

Abstract
Gliomas have substantial mortality to incidence rate ratio and a dismal clinical course. Newer molecular insights, therefore, are imperative to refine glioma diagnosis, prognosis and therapy. Meningioma 1 (MN1) gene is a transcriptional co-regulator implicated in other malignancies, albeit its significance in glioma pathology remains to be explored. IGFBP5 is regulated transcriptionally by MN1 and IGF1 and is associated with higher glioma grade and shorter survival time, prompting us to ascertain their correlation in these tumors. We quantified the expression of MN1, IGFBP5 and IGF1 in 40 glioma samples and examined their interrelatedness. MN1 mRNA-protein inter-correlation and the gene’s copy number were evaluated in these tumors. Publicly available TCGA datasets were used to examine the association of MN1 expression levels with patient survival and for validating our findings. We observed MN1 overexpression correlated with low-grade (LGGs) and not high-grade gliomas and is not determined by the copy number alteration of the gene. Notably, gliomas with upregulated MN1 have better overall survival (OS) and progression-free survival (PFS). IGFBP5 expression associated inversely with MN1 expression levels in gliomas but correlated positively with IGF1 expression in only LGGs. This suggests a potential grade-specific interplay between repressive and activating roles of MN1 and IGF1, respectively, in the regulation of IGFBP5. Thus, MN1 overexpression, a promising predictor of OS and PFS in gliomas, may serve as a prognostic biomarker in clinical practice to categorize patients with survival advantage.
Introduction
Gliomas are clinically recalcitrant and constitute one of the two most common types of primary brain and central nervous system (CNS) tumors (1,2). Epidemiological data suggest gliomas comprise <1.6% of all new cancer diagnoses but have substantially higher mortality to incidence rate ratio (MIR = 0.80) as compared with breast (0.28) and prostate (0.26) cancer that have significantly higher morbidity (Fig. 1A) (3,4). Until recently, World Health Organization (WHO) recommended the evaluation of tumor histology alone as the standard for glioma diagnosis (5). Gliomas based on their cytomorphologic similarity to non-neoplastic glial cells, namely the astrocytes, oligodendrocytes and ependymal cells, are classified as astrocytomas, oligodendrogliomas and ependymomas, respectively. Gliomas are further graded based on the WHO consensus-derived scale of I–IV (5). While the tumors of WHO grades I and II are categorized as low-grade gliomas (LGGs), those ascribed to grades III and IV are considered high-grade gliomas (HGGs) (6–9). Gliomas of all grades with the exception of WHO grade I are typically diffuse invasive tumors. They infiltrate the surrounding brain parenchyma extensively and very early in their course, making the complete surgical resection extremely difficult and unlikely (10).
Figure 1.
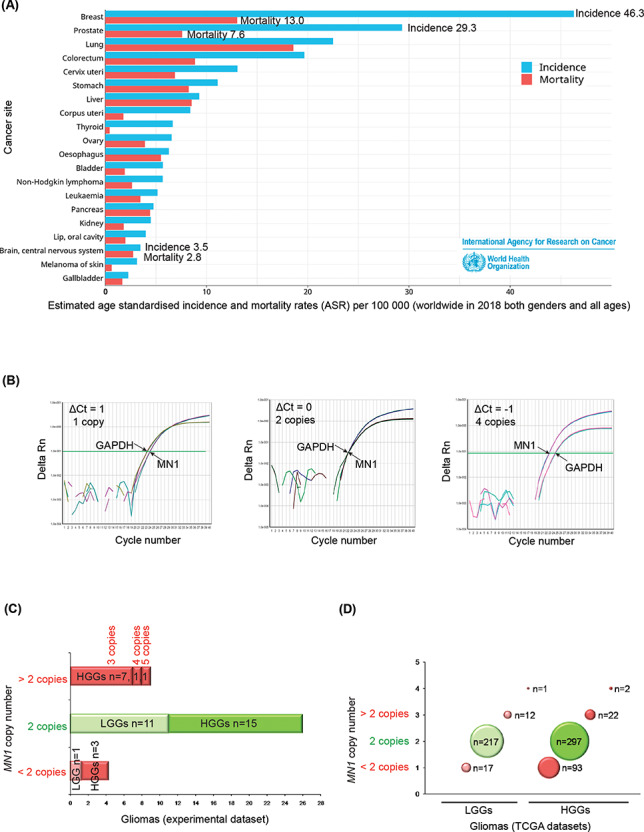
Epidemiological data and copy number analysis of MN1 gene in gliomas. (A) Bar graph showing global estimates of age standardized incidence and mortality rates (ASR) per 100 000 people, in both genders and all ages in the year 2018, and is arranged according to top 20 cancer sites (3). (B) qPCR amplification plots with MN1 as the gene of interest and RNaseP as the reference gene where ΔCt = 1 (left panel), ΔCt = 0 (middle panel) and ΔCt = −1 (right panel) correspond to one, two and four copies of MN1 gene, respectively. (C) Bar graph summarizing MN1 copy number details of gliomas examined in the present study. Red and green bars indicate cases that have altered and normal copy numbers, respectively. ‘n’ denotes the number of cases (LGGs or HGGs as indicated). (D) Bubble plot showing copy number status of MN1 gene in LGGs and HGGs from the TCGA datasets. Red and green bubbles represent cases with CNAs and normal (2) copy number, respectively. Number of cases (n) in each category is indicated along the bubble.
Besides their diverse cellular origin, gliomas show substantial molecular and genetic heterogeneity. Different studies reported certain molecular signatures to be associated with specific glioma sub-types (7,11). These molecular determinants were incorporated by WHO to revise the classification system for glioma diagnosis in 2016 (12). While the new WHO classification system is still to be widely accepted, problems are becoming evident and further revisions are being proposed (13–17). Like other cancers, gliomas are characterized by aberrations in several molecular pathways that confer a growth advantage to these tumors (10). Insulin-like growth factor (IGF) system is one such pathway that has been implicated in Gliomas (18,19). The IGF pathway involves concerted interactions of two ligands IGF1 and IGF2, three receptors comprising IGF1 receptor (IGF1R), IGF2 receptor (IGF2R) and insulin receptor (IR), and six IGF-binding proteins (IGFBP1–IGFBP6) (20). IGF ligands mediate their growth-promoting effects by binding IGF1R. This transduces the phosphatidyl inositol 3′-kinase (PI3K) and mitogen activated protein kinase (MAPK) cascades to inhibit apoptosis and elicit cellular proliferation, respectively (20,21).
The IGF system plays pleiotropic roles in the development of the CNS with certain variations between the human and murine species (22). IGF1 and its receptor-IGF1R express in the normal human brain and are known to be upregulated in gliomas (23–30). In a study comprising 39 astrocytomas (WHO grades II–IV), IGF1 expression was observed in tumors of all grades. Markedly, only the proportion of immunopositive cells and not the staining intensity correlated with the histopathological grade of gliomas (30). It is also reported that exogenous IGF1 induces proliferative and anti-apoptotic signaling through its receptor IGF1R in cultured glioma cells (28,31).
In addition to their autocrine growth promoting roles, IGFs also have characteristics of a paracrine growth factor and therefore remain in circulation. These circulating IGFs in serum are bound by IGFBPs, which prolong their half-lives and control their exit from the vascular compartment (19). In the process, IGFBPs modulate spatiotemporal distribution and bioavailability of IGFs in a cell/tissue-type and species-specific manner. Of the six IGFBPs, IGFBP5 is evolutionarily the most conserved across vertebrates and is more than 97% identical between human, mouse and rat (32). IGFBP5 and IGF1 show spatiotemporal co-expression during brain development suggesting that their interplay is crucial to the process (22,33). Also, the brain of IGF1 transgenic mice shows upregulated IGFBP5 expression (34). Independent compelling observations about opposing roles of IGFBP5 (oncogenic and anti-oncogenic) in different cancers have been reported (19,35). Nevertheless, in gliomas, elevated levels of tissue IGFBP5 were associated with higher tumor grades and poor prognosis (36–38). Silencing IGFBP5 expression impedes invasion but promotes the proliferation of GBM cells in vitro, thus having opposing effects on two cellular hallmarks of neoplastic state (39).
Expression of IGFBP5 is regulated in part by IGF1 through both IGF1R-dependent and -independent mechanisms (40,41). Besides IGF1, several other biomolecules such as retinoic acid (RA), MN1, the transcription factor-activating enhancer binding protein 2 alpha (AP-2), prostaglandin E2, cortisol and vitamin D are known to regulate the transcription of IGFBP5 in a cell-type and context-dependent manner (41,42). RA, for example, can in opposing ways either induce or inhibit IGFBP5 expression mediated by the RA receptor/retinoic x receptor (RAR/RXR) (41). MN1 (Meningioma 1 gene, 22q12.1) was initially identified due to a balanced translocation (4;22) in a meningioma patient showing absence of its expression. Therefore, the authors proposed it as a candidate tumor suppressor gene (43). MN1 is a transcriptional co-regulator that can both induce and repress RAR/RXR target genes. As a co-activator, MN1 acts synergistically with RAR/RXR to activate the RA-mediated expression of IGFBP5 (44). MN1 is also known to transcriptionally co-repress RAR/RXR target genes (45–47). Interestingly, deletions in MN1 have been implicated in patients with neurodevelopmental abnormalities (48).
