

Abstract
Fibroblasts are the primary regulator of cardiac extracellular matrix (ECM). In response to disease stimuli cardiac fibroblasts undergo cell state transitions to a myofibroblast phenotype, which underlies the fibrotic response in the heart and other organs. Identifying regulators of fibroblast state transitions would inform which pathways could be therapeutically modulated to tactically control maladaptive extracellular matrix remodeling. Indeed, a deeper understanding of fibroblast cell state and plasticity is necessary for controlling its fate for therapeutic benefit. p38 mitogen activated protein kinase (MAPK), which is part of the noncanonical transforming growth factor β (TGFβ) pathway, is a central regulator of fibroblast to myofibroblast cell state transitions that is activated by chemical and mechanical stress signals. Fibroblast intrinsic signaling, local and global cardiac mechanics, and multicellular interactions individually and synergistically impact these state transitions and hence the ECM, which will be reviewed here in the context of cardiac fibrosis.
Introduction
Indiscriminate fibrosis and scarring are a defining feature of virtually every type of cardiac disease. This remodeling of the extracellular matrix (ECM) presents a tremendous hemodynamic burden for the heart and is a strong prognostic indicator of arrhythmogenesis and the progression to failure [1–3] Given the limited regenerative capacity of the myocardium, fibrotic scarring becomes a permanent feature of the heart’s anatomy rapidly advancing it towards failure [4]. For decades, cardiac fibrosis and scarring were viewed as a secondary pathologic response to myocyte dysfunction. Now, through the use of new mouse models with inducible cardiac fibroblast-specific genetic manipulation, it is apparent that fibroblast phenotype is a primary effector of maladaptive cardiac remodeling.
Fibroblasts are the primary cell type regulating cardiac ECM, which preserves myocardial tissue integrity and organ structure [5–7]. The long-standing paradigm has been that injury and disease stimuli, such as transforming growth factor-β (TGFβ), neurohumoral activation, and/or tissue stiffness initiate fibroblast activation followed by programmed differentiation into a myofibroblast phenotype [8,9]. The gold standard phenotypic markers of myofibroblasts include: formation of alpha smooth muscle actin (αSMA) stress fibers, which give these cells contractile function, the development of super mature focal adhesions, and the secretion of large amounts of ECM containing periostin (Postn) and the EDa splice variant of fibronectin (Fn-EDa) [9–11] . Within this context, the transition between quiescence and activated myofibroblast states was viewed as a discrete progression of phenotypic changes in which quiescent progenitor fibroblasts first become proto-myofibroblasts and then transition to a more mature myofibroblast state [8,12,13].
While the gold standard markers for a myofibroblast remain useful, developments in single cell transcriptional analysis have uncovered a much broader and more heterogeneous landscape of possible fibroblast cell states through disease, which is reviewed in depth in the article, “Cardiac Fibroblast Heterogeneity after Myocardial Infarction”, published in this Matrix Biology special issue [reference article] [14–17]. Traditionally, the switch to a myofibroblast phenotype is thought to be discrete and irreversible – meaning the activation of the myofibroblast gene program is a binary switch and the process therefore was termed “differentiation” [18]. Pseudotime analysis of cardiac fibroblasts following MI challenges the notion that fibroblast activation is a discrete switch, and instead supports the existence of a continuous trajectory of fibroblast cell states on the way to becoming a myofibroblast [15,16] (Figure 1).
Figure 1. Expanding the state space for cardiac fibroblast fate post-MI.
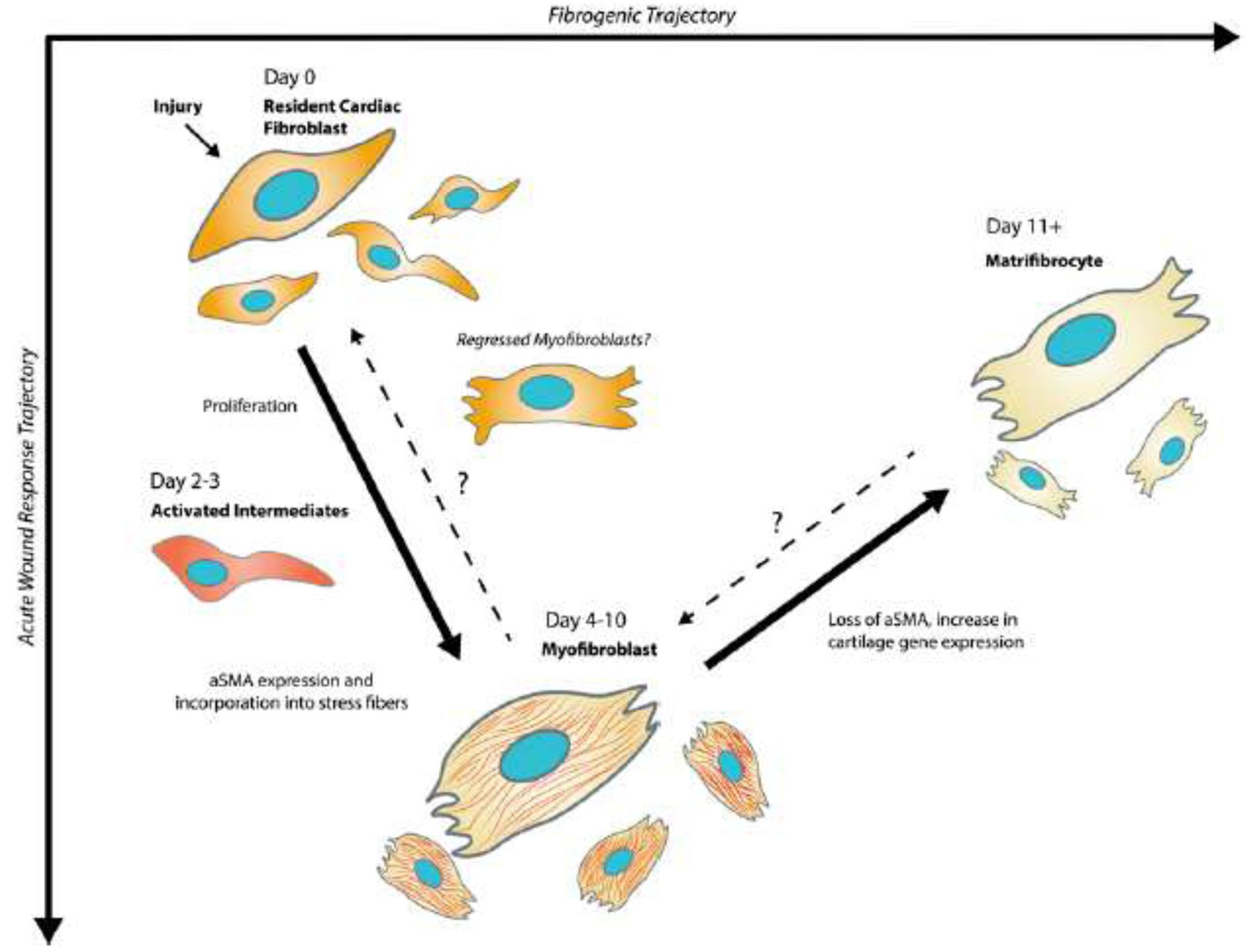
Upon injury, TCF21+ resident cardiac fibroblasts transition to a heterogeneous population of activated fibroblasts and myofibroblasts which are defined by the expression of Postn and αSMA stress fibers. Myofibroblast can also transition to a matrifibrocyte, which is defined by cartilage and osteogenic gene expression beinging around 2 weeks after myocardial infarction. Activated intermediates including fibroblasts which express periostin but not αSMA have been identified in vivo.
At 3 days after myocardial infarction, resident fibroblast populations transition toward an activated phenotype defined by intermediate levels of myofibroblast gene expression (Postn, Col1a1, and Acta2) [15]. As injury progresses through day 7, the majority of this activated fibroblast population shifts towards a myofibroblast state with 99% and 61% of these cells strongly expressing Postn and αSMA, suggesting previous myofibroblast state markers are still valid [15]. While the stability of the myofibroblast state is poorly understood, recent work in carbon tetrachloride injury of the liver, cryoinfarcted zebrafish hearts, and chronic angiotensin II-infused mouse hearts revealed that myofibroblasts become inactive rather than apoptose [19–21]. In all of these models, transition away from the myofibroblast state coincided with the regression of fibrosis and concomitant down-regulation of genes including Acta2, Postn, and type I collagen (Col1a), suggesting that the canonical myofibroblast could represent a highly transient and unstable cell state.
These results are very similar to those reported by Fu et al. that utilized Postn lineage tracing to track the genetic fate of activated cardiac fibroblasts [22]. The latter study investigated time points after the acute wound healing phase of myocardial infarction and found that myofibroblasts of the Postn lineage undergo an additional state change into matrifibrocytes – a phenotype defined by the expression of cartilage and osteogenic genes. Notably, many of these states have been validated in human cardiac tissue, suggesting that the work utilizing genetic mouse models is a powerful tool in understanding the underlying mechanisms responsible for heart disease in humans [22–24].
Collectively, these studies suggest cardiac fibroblasts rely on this ability to transit between diverse functional phenotypes to achieve structural and/or mechanical homeostasis throughout injury and its resolution. Although these studies have uncovered a larger spectrum of disease-induced fibroblast cell states, single cell studies lack longitudinal tracking of cell states over time. Because single cell RNAseq (scRNAseq) experiments only capture single snapshots in time, it is difficult to draw conclusions from the data about state stability, reversibility, and state intermediates that result as a fibroblast is on its way to becoming a myofibroblast. Also missing is the link between newly identified transcriptomic states and their impact on fibroblast function. More mature tools for spatial transcriptomics, synthetic lineage reporting, and single cell RNA velocities could help bridge the gap between fibroblast cell state and function and shed light on the relative stability or transience of the myofibroblast and its intermediate states in a disease setting [25–27].
While the field is taking advantage of next generation single cell sequencing approaches to rapidly identify a new spectrum of fibroblast and myofibroblast genomic phenotypes, our understanding of how these cell state transitions are controlled and their association with the composition and physical features of the ECM are lagging behind. Through curating the literature, the Saucerman lab has computationally derived a signaling network underlying fibroblast phenotype [28], which is being utilized to predict how dynamic signaling alters fibroblast phenotypes and fibrosis [reference Saucerman submission]. This network analysis serves as a robust predictor of master genetic regulators of myofibroblast cell state, yet each node requires in vivo validation. With the recent engineering of two different cardiac fibroblast-specific tamoxifen-inducible Cre knock-in mouse models, Tcf21-MerCreMer (resident cardiac fibroblast Cre) and Postn-MerCreMer (activated fibroblast Cre) the field has been able to interrogate the molecular regulators of cardiac fibroblast cell state and how these transitions impact both compensatory and maladaptive ECM and cardiac muscle remodeling [21,29]. Notably, these new genetic mouse models permit for the first time the interrogation of cardiac fibroblast biology in the same way the α-myosin heavy chain promoter transformed the field’s understanding of cardiac myocyte biology. So, this review will focus on the chemical and mechanical signaling pathways that regulate cardiac fibroblast cell state transitions with emphasis on those studies that validated these regulators using cardiac fibroblast-specific genetic approaches (Figure 2).
Figure 2. Nodal regulators of cardiac fibroblast cell state.
