turnover of the D30N mutant precluded protein purification and further analyses (Fig. 3A; Supplementary Material, Fig. S3B and C). Far and near-UV CD spectra were collected for wild-type PRUNE1 and the two mutants (D106N and R128Q). Qualitative differences in the far and near-UV CD spectra were observed for each mutant compared with wild-type PRUNE1 (Fig. 2B and C). Spectral deconvolution of the far-UV CD revealed differences in alpha-helical content in the D106N and R128Q muteins (45 and 48.8%, respectively) as compared with wild-type (37.3%). Near-UV CD analyses revealed differences in tertiary structure between D106N and R128Q muteins as compared with wild-type. Furthermore, increased magnitude of CD signal particularly in the region contributed by Phe and Tyr residues indicated change in overall tertiary structure. The increase in CD signal possibly resulted from a more rigid conformation of the aromatic sidechains induced by the D106N and R128Q mutations. Lastly, quantitative assessment of spectral similarity (assessed by TQ similarity match scores) of the far and near-UV CD between the wild-type and muteins demonstrated a low similarity between the spectra of the mutants compared with that of wild-type (Fig. 2D and E). Therefore, our preliminary in silico and protein biochemistry analyses predicted that these three missense mutations could result in loss of protein function, by abolishing substrate binding and catalytic activity or by destabilizing the structure of the active site.
Figure 3.
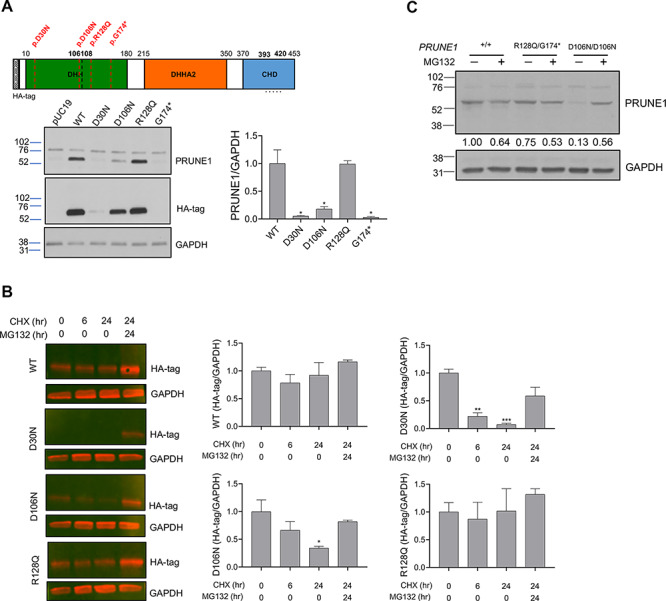
D30N and D106N variants result in reduced protein stability and proteosomal degradation, whereas R128Q variant results in stable mutant protein. (A) HEK293 cells were transfected with N-terminal HA-tagged wild-type or mutant PRUNE1 cDNA. Equal amounts of protein from whole cell lysate were used for immunoblotting using antibodies against N-terminal HA-tag, and C-terminal PRUNE1 epitope (a.a. 393-420, dotted line). GAPDH was used as a loading control. G174*mutant showed no expression, whereas D30N and D106N mutants showed significantly reduced expression as compared with wild-type and R128Q levels. (B) Immunoblots of cells overexpressing N-terminal HA-tagged wild-type or mutant PRUNE1 treated with cycloheximide (CHX) at 200 μM for 0, 6 and 24 h (with or without proteasome inhibitor MG132 at 15 μM). Bar graphs in (A) and (B) represent densitometry analyses of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, error bars: SEM. (C) Immunoblotting of endogenous PRUNE1 in patient derived fibroblasts harboring compound heterozygous R128Q; G174* variants or homozygous D106N missense variant treated with or without MG132 (15 μM). Densitometry analysis of the representative immunoblot shown in numerical format.
Missense mutations result in loss of exopolyphosphatase activity
In order to evaluate protein stability, mammalian expression constructs encoding epitope (HA)-tagged wild-type or missense PRUNE1 muteins (D30N, D106N, R128Q and G174*) were transfected into HEK293 cells (Supplementary Material, Fig. S4A). HEK293 cells have no detectable endogenous PRUNE1 expression. At steady state, immunoblotting analyses of whole cell lysates demonstrated a significant reduction of PRUNE1 levels in cells expressing D30N or D106N variants in comparison to wild-type-tagged protein. Whereas lysates from cells expressing G174* showed no discernable protein product, the R128Q variant-expressing cells showed similar expression compared with cells expressing the wild-type protein (Fig. 3A). Degradation kinetics visualized by immunoblot assays following cycloheximide-mediated translational block demonstrated rapid and significant loss of D30N and D106N mutant proteins, which was rescued by the cell-permeable proteasome inhibitor, MG132 (Fig. 3B). In contrast, the R128Q variant and wild-type PRUNE1 levels were stable after synthesis, as they were observed at >80% of baseline (0 h levels) after 24 h of cycloheximide treatment (Fig. 3B). In subsequent experiments, we evaluated PRUNE1 protein levels in patient-derived skin fibroblasts harboring the compound heterozygous R128Q/G174* variants or homozygous D106N variants (Supplementary Material, Fig. S4B). PRUNE1 levels were comparable between fibroblasts harboring compound heterozygous variants and the wild-type allele. However, cells homozygous for the D106N variant showed dramatic reduction in PRUNE1 levels at steady state (Supplementary Material, Fig. S4C). Proteasome inhibition by MG132 rescued the loss of the mutant protein in these cells (Fig. 3C). Taken together, these studies suggested that the D30N and D106N variants lead to reduced protein stability and degradation by the proteasome pathway, whereas the R128Q variant results in a stable mutant protein.
