

Abstract
TNNI3K expression worsens disease progression in several mouse heart pathology models. TNNI3K expression also reduces the number of diploid cardiomyocytes, which may be detrimental to adult heart regeneration. However, the gene is evolutionarily conserved, suggesting a beneficial function that has remained obscure. Here, we show that C57BL/6J-inbred Tnni3k mutant mice develop concentric remodeling, characterized by ventricular wall thickening and substantial reduction of cardiomyocyte aspect ratio. This pathology occurs in mice carrying a Tnni3k null allele, a K489R point mutation rendering the protein kinase-dead, or an allele corresponding to human I686T, the most common human non-synonymous TNNI3K variant, which is hypomorphic for kinase activity. Mutant mice develop these conditions in the absence of fibrosis or hypertension, implying a primary cardiomyocyte etiology. In culture, mutant cardiomyocytes were impaired in contractility and calcium dynamics and in protein kinase A signaling in response to isoproterenol, indicating diminished contractile reserve. These results demonstrate a beneficial function of TNNI3K in the adult heart that might explain its evolutionary conservation and imply that human TNNI3K variants, in particular the widespread I686T allele, may convey elevated risk for altered heart geometry and hypertrophy.
Introduction
Troponin i3 kinase (Tnni3k), a member of the mixed lineage kinase subfamily, is unique in being cardiomyocyte-specific in expression. Tnni3k has an N-terminal ankyrin repeat domain that serves as a protein–protein interaction interface, a central kinase domain and a C-terminal domain that modulates kinase activity. Tnni3k was named for its initial discovery based on physical interaction with cardiac troponin I (Tnni3) (1), but there is as yet no evidence that Tnni3 is phosphorylated by the kinase in vivo, nor have any other authentic in vivo substrates yet been identified. The Tnni3k gene is highly conserved in evolution, present in all deuterostome and protostome species with hearts, and even in some invertebrate species that are not thought to have hearts (2). This suggests a beneficial ancestral function that became co-opted specifically for heart physiology through positive selection as the gene became cardiomyocyte-specific in expression. Nonetheless, the Tnni3k gene is not essential. In rodents, some species have devolved the Tnni3k gene into a non-functional pseudogene as a presumed adaptation to their specialized ecology (2), numerous commonly-used inbred laboratory mouse strains are homozygous for a natural loss-of-function allele of the gene without suffering obvious impairment (3), and engineered germline deletion of the Tnni3k gene (null allele, no protein expression) in C57BL/6J mice similarly did not result in any externally obvious consequence (4). In the gnomAD assembly of human sequences, the observed/expected score of TNNI3K loss-of-function gene variants indicates no apparent negative selection, and although no clinical data are available, an appropriate number of individuals were identified as being homozygous for loss-of-function alleles (5).
Although the normal function of Tnni3k has remained uncertain, absence of Tnni3k has been shown to be beneficial in several types of heart injury in mice. Thus, with transgenic calsequestrin overexpression (a model of disrupted contraction) and transaortic constriction (a model of hypertension), absence of Tnni3k was protective whereas expression of Tnni3k accelerated disease progression (3,6). Similarly, in an ischemia–reperfusion injury model, absence of Tnni3k minimized the extent of cardiomyocyte death (7). Thus, there has been a dichotomy between the presumed beneficial function of the gene in normal heart biology that accounts for its evolutionary conservation and its detrimental consequences when expressed in certain heart disease contexts.
We began our study of Tnni3k from an interest in the genetics of heart regeneration. Fetal and newborn mammalian cardiomyocytes have one diploid nucleus (like most cells of the body), but then mostly become polyploid shortly after birth. Most cardiomyocytes become postmitotic and non-regenerative at the same time (8). Speculating that the relatively small population of adult diploid cardiomyocytes might have greater regenerative properties, we surveyed a large number of inbred mouse strains to identify those with a higher percentage of such cells, and by genome-wide association identified Tnni3k as one gene with natural variants that contribute to variation in this trait (4). The influence of Tnni3k alleles was highly dependent on mouse strain background, which implicates additional genes and pathways that intersect with Tnni3k, as expected for a complex phenotype. In C57BL/6J-inbred mice (which normally express the wild-type protein), engineered absence of Tnni3k resulted in an increase in the level of adult diploid cardiomyocytes and a corresponding increase in cellular regeneration after adult infarction, confirming the original hypothesis (4). By CRISPR-mediated recombination we also engineered a K489R point mutation into the Tnni3k gene in C57BL/6J mice (2). This point mutation abolishes the kinase activity of the protein without affecting protein expression and had the same consequence on adult diploid cardiomyocyte level as the null allele, demonstrating that kinase activity is required for Tnni3k function in this context.
Using an in vitro autophosphorylation assay, we previously showed (2) that several non-synonymous point mutation variants that have arisen in the human TNNI3K kinase domain substantially compromise kinase activity (i.e. are hypomorphic). Some of these are distributed in the human population at prominent frequencies. The most common (rs3737564), an isoleucine to threonine substitution at position 686 (I686T, based on ENST00000326637.3; I787T based on ENST00000370891.2), reduces in vitro autophosphorylation activity to 38% relative to the wild-type kinase (2). The I686T variant is present at an allele frequency of 1–2% worldwide (based on non-normalized gnomAD sequence entries) and approximately 4% among East Asian and South Asian populations (5), and appears to be present at much higher frequencies in specific (albeit undersampled) subpopulations (9). The physiological significance of this and other hypomorphic TNNI3K variants in the human genome has remained unknown.
Tnni3k influence in controlling postnatal cardiomyocyte ploidy occurs shortly after birth (8). The continuing cardiomyocyte-specific expression of the gene throughout life, and the evolutionary conservation of the gene even among vertebrate species such as zebrafish in which cardiomyocyte polyploidy does not occur (4), strongly suggest that the gene may have a separate and additional positive role in adult cardiomyocyte physiology. In this study, we show in mice that Tnni3k loss-of-function mutations result in impaired PKA signaling with impaired contraction and concentric remodeling. This phenotype is the consequence of deficient Tnni3k kinase activity, and we show that mice carrying the homolog of the human TNNI3K kinase domain I686T hypomorphic variant are also subject to concentric remodeling. Human patients with concentric remodeling are at higher risk for progression to heart failure (10–13). Because the I686T allele is prominent in the human population, TNNI3K and its downstream pathways and targets may have a previously unsuspected relevance to human concentric remodeling, hypertrophy and heart failure.
Results
To explore the in vivo consequences of the human TNNI3K kinase domain I686T variant, we created a new knock-in allele in C57BL/6J mice by CRISPR-mediated homologous replacement, changing the corresponding isoleucine codon in the mouse genome (at position 685 of the mouse Tnni3k protein) to encode threonine (Fig. 1A and B). Founder mice carrying the knock-in allele were crossed to wild-type C57BL/6J partners to confirm germline establishment of the allele, and heterozygous mice then intercrossed or crossed to Tnni3k null mice (also C57BL/6J-inbred) to generate mice that were used for analysis. We confirmed that the protein encoded by the I685T allele was expressed at a normal level in heart tissue (Fig. 1C).
Figure 1.