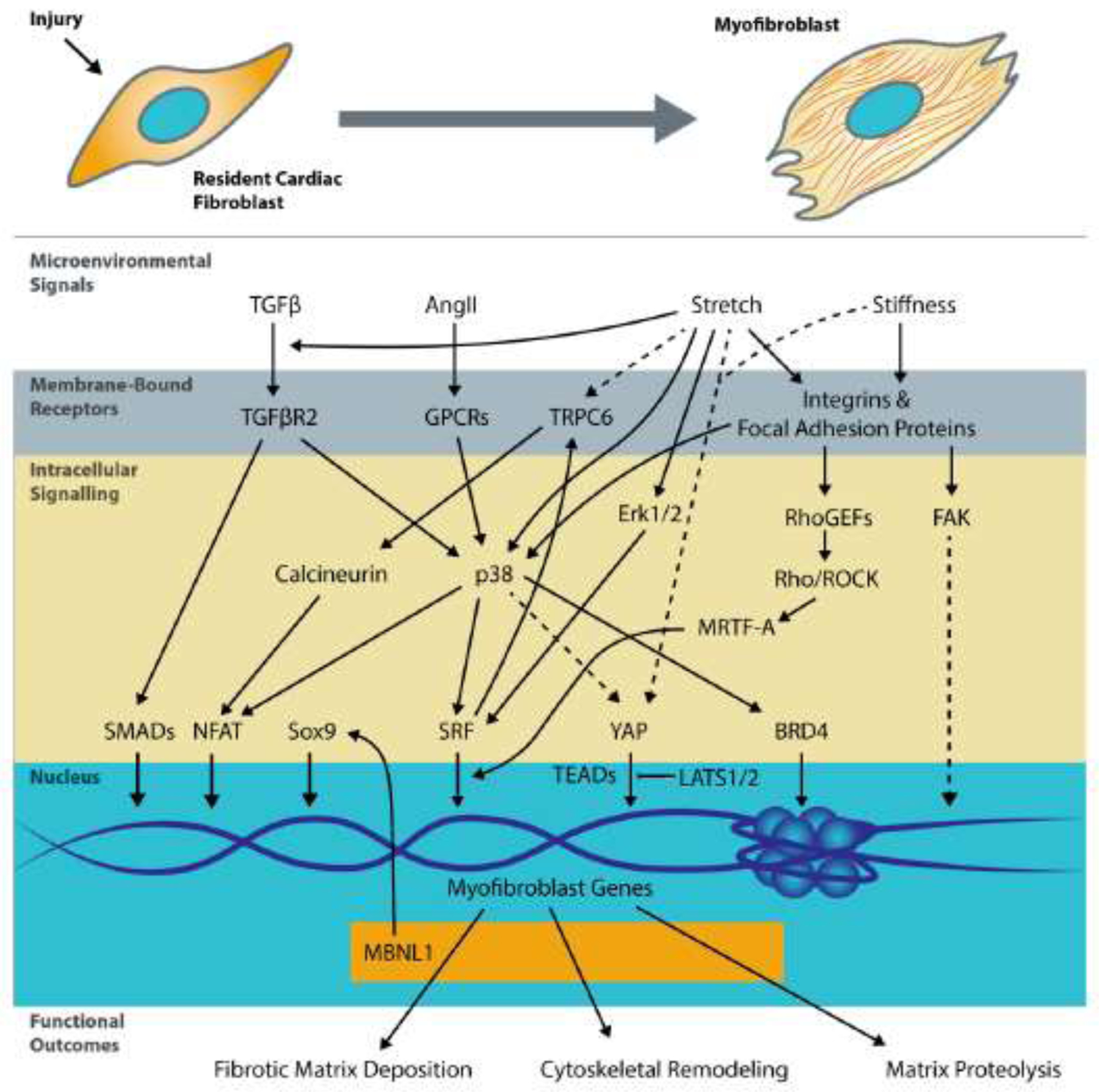
Extracellular paracrine and microenvironmental signals including TGFβ Angiotensin II, stretch, and matrix stiffness are transduced through. Following transcription, myofibroblast transcripts and hence the proteome are further regulated by RNA binding proteins like MBNL1. Solid lines denote connections directly supported by evidence in the cardiac fibroblasts, whereas dashed lines are based on data from other fibroblast types.
Transcriptional Control Through The p38 Regulatory Node
MAPK p38 has expansive function ranging from post-translational modification of transcription factors like serum response factor (SRF) and nuclear factor of activated T cells (NFAT) to the modulation of chromatin accessibility [30]. There are four p38 isoforms (α, β, δ, γ) each encoded by a different Mapk gene. In cardiac tissue, p38α is the most abundantly expressed isoform followed by low level expression of p38 δ and γ with no expression of the β isoform [31]. Similar to results from whole cardiac tissue, lineage traced Tcf21+ cardiac fibroblasts express p38α and γ but not β at the protein level, and four days after ischemic injury, when fibroblasts are activating and changing their cell state, p38α and γ are upregulated and phosphorylated in fibroblasts of the Tcf21 lineage [32]. This rise in p38α is essential to mounting an appropriate fibrotic response to myocardial infarction, as targeted deletion of p38α in Tcf21 fibroblasts significantly reduced the total area of fibrosis and scarring. These mice failed to form αSMA+ myofibroblasts due to a p38-dependent differentiation defect rather than impaired fibroblast proliferation, which would have reduced the pool of fibroblasts available to activate. Moreover, candidate analysis of myofibroblast gene expression revealed that p38α is necessary for the expression of key extracellular matrix genes like Col1a1 and Fn-EDa. Similar to ischemic injury, p38 activity is heightened in cardiac fibroblasts with 10 days of pressure overload [33]. Concomitant with the upregulation of p38 activity were myofibroblast genes like Postn, Acta2, and Col1a1 and 2. Experimentally increasing p38α activity by overexpressing a constitutively active form of MKK6 was also sufficient for changing quiescent Tcf21 fibroblasts into αSMA+ myofibroblasts and generating both interstitial and perivascular fibrosis in the absence of injury. Taken together, these data demonstrate that p38 induces a state transition from quiescent to myofibroblastic and that controlling its activity can significantly impact how cardiac fibroblasts produce ECM with and without injury.
Identifying which signals induce and maintain cell state changes is fundamentally important, yet the field is still struggling to understand whether these signals are necessary to maintain the myofibroblast state and its corresponding impact on the fibrotic outcome. In infarcted mice, loss of p38α function in activated Postn+ myofibroblasts reduced the total number of αSMA+ myofibroblasts by nearly 50% along with the total area of fibrosis, suggesting that p38 is necessary for both initiation of the myofibroblast phenotype but also its maintenance [32]. In the converse experiment where p38 activity was maintained in activated Postn+ fibroblasts by overexpressing a constitutively active MKK6 transgene both fibrosis and left ventricular hypertrophic remodeling was exacerbated in response to myocardial infarction and chronic angiotensin II exposure. Interestingly, knocking out p38α after activation was not as effective at reducing the fibrotic response or the number of myofibroblasts when compared to knocking out p38 function in quiescent Tcf21 fibroblasts prior to injury. The underlying reason for this difference is yet unclear but could be due to longevity of the ECM that was deposited between the time of myocardial infarction and turn-over of the p38 protein after recombination. An alternative explanation is that some of these cells were already transitioning to a matrifibrocyte state, which may not be regulated by p38. In spite of the explanation, these results underscore the importance of understanding the association between the timing of fibroblast cell state transitions and ECM remodeling dynamics.
Unlike myocardial infarction injury, p38α deletion in activated fibroblasts of the Postn-lineage was very effective at reducing fibrosis in an angiotensin II/phenylephrine model of interstitial fibrosis, suggesting that the association between fibroblast cell state and ECM phenotype depends on the type of injury or disease stimulus [32]. For instance, myocardial infarction initiates cardiac myocyte cell death and inflammation, which typically is not observed in a standard 2-week bout of chronic angiotensin II exposure, indicating the prolonged immune response associated with ischemic injury or lack of regenerative potential could markedly impact fibroblast state transitions and/or ECM remodeling [34,35].
Both chemical and mechanical signals in the injured myocardium trigger p38-mediated fibroblast-to myofibroblast state change. Traction forces, which are sensed by integrin receptors and the actin cytoskeleton, cause p38 to translocate to the nucleus where it initiates cytoskeletal remodeling [36,37]. Indeed, cyclic stretching of fibroblasts to supraphysiologic strains at frequencies similar to the average human heart rate initiates p38-dependent incorporation of αSMA into stress fibers, a fundamental morphologic and genomic characteristic of the myofibroblast phenotype [32]. Work in cancer cell lines also indicates that p38 activity is initiated with osmotic and environmental stress but that has yet to be confirmed in cardiac fibroblasts [38].
Chemical pathways converging on p38 include signals induced by TGFβ and angiotensin II, which work through TGFβ receptor II and G-protein coupled receptors, respectively (Figure 2). In vitro approaches using either pharmacologic inhibition or genetic silencing of p38 robustly block TGFβ and angiotensin II-mediated myofibroblast formation [32,39]. In addition, studies in lung fibroblasts also suggest that myofibroblast contraction is induced by mitochondrial biogenesis and aerobic glycolysis that require p38 signaling to induce the new metabolic program [40]. Further support that metabolic reprogramming to enhanced glycolysis transitions fibroblasts into myofibroblasts was gleaned through the deletion of the mitochondrial Ca2+ uniporter (MCU1) in Col1a2 positive fibroblasts, which increased the fibrotic response and number of myofibroblasts in the heart after myocardial infarction and chronic angiotensin II [41]. This state transition was driven by metabolic demethylases that promote the expression of myofibroblast genes. While p38 wasn’t examined in this study, evidence in the lung suggests it might drive metabolic based state transitions [40]. Taken together, p38 signaling appears to be a nodal transducer of diverse inflammatory, mechanical, and metabolic signals that turn on the myofibroblast gene program. The combinatorial actions and integrative effects of these mechanical and chemical signals on fibroblast to myofibroblast transition are not fully sorted out with respect to whether these signals are additive or synergistic, but their convergence on p38 suggests that there may be redundancy in these pathways.
Once activated, p38 utilizes diverse downstream signaling mechanisms to promote the programmed transition of cardiac fibroblasts into myofibroblasts. There are several downstream myofibroblast gene targets like Acta2 and Col1, which have profound drops in expression when p38 is silenced [42–44]. While p38’s role in regulating transcription factor activity is well known, a recent study in cardiac fibroblasts revealed that p38 also regulates TGFβ -induced recruitment of the chromatin reader BRD4 to myofibroblast genes like Postn, Acta2, and Sertad4 [45], suggesting that p38 regulates myofibroblast gene transcription by post-translationally modifying transcription factor activity and more directly through interactions with the transcriptional machinery [46]. Indeed, transcription factors like serum response factor (SRF) or MAPK-activated protein kinase 2 (MK2) require p38 function to initiate myofibroblast gene expression [40,47]. MK2’s role in cardiac fibroblast state transitions and fibrosis has not been directly tested in vivo but genetic silencing of MK2 in lung enhances the fibrotic response to bleomycin [47]. In comparison SRF has been heavily studied in cardiac fibroblasts [40,48]. SRF mediates fibroblast activation through binding of GArG boxes (CC(AT)6GG) at the promoters of myofibroblast genes like Acta2 and Col1 [59,60]. Targeted deletion of SRF in Tcf21+ resident cardiac fibroblasts prevented fibroblasts from transitioning to a myofibroblast state and significantly reduced the fibrotic response to myocardial infarction, making these mice prone to cardiac rupture.
SRF-mediated transcription of myofibroblast genes is highly sensitive to actin-dynamics and enhanced by myocardin related transcription factors (MRTF-A and B) cofactors [49]. Changes in mechanical force and cell shape activate Rho-Rho kinase (ROCK) signaling, re-organizing the actin-cytoskeleton and promoting the translocation of MRTF-A to the nucleus where it can enhance SRF-dependent gene transcription. MRTF-A knockout mice subjected to either angiotensin II stimulation or myocardial infarction had decreased scar formation, which is directly connected to Col1a2 expression [49]. Unlike mice lacking endogenous expression of p38 or SRF in the fibroblast, MRTF-A knockout mice are not prone to rupture. The reliance of MRTF-SRF signaling on actin dynamics suggests that it is important in later stages of fibroblast injury response.