PRUNE1 has been ascribed exopolyphosphatase and phosphodiesterase activity based on sequence homology to yeast exopolyphosphatase PPX1 and bacterial nuclease RecJ, respectively (4). In subsequent experiments, we assessed how missense variants in PRUNE1 influence its enzymatic function. GloSensor assays established no detectable cAMP or cGMP phosphodiesterase activity of wild-type or missense PRUNE1 variants as compared with PDE4A and PDE5A (Supplementary Material, Fig. S5A). However, consistent with the findings of Tammenkoski et al. (17), wild-type PRUNE1 catalyzed polyphosphate hydrolysis of short-chain polyphosphates P3 (kcat = 6.24 s−1; Km = 8.4 μM; Vmax = 0.06 μM/s) and P4 (kcat = 4.2 s−1; Km = 16.13 μM; Vmax = 0.04 μM/s) using Mg2+ as the metal cofactor (Fig. 4A and B) and demonstrated no expolyphosphatase against long-chain polyphosphates P45, P65 and P700 (Supplementary Material, Fig. S5C). D106N and R128Q variants completely abolished short-chain exopolyphosphatase activity compared with wild-type as assessed by BIOMOL® Green phosphate reagent using P3 (1–50 μM) and P4 (5–100 μM) as polyphosphate substrates (Fig. 4C). Similarly, Escherichia coli expressed D106N and R128Q variants had no detectable short-chain exopolyphosphatase activity as compared with wild-type (Supplementary Material, Fig. S5B).
Figure 4.
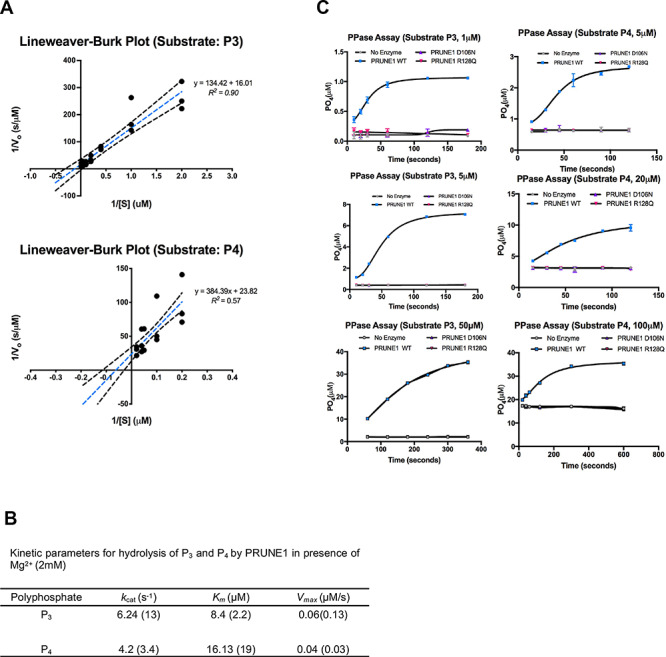
D106N and R128Q variants result in loss of short chain exopolyphosphatase activity. (A) Lineweaver−Burk plots depicting short-chain exopolyphosphatase kinetics of wild-type PRUNE1 using sodium tripolyphosphate (P3) and sodium tetrapolyphosphate (P4) as substrates. Dotted black lines represent 95% CI. (B) Kinetic parameters for hydrolysis of polyphosphates (P3 and P4) by wild-type PRUNE1 in the presence of Mg2+ (2 mM) as the cofactor. Values reported by Tammenkoski et al. are shown within parenthesis (17). (C) Short-chain exopolyphosphatase activity of wild-type, D106N and R128Q mutants on P3 and P4 determined using fixed-time BIOMOL Green phosphate detection assay. Data represented as mean ± SEM over three independent experiments with six technical replicates per sample.
Taken together, these results indicated that missense variants D30N, D106N and R128Q result in loss of PRUNE1 function either due to impaired protein stability or due to the loss of enzymatic function. These findings are consistent with the observed phenotypic concordance between patients harboring homozygous missense D30N, D106N variants and the compound heterozygous individuals (p.R128Q; p.G174*) described herein (patients 1 and 2). Importantly, these studies support the loss of PRUNE1 function as the molecular etiology of NMIHBA.
Loss of Prune1 results in embryonic lethality in mouse
In an effort to model NMIHBA in vivo, we next studied the consequences of Prune1 loss-of-function by generating Prune1−/− mice as shown in Supplementary Material, Figure S6A and B. Here, homologous recombination was employed to replace exon 2 with a lacZ reporter cassette. A stable transcript derived from this allele predicts a non-functional Prune1 protein missing amino acids 15-454. Importantly, this mutant allele provides for a lacZ reporter, enabling spatio-temporal localization of Prune1 expression. Prune1+/− animals (both sexes) are viable and appeared normal as compared with wild-type littermates, with no differences in body weight, body composition, serum chemistry and hematology (Supplementary Material, Table S4). LacZ expression profiling at E12.5 demonstrated widespread expression across multiple tissues including the brain (Supplementary Material, Fig. S6B). In adult Prune1+/− mice (postnatal day 56), strong CNS expression was observed in the hippocampus, cerebellum, amygdala, hypothalamus and the cortex (Supplementary Material, Fig. S6D).
No homozygous mutants were found among 55 pups at postnatal day 10 from Prune1+/− intercrosses (Supplementary Material, Fig. S6C). In light of this presumed embryonic lethality, embryos were collected at various developmental time points. Mendelian ratios were maintained at E9.5 (Supplementary Material, Fig. S6C); however, no Prune1−/− embryos were found beyond E12.5 suggesting lethality between E9.5 and E12.5. Whole mount examination of embryos and yolk sacs at E9.5 (~25 somites) employing endothelial-specific Pecam 1 immunochemistry revealed significant defects in vessel morphology in Prune1 mutants. First, Prune1−/− yolk sacs exhibit a profound decrease in capillary plexus formation and absence of differentiated vessels, indicative of perturbed vascular remodeling (Supplementary Material, Fig. S7B). Second, mutant embryos also show severe defects in the formation and remodeling of the cephalic vascular plexus, most strikingly revealed in 3D in Optical Projection Tomography (OPT). In addition, anomalies to the limb bud vasculature, dorsal vascular plexus (along the spinal cord) and marked cardiac hypoplasia were fully penetrant in all observed Prune1−/− embryos (Supplementary Material, Fig. S7A, Fig. 5A and B). The observed mid-gestational lethality is consistent with the phenotype reported by the International Mouse Phenotyping Consortium (https://www.mousephenotype.org/data/genes/MGI:1925152). These rodent studies demonstrate that Prune1 is essential for mouse embryonic development. Furthermore, if PRUNE1 homozygous loss of function is similarly lethal in humans, these results suggest that NMIHBA-causing PRUNE1 variants are likely hypomorphic.
