

Summary
The MRE11-RAD50-NBS1 (MRN) complex supports the synthesis of damage-induced long non-coding RNA (dilncRNA) by RNA polymerase II (RNAPII) from DNA double-strand breaks (DSBs) by an unknown mechanism. Here, we show that recombinant human MRN and native RNAPII are sufficient to reconstitute a minimal functional transcriptional apparatus at DSBs. MRN recruits and stabilizes RNAPII at DSBs. Unexpectedly, transcription is promoted independently from MRN nuclease activities. Rather, transcription depends on the ability of MRN to melt DNA ends, as shown by the use of MRN mutants and specific allosteric inhibitors. Single-molecule FRET assays with wild-type and mutant MRN show a tight correlation between the ability to melt DNA ends and to promote transcription. The addition of RPA enhances MRN-mediated transcription, and unpaired DNA ends allow MRN-independent transcription by RNAPII. These results support a model in which MRN generates single-strand DNA ends that favor the initiation of transcription by RNAPII.
Keywords: DNA damage, DNA double-strand breaks, DNA-damage induced transcription, damage-induced long non-coding RNA, dilncRNA, MRE11-RAD50-NBS1 complex, RNA polymerase II, in vitro transcription, DNA melting, single-molecule FRET
Graphical Abstract
Highlights
-
•
Purified MRE11-RAD50-NBS1 and RNAPII suffice to reconstitute dilncRNA synthesis
-
•
dilncRNA synthesis by RNAPII depends on the ability of MRN to melt DNA ends
-
•
RPA enhances MRN-dependent dilncRNA synthesis from DNA ends
-
•
Unpaired DNA ends allow MRN-independent dilncRNA synthesis
Sharma et al. show that in an in vitro reconstituted system, the DNA damage-sensing complex MRE11-RAD50-NBS1 (MRN) and RNA polymerase II are sufficient to synthesize RNA transcripts from broken DNA ends. MRN supports transcription by melting DNA ends and allowing RNA polymerase II to initiate RNA synthesis.
Introduction
DNA double-strand breaks (DSBs) are cytotoxic lesions that trigger a DNA damage response (DDR) involving a signaling cascade, culminating with the enforcement of cell-cycle checkpoints and coordination of DNA repair activities (Polo and Jackson, 2011). The MRE11-RAD50-NBS1 (MRN) complex is a DNA damage sensor and primary activator of DDR apical kinase ataxia telangectasia (ATM). ATM phosphorylates the histone variant H2AX at ser139 (γH2AX), and this and additional modifications lead to the recruitment of DDR factors and the assembly of large multi-protein complexes at DNA lesions detectable as discrete DDR foci (Blackford and Jackson, 2017; D’Alessandro and d’Adda di Fagagna, 2017; Michelini et al., 2018). The recruitment of MRN to DSBs is also important for coordinating DNA-end resection, ATR activation, DNA replication, and activation of the Fanconi anemia pathway (Carvajal-Maldonado et al., 2019; Roques et al., 2009; Yu et al., 2012).
MRN is responsible for initiating and coordinating DSB repair by non-homologous DNA-end joining (NHEJ) and homologous recombination (HR) pathways (Paull and Deshpande, 2014). HR is initiated by 5′ DNA-end resection, which involves a first slow step catalyzed by MRN generating 3′ single-stranded (ss) DNA overhangs, followed by processive DNA-end resection catalyzed by EXO1 and/or DNA2 nucleases (Cejka, 2015; Symington, 2016). MRE11 is a 3′-5′ DNA exonuclease that could in principle produce 5′ overhangs in disagreement with DSB repair models and direct observation of resection polarity in vivo (Cejka, 2015; Symington, 2016). Phosphorylated CtBP-interacting protein (CtIP) stimulates the MRN complex to cleave the 5′-terminated strand endonucleolytically, favoring the generation of 3′-ssDNA tails (Anand et al., 2016; Cannavo and Cejka, 2014). ssDNA overhangs are then bound by replication protein A (RPA) and covered by RAD51, forming a nucleoprotein filament that invades a homologous DNA template for homology-directed repair (Kowalczykowski, 2015). Biochemical and structural studies suggest additional roles for MRN in DNA-end sensing and tethering (Lavin, 2007; Paull, 2018).
Importantly, the MRN complex also has the capacity to transiently separate individual DNA strands of a duplex, although its relevance for DSB repair remains to be fully understood (Cannon et al., 2013; Gobbini et al., 2018; Liu et al., 2016; Nicolette et al., 2010; Williams et al., 2008). Specifically, it is not clear whether dsDNA melting is a prerequisite for the clipping of blocked DNA ends by MRN-CtIP. The melting capacity may also function downstream of DNA clipping to remove the short ssDNA oligonucleotide from the 5′ end. DNA melting by MRN is stimulated by ATP binding and hydrolysis by RAD50, and this is important for MRN binding to DNA (Liu et al., 2016; (Lee and Paull, 2004); Paull, 2015). The DNA melting activity of yeast homolog MRX has been shown to enhance the resection activity of ExoI in Saccharomyces cerevisiae (Gobbini et al., 2018).
The suppression of preexisting canonical transcription near DSBs has been shown to be regulated by multiple mechanisms ((Shanbhag et al., 2010); Capozzo et al., 2017). Nevertheless, the generation of damage-induced long non-coding RNA (dilncRNA) at DSBs has been reported by several groups in Neurospora crassa, Arabidopsis thaliana, Drosophila melanogaster, and human cell lines (Francia et al., 2012; Gao et al., 2014; Michelini et al., 2017; Qi et al., 2016; Wang and Goldstein, 2016; Wei et al., 2012). dilncRNAs have been proposed as important modulators of DDR activation and DDR foci formation in plants and mammals (d’Adda di Fagagna, 2014; Michelini et al., 2018) both at genomic DSBs and dysfunctional telomeres (Aguado et al., 2019; Rossiello et al., 2017). Recently, using cellular assays, we demonstrated that productive RNA polymerase II (RNAPII) is recruited to DSBs in an MRN-dependent manner that triggers the synthesis of dilncRNA at exposed DNA ends (Michelini et al., 2017; Pessina et al., 2019). MRN is part of a complex with RNAPII, as determined by co-immunoprecipitation. MRN knockdown or chemical inhibition by small-molecule mirin in cells prevents RNAPII accumulation at DSB and dilncRNA accumulation, which suggests that the nuclease activities of MRN may be involved (Michelini et al., 2017).
To address the underlying mechanism of MRN function to promote RNAPII activity at DSBs, we report here a minimal reconstituted system that results in RNA synthesis from a DSB and which includes only purified recombinant human MRN and purified native RNAPII. These experiments thus demonstrate a direct functional interplay of MRN and RNAPII at DSBs. Unexpectedly, the involvement of the MRN complex in stimulating RNAPII does not involve its nuclease activity but rather its function to melt DNA ends. Overall, our results demonstrate a non-canonical function of MRN in promoting the initiation of transcription by RNAPII at DSBs.
Results
Purified Recombinant MRE11-RAD50-NBS1 (MRN) Together with Purified Native RNAPII Are Sufficient to Support Transcription from DNA Ends
DSBs are sites of RNAPII-dependent dilncRNA synthesis (Michelini et al., 2017; Sharma and d’Adda di Fagagna, 2019). MRN is necessary for such RNA synthesis; however, whether MRN alone is sufficient to support transcription by RNAPII from DNA ends is unknown. We reconstituted these events by using a minimal set of purified components: circular or linear dsDNA, purified native core RNAPII, and purified recombinant MRN (Figure 1A). The DNA used, in its circular or blunt-ended linear form, was pUC19, a 2.7-kb-long plasmid devoid of any promoter or enhancer DNA sequences regulating transcription in mammalian systems (Figure S1A). Native core (12 subunits complex) RNAPII was purified from calf thymus (Figure S1B) (Aygün et al., 2009; Somesh et al., 2005) and tested to be free of MRN and other transcription-supporting factors (Figures S1B and S1D). Several studies have shown the properties of core RNAP to initiate transcription from naked DNA templates (Dedrick and Chamberlin, 1985; Kadesch and Chamberlin, 1982; Lorch et al., 1987; Reines et al., 1997). In particular, it was shown that eukaryotic RNAPII has some activity on linear dsDNA (Kadesch and Chamberlin, 1982). Human recombinant MRN was expressed and purified from Sf9 insect cells (Anand et al., 2016) and was tested to be free from RNAPII (Figures S1C and S1D). The purity of recombinant MRN was tested by performing nuclease assays on 5′ and 3′ labeled DNA with increasing concentrations of MRN in the presence of Mg2+ and Mn2+ (Figures S1E and S1F). No exonuclease products were observed in the presence of Mg2+, while these exonuclease products were observed in the presence of Mn2+, in agreement with established biochemical activities of the MRN complex (Figures S1E and S1F) (Anand et al., 2016). Next, to reconstitute in vitro transcription, individual purified components, or transcriptionally competent cell extracts (CFEs) (Manley et al., 1980; Sharma et al., 2015) used as a positive control (Michelini et al., 2017), were incubated with either linear or circular dsDNA in a buffer containing ribonucleotides (rNTPs) and α-32P[UTP] at 30°C for 30 min (Figure 1A). We also generated an additional DNA substrate by adding homopolymeric dC-tails to the 3′ ends of the same linear DNA (Figures 1A and S1A), since such substrates support transcription by recruiting RNAPII to the tail-duplex junction (Kadesch and Chamberlin, 1982; Reines et al., 1997). At the end of the reaction, products were purified, resolved on a denaturing agarose gel, and imaged. Under these conditions, blunt-ended DNA incubated with CFEs generated discrete RNA products, while circular DNA showed few or no transcripts, confirming that DSBs promote the initiation of transcription (Figure 1B, lanes 1 and 2). Equal amounts of dC-tailed DNA incubated with CFE showed higher levels of transcription compared to linear DNA, consistent with the notion that 3′ extended DNA supports efficient transcription (Figure 1B, lane 3) (Kadesch and Chamberlin, 1982; Reines et al., 1997). Next, we tested the activity of purified MRN on a linear or circular DNA substrate in the absence of RNAPII and confirmed that MRN preparation was devoid of RNA synthesis activity (Figure 1B, lanes 4 and 5). RNAPII alone with circular or linear DNA templates showed weak background activity (Figure 1B, lanes 6 and 7). However, when equimolar amounts of MRN and RNAPII were incubated with linear dsDNA, we observed a strong induction of RNA transcripts (Figure 1B, lanes 9 and 10). The transcripts ranged up to the full length of the DNA template (Figure 1B, lane 9). The observation that MRN did not support RNA synthesis from circular DNA (Figure 1B, lane 8) indicates that MRN is not a general positive regulator of RNAPII activity and its ability to support transcription depends on the availability of DNA ends. As expected, DNA containing 3′-dC tails was transcribed efficiently by RNAPII (Figure 1B, lane 12). MRN addition did not further increase the strong RNAPII activity on dC-tailed DNA ends (Figure 1B, compare lanes 12 and 13), indicating that this DNA-end structure can bypass the need for MRN for RNAPII activity. Overall, these observations demonstrate that MRN, together with RNAPII, represent the minimal transcriptional machinery sufficient to efficiently initiate transcription from DNA ends.
