

Abstract
Aim:
To investigate the role of epigenetics in HIV pathophysiology.
Materials & methods:
We conducted an epigenome-wide association scan on HIV infection status among people who inject drugs in the AIDS Linked to the IntraVenous Experience study with primary (n = 397) and validation samples (n = 390). DNA methylation from blood was measured by the Illumina EPIC BeadChip. We controlled for cell type heterogeneity by HIV status.
Results:
HIV infection status was associated (p < 10-8) with DNA methylation at 49 CpG sites. Sites were enriched in response to virus, interferon signaling pathway, etc. Among these sites, discovery and validation t-statistics were highly correlated (r = 0.96).
Conclusion:
In a cohort of people who inject drugs, HIV status was associated with differential DNA methylation at biologically meaningful sites.
Keywords: : DNA methylation, epigenetics, epigenome-wide association, HIV, people who inject drugs
HIV infection induces epigenetic changes, especially DNA methylation changes, in the immune system. Acute HIV infection in CD4+ and CD8+ cell lines has shown global methylation changes in vitro [1]. There is also evidence that HIV latency is regulated by DNA methylation and histone modifications mediated by PRC2 [2–4]. DNA methylation levels in viral promoters, such as the HIV long terminal repeat region, affect viral gene expression [3]. Epigenetic analyses have the potential to provide insights into biological mechanisms behind chronic HIV infection and help identify potential drug targets to remove latent viral reservoirs [5].
Recent studies have shown that there are DNA methylation (DNAm) signals associated with HIV infection in the NLRC5 and HLA genes [6–9]. The regions near the top CpG signals in the associated gene region in these studies have also presented as HIV integration sites [10]. However, previous studies using the 450K array (Illumina, CA, USA) that lacked adequate coverage of the epigenome and did not fully account for cell type heterogeneity between HIV+ and HIV- samples. Characterization of the epigenome with better genome-wide coverage of CpG sites and with careful adjustment for cell type heterogeneity between HIV+ and HIV- samples is needed.
People who inject drugs (PWID) have much higher risk of HIV infection [11], and studies have shown that injection drug use (IDU) also altered DNA methylation profiles in blood [12]. It is possible that DNA methylation changes induced by HIV infection are different between those with recent active use of injection drugs and those with no recent active use of injection drugs. Herein, we perform an epigenome-wide association analysis to examine methylation differences between HIV+ and HIV- subjects in a prospective cohort of PWID. This study also examined how recent active IDU affected the HIV-associated epigenome.
Materials & methods
Study samples
The AIDS Linked to the IntraVenous Experience study is an ongoing prospective cohort study characterizing the incidence and natural history of HIV infection among PWID in MD, USA from 1988 [13]. At each 6-month visit, clinical, behavioral and laboratory data including HIV-infection status and IDU were assessed. We analyzed 1092 unique blood samples selected from 415 unique study participants. Sample selection focused on participants who reported a change in their prior 6-month IDU status; most participants had blood samples tested after self-report of active IDU and after at least 6 months of IDU abstinence. This study has been approved by the Johns Hopkins University Institutional Review Board, and all participants provided written informed consent.
All participants had HIV serology screening at baseline. HIV serology status was assessed at each study visit for HIV- participants, whereas CD4+, CD8+ count and HIV viral load testing were performed at each study visit for HIV+ participants. There was no change in HIV status across visits in the sample selected for this analysis. Behavioral and clinical variables were self-reported and reflected exposure or activity in the prior 6 months and were obtained by interviewer or computer-administered standardized questionnaires. IDU status and type of illicit drugs used, current smoking status and antiretroviral therapy (ART) use were key covariates that could influence methylation and were incorporated into the analyses.
For the primary analyses, we randomly selected one visit per subject during a period of at least 6 months of abstinence from any IDU, yielding a total sample size of 397 subjects including 127 subjects who were HIV+ and 270 subjects who were HIV- (Table 1). For validation, we also randomly selected one visit per subject during a period of recent IDU, yielding a validation sample set of 390 subjects (386 subjects overlap with the primary analyses), with 122 HIV+ subjects and 268 HIV- subjects (Table 1).
Table 1. . AIDS Linked to the IntraVenous Experience study sample characteristics.
| Characteristic | Non injection use observations | Injection use observations |
|---|---|---|
| n | 397 | 390 |
| Mean age (SE) | 49.3 (7.5) | 48.3 (7.6) |
| Sex: | ||
| Males (%) | 271 (68.3%) | 267 (68.5%) |
| Females (%) | 126 (31.7%) | 123 (31.5%) |
| Race: | ||
| Caucasian (%) | 40 (10.1%) | 40 (10.3%) |
| African–American (%) | 357 (89.9%) | 350 (89.7%) |
| HIV status: | ||
| Positive (%) | 127 (32.0%) | 122 (31.3%) |
| CD4 (median, IQR) | 300 (226) | 258 (274) |
| Antiretroviral therapy | 92 (72.4%) | 66 (54.1%) |
| Negative (%) | 270 (68.0%) | 268 (68.7%) |
| Injection Drug type: | ||
| Heroin only (%) | N/A | 112 (28.7%) |
| Cocaine only (%) | N/A | 18 (4.6%) |
| Both drugs (%) | N/A | 179 (45.9%) |
IQR: Interquartile range; SE: Standard error.
DNA methylation measurement & preprocessing
DNA was isolated from buffy coat white blood cells using the DNeasy kit (Qiagen, Hilden, Germany) and bisulfite converted with Zymo EZ methylation gold kit at the Johns Hopkins University Center for Inherited Disease Research (MD, USA). Bisulfite treated DNA was run on the Illumina Infinium MethylationEPIC BeadChip.
