

Abstract
DNA damage tolerance relies on homologous recombination (HR) and translesion synthesis (TLS) mechanisms to fill in the ssDNA gaps generated during passing of the replication fork over DNA lesions in the template. Whereas TLS requires specialized polymerases able to incorporate a dNTP opposite the lesion and is error‐prone, HR uses the sister chromatid and is mostly error‐free. We report that the HR protein Rad52—but not Rad51 and Rad57—acts in concert with the TLS machinery (Rad6/Rad18‐mediated PCNA ubiquitylation and polymerases Rev1/Pol ζ) to repair MMS and UV light‐induced ssDNA gaps through a non‐recombinogenic mechanism, as inferred from the different phenotypes displayed in the absence of Rad52 and Rad54 (essential for MMS‐ and UV‐induced HR); accordingly, Rad52 is required for efficient DNA damage‐induced mutagenesis. In addition, Rad52, Rad51, and Rad57, but not Rad54, facilitate Rad6/Rad18 binding to chromatin and subsequent DNA damage‐induced PCNA ubiquitylation. Therefore, Rad52 facilitates the tolerance process not only by HR but also by TLS through Rad51/Rad57‐dependent and ‐independent processes, providing a novel role for the recombination proteins in maintaining genome integrity.
Keywords: DNA damage tolerance, homologous recombination, Rad52, template switching, translesion synthesis
Subject Categories: DNA Replication, Repair & Recombination
The recombination protein Rad52 promotes translesion synthesis and mutagenesis by non‐recombinogenic functions, which include the recruitment of the PCNA ubiquitylation complex Rad6/Rad18 to chromatin.

Introduction
Apart from a number of specialized DNA repair mechanisms aimed to deal with the different types of DNA damage, cells are endowed with mechanisms to tolerate DNA lesions that hinder the advance of replication forks, thus ensuring timely chromosome replication (Broomfield et al, 2001; Friedberg, 2005). The mechanisms of DNA damage tolerance (DDT) help replication forks to pass through the lesions and fill in the stretches of single‐stranded DNA (ssDNA) generated during the blocking of the fork. The DDT response involves two different evolutionarily conserved strategies to fill in the ssDNA gaps: translesion synthesis (TLS) and homologous recombination (HR) (Sale, 2013; Prado, 2014, 2018; Branzei & Psakhye, 2016). TLS mechanisms use specialized polymerases that are able to incorporate a nucleotide opposite the lesion (Fig 1A), yet this process can be mutagenic as these polymerases display low fidelity. In Saccharomyces cerevisiae, there are three TLS polymerases: Rev1, Pol ζ (formed by the catalytic subunit Rev3 and the regulatory subunits Rev7, Pol31, and Pol32), and Pol η (encoded by RAD30) (Sale, 2013; Zhao & Washington, 2017). Their contribution to DDT is influenced by the dose and type of blocking lesions, and their activity (at least for Rev1/ Pol ζ) is most dominant in G2/M (Prakash, 1981; Paulovich et al, 1997; Baynton et al, 1998; Torres‐Ramos et al, 2002; Lopes et al, 2006; Waters & Walker, 2006; Daigaku et al, 2010).
Figure 1. Persistence of MMS‐induced Rad52 foci in TS‐deficient cells.

-
AScheme for the two major mechanisms of DDT: HR and TLS. An asterisk represents a blocking DNA lesion for the replicative polymerase.
-
BRepresentative image of wild‐type cells with MMS‐induced Rad52‐YFP foci (bright field and fluorescence signal)
-
C–GssDNA repair efficiency of the indicated TS mutants as determined from the maximal and final percentages of cells with Rad52 foci during the time course (right panels). Cells were synchronized in G1 and released into S phase in the presence of 0.033% MMS for 1 h (60+), treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. They were also released into medium without MMS for 1 h to control the formation of spontaneous Rad52‐YFP foci (60‐). The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (left panels). Cell cycle progression was determined by cell sorting. The mean and SEM of 3 (wild type, rad18∆, mms2∆, rad51∆, rad57∆, rad54∆), 4 (pol30‐K164R), and 5 (rad5∆) independent experiments are shown. Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where three asterisks represent P‐values <0.001.
Source data are available online for this figure.
In marked contrast to TLS, HR mechanisms use the intact sister chromatid to fill in the ssDNA gaps and are considered error‐free (Fig 1A). These mechanisms have been extensively studied in yeast, where they require different components including Rad51, Rad52, Rad55/Rad57, and Rad54 (Prado, 2018). The recombinase Rad51 provides strand annealing and exchange activities. The mediator Rad52 competes with the ssDNA‐binding replication protein A (RPA complex; formed by Rfa1, Rfa2, and Rfa3) to load Rad51 and form the ssDNA/Rad51 nucleofilament that initiates the search for homology. The helper complex Rad55/Rad57 is involved in the stabilization of the ssDNA/Rad51 nucleofilament. Finally, the translocase Rad54 is involved in the processing of early and late recombination intermediates.
A key component in the control of DDT is the replication processivity factor PCNA (Hoege et al, 2002). In response to replicative DNA damage, but not double‐strand breaks (DSBs), the Rad6/Rad18 complex monoubiquitylates PCNA at lysine 164 (Hoege et al, 2002; Davies et al, 2008), a modification that promotes the recruitment of the TLS polymerases (Bienko et al, 2005). This ubiquitin residue is further extended with a K63‐linked polyubiquitin chain by the activity of the Mms2/Ubc13/Rad5 complex (Hoege et al, 2002; Lopes et al, 2006; Daigaku et al, 2010). The HR proteins act in concert with polyubiquitylated PCNA to form sister chromatid junctions (SCJs) as an intermediate to fill in the gaps of ssDNA (Liberi et al, 2005; Mankouri et al, 2007; Branzei et al, 2008; Minca & Kowalski, 2010; Vanoli et al, 2010; Karras et al, 2013; Giannattasio et al, 2014). HR can also deal with ssDNA gaps through an UbPCNA‐independent mechanism. This UbPCNA‐independent HR mechanism is also associated with SCJs (Branzei et al, 2008) and seems to deal with ssDNA fragments that remain unrepaired after S phase; accordingly, it is inhibited during S phase by Ubc9/Siz1‐dependent PCNA sumoylation at lysines 164 and 127 and further recruitment of the antirecombinogenic helicase Srs2 (Schiestl et al, 1990; Pfander et al, 2005; Papouli et al, 2005). In contrast to UbPCNA/HR, the UbPCNA‐independent HR pathway can lead to chromosomal rearrangements (Motegi et al, 2006). The UbPCNA‐dependent and UbPCNA‐independent HR pathways are also termed template switching (TS) and salvage pathway, respectively (Karras et al, 2013; Prado, 2014, 2018; Branzei & Psakhye, 2016).
Epistatic analyses between HR and TLS mutants on DNA damage sensitivity have genetically separated the role of HR proteins from TLS (Rattray et al, 2002; Ball et al, 2009). In fact, the frequency of UV light‐induced mutagenesis in cells defective in nucleotide‐excision repair (NER)—essential for the removal of UV‐induced photoproducts—is increased in a rad52∆ mutant. This has been interpreted as a consequence of channeling the filling of most ssDNA gaps to TLS (Paulovich et al, 1998). However, the interplay between HR proteins and TLS is more complex. Thus, the absence of Rad52 in NER proficient cells decreases the frequency and alters the type of UV‐ and ethyl methanesulfonate (EMS)‐induced mutations (Prakash & Higgins, 1982; Kunz et al, 1992; Armstrong et al, 1994), which might reflect a role for Rad52 in TLS (Armstrong et al, 1994). However, these phenotypes might also be due to the loss of recombinational repair, a process that can increase the rate and type of mutations (Hicks et al, 2010). An additional level of complexity in the interplay between HR and TLS proteins comes from their role in spontaneous and DSB‐induced events. However, a distinctive feature with the DDT mechanisms is that the repair of spontaneous and DSB lesions is not associated with PCNA ubiquitylation (Hoege et al, 2002; Stelter & Ulrich, 2003; Hirano & Sugimoto, 2006; Chen et al, 2006; Sharma et al, 2012).
Here, we show that Rad52—but not Rad51 or Rad57—acts in concert with the TLS machinery to repair methyl methanesulfonate (MMS)‐ and UV‐induced ssDNA gaps in a non‐recombinogenic manner, as inferred from the different phenotypes displayed by rad52∆ and rad54∆ mutants. Accordingly, Rad52 is required for efficient DNA damage‐induced mutagenesis. In addition, Rad52 cooperates with Rad51 and Rad57—but not Rad54—to promote efficient Rad6/Rad18 binding to chromatin and in response to replicative DNA lesions, PCNA ubiquitylation. Therefore, Rad52 can facilitate both HR and TLS at DNA repair centers, suggesting a regulatory function for this protein in the decision of tolerating DNA damage through mutagenic or recombinogenic mechanisms.
Results
Persistence of MMS‐induced Rad52 foci in TS‐deficient cells
DNA damage induces the formation of DNA repair centers that can be visualized as nuclear foci using recombination proteins fused to a fluorescent protein (Fig 1B; Lisby et al, 2001). In the case of MMS, these foci mark post‐replicative Rad52‐mediated gap filling events (González‐Prieto et al, 2013; Wong et al, 2020). We decided to follow the disappearance of Rad52 centers formed in response to MMS to explore the genetic contribution of the different DDT pathways to the post‐replicative repair of ssDNA gaps. For this, cells expressing Rad52‐YFP, which is functional in response to MMS (González‐Prieto et al, 2013), were synchronized in G1 and released into fresh medium in the presence of 0.033% MMS for 1 h, treated with sodium thiosulfate to inactivate the MMS, washed, and incubated in fresh medium for different times. All time courses included a wild type to control the experimental variation in the G1 release and cell cycle progression between different sets of experiments. The maximal fraction of wild‐type cells with Rad52 foci was ~40–60% and was reached 1–2 h after MMS inactivation, coinciding with completion of DNA replication. This percentage of wild‐type cells with Rad52‐YFP foci dropped to ~10% 5–6 h later (Fig 1, left panels; note that plotted values have been normalized to the highest value of the wild type, taken as 100). The efficiency of ssDNA repair, determined from the maximal and final percentages of cells with Rad52 foci during the time course, was ~80% (Fig 1, right panels; see Material and Methods for calculation). To address the relevance of DDT‐induced PCNA modifications on Rad52‐associated gap filling, we analyzed the effect of pol30‐K164R, which expresses a PCNA complex mutated in its major site of ubiquitylation and sumoylation (Hoege et al, 2002). The efficiency of repair in pol30‐K164R cells was reduced to ~20% (Fig 1C), suggesting that the modification of PCNA at lysine 164 is important for the resolution of Rad52 foci. Since PCNA polyubiquitylation at lysine 164 cooperates with recombination proteins in the process of TS (Prado, 2014, 2018; Branzei & Psakhye, 2016), we followed the accumulation of Rad52 foci in the absence of PCNA ubiquitylation (Rad18, Mms2, and Rad5) and HR (Rad51, Rad57, and Rad54) factors. The percentage of cells with Rad52 foci 1–2 h after MMS release was similar in these mutants to that displayed by the wild type, except for the rad57∆ and rad54∆ mutants that exhibited an unexpected lower percentage (Fig 1D–G). Importantly, in all cases the efficiency of ssDNA repair dropped to less than 20% (Fig 1D–G). These results suggest that the TS machinery is required for efficient post‐replicative ssDNA gap repair at Rad52 foci.