Independent studies have examined MN1 and IGFBP5 in varied contexts described above and associated the latter with poor prognosis in gliomas. It interested us that since MN1 transcriptionally regulates IGFBP5, are the expression levels of the two genes related and can MN1 help predict patient survival in human gliomas. Therefore, we undertook the present study to—(i) determine copy number alteration (CNA) and expression of MN1 in gliomas, and assess their correlation; (ii) discern association of MN1 expression with tumor grades and patient survival; (iii) quantify IGFBP5 and IGF1 expression in these patients and (iv) ascertain whether the expression of IGFBP5 changes as a function of MN1 and IGF1 expression levels.
Results
Gliomas show copy number alterations of MN1 gene
The gliomas in the present study (experimental dataset) were analyzed for MN1 CNA using quantitative real-time PCR (qPCR) assay. Representative qPCR amplification plots (Fig. 1B) depict the curves for MN1 (gene of interest) and RNaseP (reference gene) where the difference in cycle threshold (ΔCt) values of 1, 0 and −1 correspond to one, two and four copies of MN1 gene, respectively. We found that MN1 gene CNA occurred in gliomas (33%, i.e. n = 13/39, Fig. 1C), and it was more common in HGGs than in LGGs (P = 0.03). Specifically, in HGGs, copy number gain (n = 9/39) was observed thrice more often than loss (n = 3/39, Fig. 1C). None of the LGGs showed MN1 copy number gain.
Similar to the experimental dataset, the TCGA datasets also showed CNA of MN1 gene more frequently in HGGs than in LGGs (Fig. 1D). However, unlike the experimental dataset where copy number gain was more frequent in HGGs, in the TCGA datasets, copy number loss (n = 93) is ~4 times more frequent than gain (n = 24) in HGGs, whereas LGGs showed similar number of cases with copy number loss (n = 17) and gain (n = 13, Fig. 1D). Accordingly, MN1 copy number loss contributed considerably (P < 0.0001) to the higher overall CNA seen in HGGs as compared with LGGs. Notably, gliomas in either the experimental or TCGA datasets did not evince complete loss of the gene. To understand the role of MN1 CNA in glioma pathology, it is vital to understand the molecular significance of the gene in gliomas. As a step in this direction, we examined whether gliomas with MN1 CNA have altered gene expression and if those with normal (two) copies show expression similar to non-neoplastic brain.
Altered MN1 expression fails to conform to CNA but correlates with LGGs
Relative MN1 (mRNA) expression in the experimental dataset is represented as bar graph (Fig. 2A). MN1 expression in the non-neoplastic brain is considered as baseline/zero. Gliomas with relative MN1 expression values beyond the range of −0.3 and 0.2 are categorized as cases with downregulated and upregulated expression, respectively. We found that LGGs frequently evince MN1 overexpression as compared with HGGs (P = 0.04). Green and red numerals mentioned against the bar connote normal and altered copies, respectively, in the corresponding tumor (Fig. 2A). Tabulated summary (inset Fig. 2A) shows that ~45% gliomas (n = 18/40) have MN1 overexpression, and most gliomas, including those evaluated for protein expression, rarely show correlation between the gene’s expression and copy number status (Fig. 2A–D). To statistically assess if the observed CNA could reliably predict corresponding change in MN1 expression, we fitted the regression model to our data. High P-value and a low coefficient of determination indicate the lack of association (P = 0.17; R2 = 0.05, Fig. 2B) between gene’s copy number and mRNA expression in gliomas. Also, a negative slope (−0.2289) of the regression line suggests that with an increase in MN1 copies, gene expression tends to decrease. This signifies that alteration in MN1 copy number does not imply a relatable change in its expression.
Figure 2.
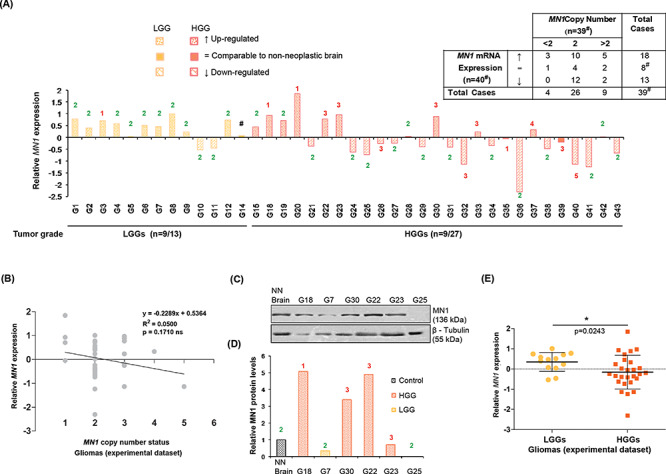
MN1 expression and its relation with the gene’s copy number alterations in gliomas. (A) Bar graph depicting relative MN1 mRNA expression (normalized to non-neoplastic brain tissues) in the experimental dataset. Numerals indicated above/below each of the bars denote copy number of MN1 gene in the corresponding glioma cases. #Case G14 showed upregulated MN1 transcripts but was not evaluable for the gene’s copy number. No. of cases showing overexpression versus total no. of cases analyzed for the two categories are mentioned below the x-axis. Inset tabulated summary of MN1 mRNA expression and copy number data in gliomas. ↑, = and ↓ signify upregulated expression, comparable and downregulated transcript levels relative to the non-neoplastic brain, respectively. (B) Scattergram shows ability of MN1 copy number to predict MN1 expression in gliomas (n = 39). Linear regression line fitted to the data is shown and ‘R2’ represents coefficient of determination. (C) Immunoblots and (D) graphical representation of densitometric evaluation of immunoblots, showing MN1 protein expression in representative glioma cases. β–tubulin was used as an experimental control. NN Brain indicates non-neoplastic sample used as control. (E) Graph summarizing relative MN1 mRNA expression in LGGs (n = 13) and HGGs (n = 27), where the central bar and whiskers indicate mean (±SD) values.
To ascertain whether gliomas with altered MN1 transcript levels have proportionate protein expression, we probed cases that had sufficient tumor tissue in excess of the diagnostic requirement, for MN1 and β-Tubulin expression. We noticed MN1 protein expression in glioma cases with discernible transcript levels (Fig. 2C and D). Nevertheless, in cases such as G7 and G23, despite observed transcript overexpression (Fig. 2A), MN1 protein levels were not upregulated (Fig. 2D). This discordance may be because commercially purchased total RNA and protein (non-neoplastic brain), used for normalizing tumor MN1 mRNA and protein expression, respectively, originated from different individuals. In the remaining cases, protein expression trend corresponds with but is not altered proportionate to the change in mRNA levels, suggesting a role of post-transcriptional regulatory mechanisms (Fig. 2A, C and D).
Interestingly in the experimental dataset, median MN1 expression is reasonably higher in LGGs than HGGs (P = 0.02, Fig. 2e). Analyses of the TCGA datasets also uncovered that LGGs have an appreciably higher MN1 expression than HGGs (P < 0.001, Fig. 3A). Further, to validate our observations of MN1 expression not correlating with its copy number status, we analyzed TCGA datasets for MN1 expression and CNA (Fig. 3B). Pairwise Wilcoxon test revealed LGGs and HGGs with single copy of the gene had markedly reduced average MN1 expression as compared with cases with normal copy number (P < 0.0001; Fig. 3B). The reverse was not true, because a gain in MN1 copy number did not signify an increased average expression in LGGs and HGGs (Fig. 3B). Regression analysis to test association between MN1 expression and the gene’s CNA yielded a significant test statistic (P < 0.0001) but had low R-squared values in both LGGs (R2 = 0.07, Fig. 3C) and HGGs (R2 = 0.12, Fig. 3D). Similar to the experimental dataset, in the TCGA dataset, examining the distribution of data points along the regression line suggests that gene’s copy number does not predict the variation in its expression levels, which is evident also in Fig. 3B.
Figure 3.
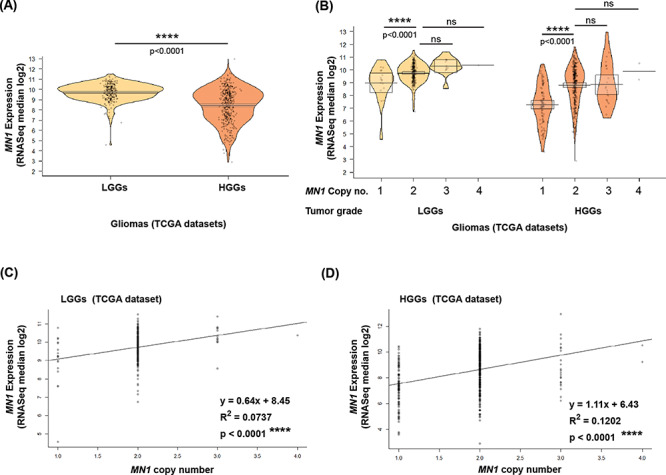
MN1 expression (RSEM) is not related to CNA in TCGA glioma datasets. Pirate plots showing MN1 expression observed in (A) LGGs (n = 249) and HGGs (n = 417), and (B) classified according to MN1 CNA in a grade wise manner (x-axis). P-values for statistical comparison of gene expression between glioma grades and cases with different copy number status are indicated above the plots. Regression analysis depicting MN1 expression (log transformed RNAseq median or RSEM values) as a function of CNA of the gene in (C) LGGs (n = 246) and (D) HGGs (n = 412). Linear regression line fitting the data is shown in black, and ‘R2’ represents the coefficient of determination.