The minfi package (Bioconductor, WA, USA) was used to process raw Illumina image files into noob preprocessed methylation beta values [14]. Cell composition of CD4+, CD8+, natural killer cells, monocytes, granulocytes and B cells were estimated based on the method described by Houseman et al. [15] and implemented in minfi [14,16]. The pipeline of preprocessing is shown in Supplementary Figure 1. Samples with low intensity (suggesting low quality, n = 0), inconsistency on predicted and observed sex (suggesting a sample swap, n = 1) and outliers in estimated cell composition (suggesting heterogeneous blood collection, n = 10) were removed for quality control. Probes with low intensity or that are known to cross-hybridize were excluded (n = 1290). Probes containing single nucleotide polymorphisms (SNPs) with minor allele frequency >0.05 were flagged. A total of 1081 samples and 864,801 probes were available for epigenome-wide association study (EWAS) analyses after quality control. Batch effects were adjusted for using the top four principal components (PCs) from negative control features that were only correlated with technical variation [17,18]. Ancestry PCs were estimated from the 59 SNP probes profiled on the EPIC array.
Accounting for CD4+/CD8+ ratio by HIV infection status
Since HIV infection is known to be associated with changes in cell population, a key covariate in peripheral blood methylation studies, we adjusted for cellular heterogeneity using both the Houseman approach [15] and an approach for specific disruptions of CD4+ and CD8+ populations. Since HIV infection leads to decreased CD4+ count and increased CD8+ count, DNAm marks that are negatively associated with CD4+ cell type and positively associated with CD8+ cell type would overshadow the true biological signal when conducting EWAS on HIV status. To fully adjust for this confounding, we ran a t-test to find the top 1000 probes that were negatively associated with CD4+ cell type and positively associated with CD8+ cell type based on the data in R package FlowSorted.Blood.450 k [16], originally from Reinius et al. [19].
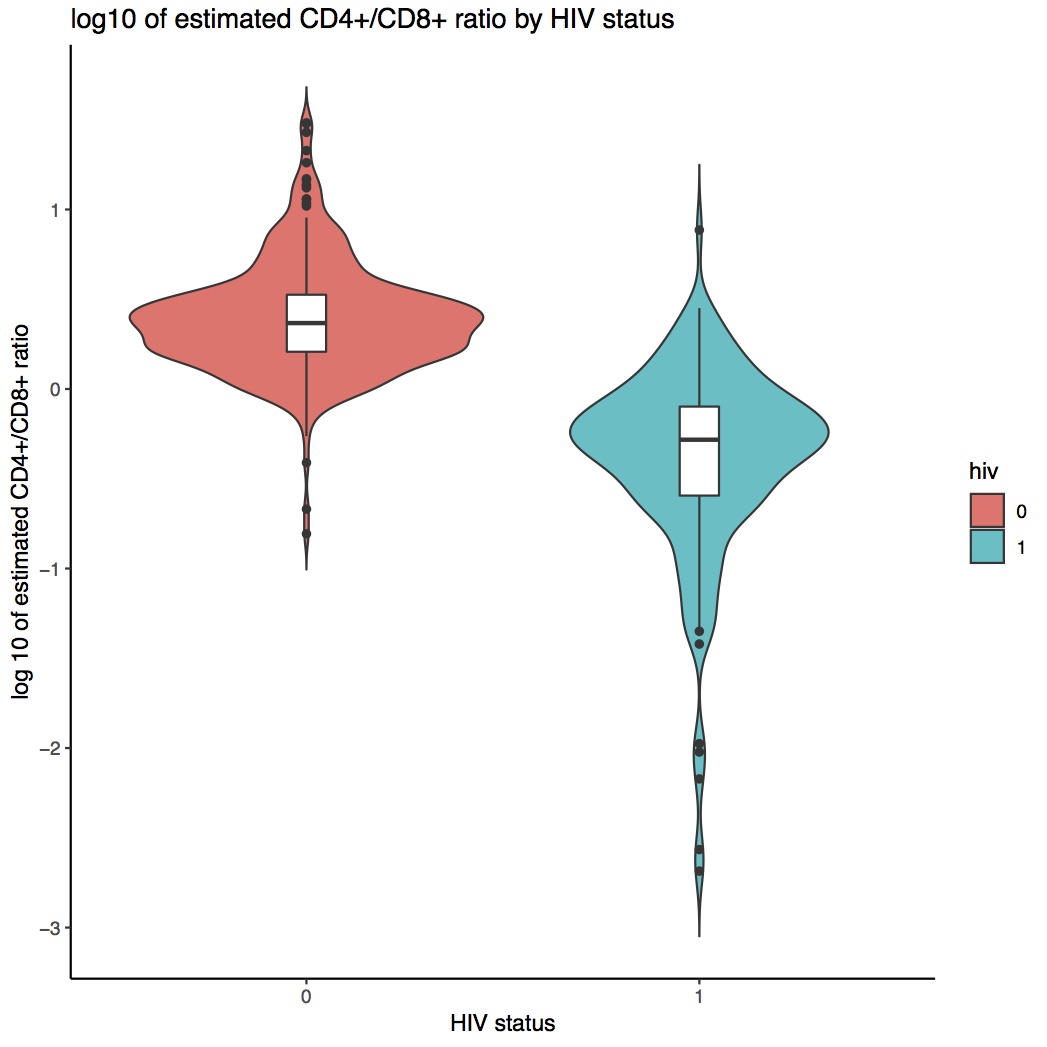
There were significant differences in the DNAm estimated CD4+/CD8+ ratio by HIV status (Supplementary Figure 2). HIV primarily infects CD4+ cells, which decreases the relative proportion of CD4+ cells and increases the proportion of CD8+ cells. These changes were reflected in the estimated log10 CD4+/CD8+ ratio in Supplementary Figure 2.
We measured CD4+ and CD8+ counts and proportions for 173 HIV+ subjects and 37 HIV- subjects, and compared estimated and measured log10 CD4+/CD8+ ratio in Supplementary Figure 2. Overall, the estimated log10 CD4+/CD8+ ratio was similar to the measured ratio with the mean difference of -0.032 among HIV+ and 0.084 among HIV-. However, there was a significant difference in HIV status between the mean difference of estimated and measured log CD4+/CD8+ ratio (p = 0.022). This indicates that the cell proportion estimation procedure may underestimate CD4+ or overestimate CD8+ among HIV+ subjects (since the sorted reference profiles used in the deconvolution algorithm were from healthy donors). Thus, we used six PCs that were reflective of CD4+/CD8+ ratio estimated in public data [16] to eliminate any additional cell type effect on our HIV EWAS analysis.