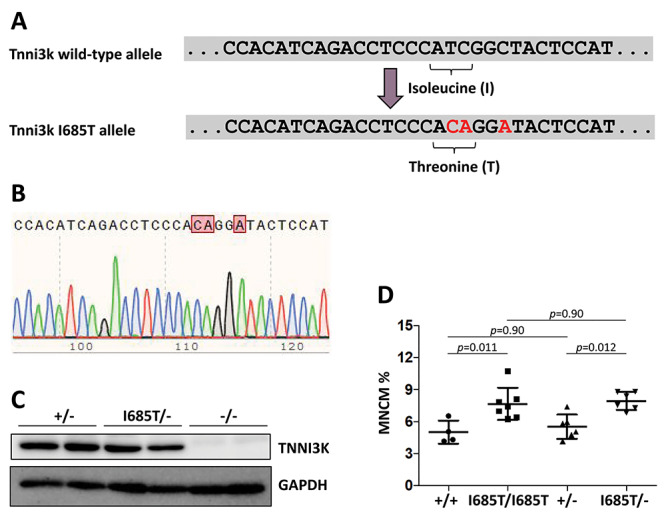
Creation and analysis of the Tnni3k I685T allele. (A) The relevant portion of the wild-type C57BL/6J Tnni3k exon 21 sequence is shown above, and below in red are the base changes made. This manipulation changed an isoleucine (ATC) codon to a threonine (ACA) codon, and introduced an EcoNI restriction site (CCTNNNNNAGG) into the sequence to facilitate genotyping. The additional base change made in the third position of the following glycine codon does not change the encoded amino acid and was introduced to suppress CRISPR/Cas9 retargeting. (B) Sequence trace of this portion of the gene in an I685T/I685T homozygous mouse. (C) Western blot showing that Tnni3k protein is expressed at normal levels in hearts of I685T/− mice. Each lane is lysate from a different mouse of the genotypes indicated above. (D) Ventricular mononuclear cardiomyocyte percentage from single cell preparations; each dot is the measurement of a different adult mouse of the indicated genotype.
Because our assay based only on in vitro autophosphorylation demonstrated that kinase activity of the I685T variant is substantially reduced but not eliminated (i.e. the variant is hypomorphic), it was uncertain if the reduction in kinase activity would have in vivo consequences. To address this, we first evaluated the percentage of ventricular mononuclear cardiomyocytes in adult mice. We previously showed that Tnni3k null mice and mice carrying the K489R allele both have an elevated level of mononuclear cardiomyocytes relative to wild-type and heterozygous mice (2,4). Here, we found a similar elevation in mononuclear cardiomyocyte percentage in I685T/I685T and in I685T/− mice (Fig. 1D; ‘—’ indicates the null allele). Although there is an indication in assays described below that I685T homozygotes are impacted less severely than I685T/− mice in certain features (as might be expected for a hypomorphic allele), we measured no difference between these genotypes in the context of cardiomyocyte mononuclear percentage. We do not know the threshold level of Tnni3k kinase activity that determines if this phenotype will or will not emerge, but the outcome with I685T mice demonstrates that this allele is sufficiently hypomorphic to compromise this Tnni3k-dependent process in vivo.
Cardiomyocyte polyploidy is a manifestation of Tnni3k kinase activity in the early newborn heart. Newborn and adult cardiomyocytes differ substantially in many molecular features (14); we therefore used the range of our available Tnni3k alleles (+, wild-type; −, full null; K489R, kinase-dead; I685T, kinase hypomorph human variant homolog) to address the consequences of Tnni3k mutant alleles on parameters of adult cardiomyocyte and heart biology. All mice were maintained on an inbred C57BL/6J background, which eliminates a major experimental variable and allowed us to focus on the specific in vivo consequences of these alleles.
In adult mice of ages 3–10 months, heart weight normalized to body weight, heart weight normalized to tibia length and left ventricular (LV) mass normalized to body weight were unchanged in Tnni3k mutant mice compared to wild-type or heterozygous controls (Fig. 2A). In histological sections (Fig. 2B), we noted that the LV and septal walls were thicker in hearts from mutant mice, although were proportionately shorter on the apex-base axis thus accounting for their normal heart weight. Measurement of the ventricular wall and septal thicknesses at the level of the papillary muscle (Supplementary Material, Fig. S1A) quantified the increase in null mice of this age (Fig. 2C). Homozygous I685T/I685T mice had an intermediate increase in myocardial dimension (Fig. 2C), although I685T/− mice were significantly increased in wall and septal thickness (as were K489R/− mice) (Supplementary Material, Fig. S1B), suggestive of a gene dosage-sensitive effect which was less severe with two copies of the hypomorphic I685T allele. Echocardiography confirmed the increase in both posterior wall and septal thickness in null mutant hearts in vivo (Fig. 2D; Table 1). These measurements also implicated a smaller ventricle chamber volume in null mutants, with a reduced stroke volume and ejection fraction (Table 1). There was no change in peripheral blood pressure in non-anesthesized mice (Fig. 2E), but heart rate was increased (Supplementary Material, Fig. S1C), presumably in a compensatory manner in order to maintain cardiac output. Because heart and ventricle mass were unchanged, this phenotype is categorized as LV concentric remodeling rather than LV hypertrophy.
Figure 2.