Since the TCGA dataset validated our findings of altered MN1 expression correlating with LGGs, we wanted to test whether MN1 overexpression can be used to predict survival and assess it in relation to the widely accepted predictors such as Isocitrate dehydrogenase (IDH) mutation and 1p19q co-deletion.
MN1 overexpression predicts favorable survival in gliomas
IDH mutations seen in gliomas (including grades II and III astrocytomas, grades II and III oligodendrogliomas and GBM) are known to confer survival advantage in patients (11,49). Also, the co-deletion of chromosome arms 1p and 19q observed in only oligodendrogliomas (grades II and III) is positively associated with survival (11,49). We tested whether MN1 overexpression can sub-type LGGs by predicting better survival. Despite HGGs having lower median MN1 expression than LGGs, we also examined the relationship between MN1 expression and HGG patient survival. Survival prediction analysis was not possible with the experimental dataset, so we used the TCGA datasets.
Kaplan–Meier estimates of survival (with log-rank test) indicate that MN1 overexpression associated with longer survival in LGGs (P = 0.0001; Fig. 4A) and HGGs (P < 0.0001; Fig. 4D). Expectedly, LGGs with IDH mutation (P < 0.0001; Fig. 4B) and 1p19q co-deletion (P < 0.0001; Fig. 4C) have prolonged overall survival (OS). Likewise, HGGs with IDH mutation (P < 0.0001; Fig. 4E) and 1p19q co-deletion (P < 0.0001; Fig. 4F) have better overall and median survival (Fig. 4G). NA values for median survival in LGGs that overexpress MN1, and those with IDH wildtype (Fig. 4G), indicate that values could not be computed because the group did not drop to 50% survival probability at the end of available data (Fig. 4A and zoomed in region of the survival curve therein, and Fig. 4B). In such a case, it implies that median OS is greater than the last point on the survival curve. Therefore, in LGGs with MN1 overexpression median OS maybe ~180 months or more, this seems better than the median OS predicted by IDH mutation or 1p19q co-deletion.
Figure 4.
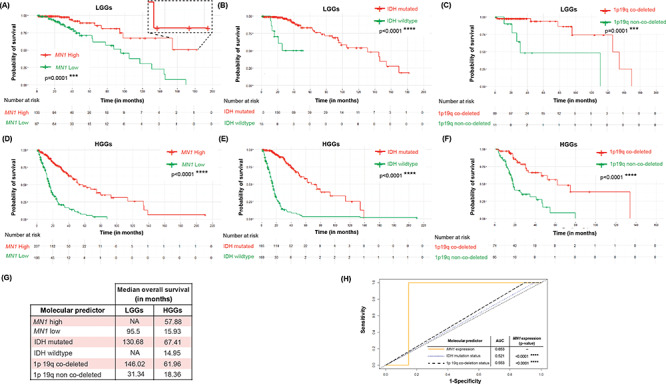
MN1 overexpression predicts better OS in gliomas. Kaplan–Meier survival curves stratify LGGs by (A) MN1 expression (n = 247), (B) IDH mutation status (n = 226) and (C) 1p19q deletion status (n = 100). HGGs are classified into sub-groups based on the association of probability of survival with (D) MN1 expression (n = 393), (E) IDH mutation status (n = 354) and (F) 1p19q deletion status (n = 139). P-values are computed using log-rank test that is used to evaluate significance. (G) Tabulated summary shows median OS for the different survival curves. ‘NA’ denotes values not available. (H) ROCs combined curve of MN1 expression, IDH mutation status and 1p19q deletion status in LGGs (n = 100). Values of AUC are indicated along with P-value for group-wise comparison with MN1 expression.
Further, we performed receiver operating characteristic (ROC) analysis to evaluate sensitivity and specificity of 3-month survival prediction for MN1 overexpression, IDH mutation and 1p19q co-deletion in the TCGA retrieved datasets. Patients with missing values for any of the three molecular predictors were excluded. This is because a comparison can be performed for the three molecular predictors only when all three values for each patient in the dataset are available. Accordingly, 100 LGGs and 131 HGGs were included in the ROC analysis. We observed that area under the ROC curve (AUC) in LGGs for MN1 expression was the largest (0.853) as compared with IDH mutation (0.521) and 1p19q co-deletion (0.553) (Fig. 4H). AUC value of ≥0.7, means that the predictor being analyzed, is a good classifier for predicting survival. However, in HGGs, AUC estimates of the 3-molecular alterations/parameters being evaluated were not available because the ROC curves were below the diagonal reference line and hence could not be computed. The AUC values suggest that MN1 overexpression has better predictive ability for making 3-month survival prediction in LGGs. Therefore, our findings suggest that MN1 overexpression may be considered as a predictor for classifying LGGs with longer OS.
Notably, Kaplan–Meier analysis also showed that glioma patients with MN1 overexpression, irrespective of low or high tumor grade (Fig. 5A and B, P < 0.0001), have a markedly longer progression-free survival (PFS).
Figure 5.
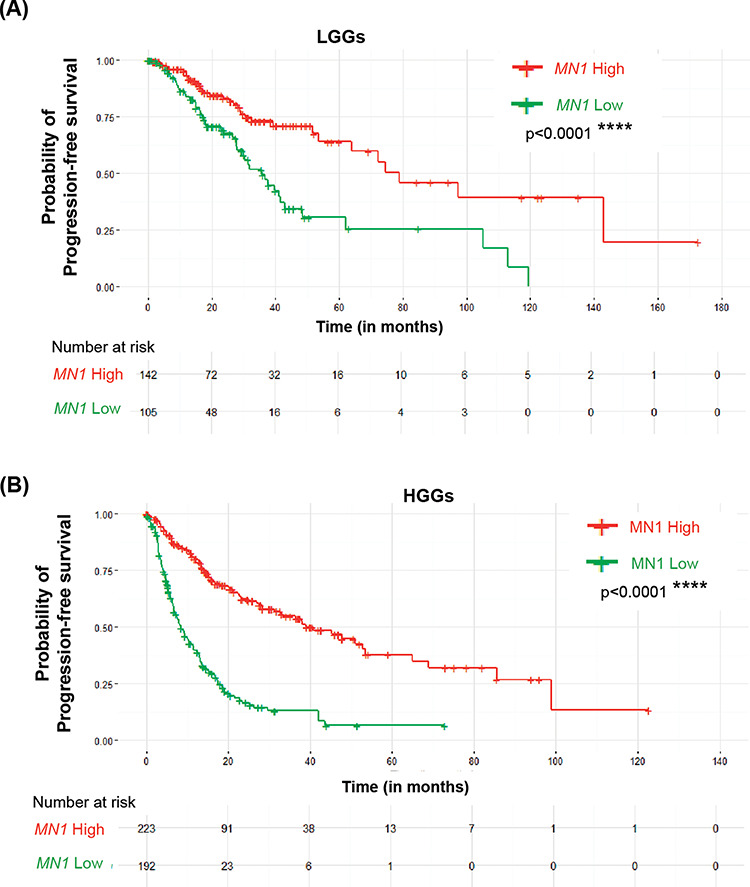
High MN1 expression correlates with the progression-free survival of glioma patients. Kaplan–Meier survival curves showing PFS in LGGs (A) and HGGs (B) based on MN1 expression levels in these tumors.
IGFBP5 and MN1 expression inversely correlate in gliomas
MN1 is a transcriptional co-regulator and is known to synergize both induction and repression of RAR/RXR target genes. IGFBP5 is also an RAR/RXR target gene; therefore, we examined its expression in gliomas. We found that IGFBP5 mRNA levels were upregulated in most gliomas (90%; n = 36/40). IGFBP5 mRNA levels seemed slightly higher in HGGs than LGGs, but the difference was not statistically significant (P = 0.58, Fig. 6A). The case-wise analysis shows the overexpression of IGFBP5 transcripts in ~84% (n = 11/13) LGGs and ~92% (n = 25/27) HGGs, and expectedly, this is not a considerable difference (P = 1.0). Further, to test whether MN1 expression affects IGFBP5 levels, we used a linear regression analysis and found that the association was not appreciable in LGGs (P = 0.99; R2 < 0.0001, Fig. 6B) or HGGs (P = 0.19; R2 = 0.07, Fig. 6C). In HGGs despite no statistical correlation, apparently IGFBP5 expression tended to decrease with an increase in the MN1 levels.
Figure 6.