Single site association analyses
DNAm M-values were used in the analysis since the M-value distribution is closer to the assumption of normality. Researchers have reported that the use of M-value usually leads to better detection rate and true positive rate compared with the DNAm beta value [20].
We used the R package limma to run the single site association analyses on HIV infection status across the epigenome for the 864,801 CpG sites, adjusting for sex, age, smoking status, cell composition, six ancestry PCs based on 59 SNPs genotyped on the EPIC array, six negative control PCs and six PCs representing CD8+/CD4+ ratio.
The model is stated below:
We also conducted a sensitivity analysis restricting our HIV+ samples to those who are under ART and assessing whether treatment status affects our EWAS results. We compared the t-statistics between sensitivity analysis and primary analysis and computed the Pearson correlation coefficient.
Differentially methylated regions by HIV infection
We used bumphunter [21] to identify differentially methylated regions by HIV infection. Regions that contained at least two CpG sites were reported. We used Bonferroni correction to adjust for multiple comparison.
Gene ontology enrichment analysis
Gene ontology enrichment analysis was performed including probes with EWAS p-value <10-5 in the GO (Gene Ontology) database [22] in the R package missMethyl, with prior correction for sampling bias [23,24]. The significantly enriched pathway was presented as a hierarchy tree structure using ShinyGO [25].
Results
Primary analysis: EWAS on HIV infection status among abstinence visits
To avoid the potential influence of recent IDU on the epigenome, we first conducted EWAS among 397 participant visits with no active injection in the prior 6 months. Among 397 subjects, we selected one ‘no recent IDU’ observation per subject. Study participants were a mean age of 49.3 years, 68.3% male, 89.9% African–American and 32% HIV+ (Table 1).
The QQ-plot among abstinence visits (Figure 1) did not show apparent inflation (λ = 1.09) after we accounted for all covariates including PCs for CpGs that were negatively associated with CD4+ and positively associated with CD8+. There were clusters of epigenome-wide significant CpG sites in chromosome 1, 2, 6, 12 and 21 as shown in the Manhattan plot (Figure 2).
Figure 1. . Quantile–quantile plot of HIV epigenome-wide association analysis among non injection visits (inflation factor = 1.090).

Figure 2. . Manhattan plot of HIV epigenome-wide association analysis among non injection visits.
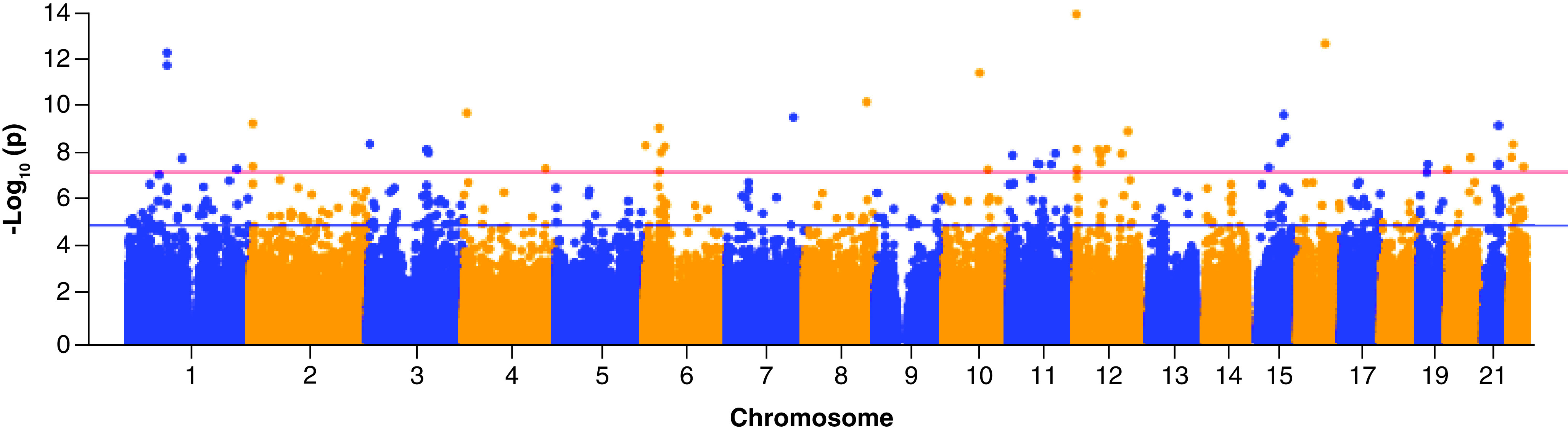
Red line indicates epigenome-wide significance threshold, while blue line indicates suggestive threshold.
After Bonferroni correction, 49 epigenome-wide significant CpG sites were associated with HIV infection status (Bonferroni p < 0.05, Table 2). Notably, cg07839457 (p = 1.53 × 10-13) in the promoter region of the NLRC5 gene was among the top ranked CpG sites. Two significant CpG sites (cg13452062, cg05696877) are located in the IFI44L gene (antiviral response), and another two significant CpG sites (cg21549285, cg17986793) are located in the MX1 gene (induced by interferon).
Table 2. . Epigenome-wide significant CpG sites among non-injection observations.