SRF’s essential functions encompass more than just the expression of phenotypic myofibroblast genes. Its activity is also required for initiating an intracellular Ca2+ signaling cascade that is both necessary and sufficient for promoting the myofibroblast phenotype and fibrosis [39]. Here, the initiation of intracellular Ca2+ signaling is due to the increased expression of transient receptor potential cation (TRPC) 6 channels at the fibroblast membrane. SRF alone induces TRPC6 transcriptional activity and expression in cardiac fibroblasts, but the addition of TGFβ boosted the occupancy of the TRPC6 promoter and synergistically increased TRPC6 promoter activity, all of which could be blocked with a p38 inhibitor. It takes 2 days for treatments with either TGFβ, angiotensin II, or constitutively active MKK6 to observe measurable differences in Ca2+ signaling and that change is only apparent in TRPC6 replete fibroblasts. Notably, TRPC6 knockout fibroblasts have no change in Ca2+ signaling and fail to convert into myofibroblasts when treated with these same profibrotic agonists or ectopic overexpression of SRF. In accordance with these data, TRPC6 knockout mice are prone to cardiac rupture and form significantly fewer myofibroblasts with less scarring in response to myocardial infarction. Interestingly, increased intracellular Ca2+ signaling alone seems insufficient for inducing a myofibroblast phenotype, as overexpression of TRPC4 and TRPC3 failed to induce myofibroblast transition equivalent to that observed with TRPC6. However, other TRP family channels like TRPM7 and TRPV4 also transit fibroblasts into a myofibroblast fate, which suggests there are channel-specific properties like the presence of Ca2+ sensitive signaling proteins within the channel microdomains that help transduce changes in intracellular Ca2+ into myofibroblast gene expression [50–52]. With TRPC6, Ca2+-activated calcineurin-NFAT signaling was essential for driving myofibroblast transformation in cardiac, embryonic, and dermal fibroblasts [39].
In cancer cell lines and drosophila, p38 has also been implicated in regulating the transcriptional co-activator yes associated protein (YAP), which causes cancer-associated fibroblasts to express fibrotic ECM [53]. While the relationship between YAP and p38 has not been studied in cardiac fibroblasts or fibrosis, it was demonstrated that retaining YAP in the cytosol is essential for maintaining cardiac fibroblast quiescence [54]. Evidence for that conclusion came from elegant studies in which the deletion of Hippo kinases, Lats 1 and 2 from mature Tcf21+ fibroblasts caused them to spontaneously transition into myofibroblasts concomitant with a fulminant fibrotic response in the absence of injury. In these mice, YAP occupied the promoters of genes associated with a myofibroblast phenotype that were also proinflammatory. YAP often works in conjunction with the TEAD family of transcription factors all of which had increased expression in myofibroblasts. Indeed, TEAD regulons were enriched and heightened occupancy of TEAD-YAP motifs were identified following myocardial infarction and in cardiac fibroblast specific loss of Lats 1/2 function. It appears from these data that the role of Lats 1 and 2 is to maintain the quiescent fibroblast state through cytosolic retention of YAP. This also fits with developmental data showing that loss of Lats 1 and 2 in epicardial progenitors prevents quiescent resident cardiac fibroblasts from fully maturing [54]. Notably, TEAD and p38 directly interact in cell lines exposed to mechanical stress [38], which further suggests that combinatorial interactions between p38 MAPK and YAP-TEAD signaling underlie cardiac fibroblast state transitions and the stability of a given fibroblast phenotype.
The convergence of so many pathways on p38 and the profound reduction of the heart’s fibrotic response when p38 is silenced suggests therapeutically targeting this pathway could effectively mitigate cardiac fibrosis. Several studies have utilized pharmacologic inhibition of p38 to rescue various animal models of heart disease including pressure-overload, myocardial infarction, and muscular dystrophy. Notably, these studies were actually targeting cardiac myocyte remodeling rather than the fibroblast or fibrosis specifically, but in all cases fibrosis was reduced with p38 inhibition [40,55,56]. For instance, mice treated with p38 inhibitors beginning 2 weeks after myocardial infarction for a duration of 14 weeks had a significant reduction in fibrosis and myocyte cross sectional area [57]. Similarly, 20 weeks of p38 inhibitor treatment reduced fibrosis and maladaptive cardiac remodeling in Bio14.6 dystrophic hamsters with dilated cardiomyopathy [56]. In two different mouse models of muscular dystrophy p38 inhibition reduced skeletal muscle fibrosis and myocyte cell death [58]. Also, a number of clinical trials have used p38 inhibitors to address human cardiovascular disease [58]. While most of these trials were aimed at reducing inflammation in atherosclerosis, in an independent trial p38 inhibitors were shown to reduce scarring in non–ST elevation myocardial infarction. These data lend support for the potential of p38 inhibition as an antifibrotic therapy, but we have yet to examine their use in explicitly targeting fibrosis.
Proteomic Control Through Transcriptome Maturation
A majority of the work on the regulation of fibroblast fate has been centrally focused on gene regulatory mechanisms with little attention given to post-transcriptional regulation of RNA fate, which ultimately controls the proteomic landscape. RNA binding proteins simultaneously regulate thousands of RNAs by dictating all facets of their biogenesis, including alternative splicing, polyadenylation and transcript localization, which in turn dramatically alters the functional proteome to match homeostatic demands as seen in cancer or cardiac remodeling [59–63]. This ability to create sweeping changes in thousands of genes in any cell type just by modulating RNA binding proteins makes them potentially powerful targets for controlling cell state. RNA binding proteins had not been investigated as regulators of cardiac fibroblast state transitions until recently, when muscleblind-like 1 (MBNL1) was identified in an unbiased gain of function genome-wide screen for inducers of myofibroblast differentiation [48]. This finding also led to the discovery that MBNL1 expression is extremely low in quiescent fibroblasts but increases 4 days after myocardial infarction or in response to TGFβ-dependent activation in vitro. Fibroblasts lacking MBNL1 expression were unable to become myofibroblasts when treated with profibrotic agonists. As expected from these data, infarcted MBNL1 knockout mice had reduced fibrotic area and numbers of αSMA+ myofibroblasts, which decreased the severity of maladaptive left ventricular remodeling. In the converse experiment, transgenic expression of MBNL1 in Tcf21 fibroblasts enhanced the level of fibrosis after 2 weeks of chronic angiotensin II infusion. RNA immunoprecipitation followed by RNAseq identified that nearly 2500 transcripts bound to MBNL1 in activated fibroblasts. These transcripts were highly enriched for functions involved in chemokine signaling, TGFβ signaling, MAPK signaling, actin cytoskeletons, focal adhesion, adherens junctions, and cell cycle regulation, suggesting MBNL1 may be a key regulatory factor of cardiac fibroblast state. The striking feature of these data were that many of the MBNL1-bound transcripts were previously identified regulators of myofibroblast differentiation including TGFβR2, SRF, and calcineurin. As reviewed above, each one of these factors plays an essential role in shifting a fibroblast toward a myofibroblast fate. Specifically, MBNL1 was stabilizing and alternatively splicing these transcripts creating a gain of function in each one of these pathways. These studies were limited in their use of a global MBNL1 knockout mouse. To combat this limitation, multiple fibroblast specific studies were performed in vitro; however, it is still unclear what the contribution of fibroblast-specific MBNL1 function is in the heart’s fibrotic response as MBNL1 is also expressed in cardiac myocytes and red blood cells [64–66].
In RNA immunoprecipitation studies, several other potential regulatory factors were identified, including the transcription factor Sox9, which was bound and upregulated in cardiac fibroblasts overexpressing MBNL1. While not further pursued in the MBNL1 studies, two independent laboratories recently showed that Sox9 is critical for the myofibroblast phenotype and might also regulate the third transition to a matrifibrocyte identity after ischemic injury [67,68]. In one of the studies, Sox9 was identified as a potent mediator of fibroblast activation via nonbiased tomo-sequencing. In the mouse heart, Sox9 has binding sites in well-known matrix genes such as Col1a2, Fn1, Lum and Vim suggesting that Sox9 aids in the transcription of key ECM transcripts involved in fibrotic remodeling post injury [67]. Sox9 expression levels are relatively low in the adult heart, but it is highly expressed during development as it promotes epithelial-to-mesenchymal transition and ECM organization [69,70]. To confirm Sox9 is required for promoting the myofibroblast phenotype in vivo, heterozygous Sox9 floxed mice were bred with a ubiquitously expressed tamoxifen-inducible Cre and the offspring subjected to ischemic injury. These mice had a decreased number of Sox9-expressing cells as well as downregulation of other downstream matrix genes including Col1a2, Col3a1, Col1a1, and Fn1, along with reduced fibrosis and scarring following ischemia-reperfusion injury. Similar results were observed when Sox9 was genetically excised from activated cardiac fibroblasts using a Postn-Cre driver. Loss of Sox9 in the myocardial infarction-activated fibroblast not only reduced fibrotic area but inhibited αSMA+, proinflammatory, and matrifibrocyte (Chad, Comp, and Cilp2) gene expression. Taken together these results suggest that Sox9 promotes a proinflammatory myofibroblast similar to the phenotype induced with dysregulated Lats-Yap signaling.
Microenvironmental Control
The cardiac fibroblast is also highly sensitive to microenvironmental cues, which are constantly changing as the heart dynamically remodels after an ischemic injury or myocardial infarction. This is both promising and challenging for uncovering a treatment of fibrosis and heart failure as interventions targeting maladaptive microenvironmental signals, such as controlling scar mechanics [71], delivering proteoglycans for chemical and mechanical control of fibroblasts and collagen structure [72,73], delivering exogenous ECMs to promote cell migration and collagen degradation [74] could be sufficient to halt or reverse fibrosis. Many of these preclinical therapies rely on uncharacterized cell adaptations to a potentially more favorable extracellular environment, one that is mechanically stabilized or promotes protease activity to disrupt the scar, in hopes of bringing the injury environment to homeostasis. Unfortunately, our knowledge about how the extracellular environment impacts fibroblast state is lacking, owing to the limited platforms for reductionist experimentation and dynamic control of microenvironmental signals. In vitro manipulation of substrate stiffness has shown that fibroblasts can be mechanically controlled to increase contractility and ECM secretion, through a variety of the mechanosensitive and integrin-linked pathways with proven in vivo relevance.
Substrate stiffness is the most extensively studied property of the cardiac microenvironment, yet varying mechanical characterization techniques produce diverse moduli to represent the stiffness of healthy cardiac muscle, and care should be taken to use consistent measurement and analysis methods when comparing an engineered microenvironment to native myocardium (Table 1). Moduli reported for tensile testing vary widely due to the lack of a consistently accepted models to fit stress-strain curves. While Atomic Force Microscopy (AFM) does not capture the directional properties of the myocardium, consistency in data acquisition and analysis has led the literature to converge on an estimated compressive elastic modulus of 12–35 kPa for intact rat, mouse, and pig myocardium [75–80]. By a functional definition, healthy myocardial ECM helps cardiac myocytes maximize work output, thus contributing to overall tissue contractility [81]. In effect, this means that the extracellular stiffness is in balance with the stiffness of a myocyte. Early biaxial studies also revealed that healthy myocardium is mechanically anisotropic, with greater stiffness in the direction of muscle fibers [82,83]. Regional gradients of stiffness throughout the layers of cardiac muscle exist, with the stiffest tissue towards the epicardium [84]. Following myocardial infarction or other acute injury, the fibrotic tissue deposited is drastically stiffer in terms of moduli above 50 kPa [75] but weaker in terms of yield strength. Both myocytes and ECM, which are likely in feedback with one another, contribute to the bulk tissue stiffness both at baseline and through disease progression. Myocytes match their stiffness as measured by AFM to the stiffness of their substrate, but the role of fibroblasts in maintaining homeostasis of matrix stiffness to match myocyte stiffness has not yet been fully explored [85]. In culture, fibroblasts appear to adapt their stiffness to match the stiffness of their substrate, causing dramatic alterations in cell morphology and behavior [86]. Hyaluronan (also referred to as hyaluronic acid, or HA) hydrogels with compressive Young’s modulus of 3–50 kPa, the threshold for adult mouse cardiac fibroblast stress fiber formation lies in the range of 3–8 kPa [87]. Fibroblasts on 50 kPa hydrogels assumed a pro-contractile phenotype with super-mature focal adhesions and well developed αSMA stress fibers. The 8 kPa “physiologically stiff” gels facilitated lower mRNA expression of an array of ECM remodeling genes, indicating that physiologic stiffness may help to maintain fibroblasts in a quiescent state. Substrate stiffening and fibroblast contractility are linked through focal adhesion size, as stiffer substrates promote larger focal adhesions, which are required for αSMA polymerization into stress fibers [88].
