

Abstract
Castration of young males is widely used in the cattle industry to improve meat quality, but the mechanism linking hypogonadism and host metabolism is not clear. Here, we use metataxonomic and metabolomic approaches to evaluate the intestinal microbiota and host metabolism in male, castrated male (CtM), and female cattle. After pubescence, the CtM cattle harbor distinct ileal microbiota dominated by the family Peptostreptococcaceae and exhibit distinct serum and muscle amino acid profiles (i.e., highly abundant branched‐chain amino acids), with increased extra‐ and intramuscular fat storage. We also evaluate the causative factor(s) that underpin the alteration of the intestinal microbiota and host metabolic phenotype in response to hypogonadism. Castration of male mice phenocopies both the intestinal microbial alterations and obese‐prone metabolism observed in cattle. Antibiotic treatment and fecal microbiota transplantation experiments in a mouse model confirm that the intestinal microbial alterations associated with hypogonadism are a key contributor to the obese phenotype in the CtM animals. Collectively, targeting the gut microbiota is a potential therapeutic strategy for the treatment of both hypogonadism and obesity.
Keywords: branched‐chain amino acids, hypogonadism, ileal microbiota, intramuscular fat, Peptostreptococcaceae
Subject Categories: Metabolism; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Primary hypogonadism caused in cattle and mouse by male castration leads to alterations of the ileal microbiota. These changes induce aberrant systemic, muscular and intestinal metabolome profiles, resulting in increased fat storage.

Introduction
Beyond its contribution as the principal male sex hormone, testosterone plays a key role in metabolism and body composition. Elderly men and patients with Klinefelter’s syndrome who usually suffer from testosterone deficiency, termed hypogonadism, often show an unfavorable body composition with increased fat mass and reduced muscle mass (Bojesen et al, 2006; Tajar et al, 2010). Conversely, the incidence of hypogonadism is increased in obese men compared with healthy men (Saboor Aftab et al, 2013; Dhindsa et al, 2018). This bidirectional relationship between obesity and male hypogonadism (meaning that primary hypogonadism promotes fat accumulation while obesity attenuates endogenous testosterone production) has been highlighted in the current obesity epidemic (Carrageta et al, 2019). Several animal model studies also support this reciprocal parallelism. For example, genetically obese (leptin‐deficient ob/ob) male mice exhibit hypogonadism‐mediated impaired reproductive function (Swerdloff et al, 1976; Batt et al, 1982) and androgen receptor knock‐out mice or castration models predispose to development of obesity (Rana et al, 2011; Harada et al, 2016). It has been proposed that the enzymatic conversion of testosterone to estradiol by adipocyte‐derived aromatase can cause hypogonadism in obese individuals (Hayes et al, 2000; Hayes et al, 2001) and that testosterone inhibits differentiation of preadipocyte to adipocyte in vitro (Singh et al, 2006). However, the mechanisms linking hypogonadism with fat accumulation are still not fully elucidated.
In the cattle industry, castration of male cattle is commonly practiced worldwide. Castration of bulls has advantages in terms of tractability and profitability. For instance, feeding male castrated bull (steer) a high‐concentrate diet results in a greater increase in the subcutaneous and intramuscular fat, and a higher meat tenderness score, than those of non‐castrated bull (Field, 1971; Marti et al, 2017). This, in turn, leads to a higher market price of the steer carcass. Modified expression patterns of the lipid metabolism genes and/or perturbed anabolic hormone status reportedly play a role in the improved quality of the steer carcass (Plouzek & Trenkle, 1991; Jeong et al, 2013), but the underlying mechanisms are poorly understood.
Castration increases the abdominal obesity of high‐fat diet (HFD)‐fed rodents (Harada et al, 2016). Intriguingly, this study also showed that the obese‐prone phenotype of castrated male mice only develops in the presence of the gut microbiota. Furthermore, several recent studies have shown a tripartite relationship between sex, metabolic/disease phenotypes, and gut microbes. Differences in the gut microbiota between male and female mice render the former less likely to develop autoimmune diseases, and male castration changes the host disease susceptibility phenotype by altering the gut microbiota (Markle et al, 2013; Yurkovetskiy et al, 2013). These sex‐related differences in the host–microbiota interaction, which has been called the “microgenderome”, are mainly dependent on the levels of androgens (Flak et al, 2013). The symbiotic gut microbiota plays a pivotal role in mammalian metabolism. Accumulating evidence indicates that the gut microbiota contributes to the development of obesity and associated metabolic disorders by regulating energy homeostasis (Turnbaugh et al, 2006; Shin et al, 2014). Accordingly, in the present study, we aimed to determine the effect of hypogonadism following male castration on the gut microbiota, serum metabolites, and the host metabolic phenotype, and the contribution of the altered microbiota to increased adiposity in hypogonadal animal models. We used metataxonomic (i.e., amplification and sequencing of bacterial 16S rRNA genes) and metabolomic approaches to analyze the gastrointestinal (rumen, ileum, cecum, and colon), serum, and striploin muscle samples of male, castrated male (CtM), and female Korean brown cattle (Bos taurus coreanae; hereafter referred to as the Hanwoo) and Holstein Friesian cattle (the Holstein). We also used a male mouse castration model to evaluate the causative role of the alteration of the intestinal microbiota in the host metabolic phenotype in response to hypogonadism.
Results
Male hypogonadism results in the slight alteration of rectal microbiota and elevated levels of the serum branched‐chain amino acids (BCAA) in post‐pubescent cattle
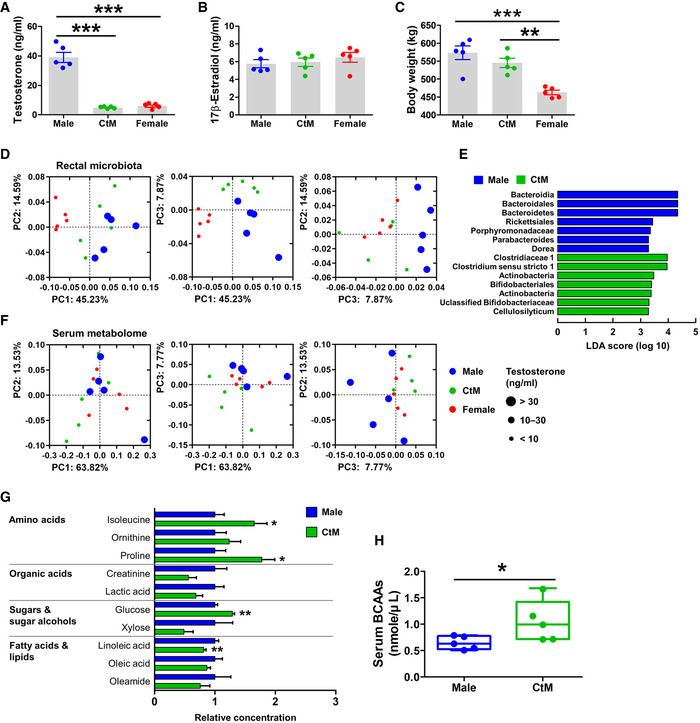
To evaluate the possible relationship between male hypogonadism and the intestinal microbiota in cattle, we investigated bacterial communities in the rectal contents of male (n = 5) and CtM Hanwoo (n = 5) at juvenile (mean age: 11.9 ± 1.1 months) and post‐pubescent stages (mean age: 19.3 ± 1.5 months). Age‐matched non‐pregnant females (n = 5) were included in the comparison of rectal microbiota. All animal groups were farmed in geographically closely located cages and fed the same diet (Appendix Table S1) to minimize any cage‐ and diet‐induced inter‐individual variation of the intestinal microbiota. At the post‐pubescent stage, but not at the juvenile stage (Appendix Fig S1A), the serum testosterone levels in the male Hanwoo were significantly higher than those in CtM and females (Fig 1A; ANOVA, P < 0.001). The serum 17β‐estradiol levels were not significantly different between groups at the juvenile and post‐pubescent stages (Appendix Figs S1B and 1B). Body weight was not significantly affected by male castration (Fig 1C). Principal coordinates analysis (PCoA) of the rectal microbiome indicated that the gut microbial communities of the female cattle at the juvenile or post‐pubescent stages were significantly separated from the other groups (PERMANOVA, P < 0.05; comparison of the female with male or CtM animals); no significant difference in rectal microbiome between the male and CtM groups was observed at the post‐pubescent stage (Appendix Figs S1C and 1D, and Appendix Table S2). At the post‐pubescent stage, however, the PCoA plots of the CtM animals separated from those of the male and grouped together with the female data on the PC3 vs. PC2 analysis (Fig 1D). We next used the linear discriminant analysis effect size (LEfSe) method (Segata et al, 2011) to analyze the patterns of bacterial taxon abundance in rectal samples from the male and CtM Hanwoo. The LEfSe plot revealed that the phylum Bacteroidetes was enriched in the male rectal samples, whereas the family Clostridiaceae and the phylum Actinobacteria were the discriminant taxa of the CtM rectal samples (Fig 1E). We additionally investigated the impact of male castration on the rectal microbiota in another breed of cattle, the Holstein. Similar to the post‐pubescent Hanwoo, PCoA of the Holstein rectal microbiota data revealed a significant bacterial community separation according to male castration (PERMANOVA, P = 0.015; comparison of the male and CtM animals), with a clustering of the CtM and female data (Appendix Fig S2A and Appendix Table S2). In terms of the alpha diversity indices, no difference was observed between the male and CtM cattle; however, the female groups in both Hanwoo and Holstein cattle showed a significantly higher value in the species diversity, richness, and evenness at the post‐pubescent stage, when compared to those of the male or CtM cattle (Appendix Tables S3–S5).
Figure 1. The rectal microbial and serum metabolome profiles of the post‐pubescent Hanwoo.
-
A–CDetermination of the body weight (A), and serum testosterone (B) and 17β‐estradiol (C) levels, in post‐pubescent male, castrated male (CtM), and female Hanwoo (n = 5 in each group).
-
DPCoA of the rectal bacterial 16S rRNA gene sequences based on the weighted UniFrac distance matrix.
-
EDiscriminant taxa of the male and CtM rectal microbiota, as determined by the linear discriminant analysis effect size (LEfSe). The different abundances are represented by LDA scores.
-
FThe serum metabolome profiles were analyzed using GC‐TOF‐MS and clustered by PCoA based on the Bray–Curtis dissimilarity matrix.