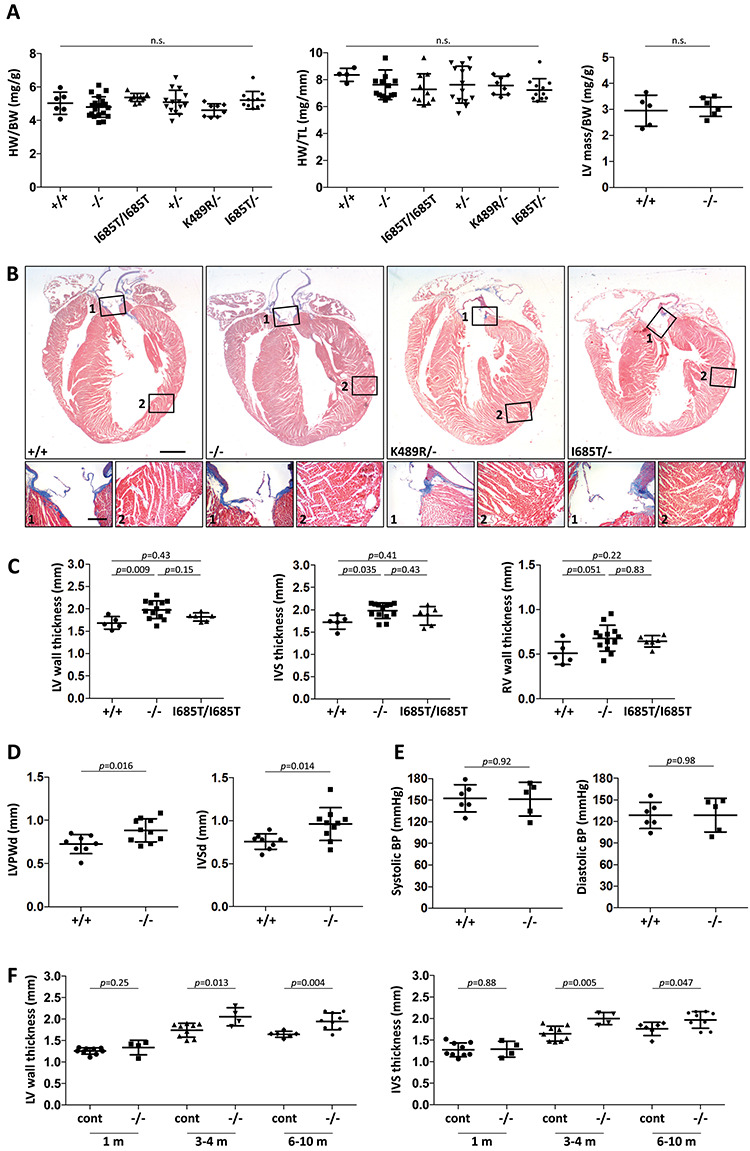
Phenotypic evaluation of Tnni3k mutant mouse hearts. (A) Heart weight to body weight ratio, heart weight to tibia length and left ventricle mass to body weight ratio measurements for hearts of the indicated genotypes; all mice were 3–10 months old, and each dot represents a unique mouse. All pair-wise comparisons were statistically non-significant (n.s.). (B) Trichrome-stained long-axis histology sections of representative hearts from 7 month old mice fixed in diastole; the magnified regions (numbered) show positive collagen staining (blue) in the aortic valve (number 1 boxes) but little if any staining in the myocardium (number 2 boxes). All corresponding images are at the same magnification. Scale bar in larger images, 1 mm; scale bar for smaller images, 200 μ. (C) Measurements of left ventricular (LV) wall, interventricular septum (IVS), and right ventricle (RV) wall thicknesses in transverse sections taken at a cross-sectional plane through the papillary muscles. LV wall thickness was measured between the two papillary muscles, and IVS and RV thicknesses directly opposite. Representative images of sections are shown in Supplementary Material, Figure S1A. Analysis of additional genotypes (+/−, K489R/−, I685T/−) is shown in Supplementary Material, Figure S1B. (D) Measurement of LV posterior wall (LVPW) and interventricular septal (IVS) diastolic diameters in live mice by echocardiography. (E) Tail-cuff measurement of systolic and diastolic blood pressure in non-anesthetized mice. (F) LV and IVS thickness in control (+/+ and +/− combined) and null (−/−) mice at different ages as measured in transverse sections.
Table 1.
Echocardiography parameters of Tnni3k wild-type and null mice
| +/+ (n = 8) | −/− (n = 10) | P-value | Significant | |
|---|---|---|---|---|
| LVPWd, mm | 0.73 ± 0.11 | 0.89 ± 0.13 | 0.016 | ✓ |
| LVPWs, mm | 1.11 ± 0.18 | 1.27 ± 0.27 | 0.16 | |
| IVSd, mm | 0.76 ± 0.09 | 0.96 ± 0.19 | 0.014 | ✓ |
| IVSs, mm | 1.15 ± 0.17 | 1.36 ± 0.27 | 0.074 | |
| LVIDd, mm | 4.07 ± 0.44 | 3.79 ± 0.29 | 0.13 | |
| LVIDs, mm | 2.83 ± 0.34 | 2.64 ± 0.33 | 0.25 | |
| FS, % | 30.31 ± 4.95 | 30.32 ± 6.06 | 0.99 | |
| LVEDV, μl | 50.17 ± 8.38 | 44.71 ± 8.50 | 0.19 | |
| LVESV, μl | 20.62 ± 6.91 | 26.60 ± 4.33 | 0.039 | ✓ |
| SV, μl | 29.55 ± 4.73 | 18.11 ± 6.85 | 0.001 | ✓ |
| EF, % | 59.27 ± 8.16 | 39.38 ± 9.54 | 0.0003 | ✓ |
Echocardiographic measurements from Tnni3k wild-type (+/+) and null (−/−) adult mice. Values are means ± standard deviations. Abbreviations: LVPWd, LVPWs, left ventricular posterior wall thickness at diastole and systole; IVSd, IVSs, interventricular septal thickness at diastole and systole; LVIDd, LVIDs, left ventricular internal diameter at diastole and systole; FS, fractional shortening; LVEDV, LVESV, left ventricular end diastolic and systolic volume; SV, stroke volume; EF, ejection fraction.
We examined ventricular wall thickness in wild-type and Tnni3k null mice of different ages (Fig. 2F). No difference was detected at 1 month, but was detected at 3–4 months and in older mice. Thus, concentric remodeling in Tnni3k mutants is an acquired phenotype that is not present from before or shortly after birth, and only becomes manifest after several months of postnatal life. Older mice were not more severely impaired than 3–4 month old mice, indicating that this is not a progressive pathology but rather one that reaches a seemingly stable point of homeostasis. Tnni3k mutant mice live to advanced age (>1 year) with no visible limitation, indicating that this alteration in heart geometry does not obviously impair their life under standard laboratory housing.
Unlike the full null Tnni3k allele, from which there is no protein expression, the I685T and K489R kinase domain point mutants express full length proteins of normal abundance that are specifically compromised in kinase activity (2). The increases in ventricular wall and septal thickness in these mutants as in null mutants demonstrates that LV concentric remodeling is the result of absent kinase activity. An additional function of the protein (e.g. interaction with other proteins through the N-terminal ankyrin repeat domain) may also be required in order to retain normal heart dimension, but at minimum, kinase activity is necessary.
Concentric remodeling is commonly considered to be a mild manifestation of the same events that lead to concentric hypertrophy (15). Furthermore, human patients with concentric remodeling are at higher risk for progression to heart failure (10–13). Several types of circumstance can lead to these conditions. Two of these include increased cardiac afterload (such as occurs in systemic hypertension or experimental aortic constriction) and alteration of the extracellular matrix of the heart. In both contexts, the change in myocardial dimension is a compensatory response to an initial extracardiac or non-cardiomyocyte perturbation. However, trichrome staining did not reveal any recognizable degree of fibrosis in the ventricular wall of mutant hearts (Fig. 2B), and measurement of peripheral blood pressure did not indicate any change (Fig. 2E), indicating an absence of hypertension. Alternatively, hypertrophy and concentric remodeling are also associated with cardiomyocyte-autonomous perturbations of contractile function and of related regulatory signaling pathways (16–19); in these cases, remodeling is a compensatory response to maintain output. Because Tnni3k is cardiomyocyte-specific in expression, the remodeling process that occurs in mutant hearts is more likely to be a primary alteration in ventricular cardiomyocyte biology rather than a secondary response to an extracardiac or non-cardiomyocyte circumstance. Indeed, ex vivo analysis of primary cardiomyocytes demonstrates a cardiomyocyte-autonomous functional deficiency, as described below.