Figure 5.
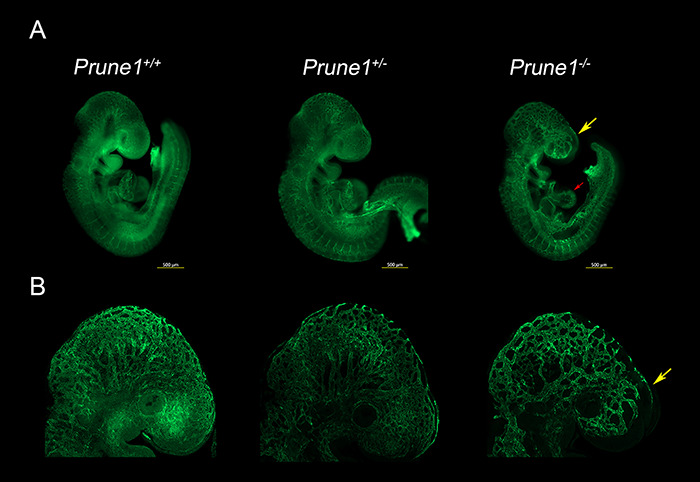
Loss of Prune1 results in vascular defects with significant disruption of the cephalic vascular plexus. (A) Representative whole mount Pecam1 staining at E9.5 demonstrated reduced plexus branching and perturbed capillary sprouting within the cephalic region (frontonasal prominence and brain, yellow arrow) in the Prune1−/− embryos as compared with wild-type and heterozygous littermates. Moreover, Prune1−/− embryos displayed cardiac defects observed as a less intricate appearance of the endocardium when compared with littermate controls (red arrow) (B) Higher magnification (6.3X) images further highlight the disruption of the cephalic vascular plexus. A total of three Prune1+/+, 2 Prune1+/− and 5 Prune1−/− embryos were analyzed in two independent experiments.
Discussion
Since our initial report of PRUNE1 as a novel candidate gene for abnormal neurodevelopment and brain malformation, 37 NMIHBA patients have been reported to date by us and others (8–11,13–15,18,19). Detailed clinical and literature review of these reported NMIHBA cases allows for a more comprehensive view of this novel neurodevelopmental disorder (Table 1,Fig. 1). The main features defining the disorder, namely profound DD, ID and brain malformations, remain consistent; however, additional clinical features such as optic nerve atrophy, gastrointestinal problems and peripheral nervous system involvement have emerged with additional reports of patients with NMIHBA. Subsequent reporting of patients with novel genetic disorders after the initial report enables a better understanding of the phenotypic spectrum and often helps in informing newly diagnosed patients and their caregivers of the natural history of disease.
The worldwide distribution of reported patients and disease-associated variants conforms with the clan genomics hypothesis where rare alleles of high effect arising in recent ancestors are more likely to contribute to disease burden versus common variants of small effect in the population (20). These rare alleles are more likely to come together in homozygosity in certain populations due to genetic mechanisms such as drift in founder populations or inbreeding in consanguineous populations. For example, the splicing variant allele c.521-2A > G has now been reported multiple times as a founder allele in the Cree population from Canada (14,21). Similarly, the D30N allele, so far reported in Saudi and Omani individuals (8,9), appears to be a founder allele in Middle Eastern populations, whereas other pathogenic rare alleles have been reported once so far in single families from different populations around the world. Interestingly, the most common pathogenic allele reported in NMIHBA patients is the D106N variant, observed in individuals from diverse ethnic backgrounds from Turkey, Italy, Lebanon, Sri Lanka and Japan. While this variant appears to be a founder allele in the Turkish population, the diversity of populations where it has been observed poses the possibility that the D106 residue is a recurrent mutational site and that the allele has arisen multiple times in different population haplotypes.
Furthermore, in this study, we investigated the molecular etiology underlying PRUNE1 dysfunction in NMIHBA. Based on our in vitro and in vivo data, we conclude that the molecular pathomechanism of this rare genetic disorder is reduced or altered function of PRUNE1 due to hypomorphic alleles. The majority of disease-associated alleles including the splice site variant within intron 4 (c.521-2A > G: IVS4-2A > G) cluster in the catalytic DHH domain of PRUNE1, underscoring the critical role of this conserved domain in NMIHBA pathogenesis. Using in silico and biochemical approaches, we explored the functional effects of four reported pathogenic PRUNE1 variants: p.D30N, p.D106N, p.R128Q and p.G174* on PRUNE1 function and examined the potential developmental and organismal functional consequences of homozygous Prune1 ablation.