Figure 1.

MRE11-RAD50-NBS1 (MRN) Is Sufficient to Support Transcription by RNAPII from DSB DNA Ends
(A) Schematic diagram depicting transcription assays, in which either circular or blunt-ended pUC19 DNA (0.1 pmol DNA ends) was incubated with MRN (0.5 pmol) and RNAPII (0.5 pmol) in a buffer containing rNTPs and α-32P[UTP] to study transcriptional events. RNA products were resolved on 2.5% formaldehyde-agarose gel.
(B) Transcription assays with circular, linear, and dC-tailed plasmid DNA with transcriptionally competent cell-free extracts (CFEs) (lanes 1–3) or purified MRN and RNAPII (lanes 4–13). Linear DNA was incubated with increasing concentrations of MRN (0.5–1.0 pmol) and RNAPII (lanes 9 and 10). m∗ indicates radioactive end-labeled single-stranded RNA ladder. MRN supports transcription from DNA ends by RNAPII. Bar plot shows the quantification of discrete band intensities of each lane in arbitrary units (a.u.). Error bars indicate means ± SEMs from 2 independent experiments.
(C) Transcription assays with MRN and RNAPII incubated with DNA DSBs containing different end configurations. m∗ indicates radioactive end-labeled single-stranded RNA ladder. All types of DNA ends can be transcribed by MRN and RNAPII. Bar plot shows quantification of discrete band intensities of each lane (a.u.). Error bars indicate means ± SEMs from 2 independent experiments.
(D) Schematic diagram depicting experimental design to study DNA:RNA hybrid formation. Transcription reactions with either purified proteins or CFEs were treated with RNase A or RNase H either at the beginning or at the end of the reaction.
(E and F) Transcription assays with CFEs or purified proteins incubated with RNase A or RNase H, during or after the reaction as indicated. m∗ indicates radioactive labeled single-stranded RNA ladder. Irrelevant lanes have been removed from the left end of the gel in (F). De novo RNA generated at the site of DSBs tend to form DNA:RNA hybrids, which are resistant to RNase A and sensitive to RNase H and are resolved in cell-free extracts. Bar plot shows the quantification of discrete band intensities of each lane of (E) and (F) (a.u.). Error bars indicate means ± SEMs from 2 independent experiments.
We next tested whether the sequence of DNA ends or structure affects transcription. Using DNA ends containing various end configurations (blunt, 5′ protruding or 3′ protruding with different sequences; Figure S1G), we observed that all DNA ends supported transcription, albeit with varying efficiencies (Figure 1C). Overall, these results suggest that MRN is sufficient to support transcription by RNAPII from DSBs with various sequences.
RNA Molecules Transcribed from DSBs Form DNA:RNA Hybrids
Recent studies have suggested the presence of DNA:RNA hybrids at DSBs (Cohen et al., 2018; D’Alessandro et al., 2018; Lu et al., 2018). Whether such hybrids are generated by a messenger RNA (mRNA) preceding DSB generation or dilncRNA is debated (Cohen et al., 2018; D’Alessandro et al., 2018; Lu et al., 2018). Our in vitro reactions, in which mRNA synthesis driven by a mammalian promoter is absent, represent an ideal system to resolve this issue. To this end, we supplemented the reactions with CFEs with either RNase A, which degrades free RNA, or RNase H, which degrades only RNA hybridized to DNA, during or after the reaction (Figure 1D). We observed that the RNA products generated in this reaction were sensitive to RNase A both during and after the reaction (Figure 1E, lanes 4 and 8). In contrast, we observed a concentration-dependent sensitivity to RNase H when it was added during the reaction (Figure 1E, lanes 5–7), which was lost when RNase H was added after the reaction (Figure 1E, lanes 9–11). We interpreted these results as suggestive that DNA:RNA hybrids are transiently formed during the transcription reaction but are promptly resolved, likely by DNA-RNA helicases present in CFEs, generating free RNA molecules. Next, we investigated these events in our minimal reconstituted system. We observed that transcripts generated on linear DNA only (Figure 1F, lane 3), were α-amanitin sensitive and thus dependent on RNAPII, as expected (Figure 1F, lane 4). These RNA species were sensitive to both RNase A and RNase H when these ribonucleases were included during the transcription reaction (Figure 1E, lanes 5 and 6). RNA products, however, were RNase A resistant but RNase H sensitive, when the respective RNases were added at the end of the reaction (Figure 1F, lanes 7 and 8). This result suggested that at the end of the reaction, most of the RNA products were in the form of DNA:RNA hybrids, even if their biogenesis involved a step in which they were not immediately paired to ssDNA, explaining the sensitivity to RNase A. Since DNA-RNA helicases were absent in this reconstituted system, most transcripts were therefore in the form of hybrids (Figures 1D–1F). We conclude that the dilncRNA molecules can form DNA:RNA hybrids.
MRN and RNAP II Proteins Stabilize Each Other at DNA Ends to Promote Transcription
We next studied the recruitment of MRN and RNAPII to DNA ends by incubating blunt-ended biotinylated DNA with purified proteins (Figure 2). The DNA-bound proteins were separated by pulldown with streptavidin beads, followed by immunoblotting (Figure 2A). We observed that, when individually tested, MRN had a stronger DNA end-binding activity compared to RNAPII (Figure 2A, lanes 5 and 6). When we tested DNA-end binding with MRN and RNAPII added sequentially (Figure 2A, lanes 7 and 8) or simultaneously (Figure 2A, lane 9), we observed that RNAPII DNA binding ability was greatly improved by the presence of MRN (Figure 2A, lanes 7–9), and, unexpectedly, MRN binding was detectably improved in the presence of RNAPII (Figure 2A, lanes 7 and 9). These results indicate that MRN and RNAP II likely stabilize each other at DSBs and, consistently with DNA-binding results, we observed enhanced de novo transcription from DSBs when MRN and RNAPII were added simultaneously (Figure 2B, lane 7).
Figure 2.

MRN and RNAPII Support Each Other in Binding to DNA Ends
(A) DNA-binding assays with 5′ end biotinylated 60-nt blunt dsDNA bound to streptavidin beads. MRN (0.5 pmol) and RNAPII (0.5 pmol) were added in different sequential order to determine their recruitment to DNA. Mutual enhancement in the binding of both MRN and RNAPII to DNA can be observed. Bar plot shows the quantification of the signal observed on each blot normalized to lanes 5 and 6 for MRE11 and RNAPII, respectively. Error bars indicate means ± SEMs of the quantified signal from 3 independent experiments.
(B) Transcription assays with MRN and RNAPII incubated with DNA in different sequential order. m∗ indicates radioactive end-labeled single-stranded RNA ladder. Higher transcription efficiency was observed when MRN and RNAPII were added together as also shown in the quantification of discrete band intensities of each lane depicted as a bar plot below. Error bars indicate means ± SEMs from 3 independent experiments.
(C) GFP-NBS1 U2OS cells were laser micro-irradiated to study early recruitment of MRN to DNA damage sites upon acute treatment with DMSO (vehicle control) or DRB (CDK7/9 inhibitor). Recruitment of MRN to DNA damage sites was reduced in the presence of DRB compared to DMSO. Scale bar, 10 μm. Scatterplot shows average intensity of NBS1-GFP signal, and each dot represents average intensity of each laser stripe. Error bars indicate means ± SEMs from 2 independent experiments (**p < 0.01).
(D) DNA-binding assay as in (A) with CFEs in the presence of DRB or DMSO (control). A reduction in binding of MRE11 to DNA ends with increasing concentrations of DRB is observed. Quantification of signal observed on each blot was normalized to lane 1. Error bars indicate means ± SEMs from 2 independent experiments.
(E) DNA-binding assay as described in (A) with MRN and RNAPII in the presence of α-amanitin. Quantification of signal observed on each blot was normalized to lane 1. Recruitment of MRN is significantly affected by transcriptional inhibition with DRB. Error bars indicate means ± SEMs from 4 independent experiments.
To test for a functional role of the observed mutually supportive activities of MRN and RNAPII in DNA-end binding and transcription, we individually interfered with them in living cells. Recruitment of RNAPII to DSB in vivo upon MRN knockdown was tested in HeLa cells exposed to ionizing irradiation (IR) by DI-PLA, a technique that allows in situ detection of a protein in proximity to exposed DNA ends (Galbiati et al., 2017) (Figure S2A). Consistent with our previous observations (Michelini et al., 2017), we detected the impaired recruitment of RNAPII to DNA ends in the absence of MRN (Figure S2A). To further test the effect of RNAPII on MRN recruitment to DSBs in vivo, we used UVA laser microirradiation on bromodeoxyuridine (BrdU)-pre-sensitized cells constitutively expressing NBS1 fused to GFP (Francia et al., 2016); lesions generated in defined nuclear spaces allow the detection of the early recruitment of MRN. To inhibit RNAPII activity, cells were acutely treated with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB, an inhibitor of RNAPII activity). Consistent with our in vitro data, we observed a significant reduction in the intensity of the NBS1-GFP signal at sites of DNA damage upon acute treatment with DRB, despite equal levels of DNA damage as measured by γH2AX (Figures 2C and S2B), thus indicating compromised recruitment or retention of MRN at damaged DNA upon RNAPII inhibition. To rule out the potential indirect effects of DRB in cells, we tested its impact in CFE in vitro. Here, we observed that DRB reduced MRN binding to DNA ends, while KU80, another DNA-end binder, showed less sensitivity (Figure 2D). We further used α-amanitin to inhibit RNAPII transcriptional activity in vitro with purified MRN and RNAPII. Also, under these conditions, we observed reduced MRN DNA binding (Figure 2E), indicating that the activity of RNAPII stabilizes MRN at DNA ends. Our results indicate that MRN and RNAPII stabilize each other at DNA ends in vitro and in vivo.