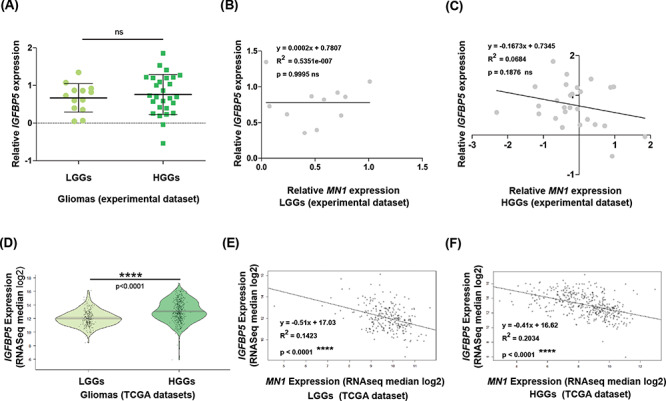
IGFBP5 expression, determining its association with MN1 levels in gliomas. (A) Graphical summary of relative IGFBP5 expression in LGGs (n = 13) and HGGs (n = 27) of the experimental dataset. The central bar and whiskers denote mean (±SD). Linear regression plots to test the association of IGFBP5 expression with MN1 expression levels in (B) LGGs (n = 11), and (C) HGGs (n = 27). (D) Pirate plots showing IGFBP5 expression (log transformed RNAseq median values) in LGGs (n = 249) and HGGs (n = 417) from the TCGA datasets. Scatter plots show the relatedness of IGFBP5 expression with MN1 expression levels in (E) LGGs (n = 249) and (F) HGGs (n = 417). The black regression line represents the regression model fitted to the data, and ‘R2’ refers to the coefficient of determination. Statistical test values comparing gene expression between the glioma grades are indicated above the respective plots.
To verify the above findings, we examined the TCGA datasets. The TCGA datasets statistically strengthened (P < 0.0001, Fig. 6D) our observation of slightly higher IGFBP5 expression in HGGs than in LGGs in the experimental dataset, and was in line with the previous reports that HGGs have higher IGFBP5 expression than LGGs. The statistical insignificance in the experimental dataset may be because of the smaller sample size. Interestingly, it emerged that IGFBP5 expression tends to decline (P < 0.0001) with an increase in MN1 expression levels in both LGGs (slope: −0.51, Fig. 6E) and HGGs (slope: −0.41, Fig. 6F). This suggests a negative correlation between MN1 and IGFBP5 expression in gliomas.
IGF1 and IGFBP5 expression correlates in LGGs
IGF1 regulates IGFBP5 expression transcriptionally and post-translationally in a cell-type specific manner (40,42,50,51). Therefore, we quantified IGF1 expression in gliomas. In LGGs, average IGF1 expression is apparently more than HGGs, but the difference is not significant (P = 0.54, Fig. 7A). Furthermore, in LGGs, a positive correlation between IGFBP5 and IGF1 expression levels was indicated by the regression line though P-value seemed barely significant (slope = 0.2637; P = 0.05; R2 = 0.33, Fig. 7B). Contrarily, IGFBP5 and IGF1 expression showed no association in HGGs (P = 0.81; R2 = 0.002, Fig. 7C).
Figure 7.
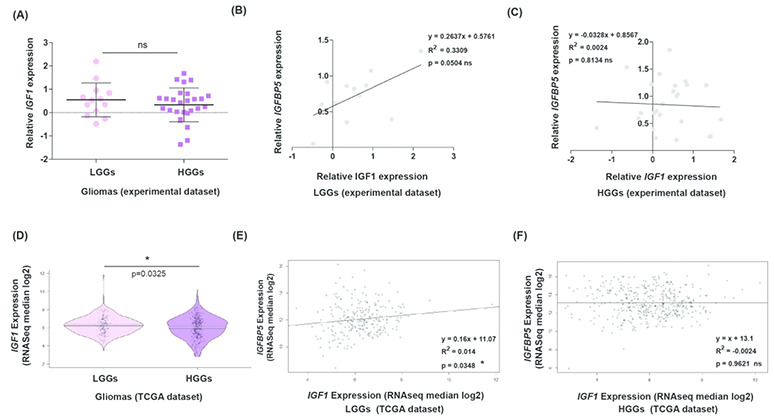
IGF1 and IGFBP5 expression does not correlate in high-grade gliomas. (A) Dot-plots representing relative IGF1 expression between LGGs (n = 13) and HGGs (n = 27). Scatter plots show linear regression model assessing relations between expression levels of IGFBP5 and IGF1 in (B) LGGs (n = 12) and (C) HGGs (n = 25). (D) In the TCGA datasets, IGF1 expression is noticeably lower in HGGs (n = 417) compared with LGGs (n = 249). Regression analysis plots assessing correlation between expression levels of IGFBP5 and IGF1 in (E) LGGs (n = 249) and (F) HGGs (n = 417). In all regression analysis plots, line fitting the data scatter is represented in black and ‘R2’ represents the coefficient of determination.
Our findings differ from the previous report that showed that IGF1 expression was higher in tumors of grades III and IV than grade II gliomas (30). Therefore, we assessed the larger sample size of TCGA datasets to verify the grade specificity of IGF1 expression. Similar to the experimental dataset, in the TCGA datasets, we found that LGGs (grade II gliomas) have higher average IGF1 expression value than HGGs (P = 0.03, Fig. 7D). Also, in the TCGA datasets, we noted IGFBP5 expression correlates with IGF1 levels in LGGs (P = 0.03; Fig. 7E) but not HGGs (P = 0.96; Fig. 7F). This suggests that in LGGs, higher expression of MN1 may repress IGFBP5 expression, while IGF1-mediated induction may contribute in maintaining discernible IGFBP5 expression.
MN1, IGF1 and IGFBP5 have different roles in meningiomas
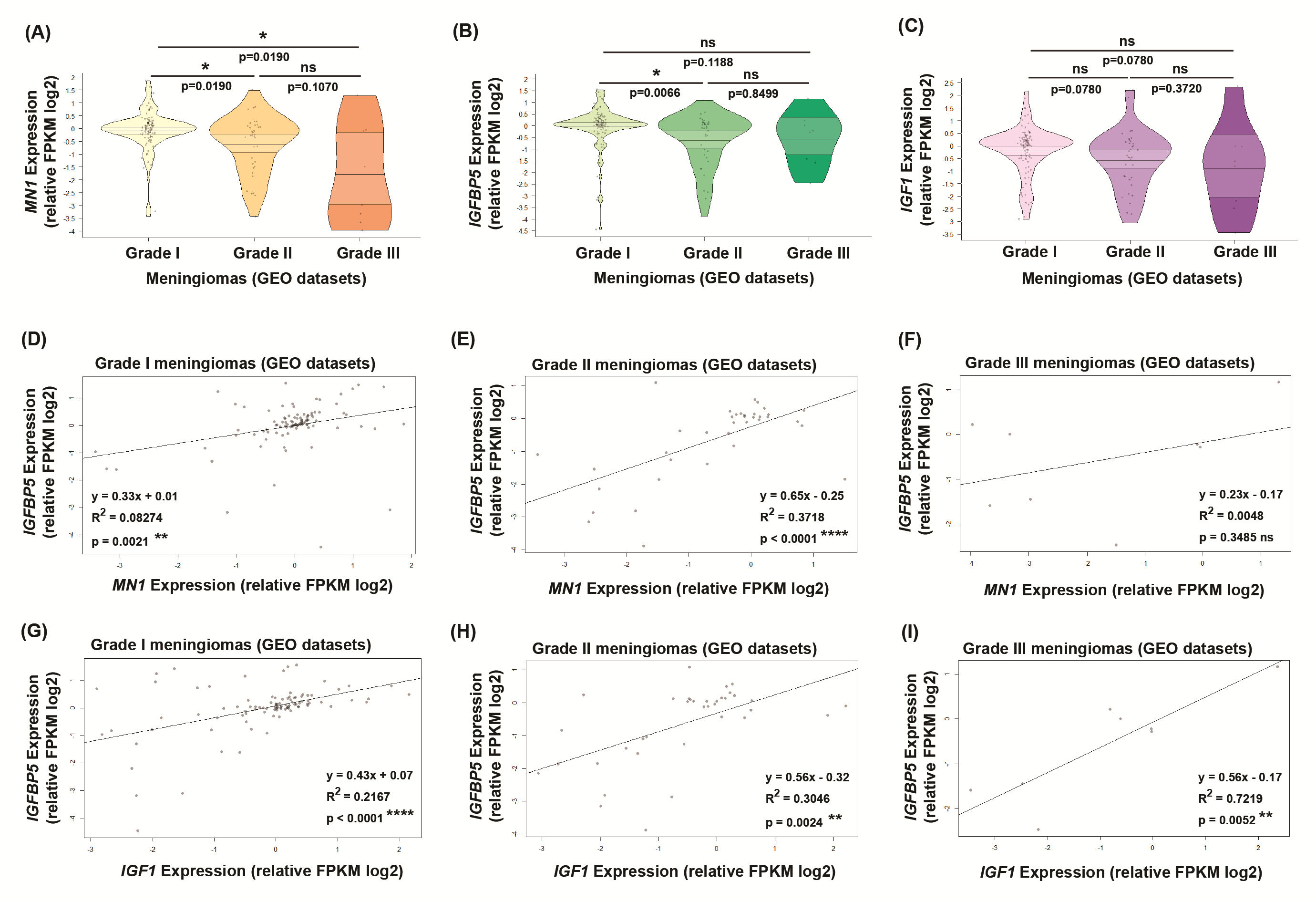
Briefly, to quantify the expression of these molecules and investigate their relationship in a different brain tumor histology, we examined 39 meningioma tumor samples. These samples were collected from meningioma patients undergoing surgical removal of their tumors at the same hospital as the glioma patients. Meningiomas in the experimental dataset showed varying expression of MN1, IGFBP5 and IGF1, but relation to grade was not possible due to limited numbers of grade II (n = 3) and grade III (n = 1) tumors (data not shown). Therefore, we analyzed meningioma datasets available in GEO datasets and the findings differed from that in gliomas (Supplementary Material, Fig. S1).