Protein homology modeling using the S. cerevisiae orthologue, PPX1 demonstrated that the D30, D106 and R128 residues fall within a metal-ion and phosphate binding interface representing the active-site for phosphate hydrolysis. Disruption of these charged residues within the active-site is predicted to impair metal coordination (D30N and D106N) and substrate binding (R128Q). Overexpression of human PRUNE1 cDNA harboring NMIHBA pathogenic variant alleles in HEK293 cells demonstrated a complete loss of protein product for the D30N mutant and a dramatic reduction in the D106N product. Both isoforms were rescued upon inhibition of the proteasome degradation pathway, suggesting that the loss of metal-binding residues destabilizes local protein structure resulting in protein degradation. In contrast, the R128Q variant was stably expressed at levels comparable to wild-type protein. Patient derived fibroblasts further confirmed these findings, implicating the metal coordinating D106 residue to be critical for maintaining active-site structure and protein stability. It has been proposed that the R128 (H149 in S.c.-PPX1) along with the binuclear metal cluster (M1 and M2 in Fig. 2A) coordinate binding of one of the phosphate moieties (PT; Fig. 2A) of the substrate, rendering the P-O bond more susceptible to nucleophilic attack. This model would suggest impaired polyphosphate processivity by the R128Q mutant. Consistent with such a model, in vitro short-chain exopolyphosphatase assays using recombinant proteins derived from mammalian or E. coli overexpression systems demonstrated the loss of exopolyphosphatase activity in R128Q and D106N mutants as compared with the wild-type. Kinetic parameters for hydrolysis of short-chain polyphosphates P3 and P4 (in the presence of 2 mM Mg2+) by wild-type PRUNE1 in our assays were comparable to those reported by Tammenkoski et al. (17). Our data related to the R128Q mutant in conjunction with enhanced kinetics of P3 hydrolysis by R128H mutant (where the positive charge is retained) demonstrated by Tammenkoski et al., further reinforce a model wherein binding of the metal ion at the active site accelerates substrate binding, and the bound substrate in turn enhances the enzyme affinity for the metal ion (17). Therefore, mutations that disrupt metal coordinating residues or substrate binding residues may result in the loss of enzymatic function. Furthermore, in line with previous reports, we demonstrate that wild-type PRUNE1 has no detectable cyclic nucleotide (cAMP or cGMP) phosphodiesterase activity or long-chain exopolyphosphatase activity (17). Taken together, these studies suggest the loss of short-chain exopolyphosphatase activity as a potential mediator of NMIHBA pathophysiology. In light of our biochemical observations on disease associated mutant proteins, it is intriguing to note that Zollo et al. (8) ascribe enhanced short-chain exopolyphosphate activity (based on P4 hydrolysis) for two NMIHBA variants they characterized including the D30N mutant which is demonstrably a loss of function variant in our analyses. Furthermore, their reported Kcat/Km ratio for P4 hydrolysis by wild-type PRUNE1 (0.014 μM−1 s−1) is an order of magnitude different than those observed in the present study (0.26 μM−1 s−1) and reported by Tammenkoski et al. (0.18 μM−1 s−1) (8,17). These discrepant results may be due to differences in cofactor concentration (10 mM Mg2+ as opposed to 2 mM Mg2+ in our assays and Tammenkoski et al.), substrate purity or other confounding factors specific to the experimental methods used.
At the time of our initial discovery of PRUNE1 as the disease causing gene, no mouse or other model organism data were available to provide further insights into the biological role of PRUNE1 in neurodevelopment and disease pathophysiology. Conservation at the protein level and similar developmental expression suggested preserved function between the mouse and human orthologues. Based on functional analyses of disease causing human alleles, we engineered a mouse model of NMIHBA by knocking out the murine Prune1 gene. Since the original publication of PRUNE1 as a novel disease gene in Karaca et al. (9), a Prune1-null mouse was developed and phenotyped by the International Mouse Phenotyping Consortium (IMPC). (http://www.mousephenotype.org/data/genes/MGI:1925152). Consistent with the IMPC data, we observed fully penetrant embryonic lethality in the null mouse prior to E12.5. Our in depth developmental phenotyping of the Prune1-null embryos demonstrated profound vascular defects manifested by poorly branched vessels within the yolk sac, cardiac hypoplasia and a disrupted cephalic vascular plexus resulting in embryonic lethality between E9 and E10. We were intrigued by the profound cardiovascular defects in the Prune1-null embryos, given the reported absence of such manifestations in NMIHBA patients. Interestingly, we observed robust LacZ reporter expression in embryonic heart (E12.5, Supplementary Material, Fig. S6E), and robust cardiac expression in both adult mouse (median TPM: 13.34; n = 6 samples) and human (median TPM: 16.39; n = 432 samples) based on in-house mouse transcriptomics and publicly available data from the Genotype-Tissue Expression (GTEx) project portal, respectively. Unfortunately, our current analyses using a germline loss of function model do not address if the cardiovascular anomalies result in lethality. Moreover, it is unclear if these represent cell-autonomous defects or a consequence of altered hemodynamics secondary to impaired vasculogenesis within the yolk sac. Conditional ablation of Prune1 in these tissues would help elucidate this further.
LacZ reporter expression in heterozygous mice demonstrates broad diffuse Prune1 expression across multiple tissues and cell lineages at E12.5 (Supplementary Material, Fig. S6B). Consistently, Prune1 expression in adult mice is observed across multiple tissues with strong expression within the CNS (Supplementary Material, Fig. S6D). Despite this widespread expression profile, the most overt phenotypes arising from loss of Prune 1 are vascular deficits most prominent in the cephalic vascular plexus possibly resulting from a local deficit in angiogenic cues or due to altered hemodynamics downstream of defective vascular development. Further analyses are required to discriminate the effect of hemodynamic defects versus the signaling perturbations downstream of Prune1 loss of function, and more importantly as to why the cephalic vessels are most affected.
It is intriguing as to why homozygous deletion results in embryonic lethality in mouse, while probands in this report and others survive past gestation. Whereas two patients have been reported to be homozygous for splice-site and frameshift variants in PRUNE1 (11,15), comprehensive analyses of protein-coding variation in PRUNE1 across several large-scale human genetic variation databases including ExAC/gnomAD and our internal (DiscovEHR) database identified no homozygous carriers of predicted loss-of-function variants despite the observation that heterozygous loss-of-function variants are well tolerated (Supplementary Material, Fig. S8) (22,23). Residual enzymatic function of the hypomorphic missense alleles beyond the range of detection of our in vitro assays, alternative non-enzymatic developmental function (in mouse) or potential compensation by other phosphatases during neurodevelopment (in human) could potentially explain the observed species-specific difference in phenotype. Future analyses using knock-in mouse models or gyrified mammals such as ferrets along with human iPSC-derived brain organoids may help discern the allelic architecture of NMIBHA and allow for molecular interrogation of PRUNE1 function in mammalian neurodevelopment.