Nucleolytic Activity of MRN Is Dispensable for Transcription by RNAP II from DSBs
Next, we determined whether MRN favors the transcriptional activity of RNAPII beyond promoting its recruitment to DSBs. Previous cellular experiments showed that mirin, a molecule that restricts the access of MRE11 to dsDNA and inhibits its nuclease activity (Moiani et al., 2018; Shibata et al., 2014), inhibited the transcription of dilncRNA (Michelini et al., 2017), suggesting that the nuclease activity of MRE11 may be involved. We hypothesized that the ability of MRNs to resect DSBs to generate 3′ protruding DNA ends could provide an ideal DNA substrate for transcription by RNAPII (Kadesch and Chamberlin, 1982). However, our standard reaction buffer lacks manganese, which is required for most MRE11-nuclease-dependent functions (Anand et al., 2016; Deshpande et al., 2016). Since our preparation of MRN lacked notable nuclease activity in buffers containing magnesium only as the metal co-factor (Figures S1E and S1F), the involvement of MRE11 nuclease activity in our system is unlikely.
To further address the role of MRN nuclease activity, we tested the potential impact of CtIP. Phosphorylated CtIP (pCtIP) is necessary for activating the endonucleolytic activity of MRN and thus its 5′-resection function (Anand et al., 2016). To test whether CtIP affects transcription by MRN and RNAP II, we included equal concentrations of phosphorylated wild-type (WT) CtIP (pCtIP) and its resection-deficient mutant pCtIP(T847A) (Anand et al., 2016) into our standard transcription assay with Mg2+ alone (Figures 3A and S2C). We observed that the addition of WT pCtIP improved transcription to a small degree, but to the same extent observed when using resection-deficient variant pCtIP (T847A) (Figure 3A, lane 6 versus lanes 7 and 8). Therefore, the observations that MRN supports efficient transcription in the absence of Mn2+ (Figure 1) and that resection-deficient pCtIP enhances transcription to same extent as WT (Figure 3A) overall suggest that the endonuclease activity of MRN per se may not be important for transcription by RNAPII from DNA ends.
Figure 3.

Integrity of Active Site of Mre11, Irrespective of Its Nuclease Activity, Is Essential for RNAPII-Mediated Transcription
(A) Transcription assay with MRN, WT, or resection-deficient pCtIP (T847A) and RNAPII as indicated. m∗ indicates radioactive end-labeled single-stranded RNA ladder. Both WT pCtIP and its resection-deficient mutant stimulate MRN to support transcription by RNAPII to a similar extent. Bar plot shows the quantification of discrete band intensities of each lane (a.u.). Error bars indicate means ± SEMs from 2 independent experiments.
(B) Nuclease assay with MRN, pCtIP, and RNAPII. Biotinylated DNA labeled at the 3′ end was incubated with MRN, pCtIP, and RNAPII in the presence of ATP or rNTPs, and the products were resolved on 15% denaturing PAGE. In lane 10, streptavidin was added as a positive control to block DNA ends. m∗ indicates radioactively labeled DNA ladder. Exonuclease products are observed when MRN and pCtIP are both present with or without rNTPs, while endonuclease products are observed only when DNA is blocked with streptavidin. Bar plot shows quantification from each lane as a percentage of the degraded substrate. Error bars indicate means ± SEMs from 2 independent experiments.
(C) Generation of dilncRNA upon DSB induction by I-PpoI-empty vector (EV) is used as negative control—in the presence of WT or mutant MRN alleles as detected by strand-specific qRT-PCR with primers flanking DAB1 locus. MRN mutations abrogate dilncRNA generation from DSBs. Bar plot shows fold change of dilncRNA levels relative to control treated with I-PpoI. Error bars indicate means ± SEMs from 3 independent experiments (∗p < 0.05 and ∗∗∗p < 0.001). Lower panel: western blot analysis of the expression levels of MRE11 mutants. HeLa cells knocked down for endogenous MRE11 with siRNA against 3′ UTR of MRE11, were transfected with plasmids expressing hemagglutinin (HA)-tagged MRE11 (WT) or MRE11 (H129N, H63D, and H63S), along with either I-PpoI-expressing plasmid or EV as control. Expression levels of WT MRE11 and its mutants are comparable.
(D) Transcription assay with WT MRN or nuclease mutant (H129LD130V) MRN and RNAPII using blunt-ended DNA DSB. Nuclease mutant MRN does not support transcription by RNAPII from DNA ends. m∗ indicates radioactively labeled single-stranded RNA ladder. Bar plot shows the quantification of discrete band intensities of each lane (a.u.) of the autoradiograph. Error bars indicate means ± SEMs from 4 independent experiments (∗p < 0.05).
Recently, the DNAPK complex was shown to specifically promote a limited DNA nuclease activity of recombinant MRN and CtIP with magnesium only (Deshpande et al., 2020). To test whether RNAPII could also direct MRN-dependent endonuclease activity at 5′ ends (Anand et al., 2016; Reginato et al., 2017; Wang et al., 2017), we performed DNA endonuclease assays in the presence of both Mg2+ and Mn2+ by incubating dsDNA 32P-labeled at 3′ ends with MRN, RNAPII, and pCtIP. We observed distinct endonuclease products when MRN and pCtIP were incubated with DNA in the presence of end-bound streptavidin, a DNA-end blocker and a positive control for our experiments (Figure 3B, lane 10). Endonucleolytic products, however, could not be observed when streptavidin was replaced by RNAPII, with or without rNTPs (Figure 3B, lanes 3 and 7) or pCtIP (Figure 3B, lanes 5 and 9). Similarly, no DNA cleavage was observed in our standard transcription reaction conditions with Mg2+ (Figure S2D). This suggests that RNAPII does not promote the nuclease activity of MRN complex under our tested experimental conditions.
To further improve the sensitivity of this system based on 32P-labeled DNA, we used qPCR to detect and quantify potentially generated small amounts of DNA-end resection catalyzed by MRN and RNAPII (Nicolette et al., 2010). Our standard DNA template carries a recognition site for restriction enzyme HindIII near one end that can be recognized and cleaved by it only if it remains unresected (Figure S2E) (Nicolette et al., 2010). By designing two sets of primers, one spanning HindIII site near one end, and another away from the ends as a control, and by performing qPCR on the DNA following an in vitro transcription with MRN and RNAP II, we could not observe reduced HindIII digestion (Figure S2E), strengthening the evidence of the absence of resection in our transcription reactions. To further test whether any DNA modification by MRN, including resection, could support RNA synthesis from DNA ends, we performed transcription reactions with purified MRN or MRN and RNAPII (reaction 1, Figure S2F) and used the DNA thus treated as a substrate for transcription reaction without MRN (reaction 2, Figure S2F). We observed no RNAPII-supported transcription from DNA ends when such pre-treated DNA was used (reactions 1 and 2, Figure S2F), indicating that no covalent modifications to the DNA by MRN were introduced under these conditions. These results indicate that MRN supports transcription by RNAPII from DNA ends in the absence of 5′ end resection.
MRE11 Active Site Mutations Impair Transcription by RNAP II by Reducing DNA Melting
To gain insights into the mechanism of RNAPII stimulation by MRN and to reconcile these data with the inhibitory capacity of mirin in vivo, we analyzed MRE11 active site mutants. Residues H129 and H63 of MRE11 are part of the same nuclease active site (Garcia et al., 2011; Moreau et al., 1999). We knocked down endogenous MRE11 with small interfering RNAs (siRNAs) against the 3′ UTR of MRE11 mRNA in HeLa cells and concomitantly transfected plasmids expressing siRNA-resistant versions of either WT or H129N, H63D, and H63S MRE11 mutants or an empty vector (Chanut et al., 2016). dilncRNA generation at an endogenous DSB site generated by I-PpoI endonuclease (Michelini et al., 2017) was tested by strand-specific qRT-PCR. We observed a significant reduction in dilncRNA expression upon MRE11 knockdown, which was robustly recovered by the expression of WT MRE11 (Figure 3C). In contrast, the expression of MRE11 mutant forms (H129N, H63D) did not support dilncRNA expression, despite equal protein levels of all MRE11 forms (Figure 3C, lower panel). Similar behavior was observed for the M(H129LD130V)RN mutant form, devoid of all nucleolytic activities (Anand et al., 2016; Arthur et al., 2004), despite equal amounts of proteins and the ability to support RNAPII recruitment to DNA ends (Figure 3D, compare lane 5 with lanes 4 and 6; Figures S3A and S3B). These results suggest that the integrity of the MRE11 active site is essential for RNAPII-mediated transcription from DNA ends. How can such results be reconciled with the lack of resection in our transcription assays?
Intriguingly, Saccharomyces cerevisiae nuclease-deficient MRX mutant was found to be impaired in DNA melting (Nicolette et al., 2010), suggesting that the integrity of the nuclease active site is important for other functions beyond nuclease activities. We next tested the role of phosphoesterase motif IV of MRE11 on dilncRNA synthesis in vitro by the use of the M(H217Y)RN mutant that shows impaired exonuclease but unaffected endonuclease activity (Déry et al., 2008; Paull and Gellert, 2000). By performing transcription and DNA-binding assays with either WT MRN or mutant M(H217Y)RN along with RNAPII, we observed no significant differences in dilncRNA synthesis or DNA-end binding (Figures S3C and S3D), indicating that the nuclease activity of MRN is not required for transcription by RNAPII (Figures S3C and S3D).
To resolve this apparent paradox, we performed single-molecule fluorescence resonance energy transfer (smFRET) assays to monitor DNA melting by WT and nuclease-deficient M(H129LD130V)RN variants. Duplex DNA was labeled with Cy3 (green:donor) and Cy5 (red:acceptor) dyes on the opposite strands with a 5-bp distance between them (Figure 4A). FRET between the two moieties was used to monitor the melting of DNA strands (Figure 4A). We first measured FRET intensity and FRET efficiency (EFRET) with a fully paired duplex DNA in the absence of MRN and we observed non-interfering trajectories of smFRET of duplex DNA showing persistent high FRET (Figure 4B). When the same measurements were performed upon the addition of recombinant human MRN in the presence of ATP, we observed transitions from a higher to a lower FRET state (Figure 4C), which is indicative of DNA melting. We next performed these measurements with WT MRN and M(H129LD130V)RN and compared normalized frequencies of smFRET distributions (Figure 4D). We observed a reduction in the normalized frequencies of M(H129LD130V)RN mutant compared to WT (Figure 4D), and the fraction of low FRET events was reduced in M(H129LD130V)RN compared to WT MRN (Figure 4E). Thus, the nuclease active site mutant exhibited reduced DNA melting, providing a mechanistic explanation for its reduced ability to support transcription mediated by RNAPII from DNA ends (Figures 3D, 4D, and 4E).