We analyzed the size and shape of primary adult (4–6 months old) cardiomyocytes isolated in diastole from control and mutant mice. Although cardiomyocytes from the entire ventricle were pooled, the analysis is heavily weighted to the left ventricle and septum which represent 80% of the ventricular cardiomyocyte population (20). Because Tnni3k influences the number of adult cardiomyocytes that are diploid or polyploid (2), we specifically focused in this analysis on binucleated cardiomyocytes, which are the vast majority (>90%) of ventricular cardiomyocytes even in Tnni3k mutants. We found that the length and width of mutant cardiomyocytes were both altered, such that mutant cells were shorter but wider (Fig. 3A; Table 2); as a result, total cardiomyocyte area was unchanged. Variance in these parameters between mice of the same genotypes was minimal (Supplementary Material, Fig. S2). The same shape alterations were observed in K489R/− and I685T/− as in null mutant cardiomyocytes, although I685T/I685T homozygotes were intermediate in phenotype, consistent with the expected impact of a hypomorphic allele. The ratio of cell length to width is the aspect ratio, which was 5.1 and 5.3 for wild-type and heterozygous cardiomyocytes, 4.0 for I685T homozygous cardiomyocytes, 3.3 for null mutant cardiomyocytes, and 3.1 and 3.5 for K489R/− and I685T/− cardiomyocytes (Table 2). A likely interpretation is that the shape changes of individual mutant cardiomyocytes collectively account for the overall change in the dimensions of the mutant myocardium, whereas the conservation of cardiomyocyte area explains the lack of change in heart and ventricular weight.
Figure 3.
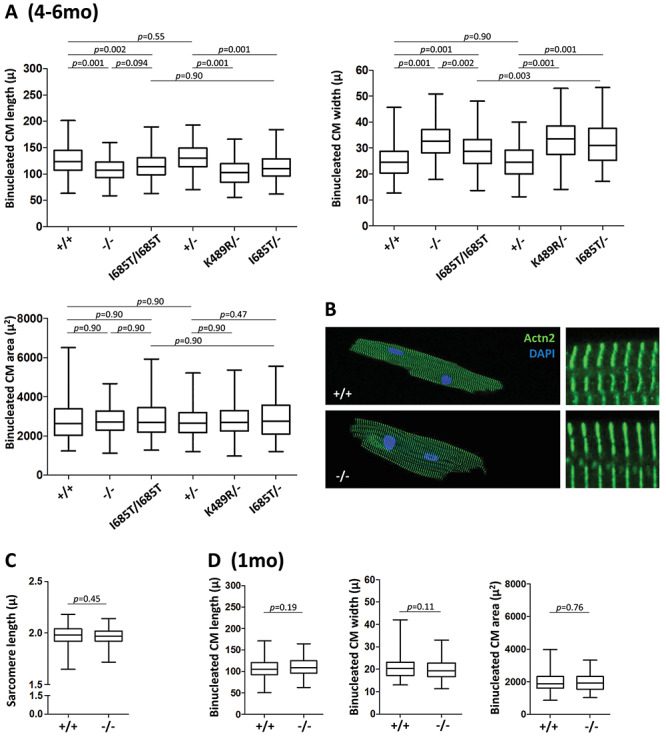
Cardiomyocyte cellular remodeling. (A) Length, width and area measurements of individual binucleated cardiomyocytes from 4 to 6 month old mice of the indicated genotypes, shown as a bars and whiskers plot (mean, middle quartiles and full range of data). Numbers of cells measured are listed in Table 2. Measurements were aggregated from roughly equal numbers of cells from 4 to 6 mice of the indicated genotypes. The (minimal) variance between cell preparations from individual mice is shown in Supplementary Material, Figure S2A. (B) Visualization of sarcomere structure and organization in cardiomyocytes from 6 month old mice by alpha-actinin2 (Actn2) staining and confocal microscopy. The cells shown correspond to the average dimensions measured in panel A. (C) Measurement of sarcomere length, obtained from the same evaluation used to measure sarcomere shortening during contraction that is shown in Figure 4A. (D) Length, width and area measurements of individual binucleated cardiomyocytes from 1 month old mice of the indicated genotypes; see also Table 2. The same analysis expressed on a per mouse basis is shown in Supplementary Material, Figure S2B.
Table 2.
Binucleated cardiomyocyte architectural parameters
| Genotype | Age (months) | n (mice); n (cells) | Length (μ) | Width (μ) | Area (μ2) | Aspect ratio |
|---|---|---|---|---|---|---|
| +/+ | 1 m | 4; 160 | 108.0 ± 21.2 | 20.8 ± 4.7 | 1981.3 ± 535.6 | 5.2 |
| −/− | 4; 177 | 110.9 ± 19.8 | 19.9 ± 4.6 | 1963.5 ± 513.0 | 5.6 | |
| +/+ | 4–6 m | 6; 147 | 126.8 ± 28.0 | 24.8 ± 6.2 | 2802.3 ± 972.2 | 5.1 |
| −/− | 5; 119 | 107.6 ± 19.9 | 32.5 ± 7.0 | 2834.1 ± 721.9 | 3.3 | |
| I685T/I685T | 4; 125 | 115.7 ± 24.2 | 28.9 ± 7.1 | 2846.2 ± 884.0 | 4.0 | |
| +/− | 4; 170 | 131.3 ± 24.7 | 24.7 ± 5.7 | 2745.0 ± 824.7 | 5.3 | |
| K489R/− | 4; 148 | 102.5 ± 22.4 | 33.5 ± 8.0 | 2827.0 ± 868.0 | 3.1 | |
| I685T/− | 4; 142 | 112.8 ± 23.7 | 32.2 ± 8.4 | 2923.8 ± 961.7 | 3.5 |
We performed immunofluorescence staining on isolated cardiomyocyte to visualize alpha-actinin 2 (Actn2), which is localized to the Z-disc in muscle cells and is commonly used as a marker to visualize sarcomere structure. No obvious sarcomere disorganization was observed in Tnni3k null cardiomyocytes (Fig. 3B). Additionally, there was no difference in sarcomere length between wild-type and mutant cardiomyocytes (Fig. 3C).
We examined cardiomyocytes from 1 month old Tnni3k wild-type and null mice in the same manner (Fig. 3D). At this age, there was no difference in cell length or width (aspect ratio 5.2 and 5.6, respectively; Table 2). Thus, changes in cardiomyocyte shape in mutant hearts, like those of the ventricular myocardium overall, are an acquired phenotype.
We next evaluated contractility in paced primary cardiomyocytes from adult 2–4 month old mice, i.e. during the early period of concentric remodeling. Under basal conditions, cardiomyocytes from wild-type and null mutant mice were equivalent in sarcomere shortening percentage as well as shortening and relaxation velocities (Fig. 4A).
Figure 4.