-
GThe relative abundances of serum metabolites displayed as bar graphs (n = 5 in each group).
-
HThe BCAA serum levels quantified enzymatically, as described in the Materials and Methods section.
Data information: The data were analyzed by using ANOVA followed by Tukey’s post hoc test (A to C) and the Mann–Whitney U‐test (G and H). The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The scattered dots in the bar graphs/box plot diagrams represent the individual replicates. *P < 0.05, **P < 0.01, and ***P < 0.001. CtM, castrated male.
To evaluate the effects of changes in the gut microbiota induced by male castration on the host metabolomic profile, we analyzed the serum metabolome profiles of post‐pubescent cattle by using gas chromatography time‐of‐flight mass spectrometry (GC‐TOF‐MS). PCoA of the Hanwoo metabolome data revealed that the male and CtM datasets clustered separately (Fig 1F; PERMANOVA, P = 0.073; comparison between the male and CtM Hanwoo). The CtM and female datasets clustered closely together. Profiling of the serum metabolomes of the male and CtM Hanwoo revealed meaningful differences in the serum amino acid levels. Specifically, the serum levels of isoleucine and proline were significantly higher in the CtM Hanwoo than in the male Hanwoo (Fig 1G; Mann–Whitney U‐test, P < 0.05). Of note, BCAAs (isoleucine, leucine, and valine) are responsible not only for the intramuscular lipid accumulation, by increasing the vascular fatty acid transport into the mouse skeletal muscle (Jang et al, 2016), but also for lipogenesis in the human adipocyte (Green et al, 2016). We accordingly quantified serum BCAA levels using an enzyme‐based assay. The serum BCAA levels were significantly higher in the post‐pubescent CtM Hanwoo than in the male controls (Fig 1H; Mann–Whitney U‐test, P < 0.05). The serum metabolite profiles of Holstein revealed that the levels of isoleucine, valine, phenylalanine, and threonine were significantly higher (Mann–Whitney U‐test, P < 0.05) in the CtM animals than in males (Appendix Fig S2B and C). Further, an enzyme‐based quantitative analysis revealed higher median serum BCAA levels in the CtM group than in the male controls, but the difference was not statistically significant (Appendix Fig S2D).
Metataxonomic analysis of different intestinal compartments reveals a marked increase in the family Peptostreptococcaceae in the ileum of CtM animals
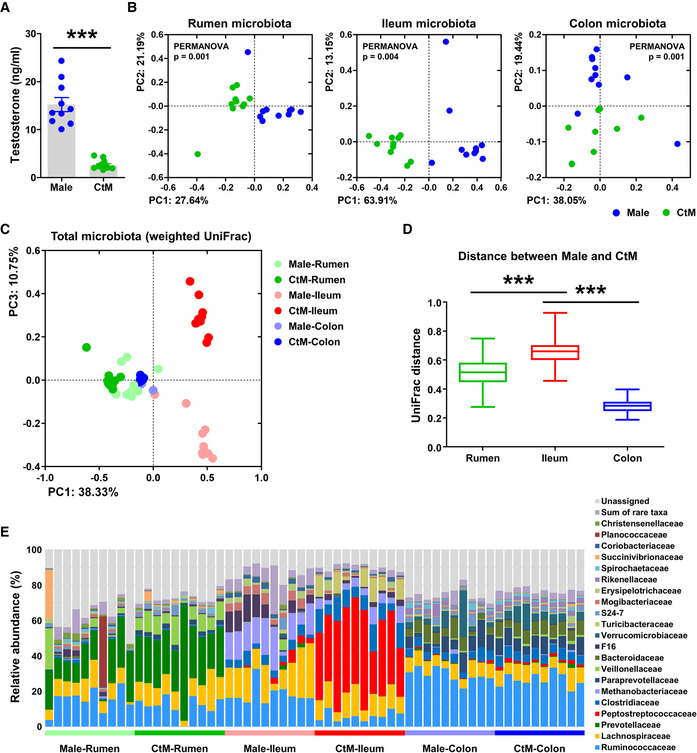
Although the analysis revealed differences in the rectal microbiota of the male and CtM cattle (Fig 1 and Appendix Fig S2), no direct link between the composition of rectal microbiota and elevated serum BCAA levels was apparent, nor any involvement of the rectal microbiota in the host amino acid metabolism (on the basis of the rectal metabolome profiles and correlation analysis of the rectal microbiota with the rectal metabolome in the post‐pubescent cattle; Appendix Figs S3 and S4). To reveal whether the high serum BCAA levels were a consequence of the gut microbial activity, we next extensively investigated microbial profiles in the different compartments of the gastrointestinal tract. We collected the luminal contents of the rumen, ileum, and colon from the adult male (mean age: 31.2 ± 5.9 months, n = 10) and CtM (mean age: 33.9 ± 1.4 months, n = 10) Hanwoo. Similar to the post‐pubescent stage, we observed a significant difference (Mann–Whitney U‐test, P < 0.001) in the serum testosterone levels between the male and CtM groups at the adult stage (Fig 2A). PCoA of the microbiota in the rumen, ileum, and colon revealed that the bacterial communities in the different segments were significantly different in the male and CtM Hanwoo (PERMANOVA, P < 0.005 for each segment; comparison of the male and CtM animals in the rumen, ileum, and colon; Fig 2B). We next evaluated the gastrointestinal segment in which the bacterial communities were mostly affected by male castration. When all gastrointestinal samples were plotted together, only the ileal microbiota in the male and CtM groups were separated on both the abundance‐weighted (weighted UniFrac, Fig 2C and Appendix Fig S5A) and abundance‐unweighted PCoA (unweighted UniFrac, Appendix Fig S5B). Similar, analysis of the weighted UniFrac distance between the male and CtM groups revealed the largest dissimilarity value for the ileal microbiota (Fig 2D).
Figure 2. Microbial metataxonomic profiles of different compartments of the intestinal tract of the adult male and castrated male (CtM) Hanwoo.
-
ASerum testosterone levels in the adult male and CtM Hanwoo (n = 10 in each group). The scattered dots in the bar graph represent the individual replicates.
-
B, CPCoA, based on the weighted UniFrac distance matrix, of the bacterial 16S rRNA gene sequence data for the rumen, ileum, and colon, shown for the different segments of the gastrointestinal tract (B) and combined data (C).
-
DMicrobial dissimilarity (calculated based on the weighted UniFrac distance matrix, 100 values in each group) between the male and CtM groups in the different intestinal compartments. The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively.
-
EThe relative abundances of abundant bacterial taxa (> 0.5% of the mean abundance).
Data information: The data were analyzed by using the Mann–Whitney U‐test (A), PERMANOVA with 999 permutations (B), or ANOVA followed by Tukey’s post hoc test (D). *P < 0.05, **P < 0.01, and ***P < 0.001. CtM, castrated male.
We then compared the relative abundances of major taxa (> 0.5% of the mean abundance) at the family level. The difference in the ileal microbiota in the male and CtM groups was apparent, with a marked increase in the family Peptostreptococcaceae in the CtM Hanwoo (Fig 2E and Appendix Fig S5C). Phylogenetic analysis of the operational taxonomic units (OTUs) assigned to the family Peptostreptococcaceae further revealed that the abundant OTUs mostly belonged to the genera Romboutsia and Paeniclostridium (Appendix Fig S6).
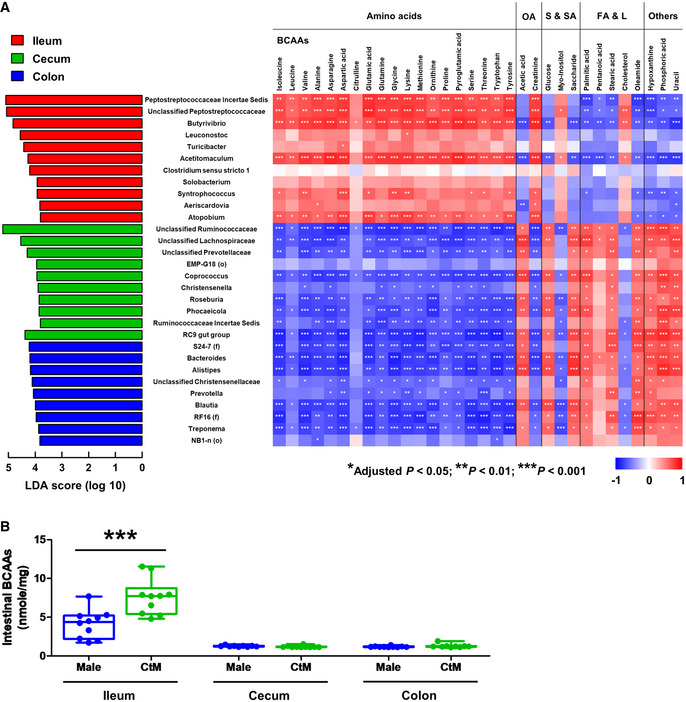
To investigate the relationship between the intestinal microbiota and the resultant microbial metabolites (especially BCAAs) in detail, we subsequently analyzed metabolomic profiles of the luminal contents of the ileum, cecum, and colon of the CtM Hanwoo. Building on the results of the LEfSe analysis and the metabolome profile data for the different intestinal compartments of the CtM Hanwoo, we attempted to identify the candidate bacterial taxa that might be responsible for the quantitative traits of the intestinal metabolites and, further, the host metabolic phenotype. In the ileum, a positive correlation (Spearman’s rank correlation analysis, adjusted P < 0.05) was detected between the BCAAs and two unclassified genera belonging to the family Peptostreptococcaceae, and the genera Butyrivibrio, Acetitomaculum, and Atopobium (Fig 3A). Furthermore, the ileal levels of intestinal BCAAs were significantly higher (Mann–Whitney U‐test, P < 0.001) in the CtM Hanwoo than those in the male controls; the BCAA levels in the cecum and colon were much lower than those in the ileum, and no quantitative difference in the BCAA levels was observed between the male and CtM groups (Fig 3B).
Figure 3. Correlation analysis of the intestinal microbial taxa and metabolites from the castrated male (CtM) Hanwoo.
-
AThe discriminant microbial taxa of the ileum, cecum, and colon of ten adult CtM Hanwoo are shown, determined to the genus level by the linear discriminant analysis (LDA) effect size (LEfSe) method. The LDA scores represent the degree of consistent difference in the relative abundance between groups. For orphan sequences (i.e., unassigned at the genus level), a high‐rank lineage is provided (f, family; o, order).