Figure 4.

Nuclease-Deficient Mutants of MRN Lack DNA Melting Activity
(A) Schematic of single-molecule FRET (smFRET) assay for probing DNA melting activity of MRN. Cy3 and Cy5 dyes are located on the opposite strands of duplex DNA, with a 5-bp distance between them.
(B) Representative smFRET trajectory of duplex DNA alone showing persistent high FRET. Top panel: donor (green) and acceptor (red) intensities; bottom panel: corresponding FRET efficiency (EFRET).
(C) Representative smFRET trajectory of duplex DNA in the presence of MRN and ATP showing transitions from higher to lower FRET state.
(D) Normalized frequency of smFRET distributions of duplex DNA alone, DNA in the presence of MRN WT + ATP, and M(H129LD130V)RN + ATP, respectively. More than 200 trajectories from at least 2 independent experiments were analyzed to generate the distributions.
(E) Quantification of population fraction (percentage) of trajectories displaying low FRET states for DNA substrate in the presence of MRN WT or M(H129LD130V)RN corresponding to the histograms shown in (D). Error bars indicate means ± SEMs from 2 independent experiments from >200 trajectories.
(F) Transcription assay with MRN and RNAPII in the presence of mirin, PFM39, and PFM01. PFM01 but not PFM39 abrogates transcription, while mirin has an intermediate effect. Extra irrelevant lanes have been removed from the gel. Bar plots show the quantification of discrete band intensities of each lane (a.u.) of the autoradiograph. Error bars indicate means ± SEMs from 2 independent experiments.
(G) Transcription assays using blunt linear DNA with MRE11(M), MRE11-RAD50 (MR), or MRE11-RAD50-NBS1(MRN) and RNAPII. Extra lane has been removed from the right end of the gel. RAD50 and NBS1 improve the efficiency of transcription mediated by MRE11 and RNAPII from DNA ends. Bar plot shows the quantification of discrete band intensities of each lane (a.u.) of the autoradiograph. Error bars indicate means ± SEMs from 3 independent experiments (*p < 0.05, **p < 0.01, ****p < 0.0001).
(H) Normalized frequency of smFRET distributions of duplex DNA alone and DNA in the presence of MR + ATP and MRN WT + ATP, respectively. More than 200 trajectories from 2 independent experiments were analyzed to generate the distributions. The data for duplex DNA and WT MRN are the same as in (D) and are replotted here for reference.
Small Molecule Inhibitors and Mutations Affecting MRN ATPase Reduce Transcription by RNAP II from DSBs
To strengthen our conclusion that MRN stimulates RNAPII activity by melting DNA using an independent approach, we used small-molecule allosteric inhibitors of MRN activities (Shibata et al., 2014). While PFM39 inhibits exonuclease activity, PFM01 attenuates the endonuclease activity of MRN (Moiani et al., 2018; Shibata et al., 2014), and mirin was shown to reduce both activities (Deshpande et al., 2016). We next performed our transcription assays with MRN and RNAPII in the presence of mirin, PFM39, or PFM01 (Shibata et al., 2014). We observed that the inclusion of PFM01 led to a complete abrogation of transcription (Figure 4F, lane 5), while PFM39 had no significant effect (Figure 4F, lane 4), compared to DMSO control (Figure 4F, lane 2). Incubation of the transcription reaction with mirin had an intermediate inhibitory effect (Figure 4F, lane 3). These findings were recapitulated in cell extracts: PFM01 significantly reduced transcription (Figure S4A, lanes 7–9), PFM39 showed an effect only at high concentrations (Figure S4A, lanes 4–6), and mirin had an intermediate effect (Figure S4A, lane 3). The specific nuclease inhibitory activities of these molecules in our assay conditions were confirmed: PFM39 and mirin inhibited exonucleolytic activity of MRN, while the addition of PFM01 had little effect as compared to a DMSO control (Figure S4B). It is believed that the endonuclease activity of the MRN ensemble requires structural changes and partial DNA helix opening to allow DNA cleavage to occur (Williams et al., 2008). The experiments with small-molecule inhibitors are thus in agreement with our model that the DNA ends melting capacity of MRN promotes transcription by RNAPII.
DNA melting activities of MRN are facilitated by RAD50 and NBS1 (Cannon et al., 2013; Liu et al., 2016; Paull and Gellert, 1999; Williams et al., 2008). To test for the potential individual contribution of RAD50 and NBS1 to MRE11 in supporting transcription by RNAPII, we performed transcription assays with equimolar amounts of MRE11 (M), MRE11-RAD50 (MR), or MRE11-RAD50-NBS1 (MRN) and RNAPII (Figure S4C). We observed that MRE11 alone could stimulate transcription from DNA ends compared to RNAPII alone, although only to a limited extent, while MR and MRN were substantially more effective (Figure 4G). Furthermore, smFRET experiments showed that normalized frequencies of FRET were lower in MR compared to MRN WT (Figures 4H and S5D). The reduced DNA melting activity by MR compared to MRN WT substantiated our findings of reduced transcription by MR and RNAPII compared to MRN and RNAPII (Figures 4G and 4H). We confirmed the absence of nuclease contamination as well as activity of MR by performing a nuclease assay with 50-bp DNA labeled at the 5′ and 3′ ends in the presence of Mg2+ and Mn2+ (Figure S4D). ATP binding and hydrolysis-deficient RAD50 variants (K42A and K42R), with no contaminating nuclease and exonucleolytically active only with Mn2+ (Figures S5A–S5C), supported transcription by RNAPII to a much reduced degree (Figure 5A) (Anand et al., 2016; Cannon et al., 2013; Liu et al., 2016). smFRET experiments revealed that K42A and K42R mutations dramatically inhibit the DNA melting capacity of MRN complex (Figures 5B and S5D).
Figure 5.

Transient DNA Melting Activity of MRN at DSBs Supports Transcription by RNAPII
(A) Transcription assay with circular or linear DNA incubated with CFEs, purified WT MRN, or ATP binding (MR(K42A)N) or hydrolysis (MR(K42R)N) RAD50 mutants together with RNAPII. Mutations in RAD50 ATPase affect transcription by MRN and RNAPII from DNA ends. m∗ indicates radioactive labeled single-stranded RNA ladder. Bar plot shows the quantification of discrete band intensities of each lane (a.u.) of the autoradiograph. Error bars indicate means ± SEMs from 2 independent experiments.
(B) Normalized frequency of smFRET distributions of duplex DNA alone, DNA in the presence of MRN WT + ATP, ATP binding (MR(K42A)N), or hydrolysis (MR(K42R)N) RAD50 mutants, respectively. More than 200 trajectories from 2 independent experiments were analyzed to generate the distributions. The data for duplex DNA and WT MRN are the same as in Figure 4D and are replotted here for reference.
(C) Transcription assay with MRN, RPA, and RNAPII as indicated. Increasing concentrations of RPA (100 and 200 nM) improves transcription by RNAPII from DNA ends by stabilizing MRN-mediated transient DNA melting. RPA has no effect on RNAPII incubated with DNA. Bar plot shows the quantification of discrete band intensities of each lane (a.u.) of the autoradiograph. Error bars indicate means ± SEMs from 2 independent experiments (*p < 0.05).
(D) Transcription assay with 100-nt oligonucleotide DNA substrates containing unpaired DNA ends. m∗ indicates 10-bp ladder. Black line indicates the removal of extra lanes from the gel. dsDNA supports transcription only when both MRN and RNAPII are added, while open DNA ends support transcription by RNAPII alone. Bar plot shows the quantification of discrete band intensities of each lane of the autoradiograph. Error bars indicate measn ± SEMs from 3 independent experiments.
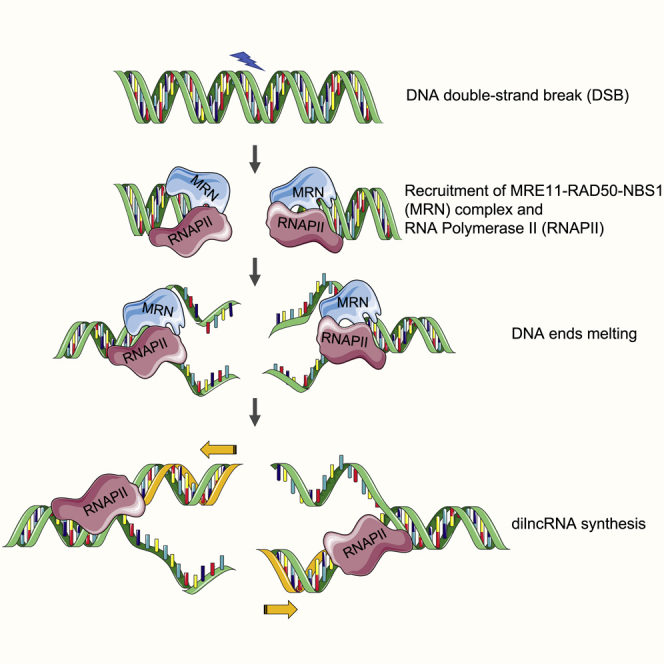
(E) Model depicting the role of MRN in supporting transcription by RNAPII from DNA ends. Upon DNA DSB, MRN is recruited to DSB, which leads to transient DNA melting, generating a structure akin to a “transcription bubble.” This unwound DNA is recognized by RNAPII and allows its transcription, leading to the generation of dilncRNA.
Stabilization of ssDNA at DSBs Supports Transcription by RNAPII
We reasoned that if the DNA melting activity of MRN is key to supporting transcription from DNA ends, any contribution to DNA melting or stabilization of ssDNA should favor transcription. RPA binds and stabilizes ssDNA (Chen and Wold, 2014). We thus included purified recombinant human RPA in our transcription assays (Figure S5E). We were excited to observe significant stimulation of transcription upon the addition of increasing amounts of RPA (Figure 5C, lanes 5–8). Notably, the addition of RPA had no effect on the dsDNA clipping activity of MRN-CtIP (Figure S5F). These results strongly suggest that ssDNA at DSBs, generated by MRN and stabilized by RPA, contributes to RNAPII-mediated transcription from DSB DNA ends. To test whether melted DNA ends structures are sufficient to support transcription by RNAPII, we used oligonucleotide substrates with unpaired DNA ends (Figure 5D), thus mimicking end structures generated by the DNA melting activity of MRN. Notably, oligonucleotide substrates containing unpaired DNA at their ends (12 bp) were transcribed efficiently, even when incubated with RNAPII alone (Figure 5D, lane 7), and the addition of MRN did not stimulate the reaction further (Figure 5D, lane 8). These data directly demonstrate that melted DNA ends favor transcription mediated by RNAPII. In conclusion, our results support a model (Figure 5E) in which MRN recruits and stabilizes RNAPII at DSB, and uses its DNA ends melting capacity to promote transcription by RNAPII.