Polyphosphate has been implicated in diverse physiological processes in higher eukaryotes, including apoptosis, mTOR activation and neuronal signaling (24–26). This pleiotropy is consistent with the essential role of inorganic diphosphate (PPi) for cellular metabolism across taxa and the potential role of polyphosphate metabolizing enzymes (including PRUNE1) in maintaining phosphate homeostasis. However, mechanistic understanding of how polyphosphate metabolism influences cellular metabolism and in turn organogenesis remains obscure. More recently, Cremers et al. proposed polyphosphate as a conserved modifier of amyloidogenic processes capable of mitigating neurotoxicity induced by amyloid fibers (27). Further studies using mouse models that closely recapitulate the NMIHBA phenotype will help uncover mechanisms by which PRUNE1 influences neurodevelopmental and neurodegenerative processes. The fact that NMIHBA-causing PRUNE1 variants lack exopolyphosphatase activity may facilitate the exploration of therapeutic avenues that would allow for preemptive rather than symptomatic intervention of NMIHBA in the future.
Materials and Methods
Cell culture
HEK293 and human fibroblast culture medium consisted of DMEM (Gibco; 11995-065) supplemented with 10% fetal bovine serum (Gibco; 16000-036), 1% non-essential amino acids (Gibco; 11140-050) and 1% Penicillin–Streptomycin (Gibco; 15140-122). Cells were dissociated using 0.05% Trypsin–EDTA (Gibco; 25300-054). HEK293 cells were seeded in 96-well plates at a density of 20 000 cells/well and in other formats (12- and 24-well) at a density of 50 000 cells/cm2. Fibroblasts were seeded at a density of 3000 cells/cm2, in all cases. Genotypes of patient-derived fibroblasts and control dermal fibroblasts (ATCC; PCS201010) were Sanger di-deoxy sequence verified using primers listed in Supplementary Material, Table S1.
Immunoblotting and protein stability analyses
N-terminal HA-tagged PRUNE1 overexpression constructs were transfected into PRUNE1 non-expressing HEK293 cells using X-tremeGENE HP DNA (Roche; 06366236001) transfection reagent. As a transfection control, CMV-driven eGFP expression construct was co-transfected at a 1:19 ratio. Two days later, whole cell lysate was collected, and total protein concentration was quantified using bicinchoninic acid (BCA) assay (ThermoFisher; 23 225). PRUNE1 levels in overexpressing HEK293 and in human fibroblasts were analyzed by immunoblotting using anti-PRUNE1 (Origene; TA344725) and/or anti-HA (Abcam; 18 181) antibodies at 1:1000 dilution, using GAPDH (CST; 3683S) as a loading control.
Stability of the various PRUNE1 muteins was evaluated by overexpressing N-terminal HA-tagged PRUNE1 constructs in HEK293 cells. After 48 h, medium containing either cycloheximide (200uM; Sigma 1810) and/or MG132 (15uM, Sigma; M8699) was added. Whole cell lysates were collected 0, 6 and 24 h of cycloheximide treatment. Whole cell lysate from cells treated with both cycloheximide and MG132 were collected after 24 h of treatment. Total protein concentration was quantified and immunoblotted using Li-COR Odyssey CLx imaging system. In complementary immunoblotting experiments, endogenous PRUNE1 levels in Sanger verified (Supplementary Material, Fig. S4B) patient-derived fibroblasts BAB3500 (PRUNE1D106N/D106N) and UDP760 (PRUNE1R128Q/G174*) was compared with control neonatal dermal fibroblasts (ATCC, PCS201010).
GloSensor cAMP and cGMP assays
HEK293 cells were plated at a density of 20 000 cells/well in poly-D-lysine-coated 96-well plates (Corning; 354 651). The following day cells were co-transfected with 12.5 ng pGloSensor-22F or pGloSensor-42F and 10 ng of PRUNE1 overexpression constructs (D30N, D106N, R128Q and 4DD) or control PS100013, PDE4A or PDE5A vectors per well. Total DNA was maintained at 100 ng per well. Two days later, cells were changed into GloSensor equilibration medium (10% fetal bovine serum and 2% GloSensor assay reagent (Promega; E1290) in CO2-independent medium (Gibco; 18 045-088). Analyses of parallel plates demonstrated no differences in cell viability (evaluated using MTT assay, ThermoFisher; V13154) and total protein concentration (evaluated using BCA) across different experimental groups. Upon equilibration at room temperature for 2 h, basal luminescence was acquired using a PerkinElmer 2104 multimode plate reader. Forskolin (Sigma; F3917) or sodium nitroprusside dihydrate (Millipore; 567 538) was added to the medium to a final concentration of 10 or 50 μM. cAMP or cGMP phosphodiesterase activity was evaluated by measuring luminescence recorded at 1-min intervals up to 45 or 60 min, respectively.
Exopolyphosphatase assay
Exopolyphosphatase activity of PRUNE1 against short-chain polyphosphate (PolyP) substrates, sodium tripolyphosphate (P3, Sigma; 72 061) and sodium tetraphosphate (P4, BOC Sciences; 7727-67-5) was determined by a fixed time assay using BIOMOL GREEN phosphate detection kit (Enzo Lifesciences; BML-AK111). Reaction was performed 15–120 s in 50 μL of 100 mM Tris/HCl, 50 μM EGTA, 2 mM MgCl2, pH 7.2 at substrate concentrations ranging from 0.5 to 100 μM and 1 to 50 μM for P3 and P4, respectively. Reactions were terminated by addition of 100 μL of BIOMOL GREEN reagent; absorbance at 620 nm was measured following a 30-min incubation at 25°C. Kinetic parameters were assessed by plotting Lineweaver–Burk plots using GraphPad PRISM 7 software.