-
BThe intestinal branched‐chain amino acid (BCAA) levels in the adult male and CtM Hanwoo (n = 10 in each group). BCAA levels in the luminal contents of the ileum, cecum, and colon were quantified by an enzymatic method using a BCAA assay kit. The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The scattered dots in the box plot diagrams represent the individual replicates.
Data information: (A) Spearman’s rank correlation coefficients and the corresponding P values were calculated based on comparisons of the relative abundances of 30 discriminant microbial taxa and 32 intestinal metabolites. Holm–Sidak correction was used for multiple comparisons. (B) The data were analyzed by the Mann–Whitney U‐test. *P < 0.05, **P < 0.01, ***P < 0.001. OA, organic acids; S and SA, sugars and sugar alcohols; FA and L, fatty acids and lipids.
Male castration results in increased intramuscular fat accumulation with high‐BCAA levels in the adult cattle
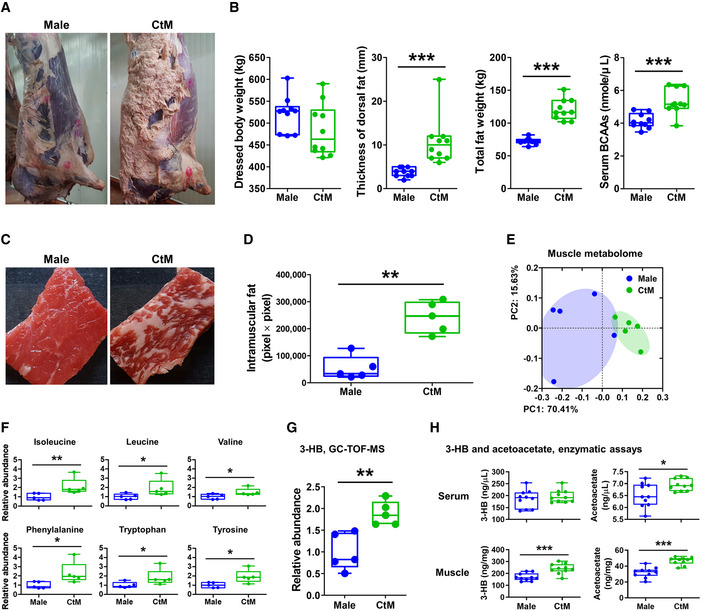
We next evaluated the consequence of the distinct metabolomic profile (i.e., high levels of BCAAs in the serum and ileum) of the CtM cattle. After the bodies were dressed (n = 10 in each group), we observed clear differences in the body composition of the male and CtM Hanwoo (Fig 4A). The thickness of the dorsal subcutaneous fat, total fat weight, and serum BCAA levels in the CtM group was significantly higher than those of the male controls (Fig 4B; Mann–Whitney U‐test, P < 0.001). We then analyzed the degree of intramuscular fat accumulation in the fresh striploin muscle in the male and CtM Hanwoo carcasses (n = 5 in each group). We observed a significantly higher (Mann–Whitney U‐test, P < 0.01) intramuscular fat accumulation in the CtM muscle than in the male muscle (Fig 4C and D). We next evaluated the intramuscular metabolite profiles. Similar to the serum metabolite profiles described above, the intramuscular metabolomes of the male and CtM carcasses clustered significantly separately by PCoA (Fig 4E; PERMANOVA, P = 0.008; comparison of the male and CtM Hanwoo). The levels of BCAAs (i.e., isoleucine and leucine), as well as phenylalanine, tryptophan, and tyrosine, were significantly higher (Mann–Whitney U‐test, P < 0.05) in the CtM muscle than in the male muscle (Fig 4F). Among the amino acids whose levels were increased in the CtM muscle, leucine, phenylalanine, tryptophan, and tyrosine can be degraded into acetyl‐CoA, a precursor of ketone bodies. We subsequently measured the intramuscular levels of β‐hydroxybutyrate (3‐HB), widely used to diagnose ketosis in dairy cow (Zhang et al, 2013), in the male and CtM striploin muscles using the GC‐TOF‐MS. The 3‐HB levels were significantly higher (Mann–Whitney U‐test, P < 0.01) in the CtM muscle than in the male muscle (Fig 4G). We also measured the levels of 3‐HB and acetoacetate in serum and muscle samples from male and CtM cattle using an enzymatic assay. We found that the serum acetoacetate level was significantly higher in the CtM cattle than in the male cattle (Mann–Whitney U‐test, P < 0.05) and that the levels of both ketone bodies were significantly higher in muscle from CtM cattle than in muscle from males (Mann–Whitney U‐test, P < 0.001; Fig 4H). Interestingly, Jang et al (2016) reported that 3‐HB secreted by the muscle cell activates endothelial fatty acid transport and promotes lipid accumulation in the muscle. This supports the data presented herein, bridging the high levels of serum BCAAs and the elevated intramuscular fat accumulation in the CtM cattle.
Figure 4. Intramuscular metabolomic profiles of the adult male and castrated male (CtM) Hanwoo.
-
ARepresentative images of dressed bodies of the adult male and CtM Hanwoo (n = 10 in each group).
-
BDressed body weight, thickness of the dorsal subcutaneous fat, total weight of the extramuscular fat (posterior subcutaneous fat, mesenteric fat, and retroperitoneal fat), and serum levels of the branched‐chain amino acids (BCAAs).
-
CRepresentative images of fresh striploin muscle from the adult male and CtM carcasses (n = 5 in each group).
-
DThe intramuscular fat area in size‐normalized muscle.
-
EThe intramuscular metabolome profiles analyzed using GC‐TOF‐MS and clustered by PCoA based on the Bray–Curtis dissimilarity matrix
-
F, GThe relative abundances of intramuscular metabolites (F) and β‐hydroxybutyrate (3‐HB) (G) in the male and CtM samples.
-
HThe serum and muscle ketone body levels, quantified enzymatically, as described in the Materials and Methods section (n = 10 in each group).
Data information: The data were analyzed by using the Mann–Whitney U‐test (B, D, F, G, and H). The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The scattered dots in the box plot diagrams represent the individual replicates. *P < 0.05, **P < 0.01, and ***P < 0.001. CtM, castrated male; BCAAs, branched‐chain amino acids; 3‐HB, β‐hydroxybutyrate.
Male hypogonadism leads to an ileal microbial alteration, adiposity, and increased serum BCAA levels in mice
The correlation‐based analysis in cattle presented above suggested an obesogenic effect(s) of the castration of young male on the subsequent host metabolic phenotype in association with altered intestinal microbiota and systemic metabolite levels. We next evaluated the causative role of the alteration of the gut microbiota following hypogonadism in the obese metabolic phenotype in a mouse model. Male mice underwent prepubertal castration by orchiectomy and were fed a HFD to promote the obese phenotype. Age‐matched male mice that underwent Sham operation and fed low‐fat diet (LFD) were included as the castration and diet controls, respectively (n = 6 in each group; Fig 5A). At the end of the experiment, the serum testosterone levels in the CtM groups were significantly lower than those in the Sham groups (unpaired Student’s t‐test, P < 0.05), confirming that hypogonadism developed after castration (Fig 5B). Weighted PCoA of both the ileal and colonic microbiota revealed a male castration‐associated separation of the intestinal microbial community structures, regardless of the intestinal compartment analyzed (PERMANOVA, P < 0.04; comparison of the Sham and CtM groups). However, the difference between the ileal microbiota was more obvious than that between the colonic microbiota in HFD‐fed mice (Fig 5C and Appendix Fig S7A and B). In both LFD‐ and HFD‐fed mice, male hypogonadism amplified the expansion of the family Peptostreptococcaceae in the ileum (Fig 5D and Appendix Fig S8A and B), and resulted in significantly increased levels of serum BCAAs (Fig 5E; unpaired Student’s t‐test, P < 0.05).
Figure 5. The intestinal microbiota and metabolic phenotypes of the male and castrated male (CtM) mice.
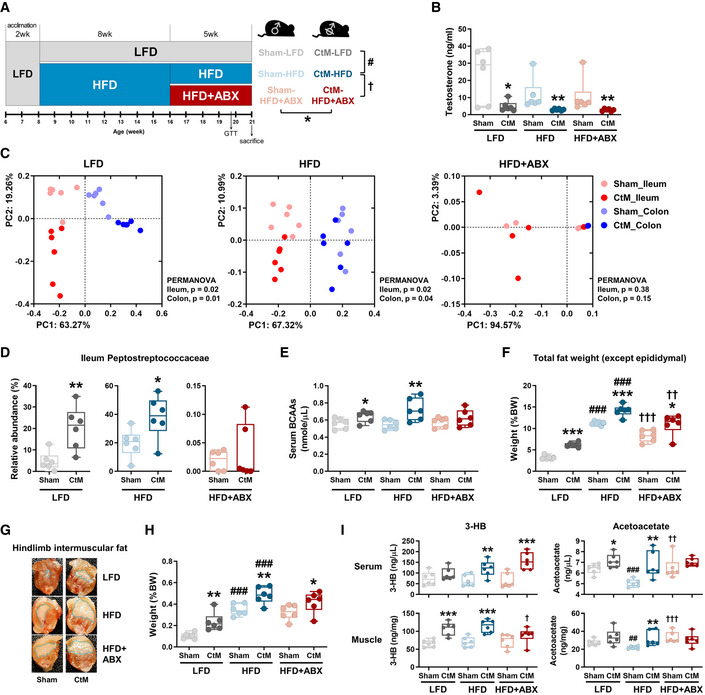
-
ASchematic design of the hypogonadism experiments in the mouse model (n = 6 in each group).
-
BSerum testosterone levels.
-
CPCoA, based on the weighted UniFrac distance matrix, of the bacterial 16S rRNA gene sequence data for the luminal contents of the ileum and colon, shown according to the different diets and antibiotic treatment.
-
DThe relative abundances of the family Peptostreptococcaceae are displayed as a box and dot plots.
-
EThe serum branched‐chain amino acid (BCAA) levels quantified enzymatically, as described in the Materials and Methods section.
-
FThe total weight of the extramuscular fat, including posterior subcutaneous fat, mesenteric fat, and retroperitoneal fat.
-
G, HRepresentative images (G) and weight (H) of the hindlimb intermuscular fat. The images are from a longitudinal section of the hind leg. The fat weight data are presented as a percentage of the body weight.