| CpG | Chr | Position | Nearest gene | Position to gene | logFC | Average M value | t | p-value | Bonferroni p-value |
|---|---|---|---|---|---|---|---|---|---|
| cg12051710 | 12 | 2989070 | C12orf32 | Body | -0.50 | 3.07 | -8.10 | 8.19E-15 | 7.08E-09 |
| cg07839457† | 16 | 57023022 | NLRC5 | TSS1500 | -1.06 | -1.13 | -7.67 | 1.53E-13 | 1.32E-07 |
| cg13452062† | 1 | 79088559 | IFI44L | 5′UTR | -2.20 | 1.33 | -7.54 | 3.78E-13 | 3.27E-07 |
| cg05696877† | 1 | 79088769 | IFI44L | 5′UTR | -1.75 | 0.41 | -7.35 | 1.26E-12 | 1.09E-06 |
| cg09801824† | 10 | 74076930 | 0.29 | -2.23 | 7.23 | 2.74E-12 | 2.37E-06 | ||
| cg12688661 | 8 | 128975417 | PVT1 | Body | -0.33 | 2.55 | -6.78 | 4.82E-11 | 4.17E-05 |
| cg09921385 | 4 | 7000882 | TBC1D14 | Body | -0.23 | 0.15 | -6.60 | 1.44E-10 | 1.24E-04 |
| cg01538263 | 15 | 74014910 | 0.16 | 1.24 | 6.57 | 1.73E-10 | 1.49E-04 | ||
| cg06756551 | 7 | 137352607 | DGKI | Body | 0.20 | 1.37 | 6.53 | 2.21E-10 | 1.91E-04 |
| cg10771443† | 2 | 7018855 | RSAD2 | Body | -0.64 | -1.63 | -6.42 | 4.11E-10 | 3.55E-04 |
| cg21549285† | 21 | 42799141 | MX1 | 5′UTR | -1.02 | 0.99 | -6.39 | 4.96E-10 | 4.29E-04 |
| cg17633074 | 6 | 30878864 | Body | 0.20 | 1.11 | 6.35 | 6.36E-10 | 5.50E-04 | |
| cg08818049 | 12 | 109085884 | CORO1C | Body | 0.31 | -2.01 | 6.29 | 8.82E-10 | 7.63E-04 |
| cg23387863† | 15 | 77472416 | Body | -0.32 | 1.33 | -6.19 | 1.58E-09 | 1.36E-03 | |
| cg23089225 | 15 | 67440902 | SMAD3 | 5′UTR; Body | 0.21 | -1.56 | 6.09 | 2.78E-09 | 2.41E-03 |
| cg12832157 | 3 | 4885491 | ITPR1 | Body | -0.27 | 2.96 | -6.07 | 3.09E-09 | 2.67E-03 |
| cg22764925 | 22 | 24979964 | GGT1 | 5′UTR | -0.41 | 1.12 | -6.07 | 3.19E-09 | 2.76E-03 |
| cg23473907 | 6 | 3523483 | 0.16 | 0.68 | 6.05 | 3.58E-09 | 3.10E-03 | ||
| cg21670513 | 6 | 43028611 | KLC4 | 5′UTR; TSS1500; Body | 0.29 | 2.81 | 6.03 | 3.97E-09 | 3.43E-03 |
| cg18544413† | 12 | 65019442 | RASSF3 | Body | 0.39 | -3.45 | 5.98 | 5.17E-09 | 4.47E-03 |
| cg18387107 | 12 | 3980774 | PARP11 | 5′UTR; Body | -0.29 | 0.55 | -5.98 | 5.33E-09 | 4.61E-03 |
| cg00959259† | 3 | 122281975 | PARP9 | 5′UTR; TSS1500 | -0.84 | 0.03 | -5.97 | 5.43E-09 | 4.70E-03 |
| cg10140784 | 12 | 48531426 | Body | 0.18 | 2.02 | 5.97 | 5.50E-09 | 4.75E-03 | |
| cg18401376 | 6 | 35141132 | 0.18 | 1.27 | 5.93 | 7.05E-09 | 6.10E-03 | ||
| cg24695011 | 3 | 125781947 | SLC41A3 | Body; 5′UTR | 0.17 | 1.37 | 5.92 | 7.24E-09 | 6.26E-03 |
| cg06798591 | 11 | 94556390 | AMOTL1 | Body | 0.14 | 0.84 | 5.90 | 8.02E-09 | 6.93E-03 |
| cg25127026 | 12 | 96606023 | ELK3 | 5′UTR | 0.39 | -2.91 | 5.90 | 8.18E-09 | 7.07E-03 |
| cg13344385 | 12 | 52642852 | AC021066.1 | 0.15 | 1.17 | 5.88 | 9.04E-09 | 7.81E-03 | |
| cg21139291 | 11 | 6425839 | APBB1 | 5′UTR; 1stExon; Body | 0.14 | 0.05 | 5.87 | 9.44E-09 | 8.16E-03 |
| cg23469117 | 22 | 21985646 | YDJC | TSS1500 | 0.16 | 1.18 | 5.84 | 1.15E-08 | 9.93E-03 |
| cg14030069 | 20 | 48037535 | Body | 0.17 | 1.07 | 5.83 | 1.21E-08 | 1.05E-02 | |
| cg20474403 | 1 | 110599939 | RP4-773N10.4 | 0.20 | 2.25 | 5.82 | 1.27E-08 | 1.10E-02 | |
| cg22485390‡ | 12 | 53089322 | RP11-641A6.3 | Body | 0.23 | 2.47 | 5.74 | 1.93E-08 | 1.67E-02 |
| cg10129948 | 11 | 57085849 | Body | 0.16 | 1.92 | 5.73 | 2.09E-08 | 1.81E-02 | |
| cg22862003†‡ | 21 | 42797588 | MX1 | TSS1500; 5′UTR | -0.56 | 0.54 | -5.73 | 2.13E-08 | 1.84E-02 |
| cg14193087 | 19 | 18614252 | Body | 0.19 | -1.96 | 5.72 | 2.25E-08 | 1.94E-02 | |
| cg11504739 | 11 | 62186060 | SCGB1A1 | TSS1500 | 0.22 | 2.15 | 5.72 | 2.25E-08 | 1.95E-02 |
| cg09381237 | 11 | 86178666 | ME3 | Body | 0.25 | 2.24 | 5.71 | 2.27E-08 | 1.97E-02 |
| cg17986793 | 21 | 42797899 | MX1 | TSS200; 5′UTR | -0.49 | -2.61 | -5.69 | 2.56E-08 | 2.21E-02 |
| cg15839328‡ | 2 | 7018885 | RSAD2 | Body | -0.60 | -1.56 | -5.68 | 2.79E-08 | 2.41E-02 |
| cg03834031 | 22 | 46465717 | RP6-109B7.4 | -0.19 | -1.79 | -5.67 | 2.90E-08 | 2.51E-02 | |
| cg16985233 | 15 | 43806249 | -0.21 | 3.93 | -5.66 | 3.11E-08 | 2.69E-02 | ||
| cg05883128‡ | 4 | 169239131 | DDX60 | 5′UTR | -0.36 | -0.38 | -5.64 | 3.44E-08 | 2.97E-02 |
| cg23389199 | 1 | 223205670 | 0.16 | 1.60 | 5.62 | 3.69E-08 | 3.19E-02 | ||
| cg17130481 | 20 | 853509 | ANGPT4 | 3′UTR | 0.12 | 0.58 | 5.62 | 3.84E-08 | 3.32E-02 |
| cg05552874 | 10 | 91153143 | Body | -0.53 | 0.83 | -5.62 | 3.85E-08 | 3.33E-02 | |
| cg05293861 | 12 | 4262708 | -0.36 | -4.68 | -5.62 | 3.85E-08 | 3.33E-02 | ||
| cg21222743† | 6 | 31543545 | TNF | 1st Exon | 0.43 | -3.26 | 5.58 | 4.55E-08 | 3.93E-02 |
| cg07613362 | 19 | 16462176 | -0.19 | 1.54 | -5.56 | 5.09E-08 | 4.40E-02 |
Also significant in injection visit EWAS.