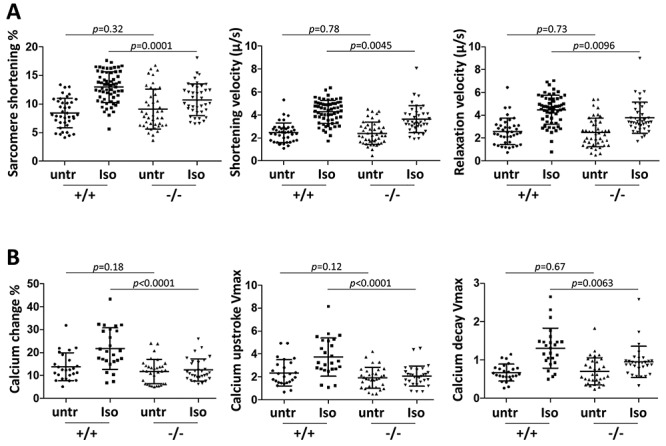
Cardiomyocyte contractility and calcium dynamics without and with isoproterenol treatment. (A) Cardiomyocyte contractility in paced cells. Sarcomere shortening percent at peak contraction, shortening velocity and relaxation velocity were measured for individual cardiomyocytes during the contraction cycle, in untreated (untr) and isoproterenol (Iso) treated cells. Cells were either untreated or treated, but not both. Single cell preparations from 11 +/+ mice and 9 −/− mice were used in this analysis. (B) Calcium dynamics in paced cells measured by Fura-2 signal. Percent change in cytoplasmic calcium fluorescence at peak amplitude, and maximum fluorescence upstroke and fluorescence decay velocities (units of fluorescence/sec) were measured for individual cardiomyocytes during the contraction cycle. Each dot indicates measurement for an individual cell from 8 +/+ mice and 8 −/− mice. See also Supplementary Material, Table S1 for statistical comparison of untreated versus treated cells by genotype.
In humans and animals, symptoms of heart failure may be mild under resting conditions but become evident with exercise, as exertion causes the release of catecholamines which drive the heart to perform more robustly. Catecholamines are agonists of beta-adrenergic receptors. Similarly, in cells in culture, an underlying contractile deficiency may be revealed by beta-adrenergic receptor activation even when basal function is normal. The difference in unstressed versus stressed contraction is termed contractile reserve. Because adrenergic response is a major feature of cardiomyocyte biology, and is associated with hypertrophy (21–23) and concentric remodeling (24–26), we addressed the possible impairment of adrenergic responsiveness in isolated Tnni3k mutant cardiomyocytes by challenging with 10 nM isoproterenol, a beta-adrenergic receptor agonist. Wild-type cardiomyocytes substantially increased all measured aspects of contraction (sarcomere shortening percent, shortening and relaxation velocities) in response to isoproterenol. In contrast, cells from null mutant mice were significantly impaired in their degree of responsiveness (Fig. 4A, Supplementary Material, Table S1).
At the subcellular level, the initiation and extent of cardiomyocyte contraction are controlled by release of calcium from the sarcoplasmic reticulum; isoproterenol and other beta-adrenergic receptor agonists stimulate contraction in part by inducing a greater level of calcium influx and release. We measured cytoplasmic calcium in paced cardiomyocytes by Fura-2 fluorescence (Fig. 4B). In untreated cells, the increase in peak fluorescence intensity, as well as the rates of calcium release and reuptake (cytoplasmic decay), were equivalent between wild-type and mutant cells. These parameters were enhanced substantially in wild-type cells in response to isoproterenol, but were severely attenuated in mutant cells (Fig. 4B, Supplementary Material, Table S1). This confirms that the impaired contractile response to isoproterenol in mutant cells is the manifestation of impaired calcium handling, including both calcium release and calcium reuptake.
Activation of beta-adrenergic receptors results in cyclic AMP production, which induces protein kinase A (PKA) activity. In cardiomyocytes, active PKA modulates (by phosphorylation) the activity of several intracellular targets that facilitate calcium handling or contraction. These targets are located in several domains of the cell, including the sarcoplasmic reticulum (e.g. the ryanodine receptor Ryr2 and phospholamban (Pln)), the nucleus (CREB1), and the sarcomere (troponin I3). In ventricular tissue lysates, there was no difference between wild-type and Tnni3k null mice (2 month old) in the expression level of these PKA targets nor of their phosphorylation levels under baseline conditions (Fig. 5A and B). This is consistent with the absence of change in contractility and calcium dynamics in isolated cardiomyocytes under basal (non-stimulated) conditions in culture, and indicates that absence of Tnni3k does not alter the abundance of these components of the contractile machinery.
Figure 5.
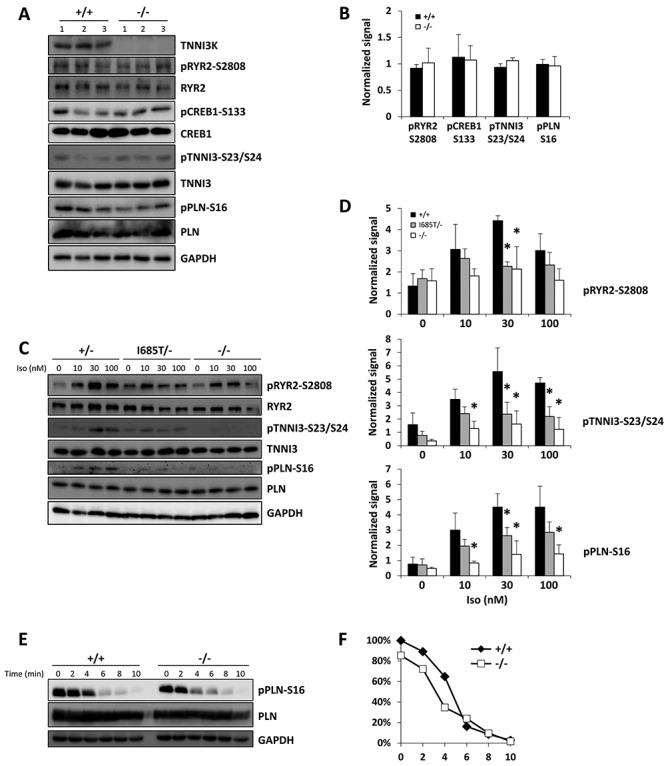
Altered PKA activity in Tnni3k mutant cardiomyocytes. (A, B) Western blot using phospho-specific and total protein antibodies with lysates from 3 +/+ and 3 −/− mouse hearts. The blots shown in A were quantified and graphed in B. (C, D) Tnni3k mutant cardiomyocytes have impaired response to isoproterenol. Cells were isolated and plated, treated with the indicated concentration of isoproterenol (Iso) for 8 min, then harvested. One western blot is shown for results with cells from individual mice of the genotypes shown; the aggregated quantified data from three independent blots was used to assemble the charts shown in (D). The control cells in the blot shown were from a heterozygous mouse (+/−); and were from +/+ mice in the other two blots; all three were aggregated in the quantitation shown in (D) (labeled as +/+). Asterisk indicates statistically significant differences (P < 0.05), a complete compilation of all statistical comparisons for western blots is provided in Supplementary Material, Table S2. A parallel analysis that includes K489R/− cardiomyocytes is shown in Supplementary Material, Figure S3A–B. (E, F) Measurement of phospholamban S16 dephosphorylation. Cells were pretreated with 300 nM isoproterenol, then washed and cultured in media without isoproterenol and supplemented instead with propranolol. The times indicated refer to time after removal of isoproterenol. Quantified data are graphed in (F). This analysis was repeated three times with comparable results.