-
ISerum and hindlimb muscle ketone body levels quantified enzymatically.
Data information: The data were analyzed by using the unpaired Student’s t‐test (B, D to F, H, and I) or PERMANOVA with 999 permutations (C). *Comparison of the Sham and CtM mice; #comparison of the LFD and HFD mice; and †comparison of the HFD and HFD + ABX mice. The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The scattered dots in the box plot diagrams represent the individual replicates. *P < 0.05, **P < 0.01, and ***P < 0.001. LFD, low‐fat diet; HFD, high‐fat diet; ABX, antibiotics; CtM, castrated male; BCAAs, branched‐chain amino acids; 3‐HB, β‐hydroxybutyrate.
To reveal the effect(s) of gut microbiota on the host metabolism, the HFD‐fed groups received a broad‐spectrum antibiotic cocktail in the drinking water for 5 weeks (HFD + ABX, Fig 5A). No signs of dehydration or anorexia were observed during the antibiotic treatment. The antibiotic treatment disrupted both the ileal and colonic microbial structures (Fig 5C and D and Appendix Figs S7C and S10C). As expected, the antibiotic treatment abolished the previously observed increase in the serum BCAA levels in the CtM groups compared with the Sham groups (Fig 5E). The proportions of both the extramuscular white adipose tissue depots (the posterior subcutaneous fat, mesenteric fat, and retroperitoneal fat) and intermuscular fat (located between the gastrocnemius and rectus femoris) were significantly increased by HFD feeding and castration (unpaired Student’s t‐test, P < 0.01; Fig 5F–H and Appendix Fig S9). We next measured the levels of 3‐HB and acetoacetate in the serum and hindlimb muscle of the mice. There were significantly higher levels of both ketone bodies in the HFD‐fed CtM group than in the HFD‐fed Sham group (unpaired Student’s t‐test, P < 0.01; Fig 5I), but the significant difference in acetoacetate level was abolished by antibiotic treatment.
Fecal microbiota transplantation (FMT) from the hypogonadal male mice donors leads to increased fat accumulation in the recipient eugonadal male mice
To determine whether the gut microbiota was responsible for the obese phenotype of castrated mice, we performed FMT from the HFD‐fed Sham or CtM donor mice to age‐matched surgery‐naïve eugonadal male recipient mice [Sham‐R (n = 4) and CtM‐R (n = 5), respectively; Fig 6A]. Intriguingly, there was a statistically significant difference in the serum testosterone levels between the Sham‐R and CtM‐R groups (Fig 6B; unpaired Student’s t‐test, P < 0.05), which suggests a possible bidirectional regulatory relationship between the gut microbiota and androgenic hormones, but further corroborating evidence is needed, because of the small number of mice studied. FMT resulted in the segregation of the ileal and colonic microbiota of the recipient mice according to treatment (PERMANOVA, P ≤ 0.05; comparison of the Sham‐R and CtM‐R groups) (Fig 6C and Appendix Fig S10A). The LEfSe analysis of the ileal microbiota revealed the family Peptostreptococcaceae as the most discriminant taxon in comparisons of the Sham‐R and CtM‐R groups (Fig 6D and E). Importantly, CtM‐R mice recapitulated the phenotype observed in the CtM‐HFD donor mice in terms of a significantly increased fat accumulation in the posterior subcutaneous, epididymal, mesenteric, and retroperitoneal depots, as well as of the intermuscular fat in the CtM‐R group compared with the Sham‐R group (Fig 6F–H and Appendix Fig S10B–G; unpaired Student’s t‐test, P < 0.05). Measurement of the ketone body levels showed significantly higher level of serum acetoacetate in the CtM‐R group than in the Sham‐R group (unpaired Student’s t‐test, P < 0.05; Fig 6I). Collectively, these observations suggest that the hypogonadism‐mediated gut microbial changes contribute to the obese‐prone metabolic phenotype per se. In terms of the gut–brain axis, the microbial shift in CtM‐R mice toward that in CtM mice might decrease serum testosterone levels by regulating a negative feedback loop between gonadotropin‐releasing hormone in the hypothalamus, luteinizing hormone/follicle‐stimulating hormone in the pituitary, and testosterone in the testis.
Figure 6. The intestinal microbiota and metabolic phenotypes of male mice in response to fecal microbiota transplantation (FMT).
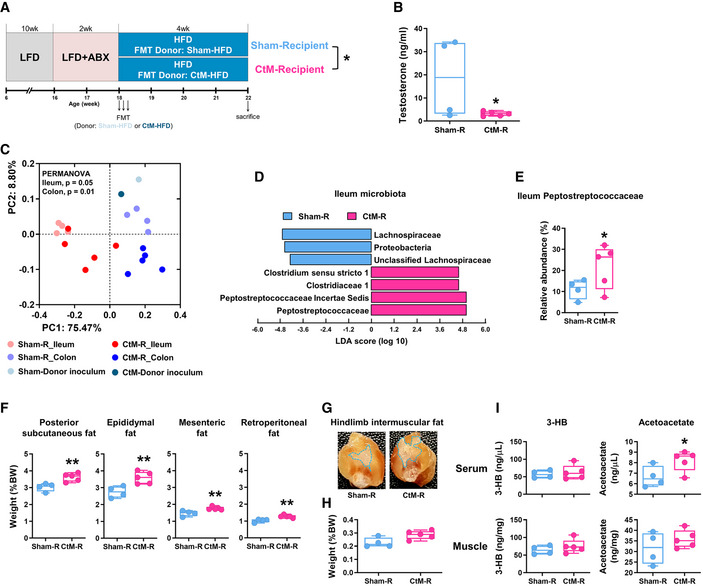
-
ASchematic design for the mouse FMT experiments (n = 4–5 in each group).
-
BThe serum testosterone levels in the Sham‐R and castrated male (CtM)‐R mice.
-
CPCoA, based on the weighted UniFrac distance matrix, of the bacterial 16S rRNA gene sequence data for the luminal contents of the ileum and colon.
-
DThe discriminant microbial taxa were determined by using the LEfSe and presented using the LDA score.
-
EThe relative abundances of the family Peptostreptococcaceae are displayed as a box and dot plots.
-
FWeights of the posterior subcutaneous fat, epididymal fat, mesenteric fat, and retroperitoneal fat in the Sham‐R and CtM‐R mice.
-
G, HRepresentative images (G) and weight (H) of the hindlimb intermuscular fat in the Sham‐R and CtM‐R mice. Images were obtained from a longitudinal section of the hind leg. The fat weight data are presented as a percentage of the body weight.
-
ISerum and hindlimb muscle ketone body levels, quantified enzymatically.
Data information: The data were analyzed by using the unpaired Student’s t‐test (B, E, F, H, and I) or PERMANOVA with 999 permutations (C). The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The scattered dots in the box plot diagrams represent the individual replicates. *P < 0.05 and **P < 0.01. LFD, low‐fat diet; HFD, high‐fat diet; ABX, antibiotics; FMT, fecal microbiota transplantation; CtM, castrated male; 3‐HB, β‐hydroxybutyrate.
Feeding BCAA‐enriched diet elicits an obese phenotype in male mice
To evaluate whether the surplus BCAAs are the key factor contributing to the obese phenotype of the castrated animal, we fed non‐castrated male mice with HFD (0% BCAA group) or customized isocaloric BCAA‐HFDs (3% and 5% BCAA groups) for 8 weeks (Fig 7A). For the BCAA‐HFD, casein in HFD was substituted by BCAAs (Appendix Table S6). Feeding BCAA‐HFD increased the body weight gain to a greater extent than feeding HFD, with a meaningful difference after 3 weeks of feeding (Fig 7B; unpaired Student’s t‐test, P < 0.05). At the end of the experiment, we observed a significantly greater adiposity (characterized by the increased percentage to body weight ratio of the posterior subcutaneous fat, epididymal fat, mesenteric fat, and retroperitoneal fat) in the BCAA‐HFD‐fed groups compared with the HFD‐fed group (Fig 7C and D, and Appendix Fig S11A–H; unpaired Student’s t‐test, P < 0.01). The study by Solon‐Biet et al (2019) showed that long‐term (> 50 weeks) consumption of a high‐BCAA diet causes hyperphagia; however, consumption of a BCAA‐HFD by mice for 8 weeks did not induce a difference in food intake between the groups (Appendix Fig S11D). Further, the serum BCAA levels were significantly increased in the mice fed diets containing over 3% of BCAAs, confirming systemic circulation of the dietary BCAAs (Fig 7E; unpaired Student’s t‐test, P < 0.01). We subsequently investigated the effect of BCAA supplementations on the gut microbiota by analyzing the ileal and colonic bacterial communities. However, no connecting evidence was apparent linking the BCAA supplementation and gut microbial alteration (Fig 7F and Appendix Fig S11I). These observations implied that the surplus BCAAs detected in the hypogonadal animals might be a consequence of the altered gut microbial activity following male castration. There were no differences in the serum or muscle levels of 3‐HB between the groups. However, there were significantly higher levels of acetoacetate in the serum and muscle of BCAA‐HFD‐fed mice (Fig 7G; unpaired Student’s t‐test, P < 0.05).
Figure 7. The metabolic phenotypes and intestinal microbiota of male mice fed branched‐chain amino acid (BCAA)‐supplemented diets.
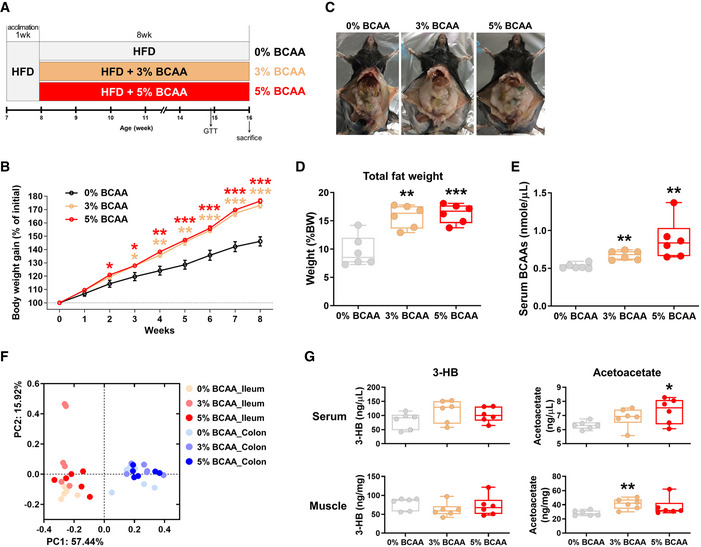
-
ASchematic design of the chronic BCAA feeding experiments (n = 6 in each group).