Discussion
MRN and RNAPII Are Sufficient to Reconstitute Minimal Machinery Sufficient to Support Transcription from DSB DNA Ends
Our results demonstrate that MRN is sufficient to promote transcription by RNAP II from DNA ends to generate RNA products up to the length of the entire linear DNA template (Figure 1B, lanes 9 and 10). The observation that CFEs incubated with the same DNA template generate transcripts of different lengths (Figure 1B, lane 2) suggests that in more complex cell extracts, transcription processivity can be further modulated. Eukaryotic core RNAPII has poor activity on dsDNA (Kadesch and Chamberlin, 1982; Vitelli et al., 2017). We observed a low level of RNAPII activity on linear DNA, supporting the notion that RNAPII needs accessory factors to catalyze transcription from DNA ends (Figure 1B). MRN, one of the first DSB responders, is recruited to DNA ends as early as 15–30 s post-damage (Ciccia and Elledge, 2010; d’Adda di Fagagna, 2014; Hartlerode et al., 2015). Our results define a direct role for MRN in promoting transcription by RNAPII, which adds to the multifaceted roles of MRN in regulating DNA damage response and repair. Perhaps related, the MRN subunit, NBS1, in association with nucleolar regulator Treacle, has been shown to regulate transcription at rDNA loci within nucleoli (Larsen et al., 2014), and an association between MRN and B-myb (a highly conserved transcription factor) has been reported (Henrich et al., 2017). Hence, it appears that the role of MRN in RNAPII-mediated transcription is emerging.
We observed an enhanced transcription activity with CFE upon incubation with DNA containing long 3′ overhangs (homopolymeric dC tails consisting of 3′ protruding DNA ends) as compared to double-stranded linear DNA (Figure 1B, lanes 2 and 3). When the same dC-tailed DNA, which to an extent mimics DNA with physiological 5′-resected ends, was incubated with purified MRN and RNAPII, the activity of MRN became dispensable (Figure 1B, lanes 12 and 13). This is consistent also with higher levels of RNA synthesis with CFEs when incubated with 3′-dC tails (Figure 1B) and a study in Schizosaccharomyces pombe proposing that 5′-resected DNA is necessary for the observed transcription from DNA ends leading to the formation of DNA:RNA hybrids (Ohle et al., 2016). We also observed that DNA:RNA hybrids can be the products of de novo transcription from DNA ends, when purified components were used, while their formation was prevented or resolved in CFEs, likely by RNA binding proteins and/or DNA-RNA helicases, respectively (Figures 1D–1F). This also suggests that our system may allow for the identification and validation of factors and mechanisms that prevent or favor DNA:RNA hybrid formation, and the absence of preexisting transcription across DSB region allows us to rule out its contribution to hybrid formation. Senataxin and DDX1 have already been shown to be involved in resolving DNA:RNA hybrids at DSBs (Cohen et al., 2018; Liao et al., 2016; Yüce and West, 2013). Recently, we and others proposed that DSB resection could favor DNA:RNA hybrid formation and play a role in DNA repair (Cohen et al., 2018; D’Alessandro et al., 2018; Lu et al., 2018). Overall, these results show that DNA:RNA hybrids at DSB can result from dilncRNA.
DSB Resection by MRN Is Not a Prerequisite for Transcription from DNA Ends
MRN possesses very little nucleolytic activity in the presence of magnesium (Cannon et al., 2013), while the requirement for manganese in MRN-dependent 5′ resection is well established (Deshpande et al., 2016; Nicolette et al., 2010; Trujillo and Sung, 2001). Since we observed that transcription from a DSB in the presence of MRN and RNAPII does not require manganese, a role for 5′ resection in this process is unlikely. Our data demonstrate that MRN may promote transcription by RNAPII independently from its nuclease and DNA-end resection activity, but by melting DNA ends. This conclusion is supported by several independent lines of evidence.
The efficiency of transcription in the presence of resection-deficient mutants of pCtIP (T847A) was observed to be similar to its WT form (Figure 3A), reinforcing the notion that 5′ resection is not required for transcription from DSBs. These results were initially in apparent contrast to the observations that the nuclease-deficient M(H129LD130V)RN mutant was also impaired in promoting RNAPII-mediated transcription. However, the equivalent yeast mutant M(H125N)RX, although nuclease deficient, was found deficient also in DNA melting (Nicolette et al., 2010). Our results obtained by smFRET strengthen our conclusions by demonstrating that also human nuclease-deficient mutants of MRN exhibit decreased DNA melting activity (Figure 4D). RAD50 has ATPase activity, which exposes the active site of MRE11 (Paull and Deshpande, 2014) and is responsible for the observed higher transcription from DSBs when either MR or MRN were incubated with RNAPII, compared to MRE11 alone. NBS1 promotes the ATP-dependent enzymatic activity of the MR complex and has been shown to promote the 5′-resection activity of MRN next to a protein block (Anand et al., 2016, 2019; Cannavo and Cejka, 2014; Deshpande et al., 2016). Accordingly, the MRN complex promotes transcription more robustly than MRE11 or MR (Figure 4G), which correlates with its enhanced DNA melting activity (Figure 4H).
Recent studies have reported specific allosteric inhibitors of MRN, which bind adjacent to active sites (Moiani et al., 2018; Shibata et al., 2014). Mirin was shown to inhibit ATM activation (Dupré et al., 2008), but MRN nuclease activity is dispensable for ATM activation (Paull, 2015). Hence, it appears that mirin inhibits MRN-dependent ATM activation independently of its nuclease activity. We observed an abrogation of transcription most strongly with PFM01 (Figures 4F and S4A). PFM01 binds to the dimer interface and blocks the ssDNA-binding groove that in turn inhibits its endonuclease activity (Moiani et al., 2018; Shibata et al., 2014). This points to the role of the ssDNA-binding activity of MRN in supporting transcription from DNA ends (Figure 4F). PFM39 that specifically inhibits exonuclease activity did not affect transcription from DNA ends (Figure 4F). Since 5′-DNA end resection depends on both the endonuclease and exonuclease activity of MRN, this result reinforces our hypothesis that 5′ resection is dispensable for transcription mediated by RNAPII. Nevertheless, we cannot exclude that DNA-end resection by MRN in cells may still play a supporting role in promoting transcription by RNAPII.
DNA Melting Activity of MRN Likely Contributes to Transcription by RNAP II from DNA Ends
During transcription, RNAPII alone can bind and maintain an open DNA region known as the “transcription bubble,” and it is sufficient to translocate, synthesize, and proofread nascent RNA (Cramer, 2004). Recent studies have shown that RPA binds to single-stranded non-template strands of a transcription bubble at promoters (Nguyen et al., 2017; Sikorski et al., 2011). We observed that RPA does not promote resection (Figure S5F), but does promote the transcription of dsDNA by MRN and RNAPII (Figure 5C). These results suggest that a transcription bubble-like structure at DNA ends is likely stabilized by RPA and exploited by RNAPII for transcription at DNA DSB ends. Crucially, we demonstrated that DNA ends containing melted DNA structures (unpaired DNA ends) can be efficiently transcribed by RNAPII in an MRN-independent manner (Figure 5D). smFRET assays further demonstrated that active site mutants of MRN, known to be deficient in nuclease activity, lack DNA melting activity (Figures 4D, 4E, 5B, and S5D), supporting our model. Hence, we propose that in response to a DSB, the DNA melting activity of MRN generates a single-stranded transcription bubble, which is used by RNAPII to support transcription from DNA ends (Figure 5E). Our recent discovery that the preinitiation complex (PIC) of RNAPII is recruited to DSB (Pessina et al., 2019) generates an additional level of control of these events.
In summary, we reconstituted a minimal system, which supports transcription at DSB. This system will be valuable for studying transcriptional events at DSBs and will help to identify novel proteins involved in regulating RNAPII activity upon DNA damage or small molecules stimulating or inhibiting these events.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal MRE11 | gift from S. P. Jackson, Wellcome Trust/Cancer Research UK Gurdon Institute, University of Cambridge | N/A |
| Mouse monoclonal RAD50 | Millipore | Cat#(13B3/2C6) 05-525; RRID:AB_309782 |
| Rabbit polyclonal NBS1 | Novus Biologicals | Cat#NB100-143; RRID:AB_10078050 |
| Mouse monoclonal RNAP II | Abcam | Cat#ab817; RRID:AB_306327 |
| Mouse monoclonal anti-γH2AX | Millipore | Cat#05-636; RRID:AB_309864 |
| CtIP | Bethyl | Cat#A300-488A; RRID: AB_2175262 |
| HA | In house facility | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mirin | Sigma-Alrich | Cat#M9948 |
| PFM39 | Sigma-Alrich | Cat#SML1839 |
| PFM01 | Sigma-Alrich | Cat#SML1735 |
| RNaseOUT | Thermo-Fisher | Cat#10777019 |
| Ribonucleotides | Promega | Cat#P1132 |
| Restriction Enzymes: SmaI | New England Biolabs | Cat#R0141S |
| α-32P[UTP] | Perkin-Elmer | Cat#NEG507H250UC |
| 5,6-Dichlorobenzimidazole 1-β-D-ribofuranoside | Sigma-Aldrich | Cat#D1916 |
| Glycogen | Thermo-Fisher | Cat#AM9510 |
| RNA loading dye | New England Biolabs | Cat#B0363S |
| MOPS-EDTA-Sodium Acetate | Sigma-Aldrich | Cat#M5755 |
| RNase H | New England Biolabs | Cat#M0297S |
| RNase A | Sigma-Aldrich | Cat#R6513 |
| Turbo DNase | Ambion | Cat#AM2238 |
| Gelatin from cold water fish skin | Sigma-Aldrich | Cat#G7765 |
| Lipofectamine® RNAiMAX Transfection Reagent | Thermo-Fisher | Cat#13778030 |
| Phenol:Chloroform:Isoamyl Alcohol 25:24:1 Saturated with 10 mM Tris, pH 8.0, 1 mM EDTA | Sigma-Aldrich | Cat#P2069 |
| Protease Inhibitor | Calbiochem | Cat#539134 |
| Mowiol® 4-88 | Sigma-Aldrich | Cat#81381 |
| Dynabeads protein G | Thermo-Fisher | Cat#10004D |
| α-amanitin | Sigma-Aldrich | Cat#A2263 |
| SsoFast EvaGreen® Supermix | Bio-Rad | Cat#1725201 |
| 19:1, acrylamide: bis-acrylamide | Sigma-Aldrich | Cat#A3449 |
| 4-OHT | Sigma-Aldrich | Cat#SML1666 |
| UltraPure BSA | Thermo-Fisher | Cat#AM2618 |
| Terminal Transferase | New England Biolabs | Cat#M0315S |
| Lipofectamine 2000 transfection reagent | Thermo-Fisher | Cat#11668019 |
| Critical Commercial Assays | ||
| Superscript IV First Strand cDNA synthesis kit | Thermo-Fisher | Cat#18091200 |
| Maxwell RSC simplyRNA tissue kit | Promega | Cat#218161 |
| Gel Extraction Kit | QIAGEN | Cat#28704 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | Cat#CCL-2 |
| U2OS NBS1-GFP | Gift from Jiri Bartek, Danish Cancer society’s Institute of Cancer Biology, Copenhagen | N/A |
| Oligonucleotides | ||
| Primers and Oligonucleotides | Sigma-Aldrich | Table S1 |
| siRNA | Sigma-Aldrich | Table S1 |
| Recombinant DNA | ||
| I-PpoI | Berkovich et al.,2007 | N/A |
| pICE-HA-MRE11-WT | Chanut et al.,2016 | AddgeneCat#82033 |
| pICE-HA-MRE11-H129N | Chanut et al.,2016 | AddgeneCat#82034 |
| pICE-HA-MRE11-H63D | Chanut et al.,2016 | AddgeneCat#82036 |
| pICE-HA-MRE11- H63S | Chanut et al.,2016 | AddgeneCat#82035 |
| pUC19 | Invitrogen | N/A |
| Software and Algorithms | ||
| Image Lab | Bio-Rad | N/A |
| ImageQuant | GE healthcare | N/A |
| ImageJ | NIH | N/A |
| Prism | Graph Pad | https://www.graphpad.com/scientificsoftware/prism/ |
Resource Availability
Lead Contact
Requests and information for resources and reagents should be directed to the lead contact Fabrizio d’Adda di Fagagna (fabrizio.dadda@ifom.eu)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any dataset or codes.