We found IGFBP5 transcript levels correlated positively with MN1 mRNA expression in meningiomas of grades I and II meningiomas (Supplementary Material, Fig. S1D and E), unlike gliomas where IGFBP5 and MN1 expressions were inversely associated in LGGs and HGGs (TCGA datasets). In grade III meningiomas also, the association seemed positive (Supplementary Material, Fig. S1F), and this may have been statistically relevant if more number of grade III samples in these GEO datasets were available for analysis. Furthermore, IGFBP5 mRNA levels associated positively with IGF1 transcript levels in all the grades of meningiomas (Supplementary Material, Fig. S1G, H and I). This suggests that in meningiomas, both MN1 and IGF1 may independently or co-operatively activate IGFBP5 expression. This is in contrast to gliomas where IGFBP5 expression is likely regulated by a grade-specific opposing (repressive and activating) interplay of MN1 and IGF1. In view of these observations, and that meningiomas are considered less aggressive with better prognosis than gliomas, it is likely that IGFBP5 expression and its regulation by MN1 and IGF1 may have distinct meningioma-specific oncogenic roles. Functional analyses along these lines will be insightful for making clinically relevant inferences.
Discussion
MN1 co-regulates gene transcription and is encoded by its gene located on chromosome 22, which is reported to show aberrations in human brain tumors including gliomas (52,53). To our knowledge, alterations in MN1 have not been assessed systematically in human gliomas earlier. We find that MN1 overexpression predicts better median OS and PFS of the patients. Also, a collective molecular dynamic of MN1, IGF1 and IGFBP5 may be a determinant of prognosis in gliomas, proving eventually to be of clinical relevance in predicting patient outcomes.
We observed a grade-specific overexpression of MN1 transcripts, with upregulated expression in 69% LGGs (n = 9/13 grades I and II) as opposed to only 33% HGGs (n = 9/27 grades III and IV) when compared with non-neoplastic/normal brain. A higher MN1 expression correlates with LGGs in both the experimental (gliomas in the current study) and validation (TCGA) datasets. Chen et al. have reported MN1 overexpression in low-risk group gliomas comprising grades II and III tumor data retrieved from the Chinese Glioma Genome Atlas array database (supplementary Fig. S1 by Chen et al.) (54).
Further, we observed that CNA in MN1 is more frequent in HGGs compared with LGGs. It was intriguing that a change in MN1 copy number did not lead to a relatable change in the gene’s expression in either the experimental or TCGA datasets. Similar discordance between gene copy number and expression is reported earlier in breast cancer and gliomas (55,56). It is likely that in such cases, altered pre-transcriptional regulation or post-transcriptional change(s) modify mRNA levels or half-life, respectively, without requiring CNA to predicate altered gene expression. Thus, change in gene dosage does not necessarily underlie altered MN1 expression in gliomas.
Survival analysis tests the ability of a molecular/biochemical parameter or treatment to predict survival probabilities in patients. It was interesting to note that within the LGG and HGG categories, patients with upregulated MN1 expression showed a survival advantage. ROC analysis showed that for a 3-month survival prediction in LGGs, high MN1 expression is a good classifier as it had greater AUC values than IDH mutation and 1p19q co-deletion. However in HGGs, ROC analysis was not informative because ROC curves were below the reference diagonal line and hence AUCs could not be computed. ROC analysis to compute time-dependent AUCs for longer time duration (more than three months) for each of the 3-molecular predictors returned AUC values <0.6. This means that the performance of the 3-predictors/classification models to predict survival for longer time had poor discriminatory power. This may be because the total number of evaluable LGGs (n = 100) and HGGs (n = 131) in the datasets was reduced owing to filtering out of data points/patients where values of any of the three-predictors were not available for comparison. Remarkably, MN1 overexpression in gliomas irrespective of tumor grade predicts longer PFS indicating that such patients were less likely to undergo disease progression. Thus, MN1 overexpression in gliomas clearly confers a survival benefit on patients and holds promise as a prognostic marker for clinical use.
MN1 possesses properties of a transcriptional regulator but lacks DNA binding domains and therefore functions as a co-regulator (46,53). It was demonstrated that cooperation between MN1 and RAR/RXR is crucial to RA-mediated transcriptional regulation of IGFBP5 (44). IGF1 is also known to regulate the expression of IGFBP5 (40,50,51). In the present study, we found higher IGFBP5 expression in HGGs, whereas similar to MN1 overexpression greater IGF1 levels correlated with LGGs. Therefore, it is likely that IGFBP5 may simultaneously be regulated by MN1 and IGF1 in gliomas.
In LGGs despite overexpression of MN1 and IGF1, the two known activators of IGFBP5, it was counter-intuitive that IGFBP5 expression correlated with HGGs instead. Therefore, we examined further and found that IGBP5 expression was inversely correlated with MN1 levels in LGGs and HGGs. This suggests that MN1 tends to repress IGFBP5 expression in gliomas. However, another modulator (IGF1) of IGFBP5 expression may oppose MN1-mediated repression to enhance IGFBP5 expression. This molecular interplay between repressive and activating regulatory forces possibly dictates grade-specific IGFBP5 expression and contributes to associated clinical attributes such as poor prognosis, disease progression and shorter survival rate (Fig. 8).
Figure 8.
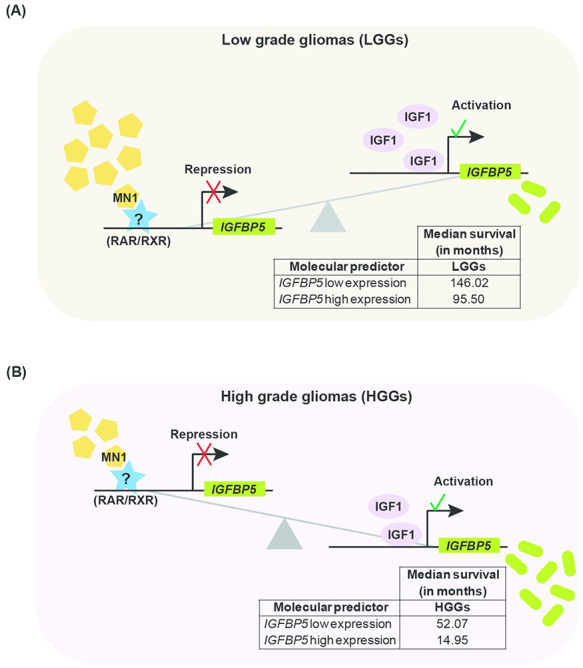
Model summarizing the association between MN1, IGFBP5 and IGF1 expression in gliomas and their possible molecular interplay. MN1, IGF1 and IGFBP5 are represented by specific symbols. In (A) LGGs and (B) HGGs, the numbers of a particular symbol denote relative abundance of the specific molecule, based on the findings of the present study. MN1 possibly synergizes with an unknown or known (RAR/RXR) repressor at regulatory elements in the IGFBP5 promoter to repress its expression in gliomas. Schematic suggests that (A) in LGGs, high MN1 levels transcriptionally repress IGFBP5, which offsets IGF1-mediated induction of IGFBP5 expression such that it is predictive of better median survival (table within the schematic), (B) whereas in HGGs, low levels of MN1 likely relieve transcriptional repression of IGFBP5, tilting the balance in favor of IGF1 mediated increase in IGFBP5 expression. This in turn estimates poor prognosis and lower median survival in HGGs than LGGs (table embedded in the schematic).
Accordingly, IGF1 levels that correlated only with LGGs seem crucial in maintaining detectable and oncogenically relevant grade-specific IGFBP5 expression (Fig. 8A). In HGGs, lower MN1 levels may result in lesser repression, causing accumulation of IGFBP5 due to IGF1-mediated induction of expression (Fig. 8B). Similar to previous report (36), IGFBP5 overexpression in LGGs and HGGs (TCGA datasets) was found to portend dismal median survival (table insets in Fig. 8A and B). Thus, IGBP5 levels in gliomas seem to be fine-tuned by the opposing actions of MN1 and IGF1 in a tumor grade-specific manner and accordingly predict patient survival.