-
BBody weight gain in response to the different levels of dietary BCAAs.
-
CRepresentative images of gross anatomy.
-
DThe total weight of the extramuscular fat (posterior subcutaneous fat, epididymal fat, mesenteric fat, and retroperitoneal fat) in the mice fed high‐fat diet (HFD) with or without BCAAs.
-
EThe serum BCAA levels in the mice fed HFD with or without BCAAs.
-
FPCoA, based on the weighted UniFrac distance matrix, of the bacterial 16S rRNA gene sequence data for the luminal contents of the ileum and colon are shown. The body weight gain data are presented as a percentage of the initial body weight. The fat weight data are presented as a percentage of the body weight.
-
GSerum and hindlimb muscle ketone body levels, quantified enzymatically.
Data information: The data were analyzed by using the unpaired Student’s t‐test (B, D, E, and G). *Comparison of the 0% BCAA and 3% BCAA or 5% BCAA‐fed mice. The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The scattered dots in the box plot diagrams represent the individual replicates. *P < 0.05, **P < 0.01, and ***P < 0.001. HFD, high‐fat diet; BCAA, branched‐chain amino acid; 3‐HB, β‐hydroxybutyrate.
Testosterone acts not only as the principal androgenic hormone but also as a metabolic hormone, affecting the hepatic urea cycle to regulate the whole‐body protein catabolism (Lam et al, 2017; Birzniece, 2018). Because the metataxonomic analyses of the ileal microbiota of castrated animals revealed a high abundance of the family Peptostreptococcaceae, we hypothesized that a positive feedback loop might exist between the hepatic urea cycle and ureolysis undertaken by the abundant gut microbiota. Accordingly, we assessed the systemic urea/ammonia metabolism activities in cattle and mice in response to male hypogonadism. In adult Hanwoo, the microbial urease activity in both the rumen and ileum in the CtM group was significantly higher (Appendix Fig S12A; Mann–Whitney U‐test, P < 0.01) than that in the male group. The intestinal microbial ureases hydrolyze urea to ammonia (Patra & Aschenbach, 2018). Hence, we subsequently measured the intestinal ammonia levels. The ileal ammonia levels in the CtM group were significantly higher (Appendix Fig S12B; Mann–Whitney U‐test, P < 0.05) than those in the male group. In the gut, ammonia produced by ureolytic bacteria is absorbed into the bloodstream and used to fuel hepatic ureagenesis (Liu et al, 2018). Accordingly, we determined ammonia levels in the mouse serum. For the HFD‐fed mice, the serum ammonia levels in the CtM group were significantly higher than those in the Sham control (Appendix Fig S12D; unpaired Student’s t‐test, P < 0.05). In both cattle and mice, however, no differences in the serum urea levels in the male and CtM groups were apparent (Appendix Fig S12C and E). This was probably because of a simple diffusion of the systemic urea into the gastrointestinal tract (Abdoun et al, 2006). Collectively, the above observations suggested that testosterone deficiency in the castrated animals might positively regulate the hepatic ureagenesis and that urea (and its hydrolysis products, i.e., ammonia and carbon dioxide) potentially affects the ileal microbial profile in the CtM animals.
Discussion
Although reciprocal parallelism between obesity and hypogonadism in men has been reported by a number of cross‐sectional studies (Bojesen et al, 2006; Saboor Aftab et al, 2013; Carrageta et al, 2019), the mechanisms linking testosterone and the obese phenotype are not fully understood. Considering the obesity epidemic, the prevalence of hypogonadism is expected to increase in the coming decades, highlighting the importance of understanding these mechanisms. For the livestock, however, animal welfare, and economical feeding and management must be considered in equal measure. In this context, understanding the metabolic features that could potentially influence the metabolism of beef cattle is important in breeding animals representing the “metabolically healthy obese” phenotype (Stefan et al, 2013) in terms of the production of high‐quality animal products. Keeping these bimodal aspects in mind, we investigated the effect of male castration on the intestinal microbiota and host metabolism in cattle and mice. We demonstrated (i) altered gut microbial profiles in the CtM animals, with a negative feedback between the serum testosterone levels and the ileal abundance of Peptostreptococcaceae, and (ii) a distinct metabolomic signature of the CtM animals with an enhanced amino acid metabolism (Fig 8). Furthermore, (iii) the obese phenotype associated with hypogonadism was transferrable via FMT in mice.
Figure 8.
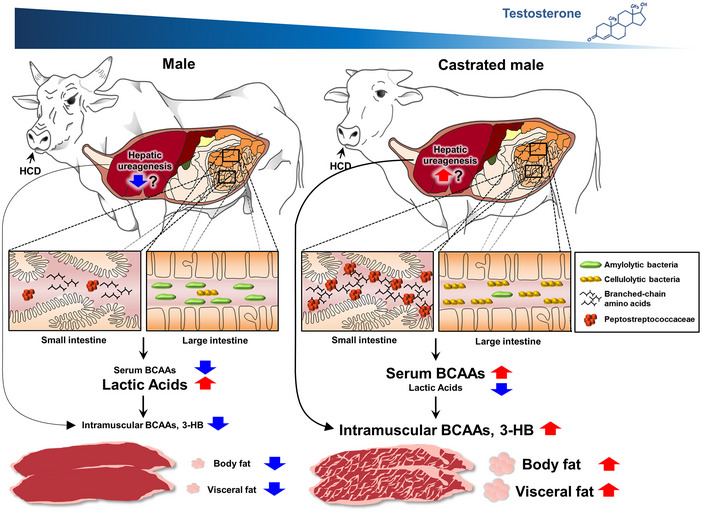
A model for the increased level of body fat storage by intestinal microbial alteration in the castrated cattle.
We used a mouse model to examine whether alterations in the gut microbiota following hypogonadism play a causative role in the obese metabolic phenotype. Generally speaking, the mouse is a monogastric animal; as such, the digestive processes and subsequent energy metabolism are different from those in ruminant animals. Importantly, however, we found a marked increase in the ileal abundance of Peptostreptococcaceae in the CtM groups of both cattle and mice, suggesting a possible link between male hypogonadism and gut microbial perturbation in other mammal species. In humans, chemical and surgical castration of the testes is used as an androgen deprivation therapy (ADT) to treat patients suffering from advanced prostate cancer (Feldman & Feldman, 2001). Recent accumulating evidence points out that an increased level of cholesterol post‐ADT is responsible for increased de novo androgen synthesis by tumor cells, which contributes to recurrence of castration‐resistant prostate cancer (Twiddy et al, 2011). The mechanism underlying ADT and further increases cholesterol levels is unclear. Intriguingly, Tsoi et al (2017) report that Peptostreptococcus anaerobius can trigger intracellular cholesterol biosynthesis in mouse colon cells. Further studies are needed to examine the hormonal perturbation–gut microbiota–cancer axis in detail.
The metabolic profiles of different compartments of the intestinal tract of the CtM Hanwoo indicated that the amino acid metabolism exclusively occurs in the small intestine (Fig 3). Amino acids that are abundant in the ileum might be processed in several ways, including absorption by the small intestinal epithelial cells to produce amino acid metabolites, and/or digestion by the colonic microbiota (Dai et al, 2015). Relatively high levels of amino acids were also detected in the serum and striploin muscle (but not in the large intestine) of the CtM cattle, suggesting that the fate of the ileal amino acids in the CtM cattle may mostly be weighted toward the small intestinal absorption. Particularly, in the current study, we demonstrated high serum BCAA levels in the castrated cattle and mice (Figs 4B and 5E). Furthermore, disruption of the gut microbiota abolished the effect of castration on BCAA levels in mice, indicating that the increased circulating BCAA levels can be attributed to altered gut microbial activity rather than hypogonadism itself. Furthermore, chronic feeding of the BCAA‐enriched HFD to male mice significantly increased its body weight and adiposity, and exacerbated glucose intolerance and insulin resistance (Fig 7D and Appendix Fig S11A–C). BCAAs are important substrates of the anaplerotic and ketogenic metabolism in various tissues, playing substantial roles in the host metabolism and subsequent metabolic disorders (i.e., insulin resistance; Lynch & Adams, 2014). Pedersen et al (2016) reported a strong association between increased levels of the serum BCAAs and insulin resistance in non‐diabetic human subjects. They also noted that specific members of the intestinal microbiota (e.g., Prevotella copri) could increase the biosynthesis of BCAAs and elicit metabolic disorders, such as insulin resistance and/or glucose intolerance. Indeed, gut‐dwelling bacteria produce highly active proteases and peptidases, enabling the hydrolysis of alimentary and/or endogenous proteins, and a subsequent absorption of amino acids in the gastrointestinal tract (Neis et al, 2015). GC‐TOF‐MS analysis in the cattle study revealed variations in the amounts of individual BCAAs (e.g., isoleucine, leucine, and valine) in intestinal luminal, serum, and striploin muscle samples from the male and CtM groups. However, the study was limited because we used a total BCAA detection kit for verification. In the mouse experiments, BCAA‐enriched diets comprised a mixture of isoleucine, leucine, and valine. Further studies will evaluate the properties and effects of each individual BCAA on host metabolism with respect to therapeutic and management purposes in humans and breeding animals, respectively.