Experimental Model and Subject Details
Culture conditions for in vitro systems
HeLa cells (ATCC) were grown under standard tissue culture conditions (37°C, 5%CO2) in MEM+Glutamax (GIBCO), 10%FBS, 1% non-essential amino acids and 1% sodium pyruvate. U2OS NBS1-GFP (a kind gift from Dr. Jiri Bartek) were grown in DMEM containing 10% FBS, 1% L-glutamine, 1% pencillin-streptomycin and Puromycin (1 μg/ml).
Authentication of cell lines used
Cell lines are authenticated at each batch freezing by STR profiling (StemElite ID System, Promega). All cell lines are are tested for mycoplasma at each batch freezing with both PCR (Uphoff and Drexler, 2004) and a biochemical test (MycoAlert, Lonza).
Method Details
Expression and Purification of Recombinant Proteins
Recombinant MRE11, MR, MRN, CtIP and their variants were expressed in Sf9 cells and purified by affinity chromatography as detailed (Anand et al., 2016, 2019). Calf thymus RNAP II was prepared as described (Thompson et al., 1990; Hu et al., 2006) with modifications. Briefly, frozen calf thymus (approx. 450 g) was obtained and cut into thin slices while semi-frozen and blended. 100 mL of pre-chilled Buffer A (50 mM Tris-HCl, pH 7.8; 10% glycerol, 10 μM ZnCl2) was added and blended again to obtain a homogeneous mixture along with protease inhibitors (Roche). Buffer A was added to the suspension to get a final volume of 1000 ml, stirred and centrifuged at 6000 RPM for 20 min. The supernatant was filtered through nylon filter-cloth. Proteins were precipitated with 10% Polyethyleneimine (PEI, pH 7.8) by gradual addition to a final concentration of 0.05% and spun at 10000 RPM for 30 min. The supernatant was discarded, and the pellet was dounce homogenized in 400 mL of Buffer B (50 mM Tris-HCl, pH 7.8; 10% glycerol, 10 μM ZnCl2; 150 mM Ammonium sulfate). The homogenate was spun in 45Ti rotor for 40 min at 40 000 rpm and the supernatant was loaded onto fast flow Q Sepharose (120 ml) pre-equilibrated in buffer B. The protein was eluted in buffer C (50 mM Tris-HCl, pH 7.8; 10% glycerol, 10 μM ZnCl2; 500 mM Ammonium sulfate), peaks were collected and loaded onto Protein A and G agarose beads pre-coupled to 8WG16 antibody (1 mg/ml) overnight. The beads were washed with buffer C and eluted in buffer D (50 mM Tris-HCl, pH7.8; 10 μM ZnCl2; 40% propylene glycol, 500 mM Ammonium sulfate) and dialyzed against dialysis buffer (50 mM Tris-HCl, pH 7.8; 10% glycerol, 10 μM ZnCl2; 150 mM Ammonium sulfate, 5 mM DTT).
In vitro transcription assay
Transcription assays were carried out in 15 μL volume in a reaction buffer containing HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) [pH 7.9], 5 mM; magnesium chloride, 5 mM; potassium chloride, 25 mM; dithiothreitol, 0.25 mM; EDTA, 0.25 mM; BSA (Invitrogen), 0.1 mg/ml and glycerol, 4%. pUC19 (Invitrogen) was digested with SmaI to generate blunt ended DNA and the linearized DNA was purified from gel using gel extraction kit. dC-tail containing DNA was synthesized by adding C nucleotides to blunt ended DNA using standard protocol for terminal transferase. Similarly, DNA substrates with different end configurations were generated by digesting pUC19 with EcoRI (5′-AATT), XbaI (5′-CTAG), SacI (AGCT-3′) and KpnI (3′-GTAC). Linearized DNA was quantified and loaded onto an agarose gel. 0.1 pmoles of pUC19 DNA ends was incubated with MRN, 0.5 pmoles and RNAP II, 0.5 pmoles in the transcription buffer containing rNTPs (ATP, GTP, CTP), 83 μM; UTP, 3 μM and α-32P[UTP], 10 μCi. Equivalent volume of protein storage buffers was added in controls. The reactions were mixed on ice and incubated for 30 min at 30°C. The reactions were stopped by the addition of RNA stop solution (Tris-HCl [pH 7.4] at 25°C, 0.3 M; sodium acetate, 0.3 M; SDS, 0.5%, EDTA, 2 mM; glycogen, 3 μg/ml) 135 μl, purified by phenol-chloroform extraction and precipitated. The products were resuspended in 1X RNA loading dye (New England Biolabs), loaded onto formaldehyde (17%)-agarose gel (2.5%) and resolved in MOPS-EDTA buffer (Sigma-Aldrich). The gels were dried on a 3 mm CHR paper (Whatman) at 65°C, exposed to storage phosphor screens (GE Healthcare) and scanned by a Typhoon Phosphor imager (FLA9000, GE HealthCare). The transcription assay with 100 nt oligonucleotide substrates were carried out in the same way, except that reactions were loaded on an 8% polyacrylamide denaturing urea gels (19:1, acrylamide: bis-acrylamide, Sigma-Aldrich). For checking resection using qPCR, the products of this reaction were subjected to HindIII digestion and qPCR assay was performed with the primers as indicated (Table S1).
DNA binding assays
20 ng of 5′- biotinylated DNA (Table S1) was incubated with 0.5 pmoles of MRN and 0.5 pmoles of RNAP II in transcription buffer containing 83 μM of rNTPs as described above for 15 min at 30°C. The reactions were then incubated with streptavidin beads pre-washed with DNA-pull down buffer (20 mM HEPES pH 8.0, 150 mM KCl, 5 mM MgCl2, 5% glycerol, 0.05% NP-40) and incubated for 10 min at 30°C with constant slow agitation. The beads were then washed 3 times with DNA pull down buffer and immunoblotted for MRE11 (rabbit polyclonal raised against recombinant human MRE11, gift from S.P. Jackson), RAD50 (Millipore), NBS1 (Novus Biologicals) and RNAP II (Abcam).
Nuclease assays
Nuclease assays were performed as previously (Anand et al., 2016). Briefly, proteins were added as indicated, in volume of 15 μL in reaction buffer containing Tris-acetate [pH 7.5], 25 mM; manganese acetate, 1 mM; magnesium acetate, 5 mM; dithiothreitol, 1 mM; ATP, 1 mM; rNTPs, 83 μM; BSA, (New England Biolabs), 0.25 mg/mL; phosphoenolpyruvate, 1 mM; pyruvate kinase, (Sigma), 80 U/mL; DNA substrate (Table S1), 1 nM and streptavidin (Sigma), 15 nM. Recombinant proteins were then added to the reactions on ice and the samples were incubated for 30 min at 37°C. Reactions were stopped and separated on 15% polyacrylamide denaturing urea gels (19:1 acrylamide: bisacrylamide, Bio-Rad). The gels were fixed for 30 min at room temperature, dried and exposed to storage phosphor screens (GE Healthcare) and scanned by a Typhoon Phosphor imager (FLA 9500, GE Healthcare).
dilncRNA expression
HeLa cells (ATCC) were grown under standard tissue culture conditions (37°C, 5% CO2) in MEM+Glutamax (GIBCO), 10% FBS, 1% non-essential amino acids and 1% sodium pyruvate. Cells were transfected with siRNA (Sigma) against 3′ UTR of MRE11 or luciferase as control (Table S1) in complete media for 48 h with the standard protocol for RNAimax. These cells were then transfected with 1 μg of either mammalian ER-I-PpoI-expressing plasmid (Berkovich et. al., 2007) or empty vector with the standard protocol for Lipofectamine 2000 (Invitrogen). In the experiment where mutants of MRE11 were expressed, pICE-HA-MRE11-WT, pICE-HA-MRE11-H129N, pICE-HA-MRE11-H63D and pICE-HA-MRE11- H63S (Addgene, Chanut et al., 2016) were co-transfected. The knockdowns and expression of MRE11 mutants was confirmed by immunoblotting. The nuclear translocation of I-PpoI was activated 24 h later by supplementing the media with 4-OHT, 2 μM for 3 h. Cells were harvested, washed and total RNA was extracted using Maxwell® RSC simplyRNA Blood Kit (Promega). RNA was quantified using Nanodrop (Thermoscientific) and 1 μg of total RNA was reverse transcribed using Superscript First strand cDNA synthesis kit with strand-specific primers (Table S1). RT-qPCR was used to determine the expression of DSB-induced transcripts using Evagreen supermix (Bio-Rad).