We then tested the functionality of adrenergic receptor-induced PKA activity in isolated primary cardiomyocytes from 2 month old mice untreated or treated with varying doses of isoproterenol. For all PKA targets, phosphorylation was compromised in cells from mice of all mutant genotypes (null, K489R, I685T) compared to wild-type or heterozygous cells (Figs. 5C, D and S3A, B). Because these targets are in different domains of the cell, this outcome implies that overall PKA signaling, rather than a specific PKA interaction domain or substrate, was blunted in mutant cells. The same degree of compromised phosphorylation of these targets was observed after treatment of cells with 8-Br-cAMP, a membrane-permeable non-degradable cAMP analog that bypasses the involvement of adrenergic receptors to directly activate PKA activity (Supplementary Material, Fig. S3C and D). The level of the PKA catalytic subunit (PRKACA) was not affected by Tnni3k mutation (Supplementary Material, Fig. S3A and C), implying that Tnni3k modulates PKA activity rather than protein expression.
We next tested PKA activity in response to isoproterenol in 1 month old cardiomyocytes (Supplementary Material, Fig. S3E); the 1-month timepoint is before the onset of morphological changes associated with concentric remodeling (Figs. 2F and 3D). We observed the same impairment at 1 month as we observed in cells from older mice, indicating at minimum that altered PKA signaling is not a consequence of altered cardiomyocyte geometry associated with Tnni3k mutation.
Phosphorylation levels of kinase substrates represent the combined activities of kinase phosphorylation and phosphatase dephosphorylation. To address whether phosphatase activity was altered by Tnni3k, we fully activated PKA signaling by treating cells with a maximum dose of isoproterenol (300 nM), then replaced the culture media with fresh media lacking isoproterenol and instead supplemented with propranolol, which is a beta-adrenergic receptor antagonist. We analyzed Pln phosphorylation at various times after media replacement. As expected, Pln phosphorylation decreased in a time-dependent manner, which under these conditions is a reflection of phosphatase activity. There was no difference in the rate of dephosphorylation in control and mutant cells (Fig. 5E and F), and thus no indication with this assay of any influence of Tnni3k on phosphatase abundance or activity. These data implicate Tnni3k as a positive modulator of PKA signaling in cardiomyocytes, such that adrenergic responsiveness is compromised in Tnni3k mutant cells.
Discussion
Despite its evolutionary conservation, prior observations of Tnni3k function in heart physiology have only reported a maladaptive role when expressed, causing a worsening of outcome in the context of ischemia–reperfusion, calsequestrin overexpression and transaortic constriction. In this analysis, we have shown in C57BL/6J mice that Tnni3k loss-of-function mutations result in concentric remodeling and in impaired contraction under isoproterenol challenge. These studies are the first demonstration that Tnni3k has a beneficial role in normal heart biology, which provides a rationale for the conservation of the gene through evolution.
Concentric remodeling as occurs in Tnni3k null mice was replicated in mice expressing the K489R kinase-dead variant, demonstrating the necessity of Tnni3k kinase activity in maintaining normal cardiac physiology. The same phenotype was observed in mice carrying the homolog of the human I686T variant with hypomorphic kinase activity. Clearly, this variant is sufficiently compromised in activity to replicate the consequences to myocardium dimension, cardiomyocyte aspect ratio, PKA signaling and percentage of diploid cardiomyocytes as seen in null mice. Human heart geometry is known to be variable even among patients with comparable systolic blood pressure (27,28); the I686T allele and other TNNI3K hypomorphic or null alleles distributed in the human population (5) might therefore contribute to this variation. Individuals with concentric remodeling, like those with concentric hypertrophy, are at heightened risk for progression to dilated cardiomyopathy and heart failure (10–13). This suggests that TNNI3K alleles, as well as TNNI3K downstream targets and pathways, may also have a previously unsuspected contribution to human heart disease.
In previous studies, families with private heterozygous TNNI3K variants have been characterized for conduction system disease. In at least several cases, the variants are almost certainly hypomorphic or loss-of-function alleles (29–32), implying haploinsufficiency rather than gain-of-function (although one gain-of-function allele with similar conduction system disease has also been described (33)). Interestingly, a common observation in these families was progression to dilated cardiomyopathy and heart failure, which might have been a consequence of decades of abnormal heart rhythm, but alternatively might instead reflect a suboptimal level of TNNI3K kinase activity in ventricular cardiomyocytes that over time progressed beyond concentric remodeling. The I686T allele has not to date been associated with either conduction system disease or heart failure. We speculate that these phenotypes might be accelerated under specific (but currently unknown) environmental conditions or in conjunction with additional genetic burden; for example, in human hypertrophy, disease severity with a primary gene mutation is worsened when additional gene variant alleles or additional disease comorbidities are present (34,35). In our analysis, mice were never evaluated beyond 1 year of age nor challenged in a manner that might have elicited a more extreme pathology spectrum.
In a prior study, cardiomyocyte-specific transgenic expression of wild-type TNNI3K in DBA/2J mice (which do not express endogenous Tnni3k) resulted in hypertrophy, fibrosis and reduced sarcomere length; this was not seen with expression of a kinase-dead version of the protein (6). One possible explanation is that the DBA/2J background is particularly sensitive to TNNI3K kinase activity such that these outcomes occurred. Alternatively, because the transgene was overexpressed relative to the natural level of expression in C57BL/6J mice, another possibility is that normal cardiomyocyte physiology in any mouse strain requires a moderate range of TNNI3K kinase activity, such that too much (as in the DBA/2J transgenic study) or too little (as in our C57BL/6J loss-of-function studies) both cause detrimental consequences, albeit not of the exact same type.
Because a natural Tnni3k loss-of-function allele is homozygous in numerous commonly-used inbred mouse strains (3), it might be thought surprising that the concentric remodeling phenotype as reported here has not been previously recognized. All inbred mouse strains carry a unique range of gene variants that positively or negatively modify any phenotype of interest, including the tendency to hypertrophy (27). Without direct genetic manipulation, it would be difficult to recognize TNNI3K involvement in concentric remodeling or in any manifestation of hypertrophy through an evaluation between strains, nor would this be easy to detect by genome-wide association in mouse or human studies. In our analysis we kept all mice on a single inbred background, which allowed us to visualize the morphological and functional consequences of Tnni3k mutant alleles in the C57BL/6J context in a fully controlled manner.
Although the phenotypic consequences of Tnni3k mutation are evident, the pathways through which Tnni3k function supports normal cardiomyocyte physiology are uncertain. PKA signaling after isoproterenol treatment was attenuated in mutant cardiomyocytes, and in vivo over the course of months, diminished adrenergic response may cause or contribute to the onset of concentric remodeling. We note that impaired PKA signaling was observed in cardiomyocytes from 1 month old mice, preceding the onset of concentric remodeling although not necessarily causing this phenotype. As with all major intracellular signaling pathways, it is likely that Tnni3k influences multiple pathways, including but not limited to PKA. The primary limitation to understanding Tnni3k function is that no authentic in vivo kinase substrates are yet known. We have initiated a phosphoproteomic screen that may reveal primary substrates and components of downstream pathways that will clarify how Tnni3k function contributes to normal heart homeostasis and how disease pathology emerges in its absence.