Table 1.
Stiffness measurements of myocardium and cardiac ECM
| Paper | Sample Tested | Measurement | Analysis | Reported stiffness | Key Findings |
|---|---|---|---|---|---|
| Exponential stress-strain function | |||||
| A = 3.9±2.7kPa (cross-fiber direction) | directions. | ||||
| Halperin 1987 [91] | Canine IVS | Triaxial; Tensile in-plane with transverse compression | Slope of indentation peak stress-strain curve | E = 14.8kPa (Transverse Compressive) | In-plane stress and strain indices positively correlated with transverse stiffness. |
| Przyklenk 1987 [92] | Canine LVFW | Uniaxial tensile | TM at 2g/mm2 | TM = 2–4kPa | Tensile strength and stiffness correlated positively with hydroxyproline content. Epicardium and visceral pericardium were the stiffest and most collagenous. |
| Yin 1987 [93] | Canine LV | Biaxial tensile | Fit three strain-energy functions | See paper for functions and parameters | |
| Humphrey 1990 [94] | Canine LV, RV walls | Biaxial tensile | Stress strain curves but no moduli reported | ||
| Sacks & Chuong 1993 [83] | Canine RVFW | Biaxial tensile | Reported the maximum TM of generated stress-strain curves | ||
| TM = ~50kPa (LVFW, both) | |||||
| Novak 1994 [84] | Canine IVS and LVFW | Biaxial tensile | Five parameter pseudostrain-energy function | Several fitted parameters | Inner and outer regions of the LVFW were stiffer than the middle sections tested. |
| Kag 1996 [95] | Bovine endo, myo-, epicardium | Biaxial tensile | Pseudostrain -energy functions | Several fitted parameters | Endocardium was stiffer at low strains. |
| Rat isolated myocytes | Classical Infinitesimal Strain Theory – Conical Geometry | Myocytes stiffened with age, and intracellular stiffness was independent of myocyte dimensions. | |||
| E = 42.5±1kPa (30 months age) | |||||
| Berry 2006 [75] | Rat LV; infarcted and control | AFM indentation | Hertz Model | Infarcted rat hearts were stiffer than control/healthy tissues. | |
| E = 55±15kPa (infarct) | |||||
| Engler 2008 [81] | Quail, embryonic | AFM indentation | Hertz model | E = 11 kPa | |
| Fomovsky, Holmes 2010 [76] | Rat LV; infarcted | Biaxial tensile | Strain-energy function quadratic fit | C1 = 400kPa stiffening to ~1mPa | Infarcted rat hearts increased in collagen content, collagen crosslinking, and biaxial tensile strength over time from 3 to 6 weeks after Ml. Infarcts were mechanically isotropic rather than anisotropic. |
| Jacot 2010 [77] | Murine epicardial surface E13.5 to P14 | AFM indentation | Hertz Model | ||
| E = 39±7kPa (P14) | |||||
| 2013 [97] | tensile | modulus (60–70% strain; linear region) | E = 20kPa (adult) | ||
| Kichula 2014 [98] | Explanted Ovine LV | Biaxial tensile | Longitudinal stiffness was significantly higher than circumferential stiffness in passive measurements of control heart tissue, highlighting the importance of the method choice for reporting overall myocardial stiffness. | ||
| E at strains 0.05–0.1; 0.1–0.15; 0.15–0.2 | |||||
| E = 70kPa (circumferential) | |||||
| Polyacrylamide gels of myocardial-like stiffness were optimal for the In vitro culture of rat cardiomyocytes. | |||||
| E = 25.6±15.9kPa (adult) | |||||
| Human heart transplant biopsies | |||||
| E~26±12kPa (cross-fiber) (5% E calculated from table 10) LVFW: 54kPa Septum: 48kPa RVFW: 44kPa | |||||
| Perea Gil 2015 [79] | Porcine LV, intact vs. dECM | AFM indentation | Hertz Model | Native tissue: 26.1 ±3.6kPa, | Decellularization caused no significant changes to E. There were no significant differences in stiffness between heart layers. |
| Quinn 2016 [101] | Rat LV ECM | Uniaxial tensile | Microstructur al fiber recruitment model: E at full fiber recruitment and stretch of 1.25 | ECM elastic modulus did not differ between circumferential and longitudinal stretching. In the postinfarct scar, collagen content increased but tensile elastic modulus decreased. The non-linear toe regions of the stress-strain curves were elongated following Ml. | |
| E1.25 = 38 ±26kPa (8wks post Ml) | |||||
| from Chen et al, 1996[103] | E = 63±23kPa (porcine) | (PDMS) | |||
| Gluck 2017 [104] | Porcine dECM LV and SAN | AFM indentation | |||
| E = 16.69 ±0.32kPa (SAN) | |||||
| Spreeuwel 2017 [80] | Murine LV and RV, mdx and TAC models | Microindentati on, 2mm probe | Indentation model for planar anisotropic soft tissue from Cox et al 2006 [105] | Mdx mutation significantly lowered myocardial LV modulus but not RV modulus. TAC did not have a significant impact on overall LV modulus. | |
| ERV = 11.1±3.9 kPa (healthy) | |||||
| Notari 2018 [106] | Murine neonatal dECM LV, P1/P2 | AFM indentation | Hertz Model | ||
| E~35–45kPa (P2) | |||||
| Fujita 2018 [107] | Goat dECM ventricle | Uniaxial compression | Nonlinear Kelvin Model | ||
| K2=0.1kPa |
Heart Anatomy Abbreviations: LV = left ventricle; IVS = interventricular septum; LVFW = left ventricular free wall; RVFW = right ventricular free wall; SAN = sinoatrial node; dECM = decellularized extracellular matrix; MI = myocardial infarction; TAC = trans-aortic constriction
Mechanics Abbreviations: A = stress-strain amplitude parameter; TM = tangent modulus; E = Young’s/elastic modulus; K1, K2 = spring stiffness elements; C1 = Cauchy-Green fitted coefficient; DMA = dynamic mechanical analysis
The role for mechanosensitive cues alone is exemplified in a simple model utilizing ring-shaped collagen gels seeded with rat neonatal cardiac fibroblasts. Tensile stiffness of these gels can be increased by activating fibroblasts through the Rho/ROCK pathway [89]. Specifically, p63RhoGEF activation upstream of Rho/ROCK potentiated the pro-fibrotic transcriptional response of fibroblasts to angiotensin II [89]. This aligns well with in vivo data proposing this pathway is more related to the maintenance of the myofibroblast phenotype than initial activation, although this has not been directly examined in vivo. More recently, the same hydrogel system was used to show an increase in engineered tissue stiffness and compaction with TGFβ activation, which was prevented by ROCK inhibition [90]. These findings are particularly interesting as they show that inhibition of a mechanosensitive signaling pathway is sufficient to prevent fibrotic outcomes from biochemical activation, opening up questions regarding the hierarchical control of fibroblast state transitions [89,90].
Dynamic stiffening of biomaterials has also been harnessed to explore fibroblast response to the evolution of scar mechanics [108–110]. Fibroblasts activated by TGFβ on stiffening silk hydrogels assumed a pro-contractile phenotype highlighted by increased protein levels of αSMA and vinculin, integrins, and integrin-linked kinases (ILK) [108]. Murine adult cardiac fibroblasts have also been activated on photostiffening HA hydrogels in the absence of exogenous fibrotic agonists [87]. While Col1a1 and 2 transcription increased and Col3a1 transcription decreased, no changes in TGFβ expression levels were observed, suggesting cardiac fibroblasts on these synthetic platforms could be activated along pathways independent of canonical TGFβ signaling. Photostiffening PEG hydrogels have also been employed to dynamically stiffen from a Young’s modulus of 10 kPa to 90 kPa, mimicking a preexisting AFM study of the mechanics of postinfarct scar [75,110]. Rat cardiac fibroblasts on dynamically stiffened gels exhibited increased cell area and decreased nuclear roundness, as well as enhanced nuclear localization of NFAT over 5hrs on the dynamically stiffened gels compared to the static 10 kPa and 50 kPa controls. Calcineurin/NFAT signaling has been implicated in fibroblast activation, but prior to the development of dynamic biomaterials, study of this translocation event in response to mechanics has not been possible on an acute timescale [39]. Altogether, in vitro studies of dynamic stiffening suggest that mechanical changes alone can induce contractile and secretory behaviors in the cardiac fibroblast independent to TGFβ signaling.
Softening materials have also been explored, with the hopes of reversing pro-fibrotic fibroblast trajectories. For example, global softening of a photosensitive polyacrylamide dramatically reduced the spread area of 3T3 fibroblasts in culture, while regional softening also revealed a propensity for fibroblasts to migrate away from the softened area [111]. Light irradiation of photo-softening PEG has been used to reverse the αSMA-positive contractile cell state of activated valvular interstitial cells, with no changes to proliferation markers or cell count [112].
Studies employing substrates with reversible mechanics are key to understanding whether stiffness induced changes to fibroblast cell state are also reversible. Cyclic stiffening of a photosensitive PEG hydrogel resulted in an increase in luciferase activity for 3T3 fibroblasts transfected with αSMA and Postn-luciferase reporter plasmids encapsulated within the gel [113]. Despite a lower average modulus of the photocycled material, αSMA and Postn activity were enhanced, suggesting that material dynamics rather than absolute stiffness dictated the fibroblast response. In a similar study of reversibility, a magnetic field was used to stiffen iron-particle laden PDMS on demand, enabling changes in the material’s compressive Young’s modulus to 10–55 kPa [114]. Human induced pluripotent stem cell derived (iPSC) fibroblasts were seeded on gels and subject to dynamic mechanical changes, either from soft-to-stiff or stiff-to-soft. Dynamic stiffening of the material promoted cell spreading, αSMA stress fiber formation, and YAP nuclear translocation. Interestingly, on substrates of intermediate stiffness, fibroblasts were more spread and had greater αSMA stress fiber formation transitioning from stiff-to-soft than vice versa. As none of the reversible mechanics studies have been performed in cardiac fibroblasts, a thorough study of the response of these cells to these engineered substrates would be useful in probing the cardiac microenvironmental cues on the fibroblast during the progression of or during the resolution of injury. These tools allow for spatially and mechanically, albeit not necessarily temporally, relevant disease models in a dish that better mimic the complexity of the in vivo environment.