Micro-irradiation for studying the recruitment of MRN at the site of DNA damage
U2OS NBS1-GFP (a kind gift from Dr. Jiri Bartek) were grown in DMEM containing 10% FBS, 1% L-glutamine, 1% pencillin-streptomycin and Puromycin (1 μg/ml). Leica TCS SP5 point scanning confocal microscope equipped with a Leica HCX PL APO 63X/1.4 NA oil immersion objective, was used to perform live-cell imaging and laser-induced DNA damage (Francia et al., 2016). Cells were cultured in BrdU (10 μM, 72 h) and treated with DRB (Sigma, 100 μM) for 2 h and equivalent volume of DMSO as control. Laser micro-irradiation was performed using 50 mW 405 nm diode laser. ROI were selected for each nuclei and 405 nm laser scanned the ROIs for 50 iterations. Intensity of NBS1-GFP was calculated and presented as a scatterplot and statistical analysis was performed.
Single-molecule FRET (smFRET) Assay
The smFRET experiments were carried out on a customized inverted microscope (IX51, Olympus), with a high NA TIRF objective (100X, 1.49 NA, oil immersion, Olympus). Flow chambers were passivated with PEG and 30 mg/mL BSA before immobilizing 50 pM duplex DNA through biotin/neutravidin interactions, as previously described (Zhao et al., 2019). Then chambers were washed with reaction buffer to remove any free DNA not specifically immobilized to the surface. The reaction buffer contained 20 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 25 mM KCl, 0.25 mM DTT, 0.25 mM EDTA, 0.1 mg/ml BSA and an oxygen scavenging system composed of 0.8% (w/v) glucose, 0.5 mg/mL glucose oxidase, 0.4 μg/mL catalase, and 5 mM Trolox. The Cy3/Cy5 labeled duplex DNA were excited by a 532 solid-state laser (OEM Laser Systems), and the emission from the two dyes was split using a dichroic (FF660, Semrock) and narrow-band bandpass filters (582/75 and 680/42, Semrock) inside an Optosplit II (Cairn Research). Thousand-frame movies were recorded with an exposure time of 30 ms via an EMCCD camera (iXon+, Andor) at room temperature. Custom written MATLAB scripts were applied to view and analyze the fluorescence time trajectories and then to extract the FRET efficiency. Each normalized smFRET distribution was generated from a minimum of 200 trajectories obtained from at least two independent experiments.
Quantification and Statistical Analysis
Quantification of radioactive gels was performed with ImageQuant software. For DNA binding assays, blots were quantified using Image Lab software. Values are plotted after subtracting the background signal above the DNA or protein bands. For the quantification of GFP signal in laser-damage-induced stripes in U2OS NBS1-GFP cells, ImageJ software was used by drawing the ROI of laser damage. The mean fluorescence intensity was then calculated by measuring it in each damaged area and an identical area in an undamaged region of the same nucleus which is then subtracted as background. The bar graphs are plotted using GraphPad Prism software and statistical significance was calculated using two-tailed Student’s t test. Further details are described in Figure Legends and Method Details.
Acknowledgments
We thank Manuel Stucki for sharing with us reagents in the initial steps of this project and Yan Coulombe for technical help. We thank Elena Petricci for providing us MRN inhibitors; Mohamed Fareh and Chirlmin Joo of the Delft University of Technology, the Netherlands and Mohamed A. Sayed and Samir M. Hamdan, KAUST, Saudi Arabia, for the preliminary FRET studies. We thank Valentina Matti for technical help and all of the members of the d’Adda di Fagagna group for helpful comments on the manuscript. S.S. was supported by a SIPOD postdoctoral fellowship (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7 under grant agreement no. 600399. J.-Y.M. is FRQS Chercheur National Investigator and this research was supported by the CIHR. Work in the d’Adda di Fagagna laboratory is supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) (application 12971), the Cariplo Foundation (grant nos. 2010.0818 and 2014-0812), the Fondazione Telethon (GGP12059 and GGP17111), the Association for International Cancer Research (AICR-Worldwide Cancer Research Rif. no. 14-1331), Progetti di Ricerca di Interesse Nazionale (PRIN) 2010–2011 and 2005, the Italian Ministry of Education Universities and Research EPIGEN Project, the European Research Council advanced grant (322726), AriSLA (project “DDRNA and ALS”), AIRC Special Program 5 per Mille Metastases project no. 21091, the AMANDA and InterSLA projects Accordo Quadro Regione Lombardia–CNR and flagship progetto InterOmics. The research in the Cejka laboratory was supported by Swiss National Science Foundation (SNCF) grant no. 31003A_175444 and European Research Council (ERC) grant no. 681630.
Author Contributions
S.S. and F.d’A.d.F. conceived and designed the experiments. R.A. carried out the nuclease assay and produced the proteins. S.F., F.M., and A.G. carried out the micro-irradiation and DIPLA assays. H.W. purified RNAPII. X.Z. and E.R. performed the smFRET assays; all of the other experiments were carried out by S.S. J.-Y.M. provided the exo mutant and corresponding WT MRN. P.C. supervised R.A. and provided critical input in the experimental design and result interpretation. S.S. and F.d’A.d.F. wrote the paper, and all of the authors commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: January 5, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108565.
Contributor Information
Petr Cejka, Email: petr.cejka@irb.usi.ch.
Fabrizio d’Adda di Fagagna, Email: fabrizio.dadda@ifom.eu.
Supplemental Information
References
- Aguado J., Sola-Carvajal A., Cancila V., Revêchon G., Ong P.F., Jones-Weinert C.W., Wallén Arzt E., Lattanzi G., Dreesen O., Tripodo C. Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson-Gilford Progeria Syndrome. Nat. Commun. 2019;10:4990. doi: 10.1038/s41467-019-13018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R., Ranjha L., Cannavo E., Cejka P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell. 2016;64:940–950. doi: 10.1016/j.molcel.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Anand R., Jasrotia A., Bundschuh D., Howard S.M., Ranjha L., Stucki M., Cejka P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019;38:e101005. doi: 10.15252/embj.2018101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur L.M., Gustausson K., Hopfner K.P., Carson C.T., Stracker T.H., Karcher A., Felton D., Weitzman M.D., Tainer J., Carney J.P. Structural and functional analysis of Mre11-3. Nucleic Acids Res. 2004;32:1886–1893. doi: 10.1093/nar/gkh343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygün O., Xu X., Liu Y., Takahashi H., Kong S.E., Conaway R.C., Conaway J.W., Svejstrup J.Q. Direct inhibition of RNA polymerase II transcription by RECQL5. J. Biol. Chem. 2009;284:23197–23203. doi: 10.1074/jbc.M109.015750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E., Monnat R.J., Jr., Kastan M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Cannavo E., Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Cannon B., Kuhnlein J., Yang S.H., Cheng A., Schindler D., Stark J.M., Russell R., Paull T.T. Visualization of local DNA unwinding by Mre11/Rad50/Nbs1 using single-molecule FRET. Proc. Natl. Acad. Sci. USA. 2013;110:18868–18873. doi: 10.1073/pnas.1309816110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzo I., Iannelli F., Francia S., d’Adda di Fagagna F. Express or repress? The transcriptional dilemma of damaged chromatin. FEBS J. 2017;284:2133–2147. doi: 10.1111/febs.14048. [DOI] [PubMed] [Google Scholar]
- Carvajal-Maldonado D., Byrum A.K., Jackson J., Wessel S., Lemaçon D., Guitton-Sert L., Quinet A., Tirman S., Graziano S., Masson J.Y. Perturbing cohesin dynamics drives MRE11 nuclease-dependent replication fork slowing. Nucleic Acids Res. 2019;47:1294–1310. doi: 10.1093/nar/gky519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P. DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J. Biol. Chem. 2015;290:22931–22938. doi: 10.1074/jbc.R115.675942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut P., Britton S., Coates J., Jackson S.P., Calsou P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016;7:12889. doi: 10.1038/ncomms12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Wold M.S. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. BioEssays. 2014;36:1156–1161. doi: 10.1002/bies.201400107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S., Puget N., Lin Y.L., Clouaire T., Aguirrebengoa M., Rocher V., Pasero P., Canitrot Y., Legube G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018;9:533. doi: 10.1038/s41467-018-02894-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P. Structure and function of RNA polymerase II. In: Conaway R.C., Conaway J.W., editors. Proteins in Eukaryotic Transcription. Volume 67. Elsevier; 2004. pp. 1–42. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F. A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol. 2014;24:171–178. doi: 10.1016/j.tcb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- D’Alessandro G., d’Adda di Fagagna F. Transcription and DNA Damage: Holding Hands or Crossing Swords? J. Mol. Biol. 2017;429:3215–3229. doi: 10.1016/j.jmb.2016.11.002. [DOI] [PubMed] [Google Scholar]
- D’Alessandro G., Whelan D.R., Howard S.M., Vitelli V., Renaudin X., Adamowicz M., Iannelli F., Jones-Weinert C.W., Lee M., Matti V. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 2018;9:5376. doi: 10.1038/s41467-018-07799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedrick R.L., Chamberlin M.J. Studies on transcription of 3′-extended templates by mammalian RNA polymerase II. Parameters that affect the initiation and elongation reactions. Biochemistry. 1985;24:2245–2253. doi: 10.1021/bi00330a019. [DOI] [PubMed] [Google Scholar]
- Déry U., Coulombe Y., Rodrigue A., Stasiak A., Richard S., Masson J.Y. A glycine-arginine domain in control of the human MRE11 DNA repair protein. Mol. Cell. Biol. 2008;28:3058–3069. doi: 10.1128/MCB.02025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande R.A., Lee J.H., Arora S., Paull T.T. Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol. Cell. 2016;64:593–606. doi: 10.1016/j.molcel.2016.10.010. [DOI] [PubMed] [Google Scholar]