Tnni3k mutation in C57BL/6J mice results in an increase in the percentage of diploid cardiomyocytes in the postnatal heart (4). Theoretically, concentric remodeling and impaired contraction in Tnni3k mutant hearts could be a consequence of elevated diploid cardiomyocyte content rather than a manifestation of Tnni3k in the contractile function of all adult cardiomyocytes. However, mouse strains with wild-type Tnni3k with a level of mononuclear cardiomyocytes higher than C57BL/6J-inbred Tnni3k mutants were identified in our previous study (4) and these are not known to have remodeling or hypertrophy, implying that having more diploid cardiomyocytes per se does not lead to this response. A fuller resolution will come as we introduce different gene manipulations into C57BL/6J mice; we predict these will increase diploid cardiomyocyte content without a contractile and remodeling phenotype, thus demonstrating the latter to be a specific consequence of Tnni3k action.
Tnni3k influences the percentage of diploid cardiomyocytes by influencing the degree to which cell cycle-activated cardiomyocytes are able to complete cytokinesis (4). This property, in which absence of Tnni3k results in a higher frequency of completed cell cycle events, is manifest naturally only shortly after birth, the time when most cardiomyocytes become polyploid; cell cycle activity in the normal heart is minimal for the remainder of life. However, reactivation of cell cycle and the same influence of Tnni3k in modulating cell cycle completion are reiterated in adulthood after infarction injury. Pharmacological TNNI3K inhibitors have been developed recently (36,37); our analysis suggests that pharmacological inhibition over an extended period might promote concentric remodeling and hypertrophy. However, cardiomyocyte cell cycle activity after adult heart injury in mice is mostly confined to a 1–2 week window, such that pharmacological TNNI3K inhibition in an infarction patient during this period might support more efficient heart regeneration and yet avoid adverse consequences to cardiomyocyte contraction and geometry.
Materials and Methods
An expanded Materials and Methods section is included as Supplemental materials. Tnni3k null and K489R alleles used in this study were described in our prior work (2,4). The I685T allele was made by standard CRISPR-mediated replacement in fertilized C57BL/6J eggs. All mice originated on and were maintained on a C57BL/6J background; wild-type and heterozygous mice came from the same breeding colony. Freshly isolated hearts were immersed in KCl to arrest in diastole prior to fixation and sectioning. Cardiomyocytes were isolated by Langendorff collagenase perfusion, then either fixed for geometric measurement or restored to normal calcium for physiological studies. Contractility and calcium dynamics were visualized in Fura2-loaded cells at 1 Hz pacing. Treatment of cultured cells and western blotting of cell lysate followed standard procedures.
Supplementary Material
Acknowledgements
The authors acknowledge Martin Morad for discussions and for supervising early patch clamp experiments that were not included in this manuscript, Monica Sentmanat of the Genome Engineering & iPSC Center (GEiC), Washington University, St. Louis for design and testing of CRISPR reagents, and Alexander Awgulewitsch and Jan Guz of the MUSC Transgenic and Genome Editing Core for derivation of the Tnni3k I685T allele.
Conflict of Interest statement. D.P.J. has received personal payments as a scientific advisor from 4d Molecular Therapeutics, ADRx Therapeutics and Pfizer; MUSC receives payments for his clinical trials from Array Biopharma and Eidos Therapeutics. There is no overlap between the work described in the current manuscript and these activities. The remaining authors declare that no conflicts of interests exist.
Contributor Information
Peiheng Gan, Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, SC, USA; Department of Stem Cell Biology and Regenerative Medicine, University of Southern California Keck School of Medicine, Los Angeles, CA, USA.
Catalin Baicu, Department of Medicine Division of Cardiology, Medical University of South Carolina, Charleston, SC, USA.
Hirofumi Watanabe, Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, SC, USA.
Kristy Wang, Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, SC, USA.
Ge Tao, Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, SC, USA.
Daniel P Judge, Department of Medicine Division of Cardiology, Medical University of South Carolina, Charleston, SC, USA.
Michael R Zile, Department of Medicine Division of Cardiology, Medical University of South Carolina, Charleston, SC, USA.
Takako Makita, Darby Children’s Research Institute, Department of Pediatrics, Medical University of South Carolina, Charleston, SC, USA.
Rupak Mukherjee, Department of Medicine Division of Cardiology, Medical University of South Carolina, Charleston, SC, USA.
Henry M Sucov, Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, SC, USA; Department of Medicine Division of Cardiology, Medical University of South Carolina, Charleston, SC, USA.
Funding
National Institutes of Health (HL144938 to H.M.S.). Predoctoral fellowship from the American Heart Association (P.G.).
References
- 1. Zhao Y., Meng X.M., Wei Y.J., Zhao X.W., Liu D.Q., Cao H.Q., Liew C.C. and Ding J.F. (2003) Cloning and characterization of a novel cardiac-specific kinase that interacts specifically with cardiac troponin I. J. Mol. Med. (Berl), 81, 297–304. [DOI] [PubMed] [Google Scholar]
- 2. Gan P., Patterson M., Velasquez A., Wang K., Tian D., Windle J.J., Tao G., Judge D.P., Makita T., Park T.J. et al. (2019) Tnni3k alleles influence ventricular mononuclear diploid cardiomyocyte frequency. PLoS Genet., 15, e1008354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wheeler F.C., Tang H., Marks O.A., Hadnott T.N., Chu P.L., Mao L., Rockman H.A. and Marchuk D.A. (2009) Tnni3k modifies disease progression in murine models of cardiomyopathy. PLoS Genet., 5, e1000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patterson M., Barske L., Van Handel B., Rau C.D., Gan P., Sharma A., Parikh S., Denholtz M., Huang Y., Yamaguchi Y. et al. (2017) Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet., 49, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang H., Xiao K., Mao L., Rockman H.A. and Marchuk D.A. (2013) Overexpression of TNNI3K, a cardiac-specific MAPKKK, promotes cardiac dysfunction. J. Mol. Cell. Cardiol., 54, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vagnozzi R.J., Gatto G.J. Jr., Kallander L.S., Hoffman N.E., Mallilankaraman K., Ballard V.L., Lawhorn B.G., Stoy P., Philp J., Graves A.P. et al. (2013) Inhibition of the cardiomyocyte-specific kinase TNNI3K limits oxidative stress, injury, and adverse remodeling in the ischemic heart. Sci. Transl. Med., 5, 207ra141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gan P., Patterson M. and Sucov H.M. (2020) Cardiomyocyte polyploidy and implications for heart regeneration. Annu. Rev. Physiol., 82, 45–61. [DOI] [PubMed] [Google Scholar]