Probe that contains a common SNP.
EWAS: Epigenome-wide association study; SNP: Single-nucleotide polymorphism.
We also conducted a sensitivity analysis restricting to the comparison of the epigenome profiles between 92 HIV+ participants who were on ART and 270 HIV- participants (Supplementary Table 1). Among the identified 49 CpG sites, the correlation of t-statistics between the primary analysis and sensitivity analysis is 0.992.
Differentially methylated regions of HIV infection
We identified three differentially methylated regions in IFI44L, HKR1 and PARP9 after Bonferroni correction (Table 4). The differentially methylated regions result is generally consistent with our single-site analysis. Two significant CpG sites are located near IFI44L and one significant CpG site is near PARP9 (Table 2).
Table 4. . Differentially methylated regions by HIV infection status.
| Chromosome | Start | End | Gene | Number of CpG | p-value | Bonferroni p-value |
|---|---|---|---|---|---|---|
| 1 | 79088559 | 79091177 | IFI44L | 3 | <1E-05 | <0.005 |
| 19 | 37825009 | 37826008 | HKR1 | 12 | 6.77E-05 | 2.86E-02 |
| 3 | 122281881 | 122282157 | PARP9 | 4 | 8.05E-05 | 3.40E-02 |
Gene ontology enrichment analysis
To identify the biological pathways associated with the top hits from our EWAS analysis, we conducted gene ontology enrichment analysis among probes with p < 10-5. The top enriched pathways were highly related to HIV infection (Figure 3). Three significant pathways were type I interferon response pathways (p1 = 5.7 × 10-7, p2 = 5.7 × 10-7, p3 = 6.3 × 10-7). A cluster of six enriched pathways were viral life cycle-related pathways with p values ranging from 3.8 × 10-5 to 6.1 × 10-4 (Figure 3). Other enriched pathways were major immune response pathways including defense response to virus and responses to cytokines, external biotic stimulus and to other organisms (Figure 3). These results showed that the EWAS analysis identified DNAm marks located in biologically meaningful genes involved in HIV pathophysiology.
Figure 3. . Gene ontology analysis for CpG sites with p-value <10-5.

p-values are shown on the figure, and the hierarchy tree structure is based on ShinyGO.
Validation: EWAS on HIV among injection visits
To test for validation of our previous analysis, we selected another study visit where essentially the same study participants (386 of the 390 included in the primary analysis) were sampled when self-reporting actively injecting drugs in the prior 6 months. The sample characteristics of the validation set were highly comparable to the primary analysis (Table 1). Almost all participants reported injecting heroin (95%), either with (46%) or without cocaine (29%); only 5% of participants reported only injecting cocaine.
In the validation EWAS, there were 28 epigenome-wide significant CpG sites after Bonferroni correction (Bonferroni p < 0.05, Table 3). 11 CpG sites were significant among EWAS in both the abstinence and the injection visits (Tables 2 & 3). cg07839457 (p = 6.88 × 10-12) in the NLRC5 gene, cg13452062 (p = 1.69 × 10-12) and cg05696877 (p = 8.02 × 10-13) in the IFI44L gene were among the top epigenome-wide significant signals in both the primary and the validation analyses. In addition, CpG sites in genes associated with viral and immune response pathways were also replicated – two CpG sites in the MX1 gene (cg21549285, p = 7.39 × 10-10; cg22862003, p = 3.66 × 10-8), one CpG site in the PARP9 gene (cg00959259, p = 3.12 × 10-8), one CpG site in the RSAD2 gene (cg10771443, p = 5.74 × 10-9) and one CpG site in the TNF gene (cg21222743, p = 6.75 × 10-9).
Table 3. . Epigenome-wide significant CpG sites among injection observations.