Altered hemodynamic loading is a key factor contributing to cardiac hypertrophy and associated fibrosis during disease. Mechanical manipulation of fibroblasts has been studied on flexible 2D substrates for over a decade, yielding novel insights into the role of cardiac mechanics in mediating the progression of fibrosis. Adult rat cardiac fibroblasts on collagen-coated membranes subject to 10% strain upregulated transcription of Col3a1 and fibronectin three-fold and increased TGFβ protein activity [115]. Thus, it would appear that fibroblasts have enhanced secretory behaviors when under active stretch. Compression of fibroblasts can also induce a fibrotic response. Neonatal rat cardiac fibroblasts subject to cyclic compression assume a longer aspect ratio with elevated expression of αSMA and an increase in transcription of matrix and matrix metalloproteinase (MMP) genes, demonstrating that, in this system, the fibroblasts not only become contractile, but also secretory and proteolytic [116]. These effects were exacerbated by TGFβ activation of the 3D culture or inhibited by addition of a TGFβ inhibitor, again alluding to the potential hierarchy or additive effects of mechanics and biochemical signals. Studies including both individual mechanical perturbations as well as combined chemical and mechanical stimuli for fibroblasts allow for more complex conclusions to be made about microenvironmental factors affecting fibroblast cell state. However, it is rare in studies to date to include integrated input perturbations beyond mechanical manipulation with or without TGFβ. Cells isolated from genetic mouse models hijacking known fibroblast pathways should be combined with cytokine and growth factor stimulation in these various static and dynamic mechanical setups to further understand the synergistic effects of mechanics and biochemical signals in the injured tissue environment.
Fibroblasts sense stretch through a variety of pathways over fairly short timescales, and these pathways have been explored therapeutically in cardiac disease models. On membranes coated with various matrix proteins, neonatal rat cardiac fibroblasts cyclically stretched to 5% strain demonstrated phosphorylation changes in p42/44 MAPK (Erk1/2) over timescales as short as ten minutes [117]. This response was augmented by culture on aligned collagen or fibronectin. In vivo, small molecule inhibition of Erk1/2 phosphorylation suppressed MMP9 levels post-myocardial infarction, leading to a reduction in fibrotic scar area and improved cardiac function [118]. Genetic deletion of MMP9 attenuated post-myocardial infarction ECM remodeling, but Erk1/2 are essential for development and inducible, cardiac fibroblast specific knockouts have not yet been created [119].
Collectively, these data suggest that cyclic stretch and topography can synergistically effect fibroblast activation in vitro, which, in turn, activates pathways known in vivo to drive pathological remodeling. In stretched cultures of fetal rat CFs at non-physiologic strains of 20%, transient activation of p38 MAPK has also been observed within 10–15 minutes, tapering off after 60 minutes of continuous stimulation. Interestingly, in vitro activation of p38 through MKK6 was shown to negatively regulate procollagen gene expression, in contrast to the profibrotic in vivo effects reported for this pathway [32,120]. Thus, reductionist in vitro studies may not capture the full context-dependency of MAPK signaling driving fibroblast activation.
Downstream pathways of activation, such as MAPKs, are triggered by activation of integrins and their associated kinases. In accordance with in vitro data identifying FAK as a mechanically sensitive mediator of fibroblast state, systemic in vivo siRNA inhibition of FAK in mice diminished the hypertrophic and fibrotic response to transaortic constriction (TAC) [121]. Strain of atrial human cardiac fibroblasts also triggers the nuclear translocation of YAP after 72 hours of culture, with an enhancement in proliferation at lower levels of strain [122], and as reviewed above YAP activity in the nucleus spontaneously transitions cardiac fibroblasts into myofibroblasts [123]. Stretch can also act in synergy with stiffness and Ca2+- mediated pathways [87]. Polyacrylamide gels of varying stiffness attached to a PDMS membrane were also stretched to distinguish fibroblast responses differentially mediated by substrate stiffness and stretch. While stretch appeared to directly regulate the expression of αSMA mRNA and the phosphorylation of FAK, it did not facilitate the incorporation of αSMA into stress fibers [87]. The combined contributions of substrate stiffness and topographical cues have also been studied on polyacrylamide gels seeded with neonatal rat cardiac fibroblasts, demonstrating synergistic effects of these two microenvironmental factors in increasing cell elongation [124]. Stretch can also open stretch sensitive ion channels in the TRP channel family, causing Ca2+ influx to the fibroblast and a contractile, αSMA positive state [125].
Material considerations extending beyond elastic modulus include topology, viscoelasticity, and nonlinear mechanics. Fibroblasts cultured on very soft (100 Pa) fibrin gels can behave similarly to those cultured on a stiffer polyacrylamide gel due to local stiffness gradients generated by cell traction forces on a strain-stiffening fibrillar matrix [126]. Encapsulation of fibroblasts in a matrix can also increase the bulk shear moduli of these hydrogels, further complicating comparisons between mechanical measurements on acellular biomaterials and the highly cellular myocardium. The extracellular environment can influence cell behavior not just mechanically, but in synergy with other established chemical routes for fibroblast activation [127]. For example, TGFβ can be sequestered in the ECM and later released with pathological loading on the ECM. Computational models of fibroblast activation have also predicted mechanical biochemical crosstalk through the TGFβR1 receptor, in which inhibiting this receptor can attenuate mechanically-induced activation [28]. Reversible TGFβ patterning has been accomplished on a PEG hydrogel using bio-orthogonal photochemistries [128]. Seeding fluorescent Smad-reporter mouse embryonic fibroblasts onto TGFβ -patterned gels resulted in differential SMAD activity in the patterned areas.
An interesting method to distinguish local from global contraction in cell-laden 3D collagen gels was developed with fluorescent beads for cell-level measurements and whole gel contraction for global collagen compaction. Human foreskin fibroblasts cultured in 3D matrices demonstrated contractile cell morphology and focal adhesion organization in significantly compacted collagen gels compared to dendritic phenotypes with punctate focal adhesions in less compacted collagen. Rho kinase inhibition prevented cell morphology changes but did not prevent collagen matrix remodeling, indicating a distinction between fibroblast cytoskeletal rearrangement and collagen fibril translocation [129]. Additional studies could incorporate varying concentrations and/or crosslinking of input collagen as well as external stretch/strain. This would allow better understanding of the combined effects of 3D mechanics and dynamic stretching, which would more adequately represent the cardiac environment. In studies of cell alignment in 3D gels, dermal fibroblasts exhibit strain-dependent alignment, with parallel fibers in static and perpendicular in cyclic/dynamic strain [130,131]. The 3D environment makes controlling multifactorial perturbations more difficult than conventional culture.
Dermal fibroblasts cultured in 2D on various stiffnesses (E =1.2 kPa or E = 8.5 kPa) and under cyclic axial compression demonstrated ECM gene upregulation under mechanical loading compared to increased MMP secretion and contraction in response to the scaffold stiffness at initial time points, indicating a discrete biochemical response to scaffold mechanics vs. applied strain. The cyclic mechanical loading produced a preserved cell response over time – tissues reached converged Young’s moduli, compression force, and elasticity regardless of initial seeding scaffold stiffness [132]. It is unknown if cardiac fibroblasts respond similarly with this preference to respond to the cyclic force and remodel the environment to meet those demands rather than remembering the stiffness of the tissue from which they were derived. It would be useful to test cardiac fibroblasts in this system with physiological scaffold stiffness (12–35kPa) compared to an infarct environment (50+ kPa).
Fibroblasts from various organs may perform similar overall functions, but they may have different progenitors/origins and the environments in which they reside confer unique phenotypes [133,134]. Cardiac fibroblasts that experience loading and unloading thousands (up to hundreds of thousands) of times per day may be quite distinct from fibroblasts or stromal cells in less mechanically dynamic tissue such as the liver [133]. Upon injury, fibroblast activation and matrix secretion are common among all organs for wound healing, but, to our knowledge, direct comparisons of how the local biochemical and mechanical environments influence fibroblast state trajectories have not been performed. In the liver, metabolism has been studied most extensively due to the metabolic demands of that particular organ, and inhibition of glutamine breakdown has shown to reduce liver fibrosis [135]. In the heart, the cardiac fibroblast is often studied in the context of mechanics, again an intuitive choice for study. Interestingly, mouse embryonic fibroblasts become more activated in both 2D and 3D under cyclic stretching compared to a static substrate stiffness cue [32,113]. Given this different response to mechanical stimuli in a heterogenous, embryonic cell population [136], fibroblasts in all organ systems might be originally primed for response to cyclic stress and strain. In human periodontal ligament fibroblasts, osteoblastic transcription factors, MAP kinase, P38, and JNK were all upregulated in both static and cyclic stretching, with an exacerbated response under the cyclic conditions and perpendicular alignment of cells with respect to applied force only in cyclic stretching [137]. It is unknown when fibroblasts in less mechanically active organs develop (and if they change) this more extreme response to cyclic loading due to their homeostasis in an otherwise soft, more static environment. Conversely, fibroblasts in the lung and heart, which are regularly subject to cyclic stretching, may adapt by lessening this response to avoid an activation that might otherwise happen with each breath or heartbeat. Neonatal cardiac fibroblasts may need disorganized matrix topography cues in addition to cyclic stretching to result in significant MAPK activation, indicating the dependence on cooperative, more complex inputs than dynamic substrate stretching [117]. It would benefit the field to make direct comparisons between tissue-specific fibroblasts and to more carefully analyze them in controlled mechanical, structural, and biochemical microenvironments to glean from data in other systems in combating fibrotic disease.
Future studies mimicking fibrosis in vitro should carefully consider what set of material parameters are relevant to the tissue of interest and choose material characterization techniques that are consistent with available in vivo data. To guide better in vitro modelling, a consistent standard for stiffness measurements should be established to compare in vivo tissues with their in vitro limitations. Future in vivo studies should more clearly separate the contributions of muscle and matrix to overall myocardial mechanics by direct comparison of decellularized and intact preparations and quantification of how relative contributions change through the course of disease.
Recent work comparing 3D to 2D culture of unpurified as well as pure, lineage-traced populations of murine cardiac fibroblasts revealed 3D aggregate culture increased expression of genes like metalloproteinases and cytokines that have been previously shown to become upregulated following isoproterenol infusion. However, 3D culture also decreased αSMA and collagen expression in fibroblasts compared to 2D plastic culture. Here the authors speculated that the 3D fibroblast aggregates modelled myofibroblast dedifferentiation from a secretory phenotype into a proteolytic one. While the phenotypic driver was assumed to be topology due to cell-cell aggregate morphology compared to monolayer culture, the discrepancy in local mechanics and the “tissue” surrounding the cells in each condition could be contributing to these outcomes [138]. As previously mentioned, topology and mechanics in stretch activated pathways of the fibroblast have synergistic effects on FAK-mediated cell elongation and expression of “activation” markers but do not always lead to functional αSMA+ stress fibers. This in vitro system was used as a model of the in vivo changes from dispersed cardiac fibroblasts, which have heterocellular contacts in healthy tissue to the aggregation of activated and/or proliferative cardiac fibroblasts during the injury response. These set of studies provided a robust characterization of the fibroblast phenotypes in vitro in both 2D and 3D but did not include other cell types or characterize the secreted matrix microenvironment influencing the cells in each condition. Future work in this area could involve expansion to genetically controlled fibroblasts from aforementioned mouse models in heterocellular cultures. For example, this culture system with fibroblast-specific knockdown or overexpression of TGFβ family proteins compared to mechanically sensitive, late-phase activation pathways (MRFT-A) might allow for better insight on the dynamics and mechanisms of mechanical, structural, and chemical fibroblast activation.