Exopolyphosphatase activity of wild-type PRUNE1 on medium and long-chain PolyP substrates (consisting of 45, 65 and 700 orthophosphate monomers, Sigma S4379, Sigma; S6253, Kerafast; EUI002) was assessed by comparing to S. cerevesia PPX1 (Bioorbyt; ORB419100). About 50 nM of PRUNE1 or 100 nM PPX1 were incubated with polyphosphates at a final concentration of 2 mM (in terms of phosphate residues) in 100 mM Tris–HCl, pH 7.2, 50 μM EGTA, 2 mM MgCl2 or 50 mM HEPES/KOH, pH 8.0, 1 mM MgCl2, 125 mM KCl, respectively. Reactions were allowed to proceed at 37°C for 150 min. Reactions were terminated at 15, 30, 60 and 150 min. Negative control reactions, containing no enzyme in both buffer systems, were also prepared as above.
Polyphosphate hydrolysis was tracked by running fractions of the reactions on TBE gels and staining polyphosphates present using toluidine blue O, as previously described (28). Briefly, reactions were diluted with Novex Hi-Density TBE Sample Buffer (Thermo Fisher; LC6678), and run on precast TBE gels, either 20% (EC63155; Novex) for P45 and P65 reactions or 6% (EC62655; Novex) for P700 reactions. Following electrophoresis, the gels were stained in 0.05% toluidine blue O in fixative solution (an aqueous solution of 25% methanol and 5% glycerol) for 15 min, with agitation, followed by 3 h of washing in fixative solution and imaging. In concurrent analyses, released phosphate was quantified using BIOMOL GREEN phosphate detection kit as described above. All experiments were repeated at least three times with a minimum of four technical replicates per sample. Data were analyzed using PRISM 7 (GraphPad) software.
CD and in silico modeling of PRUNE1 mutations
PRUNE1 wild-type, D106N and R128Q muteins were analyzed by CD spectroscopy by collecting far-UV (<250 nm) and near-UV (260–320 nm) CD spectra. Prior to CD measurements, all samples were dialyzed twice in a base buffer, 20 mM Tris–HCl, pH 7.8 to eliminate DTT that could potentially interfere with far-UV CD measurements. Following overnight dialysis, concentrations were determined by A280 measurements, using a theoretical extinction coefficient based on amino acid sequence of each protein. Near-UV CD measurements were obtained at 0.75, 0.7 and 0.45 mg/ml for wild-type, D106N and R128Q samples, respectively. All samples were further diluted (2-fold) using 0.02 μM filtered dialysate for far-UV CD measurements. Far-UV CD spectra were evaluated at 0.38, 0.35 and 0.21 mg/ml for wild-type, D106N and R128Q samples, respectively. CD measurements were performed on a Jasco 1500 spectropolarimeter at 25°C using a 1 cm cell (Jasco; Type J3) for near UV-CD and 0.1 cm cell (Jasco; Type J10559) for far-UV CD. The raw data were baseline subtracted and converted to units of molar ellipticity to correct for protein concentration differences. Degree of spectral difference between wild-type and D106N or R128Q muteins was assessed using a spectral classification technique (TQ Analyst®). The reported TQ Analyst® similarity match score indicates how closely the spectrum of each mutant matches that of the wild-type protein. Highly comparable far-UV CD spectra result in scores >92.6, whereas highly comparable near-UV CD give TQ similarity match scores >98.
The RCSB Protein Data Bank was searched for a structure with high sequence homology to human PRUNE1. Saccharomyces cerevesiae cytosolic exopolyphosphatase (PPX1) (PDB ID: 2QB7), with a Blast e-value of 2.3E−19 and 25% sequence identity (45% similarity), was the best hit. Since the active site residues were conserved, we did not attempt to generate a PRUNE1 homology model. PPX1 structure (shown in Fig. 2A) was rendered by exchanging orthologous active site residues: Asp41 to Asn, Asp127 to Asn, and H149 to Gln. Three bound phosphate moieties PT, PE1 and PE3 shown in ball-and-stick, and the two catalytic metal ions as M1 and M2 as pink spheres.
Animal studies
Prune1−/− mice were generated by replacing exon 2 (ENSMUSE00001210512) of the Prune1 gene with a β-galactosidase (lacZ) reporter cassette using VelociGene technology (29). Mice carrying the deletion were genotyped by a loss of allele assay as described previously (30). Targeted, cassette-deleted heterozygous mice were bred to obtain desired genotypes. Homozygous and heterozygous knockout mice cohorts derived from F1 breeding were used for phenotypic evaluation. All mice used in this study were in a 75% C57Bl/6NTac, 25% 129S6/SvEvTac genetic background, and housed in a pathogen free environment. Autoclaved water and sterile mouse chow were provided ad libitum.
Ethics statement
All experimental protocols in mice, including anesthesia, imaging procedures and tissue sampling procedures performed in this study, were approved by the Regeneron Pharmaceuticals Inc. Institutional Animal Care and Use Committee (IACUC) under protocol number 430 and the US Animal Welfare Act.
Anti-PECAM1 staining and imaging
Embryo processing, immunostaining and imaging were performed as described previously (31). Briefly, embryos were collected at embryonic day (E) 9.5 and fixed for 3 h in 4% paraformaldehyde at 4°C. Prior to staining, endogenous peroxidase activity was quenched with 3% H2O2. Non-specific antibody binding was blocked by preincubating embryos in 10% normal goat serum (Vector Labs; S-1000) and 0.5% Bovine Serum Albumin (Fisher Scientific; BP1600). Embryos were stained overnight at 4°C with rat anti-PECAM1 antibody at 1:300 dilution (Pharmingen; 553 370,). After primary antibody incubation, embryos were incubated overnight with donkey anti-rat HRP secondary antibody at 1:1000 dilution (Jackson ImmunoResearch; 712-036-153) followed by incubation with 488-tyramide reagent at 1:25 dilution made using Dylight 488 NHS ester (Thermo Scientific; 46 402). Images were acquired through optical sectioning with an ApoTome.2 on a ZEISS Axio Zoom. Extended Depth of Focus images were generated by Zen software. Imaging was performed using Leica MZFLIII stereozoom microscope quipped with a 0.5x objective and 1.0x camera lens, with a zoom between 4.0x and 6.3x. Alternatively, stained embryos were imaged using OPT as described previously (31). For OPT, samples were embedded in low melting agarose and subsequently cleared using a mixture of benzyl alcohol and benzyl benzoate (1:2 ratio). Sample was rotated stepwise (with a 0.9o step size) for one revolution, and a GFP1 filtered autofluorescence view and a Cy3 fluorescence view (captured using a Cy3 filter) were acquired using Retiga Exi CCD camera. Reconstruction of all image slices yielded a 3D volumetric rendering of the specimen as described previously (32).