| CpG | Chr | Position | Nearest gene | Position to gene | logFC | Average M value | t | p-value | Bonferroni p-value |
|---|---|---|---|---|---|---|---|---|---|
| cg05696877† | 1 | 79088769 | IFI44L | 5′UTR | -1.83 | 0.36 | -7.43 | 8.02E-13 | 6.93E-07 |
| cg13452062† | 1 | 79088559 | IFI44L | 5′UTR | -2.26 | 1.17 | -7.31 | 1.69E-12 | 1.46E-06 |
| cg07839457† | 16 | 57023022 | NLRC5 | TSS1500 | -1.02 | -1.20 | -7.09 | 6.88E-12 | 5.95E-06 |
| cg13857933 | 1 | 208046539 | 0.41 | -3.97 | 6.80 | 4.28E-11 | 3.71E-05 | ||
| cg10717214 | 6 | 31543557 | TNF | 1stExon | 0.41 | -2.52 | 6.63 | 1.20E-10 | 1.04E-04 |
| cg23387863† | 15 | 77472416 | Body | -0.32 | 1.33 | -6.59 | 1.58E-10 | 1.37E-04 | |
| cg16998839 | 9 | 134531983 | RAPGEF1 | Body | 0.31 | -2.47 | 6.47 | 3.17E-10 | 2.74E-04 |
| cg09801824† | 10 | 74076930 | 0.27 | -2.19 | 6.38 | 5.36E-10 | 4.64E-04 | ||
| cg03074874 | 6 | 47172552 | 0.19 | 1.07 | 6.36 | 6.23E-10 | 5.38E-04 | ||
| cg21549285† | 21 | 42799141 | MX1 | 5′UTR | -1.16 | 0.88 | -6.33 | 7.39E-10 | 6.39E-04 |
| cg22012079 | 1 | 79091177 | IFI44L | 5′UTR | -0.51 | 0.29 | -6.33 | 7.42E-10 | 6.42E-04 |
| cg06716655 | 1 | 154579727 | ADAR | TSS1500; Body; 5′UTR | -0.80 | -2.71 | -6.19 | 1.65E-09 | 1.43E-03 |
| cg00574958 | 11 | 68607622 | CPT1A | 5′UTR | 0.47 | -3.28 | 5.98 | 5.33E-09 | 4.61E-03 |
| cg10771443† | 2 | 7018855 | RSAD2 | Body | -0.66 | -1.64 | -5.97 | 5.74E-09 | 4.96E-03 |
| cg21222743† | 6 | 31543545 | TNF | 1stExon | 0.50 | -3.20 | 5.94 | 6.75E-09 | 5.84E-03 |
| cg08821232 | 8 | 134074665 | SLA | Body; 5′UTR | 0.24 | -1.88 | 5.90 | 8.42E-09 | 7.29E-03 |
| cg14129477 | 17 | 53422220 | -0.48 | 1.76 | -5.89 | 8.69E-09 | 7.51E-03 | ||
| cg19789466 | 12 | 113344923 | OAS1 | 1stExon | -0.42 | -4.10 | -5.78 | 1.62E-08 | 1.40E-02 |
| cg04425624 | 6 | 31543565 | TNF | 1stExon | 0.41 | -1.69 | 5.76 | 1.82E-08 | 1.57E-02 |
| cg18544413† | 12 | 65019442 | RASSF3 | Body | 0.38 | -3.41 | 5.74 | 1.99E-08 | 1.72E-02 |
| cg09246203 | 3 | 114018592 | Body | -0.31 | 2.42 | -5.71 | 2.29E-08 | 1.98E-02 | |
| cg00959259† | 3 | 122281975 | PARP9 | 5′UTR | -0.82 | -0.13 | -5.66 | 3.12E-08 | 2.70E-02 |
| cg07815522 | 3 | 122282157 | PARP9 | 5′UTR; TSS1500 | -0.73 | 0.61 | -5.66 | 3.15E-08 | 2.73E-02 |
| cg24319850 | 11 | 74975246 | ARRB1 | 3′UTR | 0.31 | -3.55 | 5.65 | 3.21E-08 | 2.78E-02 |
| cg22862003†‡ | 21 | 42797588 | MX1 | TSS1500; 5′UTR | -0.64 | 0.46 | -5.63 | 3.66E-08 | 3.16E-02 |
| cg03607951 | 1 | 79085586 | IFI44L | TSS1500 | -0.73 | -0.25 | -5.60 | 4.15E-08 | 3.58E-02 |
| cg13029130 | 2 | 68593246 | AC015969.3 | Body | 0.25 | -2.78 | 5.58 | 4.81E-08 | 4.16E-02 |
| cg10782071 | 17 | 75417278 | SEPTIN9 | Body | 0.34 | -3.10 | 5.55 | 5.59E-08 | 4.84E-02 |
Also significant in non-injection visit EWAS.
Probe that contains a common SNP.
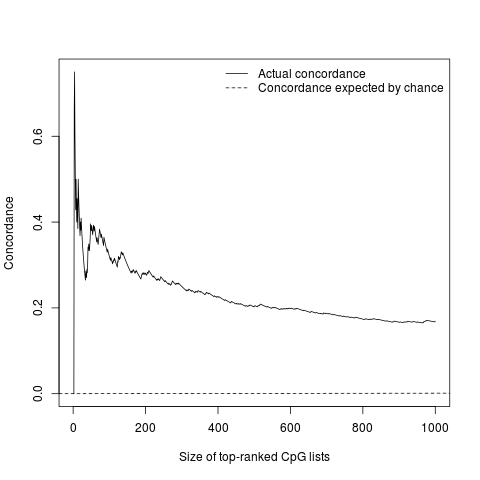
We compared the t-statistics from the 49 epigenome-wide significant CpG sites from the primary analysis and the validation analysis (Figure 4). The correlation of the top 49 CpGs’ t-statistics between two analyses was 0.962 (95% CI: 0.933–0.978). We also examined the concordance of the top ranked CpG sites between the two analyses by CATplot in Supplementary Figure 3, 47% of the top 15 ranked HIV associated CpG sites between primary and validation observations were the same, indicating a moderately high concordance across the two analyses.
Figure 4. . Comparison of top 49 sites' t-statistics between primary and validation analysis testing for an association between DNA methylation and HIV status.