Fibroblast-Cell Contact Induced State Transitions
There are inherent difficulties in dissociating the effects of paracrine signaling versus direct fibroblast-cell contact on fibroblast state transitions, but scRNAseq studies have illuminated the cellular heterogeneity within the cardiac fibroblast population. We would predict this is partially associated with direct cellular interactions within an environmental niche. The most obvious mechanism is through paracrine signaling, and work from several labs shows that quiescent cardiac fibroblasts differentially express hundreds of genes relevant to fibrosis when compared to fibroblasts derived from other types of tissue [139,140]. Through paracrine communication, cardiac fibroblasts can receive cues from cardiac myocytes, endothelial cells, smooth muscle cells, and immune cells, which bidirectionally inform cell state and tissue function in the heart [14,139,141]. For example, platelet derived growth factors (PDGF) promote vascularization, endothelial cell proliferation, fibroblast homeostasis, and changes in fibroblast cell state indicative of a “priming” toward fibroblast activation in injury [142,143]. Furthermore, inhibiting connective tissue growth factor in injury models and in culture decreased fibroblast activation and scar formation [144]. Both growth factors can be secreted by several cell types in the cardiac milieu, and fibroblasts express receptors for them throughout development, adulthood, and disease. Another review in this Matrix Biology SI by Reese-Peterson et al provides a more extensive description of the impacts of specific growth factors on fibroblast activation and fibrogenesis [reference article]. Cardiac fibroblasts can also communicate through direct cell-cell contact mediated by gap junctions, adherens junctions, and even nanotubes, which allow for the exchange ions, metabolites, and possibly organelle transfer [145,146]. Fibroblast-myocyte contact plays an important role for the electrical coupling of cells in the heart [147]. However, to date, there have been limited attempts at controlling cardiac fibroblast state through direct cell communication, leaving open whether direct cell contact plays a role in regulating cardiac fibroblast cell state at the tissue scale.
Generalizability of ECM Remodeling Models To Human Disease.
Most manipulations on fibroblast cell state have been performed in transgenic mouse models of disease, as the dominant narrative of the field remains that ECM proteins are highly conserved between mammals and even beyond [148,149]. In addition, collagen knockout mice have largely produced phenotypes that mimic human diseases, with the notable exception of type X collagen, which is not a major component of myocardial matrix [150]. Components of the ECM are subject to complex transcriptional, translational, and post-translational regulation, any of which could vary between species. It has been suggested by gene network analysis from mouse and human datasets that there may be differences in the regulation of type III collagen between mammals [151]. It is unclear whether this regulatory difference leads to any functional changes in transcription. From an existing transcriptomic dataset comparing the mouse and human heart through development, collagen types VI and XV diverge most significantly between adult mice and humans at the transcriptional level [152]. The higher levels of collagen type VI found in the human transcriptome could be clinically significant as this collagen type is elevated following myocardial infarction in humans and ColVI deletion improves outcomes in a mouse MI model [153]. Though transcriptional levels of collagens between species lends some insights worth exploring in future work, the translational and post-translational regulation of ECM proteins cannot be ignored. At the proteome level, the cardiac ECM of a variety of species has been profiled in an unbiased manner, including mice, rats, pigs, and human patients [154–157]. Such studies of the cardiac matrisome are often limited by the relative insolubility and extensive crosslinking of ECM proteins [158]. Extraction and solubilization methods vary, and different methods used on the same sample can yield different proteins detected by mass spec [155,159]. For example, proteomics studies on rodent cardiac ECM come to a consensus that Collagens type I, VI, IV, and III are all abundant, but the relative proportions of these components vary by study, likely due to methodological differences [154,157]. One study has directly compared components of human and porcine myocardial matrix, finding several proteins including periostin and type XII collagen that were not present in porcine matrix [160]. The human cardiac matrisome has also been profiled from patient samples, confirming the presence and relative abundance of these collagens [161]. To better understand the potential differences in ECM composition between species, the field needs well controlled proteomic studies that make a direct comparison between murine and human samples.
Conclusions
To better understand and then control cardiac fibroblast cell state and hence the ECM, combinatorial studies using cardiac fibroblast-specific genetic approaches and 3D mechanical platforms for interrogating mechanical signaling will be critical to illuminating the signaling network underlying fibroblast state transitions. With the ability to monitor ECM reorganization with nondestructive optical imaging as well as fluorescent reporters, the dynamic effects of cardiac fibroblast cell state transitions can be measured in cardiac tissues engineered with the full spectrum of cardiac cell types to achieve greater insight to the homeostatic role of fibroblasts in the heart. As the community continues to learn more about the regulators of the fibroblast cell state along several axes of behaviors - secretion, proteolysis, migration, proliferation, chemical signaling, and contraction – we can begin to control these functions independently or in combination with an eye toward tactically modifying the extracellular matrix for a given function that could even promote myocardial repair.
Highlights.
Fibroblasts undergo multiple state transitions during the cardiac injury response.
Chemical and mechanical signals in the injured heart converge on p38 MAP kinase, which initiates myofibroblast gene expression.
Maturation of the fibroblast transcriptome promotes the myofibroblast state transition.
Microenvironmental signals significantly impact fibroblast cell state.
Sources of funding
This work was supported by grants from the National Institutes of Health (HL141187 & HL142624).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors declare no competing financial interests.
References
- 1.Loring Z; Zareba W; McNitt S; Strauss DG; Wagner GS; Daubert JP ECG quantification of myocardial scar and risk stratification in MADIT-II. Ann. Noninvasive Electrocardiol 2013, 18, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki T; Fukumoto Y; Sugimura K; Oikawa M; Satoh K; Nakano M; Nakayama M; Shimokawa H Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure: Comparison between preserved and reduced ejection fraction heart failure. Circ. J 2011, 75, 2605–2613. [DOI] [PubMed] [Google Scholar]
- 3.Gulati A; Japp AG; Raza S; Halliday BP; Jones DA; Newsome S; Ismail NA; Morarji K; Khwaja J; Spath N; et al. Absence of Myocardial Fibrosis Predicts Favorable Long-Term Survival in New-Onset Heart Failure. Circ. Cardiovasc. Imaging 2018, 11, e007722. [DOI] [PubMed] [Google Scholar]
- 4.Bergmann O; Bhardwaj RD; Bernard S; Zdunek S; Walsh S; Zupicich J; Alkass K; Buchholz BA; Jovinge S; Frisén J; et al. Renewal in Humans Evidence for Cardiomyocyte. 2017, 324, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan D; Takawale A; Lee J; Kassiri Z Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travers JG; Kamal FA; Robbins J; Yutzey KE; Blaxall BC Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res 2016, 118, 1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khalil H; Kanisicak O; Vagnozzi RJ; Johansen AK; Maliken BD; Prasad V; Boyer JG; Brody MJ; Schips T; Kilian KK; et al. Cell-specific ablation of Hsp47 defines the collagen producing cells in the injured heart. JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tallquist MD; Molkentin JD Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol 2017, 14, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis J; Molkentin JD Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol 2014, 70, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stempien-Otero A; Kim DH; Davis J Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell. Cardiol 2016, 97, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobaczewski M; De Haan JJ; Frangogiannis NG The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J. Cardiovasc. Transl. Res 2012, 5, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falke LL; Gholizadeh S; Goldschmeding R; Kok RJ; Nguyen TQ Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat. Rev. Nephrol 2015, 11, 233–244. [DOI] [PubMed] [Google Scholar]
- 13.Tomasek JJ; Gabbiani G; Hinz B; Chaponnier C; Brown RA Myofibroblasts and mechanoregulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol 2002, 3, 349–363. [DOI] [PubMed] [Google Scholar]
- 14.Skelly DA; Squiers GT; McLellan MA; Bolisetty MT; Robson P; Rosenthal NA; Pinto AR Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [DOI] [PubMed] [Google Scholar]
- 15.Farbehi N; Patrick R; Dorison A; Xaymardan M; Janbandhu V; Wystub-Lis K; Wk Ho J; Nordon RE; Harvey RP Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. 2019. [DOI] [PMC free article] [PubMed]
- 16.Mouton AJ; Ma Y; Rivera Gonzalez OJ; Daseke MJ; Flynn ER; Freeman TC; Garrett MR; DeLeon-Pennell KY; Lindsey ML Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol 2019, 114, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schafer S; Viswanathan S; Widjaja AA; Lim WW; Moreno-Moral A; DeLaughter DM; Ng B; Patone G; Chow K; Khin E; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Souders CA; Bowers SLK; Baudino TA Cardiac fibroblast: the renaissance cell. Circ. Res 2009, 105, 1164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kisseleva T; Cong M; Paik Y; Scholten D; Jiang C; Benner C; Iwaisako K; Moore-Morris T; Scott B; Tsukamoto H; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 9448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sánchez-Iranzo H; Galardi-Castilla M; Sanz-Morejón A; González-Rosa JM; Costa R; Ernst A; Sainz de Aja J; Langa X; Mercader N Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc. Natl. Acad. Sci 2018, 115, 4188–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanisicak O; Khalil H; Ivey MJ; Karch J; Maliken BD; Correll RN; Brody MJ; J Lin S-C; Aronow BJ; Tallquist MD; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun 2016, 7, 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu X; Khalil H; Kanisicak O; Boyer JG; Vagnozzi RJ; Maliken BD; Sargent MA; Prasad V; Valiente-Alandi I; Blaxall BC; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest 2018, 128, 2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willems IEMG; Havenith MG; De Mey JGR; Daemen MJAP The a-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol 1994, 145, 868–875. [PMC free article] [PubMed] [Google Scholar]
- 24.Furtado MB; Costa MW; Pranoto EA; Salimova E; Pinto AR; Lam NT; Park A; Snider P; Chandran A; Harvey RP; et al. Cardiogenic Genes Expressed in Cardiac Fibroblasts Contribute to Heart Development and Repair. Circ. Res 2014, 114, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frieda KL; Linton JM; Hormoz S; Choi J; Chow KHK; Singer ZS; Budde MW; Elowitz MB; Cai L Synthetic recording and in situ readout of lineage information in single cells. Nature 2017, 541, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Manno G; Soldatov R; Zeisel A; Braun E; Hochgerner H; Petukhov V; Lidschreiber K; Kastriti ME; Lönnerberg P; Furlan A; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess DJ Spatial transcriptomics coming of age. Nat. Rev. Genet 2019, 20, 317. [DOI] [PubMed] [Google Scholar]