Statistical analysis
Statistical analyses were conducted using Prism 7 (GraphPad) software. Unpaired Student’s t-test was used unless indicated otherwise. Statistical significance reported when P < 0.05. Where appropriate sample sizes, the statistical test used and significance are indicated in each figure legend.
Supplementary Material
Acknowledgements
We wish to thank the patients and families that consented to be part of this study and acknowledge Kristy Neiman and William Poueymirou for their assistance with mouse colony management.
Conflict of Interest statement. H.N., C.G.-J., S.M.C., S.R., S.N., D.D., M.C.F., P.S., M.P., Y.T., M.G.D., R.A.D., C.J.S., B.Z., N.W.C. and A.N.E. are full-time employees of the Regeneron Genetics Center or Regeneron Pharmaceuticals Inc. and receive stock options/restricted stock as part of compensation. J.R.L. is a paid consultant for Regeneron Pharmaceuticals, Inc., has stock ownership in 23andMe and is a co-inventor on multiple USA and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The remaining authors declare no conflict of interest.
Contributor Information
Harikiran Nistala, Regeneron Genetics Center, Tarrytown, NY 10591, USA.
John Dronzek, Regeneron Genetics Center, Tarrytown, NY 10591, USA.
Claudia Gonzaga-Jauregui, Regeneron Genetics Center, Tarrytown, NY 10591, USA.
Shek Man Chim, Regeneron Genetics Center, Tarrytown, NY 10591, USA.
Saathyaki Rajamani, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Samer Nuwayhid, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Dennis Delgado, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Elizabeth Burke, Undiagnosed Diseases Program Translational Laboratory, NHGRI, National Institutes of Health, Bethesda, MD 20892, USA.
Ender Karaca, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Department of Genetics, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Matthew C Franklin, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Prasad Sarangapani, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Michael Podgorski, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Yajun Tang, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Melissa G Dominguez, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Marjorie Withers, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Ron A Deckelbaum, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Christopher J Scheonherr, Regeneron Genetics Center, Tarrytown, NY 10591, USA.
William A Gahl, Undiagnosed Diseases Program Translational Laboratory, NHGRI, National Institutes of Health, Bethesda, MD 20892, USA.
May C Malicdan, Undiagnosed Diseases Program Translational Laboratory, NHGRI, National Institutes of Health, Bethesda, MD 20892, USA.
Brian Zambrowicz, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Nicholas W Gale, Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Richard A Gibbs, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Wendy K Chung, Columbia University Medical Center, New York, NY 10032, USA.
James R Lupski, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA; Texas Children’s Hospital, Houston, TX 77030, USA.
Aris N Economides, Regeneron Genetics Center, Tarrytown, NY 10591, USA; Regeneron Pharmaceuticals, Inc., Tarrytown, NY 10591, USA.
Funding
Regeneron Pharmaceuticals, Inc.; joint National Human Genome Research Institute (NHGRI)/National Heart Blood and Lung Institute (NHLBI) grant UM1 HG006542 of the National Institutes of Health to the Baylor-Hopkins Center for Mendelian Genomics and NINDS R35 grant to J.R.L.
References
- 1. Lovett S.T. and Kolodner R.D. (1989) Identification and purification of a single-stranded-DNA-specific exonuclease encoded by the recJ gene of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A., 86, 2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shintani T., Uchiumi T., Yonezawa T., Salminen A., Baykov A.A., Lahti R. and Hachimori A. (1998) Cloning and expression of a unique inorganic pyrophosphatase from Bacillus subtilis: evidence for a new family of enzymes. FEBS Lett., 439, 263–266. [DOI] [PubMed] [Google Scholar]
- 3. Wurst H. and Kornberg A. (1994) A soluble exopolyphosphatase of Saccharomyces cerevisiae. Purification and characterization. J. Biol. Chem., 269, 10996–11001. [PubMed] [Google Scholar]
- 4. Aravind L. and Koonin E.V. (1998) A novel family of predicted phosphoesterases includes drosophila prune protein and bacterial RecJ exonuclease. Trends Biochem. Sci., 23, 17–19. [DOI] [PubMed] [Google Scholar]
- 5. Carotenuto P., Marino N., Bello A.M., D'Angelo A., Di Porzio U., Lombardi D. and Zollo M. (2006) PRUNE and NM23-M1 expression in embryonic and adult mouse brain. J. Bioenerg. Biomembr., 38, 233–246. [DOI] [PubMed] [Google Scholar]
- 6. D'Angelo A., Garzia L., Andre A., Carotenuto P., Aglio V., Guardiola O., Arrigoni G., Cossu A., Palmieri G., Aravind L. et al. (2004) Prune cAMP phosphodiesterase binds nm23-H1 and promotes cancer metastasis. Cancer Cell, 5, 137–149. [DOI] [PubMed] [Google Scholar]
- 7. Garzia L., Roma C., Tata N., Pagnozzi D., Pucci P. and Zollo M. (2006) H-prune-nm23-H1 protein complex and correlation to pathways in cancer metastasis. J. Bioenerg. Biomembr., 38, 205–213. [DOI] [PubMed] [Google Scholar]