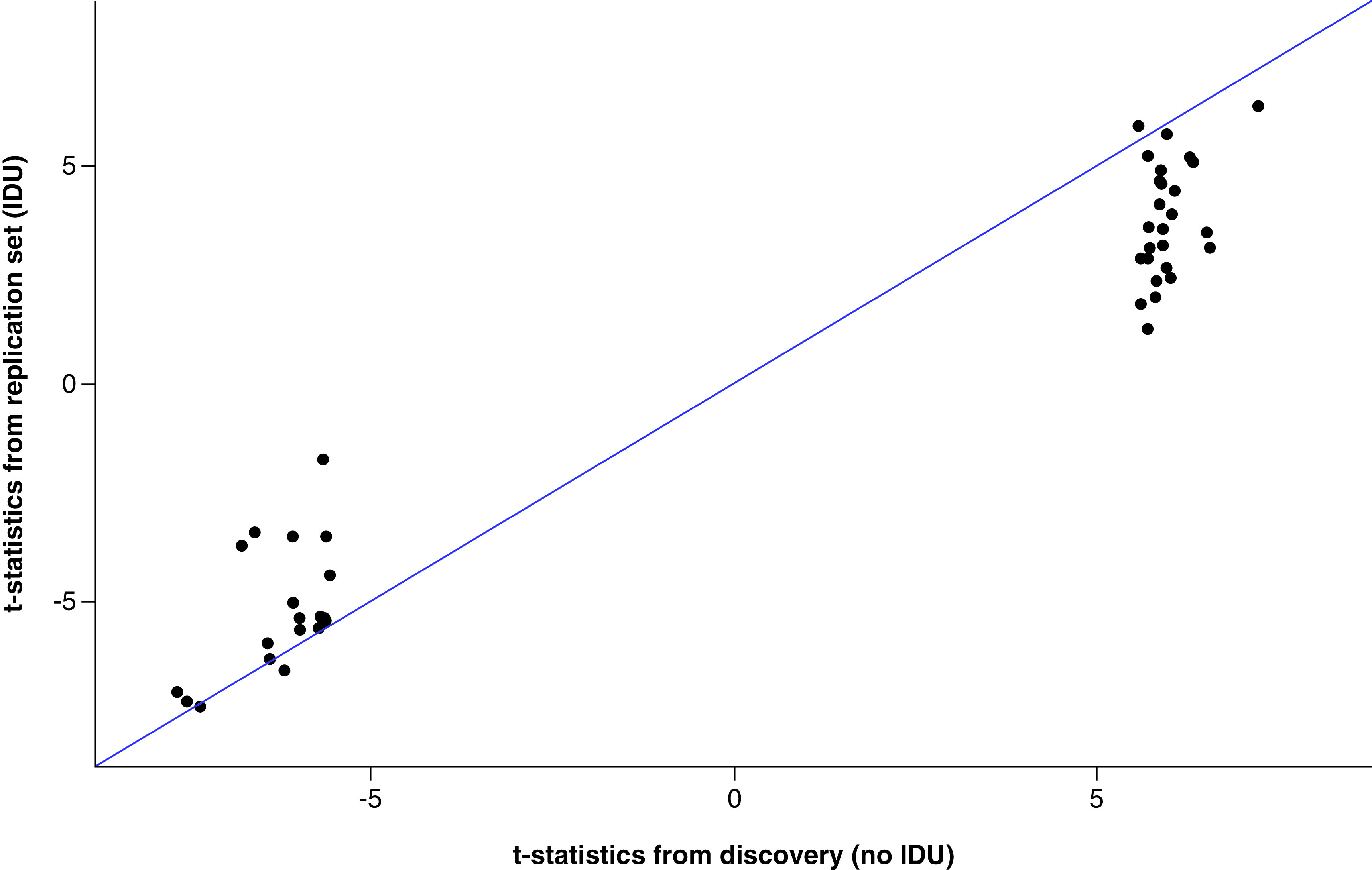
Dots falling on the blue line indicate that the t-statistics are exactly the same between the primary and the validation analysis.
IDU: Injection drug use.
Discussion
We performed an epigenome-wide scan on HIV infection in peripheral blood with rigorous adjustment for cellular composition tailored to HIV infection. We found 49 CpG sites associated with HIV infection in a cohort of PWID during periods of abstinence from drug injection. Validation EWAS during periods of drug injection showed good concordance with the primary analysis; 11 CpG sites were replicated at both time points. The top CpG sites identified were enriched in immune and viral response pathways biologically relevant to HIV pathogenesis.
The CpG site cg07839457 in the promoter region of the NRLC5 gene was the top ranked gene associated with HIV status in both primary and validation analyses. This gene has also been reported in other independent HIV EWAS studies [7–9]. Interestingly, the NRLC5 gene has been reported to be one of the persistent HIV integration sites in infected patients [10]. This change in DNA methylation therefore may be a result of either chronic infection by HIV virus, or HIV integration near this gene. Further observational and experimental studies on this gene are needed to shed light on its role in chronic HIV infection and downstream impacts on HIV clinical course.
Concordance between the primary and validation analysis was moderately good suggesting that active IDU status in the prior 6 months does not significantly alter the HIV-associated epigenome. The biologically relevant period of exposure to illicit drug use that affects the epigenome is unknown. As our cohort largely represents PWID with long-term injection careers, our findings may reflect that DNA methylation changes may not occur after only a 6-month period of abstinence. However, the t-statistics of validation analysis highly correlated with the primary analysis, but the t-statistics of validation analyses are generally less significant. Another potential explanation is that recent IDU may induce changes in epigenome and introduce noise to the HIV EWAS, leading to lower power.
There are several unique strengths of this study. First, to avoid confounding by differences in cell type composition due to HIV infection, we constructed latent variables positively associated with CD4+ cells and negatively associated with CD8+ cells. Second, we performed primary and validation analysis in the same participants during periods of abstinence and of active drug injection, respectively. Third, we used the Illumina Methylation EPIC assay that doubled the epigenome-wide coverage compared with Illumina Methylation 450K assay used in previous HIV EWAS.
Although we took extra measures to control for cell type heterogeneity by HIV status, our study may remain limited by the fact that chronic HIV infection is closely associated with the levels and proportion of CD4+ and CD8+ T cells and potential residual confounding may have occurred. However, inspection of QQ-plots did indicate an absence of genome-wide inflation in test statistics. Another limitation is study generalizability, as our analyses were performed in a sample of largely male and African–American PWID. Although this focused population is unlikely to negate any observed biological findings, validation is warranted in other populations.
Additional work is needed to explore how IDU modifies the effects of chronic HIV infection on DNA methylation profile in immune system genes. There was approximately 30% concordance between the EWAS results on non injection visits and injection visits, suggesting a potential interaction effect of IDU and chronic HIV infection on DNA methylation profiles. Previous literature has also reported potential epigenetic changes by IDU [12].
Other factors that may potentially affect the epigenome include HIV treatment status, HIV progression and HIV integration sites. Exposure to those medications, removal of circulating virus and associated inflammation can lead to potential epigenetic changes.