The high levels of global BCAAs in cattle and mice might be attributed to high production of BCAAs by gut microbiota. However, we were not able to obtain clear evidence of causation to bridge the gap between high‐BCAA levels and the marked increase in microbial taxa (i.e., Peptostreptococcaceae) in CtM animals. Rather, a high rate of protein breakdown may contribute to increased levels of circulating amino acids, including BCAAs. Testosterone, as a metabolic hormone, affects whole‐body anabolism by negatively regulating the hepatic urea cycle (Kelly & Jones, 2013; Birzniece, 2018). The opposite is also true, as testosterone deficiency may lead to increased hepatic ureagenesis, resulting in whole‐body nitrogen loss. Indeed, several lines of evidence (i.e., increased serum ammonia levels and intestinal ammonia and urease activity) support the notion of increased hepatic ureagenesis in CtM animals (Appendix Fig S12). Mammals cannot hydrolyze urea but urease‐producing bacteria cleave urea into ammonia in the gut (Walser & Bodenlos, 1959). From the perspective of the hologenome theory (namely, that the host and all of its associated microbiota form a unit of selection during the evolutionary change; Zilber‐Rosenberg & Rosenberg, 2008), the ileal microbial alteration and enhanced microbial urease activity can both conceivably be used to cope with nitrogen loss. Wiebler et al (2018) described the role of an enhanced gut microbial urea hydrolysis in the urea–nitrogen recycling in a hibernating frog. Metataxonomic analysis presented in the current study revealed the presence of abundant sequences assigned to the genera Romboutsia and Paeniclostridium in the ileal samples of the CtM Hanwoo (Fig 2E and Appendix Fig S6). Interestingly, recent genomic analyses of species belonging to the genera Romboutsia (e.g., R. ilealis CRIBT) and Paeniclostridium (e.g., P. sordellii ATCC 9714T) revealed the presence of a highly conserved urease gene cluster in the respective genomes (Couchman et al, 2015; Gerritsen et al, 2017); this suggests that these two bacterial taxa can utilize urea as a nitrogen source. The metataxonomic data reveal the gut microbial communities in cattle and mice at the genus level of taxonomic resolution. Given that different microbial species within a genus have different metabolic functions, isolation of key species from the gut of CtM animals to evaluate the effects of microbial signatures for host metabolism will be of interest. In addition, several sex steroid hormones regulate the secretion of digestive enzymes by the pancreas to the small intestine (Chen et al, 1995). In a rodent, an impaired pancreatic secretion (i.e., antimicrobial peptide secretion to the small intestine) affects the intestinal microbiota and, consequently, the fitness of the host, e.g., the weight and survival rate (Ahuja et al, 2017). The results of the current study, together with those of recently published studies, warrant further investigation to identify the mechanism linking hypogonadism and alteration of the intestinal microbiota.
Taken together, the observations presented herein indicate that primary hypogonadism as an outcome of castration leads to alterations in the ileal microbiota (i.e., dominance of the family Peptostreptococcaceae); changes in the systemic, muscular, and intestinal metabolome profiles (i.e., high abundance of BCAAs); and increased fat storage in the cattle and mice. We also uncovered bidirectional regulation of the circulating testosterone levels by the gut microbiota as revealed by the FMT experiment. These findings suggest that hypogonadism may integrate microbiota‐derived cues to modulate the host metabolism, suggesting that targeting the gut microbiota could constitute a potential therapeutic approach for the treatment of hypogonadism, as well as the associated metabolic disorders. Further studies regarding the effects of the spatiotemporal dynamics of the microbiome in other segments of the gastrointestinal tract, and the small intestine in particular, on host metabolic phenotypes should be performed. Some of the parameters measured during the mouse experiments [e.g., the insignificant effect of antibiotic treatment on castration‐induced adiposity (Fig 5F–H) and the high total fat mass in the absence of high serum BCAA levels in the FMT experiments (Appendix Fig S10G and H)] are not consistent with the findings of the cattle study. These discrepancies between the cattle and mouse models might be attributable to the small sample size and/or intrinsic physiological differences in the two animal species, for example, host genetic factors, diet, and/or their digestive systems.
Materials and Methods
Ethics approval
The study protocol was approved by the Institutional Animal Care and Use Committee of Kyung Hee University [KHUASP(SE)‐17‐026] for cattle study and [KHUASP(SE)‐18‐010] for mice study. The experiments were performed in agreement with the ARRIVE guidelines.
Cattle studies
To obtain the CtM animals, male cattle were orchiectomized by a veterinarian (both testicles were removed). Prior to sample collection, the cattle were examined for symptoms of diarrhea, and antibiotic or other medication treatment within 1 month of sampling. Animals with the above conditions were excluded for further analysis. Rectal material and the blood were sampled from 45 healthy animals: juvenile (male, CtM, and female; mean age: 11.9 ± 1.1 months; n = 5 of each) and post‐pubescent (male, CtM, and female; mean age: 19.3 ± 1.5 months; n = 5 of each) Hanwoo, and post‐pubescent (male, CtM, and female; mean age: 12.1 ± 1.6 months; n = 5 of each) Holsteins. Sample size was determined according to the resource equation method (Charan & Kantharia, 2013). The cattle are the property of a farm owner, and the number of researchers with access to the farm was limited. Consequently, no randomization and blinding to minimize the effects of subjective bias were applied. The rectal luminal content was collected by rectal enema using clean disposable latex gloves. The blood samples (5 ml) were collected from the jugular vein by a veterinarian and immediately centrifuged in BD Microtainer chemistry tubes (Becton Dickinson, Franklin Lakes, NJ, USA) for serum collection. The blood and striploin muscle samples, and the luminal content of the rumen, ileum, cecum, and colon of the adult male (mean age: 31.2 ± 5.9 months) and CtM Hanwoo (mean age: 33.9 ± 1.4 months, n = 10 in each group) were collected from a local slaughterhouse (Gunwi‐Gun, South Korea) under the supervision of the official veterinarian; the serum was collected from whole‐blood samples as described above. To assess fat mass, the posterior subcutaneous fat, mesenteric fat, and retroperitoneal fat were dissected and weighed during the slaughtering process. The collected samples were transported to the laboratory on dry ice and stored at −80°C until use.
Mouse studies
C57BL/6J mice were purchased from the Japan SLC Inc. (Hamamatsu, Japan) and supplied by Central Lab. Animal Inc. (Seoul, South Korea). All mice were housed in individually ventilated cages in a temperature‐controlled room with a 12‐h light/12‐h dark cycle, and given sterilized bedding and food and water ad libitum. At the end of the experiments, mice were fasted (6 h), anesthetized by isoflurane inhalation and euthanized by cardiac exsanguination. To sample the intestinal (luminal) contents, the target regions of the intestinal tract were flushed with sterile phosphate‐buffered saline (PBS). To measure the fat mass, we dissected and weighed posterior subcutaneous fat, mesenteric fat, retroperitoneal fat, and epididymal fat depots.
Castrated male mouse studies
To obtain the CtM animals, 4‐week‐old male mice were castrated by orchiectomy. For the Sham groups, age‐matched male mice underwent a Sham operation (i.e., the muscle layer and skin incision). After the surgery, both groups were given antibiotics (enrofloxacin) and an analgesic (Repellent). To reverse the effect of enrofloxacin on the gut microbiota, the Sham and CtM mice underwent FMT (the fecal samples were collected from the strain‐, age‐, and sex‐matched non‐operated donors, n = 10) once a day for 2 continuous days prior to the dietary intervention. Subsequently, the mice were provided either a LFD (10% fat; D12450H, Research Diets, New Brunswick, NJ, USA) or a HFD (45% fat; D12451, Research Diet). For the antibiotic‐treated groups, the HFD‐fed mice were given a combination of ampicillin (1 g/l), neomycin (1 g/l), metronidazole (1 g/l), and vancomycin (0.5 g/l) in drinking water for 5 weeks (n = 6 per group; Fig 5A). The antibiotic cocktail was renewed every 4 days.
Mouse FMT studies
Non‐castrated male mice (6 weeks old) were maintained on LFD for 10 weeks. Prior to FMT, the mice received an antibiotic cocktail in drinking water. After a 12‐h washout period, the mice were subjected to FMT (once a day for 3 continuous days) and fed HFD for 4 weeks. Fecal pellets from the Sham‐HFD and CtM‐HFD mouse donors (n = 6 in each group) were freshly collected and transferred immediately to an anaerobic chamber (Bactron II‐2, Sheldon Manufacturing) filled with an atmosphere of 5% H2, 5% CO2, and 90% N2. The pellets were resuspended in sterile and reduced PBS (100 mg feces/ml PBS), vortex‐mixed for 3 min, and sieved through a 70‐μm cell strainer. The fecal suspension (inoculum) was kept in Hungate anaerobic culture tubes and administered to the recipient mice (Sham‐R and CtM‐R, n = 5 in each group; Fig 6A). One mouse belonging to the Sham‐R group (Sham‐R #2) exhibited an abnormal anatomical finding (splenomegaly) at sacrifice, and therefore, it was excluded from further analysis.
BCAA‐enriched diet feeding studies
Non‐castrated male mice (7 weeks old) were acclimated on HFD for 1 week. The mice were fed HFD (0% BCAA) or a customized isocaloric BCAA‐HFD (3% and 5% BCAA) for 8 weeks (n = 6 in each group; Fig 7A). The amount of each BCAA in the BCAA‐enriched HFD is set out in Appendix Table S6.
DNA extraction and 16S rRNA gene sequencing
Bacterial genomic DNA was extracted from the intestinal luminal content of cattle and mice using a method involving the Repeated Bead‐Beating plus column (QIAamp DNA stool mini kit; Qiagen, Valencia, CA, USA; Yu & Morrison, 2004). In preparation for the GS‐FLX sequencing of the rectal samples from the juvenile and post‐pubescent cattle, a fragment of the 16S rRNA gene spanning the hypervariable V1–V2 regions was amplified by polymerase chain reaction (PCR) using bacterial universal primers 8F (5′‐X‐GAG TTT GAT CMT GGC TCA G‐3′) and 338R (5′‐TGC TGC CTC CCG TAG GAG T‐3′); 10 b sample‐specific barcodes (designated as X) were added to the 5′ primer terminus to tag the PCR products. In preparation for the Illumina MiSeq sequencing of the gastrointestinal samples from the adult Hanwoo and mice, a fragment of the 16S rRNA gene spanning the hypervariable V3–V4 regions was amplified by PCR using the forward primer 5′‐TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG‐3′ and the reverse primer 5′‐GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C‐3′. The PCR was performed in a C 1000 thermal cycler (Bio‐Rad, Hercules, CA, USA). The PCR conditions were as follows: initial denaturation at 96°C for 6 min; followed by repeated cycles of denaturation at 94°C for 1 min, annealing at 50°C for 30 s, and extension at 72°C for 90 s; and a final extension step at 72°C for 10 min. For the PCR of DNA extracted from the rectum, rumen, cecum, and colon contents, 18 repeated cycles were used; for the PCR of DNA extracted from the ileum content, 25 repeated cycles were used. Three PCRs with the same template were pooled, and the products were purified using the QIAquick PCR Purification Kit (Qiagen). Equimolar amounts of the purified amplicons were then combined in a single tube. For the GS‐FLX sequencing, the amplicons were pyrosequenced by Macrogen (Seoul, Republic of Korea) using the Genome Sequencer FLX Titanium (Roche, Branford, CT, USA). For the Illumina MiSeq sequencing, a 16S V3–4 PCR product library was prepared using the Nextera XT Index (Illumina). The library was then sequenced using an Illumina MiSeq platform and the paired‐end 2 × 300 bp reagent kit, according to the manufacturer’s instructions.