- 8. Zollo M., Ahmed M., Ferrucci V., Salpietro V., Asadzadeh F., Carotenuto M., Maroofian R., Al-Amri A., Singh R., Scognamiglio I. et al. (2017) PRUNE is crucial for normal brain development and mutated in microcephaly with neurodevelopmental impairment. Brain, 140, 940–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Coban Akdemir Z., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I.M. et al. (2015) Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron, 88, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alfadhel M., Nashabat M., Hundallah K., Al Hashem A., Alrumayyan A. and Tabarki B. (2018) PRUNE syndrome is a new neurodevelopmental disorder: report and review. Child Neurol. Open, 5, 2329048X17752237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Costain G., Shugar A., Krishnan P., Mahmutoglu S., Laughlin S. and Kannu P. (2017) Homozygous mutation in PRUNE1 in an Oji-Cree male with a complex neurological phenotype. Am. J. Med. Genet. A, 173, 740–743. [DOI] [PubMed] [Google Scholar]
- 12. Kobayashi T., Hino S., Oue N., Asahara T., Zollo M., Yasui W. and Kikuchi A. (2006) Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol. Cell. Biol., 26, 898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alhaddad B., Schossig A., Haack T.B., Kovacs-Nagy R., Braunisch M.C., Makowski C., Senderek J., Vill K., Muller-Felber W., Strom T.M. et al. (2018) PRUNE1 deficiency: expanding the clinical and genetic spectrum. Neuropediatrics, 49, 330–338. [DOI] [PubMed] [Google Scholar]
- 14. Lacomino M., Fiorillo C., Torella A., Severino M., Broda P., Romano C., Falsaperla R., Pozzolini G., Minetti C., Striano P. et al. (2018) Spinal motor neuron involvement in a patient with homozygous PRUNE mutation. Eur. J. Paediatr. Neurol., 22, 541–543. [DOI] [PubMed] [Google Scholar]
- 15. Karakaya M., Yilmaz S., Storbeck M., Hoelker I., Heller R., Serdaroglu G., Gokben S., Yis U. and Wirth B. (2017) PRUNE1: a disease-causing gene for secondary microcephaly. Brain, 140, e61. [DOI] [PubMed] [Google Scholar]
- 16. Ugochukwu E., Lovering A.L., Mather O.C., Young T.W. and White S.A. (2007) The crystal structure of the cytosolic exopolyphosphatase from Saccharomyces cerevisiae reveals the basis for substrate specificity. J. Mol. Biol., 371, 1007–1021. [DOI] [PubMed] [Google Scholar]
- 17. Tammenkoski M., Koivula K., Cusanelli E., Zollo M., Steegborn C., Baykov A.A. and Lahti R. (2008) Human metastasis regulator protein H-prune is a short-chain exopolyphosphatase. Biochemistry, 47, 9707–9713. [DOI] [PubMed] [Google Scholar]
- 18. Fujii H., Sato N., Takanashi J.I., Kimura Y., Morimoto E., Shigemoto Y., Suzuki F., Sasaki M. and Sugimoto H. (2020) Altered MR imaging findings in a Japanese female child with PRUNE1-related disorder. Brain Dev., 42, 302–306. [DOI] [PubMed] [Google Scholar]
- 19. Papuc S.M., Abela L., Steindl K., Begemann A., Simmons T.L., Schmitt B., Zweier M., Oneda B., Socher E., Crowther L.M. et al. (2019) The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur. J. Hum. Genet., 27, 408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lupski J.R., Belmont J.W., Boerwinkle E. and Gibbs R.A. (2011) Clan genomics and the complex architecture of human disease. Cell, 147, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartley J.N., Simard L.R., Ly V., Del Bigio M.R. and Frosk P. (2019) A homozygous canonical splice acceptor site mutation in PRUNE1 is responsible for a rare childhood neurodegenerative disease in Manitoba Cree families. Am. J. Med. Genet. A, 179, 206–218. [DOI] [PubMed] [Google Scholar]
- 22. Dewey F.E., Murray M.F., Overton J.D., Habegger L., Leader J.B., Fetterolf S.N., O'Dushlaine C., Van Hout C.V., Staples J., Gonzaga-Jauregui C. et al. (2016) Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science, 354, aaf6814. [DOI] [PubMed] [Google Scholar]
- 23. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holmstrom K.M., Marina N., Baev A.Y., Wood N.W., Gourine A.V. and Abramov A.Y. (2013) Signalling properties of inorganic polyphosphate in the mammalian brain. Nat. Commun., 4, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moreno S.N. and Docampo R. (2013) Polyphosphate and its diverse functions in host cells and pathogens. PLoS Pathog., 9, e1003230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith S.A., Choi S.H., Davis-Harrison R., Huyck J., Boettcher J., Rienstra C.M. and Morrissey J.H. (2010) Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood, 116, 4353–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cremers C.M., Knoefler D., Gates S., Martin N., Dahl J.U., Lempart J., Xie L., Chapman M.R., Galvan V., Southworth D.R. and Jakob U. (2016) Polyphosphate: a conserved modifier of amyloidogenic processes. Mol. Cell, 63, 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith S.A. and Morrissey J.H. (2007) Sensitive fluorescence detection of polyphosphate in polyacrylamide gels using 4′,6-diamidino-2-phenylindol. Electrophoresis, 28, 3461–3465. [DOI] [PubMed] [Google Scholar]
- 29. Valenzuela D.M., Murphy A.J., Frendewey D., Gale N.W., Economides A.N., Auerbach W., Poueymirou W.T., Adams N.C., Rojas J., Yasenchak J. et al. (2003) High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol., 21, 652–659. [DOI] [PubMed] [Google Scholar]
- 30. Frendewey D., Chernomorsky R., Esau L., Om J., Xue Y., Murphy A.J., Yancopoulos G.D. and Valenzuela D.M. (2010) The loss-of-allele assay for ES cell screening and mouse genotyping. Methods Enzymol., 476, 295–307. [DOI] [PubMed] [Google Scholar]
- 31. Walls J.R., Coultas L., Rossant J. and Henkelman R.M. (2008) Three-dimensional analysis of vascular development in the mouse embryo. PLoS One, 3, e2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong M.D., Dazai J., Walls J.R., Gale N.W. and Henkelman R.M. (2013) Design and implementation of a custom built optical projection tomography system. PLoS One, 8, e73491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.