Our study shows for the first time that MN1 overexpression correlates with LGGs, is not determined by the changes in the gene’s copy number, instead associates inversely with IGFBP5 expression and thus predicts better median OS and PFS in gliomas. Furthermore, based on our analyses, we propose that grade-specific expressions of MN1, IGF1 and IGFBP5 underlie their mutual regulatory dynamic to govern the clinical course and characteristics of different glioma grades. Future functional studies should help validate the proposed molecular interplay and its significance to glioma pathology. Notwithstanding the significance of studies needed on this line, we find that MN1 overexpression with varying tumor grade may be leveraged as a predictor of better OS and PFS in glioma patients.
Materials and Methods
Patients and sample collection
This study was approved by the Institutional Human Ethics Committee of the National Institute of Immunology (IHEC/VB/24/2008) and the Max Healthcare Ethics Committee and was performed in accordance with the Declaration of Helsinki. Samples were collected with due informed written consents from the concerned patients. A part of the resected tumor tissues along with matched peripheral blood samples were collected with due informed written consents of the concerned patients, who were consecutively operated for intracranial gliomas during May 2008–August 2009. All the tumors were examined histopathologically and graded following the WHO criteria 2007 (5). Clinical details of samples analyzed in the experimental dataset are presented in Table 1. The glioma cases were grouped and numbered such that the progressing numbers correlate with increasing grades. Forty glioma (G) patients included in the study represent primary tumors, except G15 and G20, that represented recurrent cases. The glioma patient G20 had surgical resections for astrocytoma (II), oligodendroglioma (III) and oligodendroglioma (II) in 1999, 2005 and 2006, respectively. Tumor tissues of earlier resections for both G15 and G20 were not available for analyses. All GBMs analyzed in the study were de novo tumors.
Table 1.
Clinical details of glioma cases analyzed in this study
| Glioma cases | Histological sub-types | Total no. cases per grade | ||
|---|---|---|---|---|
| sub-type | No. of cases | |||
| Grade I (G1–G3) | PA | 2 | n = 3 | |
| LGGs (n = 13) | SEGA | 1 | ||
| Grade II (G4–G12, G14) | DA | 5 | n = 10 | |
| O | 2 | |||
| OA | 3 | |||
| HGGs (n = 27) | Grade III (G15, G18–G23) | AA | 3 | n = 7 |
| AO | 2 | |||
| AOA | 2 | |||
| Grade IV (G24–G43) | GBM | 20 | n = 20 | |
AA, Anaplastic astrocytoma; AO, Anaplastic oligodendroglioma; AOA, Anaplastic oligoastrocytoma; DA, Diffuse astrocytoma; GBM, Glioblastoma multiforme; HGGs, High-grade gliomas; LGGs, Low-grade gliomas; O, Oligodendroglioma; OA, Oligo astrocytoma; PA, Pilocytic astrocytoma; SEGA, sub-ependymal giant cell astrocytoma.
Sources of publicly available datasets
We assessed publicly available glioma datasets for validating our findings. Molecular data and associated clinical information pertaining to diffuse glioma (grades II and III) and GBM (grade IV) datasets of The Cancer Genome Atlas (TCGA) were retrieved in March 2020 from cBioPortal for cancer genomics (57–59). Like grade II and III gliomas, GBM is also a diffuse tumor entity, but TCGA classifies these into two different cohorts. Nevertheless, according to accepted method of grouping gliomas as LGGs (grades I and II) and HGGs (grades III and IV) (6–9), and similar to the experimental dataset, we extracted grade III glioma data from the TCGA diffuse glioma dataset and collated it with that of the GBM cohort. RSEM data were not available for TCGA grade I (Pilocytic astrocytomas) dataset and hence could not be analyzed as part of TCGA LGGs. For analyses, relevant data were cleaned by eliminating missing values. We also analyzed meningioma GEO datasets—GSE88720; GSE16581; GSE43290; GSE85133 and GSE77259.
Isolation of genomic DNA and total RNA from samples
DNA was isolated from tumor tissues, which were collected in RNA later (Ambion, Austin, TX), following standard phenol chloroform extraction method. DNA extraction kit (Qiagen, Valencia, CA) was used for isolating DNA from peripheral blood leukocytes (PBLs), as per the manufacturer’s protocol. Total RNA from tumor tissues was isolated using Tri Reagent RT (Molecular Research Centre Cincinnati, OH), according to manufacturer’s recommendations. Potential genomic DNA contamination of total RNA was checked using GAPDH primers in a 20μL reaction volume of PCR. Total RNA was reverse transcribed to cDNA using High Capacity archive kit (ABI, Carlsbad, CA).
Real-time quantitation of gene expression
mRNA transcripts of genes of interest (GIs—MN1, IGFBP5 and IGF1) in the tumor tissues were quantified by qPCR. Commercially purchased total RNA samples from normal human adult ♂ and ♀ brains (Stratagene, La Jolla, CA) were used as controls. Primers specific for GIs and gene of reference (GR) were designed using Primer Express 3.0 software (ABI) and are detailed in Table 2. GAPDH was used as GR, as it showed stable expression in both control and test samples. Specificity of the primers was confirmed by BLAST search, and their efficiency was tested. Amplification was carried out with SYBR® green (ABI) using 7500 Real-Time PCR System (ABI). All the assays were performed with 100 nM of forward and reverse primers each, of the respective gene, in a final reaction volume of 20 μL, employing universal cycling conditions recommended by ABI. GIs and GR were amplified in separate wells. The results were ratified, when from the triplicate cycle threshold (Ct) values, two were concordant. Relative expression levels of GIs in patient samples were calculated as described previously (56,60). Data were analyzed on SDS 7500™ Software v2.0.3 and Data Assist™ Software v2.0 (ABI).
Table 2.
Details of primers used for qPCR-based mRNA expression analysis
| Gene | Primer sequencesa | Amplicon size(bp) |
|---|---|---|
| GAPDH | FP 5′ GCCACATCGCTCAGACACCAT 3′ | 72 |
| RP 5′ ACCAGGCGCCCAATACG 3′ | ||
| MN1 | FP 5′ CAGAACCCCAACAGCAAAGAA 3′ | 90 |
| RP 5′ GACAGACAGGCACTGCAAGTG 3′ | ||
| IGFBP5 | FP 5′ CTACAAGAGAAAGCAGTGCAAACC 3′ | 62 |
| RP 5′ TCCACGCACCAGCAGATG 3′ | ||
| IGF1 | FP 5′ GTGCTGCTTTTGTGATTTCTTGA 3′ | 76 |
| RP 5′ GCACAGCGCCAGGTAGAAG 3′ |
aForward and reverse primers are located in different exons, thus eliminating chance amplification from potential contaminating genomic DNA. FP and RP represent forward and reverse primers, respectively.
Immunoblotting
Glioma cases having adequate tumor tissue were subjected to protein expression analysis by western blotting. Total protein was extracted by homogenizing the tumor tissue in cold RIPA lysis buffer and clarified by centrifugation. Commercially purchased total proteins of non-neoplastic brain served as control (Biochain, Hayward, CA). Equal amounts of proteins were resolved on 12% SDS polyacrylamide gels and transferred to nitrocellulose membranes (Millipore, Billerica, MA). The membranes were blocked in 3% non-fat dried milk and 2% BSA (Cell Signaling Technology, Boston, MA) in PBS. Blots were incubated with goat polyclonal anti-MN1 (sc-27 349; Santa Cruz Biotechnology, Dallas, TX), rabbit polyclonal anti-β-Tubulin (RB-9249-P1; Neo Markers) primary antibodies and secondary horse-radish peroxidase conjugated mouse anti-goat (sc-2354; Santa Cruz Biotechnology) and anti-rabbit (111–036-045; Jackson Immuno Research Laboratories Inc., West Grove, PA) antibodies. Subsequently, the immunoreactive signals were detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore), as per manufacturer’s instructions. The relative abundance of the proteins was quantified from scanned images of immunoblots using Labworks™ image acquisition and analysis software (v.4.0.0.8 UVP). Levels of β-Tubulin were used to normalize the protein expression.
Copy number analysis by qPCR
Copy number of MN1 was assessed using TaqMan® assay (Assay Id. Hs02424444_cn; ABI), and TaqMan® RNase P detection (P/N: 4316831; ABI) as the reference gene, on 7500 Real-Time PCR System (ABI) using tumor genomic DNA as the test samples. Genomic DNA from matched PBLs of the respective patient was also assayed to determine whether copy number variations detected were de novo somatic events or inherited ones. Commercially purchased human brain genomic DNA (BioChain) and blood genomic DNA from two healthy volunteers served as non-neoplastic tissue and healthy controls, respectively. All samples and controls were run in triplicates, in 20μL singleplex reactions, comprising 20 ng of genomic DNA, 1x TaqMan® Universal PCR Master Mix (ABI) and 1x primer probe mix, using universal cycling conditions recommended by ABI. The gene copy number per diploid genome was calculated as described previously (60). All possible precautions were taken to avoid the presence of non-tumor genomic DNA in the test samples.