However, our sensitivity analysis showed that restricting our analysis to HIV+ participants with ART treatment did not substantively change our findings. We did not have enough samples to assess epigenome changes for participants without ART treatment in the primary analysis and will address this in future studies. It is also possible that there are epigenetic changes due to HIV disease progression and HIV integration sites. NRLC5 is an HIV integration site is of critical interest and may highlight a general mechanism by which the virus acts through the epigenome to impact disease course. This is an area that deserves additional attention. Currently, we do not have data to assess epigenetic changes with regard to HIV disease progression, or information on HIV integration sites for each HIV+ participant. Future studies incorporating HIV disease progression and HIV integration sites are warranted.
Overall, we identified several biologically relevant DNA methylation markers associated with HIV infection in a long-term cohort of PWID. Further biological studies on the identified epigenetics marks and genes are warranted.
Conclusion
In conclusion, we identified 49 CpG sites associated with HIV infection in a cohort of PWID. The top CpG sites were enriched in immune and viral response pathways that are biologically relevant to HIV pathogenesis. Of note, the top site in NLRC5 is a known site of HIV viral integration. An important hypothesis for future investigation is whether this is a common mechanism by which HIV infection causes chronic health conditions in people living with HIV infections.
Future perspective
Future studies should focus on the association of specific DNA methylation sites with adherence to antiretroviral therapies and with indicators of disease course, such as CD4+ count and HIV viral load. Such studies would highlight the biological pathways by which treatment succeeds or fails. Also, given the high frequency of IDU among people living with HIV, future work should also investigate the interaction effect of IDU, such as heroin and cocaine, with HIV serostatus on DNA methylation changes. At last, given our finding of a strong association between methylation at an HIV integration site and HIV status, future work should include a systematic look at whether HIV impacts methylation directly at known integration sites throughout the genome.
Summary points.
We identified 49 epigenome-wide significant CpG sites associated with HIV infection in a cohort of persons who inject drugs during periods of abstinence from drug injection.
These CpG sites are located near genes related with HIV pathophysiology and immune response such as NLRC5, IFI44L, PARP9 and MX1.
We replicated HIV-related CpG site in NLRC5 consistent with previous reports.
We also found three differentially methylated regions in IFI44L, HKR1 and PARP9.
Sensitivity analysis restricting to HIV+ participants with antiretroviral therapy showed great consistency in t-statistics between primary and sensitivity analysis (r = 0.992).
Validation epigenome-wide association showed that the correlation of t-statistics between the two analyses for the 49 CpG sites was 0.962 (95% CI: 0.933–0.978).
The identified CpG sites were enriched in immune and viral response pathways such as response to virus and interferon signaling pathway.
Future studies on the epigenetic changes by HIV treatment, HIV progression and HIV integration sites are warranted.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the participants and staff of the AIDS Linked to the IntraVenous Experience (ALIVE) cohort study.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/epi-2020-0123
Financial & competing interests disclosure
This work was supported by NIDA grants R01DA039408, R01DA047064, and U01DA036297 and NIAID K24-AI118591.
SH Mehta has the following disclosures: PI for research grants - funds paid to Johns Hopkins University: AbbVie, Assembly Bio, Gilead, Proteus Digital Health. Scientific advisor/Consultant: The terms of these arrangements are being managed by the Johns Hopkins University in accordance with its conflict of interest policies: AbbVie, Arbutus, Gilead. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
- 1.Maricato JT, Furtado MN, Takenaka MC. et al. Epigenetic modulations in activated cells early after HIV-1 infection and their possible functional consequences. PLoS ONE 10(4), e0119234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology Apr(454–455), 328–339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma M. Epigenetic regulation of HIV, AIDS, and AIDS-related malignancies. Methods Mol. Biol. 1238, 381–403 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Mbonye U, Karn J. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr. HIV Res. 9(8), 554–567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ay E, Banati F, Mezei M. et al. Epigenetics of HIV infection: promising research areas and implications for therapy. AIDS Rev. 15(3), 181–188 (2013). [PubMed] [Google Scholar]
- 6.Gross AM, Jaeger PA, Kreisberg JF. et al. Methylome-wide analysis of chronic HIV infection reveals five-year increase in biological age and epigenetic targeting of HLA. Mol. Cell 62(2), 157–168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Justice AC, Hu Y. et al. Epigenome-wide differential DNA methylation between HIV-infected and uninfected individuals. Epigenetics 11(10), 750–760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson KN, Hui Q, Rimland D. et al. Identification of HIV infection-related DNA methylation sites and advanced epigenetic aging in HIV-positive, treatment-naive US veterans. AIDS 31(4), 571–575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiau S, Strehlau R, Wang S. et al. Distinct epigenetic profiles in children with perinatally-acquired HIV on antiretroviral therapy. Sci. Rep. 9(1), 10495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 17(8), 1186–1194 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoenbaum EE, Hartel D, Selwyn PA. et al. Risk factors for human immunodeficiency virus infection in intravenous drug users. N. Engl. J. Med. 321(13), 874–879 (1989). [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Hu Y, Justice AC. et al. DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty. Nature Commun. 8(1), 2243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vlahov D, Anthony JC, Munoz A. et al. The ALIVE study, a longitudinal study of HIV-1 infection in intravenous drug users: description of methods and characteristics of participants. NIDA Res. Monogr. 109, 75–100 (1991). [PubMed] [Google Scholar]
- 14.Aryee MJ, Jaffe AE, Corrada-Bravo H. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30(10), 1363–1369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houseman EA, Accomando WP, Koestler DC. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13(1), 86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 15(2), R31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gagnon-Bartsch JA, Speed TP. Using control genes to correct for unwanted variation in microarray data. Biostatistics 13(3), 539–552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fortin J-P, Labbe A, Lemire M. et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15(11), 503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinius LE, Acevedo N, Joerink M. et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE 7(7), e41361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du P, Zhang X, Huang C-C. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11(1), 587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaffe AE, Murakami P, Lee H. et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 41(1), 200–209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashburner M, Ball CA, Blake JA. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25(1), 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics 29(15), 1851–1857 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics 32(2), 286–288 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36(8), 2628–2629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.