- 28.Zeigler A; Richardson W; Holmes JW; Saucerman J A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J. Mol. Cell. Cardiol 2016, 94, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acharya A; Baek ST; Huang G; Eskiocak B; Goetsch S; Sung CY; Banfi S; Sauer MF; Olsen GS; Duffield JS; et al. The bHLH transcription factor Tcf21 is required for lineage specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner; Blythe Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis 2019, 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren J; Zhang S; Kovacs A; Wang Y; Muslin AJ Role of p38α MAPK in cardiac apoptosis and remodeling after myocardial infarction. J. Mol. Cell. Cardiol 2005, 38, 617–623. [DOI] [PubMed] [Google Scholar]
- 32.Molkentin JD; Bugg D; Ghearing N; Dorn LE; Kim P; Sargent MA; Gunaje J; Otsu K; Davis J Fibroblast-Specific Genetic Manipulation of p38 MAPK in vivo Reveals its Central Regulatory Role in Fibrosis. Circulation 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lighthouse JK; Burke RM; Velasquez LS; Dirkx RA; Aiezza A; Moravec CS; Alexis JD; Rosenberg A; Small EM Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai SL; Marín-Juez R; Stainier DYR Immune responses in cardiac repair and regeneration: a comparative point of view. Cell. Mol. Life Sci 2019, 76, 1365–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez A; Yin V Emerging Roles for Immune Cells and MicroRNAs in Modulating the Response to Cardiac Injury. J. Cardiovasc. Dev. Dis 2019, 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J; Seth A; Mcculloch AG Force regulates smooth muscle actin in cardiac fibroblasts. Am. J. Physiol. - Hear. Circ. Physiol 2000, 279. [DOI] [PubMed] [Google Scholar]
- 37.Lew AM; Glogauer M; McCulloch CAG Specific inhibition of skeletal α-actin gene transcription by applied mechanical forces through integrins and actin. Biochem. J 1999, 341, 647–653. [PMC free article] [PubMed] [Google Scholar]
- 38.Lin KC; Moroishi T; Meng Z; Jeong HS; Plouffe SW; Sekido Y; Han J; Park HW; Guan KL Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol 2017, 19, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis Jennifer, Davis F. Gregory, Birnbaumer Lutz, Molkentin D. Jeffery. B.R.A.; Davis J; Burr AR; Davis GF; Birnbaumer L; Molkentin JD A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2014, 19, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernard K; Logsdon NJ; Ravi S; Xie N; Persons BP; Rangarajan S; Zmijewski JW; Mitra K; Liu G; Darley-Usmar VM; et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem 2015, 290, 25427–25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lombardi AA; Gibb AA; Arif E; Kolmetzky DW; Tomar D; Luongo TS; Jadiya P; Murray EK; Lorkiewicz PK; Hajnóczky G; et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stambe C The Role of p38 Mitogen-Activated Protein Kinase Activation in Renal Fibrosis. J. Am. Soc. Nephrol 2004, 15, 370–379. [DOI] [PubMed] [Google Scholar]
- 43.Kompa AR; See F; Lewis DA; Adrahtas A; Cantwell DM; Wang BH; Krum H Long-Term but Not Short-Term p38 Mitogen-Activated Protein Kinase Inhibition Improves Cardiac Function and Reduces Cardiac Remodeling Post-Myocardial Infarction. Pharmacology 2008, 325, 741–750. [DOI] [PubMed] [Google Scholar]
- 44.See F; Thomas W; Way K; Tzanidis A; Kompa A; Lewis D; Itescu S; Krum H P38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J. Am. Coll. Cardiol 2004, 44, 1679–1689. [DOI] [PubMed] [Google Scholar]
- 45.Stratton MS; Bagchi RA; Felisbino MB; Hirsch RA; Smith HE; Riching AS; Enyart BY; Koch KA; Cavasin MA; Alexanian M; et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res 2019, 125, 662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voss TC; Hager GL Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet 2014, 15, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu T; Warburton RR; Guevara OE; Hill NS; Fanburg BL; Gaestel M; Kayyali US Lack of MK2 Inhibits Myofibroblast Formation and Exacerbates Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol 2007, 37, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis J; Salomonis N; Ghearing N; Lin S-CJ; Kwong JQ; Mohan A; Swanson MS; Molkentin JD MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun 2015, 6, 10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Small EM; Thatcher JE; Sutherland LB; Kinoshita H; Gerard RD; Richardson JA; Dimaio JM; Sadek H; Kuwahara K; Olson EN Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res 2010, 107, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adapala RK; Thoppil RJ; Luther DJ; Paruchuri S; Meszaros JG; Chilian WM; Thodeti CK TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol 2013, 54, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu QF; Dong Q; Du YM Transient receptor potential (TRP) channels and cardiac fibrosis. Sheng Li Ke Xue Jin Zhan 2014, 45, 416–420. [PubMed] [Google Scholar]
- 52.Du J; Xie J; Zhang Z; Tsujikawa H; Fusco D; Silverman D; Liang B; Yue L TRPM7- mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res 2010, 106, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobrokhotov O; Samsonov M; Sokabe M; Hirata H Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin. Transl. Med 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao Y; Hill MC; Zhang M; Martin TJ; Morikawa Y; Wang S; Moise AR; Wythe JD; Martin JF Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Auger-Messier M; Accornero F; Goonasekera SA; Bueno OF; Lorenz JN; van Berlo JH; Willette RN; Molkentin JD Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res 2013, 112, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kyoi S; Otani H; Matsuhisa S; Akita Y; Tatsumi K; Enoki C; Fujiwara H; Imamura H; Kamihata H; Iwasaka T Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res 2006, 69, 888–898. [DOI] [PubMed] [Google Scholar]
- 57.Liu YH; Wang D; Rhaleb NE; Yang XP; Xu J; Sankey SS; Rudolph AE; Carretero OA Inhibition of p38 mitogen-activated protein kinase protects the heart against cardiac remodeling in mice with heart failure resulting from myocardial infarction. J. Card. Fail 2005, 11, 74–81. [DOI] [PubMed] [Google Scholar]
- 58.Wissing ER; Boyer JG; Kwong JQ; Sargent MA; Karch J; Mcnally EM; Otsu K; Molkentin JD P38a MAPK underlies muscular dystrophy and myofiber death through a Bax dependent mechanism. [DOI] [PMC free article] [PubMed]
- 59.Janga SC From specific to global analysis of posttranscriptional regulation in eukaryotes: Posttranscriptional regulatory networks. Brief. Funct. Genomics 2012, 11. [DOI] [PubMed] [Google Scholar]
- 60.Licatalosi DD; Darnell RB RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet 2010, 11, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang ET; Cody NAL; Jog S; Biancolella M; Wang TT; Treacy DJ; Luo S; Schroth GP; Housman DE; Reddy S; et al. Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell 2012, 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blech-Hermoni Y; Ladd AN RNA binding proteins in the regulation of heart development. Int. J. Biochem. Cell Biol 2013, 45, 2467–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Truitt ML; Ruggero D New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanadia RN; Johnstone KA; Mankodi A; Lungu C; Thornton CA; Esson D; Timmers AM; Hauswirth WW; Swanson MS A Muscleblind Knockout Model for Myotonic Dystrophy. Science (80-. ) 2003, 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- 65.Kalsotra A; Xiao X; Ward AJ; Castle JC; Johnson JM; Burge CB; Cooper TA A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci 2008, 105, 20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng AW; Shi J; Wong P; Luo KL; Trepman P; Wang ET; Choi H; Burge CB; Lodish HF Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood 2014, 124, 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lacraz GPA; Junker JP; Gladka MM; Molenaar B; Scholman KT; Vigil-Garcia M; Versteeg D; De Ruiter H; Vermunt MW; Creyghton MP; et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis during Ischemic Injury. Circulation 2017, 136, 1396–1409. [DOI] [PubMed] [Google Scholar]
- 68.Scharf GM; Kilian K; Cordero J; Wang Y; Grund A; Hofmann M; Froese N; Wang X; Kispert A; Kist R; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Akiyama H; Chaboissier M-C; Behringer RR; Rowitch DH; Schedl A; Epstein JA; de Crombrugghe B Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci 2004, 101, 6502–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lincoln J; Kist R; Scherer G; Yutzey KE Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol 2007, 305, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clarke SA; Goodman NC; Ailawadi G; Holmes JW Effect of Scar Compaction on the Therapeutic Efficacy of Anisotropic Reinforcement Following Myocardial Infarction in the Dog. J. Cardiovasc. Transl. Res 2015, 8, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan W; Wang P; Zhao CX; Tang J; Xiao X; Wang DW Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor beta/Smad and p38 mitogen-activated protein kinase signaling pathways. Hum. Gene Ther 2009, 20, 1190–1200. [DOI] [PubMed] [Google Scholar]
- 73.Christensen G; Herum KM; Lunde IG Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. 2019, 75–76, 286–299. [DOI] [PubMed] [Google Scholar]
- 74.Wang RM; Christman KL Decellularized myocardial matrix hydrogels: In basic research and preclinical studies. Adv. Drug Deliv. Rev 2016, 96, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berry MF; Engler AJ; Woo YJ; Pirolli TJ; Bish LT; Jayasankar V; Morine KJ; Gardner TJ; Discher DE; Sweeney HL Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am. J. Physiol. Circ. Physiol 2006, 290, H2196–H2203. [DOI] [PubMed] [Google Scholar]
- 76.Fomovsky GM; Holmes JW Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am. J. Physiol. Circ. Physiol 2010, 298, H221–H228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jacot JG; Martin JC; Hunt DL Mechanobiology of cardiomyocyte development. J. Biomech 2010, 43, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhana B; Iyer RK; Chen WLK; Zhao R; Sider KL; Likhitpanichkul M; Simmons CA; Radisic M Influence of substrate stiffness on the phenotype of heart cells. Biotechnol. Bioeng 2010, 105, 1148–1160. [DOI] [PubMed] [Google Scholar]
- 79.Perea-Gil I; Uriarte JJ; Prat-Vidal C; Gálvez-Montón C; Roura S; Llucià-Valldeperas A; Soler-Botija C; Farré R; Navajas D; Bayes-Genis A In vitro comparative study of two decellularization protocols in search of an optimal myocardial scaffold for recellularization. Am. J. Transl. Res 2015, 7, 558–73. [PMC free article] [PubMed] [Google Scholar]
- 80.van Spreeuwel ACC; Bax NAMM; van Nierop BJ; Aartsma-Rus A; Goumans M-JTHH; Bouten CVCC; Spreeuwel A.C.C. van; Bax NAMM; Nierop B.J. van; Aartsma-Rus A; et al. Mimicking Cardiac Fibrosis in a Dish: Fibroblast Density Rather than Collagen Density Weakens Cardiomyocyte Function. J. Cardiovasc. Transl. Res 2017, 10, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Engler AJ; Carag-Krieger C; Johnson CP; Raab M; Tang H-Y; Speicher DW; Sanger JW; Sanger JM; Discher DE Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J. Cell Sci 2008, 121, 3794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Demer LL; Yin FC Passive biaxial mechanical properties of isolated canine myocardium. J. Physiol 1983, 339, 615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sacks MS; Chuong CJ Biaxial Mechanical Properties of Passive Right Ventricular Free Wall Myocardium. J. Biomech. Eng 1993, 115, 202. [DOI] [PubMed] [Google Scholar]
- 84.Novak VP; Yin FC; Humphrey JD Regional mechanical properties of passive myocardium. J. Biomech 1994, 27, 403–12. [DOI] [PubMed] [Google Scholar]
- 85.Engler AJ; Griffin MA; Sen S; Bönnemann CG; Sweeney HL; Discher DE Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J. Cell Biol 2004, 166, 877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Solon J; Levental I; Sengupta K; Georges PC; Janmey PA Fibroblast Adaptation and Stiffness Matching to Soft Elastic Substrates. Biophys. J 2007, 93, 4453–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herum KM; Choppe J; Kumar A; Engler AJ; McCulloch AD Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goffin JM; Pittet P; Csucs G; Lussi JW; Meister J-J; Hinz B Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol 2006, 172, 259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ongherth A; Pasch S; Wuertz CM; Nowak K; Kittana N; Weis CA; Jatho A; Vettel C; Tiburcy M; Toischer K; et al. p63RhoGEF regulates auto- and paracrine signaling in cardiac fibroblasts. J. Mol. Cell. Cardiol 2015, 88, 39–54. [DOI] [PubMed] [Google Scholar]