Data plotting and statistical analyses
GraphPad Prism (version 5.03), R studio (version 1.2.5033) and YaRrr, survminer, timeROC were used for making various plots and performing statistical analyses (61). Missing values were filtered out by R-packages. Therefore, the number of cases in a particular TCGA dataset may vary between different analyses and the number of cases in a specific analysis are indicated where related data are presented.
Survival data including patients’ survival time and vital status (death/progression/event = 1, living/censored = 0 to censor data) were used from the TCGA datasets. The censored observations (patients that are alive at last follow-up) are shown as marks on the survival curve for different time intervals (x-axis). The risk table depicts the number of patients under observation for a particular time interval. The survival curve is unchanged for a time interval when censored observations are recorded, but at the next time interval, the number of patients at risk is reduced by the number censored between the two time intervals as reflected in the risk table.
To determine grade-specific over-expression of MN1, IGFBP5 and IGF1 between LGGs and HGGs, a two-tailed non-parametric Mann–Whitney test was performed and contingency tables were also analyzed using two-tailed Fisher’s exact test. The P-value, slope and coefficient of determination (R2) from linear regression analysis were used to discern association, nature of association (positive/negative) between parameters and whether the model explains the variability in the response data, respectively. In the pirate plots for the RSEM data (TCGA datasets), dots, bar/line and band represent data points/cases, mean and confidence interval, respectively. The level of significance is indicated as *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
{kind=link}
Acknowledgements
The authors thank the patients for their consent to participate in this study, Dr Urmi Mukherjee and Dr Andleeb Abrari for histopathological diagnosis of patient samples and Khem Singh Negi and Pankaj Kumar Sharma for technical help. Equipment donation from the Alexander Von Humboldt Foundation, Bonn, Germany to S.A. is gratefully acknowledged. M.S. acknowledges the support of the Department of Biotechnology e-Library Consortium (DeLCON) at the current institute- Regional Centre for Biotechnology, for providing access to e-resources.
Conflict of Interest statement. None declared.
Contributor Information
Masum Saini, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110067, India; Regional Centre for Biotechnology, NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurgaon Expressway, Faridabad 121001, India.
Ajaya Nand Jha, Max Super Specialty Hospital, 1, Press Enclave Road, Saket, New Delhi 110017, India.
Rajiv Tangri, Max Super Specialty Hospital, 1, Press Enclave Road, Saket, New Delhi 110017, India; Dr. Lal PathLabs, National Reference Laboratory, Sector 18, Rohini, New Delhi 110085, India.
Md Qudratullah, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110067, India.
Sher Ali, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110067, India; Department of Life Sciences, SBSR, Sharda University, KP-III, Greater Noida 201310, India.
Funding
Core grant from the Department of Biotechnology (DBT), New Delhi, Government of India to National Institute of Immunology (NII), New Delhi, a DBT research grant (BT/PR14102/AAQ/01/438/2010) and J. C. Bose National Award from Science and Engineering Research Board- Department of Science and Technology, Government of India, New Delhi to S.A.; DBT Senior Research Fellowship (2012) to M.S.; NII core grant support to Md.Q.; Early Career Fellowship by the DBT/Wellcome Trust India Alliance (IA/E/16/1/503028) currently to M.S.
References
- 1. Ostrom Q.T., Gittleman H., Truitt G., Boscia A., Kruchko C. and Barnholtz-Sloan J.S. (2018) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro-Oncology, 20, iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ostrom Q.T., Bauchet L., Davis F.G., Deltour I., Fisher J.L., Langer C.E., Pekmezci M., Schwartzbaum J.A., Turner M.C., Walsh K.M. et al. (2014) The epidemiology of glioma in adults: a "state of the science" review. Neuro-Oncology, 16, 896–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Globocan (2018) Global Cancer Obervatory. https://gco.iarc.fr.
- 4. Ferlay J., Colombet M., Soerjomataram I., Mathers C., Parkin D.M., Pineros M., Znaor A. and Bray F. (2019) Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer, 144, 1941–1953. [DOI] [PubMed] [Google Scholar]
- 5. Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., Burger P.C., Jouvet A., Scheithauer B.W. and Kleihues P. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol., 114, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Venkatesh H.S., Tam L.T., Woo P.J., Lennon J., Nagaraja S., Gillespie S.M., Ni J., Duveau D.Y., Morris P.J., Zhao J.J. et al. (2017) Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature, 549, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Claus E.B., Walsh K.M., Wiencke J.K., Molinaro A.M., Wiemels J.L., Schildkraut J.M., Bondy M.L., Berger M., Jenkins R. and Wrensch M. (2015) Survival and low-grade glioma: the emergence of genetic information. Neurosurg. Focus., 38, E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nikiforova M.N. and Hamilton R.L. (2011) Molecular diagnostics of gliomas. Arch. Pathol. Lab. Med., 135, 558–568. [DOI] [PubMed] [Google Scholar]
- 9. Sanai N., Chang S. and Berger M.S. (2011) Low-grade gliomas in adults. J. Neurosurg., 115, 948–965. [DOI] [PubMed] [Google Scholar]
- 10. Furnari F.B., Fenton T., Bachoo R.M., Mukasa A., Stommel J.M., Stegh A., Hahn W.C., Ligon K.L., Louis D.N., Brennan C. et al. (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev., 21, 2683–2710. [DOI] [PubMed] [Google Scholar]
- 11. Molinaro A.M., Taylor J.W., Wiencke J.K. and Wrensch M.R. (2019) Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol., 15, 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Louis D.N., Perry A., Reifenberger G., Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P. and Ellison D.W. (2016) The 2016 World Health Organization classification of Tumors of the central nervous system: a summary. Acta Neuropathol., 131, 803–820. [DOI] [PubMed] [Google Scholar]
- 13. Louis D.N., Wesseling P., Paulus W., Giannini C., Batchelor T.T., Cairncross J.G., Capper D., Figarella-Branger D., Lopes M.B., Wick W. et al. (2018) cIMPACT-NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol., 135, 481–484. [DOI] [PubMed] [Google Scholar]
- 14. Louis D.N., Giannini C., Capper D., Paulus W., Figarella-Branger D., Lopes M.B., Batchelor T.T., Cairncross J.G., Bent M., Wick W. et al. (2018) cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol., 135, 639–642. [DOI] [PubMed] [Google Scholar]
- 15. Brat D.J., Aldape K., Colman H., Holland E.C., Louis D.N., Jenkins R.B., Kleinschmidt-DeMasters B.K., Perry A., Reifenberger G., Stupp R. et al. (2018) cIMPACT-NOW update 3: recommended diagnostic criteria for diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV. Acta Neuropathol., 136, 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellison D.W., Hawkins C., Jones D.T.W., Onar-Thomas A., Pfister S.M., Reifenberger G. and Louis D.N. (2019) cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol., 137, 683–687. [DOI] [PubMed] [Google Scholar]
- 17. Nandakumar P., Mansouri A. and Das S. (2017) The role of ATRX in Glioma biology. Front. Oncol., 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trojan J., Cloix J.F., Ardourel M.Y., Chatel M. and Anthony D.D. (2007) Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience, 145, 795–811. [DOI] [PubMed] [Google Scholar]
- 19. Baxter R.C. (2014) IGF binding proteins in cancer: mechanistic and clinical insights. Nat. Rev. Cancer, 14, 329–341.24722429 [Google Scholar]
- 20. Samani A.A., Yakar S., LeRoith D. and Brodt P. (2007) The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr. Rev., 28, 20–47. [DOI] [PubMed] [Google Scholar]
- 21. LeRoith D. and Roberts C.T. Jr. (2003) The insulin-like growth factor system and cancer. Cancer Lett., 195, 127–137. [DOI] [PubMed] [Google Scholar]
- 22. Russo V.C., Gluckman P.D., Feldman E.L. and Werther G.A. (2005) The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev., 26, 916–943. [DOI] [PubMed] [Google Scholar]
- 23. Han V.K., D'Ercole A.J. and Lund P.K. (1987) Cellular localization of somatomedin (insulin-like growth factor) messenger RNA in the human fetus. Science, 236, 193–197. [DOI] [PubMed] [Google Scholar]
- 24. Gammeltoft S., Ballotti R., Nielsen F.C., Kowalski A. and Van Obberghen E. (1988) Receptors for insulin-like growth factors in the central nervous system: structure and function. Horm. Metab. Res., 20, 436–442. [DOI] [PubMed] [Google Scholar]