- 9. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M. and Sirotkin K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gaasch W.H., Delorey D.E., St John Sutton M.G. and Zile M.R. (2008) Patterns of structural and functional remodeling of the left ventricle in chronic heart failure. Am. J. Cardiol., 102, 459–462. [DOI] [PubMed] [Google Scholar]
- 11. Lavie C.J., Patel D.A., Milani R.V., Ventura H.O., Shah S. and Gilliland Y. (2014) Impact of echocardiographic left ventricular geometry on clinical prognosis. Prog. Cardiovasc. Dis., 57, 3–9. [DOI] [PubMed] [Google Scholar]
- 12. Pierdomenico S.D., Di Nicola M., Pierdomenico A.M., Lapenna D. and Cuccurullo F. (2011) Cardiovascular risk in subjects with left ventricular concentric remodeling at baseline examination: a meta-analysis. J. Hum. Hypertens., 25, 585–591. [DOI] [PubMed] [Google Scholar]
- 13. Zile M.R., Gaasch W.H., Patel K., Aban I.B. and Ahmed A. (2014) Adverse left ventricular remodeling in community-dwelling older adults predicts incident heart failure and mortality. JACC Heart Fail, 2, 512–522. [DOI] [PubMed] [Google Scholar]
- 14. Guo Y. and Pu W.T. (2020) Cardiomyocyte maturation: new phase in development. Circ. Res., 126, 1086–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuroda K., Kato T.S. and Amano A. (2015) Hypertensive cardiomyopathy: a clinical approach and literature review. World J. Hypertension, 5, 41–52. [Google Scholar]
- 16. Clemente C.F., Xavier-Neto J., Dalla Costa A.P., Consonni S.R., Antunes J.E., Rocco S.A., Pereira M.B., Judice C.C., Strauss B., Joazeiro P.P. et al. (2012) Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J. Mol. Cell. Cardiol., 52, 493–501. [DOI] [PubMed] [Google Scholar]
- 17. Kehat I., Davis J., Tiburcy M., Accornero F., Saba-El-Leil M.K., Maillet M., York A.J., Lorenz J.N., Zimmermann W.H., Meloche S. et al. (2011) Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res., 108, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ver Heyen M., Heymans S., Antoons G., Reed T., Periasamy M., Awede B., Lebacq J., Vangheluwe P., Dewerchin M., Collen D. et al. (2001) Replacement of the muscle-specific sarcoplasmic reticulum ca(2+)-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ. Res., 89, 838–846. [DOI] [PubMed] [Google Scholar]
- 19. Burke M.A., Cook S.A., Seidman J.G. and Seidman C.E. (2016) Clinical and mechanistic insights into the genetics of cardiomyopathy. J. Am. Coll. Cardiol., 68, 2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerdes A.M., Moore J.A., Hines J.M., Kirkland P.A. and Bishop S.P. (1986) Regional differences in myocyte size in normal rat heart. Anat. Rec., 215, 420–426. [DOI] [PubMed] [Google Scholar]
- 21. Rababa'h A., Singh S., Suryavanshi S.V., Altarabsheh S.E., Deo S.V. and McConnell B.K. (2014) Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase a (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci., 16, 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu J., Han Q., Shi H., Liu W., Chu T. and Li H. (2017) Role of PKA in the process of neonatal cardiomyocyte hypertrophy induced by urotensin II. Int. J. Mol. Med., 40, 499–504. [DOI] [PubMed] [Google Scholar]
- 23. Yamazaki T., Komuro I. and Yazaki Y. (1998) Signalling pathways for cardiac hypertrophy. Cell. Signal., 10, 693–698. [DOI] [PubMed] [Google Scholar]
- 24. Donaldson C., Palmer B.M., Zile M., Maughan D.W., Ikonomidis J.S., Granzier H., Meyer M., VanBuren P. and LeWinter M.M. (2012) Myosin cross-bridge dynamics in patients with hypertension and concentric left ventricular remodeling. Circ. Heart Fail., 5, 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patel B.B., Raad M., Sebag I.A. and Chalifour L.E. (2013) Lifelong exposure to bisphenol a alters cardiac structure/function, protein expression, and DNA methylation in adult mice. Toxicol. Sci., 133, 174–185. [DOI] [PubMed] [Google Scholar]
- 26. Yingxin P., Jiang S., Xiaoyong Q., Hao X., Chunli R., Dongmei Y., Zhiqin G., Shiling Z. and Min W. (2004) Regulation of the catecholamine beta-adrenergic system in ventricular remodeling of hypertension. Jpn. Heart J., 45, 285–296. [DOI] [PubMed] [Google Scholar]
- 27. Barrick C.J., Rojas M., Schoonhoven R., Smyth S.S. and Threadgill D.W. (2007) Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol., 292, H2119–H2130. [DOI] [PubMed] [Google Scholar]
- 28. Liebson P.R., Grandits G., Prineas R., Dianzumba S., Flack J.M., Cutler J.A., Grimm R. and Stamler J. (1993) Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the treatment of mild hypertension study (TOMHS). Circulation, 87, 476–486. [DOI] [PubMed] [Google Scholar]
- 29. Fan L.L., Huang H., Jin J.Y., Li J.J., Chen Y.Q., Zhao S.P. and Xiang R. (2018) Whole exome sequencing identifies a novel mutation (c.333+2T>C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene, 648, 63–67. [DOI] [PubMed] [Google Scholar]
- 30. Theis J.L., Zimmermann M.T., Larsen B.T., Rybakova I.N., Long P.A., Evans J.M., Middha S., Andrade M., Moss R.L., Wieben E.D. et al. (2014) TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy. Hum. Mol. Genet., 23, 5793–5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xi Y., Honeywell C., Zhang D., Schwartzentruber J., Beaulieu C.L., Tetreault M., Hartley T., Marton J., Vidal S.M., Majewski J. et al. (2015) Whole exome sequencing identifies the TNNI3K gene as a cause of familial conduction system disease and congenital junctional ectopic tachycardia. Int. J. Cardiol., 185, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu J., Liu D., Li M., Wu K., Liu N., Zhao C., Shi X. and Liu Q. (2020) Identification of a nonsense mutation in TNNI3K associated with cardiac conduction disease. J. Clin. Lab. Anal., 34, e23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Podliesna S., Delanne J., Miller L., Tester D.J., Uzunyan M., Yano S., Klerk M., Cannon B.C., Khongphatthanayothin A., Laurent G. et al. (2019) Supraventricular tachycardias, conduction disease, and cardiomyopathy in 3 families with the same rare variant in TNNI3K (p.Glu768Lys). Heart Rhythm, 16, 98–105. [DOI] [PubMed] [Google Scholar]
- 34. Burns C., Bagnall R.D., Lam L., Semsarian C. and Ingles J. (2017) Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ. Cardiovasc. Genet., 10, e001666. [DOI] [PubMed] [Google Scholar]
- 35. Claes G.R., Tienen F.H., Lindsey P., Krapels I.P., Helderman-van den Enden A.T., Hoos M.B., Barrois Y.E., Janssen J.W., Paulussen A.D., Sels J.W. et al. (2016) Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur. Heart J., 37, 1815–1822. [DOI] [PubMed] [Google Scholar]
- 36. Asquith C.R.M., Laitinen T., Wells C.I., Tizzard G.J. and Zuercher W.J. (2020) New insights into 4-Anilinoquinazolines as inhibitors of cardiac troponin I-interacting kinase (TNNi3K). Molecules, 25, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Philp J., Lawhorn B.G., Graves A.P., Shewchuk L., Rivera K.L., Jolivette L.J., Holt D.A., Gatto G.J. Jr. and Kallander L.S. (2018) 4,6-Diaminopyrimidines as highly preferred troponin I-interacting kinase (TNNI3K) inhibitors. J. Med. Chem., 61, 3076–3088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.