- Deshpande R.A., Myler L.R., Soniat M.M., Makharashvili N., Lee L., Lees-Miller S.P., Finkelstein I.J., Paull T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020;6:eaay0922. doi: 10.1126/sciadv.aay0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré A., Boyer-Chatenet L., Sattler R.M., Modi A.P., Lee J.H., Nicolette M.L., Kopelovich L., Jasin M., Baer R., Paull T.T., Gautier J. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S., Michelini F., Saxena A., Tang D., de Hoon M., Anelli V., Mione M., Carninci P., d’Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–235. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S., Cabrini M., Matti V., Oldani A., d’Adda di Fagagna F. DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J. Cell Sci. 2016;129:1468–1476. doi: 10.1242/jcs.182188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati A., Beauséjour C., d’Adda di Fagagna F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell. 2017;16:422–427. doi: 10.1111/acel.12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Wei W., Li M.M., Wu Y.S., Ba Z., Jin K.X., Li M.M., Liao Y.Q., Adhikari S., Chong Z. Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 2014;24:532–541. doi: 10.1038/cr.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V., Phelps S.E., Gray S., Neale M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011;479:241–244. doi: 10.1038/nature10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbini E., Vertemara J., Longhese M.P. Local unwinding of double-strand DNA ends by the MRX complex promotes Exo1 processing activity. Mol. Cell. Oncol. 2018;5:e1511208. doi: 10.1080/23723556.2018.1511208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlerode A.J., Morgan M.J., Wu Y., Buis J., Ferguson D.O. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat. Struct. Mol. Biol. 2015;22:736–743. doi: 10.1038/nsmb.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich S.M., Usadel C., Werwein E., Burdova K., Janscak P., Ferrari S., Hess D., Klempnauer K.H. Interplay with the Mre11-Rad50-Nbs1 complex and phosphorylation by GSK3β implicate human B-Myb in DNA-damage signaling. Sci. Rep. 2017;7:41663. doi: 10.1038/srep41663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Malik S., Negroiu C.C., Hubbard K., Velalar C.N., Hampton B., Grosu D., Catalano J., Roeder R.G., Gnatt A. A Mediator-responsive form of metazoan RNA polymerase II. Proc. Natl. Acad. Sci. USA. 2006;103:9506–9511. doi: 10.1073/pnas.0603702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch T.R., Chamberlin M.J. Studies of in vitro transcription by calf thymus RNA polymerase II using a novel duplex DNA template. J. Biol. Chem. 1982;257:5286–5295. [PubMed] [Google Scholar]
- Kowalczykowski S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015;7:a016410. doi: 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen D.H., Hari F., Clapperton J.A., Gwerder M., Gutsche K., Altmeyer M., Jungmichel S., Toledo L.I., Fink D., Rask M.B. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 2014;16:792–803. doi: 10.1038/ncb3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–7758. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- Lee J.H., Paull T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- Liao S., Tammaro M., Yan H. The structure of ends determines the pathway choice and Mre11 nuclease dependency of DNA double-strand break repair. Nucleic Acids Res. 2016;44:5689–5701. doi: 10.1093/nar/gkw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Sung S., Kim Y., Li F., Gwon G., Jo A., Kim A.K., Kim T., Song O.K., Lee S.E., Cho Y. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J. 2016;35:743–758. doi: 10.15252/embj.201592462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch Y., LaPointe J.W., Kornberg R.D. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell. 1987;49:203–210. doi: 10.1016/0092-8674(87)90561-7. [DOI] [PubMed] [Google Scholar]
- Lu W.T., Hawley B.R., Skalka G.L., Baldock R.A., Smith E.M., Bader A.S., Malewicz M., Watts F.Z., Wilczynska A., Bushell M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018;9:532. doi: 10.1038/s41467-018-02893-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley J.L., Fire A., Cano A., Sharp P.A., Gefter M.L. DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc. Natl. Acad. Sci. USA. 1980;77:3855–3859. doi: 10.1073/pnas.77.7.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F., Pitchiaya S., Vitelli V., Sharma S., Gioia U., Pessina F., Cabrini M., Wang Y., Capozzo I., Iannelli F. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017;19:1400–1411. doi: 10.1038/ncb3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F., Jalihal A.P., Francia S., Meers C., Neeb Z.T., Rossiello F., Gioia U., Aguado J., Jones-Weinert C., Luke B. From “Cellular” RNA to “Smart” RNA: Multiple Roles of RNA in Genome Stability and Beyond. Chem. Rev. 2018;118:4365–4403. doi: 10.1021/acs.chemrev.7b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiani D., Ronato D.A., Brosey C.A., Arvai A.S., Syed A., Masson J.Y., Petricci E., Tainer J.A. Targeting Allostery with Avatars to Design Inhibitors Assessed by Cell Activity: Dissecting MRE11 Endo- and Exonuclease Activities. Methods Enzymol. 2018;601:205–241. doi: 10.1016/bs.mie.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S., Ferguson J.R., Symington L.S. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.D., Yadav T., Giri S., Saez B., Graubert T.A., Zou L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell. 2017;65:832–847.e4. doi: 10.1016/j.molcel.2017.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolette M.L., Lee K., Guo Z., Rani M., Chow J.M., Lee S.E., Paull T.T. Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2010;17:1478–1485. doi: 10.1038/nsmb.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell. 2016;167:1001–1013.e7. doi: 10.1016/j.cell.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Paull T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015;84:711–738. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- Paull T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell. 2018;71:419–427. doi: 10.1016/j.molcel.2018.06.033. [DOI] [PubMed] [Google Scholar]
- Paull T.T., Deshpande R.A. The Mre11/Rad50/Nbs1 complex: recent insights into catalytic activities and ATP-driven conformational changes. Exp. Cell Res. 2014;329:139–147. doi: 10.1016/j.yexcr.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull T.T., Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull T.T., Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc. Natl. Acad. Sci. USA. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessina F., Giavazzi F., Yin Y., Gioia U., Vitelli V., Galbiati A., Barozzi S., Garre M., Oldani A., Flaus A. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 2019;21:1286–1299. doi: 10.1038/s41556-019-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Zhang Y., Baller J.A., Voytas D.F. Histone H2AX and the small RNA pathway modulate both non-homologous end-joining and homologous recombination in plants. Mutat. Res. 2016;783:9–14. doi: 10.1016/j.mrfmmm.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Reginato G., Cannavo E., Cejka P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017;31:2325–2330. doi: 10.1101/gad.308254.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines D., Dvir A., Conaway J.W., Conaway R.C. Assays for investigating transcription by RNA polymerase II in vitro. Methods. 1997;12:192–202. doi: 10.1006/meth.1997.0471. [DOI] [PubMed] [Google Scholar]
- Roques C., Coulombe Y., Delannoy M., Vignard J., Grossi S., Brodeur I., Rodrigue A., Gautier J., Stasiak A.Z., Stasiak A. MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J. 2009;28:2400–2413. doi: 10.1038/emboj.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiello F., Aguado J., Sepe S., Iannelli F., Nguyen Q., Pitchiaya S., Carninci P., d’Adda di Fagagna F. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat. Commun. 2017;8:13980. doi: 10.1038/ncomms13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag N.M., Rafalska-Metcalf I.U., Balane-Bolivar C., Janicki S.M., Greenberg R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010 Jun;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., d’Adda di Fagagna F. In Vitro Detection of Long Noncoding RNA Generated from DNA Double-Strand Breaks. In: Badrinarayanan A., editor. SMC Complexes: Methods and Protocols. Springer; 2019. pp. 209–219. [DOI] [PubMed] [Google Scholar]
- Sharma S., Javadekar S.M., Pandey M., Srivastava M., Kumari R., Raghavan S.C. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015;6:e1697. doi: 10.1038/cddis.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A., Moiani D., Arvai A.S., Perry J., Harding S.M., Genois M.M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski T.W., Ficarro S.B., Holik J., Kim T., Rando O.J., Marto J.A., Buratowski S. Sub1 and RPA associate with RNA polymerase II at different stages of transcription. Mol. Cell. 2011;44:397–409. doi: 10.1016/j.molcel.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somesh B.P., Reid J., Liu W.F., Søgaard T.M., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell. 2005;121:913–923. doi: 10.1016/j.cell.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Symington L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016;51:195–212. doi: 10.3109/10409238.2016.1172552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson N.E., Aronson D.B., Burgess R.R. Purification of eukaryotic RNA polymerase II by immunoaffinity chromatography. Elution of active enzyme with protein stabilizing agents from a polyol-responsive monoclonal antibody. J. Biol. Chem. 1990;265:7069–7077. [PubMed] [Google Scholar]
- Trujillo K.M., Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50∗Mre11 complex. J. Biol. Chem. 2001;276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- Uphoff C.C., Drexler H.G. Detecting mycoplasma contamination in cell cultures by polymerase chain reaction. In: Langdon S.P., editor. Cancer Cell Culture: Methods and Protocols. Springer; 2004. pp. 319–326. [DOI] [PubMed] [Google Scholar]
- Vitelli V., Galbiati A., Iannelli F., Pessina F., Sharma S., d’Adda di Fagagna F. Recent Advancements in DNA Damage-Transcription Crosstalk and High-Resolution Mapping of DNA Breaks. Annu. Rev. Genomics Hum. Genet. 2017;18:87–113. doi: 10.1146/annurev-genom-091416-035314. [DOI] [PubMed] [Google Scholar]
- Wang Q., Goldstein M. Small RNAs Recruit Chromatin-Modifying Enzymes MMSET and Tip60 to Reconfigure Damaged DNA upon Double-Strand Break and Facilitate Repair. Cancer Res. 2016;76:1904–1915. doi: 10.1158/0008-5472.CAN-15-2334. [DOI] [PubMed] [Google Scholar]
- Wang W., Daley J.M., Kwon Y., Krasner D.S., Sung P. Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes Dev. 2017;31:2331–2336. doi: 10.1101/gad.307900.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W., Ba Z., Gao M., Wu Y., Ma Y., Amiard S., White C.I., Rendtlew Danielsen J.M., Yang Y.G., Qi Y. A role for small RNAs in DNA double-strand break repair. Cell. 2012;149:101–112. doi: 10.1016/j.cell.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Williams R.S., Moncalian G., Williams J.S., Yamada Y., Limbo O., Shin D.S., Groocock L.M., Cahill D., Hitomi C., Guenther G. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Vogel G., Coulombe Y., Dubeau D., Spehalski E., Hébert J., Ferguson D.O., Masson J.Y., Richard S. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012;22:305–320. doi: 10.1038/cr.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yüce Ö., West S.C. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 2013;33:406–417. doi: 10.1128/MCB.01195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Watanabe G., Morten M.J., Reid D.A., Rothenberg E., Lieber M.R. The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nat. Commun. 2019;10:3588. doi: 10.1038/s41467-019-11507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any dataset or codes.