Persistence of MMS‐induced Rad52 foci in TLS‐deficient cells
TS operates mostly in S phase (Karras & Jentsch, 2010; Daigaku et al, 2010; Ortiz‐Bazán et al, 2014). Thus, PCNA polyubiquitylation should be dispensable for HR after completion of DNA replication. To address this point, the RAD18 promoter was replaced by the GAL1 promoter (Gp::RAD18), which is expressed in the presence of galactose and repressed in the presence of glucose. Accordingly, the kinetics of Rad52 foci accumulation in Gp::RAD18 cells grown in galactose was similar to that displayed by wild‐type cells (Fig EV1A), indicating that Rad18 expression from the GAL1 promoter sustained DNA repair. Interestingly, if Rad18 expression was repressed after completion of DNA replication (1 h after MMS inactivation) by shifting the cells from galactose to glucose‐containing medium, a significant percentage of Rad52 foci persisted at the end of the time course compared to the wild type (Fig 2A). This suggests that PCNA ubiquitylation is required for efficient ssDNA gap repair at Rad52 centers after completion of S phase. To confirm that this requirement was for PCNA monoubiquitylation and not polyubiquitylation, we repeated the analysis using a strain in which the MMS2 gene was under control of the GAL1 promoter (Gp::MMS2). In this case, cells expressing MMS2 from the GAL1 promoter in galactose repaired the MMS‐induced ssDNA gaps with lower efficiency (~50%; Fig 2B, condition a) than wild‐type cells (~80%; Fig EV1B). Repression of Mms2 expression for only 1 h before release into S phase was sufficient to cause a complete defect in Rad52‐associated ssDNA repair (Fig 2B; compare conditions a and b). In contrast, repression of Mms2 expression after completion of DNA replication (2 h after MMS inactivation) did not compromise repair efficiency as compared with cells continuously expressing Mms2 (Fig 2B; compare conditions a and c), indicating that PCNA polyubiquitylation is unlikely to be important in G2/M. These results are consistent with a specific role for PCNA monoubiquitylation in the resolution of Rad52 foci.
Figure EV1. Time course analyses of Rad52‐YFP foci in DDT mutants.

-
AssDNA repair efficiency of cells expressing RAD18 from the GAL1 promoter as determined from the maximal and final percentages of cells with Rad52 foci during the time course (right panel). Cells were synchronized in G1 from galactose‐containing medium and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. Cells were grown in galactose‐containing medium during the whole time course. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (left panel). See Fig 2A for more details. The mean and SEM of four independent experiments are shown.
-
BssDNA repair efficiency of mms2∆ (right) and wild‐type (left) cells as determined from the maximal and final percentages of cells with Rad52 foci during the time course (right panels). Cells were synchronized in G1 from galactose‐containing medium and shifted to glucose (a) or maintained in galactose (b, c) for 1 h in G1; then, cells were released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. Cells were maintained either in galactose (a), glucose (b), or shifted to glucose 2 h after MMS inactivation (c). The percentage of cells with foci at each point was normalized to the highest value in galactose, taken as 100 (left panels). See Fig 2B for more details. The mean and SEM of three independent experiments are shown.
Data information: Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where three asterisks represents a P‐value < 0.001.
Figure 2. Persistence of MMS‐induced Rad52 foci in TLS‐deficient cells.

-
AssDNA repair efficiency of cells expressing RAD18 from the GAL1 promoter as determined from the maximal and final percentages of cells with Rad52 foci during the time course (right panel). Cells were synchronized in G1 from galactose‐containing medium and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. Cells were shifted to glucose‐containing medium 1 h after MMS inactivation. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (left panel). See Fig EV1A for control cells released in galactose‐containing medium during the entire time course.
-
BssDNA repair efficiency of cells expressing MMS2 from the GAL1 promoter as determined from the maximal and final percentages of cells with Rad52 foci during the time course (right panel). Cells were synchronized in G1 from galactose‐containing medium and maintained in galactose (a, c) or shifted to glucose (b) for 1 h in G1; subsequently, cells were released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. Cells were maintained either in galactose (a), glucose (b) or shifted to glucose 2 h after MMS inactivation (c). The percentage of cells with foci at each point was normalized to the highest value in galactose, taken as 100 (left panel). Wild‐type and mms2∆ controls were analyzed in parallel (Fig EV1B).
-
C–EssDNA repair efficiency of the indicated TLS mutants as determined from the maximal and final percentages of cells with Rad52 foci during the time course (D and E, right panel). Cells were synchronized in G1 and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (C and E, left panel).
Data information: The mean and SEM of 4 (A), 3 (B, E) and 3–12 (C, D; 12 (wild type), 6 (rev1∆, rev3∆, rad30∆, rev1∆ rad30∆, rev3∆ rad30∆), and 3 (rev1∆ rev3∆ and rev1∆ rev3∆ rad30∆) independent experiments are shown. Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where one, two, and three asterisks represent P‐values < 0.05, < 0.01, and < 0.001, respectively.
Source data are available online for this figure.
In addition to priming PCNA polyubiquitylation for TS, Rad18‐mediated PCNA monoubiquitylation is required for the recruitment of TLS polymerases to DNA lesions (Hoege et al, 2002; Stelter & Ulrich, 2003; Bienko et al, 2005). Moreover, Rev1‐dependent TLS occurs preferentially in G2/M (Waters & Walker, 2006). Thus, we examined if blocking TLS by eliminating the TLS polymerases also impairs ssDNA processing at Rad52 repair centers. Remarkably, the absence of any of the highly mutagenic polymerases, Rev1 or Pol ζ, in rev1∆ or rev3∆ cells reduced the efficiency of ssDNA repair at Rad52 foci (~35–40%), whereas the absence of Pol η in rad30∆ had a much weaker effect (Fig 2C and D). Interestingly, the lack of a regulatory subunit of Pol ζ in pol32∆ cells caused a more severe defect (~20%; Fig 2E). This might be related to the role of Pol32 as part of the replicative polymerase Pol δ in the formation of SCJ during TS (Vanoli et al, 2010).
The effects of rev1∆ and rev3∆ were not additive (Fig 2C and D), consistent with a cooperation between Rev1 and Pol ζ (Haracska et al, 2001). In fact, the double mutant rev1∆ rev3∆ increased ssDNA repair efficiency as compared to the single mutants, and this increase required the activity of the polymerase Pol η (Fig 2D; compare rev1∆ rev3∆ rad30∆ with rev1∆ rev3∆). These results indicate that Pol η can operate in the absence of the entire Rev1/Pol ζ complex but not of just either Rev1 or Pol ζ, suggesting that the presence of a non‐functional Rev1/Pol ζ complex hampers the entry and/or activity of Pol η at the ssDNA gap. In sum, these results suggest that Rad6/Rad18‐mediated PCNA monoubiquitylation, the activities of Rev1/Pol ζ, and to a lesser extent Pol η are required for efficient post‐replicative processing of the ssDNA gaps that occurs at the Rad52 repair centers in response to MMS.
Persistence of Rad52 foci in TLS mutants is not due to a demand for TLS polymerases during HR
Since cells are released into S phase in the presence of MMS in our experimental setup, the blocking lesions accumulate not only ahead of the fork but also within the nascent strands. Thus, one possibility to explain the genetic connection between the processing of ssDNA gaps at Rad52 repair centers and the TLS machinery would be that DNA synthesis at the invaded sister chromatid during HR was blocked by additional lesions and therefore required TLS polymerases (Fig 3A; MMS in S/G2). To address this possibility, cells were synchronized in G1 and treated with MMS for 1 h; after that, the MMS was inactivated, and then, cells were released into S phase in the absence of DNA damage. Under these conditions, the MMS‐induced blocking lesions are restricted to the parental molecule, leaving intact the nascent DNA molecules (Fig 3A; MMS in G1 only). In this case, Rad52 foci also persisted in the absence of Rev1 (Fig 3A). Therefore, the requirement of Rev1 for ssDNA processing at Rad52 repair centers is not due to a demand for TLS polymerases to bypass lesions in the newly synthesized DNA. Hence, TLS polymerases are unlikely to be required for the HR process. To confirm this, we analyzed HR using an unequal sister chromatid exchange (uSCE) recombination system (Fasullo & Davis, 1987) in cells growing on MMS‐containing solid medium. Neither Rev1 nor Rev3 was required for recombination; actually, the frequency of MMS‐induced HR increased in both rev1∆ and rev3∆ mutants (Fig 3B), consistent with the channeling of ssDNA gaps to HR in the absence of TLS.
Figure 3. Persistence of Rad52 foci in TLS mutants is not due to a demand for TLS polymerases during HR.

-
AssDNA repair efficiency of the rev1∆ mutant treated with MMS only in G1 as determined from the maximal and final percentages of cells with Rad52 foci during the time course (right panel). Cells were synchronized in G1 and treated with 0.033% MMS for 1 h; after that the MMS was inactivated with 2.5% sodium thiosulfate and cells were released into fresh medium in the absence of DNA damage. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (middle panel). A scheme on the left shows the experimental set up followed in this (MMS in G1 only) and the other experiments (MMS in S/G2). An asterisk represents a blocking DNA lesion for the replicative polymerase.
-
BEffect of the rev1∆ and rev3∆ mutations in spontaneous and MMS‐induced uSCE. The frequency of spontaneous and MMS‐induced recombinants was determined from colonies grown in the absence or presence of 0.01% MMS. The mean and SEM of four independent fluctuation tests are shown.
-
C, DssDNA repair efficiency of the rad5∆ and rev1∆ mutants as determined from the maximal and final percentages of cells with Rad54 foci during the time course (right panels). Cells were synchronized in G1 and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (left panels).
Data information: The mean and SEM of 4 (A) and 3 (C, D) independent experiments are shown. Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where one, two, and three asterisks represent P‐values < 0.05, < 0.01, and < 0.001, respectively.
Source data are available online for this figure.
The failure of TLS mutants to resolve Rad52 foci might be a consequence of HR being the only operative DNA repair process to deal with ssDNA gaps. Rad54 plays critical roles in HR, but contrary to Rad52 that functions early during the formation of the ssDNA/Rad51 nucleofilament, Rad54 also plays a role at a later stage during the processing of recombination intermediates (Ceballos & Heyer, 2011). We performed the time course analyses of foci accumulation in cells expressing Rad54‐YFP, which was functional in the response to MMS (Appendix Fig S1A). Wild‐type cells with Rad54 foci reached a peak (~40% of cells) ~1–2 h after MMS inactivation that dropped to ~5–10% ~5 h later (Fig 3C and D, left panels; note that plotted values have been normalized to the highest value of the wild type, taken as 100). As expected by the cooperation between HR proteins and PCNA polyubiquitylation, the efficiency of ssDNA repair at Rad54‐YFP foci was dependent on Rad5 (Fig 3C). However, and in contrast to what we had observed with Rad52 foci, the elimination of Rev1 did not affect the accumulation or resolution of Rad54 foci during the time course (Fig 3D), despite the fact that Rad54 operates downstream of Rad52 in HR and accordingly did not form foci in rad52∆ cells (Appendix Fig S1B). This suggests that the absence of TLS does not affect the kinetics of ssDNA filling by HR.