Evaluation of contaminating DNA
Possible DNA contamination of all reagents used for DNA extraction and sequencing was investigated. PCR analysis for the presence of contaminating bacterial 16S rRNA genes (in a 30 cycle reaction) revealed no apparent contamination of any reagent used.
Sequence analysis
To generate the GS‐FLX sequencing datasets, the raw sequences were preprocessed using the QIIME software package 1.9.0 (Caporaso et al, 2010) to exclude poor‐quality sequences and/or sequencing errors. Briefly, raw sequences containing more than one ambiguous base call, with errors in the barcode or primer regions, with average quality scores < 25, or shorter than 200 bp were removed. To generate the Illumina MiSeq sequencing datasets, paired‐end reads (forward and reverse fastq files) were joined and quality‐filtered using the QIIME software. Chimeric sequences were excluded from the quality‐filtered sequence sets using USEARCH software (version 7.0.1090). Unless noted otherwise in the text, default parameters were used. UCLUST software (version 1.2.21) clustered OTUs (at 97% sequence similarity) using the open reference OTU picking method. Singleton OTUs were excluded from further analyses. A representative sequence was selected for each OTU and aligned with reference sequences deposited in the Greengenes database using PyNAST (version 1.2.2). A phylogenetic tree of the aligned sequences was then constructed using Fasttree. Overviews of the 16S rRNA gene sequence datasets are provided in Appendix tables, as follows: the GS‐FLX‐generated datasets: the juvenile Hanwoo (15 samples) in Appendix Table S3; post‐pubescent Hanwoo (15 samples) in Appendix Table S4; and post‐pubescent Holstein (15 samples) in Appendix Table S5. The Illumina MiSeq‐generated datasets: the adult Hanwoo (60 samples) in Appendix Table S7; castrated or Sham‐operated male mice (72 samples) in Appendix Table S8; FMT recipient mice (22 samples) in Appendix Table S9; and BCAA‐HFD‐fed mice (36 samples) in Appendix Table S10.
For phylogenetic analysis of abundant OTUs assigned to the family Peptostreptococcaceae, representative sequences were aligned with those of closely related species using the multiple sequence alignment program ClustalW (Bioedit 7.2.5; Thompson et al, 1997). A phylogenetic consensus tree was reconstructed using MEGA version 7 (Kumar et al, 2016) and the neighbor‐joining algorithm (Saitou & Nei, 1987) based on 1,000 randomly chosen bootstrap replications.
Metabolite extraction
Both extracellular and intracellular extracts of intestinal luminal contents, serum, and striploin muscle were prepared for metabolite profiling as follows: The luminal contents (250–500 mg) of the ileum, cecum, and colon were extracted into 1 ml water using the MM400 mixer mill (Retsch®, Haan, Germany) at a frequency of 30 s−1 for 5 min. After sonication (10 min) and centrifugation (12,578 g, 4°C, 10 min), the supernatant was filtered through a 0.2‐μm polytetrafluoroethylene (PTFE) filter and concentrated using a speed‐vacuum concentrator. For the serum extractions, 1 ml methanol was added to 200 μl of the serum, and then extracted by sonication and shaking for 10 min. After centrifugation (12,578 g, 4°C, 10 min), the supernatant was filtered through a 0.2‐μm PTFE filter and dried using a speed‐vacuum concentrator. The muscle samples (5 g) were finely chopped before extraction, following which 10 ml methanol was added; the samples were then sonicated (10 min) and agitated (1 h). After centrifugation (12,578 g, 4°C, 10 min), the supernatant was filtered through a 0.2‐μm PTFE filter and dried using a speed‐vacuum concentrator.
Metabolite profiling and data processing
The dried samples were oximated in 50 μl methoxyamine hydrochloride (20 mg/ml in pyridine) for 90 min at 30°C, silylated in 50 μl N‐methyl‐N‐(trimethylsilyl) trifluoroacetamide for 30 min at 37°C, and analyzed by GC‐TOF‐MS, as previously described (Jung et al, 2015). The analysis was performed using an Agilent 7890 gas chromatography system (Agilent Technologies, Palo Alto, CA, USA), an Agilent 7693 auto‐sampler (Agilent Technologies), and a Pegasus HT TOF MS (LECO, St. Joseph, MI, USA) system.
The GC‐TOF‐MS data were acquired and preprocessed using the LECO Chroma TOF™ software (version 4.44; LECO Corp., St. Joseph, MI, USA) and converted to the NetCDF format (*.cdf) using the LECO Chroma TOF™ software. After conversion, peak detection, retention time correction, and alignment were assessed using MetAlign 3.0 (http://www.metalign.nl). Data were exported to an Excel file. Multivariate statistical analyses were conducted using the SIMCA‐P+ program (version 12.0, Umetrics, Umea, Sweden). The datasets were auto‐scaled and mean‐centered in a column‐wise fashion. The discriminant variables were selected based on the variable importance in projection (VIP) value (VIP > 0.7).
Enzymatic assays for BCAA and ketone body quantifications
Branched‐chain amino acid levels were determined in the serum samples from cattle and mice, and from the intestinal luminal contents from cattle. The BCAA concentrations were quantified using the BCAA assay kit (Sigma, St. Louis, MO, USA). A standard curve was constructed for each experiment using a leucine standards series (MAK003D, Sigma). The BCAA levels were then calculated based on the standard curves. The levels of ketone bodies were determined in serum and muscle samples (the striploin and hindlimb muscles of cattle and mice, respectively) from cattle and mice. The 3‐HB and acetoacetate concentrations were quantified using a β‐hydroxybutyrate assay kit (MAK041, Sigma) and an acetoacetate colorimetric assay kit (MAK199, Sigma), respectively. The 3‐HB and acetoacetate levels in each sample were calculated using standard curves.
Hormone measurements
Serum samples from cattle and mice were prepared as described above. The concentrations of serum testosterone and 17β‐estradiol were determined by enzyme‐linked immunosorbent assay (ELISA) using a testosterone ELISA kit (Enzo Life Sciences, Farmingdale, NY, USA) and a 17β‐estradiol high sensitivity ELISA kit (Enzo Life Sciences), respectively. Concentration of the serum insulin was determined by using a mouse ultrasensitive insulin ELISA kit (ALPCO, Salem, NH, USA).
Analysis of gastrointestinal urease activity
Urease activity was assayed in the ruminal and ileal luminal contents of the adult Hanwoo. The luminal contents were suspended in 10 volumes of PBS. The samples were homogenized in an assay buffer using a bead‐beating method. After centrifugation, urease activity in the clear lysate solutions was determined by using a urease activity assay kit (MAK120, Sigma). A standard curve was constructed for each experiment using a series of ammonium chloride standards (MAK120C, Sigma). The amount of urease enzymatic activity in the samples was calculated according to the manufacturer’s instructions.
Ammonia and urea measurements
The ammonia levels in the rumen and ileum of the adult Hanwoo were determined using the urease activity assay kit as described above. Ammonia levels in the mouse serum were determined using an ammonia assay kit (AA0100, Sigma), according to the manufacturer’s instructions. A standard curve was constructed for each experiment using a series of ammonium sulfate standards (A0978, Sigma). To determine urea levels in the mouse serum, a urea assay kit (MAK006) was used. A standard curve for each experiment was constructed using a series of urea standards (MAK006F, Sigma).
Glucose tolerance test
Glucose tolerance test was performed using overnight (16 h)‐fasted mice by an intraperitoneal injection of glucose (1 g/kg body weight). Blood glucose concentrations were measured with an Accu‐Chek glucometer (Roche) before (0 min), and 15, 30, 60, and 120 min after glucose injection.
Intramuscular fat image analysis
Striploin muscle images were size‐normalized. The sizes of intramuscular fat areas were calculated using the i‐Solution image analyzer (IMT i‐Solution Inc., Vancouver, BC, Canada).
Statistics
The statistical analyses were performed using GraphPad Prism version 5.0 for Windows (GraphPad Software, La Jolla, CA, USA). Statistical analysis was initially performed using the Shapiro–Wilk normality test. Comparisons between two samples were made using the non‐parametric Mann–Whitney U‐test for the cattle dataset and the unpaired Student’s t‐test (one‐tailed) for the mouse dataset. Comparisons between multiple samples were conducted by the analysis of variance (ANOVA), followed by Tukey’s post hoc test (*P < 0.05, **P < 0.01, and ***P < 0.001). The differences in the bacterial community composition between groups were assessed by pairwise distance using presence/absence (e.g., unweighted UniFrac analysis) or quantitative species abundance data (e.g., weighted UniFrac and Bray–Curtis analyses). The statistical significance for the observed variations was assessed using the PERMANOVA function with 999 permutations. The lines, boxes, and whiskers in the box plot diagrams represent the median, first, and third quartiles, and min‐to‐max distribution of replicate values, respectively. The values and scattered dots in the bar graphs represent the means ± SEM and the individual replicates, respectively.
Author contributions
J‐WB and TWW designed the experiments. TWW, HSK, and N‐RS performed the majority of the experiments and analyzed the data. EJT, HS, M‐JJ, Y‐SJ, D‐WH, and PSK helped with sample collection and data presentation. ESJ, YKJ, and CHL performed the metabolomic experiments and analyzed the data. TWW, HSK, N‐RS, and J‐WB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank the farm owners, veterinarians, and slaughterhouse staff for helping with sample collection. We especially thank Hong Gil Kim (President of the National Hanwoo Association) and Sang Uk Han (CJ CheilJedang Corp.) for advising on the sampling strategy and helpful discussions. This study was supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, and Forestry (IPET) through the Agricultural Microbiome R&D Program, funded by the Ministry of Agriculture, Food, and Rural Affairs (MAFRA) (918011‐04‐1‐SB010); the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (grant number: NRF‐2018R1A5A1025077); the Mid‐Career Researcher Program (NRF‐2020R1A2C3012797); and the Bio & Medical Technology Development Program (NRF‐2017M3A9F3046549).
EMBO Reports (2021) 22: e50663.