- 25. Gammeltoft S., Ballotti R., Kowalski A., Westermark B. and Van Obberghen E. (1988) Expression of two types of receptor for insulin-like growth factors in human malignant glioma. Cancer Res., 48, 1233–1237. [PubMed] [Google Scholar]
- 26. Sandberg A.C., Engberg C., Lake M., Holst H. and Sara V.R. (1988) The expression of insulin-like growth factor I and insulin-like growth factor II genes in the human fetal and adult brain and in glioma. Neurosci. Lett., 93, 114–119. [DOI] [PubMed] [Google Scholar]
- 27. Glick R.P., Gettleman R., Patel K., Lakshman R. and Tsibris J.C. (1989) Insulin and insulin-like growth factor I in brain tumors: binding and in vitro effects. Neurosurgery, 24, 791–797. [DOI] [PubMed] [Google Scholar]
- 28. Merrill M.J. and Edwards N.A. (1990) Insulin-like growth factor-I receptors in human glial tumors. J. Clin. Endocrinol. Metab., 71, 199–209. [DOI] [PubMed] [Google Scholar]
- 29. Sandberg-Nordqvist A.C., Stahlbom P.A., Reinecke M., Collins V.P., von Holst H. and Sara V. (1993) Characterization of insulin-like growth factor 1 in human primary brain tumors. Cancer Res., 53, 2475–2478. [PubMed] [Google Scholar]
- 30. Hirano H., Lopes M.B., Laws E.R. Jr., Asakura T., Goto M., Carpenter J.E., Karns L.R. and VandenBerg S.R. (1999) Insulin-like growth factor-1 content and pattern of expression correlates with histopathologic grade in diffusely infiltrating astrocytomas. Neuro-Oncology, 1, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang B.C., Chang H.M., Wang Y.S., Chen R.F. and Lin S.J. (1996) Transient induction of apoptosis in serum-starved glioma cells by insulin and IGF-1. Biochim. Biophys. Acta, 1314, 83–92. [DOI] [PubMed] [Google Scholar]
- 32. James P.L., Jones S.B., Busby W.H. Jr., Clemmons D.R. and Rotwein P. (1993) A highly conserved insulin-like growth factor-binding protein (IGFBP-5) is expressed during myoblast differentiation. J. Biol. Chem., 268, 22305–22312. [PubMed] [Google Scholar]
- 33. Bondy C. and Lee W.H. (1993) Correlation between insulin-like growth factor (IGF)-binding protein 5 and IGF-I gene expression during brain development. J. Neurosci., 13, 5092–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ye P. and D'Ercole J. (1998) Insulin-like growth factor I (IGF-I) regulates IGF binding protein-5 gene expression in the brain. Endocrinology, 139, 65–71. [DOI] [PubMed] [Google Scholar]
- 35. Wang J., Ding N., Li Y., Cheng H., Wang D., Yang Q., Deng Y., Yang Y., Ruan X., Xie F. et al. (2015) Insulin-like growth factor binding protein 5 (IGFBP5) functions as a tumor suppressor in human melanoma cells. Oncotarget, 6, 20636–20649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang H., Zhang W. and Fuller G.N. (2006) Overexpression of IGFBP5, but not IGFBP3, correlates with the histologic grade of human diffuse glioma: a tissue microarray and immunohistochemical study. Technol. Cancer Res. Treat, 5, 195–199. [DOI] [PubMed] [Google Scholar]
- 37. Jiang R., Mircean C., Shmulevich I., Cogdell D., Jia Y., Tabus I., Aldape K., Sawaya R., Bruner J.M., Fuller G.N. et al. (2006) Pathway alterations during glioma progression revealed by reverse phase protein lysate arrays. Proteomics, 6, 2964–2971. [DOI] [PubMed] [Google Scholar]
- 38. Santosh V., Arivazhagan A., Sreekanthreddy P., Srinivasan H., Thota B., Srividya M.R., Vrinda M., Sridevi S., Shailaja B.C., Samuel C. et al. (2010) Grade-specific expression of insulin-like growth factor-binding proteins-2, −3, and −5 in astrocytomas: IGFBP-3 emerges as a strong predictor of survival in patients with newly diagnosed glioblastoma. Cancer Epidemiol. Biomark. Prev., 19, 1399–1408. [DOI] [PubMed] [Google Scholar]
- 39. Dong C., Zhang J., Fang S. and Liu F. (2020) IGFBP5 increases cell invasion and inhibits cell proliferation by EMT and Akt signaling pathway in Glioblastoma multiforme cells. Cell Div., 15, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shemer J., Yaron A., Werner H., Shao Z.M., Sheikh M.S., Fontana J.A., LeRoith D. and Roberts C.T.J. (1993) Regulation of insulin-like growth factor (IGF) binding protein-5 in the T47D human breast carcinoma cell line by IGF-I and retinoic acid. J. Clin. Endocrinol. Metab., 77, 1246–1250. [DOI] [PubMed] [Google Scholar]
- 41. Akkiprik M., Feng Y., Wang H., Chen K., Hu L., Sahin A., Krishnamurthy S., Ozer A., Hao X. and Zhang W. (2008) Multifunctional roles of insulin-like growth factor binding protein 5 in breast cancer. Breast Cancer Res., 10, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider M.R., Wolf E., Hoeflich A. and Lahm H. (2002) IGF-binding protein-5: flexible player in the IGF system and effector on its own. J. Endocrinol., 172, 423–440. [DOI] [PubMed] [Google Scholar]
- 43. Lekanne Deprez R.H., Groen N.A., Biezen N.A., Hagemeijer A., Drunen E., Koper J.W., Avezaat C.J., Bootsma D. and Zwarthoff E.C. (1991) A t(4;22) in a meningioma points to the localization of a putative tumor-suppressor gene. Am. J. Hum. Genet., 48, 783–790. [PMC free article] [PubMed] [Google Scholar]
- 44. Meester-Smoor M.A., Molijn A.C., Zhao Y., Groen N.A., Groffen C.A., Boogaard M., Dalsum-Verbiest D., Grosveld G.C. and Zwarthoff E.C. (2007) The MN1 oncoprotein activates transcription of the IGFBP5 promoter through a CACCC-rich consensus sequence. J. Mol. Endocrinol., 38, 113–125. [DOI] [PubMed] [Google Scholar]
- 45. Cesi V., Giuffrida M.L., Vitali R., Tanno B., Mancini C., Calabretta B. and Raschella G. (2005) C/EBP alpha and beta mimic retinoic acid activation of IGFBP-5 in neuroblastoma cells by a mechanism independent from binding to their site. Exp. Cell Res., 305, 179–189. [DOI] [PubMed] [Google Scholar]
- 46. Meester-Smoor M.A., Janssen M.J., Grosveld G.C., Klein A., van I.W.F., Douben H. and Zwarthoff E.C. (2008) MN1 affects expression of genes involved in hematopoiesis and can enhance as well as inhibit RAR/RXR-induced gene expression. Carcinogenesis, 29, 2025–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heuser M., Yung E., Lai C., Argiropoulos B., Kuchenbauer F., Park G., Lin G., Jessica P., Vollett S., Leung M. et al. (2010) MN1 inhibits myeloid differentiation by transcriptional repression of EGR2. Blood, 116, 229. [Google Scholar]
- 48. Mak C.C.Y., Doherty D., Lin A.E., Vegas N., Cho M.T., Viot G., Dimartino C., Weisfeld-Adams J.D., Lessel D., Joss S. et al. (2020) MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain, 143, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee S.C. (2018) Diffuse gliomas for nonneuropathologists: the new integrated molecular diagnostics. Arch. Pathol. Lab. Med., 142, 804–814. [DOI] [PubMed] [Google Scholar]
- 50. Duan C., Hawes S.B., Prevette T. and Clemmons D.R. (1996) Insulin-like growth factor-I (IGF-I) regulates IGF-binding protein-5 synthesis through transcriptional activation of the gene in aortic smooth muscle cells. J. Biol. Chem., 271, 4280–4288. [DOI] [PubMed] [Google Scholar]
- 51. Kiepe D., Ciarmatori S., Hoeflich A., Wolf E. and Tonshoff B. (2005) Insulin-like growth factor (IGF)-I stimulates cell proliferation and induces IGF binding protein (IGFBP)-3 and IGFBP-5 gene expression in cultured growth plate chondrocytes via distinct signaling pathways. Endocrinology, 146, 3096–3104. [DOI] [PubMed] [Google Scholar]
- 52. Rey J.A., Bello M.J., Campos J.M., Vaquero J., Kusak M.E., Sarasa J.L. and Pestana A. (1993) Abnormalities of chromosome 22 in human brain tumors determined by combined cytogenetic and molecular genetic approaches. Cancer Genet. Cytogenet., 66, 1–10. [DOI] [PubMed] [Google Scholar]
- 53. Lekanne Deprez R.H., Riegman P.H., Groen N.A., Warringa U.L., Biezen N.A., Molijn A.C., Bootsma D., Jong P.J., Menon A.G., Kley N.A. et al. (1995) Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene, 10, 1521–1528. [PubMed] [Google Scholar]
- 54. Chen B., Liang T., Yang P., Wang H., Liu Y., Yang F. and You G. (2016) Classifying lower grade glioma cases according to whole genome gene expression. Oncotarget, 7, 74031–74042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Myhre S., Lingjaerde O.C., Hennessy B.T., Aure M.R., Carey M.S., Alsner J., Tramm T., Overgaard J., Mills G.B., Borresen-Dale A.L. et al. (2013) Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins. Mol. Oncol., 7, 704–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saini M., Jha A.N., Abrari A. and Ali S. (2012) A subset of human gliomas shows over-expression of KIT without its amplification. Gene, 497, 155–163. [DOI] [PubMed] [Google Scholar]
- 57. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov., 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal., 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. (2013) NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res., 41, D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Livak K.J. and Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 61. Team R.C. (2020), R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.