Rad52 is required for post‐replicative ssDNA gap filling through a non‐recombinogenic process
The fact that MMS‐induced Rad54 foci displayed a wild‐type kinetics in the absence of Rev1 raised the possibility that the requirement of the TLS machinery for the resolution of Rad52 foci reflected a non‐recombinogenic role of Rad52 in TLS. To test this possibility, we determined the contribution of the HR and TLS processes to Rad52‐mediated ssDNA repair. To analyze the impact of disrupting HR, we used a rad54∆ mutant, as Rad54 foci kinetics was not affected in the absence of Rev1 (Fig 3D).
First, we confirmed that Rad54 is essential for MMS‐induced HR. We used the uSCE system to quantitatively determine the impact of the lack of Rad52 and Rad54 on MMS‐induced HR. Since rad52∆ and rad54∆ mutants cannot grow on MMS‐containing solid medium, the analyses were performed with cell cultures grown until mid‐log phase and then treated with 0.02% MMS for 4 h. The frequency of uSCE was determined before and after DNA damage by measuring the frequency of His+ recombinants. Thus, DNA damage‐induced HR includes the recombinants that arose spontaneously before MMS addition. In wild‐type cells, MMS treatment increased the frequency of recombinants by about 6‐fold (Fig 4A). As expected, Rad52 was required for spontaneous and MMS‐induced HR, whereas Rad54 was dispensable for spontaneous HR. This is likely due to the fact that spontaneous HR arises from multiple lesions that are processed by different mechanisms, including DSB‐induced SCE events that depend totally on Rad52 and partially on Rad54 (Cortes‐Ledesma et al, 2007). In contrast, MMS‐induced HR was completely abolished in the absence of Rad54 (Fig 4A), consistent with previous studies showing that Rad54 is strictly required for SCJ accumulation (Mankouri et al, 2007) and salvage pathway functionality (Schiestl et al, 1990; Pfander et al, 2005) in response to MMS. In agreement with the importance of HR in the response to MMS, cell viability was severely reduced in the rad54∆ mutant as compared to the wild type (~100 times). Interestingly, the loss of viability was more severe in the rad52∆ mutant (~1,000 times), suggesting that Rad52 was preventing MMS‐induced DNA damage through additional mechanisms.
Figure 4. Rad52 is required for post‐replicative ssDNA gap filling through a non‐recombinogenic process.

-
AEffect of the rad54∆ and rad52∆ mutations in spontaneous and MMS‐induced uSCE. The frequency of spontaneous and MMS‐induced recombinants was determined in mid‐log phase cell cultures before (spontaneous uSCE) and after treatment with 0.02% MMS for 4 h (DNA damage‐induced uSCE). Thus, the frequency of DNA damage‐induced uSCE includes the recombinants that arose spontaneously before MMS addition. The frequency of uSCE was determined at 0.02% MMS because at higher MMS concentrations the loss of viability of rad52∆ and rad54∆ mutants did not allow us to select for His+ recombinants. The mean and SEM of five independent fluctuation tests are shown; n.a., not applicable.
-
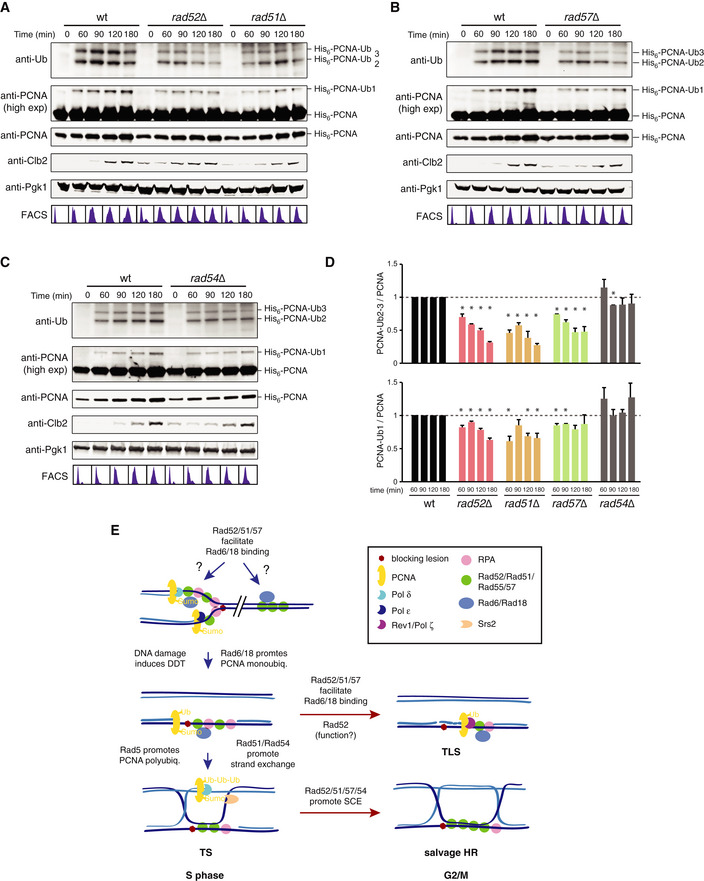
BssDNA repair efficiency of the indicated mutants as determined from the maximal and final percentages of cells with RPA foci during the time course (see Fig EV2D and E for the time course analyses; cells were synchronized in G1 and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times).
-
C–ENumber of foci per cell in cells with foci (C), fluorescence signal per focus (D) and fluorescence signal of RPA foci per cell (E) at times 1 or 2 (peak) and 7 h from the time course shown in (B). Counting and quantification were done with the MetaMorph software. Representative images of wild‐type and mutant cells with MMS‐induced RPA foci (bright field and fluorescence signal) are shown (D; left panels).
-
FssDNA repair efficiency of the indicated mutants as determined by ChIP against Rfa1‐YFP at the ARS305 replication origin. Cells were synchronized in G1 and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium. The amount of Rfa1 at ssDNA was determined at 1 and 7 h after MMS release. RPA enrichment was calculated as the ratio between immunoprecipitate and input values. All values were normalized to the wild type at 7 h, taken as 1.
Data information: (B–F) The mean and SEM of three independent experiments are shown. (A–F) Asterisks indicate statistically significant differences according to an unpaired two‐tailed Student’s t‐test. One, two, and three asterisks represent P‐values < 0.05, < 0.01, and < 0.001, respectively. In (F), all values were significant relative to the untagged controls (not shown for clarity).
Source data are available online for this figure.
Next, we addressed the effect of eliminating HR and TLS on the resolution of MMS‐induced Rad52 foci. The defect in the efficiency of Rad52 foci disappearance in the rad54∆ mutant was not increased in the double mutant rad54∆ rev1∆ (Fig EV2A). A more exhaustive quantitative analysis showed that the number of foci per cell in cells with foci was reduced from ~2 at 1–2 h to ~1 at 7 h after MMS release in wild‐type and rev1∆ cells, but not in rad54∆ and rad54∆ rev1∆ cells (Fig EV2B). This is consistent with our previous results showing that the repair defect in the absence of TS was not further increase by the absence of TLS (compare rad18∆ with mms2∆ in Fig 1D and E). These results suggest that HR is the main pathway to repair MMS‐induced ssDNA gaps. However, the Rad52‐YFP foci intensity was similar during the time course analysis in rad54∆ and wild‐type cells (Fig EV2C). Thus, we cannot discard the possibility that ssDNA filling occurs to some extent at the Rad52‐associated centers in the rad54∆ mutant. Actually, the Rad52 foci signal was significantly increased in the rad54∆ rev1∆ double mutant as compared to the rad54∆ and rev1∆ single mutants (Fig EV2C).
Figure EV2. Contribution of Rad54 and Rev1 to MMS‐induced ssDNA repair.

-
A–CssDNA repair efficiency (A), number of foci per cell in cells with foci (B) and fluorescence signal per focus (C) of the indicated mutants. ssDNA repair efficiency was determined from the maximal and final percentages of cells with Rad52 foci during the time course (A; right panel). Cells were synchronized in G1 and released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (A; middle panel). Cell cycle progression was determined by cell sorting (A; left panel). Counting and quantification were done directly on the processed samples (A) or with the MetaMorph software (B, C).
-
D, EssDNA repair efficiency of the indicated mutants as determined by analyzing RPA foci in cells synchronized in G1, released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (D; right panel and E). Cell cycle progression was determined by cell sorting (D; left panel). Counting and quantification were done directly on the processed samples (D) or with the MetaMorph software (E).
-
FssDNA repair efficiency of the indicated mutants as determined by analyzing RPA foci in cells synchronized in G1, released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. The percentage of cells with foci (lower panel) and the fluorescence signal at RPA foci per cell (right panel) at times 1 or 2 (the maximal percentage of cells with foci during the time course) and 7 h are shown. The percentage of cells with foci at each point was normalized to the highest value of the rad54∆ mutant, taken as 100. Cell cycle progression was determined by cell sorting (upper panel). Counting and quantification were done with the MetaMorph software.
Data information: The mean and SEM of three independent experiments are shown. Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where one, two, and three asterisks represent P‐values < 0.05, < 0.01, and < 0.001, respectively.
To better address ssDNA processing at DNA repair centers, we followed the kinetics of MMS‐induced RPA foci in cells expressing Rfa1‐YFP. RPA foci mark post‐replicative ssDNA filling events (González‐Prieto et al, 2013; Wong et al, 2020). Importantly, RPA binds to and signals all ssDNA lesions irrespective of the gap filling mechanism and is therefore an excellent readout for the accumulation of unrepaired ssDNA gaps. After MMS treatment, RPA foci were detected in more cells (~80–90%) and slightly earlier than Rad52 foci, but were also efficiently resolved 7 h after MMS release in wild‐type cells (Figs 4B and EV2D and E; note that plotted values have been normalized to the highest value of the wild type, taken as 100) (González‐Prieto et al, 2013). As previously reported, Rad52 was essential for the repair of MMS‐induced ssDNA gaps as inferred by the percentage of cells that retained RPA foci at the end of the time course (González‐Prieto et al, 2013). The lack of Rad54 had a similar effect, regardless of the presence or absence of Rev1, further supporting a major role of HR in MMS‐induced ssDNA repair (Figs 4B and EV2D and E).
The number of foci per cell in cells with foci was also similar for RPA and Rad52 (Figs 4C and EV2B), consistent with their overlapping at the same repair centers (Wong et al, 2020). However, the kinetics of RPA accumulation in response to MMS was different from that observed for Rad52. RPA intensity at foci was higher at the end of the time course (7 h) than at the peak of cells with foci (1–2 h after MMS release; Fig 4D), suggesting an accumulation of unrepaired ssDNA fragments in these foci. The highest accumulation was observed in the rad52∆ mutant; the rad54∆ and rev1∆ mutants displayed wild‐type levels of RPA signal per focus, and only the double mutant rad54∆ rev1∆ reached a signal close to that observed in the absence of Rad52 (Fig 4D). Therefore, we calculated the fluorescence signal at RPA foci per cell to determine more accurately the repair efficiency of MMS‐induced ssDNA gaps (Fig 4E). This analysis revealed a stronger effect of rad52∆ relative to rad54∆ on the accumulation of RPA signal in response to MMS. This milder effect of rad54∆ was not due to the activity of its homolog Rdh54 as the double mutant rad54∆ rdh54∆ displayed no significant differences with the rad54∆ mutant (Fig EV2F), consistent with the complete requirement of Rad54 for MMS‐induced HR (Fig 4A). Only the elimination of Rad54 and Rev1 together caused an accumulation of RPA signal similar to that observed in rad52∆ cells (Fig 4E).