Data availability
The raw sequences of 16S rRNA genes obtained from the intestinal luminal content of cattle and mice have been deposited in the European Nucleotide Archive and are available under the accession number PRJEB19502.
References
- Abdoun K, Stumpff F, Martens H (2006) Ammonia and urea transport across the rumen epithelium: a review. Anim Health Res Rev 7: 43–59 [DOI] [PubMed] [Google Scholar]
- Ahuja M, Schwartz DM, Tandon M, Son A, Zeng M, Swaim W, Eckhaus M, Hoffman V, Cui Y, Xiao B et al (2017) Orai1‐mediated antimicrobial secretion from pancreatic acini shapes the gut microbiome and regulates gut innate immunity. Cell Metab 25: 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt RA, Everard DM, Gillies G, Wilkinson M, Wilson CA, Yeo TA (1982) Investigation into the hypogonadism of the obese mouse (genotype ob/ob). J Reprod Fertil 64: 363–371 [DOI] [PubMed] [Google Scholar]
- Birzniece V (2018) Hepatic actions of androgens in the regulation of metabolism. Curr Opin Endocrinol Diabetes Obes 25: 201–208 [DOI] [PubMed] [Google Scholar]
- Bojesen A, Kristensen K, Birkebaek NH, Fedder J, Mosekilde L, Bennett P, Laurberg P, Frystyk J, Flyvbjerg A, Christiansen JS et al (2006) The metabolic syndrome is frequent in Klinefelter's syndrome and is associated with abdominal obesity and hypogonadism. Diabetes Care 29: 1591–1598 [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI et al (2010) QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7: 335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrageta DF, Oliveira PF, Alves MG, Monteiro MP (2019) Obesity and male hypogonadism: tales of a vicious cycle. Obes Rev 20: 1148–1158 [DOI] [PubMed] [Google Scholar]
- Charan J, Kantharia ND (2013) How to calculate sample size in animal studies? J Pharmacol Pharmacother 4: 303–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TS, Doong ML, Chang FY, Lee SD, Wang PS (1995) Effects of sex steroid hormones on gastric emptying and gastrointestinal transit in rats. Am J Physiol 268: G171–G176 [DOI] [PubMed] [Google Scholar]
- Couchman EC, Browne HP, Dunn M, Lawley TD, Songer JG, Hall V, Petrovska L, Vidor C, Awad M, Lyras D et al (2015) Clostridium sordellii genome analysis reveals plasmid localized toxin genes encoded within pathogenicity loci. BMC Genom 16: 392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai ZL, Wu ZL, Hang SQ, Zhu WY, Wu GY (2015) Amino acid metabolism in intestinal bacteria and its potential implications for mammalian reproduction. Mol Hum Reprod 21: 389–409 [DOI] [PubMed] [Google Scholar]
- Dhindsa S, Ghanim H, Batra M, Dandona P (2018) Hypogonadotropic hypogonadism in men with diabesity. Diabetes Care 41: 1516–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D (2001) The development of androgen‐independent prostate cancer. Nat Rev Cancer 1: 34–45 [DOI] [PubMed] [Google Scholar]
- Field RA (1971) Effect of castration on meat quality and quantity. J Anim Sci 32: 849–858 [DOI] [PubMed] [Google Scholar]
- Flak MB, Neves JF, Blumberg RS (2013) Immunology. Welcome to the microgenderome. Science 339: 1044–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen J, Hornung B, Renckens B, van Hijum S, Martins Dos Santos VAP, Rijkers GT, Schaap PJ, de Vos WM, Smidt H (2017) Genomic and functional analysis of Romboutsia ilealis CRIB(T) reveals adaptation to the small intestine. PeerJ 5: e3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, Metallo CM (2016) Branched‐chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol 12: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Hanaoka R, Horiuchi H, Kitakaze T, Mitani T, Inui H, Yamaji R (2016) Castration influences intestinal microflora and induces abdominal obesity in high‐fat diet‐fed mice. Sci Rep 6: 23001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF Jr (2001) Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle‐stimulating hormone secretion. J Clin Endocrinol Metab 86: 53–58 [DOI] [PubMed] [Google Scholar]
- Hayes FJ, Seminara SB, Decruz S, Boepple PA, Crowley WF Jr (2000) Aromatase inhibition in the human male reveals a hypothalamic site of estrogen feedback. J Clin Endocrinol Metab 85: 3027–3035 [DOI] [PubMed] [Google Scholar]
- Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A et al (2016) A branched‐chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 22: 421–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Bong J, Kim GD, Joo ST, Lee HJ, Baik M (2013) Transcriptome changes favoring intramuscular fat deposition in the longissimus muscle following castration of bulls. J Anim Sci 91: 4692–4704 [DOI] [PubMed] [Google Scholar]
- Jung ES, Park HM, Lee KE, Shin JH, Mun S, Kim JK, Lee SJ, Liu KH, Hwang JK, Lee CH (2015) A metabolomics approach shows that catechin‐enriched green tea attenuates ultraviolet B‐induced skin metabolite alterations in mice. Metabolomics 11: 861–871 [Google Scholar]
- Kelly DM, Jones TH (2013) Testosterone: a metabolic hormone in health and disease. J Endocrinol 217: R25–45 [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33: 1870–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T, Poljak A, McLean M, Bahl N, Ho KK, Birzniece V (2017) Testosterone prevents protein loss via the hepatic urea cycle in human. Eur J Endocrinol 176: 489–496 [DOI] [PubMed] [Google Scholar]
- Liu J, Lkhagva E, Chung HJ, Kim HJ, Hong ST (2018) The pharmabiotic approach to treat hyperammonemia. Nutrients 10: 140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CJ, Adams SH (2014) Branched‐chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol 10: 723–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markle JG, Frank DN, Mortin‐Toth S, Robertson CE, Feazel LM, Rolle‐Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS (2013) Sex differences in the gut microbiome drive hormone‐dependent regulation of autoimmunity. Science 339: 1084–1088 [DOI] [PubMed] [Google Scholar]
- Marti S, Jackson JA, Slootmans N, Lopez E, Hodge A, Perez‐Juan M, Devant M, Amatayakul‐Chantler S (2017) Effects on performance and meat quality of Holstein bulls fed high concentrate diets without implants following immunological castration. Meat Sci 126: 36–42 [DOI] [PubMed] [Google Scholar]
- Neis EP, Dejong CH, Rensen SS (2015) The role of microbial amino acid metabolism in host metabolism. Nutrients 7: 2930–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra AK, Aschenbach JR (2018) Ureases in the gastrointestinal tracts of ruminant and monogastric animals and their implication in urea‐N/ammonia metabolism: a review. J Adv Res 13: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, Forslund K, Hildebrand F, Prifti E, Falony G et al (2016) Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535: 376–381 [DOI] [PubMed] [Google Scholar]
- Plouzek CA, Trenkle A (1991) Insulin‐like growth factor‐I concentrations in plasma of intact and castrated male and female cattle at four ages. Domest Anim Endocrinol 8: 73–79 [DOI] [PubMed] [Google Scholar]
- Rana K, Fam BC, Clarke MV, Pang TP, Zajac JD, MacLean HE (2011) Increased adiposity in DNA binding‐dependent androgen receptor knockout male mice associated with decreased voluntary activity and not insulin resistance. Am J Physiol Endocrinol Metab 301: E767–E778 [DOI] [PubMed] [Google Scholar]
- Saboor Aftab SA, Kumar S, Barber TM (2013) The role of obesity and type 2 diabetes mellitus in the development of male obesity‐associated secondary hypogonadism. Clin Endocrinol (Oxf) 78: 330–337 [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M (1987) The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425 [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome biol 12: R60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, Bae JW (2014) An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet‐induced obese mice. Gut 63: 727–735 [DOI] [PubMed] [Google Scholar]
- Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez‐Cadavid NF, Bhasin S (2006) Testosterone inhibits adipogenic differentiation in 3T3‐L1 cells: nuclear translocation of androgen receptor complex with beta‐catenin and T‐cell factor 4 may bypass canonical Wnt signaling to down‐regulate adipogenic transcription factors. Endocrinology 147: 141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon‐Biet SM, Cogger VC, Pulpitel T, Wahl D, Clark X, Bagley E, Gregoriou GC, Senior AM, Wang QP, Brandon AE et al (2019) Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat Metab 1: 532–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan N, Haring HU, Hu FB, Schulze MB (2013) Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol 1: 152–162 [DOI] [PubMed] [Google Scholar]
- Swerdloff RS, Batt RA, Bray GA (1976) Reproductive hormonal function in the genetically obese (ob/ob) mouse. Endocrinology 98: 1359–1364 [DOI] [PubMed] [Google Scholar]
- Tajar A, Forti G, O'Neill TW, Lee DM, Silman AJ, Finn JD, Bartfai G, Boonen S, Casanueva FF, Giwercman A et al (2010) Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab 95: 1810–1818 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, Chan AWH, Chan FKL, Sung JJY, Yu J (2017) Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology 152: 1419–1433.e1415 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031 [DOI] [PubMed] [Google Scholar]
- Twiddy AL, Leon CG, Wasan KM (2011) Cholesterol as a potential target for castration‐resistant prostate cancer. Pharm Res‐Dordr 28: 423–437 [DOI] [PubMed] [Google Scholar]
- Walser M, Bodenlos LJ (1959) Urea metabolism in man. J Clin Invest 38: 1617–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebler JM, Kohl KD, Lee RE Jr, Costanzo JP (2018) Urea hydrolysis by gut bacteria in a hibernating frog: evidence for urea‐nitrogen recycling in Amphibia. Proc Biol Sci 285: 20180241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Morrison M (2004) Improved extraction of PCR‐quality community DNA from digesta and fecal samples. Biotechniques 36: 808–812 [DOI] [PubMed] [Google Scholar]
- Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, Antonopoulos D, Umesaki Y, Chervonsky AV (2013) Gender bias in autoimmunity is influenced by microbiota. Immunity 39: 400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Wu L, Xu C, Xia C, Sun LW, Shu S (2013) Plasma metabolomic profiling of dairy cows affected with ketosis using gas chromatography/mass spectrometry. BMC Vet Res 9: 186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilber‐Rosenberg I, Rosenberg E (2008) Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol Rev 32: 723–735 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Data Availability Statement
The raw sequences of 16S rRNA genes obtained from the intestinal luminal content of cattle and mice have been deposited in the European Nucleotide Archive and are available under the accession number PRJEB19502.