To address more directly the accumulation of DNA damage, we measured the amount of RPA bound to ssDNA by chromatin immunoprecipitation (ChIP). For this, we repeated the kinetics of MMS‐induced DNA damage and repair and immunoprecipitated Rfa1‐YFP at 1 and 7 h after MMS released. The kinetics of RPA binding to the ssDNA generated at the ARS305 replication origin determined by ChIP was similar to that observed for RPA intensity by fluorescence microscopy, with the rad52∆ and rad54∆ rev1∆ mutants accumulating more RPA at ssDNA gaps than the rad54∆ and rev1∆ mutants (Fig 4F). Altogether, these results indicate that Rad52 contribute through independent mechanisms to both HR and TLS in the processing of MMS‐induced DNA damage.
Rad52 is required for efficient MMS‐induced mutagenesis
To genetically demonstrate that Rad52 is required for TLS, we studied DNA damage‐induced mutagenesis. We followed the same strategy used to measure uSCE; cell cultures were grown until mid‐log phase and then treated with MMS for 4 h. The frequency of mutants was determined before and after DNA damage by measuring the frequency of forward mutagenesis at the CAN1 locus (selected as canavanine‐resistant cells). Thus, the frequency of DNA damage‐induced mutagenesis includes the mutants that arose spontaneously before MMS addition. MMS treatment increased the frequency of mutants by about 50‐fold in the wild type (Fig 5). The frequency of MMS‐induced mutants was similar in wild‐type and rad52∆ cells. However, the absence of Rad52 augmented the frequency of spontaneous mutagenesis (Borstel et al, 1971; Roche et al, 1995; Paulovich et al, 1998; Rattray et al, 2002) to similar levels (Fig 5), hiding a possible effect of rad52∆ on MMS‐induced mutagenesis. In any case, a direct comparison of wild‐type and rad52∆ cells does not allow us to determine the effects of the absence of Rad52 in MMS‐induced mutagenesis, because the error‐free HR mechanisms are operative in the wild type and not in the rad52∆ mutant, and consequently, the amount of DNA gaps that are channeled to the mutagenic TLS pathway is different in each strain. To avoid this problem, we repeated the assay in a rad54∆ background to completely eliminate HR. The absence of Rad54 augmented the frequency of spontaneous mutagenesis to the same levels than the absence of Rad52, further supporting a role for HR in preventing the accumulation of spontaneous mutations. Furthermore, the absence of Rad54 caused a 4‐fold increase in the frequency of MMS‐induced mutagenesis as compared to the wild type, in accordance with the elimination of the HR mechanisms and the channeling of the MMS‐induced DNA gaps to TLS. More importantly, this increase depended on Rad52 (Fig 5; compare rad54∆ with rad54∆ rad52∆), indicating that Rad52 is required for efficient DNA damage‐induced mutagenesis. Note that we cannot determine to what extent Rad52 is required for MMS‐induced mutagenesis due to the high level of spontaneous mutagenesis in the HR mutants, which arises through different and PCNA ubiquitylation‐independent mechanisms (Stelter & Ulrich, 2003; Davies et al, 2008).
Figure 5. Rad52 is required for MMS‐induced mutagenesis.

Effect of the lack of HR factors in spontaneous and MMS‐induced mutagenesis. The frequency of spontaneous and MMS‐induced forward mutagenesis at the CAN1 locus was determined in mid‐log phase cell cultures before (spontaneous mutagenesis) and after treatment with 0.01% MMS for 4 h (DNA damage‐induced). Thus, the frequency of DNA damage‐induced mutagenesis includes the mutants that arose spontaneously before MMS addition. The frequency of mutagenesis was determined at 0.01% MMS because at higher MMS concentrations the loss of viability of the rad mutants did not allow us to select for canavanine‐resistant cells. Survival (%) after 4 h in MMS relative to the wild type (taken as 100) is shown below the genotypes. The mean and SEM of 6 (wild type), 9 (rad54∆), and 3 (rest) independent fluctuation tests are shown. Asterisks indicate statistically significant differences according to an unpaired two‐tailed Student’s t‐test (one, two, and three asterisks represent P‐values < 0.05, < 0.01 and < 0.001, respectively). Only the statistical analysis of the effect of rad52∆, rad57∆, and rad51∆ on rad54∆‐mediated mutagenesis is included for clarity. All mutants displayed statistically significant differences with the wild type and not with each other in the absence of MMS.Source data are available online for this figure.
Next, we asked whether the early HR proteins Rad51 and Rad57 cooperate with Rad52 in TLS. First, we calculated the fluorescence signal at RPA foci per cell upon MMS‐induced DNA damage and repair and observed that the rad51∆ and rad57∆ mutants displayed a defect similar to that of the rad54∆ mutant (Fig EV3). Next, we analyzed the frequency of MMS‐induced mutagenesis as indicated above. The absence of Rad57, but not of Rad51, caused a slight reduction in the frequency of rad54∆‐associated mutagenesis. However, the absence of Rad57 alone did not significantly reduce mutagenesis as compared to the rad54∆ mutants (Fig 5).
Figure EV3. ssDNA repair efficiency in HR mutants.
ssDNA repair efficiency of the indicated mutants as determined by analyzing RPA foci in cells synchronized in G1, released into S phase in the presence of 0.033% MMS for 1 h, treated with 2.5% sodium thiosulfate to inactivate the MMS, washed, and released into fresh medium for different times. The percentage of cells with foci (normalized at each point to the highest value of the wild type, taken as 100; middle panel) and the fluorescence signal at RPA foci per cell (right panel) at times 1 or 2 (the maximal percentage of cells with foci during the time course) and 7 h are shown. Cell cycle progression was determined by cell sorting (left panel). Counting and quantification were done with the MetaMorph software. The mean and SEM of three independent experiments are shown. Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where one, two, and three asterisks represent P‐values <0.05, <0.01, and <0.001, respectively.
Rad52 is required for efficient UV‐induced TLS
We used UV light to test whether the role of Rad52 on TLS was specific for MMS or could also be observed with another agent that induced the DDT response. First, we confirmed that Rad54 was also essential for UV‐induced HR using the uSCE system (Fig 6A). Next, we analyzed the kinetics of RPA accumulation by fluorescence microscopy in cells synchronized in G1, irradiated with 20 J/m2, and released into S phase for different times. The peak of wild‐type cells with RPA foci was ~60% and was reached 2 h after UV irradiation (Fig 6B, middle panel; note that plotted values have been normalized to the highest value of the wild type, taken as 100). The fraction of wild‐type cells with RPA foci dropped to ~20% 3 h later, suggesting that the efficiency of ssDNA repair was ~70% (Fig 6B, right panel). In contrast to MMS, RPA foci persisted in most cells until the end of the time course not only in rad52∆ and rad54∆ but also in the rev1∆ mutant (Fig 6B). In fact, the analysis of the fluorescence signal at RPA foci showed a stronger DNA repair defect in the rev1∆ mutant than in the rad54∆ mutant (Figs 6C and EV4), consistent with the predominant role of TLS over HR in the response to UV light (Daigaku et al, 2010). However, the repair defect of both rad54∆ and rev1∆ mutants was milder than that displayed by the rad52∆ mutant, and only the double mutant rad54∆ rev1∆ reached a signal close to that observed in the absence of Rad52 (Fig 6C). These results suggest that Rad52 acts through independent mechanisms with HR and TLS in the processing of both MMS and UV‐induced ssDNA gaps.
Figure 6. Rad52 is required for efficient UV‐induced TLS.

-
AEffect of the rad54∆ and rad52∆ mutations in spontaneous and UV‐induced uSCE. The frequency of spontaneous and UV‐induced recombinants was determined by irradiating or not cells plated onto solid medium after the corresponding dilutions (SMM and SMM without histidine to calculate total and recombinant cells, respectively). The mean and SEM of 3–5 independent fluctuation tests are shown. Asterisks indicate statistically significant differences according to an unpaired two‐tailed Student’s t‐test. Two and three asterisks represent P‐values < 0.01 and < 0.001, respectively.
-
B, CssDNA repair efficiency (B) and fluorescence signal at RPA foci per cell (C) of the indicated mutants. ssDNA repair efficiency was determined from the maximal and final percentages of cells with RPA foci during the time course (B, right panel). Cells were synchronized in G1, irradiated with 20 J/m2, and released into S phase for different times. The percentage of cells with foci at each point was normalized to the highest value of the wild type, taken as 100 (B, middle panel). Cell cycle progression was determined by cell sorting (B, left panel). Counting and quantification were done with the MetaMorph software. The mean and SEM of four independent experiments are shown. Statistically significant differences according to a paired two‐tailed Student’s t‐test are shown, where two and three asterisks represent P‐values < 0.01 and < 0.001, respectively.
-
DEffect of the rad54∆ and rad52∆ mutations in spontaneous and UV‐induced mutagenesis. The frequency of spontaneous and UV‐induced forward mutagenesis at the CAN1 locus was determined by irradiating or not cells plated onto solid medium after the corresponding dilutions (SMM and SMM with canavanine to calculate total and mutant cells, respectively). Cell viability (right panel) was calculated from total irradiated cells relative to non‐irradiated cells. The mean and SEM of six independent fluctuation tests are shown. Two asterisks represent a P‐value < 0.01, according to a two‐way ANOVA followed by a Bonferroni test.
Source data are available online for this figure.
Figure EV4. Contribution of HR and TLS to the number and signal of UV‐induced RPA foci.

Number of foci per cell in cells with foci (left) and fluorescence signal per focus (right) at times 2 (the maximal peak) and 5 h from the time course shown in Fig 6B. Counting and quantification were done with the MetaMorph software. The mean and SEM of four independent experiments are shown. Statistically significant differences according to an unpaired two‐tailed Student’s t‐test are shown, where one and two asterisks represent P‐values <0.05 and <0.01, respectively.
Finally, we analyzed the effect of the rad52∆ mutation on UV‐induced mutagenesis in the presence and absence of Rad54. This study showed two differences with the MMS treatment. First, the absence of HR in the rad52∆ and rad54∆ mutants did not significantly affect cell viability upon UV light irradiation (Fig 6D, right panel); second, the absence of Rad54 did not increase the frequency of UV‐induced mutagenesis (Fig 6D, left panel). Both observations are consistent with a major role of TLS in the response to UV light (Daigaku et al, 2010). The absence of Rad52 partially suppressed UV‐induced mutagenesis as previously reported (Fig 6D, left panel; Armstrong et al, 1994). Importantly, the absence of Rad54 did not reduce the frequency of mutagenesis, indicating that the effect of the rad52∆ mutation is not due to the elimination of HR. Therefore, we conclude that Rad52 is partially required for TLS in response to agents that activate DDT. It is worth noting that UV‐induced mutagenesis was still high in the absence of Rad52, which might explain why cell viability was not significantly affected in the rad52∆ mutant as compared to the wild type.
Rad52, Rad51, and Rad57, but not Rad54, promote efficient Rad6/Rad18 binding to chromatin and PCNA ubiquitylation
To understand how Rad52 promotes TLS during DDT, we explored the possibility that it was required for Rad6/Rad18 DNA binding. It was previously shown by ChIP analyses that Rad18 accumulates at early replication origins in cells released into S phase in the presence of the replication inhibitor hydroxyurea (HU) (Davies et al, 2008). We analyzed the accumulation of a functional chimera of Rad18 (Rad18‐YFP; Appendix Fig S2A) at the replication origin ARS305 in cells released into S phase in the presence or absence of 0.05% MMS for 1 h. Under these conditions, we did not detect Rad18 even slowing down replication at 22°C (Appendix Fig S2B). Thus, we repeated the experiment releasing the cells in the presence of both 0.05% MMS and 200mM HU to further increase the amount of ssDNA. In this case, Rad18 was detected at the replication origin independently of the presence of Rad52 (Appendix Fig S2C). However, HU triggers a fork‐specific checkpoint response that differs from that elicited by lesions in the replication template (García‐Rodríguez et al, 2018).
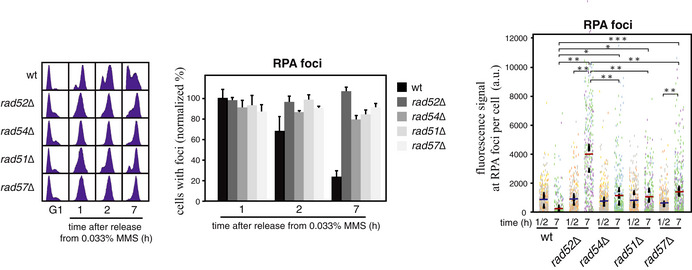
An alternative method to detect protein chromatin binding is the chromatin endogenous cleavage (ChEC) method (Schmid et al, 2004). In this assay, cells expressing a chimera of the protein of interest fused to MNase are permeabilized with digitonin and treated with Ca2+ ions for different periods to activate the nuclease; then, total DNA is extracted and analyzed for the presence of specific cuts. We have used this method to follow the binding of repair factors to ssDNA gaps scattered throughout the genome as those generated by MMS (González‐Prieto et al, 2013, 2020). Here, total DNA is analyzed on an agarose gel. The rationale behind this approach is that the chimera will generate a detectable cut only if it is targeted to a lesion that is not a DSB (Fig 7A). Therefore, we can infer the binding of a protein to non‐DSB DNA from the extent of DNA digestion.
Figure 7. Rad52, Rad51, and Rad57, but not Rad54, are required for efficient Rad6/Rad18 binding to chromatin.
-
AScheme with the rationale of the ChEC approach for non‐DSB lesions. Rad6 fused to MNaseI will induce a detectable cut only if it is bound to a DNA lesion other than a DSB.
-
BRad6 binds to DNA in the absence and presence of MMS. ChEC analysis of G1‐synchronized cells released in the absence or the presence of 0.033% MMS for the indicated times.
-
C–FRad6 binding to DNA depends on Rad18, Rad52 (C), Rad51 (D), and Rad57 (E) but not Rad54 (F) as determined by ChEC analyses of asynchronous cell cultures without or with 0.05% MMS for 2 h.
Data information: (B–F) DNA gel electrophoresis, DNA content (cell sorting), and quantification of DNA digestion profiles are shown. The experiments were repeated twice with similar results.
Source data are available online for this figure.
We applied this assay to follow Rad6 binding to chromatin in the absence and presence of MMS (Rad6‐MN but not Rad18‐MN was functional; Fig EV5A). A wild‐type strain was included in all time course analyses to control the approach‐intrinsic variability in the extent of DNA digestion among different experiments. A time course analysis of cells synchronized in G1 and released into fresh medium showed that Rad6‐MN was bound to DNA in the presence of 0.033% MMS during the S phase and that this binding was reduced as cells reached G2/M (Fig 7B). Intriguingly, the extent of DNA digestion was larger in the absence of MMS, suggesting that Rad6 was bound to chromatin under unperturbed conditions and partially released upon DNA damage. To validate this result, we analyzed the binding of Rad18 to DNA by chromatin fractionation followed by Western blot. Rad18 was bound to chromatin under unperturbed conditions and displayed a slight increase in the presence of MMS (Fig EV5B). An explanation for this discrepancy is that chromatin‐associated factors sterically hinder the ability of Rad6‐MN to cut the DNA in the presence of MMS.
Figure EV5. Rad6/Rad18 binding to chromatin.

-
ARad6‐MN is functional in MMS‐induced DDT as determined by cell growth analysis.
-
BRad18‐YFP is bound to chromatin in the absence and presence of MMS, as determined by chromatin fractionation and Western blot from asynchronous cell cultures treated or not with 0.05% MMS for 2 h (left and middle panels). Pgk1 and histone H4 were used as controls for soluble and chromatin fractions, respectively (right panel). The amount of Rad18‐YFP was normalized to the amount of Pgk1 (note that histones are partially released from chromatin after DNA damage; (Hauer et al, 2017)). The mean and SEM from three independent experiments are shown. The amount of Rad18 was not significantly increased according to an unpaired two‐tailed Student’s t‐test.
-
CRad6 binding to DNA is not significantly affected in the absence of Bre1, as determined by ChEC analysis of asynchronous cultures. DNA gel electrophoresis, DNA content (cell sorting), and quantification of DNA digestion profiles are shown. The experiments were repeated twice with similar results.
-
DChEC repetition of the binding of Rad6‐MN to chromatin in the absence of Rad54 (See Fig 7F for more details).
Importantly, DNA digestion by Rad6‐MN, both with and without MMS, was partially prevented in the absence of Rad18 (Fig 7C), which provides the DNA binding activity of the complex (Bailly et al, 1997; Davies et al, 2008), demonstrating that DNA digestion by the Rad6‐MN chimera (i) requires Rad6 binding to chromatin, and (ii) is specific to the Rad6/Rad18 complex. Since some DNA remained digested in the absence of Rad18 (especially at longer incubation times), we analyzed the effect of the absence of Bre1, which can also recruit Rad6 to chromatin (Wood et al, 2003). The absence of Bre1 did not affect the binding of Rad6‐MN to chromatin (Fig EV5C). Thus, the remaining DNA digestion in the rad18∆ mutant may reflect the binding of Rad6 through a different mechanism and/or the DNA affinity of the MNase domain of the chimera (Schmid et al, 2004). Importantly, DNA digestion by Rad6‐MN was partially prevented in rad52∆ cells in the absence and presence of MMS (Fig 7C), indicating that Rad52 promotes efficient Rad6/Rad18 binding to chromatin.
This function was not specific to Rad52, as both the recombinase Rad51 and its helper Rad57 facilitated Rad6/Rad18 binding to DNA with and without MMS, as inferred from the partial suppression of the Rad6‐MN‐mediated DNA digestion provoked by rad51∆ and rad57∆ (Fig 7D and E). Therefore, we asked whether HR was required to facilitate Rad6/Rad18 chromatin binding. For this, we tested the role of Rad54, previously shown to be essential for MMS‐induced HR (Fig 4A; Schiestl et al, 1990; Pfander et al, 2005; Mankouri et al, 2007). The lack of Rad54 did not affect Rad6 binding to chromatin either in the absence or presence of MMS (Figs 7F and EV5D). Therefore, the reduced efficiency of Rad6 binding to chromatin in rad52∆, rad51∆, and rad57∆ cells is not a consequence of defective HR.
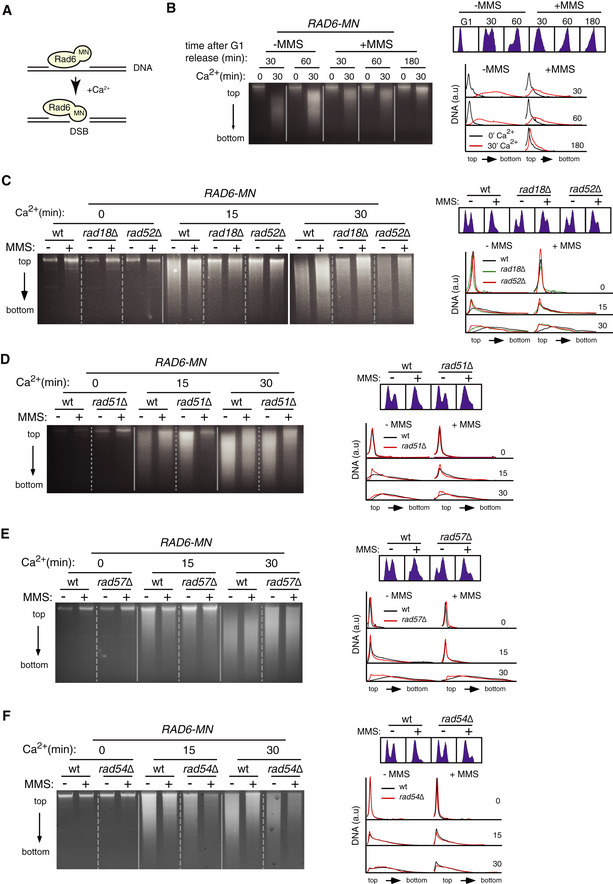
To validate the ChEC results, we analyzed PCNA ubiquitylation, which is carried out by Rad6/Rad18 at chromatin during DDT (Hoege et al, 2002; Garg & Burgers, 2005; Davies et al, 2008). An expectation from our previous results was that rad52∆, rad51∆, and rad57∆, but not rad54∆ cells, were affected in PCNA ubiquitylation. DDT activation in wild‐type cells led to PCNA mono‐ and polyubiquitylation in S phase; PCNA polyubiquitylation decreased as cells completed replication, whereas PCNA monoubiquitylation reached its maximal peak in G2/M (Fig 8A–D). Importantly, consistent with the ChEC results, the absence of Rad52, Rad51, or Rad57, but not that of Rad54, reduced the amount of both mono‐ and polyubiquitylated PCNA to similar levels (Fig 8A–D). The effect was more severe on PCNA polyubiquitylation than monoubiquitylation, although this might be related to the fact that they are detected with different antibodies. In sum, the early HR factors Rad51, Rad52, and Rad57 promote efficient Rad6/Rad18 binding to chromatin and DNA damage‐dependent PCNA ubiquitylation through a non‐recombinogenic mechanism.
Figure 8. Rad52, Rad51, and Rad57, but not Rad54, are required for efficient PCNA ubiquitylation.
-
A–DEffect of rad52∆, rad51∆ (A), rad57∆ (B), and rad54∆ (C) on PCNA ubiquitylation in cells synchronized in G1 and released into fresh medium in the presence of 0.025% MMS. Cell cycle progression was followed by cell sorting analysis and Western blot against Clb2. Pgk1 was used as loading control. The amounts of mono‐ and polyubiquitylated PCNA were normalized to the total amount of PCNA. For each time point, rad mutant values were relativized to those of the wild type, taken as 1. The mean and SEM of three independent experiments are shown (D). An asterisk indicates a mean significantly different than 1, according to a one‐sample t‐test.
-
EModel for Rad52‐mediated tolerance during DDT. The recombination factors Rad52, Rad51, and Rad57 promote efficient Rad6/Rad18 binding to chromatin under unperturbed conditions. In response to blocking lesions that impair replication fork advance, the Rad6/Rad18 complex is mobilized to ssDNA gaps where, together with Mms2/Ubc13/Rad5, ubiquitylates PCNA, and promotes TS. The ssDNA gaps that escape from this error‐free pathway during S phase accumulate in G2/M, where they are filled in by two additional, less accurate mechanisms regulated by Rad52: UbPCNA‐independent HR (recombinogenic) and TLS (mutagenic). Apart from facilitating Rad6/Rad18 chromatin recruitment, Rad52 plays an additional uncharacterized Rad51/Rad57‐independent role in TLS.
Source data are available online for this figure.
Discussion
A major role for recombination proteins in the DDT response was established long time ago, but it was assumed to be restricted to HR mechanisms (TS and salvage pathways). Here, we show that Rad52 deals with MMS‐ and UV light‐induced ssDNA gaps together with the TLS machinery through a non‐recombinogenic process; accordingly, Rad52 is required for efficient MMS‐ and UV light‐induced mutagenesis. We also show that Rad52, Rad51, and Rad57—but not Rad54—facilitate Rad6/Rad18 binding to chromatin and in response to DNA damage the subsequent ubiquitylation of PCNA. Therefore, Rad52 plays recombinational and non‐recombinational roles in DDT that might impact the choice between TLS and HR and therefore genome integrity.
Rad52 is required for efficient TLS
HR proteins are required for the post‐replicative filling of the ssDNA gaps generated during DDT, and genetic and molecular evidence demonstrate that they operate in the recombinogenic TS and salvage pathways (Prado, 2014, 2018; Branzei & Psakhye, 2016). The partial requirement of Rad52 in UV‐ and EMS‐induced mutagenesis led to hypothesize a putative role for this HR factor in TLS (Prakash & Higgins, 1982; Kunz et al, 1992; Armstrong et al, 1994). This idea was later supported by the finding of Rad51 at unperturbed replication forks in both yeast and Xenopus (Hashimoto et al, 2010). However, a role for recombination proteins in TLS was unexpected as inferred from epistatic analyses of DNA damage sensitivity and mutagenesis between HR and TLS mutants (Paulovich et al, 1998; Rattray et al, 2002; Ball et al, 2009). Here, we show that Rad52 acts in concert with the TLS machinery in the repair of ssDNA gaps induced during DDT. Several mechanisms could be invoked to explain this cooperation. TLS polymerases have been implicated in DSB‐induced HR (McVey et al, 2016); however, the amount of MMS used in our study does not lead to the formation of DSBs (González‐Prieto et al, 2013). Moreover, PCNA ubiquitylation is not required for the function of Pol ζ or Pol η in DSB‐induced HR (Hirano & Sugimoto, 2006; Sharma et al, 2012). Therefore, a contribution of the TLS polymerases to DSB repair appears unlikely in this context. Alternatively, PCNA ubiquitylation and TLS polymerases might operate at the newly synthesized invaded strand during HR. However, Rev1 is required for repairing ssDNA gaps even if the generation of blocking lesions in the newly synthesized DNA is prevented (Fig 3A). Furthermore, Pol ζ and Pol η are required neither for SCJ formation (Vanoli et al, 2010) nor MMS‐induced HR (Fig 3B). We therefore propose an alternative mechanism, based on the following observations: (i) Rad54, which is essential for HR during DDT (Figs 4A and 6A; Schiestl et al, 1990; Pfander et al, 2005; Mankouri et al, 2007; Ceballos & Heyer, 2011; Prado, 2014, 2018), does not cooperate with the TLS machinery in the filling of ssDNA gaps (Fig 3D); (ii) Rad52 is required for the filling of MMS‐ and UV‐induced ssDNA gaps through a Rev1‐dependent, Rad54‐independent mechanism (Figs 2D, 4C–F and 6B and C); and (iii) Rad52, but not Rad54, is required for MMS‐ and UV‐induced mutagenesis (Figs 5 and 6D). Altogether, these results support a novel non‐recombinogenic function for Rad52 in TLS during DDT.
What is the function of Rad52 in TLS? We provide evidence that Rad52 facilitates Rad6/Rad18 chromatin binding (Figs 7 and 8). However, this function alone cannot explain the observed defects in TLS‐mediated ssDNA filling and mutagenesis of the rad52∆ mutant, because Rad51 and Rad57 cooperate with Rad52 in the recruitment of Rad6/Rad18 but neither rad51∆ nor rad57∆ display substantial defects in Rad52 foci resolution and mutagenesis. The function of Rad52 in TLS is expected to be independent of its recombinogenic ssDNA annealing and mediator activities and could be rather related to the recruitment of factors that promote TLS. Future studies will be required to decipher this specific Rad51/Rad57‐independent role of Rad52 in TLS during DDT, which underlies the complex interdependence of the DNA tolerance pathways. It is worth noting that the absence of Rad52 reduces the efficiency but does not eliminate TLS, as inferred from the up to ~40‐fold increase in mutagenesis induced by UV light in rad52∆ cells (Fig 6D, 40 J/m2; the levels of spontaneous mutagenesis mask the real contribution of Rad52 to MMS‐induced TLS). Therefore, DNA damage‐induced TLS is still partially operative in the rad52∆ mutant, which might explain the synergistic sensitivity to MMS and UV light previously observed in the absence of both Rad52 and TLS polymerases (Zgaga, 1991; Ball et al, 2009).
Rad52, Rad51, and Rad57 facilitate Rad6/Rad18 chromatin binding and DNA damage‐induced PCNA ubiquitylation through a non‐recombinogenic process
TLS during DDT relies on PCNA monoubiquitylation by the Rad6/Rad18 complex, an evolutionarily conserved modification that facilitates the recruitment of TLS polymerases (Hoege et al, 2002). Rad6 and Rad18 form a complex with ubiquitin‐conjugating (Rad6) and ubiquitin‐ligase (Rad18) activities (Bailly et al, 1997; Hoege et al, 2002) that ubiquitylates the pool of PCNA that is bound to DNA in response to replicative DNA damage (Garg & Burgers, 2005; Davies et al, 2008). Rad6/Rad18 is targeted to DNA through different mechanisms: Rad18 binding to ssDNA (Bailly et al, 1997) and Rad18 interactions with RPA and sumoylated PCNA (Davies et al, 2008; Parker & Ulrich, 2012). RPA binds to and protects ssDNA fragments that result during DNA replication and repair, but it has to be replaced with Rad51 with the help of Rad52 and Rad55/Rad57 for HR to occur (Heyer et al, 2010). It is unlikely that this replacement completely removes RPA from the ssDNA fragments generated during DDT, as PCNA ubiquitylation requires the continuous presence of RPA (Davies et al, 2008). Therefore, RPA and early HR proteins are expected to coexist at ssDNA fragments, and according to our results, they both contribute to Rad6/Rad18 recruitment to ssDNA gaps and PCNA ubiquitylation. Indeed, the competition between RPA and Rad51 at the ssDNA might explain why the PCNA ubiquitylation defect is modest and had no significant effects on mutagenesis in the HR mutants.
Importantly, Rad54, which is essential for MMS‐induced HR (Fig 4A; Schiestl et al, 1990; Pfander et al, 2005; Mankouri et al, 2007; Ceballos & Heyer, 2011; Prado, 2014, 2018), was dispensable for Rad6/Rad18 recruitment and PCNA ubiquitylation, indicating that they occur through a non‐recombinogenic process. Moreover, the lack of effect of the rad54∆ mutant rules out the possibility that the defects in Rad6/Rad18 recruitment to chromatin and PCNA ubiquitylation observed in the rad52∆, rad51∆, and rad57∆ mutants were due to replication defects, which were similar or even more severe in cells lacking Rad54 (Appendix Fig S3).
Interestingly, Rad6/Rad18 was already detected at chromatin in the absence of MMS, although PCNA ubiquitylation occurs specifically in response to DNA damage (Hoege et al, 2002). This observation is consistent with the fact that the physical interaction between Rad18 and RPA is not damage‐dependent (Davies et al, 2008). The contribution of the same HR factors to Rad6/Rad18 binding to chromatin before and after MMS opens the possibility that the complex is present under unperturbed conditions at specific chromatin regions from where it is mobilized to ssDNA gaps in response to DNA damage. In accordance with this model, we have observed by chromatin fractionation that Rad52 and Rad51 accumulate at the DNA in G1 and are released during S phase; however, they remain at chromatin in the presence of MMS to facilitate the repair of ssDNA gaps through a non‐recombinogenic process (Cabello‐Lobato et al, manuscript in preparation).
How Rad52, Rad51, and Rad57 facilitate Rad6/Rad18 binding to chromatin is currently unknown. One possibility is that some of the recombination proteins interact directly or indirectly with Rad6/Rad18. The Rad55/Rad57 complex is the main candidate, as its absence does not prevent Rad52 or Rad51 binding to ssDNA (Sugawara et al, 2003), and the human homolog of Rad57—Rad51C—interacts physically with human Rad18 (Huang et al, 2009). However, we have so far been unable to detect physical interactions between Rad18 and Rad57, suggesting that either the interactions are weak/transient, another recombination factor interacts with Rad6/Rad18, or Rad52, Rad51, and Rad57 promote Rad6/Rad18 recruitment by different means.
Rad52 as a molecular switch for HR and TLS?
In sum, we provide evidence for non‐recombinogenic roles of Rad52 in TLS during DDT. Rad52, together with Rad51 and Rad57, facilitates Rad6/Rad18 binding to chromatin in the absence of DNA damage. This accumulation might take place at replication forks and/or not‐yet defined regions (Fig 8E). In response to DNA lesions that trigger DDT, Rad6/Rad18 would be mobilized to ssDNA gaps to ubiquitylate PCNA (Hoege et al, 2002). Rad5, which interacts physically with Rad6/Rad18 (Ulrich & Jentsch, 2000) and peaks in S phase (Ortiz‐Bazán et al, 2014), assembles a polyubiquitin chain that leads to TS (Hoege et al, 2002; Stelter & Ulrich, 2003) through a mechanism that additionally requires recombinogenic activities provided by Rad51 and Rad54 (Liberi et al, 2005; Mankouri et al, 2007; Branzei et al, 2008; Vanoli et al, 2010; Minca & Kowalski, 2010; Karras et al, 2013). We propose that the ssDNA gaps that escape from the error‐free TS pathway during S phase can be repaired in G2/M by two additional, less accurate mechanisms regulated by Rad52 through Rad51/Rad57‐dependent and ‐independent mechanisms: the salvage pathway and TLS. An important implication of this model is that Rad52 might potentially exert an additional control on genome integrity by influencing the choice between the different mechanisms of DDT at the repair centers: Rad6/Rad18 recruitment together with not‐yet known functions would promote TLS and mutagenesis, whereas DNA strand invasion and exchange would lead to HR.
Materials and Methods
Yeast strains, plasmids, and growth conditions
Yeast strains used in this study are listed in Table EV1. Tagged, deleted, and GAL1pr‐driven genes were constructed by a PCR‐based strategy (Longtine et al, 1998). The integrative plasmid pRS306CAN1 was used to replace can1–100 with CAN1 in the W303 strain. Briefly, the strain was transformed with pRS306CAN1 (cut with HindIII) and grown first in medium without uracil to select the integration event and then in medium with 5‐fluoroorotic acid to select strains that had lost one of the two can1 copies and the intervening sequence. CAN1 was confirmed by DNA sequencing and canavanine sensitivity. pRS306CAN1 was constructed by inserting a PCR‐amplified fragment of CAN1 containing the wild‐type region corresponding to the can1–100 mutation at the XhoI‐XbaI site of pRS306. Oligonucleotide sequences are available upon request. pWJ1213 (Feng et al, 2007) and pRS314R52YFP (Murillo‐Pineda et al, 2014) are centromeric plasmid expressing RAD52‐YFP and HIS3 or TRP1, respectively. Yeast cells were grown at 30°C in supplemented minimal medium (SMM), except for the analyses of PCNA ubiquitylation, which were performed in YPAD rich medium. Cell cultures were synchronized in G1 with α‐factor at 250 ng/ml (W303 and BY strains) or 50 ng/ml (DF5 strains) for 2.30 h. Then, cells were washed and released into fresh medium with 50 μg/ml pronase in the absence or presence of MMS at the indicated concentrations. MMS sensitivity was determined by spotting ten‐fold serial dilutions of the same number of mid‐log growing cells onto medium with or without the drug.
Flow cytometry
DNA content analysis was performed by flow cytometry as reported previously (Prado & Aguilera, 2005). Cells were fixed with 70% ethanol, washed with phosphate‐buffered saline (PBS), incubated with 1 mg of RNaseA/ml PBS, and stained with 5 μg/ml propidium iodide. Samples were sonicated to separate single cells and analyzed in a FACSCalibur flow cytometer.
DNA repair foci
For MMS kinetics, cells were grown in liquid cultures under the indicated conditions and processed as indicated below. For UV light kinetics, G1 synchronized cells were washed twice with water, resuspended in water, and UV irradiated in a petri dish. Then, cells were collected, resuspended in SMM medium, and grown in darkness to avoid photolyase activation. To visualize the foci, the cells were fixed with 2.5% formaldehyde in 0.1 M potassium phosphate pH 6.4 for 10 min, washed twice with 0.1 M potassium phosphate pH 6.6, and resuspended in 0.1 M potassium phosphate pH 7.4. Finally, cells were fixed with 80% ethanol for 10 min, resuspended in H2O, and visualized with a Leica CTR6000 fluorescence microscope. The percentage of cells with foci was counted directly on the processed samples under the microscope, except for Figs 4B–E, 6B and C, EV2B, C, E and F, EV3, and EV4, in which six contrast and fluorescence images along the z‐axis (at 0.49 μm steps) were acquired to find well‐defined foci. Images were processed and analyzed with the MetaMorph software (Molecular devices). A total number of approximately 100 cells were analyzed for each time point and experiment. The efficiency of ssDNA repair was inferred from the foci kinetics and calculated as [100 ‐ percentage of cells with foci at the end of the time course x 100/ maximal percentage of cells with foci during the time course].
Genetic recombination and mutagenesis assays
HR was determined by measuring the frequency of His+ recombinants generated by uSCE in a chromosomal‐integrated system (Fasullo & Davis, 1987), whereas mutagenesis was determined by measuring the frequency of forward mutagenesis at the CAN1 locus (selected as canavanine‐resistant cells). Recombination frequencies were determined by fluctuation tests as previously reported (Prado & Aguilera, 1995). Briefly, for rev1∆ and rev3∆ mutants, cells from six independent colonies of similar size and isolated on medium with or without MMS were plated with the appropriate dilutions onto SMM without histidine and SMM to calculate recombinants and total viable cells (as colony‐forming units), respectively. The frequency of HR was calculated using the median of recombinants and the mean of total cells. To have a more accurate value, the mean and SEM of at least three independent fluctuation tests are given. Since rad mutants do not form colonies in the presence of MMS, MMS‐induced HR in these strains was calculated from six independent cell cultures (started with a colony) grown to the same mid‐log phase and then treated with MMS for 4 h. Recombinants and total cells were determined as previously mentioned by plating on specific media from the liquid cultures just before and after MMS addition, and the HR frequencies calculated as the mean of at least three independent fluctuation tests. The same approach was used to calculate MMS‐induced mutagenesis in the rad strains, but mutants were selected on SMM plates lacking arginine but containing 60 μg/ml canavanine. Therefore, MMS‐induced recombination/mutagenesis frequencies from liquid cultures also include the recombinants/mutants that arose spontaneously before MMS addition.
The frequencies of UV‐induced HR and mutagenesis were determined with the same genetic systems. For fluctuation tests, cells from six independent colonies of similar size were plated with the appropriate dilutions onto specific media, irradiated or not with different doses of UV‐C light, and grown for 3–4 days to isolate His+ recombinants (or canavanine‐resistant mutants) and total viable cells. Cells were grown in darkness to avoid photolyase activation.
For each test, the survival percentage was determined from total viable cells in the presence of DNA damage relative to total viable cells in the absence of DNA damage.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation analyses were performed as described previously with some modifications (Maya‐Miles et al, 2019). Cells were fixed for 15 min with 1% formaldehyde. Glycine was added to quench the reaction at a final concentration of 125 mM. Cells were sedimented by centrifugation, washed twice with cold PBS, and stored at −80°C until use. ChIP samples were obtained by breaking cells with a homogenizer (Multibeads shocker, Yasui Kikai) for 1 h at 2,500 rpm (60 s on/60 s off intervals) in lysis buffer (50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 1% Triton, 0.1% sodium deoxycholate) supplemented with protease cocktail inhibitors (Roche) and 3 mM PMSF. Supernatant was transferred to new tubes by soft centrifugation piercing the bottom of the tube with a G25 needle. Chromatin was further concentrated by centrifugation and then resuspended in lysis buffer (supplemented with protease cocktail inhibitors). Chromatin was sheared via sonication to a size between 200 and 600 bp using a sonicator (Branson Digital Sonifier, Branson Ultrasonics). About 10 µl of supernatant was kept on ice and used as the input DNA control; the rest was incubated overnight with gtma‐20 GFP‐trap_MA magnetic beads (Chromotek) to immunoprecipitate the GFP‐tagged proteins. After immunoprecipitation, samples were washed twice with 1 ml of the following solutions: lysis buffer, lysis buffer plus 0.5 M NaCl, wash buffer (0.25 M LiCl, 10mM Tris–HCl pH 8, 1mM EDTA, 0.5% NP‐40, 0.5% sodium deoxycholate), and TE 1×, all supplemented with protease cocktail inhibitors (Roche), and then eluted from magnetic beads with a 1% SDS‐TE solution. Input and immunoprecipitated samples were incubated 2 h at 42°C and 6 h at 65°C to reverse crosslinking, treated with 1.5 mg/ml of pronase, and extracted using a standard phenol/chloroform DNA purification. ChIP data were obtained by real‐time qPCR using iTaq™ Universal SYBR Green (Bio‐Rad).
In vivo ChEC
Chromatin endogenous cleavage (ChEC) of Rad6‐MN cells was performed as reported (González‐Prieto et al, 2013, 2020). For cleavage induction, digitonin‐permeabilized cells were incubated with 2 mM CaCl2 at 30°C under gentle agitation. Total DNA was isolated and resolved into 0.8% TAE 1× agarose gels. Gels were scanned in a Fuji FLA5100, and the signal profile quantified using ImageGauge. The area of the DNA digestion profiles was equalized to eliminate DNA loading differences.
Chromatin fractionation
Chromatin fractionation was performed as described for young yeast cells (Feser et al, 2010) with some modifications. Samples (15–30 ml) from mid‐log phase cultures were collected by centrifugation, washed with cold 0.1 mM Tris pH 9.4, 10 mM DTT, and incubated for 15 min in 1 ml of the same buffer on ice. Cells were then washed with cold spheroplasting buffer (20 mM Hepes pH 7.4, 1.2 M sorbitol, Roche Complete EDTA free protease inhibitor cocktail) and incubated with 1 ml of the same buffer with 210 μg zymolyase 20T for 1 h at 30°C. The spheroplasts were collected, washed twice with cold washing buffer (20 mM Tris pH 7.4, 20 mM KCl, 0.4M sorbitol, 0.1 μM spermine, 0.25 μM spermidine, protease inhibitors), and resuspended in 1 ml lysis buffer (20 mM Tris pH 7.4, 20 mM KCl, 0.4 M sorbitol, 0.1 μM spermine, 0.25 μM spermidine, 1% Triton X‐100, protease inhibitors) for 5 min on ice. An aliquot (80 μl) was removed for the total sample, and the remaining sample was centrifuged for 15 min at 13,000 g at 4°C to separate soluble (supernatant) and chromatin‐enriched (pellet) fractions. Each pellet was washed with 0.5 ml cold lysis buffer and resuspended in 160 μl of water. Chromatin, soluble, and total samples were mixed with SDS buffer for Western blot analyses. Similar volumes were loaded from each time point for each kinetics, and similar cell equivalents of the chromatin and soluble fractions were loaded for the fractionation controls. Rad18‐YFP, histone H4, and Pgk1 were analyzed by SDS–PAGE and Western blot using antibodies against GFP (632381, Clontech), Pgk1 (22C5D8, Invitrogen), or H4 (ab10158, Abcam). All signals were acquired and quantified in a ChemiDoc MP image system (Bio‐Rad).
PCNA ubiquitylation
PCNA ubiquitylation was detected by His6‐PCNA isolation and analysis as previously reported (Daigaku et al, 2010). Briefly, total cell extracts were prepared under denaturing conditions and subjected to Ni‐NTA affinity purification, and samples were analyzed by SDS–PAGE and Western blot using antibodies against PCNA (H. Ulrich Lab) and ubiquitin (P4D1; Cell Signaling). Polyclonal anti‐Clb2 (y‐180; Santa Cruz) was used to monitor cell cycle progression. Monoclonal anti‐Pgk1 antibody 22C5D8 (Invitrogen) was used for loading controls. Proteins were detected and quantified using either fluorophore‐conjugate secondary antibodies with an ImageSudio system (Odyssey; for PCNA, Clb2 and Pgk1) or a peroxidase‐conjugate secondary antibody and the ECL Prime detection kit (GE Healthcare) in a ChemiDoc MP system (Bio‐Rad; for ubiquitin).
Statistical analyses
Statistical analyses were performed using the Prism software (GraphPad). Mean, SEM, sample size, and statistical tests are indicated in the Figure legends. Sample size was not predetermined using statistical methods. Given the reduced sample size, the analyses were performed assuming that they follow normal distributions. Scatter SuperPlots were done as recently reported (Lord et al, 2020).
Author contributions
MICL, AYV, NGR, MBM, and RGP investigated the study. FP conceptualized and wrote the original draft. FP, HDU, and PSS wrote, reviewed, and edited the manuscript and acquired funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank Michael Fasullo, Avelino Bueno, and José Antonio Tercero for various strains and reagents, and Felipe Cortés Ledesma for critical reading of the manuscript. MICL, AYV, and RGP were recipients of predoctoral training grants from the Spanish government. NGR is supported by the Marie Sklodowska‐Curie Fellowship MSCA‐IF‐2017‐794054 from the European Union’s Horizon 2020 Programme. This work was funded by grants BFU2015‐63698‐P and PGC2018‐099182‐B‐I00 (to FP), BFU2015‐65417‐R and RTI2018‐099055‐B‐I00 (to PSS) and BFU2015‐69142‐REDT (to FP and PSS) from the Spanish Government, and ERC AdG323179 (to HDU) from the European Research Council.
EMBO Reports (2021) 22: e50410.
Data availability
No datasets have been produced. Data are uploaded as source data or available upon request.
References
- Armstrong JD, Chadee DN, Kunz BA (1994) Roles for the yeast RAD18 and RAD52 DNA repair genes in UV mutagenesis. Mutat Res 315: 281–293 [DOI] [PubMed] [Google Scholar]
- Bailly V, Lauder S, Prakash S, Prakash L (1997) Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J Biol Chem 272: 23360–23365 [DOI] [PubMed] [Google Scholar]
- Ball LG, Zhang K, Cobb JA, Boone C, Xiao W (2009) The yeast Shu complex couples error‐free post‐replication repair to homologous recombination. Mol Microbiol 73: 89–102 [DOI] [PubMed] [Google Scholar]
- Baynton K, Bresson‐Roy A, Fuchs RP (1998) Analysis of damage tolerance pathways in Saccharomyces cerevisiae: a requirement for Rev3 DNA polymerase in translesion synthesis. Mol Cell Biol 18: 960–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR et al (2005) Ubiquitin‐binding domains in Y‐family polymerases regulate translesion synthesis. Science 310: 1821–1824 [DOI] [PubMed] [Google Scholar]
- von Borstel RC, Cain KT, Steimberg CM (1971) Inheritance of spontaneous mutability in yeast. Genetics 69: 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Vanoli F, Foiani M (2008) SUMOylation regulates Rad18‐mediated template switch. Nature 456: 915–920 [DOI] [PubMed] [Google Scholar]
- Branzei D, Psakhye I (2016) DNA damage tolerance. Curr Op Cell Biol 40: 137–144 [DOI] [PubMed] [Google Scholar]
- Broomfield S, Hryciw T, Xiao W (2001) DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae . Mutat Res 486: 167–184 [DOI] [PubMed] [Google Scholar]
- Ceballos SJ, Heyer W‐D (2011) Functions of the Snf2/Swi2 family Rad54 motor protein in homologous recombination. BBA – Gene Regulatory Mechanisms 1809: 509–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Motegi A, Hasegawa Y, Myung K, Kolodner R, D'Andrea A (2006) Genetic analysis of ionizing radiation‐induced mutagenesis in Saccharomyces cerevisiae reveals TransLesion Synthesis (TLS) independent of PCNA K164 SUMOylation and ubiquitination. DNA Repair 5: 1475–1488 [DOI] [PubMed] [Google Scholar]
- Cortes‐Ledesma F, Tous C, Aguilera A (2007) Different genetic requirements for repair of replication‐born double‐strand breaks by sister‐chromatid recombination and break‐induced replication. Nucleic Acids Res 35: 6560–6570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD (2010) Ubiquitin‐dependent DNA damage bypass is separable from genome replication. Nature 465: 951–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD (2008) Activation of ubiquitin‐dependent DNA damage bypass is mediated by replication protein A. Mol Cell 29: 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasullo MT, Davis RW (1987) Recombinational substrates designed to study recombination between unique and repetitive sequences in vivo. Proc Natl Acad Sci USA 84: 6215–6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Düring L, de Mayolo AA, Lettier G, Lisby M, Erdeniz N, Mortensen UH, Rothstein R (2007) Rad52 and Rad59 exhibit both overlapping and distinct functions. DNA Repair 6: 27–37 [DOI] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK (2010) Elevated histone expression promotes life span extension. Mol Cell 39: 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC (2005) Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol 6: 943–953 [DOI] [PubMed] [Google Scholar]
- García‐Rodríguez N, Morawska M, Wong RP, Daigaku Y, Ulrich HD (2018) Spatial separation between replisome‐ and template‐induced replication stress signaling. EMBO J 37: 779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P, Burgers PM (2005) Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci USA 102: 18361–18366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Zwicky K, Lopes M, Branzei D (2014) Visualization of recombination‐mediated damage bypass by template switching. Nature Struct Mol Biol 21: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Prieto R, Muñoz‐Cabello AM, Cabello‐Lobato MJ, Prado F (2013) Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J 32: 1307–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Prieto R, Cabello‐Lobato MJ, Prado F (2020) In vivo binding of recombination proteins to non‐DSB DNA lesions and to replication forks. Methods Mol Biol 2153: 447–458 [DOI] [PubMed] [Google Scholar]
- Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S, Prakash L (2001) Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev 15: 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen‐Hughes T et al (2017) Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nature 24: 99–107 [DOI] [PubMed] [Google Scholar]
- Heyer W‐D, Ehmsen KT, Liu J (2010) Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44: 113–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WM, Kim M, Haber JE (2010) Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Sugimoto K (2006) ATR homolog Mec1 controls association of DNA polymerase ζ‐Rev1 complex with regions near a double‐strand break. Curr Biol 16: 586–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan G‐L, Pyrowolakis G, Jentsch S (2002) RAD6‐dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- Huang J, Huen MSY, Kim H, Leung CCY, Glover JNM, Yu X, Chen J (2009) RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat Cell Biol 11: 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras GI, Jentsch S (2010) The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141: 255–267 [DOI] [PubMed] [Google Scholar]
- Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, Jentsch S (2013) Noncanonical role of the 9‐1‐1 clamp in the error‐free DNA damage tolerance pathway. Mol Cell 49: 536–546 [DOI] [PubMed] [Google Scholar]
- Kunz BA, Gabriel M, Kang X, Kohalmi SE, Terrick KA (1992) DNA repair modifies the site and strand specificity of ethyl methanesulfonate mutagenesis in yeast. Mutagenesis 7: 461–469 [DOI] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta‐Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51‐dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA 98: 8276–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, Mckenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21: 15–27 [DOI] [PubMed] [Google Scholar]
- Lord SJ, Velle KB, Mullins RD, Fritz‐Laylin LK (2020) SuperPlots: Communicating reproducibility and variability in cell biology. J Cell Biol 219: e202001064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Ngo H‐P, Hickson ID (2007) Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1‐Rmi1‐Top3. Mol Biol Cell 18: 4062–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya‐Miles D, Andújar E, Pérez‐Alegre M, Murillo‐Pineda M, Barrientos‐Moreno M, Cabello‐Lobato MJ, Gómez‐Marín E, Morillo‐Huesca M, Prado F (2019) Crosstalk between chromatin structure, cohesin activity and transcription. Epigenetics Chromatin 12: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer W‐D (2016) Eukaryotic DNA polymerases in homologous recombination. Annu Rev Genet 50: 393–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minca EC, Kowalski D (2010) Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell 38: 649–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A, Kuntz K, Majeed A, Smith S, Myung K (2006) Regulation of gross chromosomal rearrangements by ubiquitin and SUMO ligases in Saccharomyces cerevisiae . Mol Cell Biol 26: 1424–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo‐Pineda M, Cabello‐Lobato MJ, Clemente‐Ruiz M, Monje‐Casas F, Prado F (2014) Defective histone supply causes condensin‐dependent chromatin alterations, SAC activation and chromosome decatenation impairment. Nucleic Acids Res 42: 12469–12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz‐Bazán MÁ, Gallo‐Fernández M, Saugar I, Jiménez‐Martín A, Vázquez MV, Tercero JA (2014) Rad5 plays a major role in the cellular response to DNA damage during chromosome replication. Cell Rep 9: 460–468 [DOI] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA Is mediated by recruitment of the helicase Srs2p. Mol Cell 19: 123–133 [DOI] [PubMed] [Google Scholar]
- Parker JL, Ulrich HD (2012) A SUMO‐interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO‐modified PCNA. Nucleic Acids Res 40: 11380–11388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH (1997) RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics 145: 45–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Armour CD, Hartwell LH (1998) The Saccharomyces cerevisiae RAD9, RAD17, RAD24 and MEC3 genes are required for tolerating irreparable, ultraviolet‐induced DNA damage. Genetics 150: 75–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander B, Moldovan G‐L, Sacher M, Hoege C, Jentsch S (2005) SUMO‐modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436: 428–433 [DOI] [PubMed] [Google Scholar]
- Prado F, Aguilera A (1995) Role of reciprocal exchange, one‐ended invasion crossover and single‐strand annealing on inverted and direct repeat recombination in yeast: different requirements for the RAD1, RAD10, and RAD52 genes. Genetics 139: 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A (2005) Partial depletion of histone H4 increases homologous recombination‐mediated genetic instability. Mol Cell Biol 25: 1526–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F (2014) Homologous recombination maintenance of genome integrity during DNA damage tolerance. Mol Cell Oncol 1: e957039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F (2018) Homologous recombination: to fork and beyond. Genes (Basel) 9: 603–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash L (1981) Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol Gen Genet 184: 471–478 [DOI] [PubMed] [Google Scholar]
- Prakash L, Higgins D (1982) Role of DNA repair in ethyl methanesulfonate‐induced mutagenesis in Saccharomyces cerevisiae . Carcinogenesis 3: 439–444 [DOI] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, McGill CB, Strathern JN (2002) The roles of REV3 and RAD57 in double‐strand‐break‐repair‐induced mutagenesis of Saccharomyces cerevisiae . Genetics 162: 1063–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche H, Gietzt RD, Kunz BA (1995) Specificities of the Saccharomyces cerevisiae rad6, radl8, and rad52 Mutators exhibit different degrees of dependence on the REV3 gene product, a putative nonessential DNA polymerase. Genetics 140: 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE (2013) Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harbor Perspectives in Biology 5: a012708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Prakash S, Prakash L (1990) The SRS2 suppressor of rad6 mutations of Saccharomyces cerevisiae acts by channeling DNA lesions into the RAD52 DNA repair pathway. Genetics 124: 817–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Durussel T, Laemmli UK (2004) ChIC and ChEC. Mol Cell 16: 147–157 [DOI] [PubMed] [Google Scholar]
- Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn J‐Y, Glover TW, Canman CE (2012) REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res 40: 682–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter P, Ulrich HD (2003) Control of spontaneous and damage‐induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425: 188–191 [DOI] [PubMed] [Google Scholar]
- Sugawara N, Wang X, Haber JE (2003) In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51‐mediated recombination. Mol Cell 12: 209–219 [DOI] [PubMed] [Google Scholar]
- Torres‐Ramos CA, Prakash S, Prakash L (2002) Requirement of RAD5 and MMS2 for postreplication repair of UV‐damaged DNA in Saccharomyces cerevisiae . Mol Cell Biol 22: 2419–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD, Jentsch S (2000) Two RING finger proteins mediate cooperation between ubiquitin‐conjugating enzymes in DNA repair. EMBO J 19: 3388–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D (2010) Replication and recombination factors contributing to recombination‐dependent bypass of DNA lesions by template switch. PLoS Genet 6: e1001205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Walker GC (2006) The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci USA 103: 8971–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RP, García Rodríguez N, Zilio N, Hanulová M, Ulrich HD (2020) Processing of DNA polymerase‐blocking lesions during genome replication is spatially and temporally segregated from replication forks. Mol Cell 77: 3–16.e4 [DOI] [PubMed] [Google Scholar]
- Wood A, Krogan NJ, Dover J, Schneider J, Heidt J, Boateng MA, Dean K, Golshani A, Zhang Y, Greenblatt JF et al (2003) Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell 11: 267–274 [DOI] [PubMed] [Google Scholar]
- Zgaga Z (1991) Transformation of Saccharomyces cerevisiae with UV‐irradiated single‐stranded plasmid. Mutat. Res. 263: 211–215 [DOI] [PubMed] [Google Scholar]
- Zhao L, Washington M (2017) Translesion synthesis: insights into the selection and switching of DNA polymerases. Genes (Basel) 8: 24–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Data Availability Statement
No datasets have been produced. Data are uploaded as source data or available upon request.