- 90.Santos GL; Hartmann S; Zimmermann W-H; Ridley A; Lutz S Inhibition of Rho-associated kinases suppresses cardiac myofibroblast function in engineered connective and heart muscle tissues. J. Mol. Cell. Cardiol 2019, 134, 13–28. [DOI] [PubMed] [Google Scholar]
- 91.Halperin HR; Chew PH; Weisfeldt ML; Sagawa K; Humphrey JD; Yin FC Transverse stiffness: a method for estimation of myocardial wall stress. Circ. Res 1987, 61, 695–703. [DOI] [PubMed] [Google Scholar]
- 92.Przyklenk K; Connelly CM; McLaughlin RJ; Kloner RA; Apstein CS Effect of myocyte necrosis on strength, strain, and stiffness of isolated myocardial strips. Am. Heart J 1987, 114, 1349–1359. [DOI] [PubMed] [Google Scholar]
- 93.Yin FC; Strumpf RK; Chew PH; Zeger SL Quantification of the mechanical properties of noncontracting canine myocardium under simultaneous biaxial loading. J. Biomech 1987, 20, 577–89. [DOI] [PubMed] [Google Scholar]
- 94.Humphrey JD; Strumpf RK; Yin FC Biaxial mechanical behavior of excised ventricular epicardium. Am. J. Physiol. Circ. Physiol 1990, 259, H101–H108. [DOI] [PubMed] [Google Scholar]
- 95.Kang T; Humphrey JD; Yin FC Comparison of biaxial mechanical properties of excised endocardium and epicardium. Am. J. Physiol. Circ. Physiol 1996, 270, H2169–H2176. [DOI] [PubMed] [Google Scholar]
- 96.Lieber SC; Aubry N; Pain J; Diaz G; Kim S-J; Vatner SF Aging increases stiffness of cardiac myocytes measured by atomic force microscopy nanoindentation. Am. J. Physiol. Circ. Physiol 2004, 287, H645–H651. [DOI] [PubMed] [Google Scholar]
- 97.Gershlak JR; Resnikoff JI; Sullivan KE; Williams C; Wang RM; Black LD Mesenchymal stem cells ability to generate traction stress in response to substrate stiffness is modulated by the changing extracellular matrix composition of the heart during development. Biochem. Biophys. Res. Commun 2013, 439, 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kichula ET; Wang H; Dorsey SM; Szczesny SE; Elliott DM; Burdick JA; Wenk JF Experimental and Computational Investigation of Altered Mechanical Properties in Myocardium after Hydrogel Injection. Ann. Biomed. Eng 2014, 42, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Theret DP; Levesque MJ; Sato M; Nerem RM; Wheeler LT The Application of a Homogeneous Half-Space Model in the Analysis of Endothelial Cell Micropipette Measurements. J. Biomech. Eng 1988, 110, 190. [DOI] [PubMed] [Google Scholar]
- 100.Sommer G; Schriefl AJ; Andrä M; Sacherer M; Viertler C; Wolinski H; Holzapfel GA Biomechanical properties and microstructure of human ventricular myocardium. Acta Biomater. 2015, 24, 172–192. [DOI] [PubMed] [Google Scholar]
- 101.Quinn KP; Sullivan KE; Liu Z; Ballard Z; Siokatas C; Georgakoudi I; Black LD Optical metrics of the extracellular matrix predict compositional and mechanical changes after myocardial infarction. Sci. Rep 2016, 6, 35823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramadan S; Paul N; Naguib HE Standardized static and dynamic evaluation of myocardial tissue properties. Biomed. Mater 2017, 12, 025013. [DOI] [PubMed] [Google Scholar]
- 103.Chen EJ; Novakofski J; Jenkins WK; O’Brien WD Young’s modulus measurements of soft tissues with application to elasticity imaging. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 1996, 43, 191–194. [Google Scholar]
- 104.Gluck JM; Herren AW; Yechikov S; Kao HKJ; Khan A; Phinney BS; Chiamvimonvat N; Chan JW; Lieu DK Biochemical and biomechanical properties of the pacemaking sinoatrial node extracellular matrix are distinct from contractile left ventricular matrix. PLoS One 2017, 12, e0185125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cox MA; Driessen NJ; Bouten CV; Baaijens FP Mechanical Characterization of Anisotropic Planar Biological Soft Tissues Using Large Indentation: A Computational Feasibility Study. J. Biomech. Eng 2005, 128, 428. [DOI] [PubMed] [Google Scholar]
- 106.Notari M; Ventura-Rubio A; Bedford-Guaus SJ; Jorba I; Mulero L; Navajas D; Martí M; Raya Á The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci. Adv 2018, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fujita K; Feng Z; Sato D; Kosawada T; Nakamura T; Shiraishi Y; Umezu M Modulation of the mechanical properties of ventricular extracellular matrix hydrogels with a carbodiimide crosslinker and investigation of their cellular compatibility. AIMS Mater. Sci 2018, 5, 54–74. [Google Scholar]
- 108.Stoppel WLWL; Gao AEAE; Greaney AMAM; Partlow BPBP; Bretherton RCRC; Kaplan DLDL; Black LDLD Elastic, silk-cardiac extracellular matrix hydrogels exhibit time dependent stiffening that modulates cardiac fibroblast response. J. Biomed. Mater. Res. - Part A 2016, 104, 3058–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Herum KM; Choppe J; Kumar A; Engler AJ; McCulloch AD Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Günay KA; Ceccato TL; Silver JS; Bannister KL; Bednarski OJ; Leinwand LA; Anseth KS PEG-Anthracene Hydrogels as an on-Demand Stiffening Matrix to Study Mechanobiology. Angew. Chemie Int. Ed 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Frey MT; Wang Y A photo-modulatable material for probing cellular responses to substrate rigidity. Soft Matter 2009, 5, 1918–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kloxin AM; Benton JA; Anseth KS In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials 2010, 31, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu L; Shadish JA; Arakawa CK; Shi K; Davis J; DeForest CA Cyclic Stiffness Modulation of Cell‐Laden Protein–Polymer Hydrogels in Response to User‐Specified Stimuli Including Light. Adv. Biosyst 2018, 2, 1800240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Corbin EA; Vite A; Peyster EG; Bhoopalam M; Brandimarto J; Wang X; Bennett AI; Clark AT; Cheng X; Turner KT; et al. Tunable and Reversible Substrate Stiffness Reveals a Dynamic Mechanosensitivity of Cardiomyocytes. ACS Appl. Mater. Interfaces 2019, 11, 20603–20614. [DOI] [PubMed] [Google Scholar]
- 115.Lee AA; Delhaas T; McCulloch AD; Villarreal FJ Differential responses of adult cardiac fibroblasts to in vitro biaxial strain patterns. J. Mol. Cell. Cardiol 1999, 31, 1833–43. [DOI] [PubMed] [Google Scholar]
- 116.Kong M; Lee J; Yazdi IK; Miri AK; Lin Y-D; Seo J; Zhang YS; Khademhosseini A; Shin SR Cardiac Fibrotic Remodeling on a Chip with Dynamic Mechanical Stimulation. Adv. Healthc. Mater 2019, 8, 1801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Atance J; Yost MJ; Carver W Influence of the extracellular matrix on the regulation of cardiac fibroblast behavior by mechanical stretch. J. Cell. Physiol 2004, 200, 377–386. [DOI] [PubMed] [Google Scholar]
- 118.Luo S; Hieu TB; Ma F; Yu Y; Cao Z; Wang M; Wu W; Mao Y; Rose P; Law BY-K; et al. ZYZ-168 alleviates cardiac fibrosis after myocardial infarction through inhibition of ERK1/2-dependent ROCK1 activation. Sci. Rep 2017, 7, 43242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ducharme A; Frantz S; Aikawa M; Rabkin E; Lindsey M; Rohde LE; Schoen FJ; Kelly RA; Werb Z; Libby P; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest 2000, 106, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Papakrivopoulou J; Lindahl GE; Bishop JE; Laurent GJ Differential roles of extracellular signal-regulated kinase 1/2 and p38MAPK in mechanical load-induced procollagen α1(I) gene expression in cardiac fibroblasts. Cardiovasc. Res 2004, 61, 736–744. [DOI] [PubMed] [Google Scholar]
- 121.Clemente CFMZ; Tornatore TF; Theizen TH; Deckmann AC; Pereira TC; Lopes-Cendes I; Souza JRM; Franchini KG Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ. Res 2007, 101, 1339–48. [DOI] [PubMed] [Google Scholar]
- 122.Ugolini GS; Rasponi M; Pavesi A; Santoro R; Kamm R; Fiore GB; Pesce M; Soncini M On-chip assessment of human primary cardiac fibroblasts proliferative responses to uniaxial cyclic mechanical strain. Biotechnol. Bioeng 2016, 113, 859–869. [DOI] [PubMed] [Google Scholar]
- 123.Xiao Y; Hill MC; Li L; Deshmukh V; Martin TJ; Wang J; Martin JF Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019, 33, 1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Al-Haque S; Miklas JW; Feric N; Chiu LLY; Chen WLK; Simmons CA; Radisic M Hydrogel Substrate Stiffness and Topography Interact to Induce Contact Guidance in Cardiac Fibroblasts. Macromol. Biosci 2012, 12, 1342–1353. [DOI] [PubMed] [Google Scholar]
- 125.Spassova MA; Hewavitharana T; Xu W; Soboloff J; Gill DL A common mechanism underlies stretch activation and activation of TRPC6 channels. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 16586–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Winer JP; Oake S; Janmey PA Non-Linear Elasticity of Extracellular Matrices Enables Contractile Cells to Communicate Local Position and Orientation. PLoS One 2009, 4, 6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hinz B The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [DOI] [PubMed] [Google Scholar]
- 128.Grim JC; Brown TE; Aguado BA; Chapnick DA; Viert AL; Liu X; Anseth KS A Reversible and Repeatable Thiol–Ene Bioconjugation for Dynamic Patterning of Signaling Proteins in Hydrogels. ACS Cent. Sci 2018, 4, 909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tamariz E; Grinnell F Modulation of fibroblast morphology and adhesion during collagen matrix remodeling. Mol. Biol. Cell 2002, 13, 3915–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Curtis MW; Russell B Micromechanical regulation in cardiac myocytes and fibroblasts: Implications for tissue remodeling. Pflugers Arch. Eur. J. Physiol 2011, 462, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Balestrini JL; Skorinko JK; Hera A; Gaudette GR; Billiar KL Applying controlled non- uniform deformation for in vitro studies of cell mechanobiology. Biomech. Model. Mechanobiol 2010, 9, 329–344. [DOI] [PubMed] [Google Scholar]
- 132.Petersen A; Joly P; Bergmann C; Korus G; Duda GN The impact of substrate stiffness and mechanical loading on fibroblast-induced scaffold remodeling. Tissue Eng. - Part A 2012, 18, 1804–1817. [DOI] [PubMed] [Google Scholar]
- 133.Czubryt Cardiac Fibroblast to Myofibroblast Phenotype Conversion—An Unexploited Therapeutic Target. J. Cardiovasc. Dev. Dis 2019, 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lynch MD; Watt FM Fibroblast heterogeneity: implications for human disease. J. Clin. Invest 2018, 128, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Du K; Hyun J; Premont RT; Choi SS; Michelotti GA; Swiderska-Syn M; Dalton GD; Thelen E; Rizi BS; Jung Y; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Singhal PK; Sassi S; Lan L; Au P; Halvorsen SC; Fukumura D; Jain RK; Seed B Mouse embryonic fibroblasts exhibit extensive developmental and phenotypic diversity. Pnas 2015, 113, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Papadopoulou A; Iliadi A; Eliades T; Kletsas D Early responses of human periodontal ligament fibroblasts to cyclic and static mechanical stretching. 2016. [DOI] [PubMed]
- 138.Yu J; Seldin MM; Fu K; Li S; Lam L; Wang P; Wang Y; Huang D; Nguyen TL; Wei B; et al. Topological Arrangement of Cardiac Fibroblasts Regulates Cellular Plasticity. Circ. Res 2018, 123, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang J; Tao R; Campbell KF; Carvalho JL; Ruiz EC; Kim GC; Schmuck EG; Raval AN; da Rocha AM; Herron TJ; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun 2019, 10, 2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Iwamiya T; Matsuura K; Masuda S; Shimizu T; Okano T Cardiac fibroblast-derived VCAM-1 enhances cardiomyocyte proliferation for fabrication of bioengineered cardiac tissue. Regen. Ther 2016, 4, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kofron CM; Mende U In vitro models of the cardiac microenvironment to study myocyte and non-myocyte crosstalk: bioinspired approaches beyond the polystyrene dish: Cardiac in vitro models for myocyte and non-myocyte crosstalk. J. Physiol 2017, 595, 3891–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ivey MJ; Kuwabara JT; Riggsbee KL; Tallquist MD Platelet-derived growth factor receptor-α is essential for cardiac fibroblast survival. Am. J. Physiol. Circ. Physiol 2019, 317, H330–H344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Asli NS; Xaymardan M; Forte E; Waardenberg AJ; Cornwell J; Janbandhu V; Kesteven S; Chandrakanthan V; Malinowska H; Reinhard H; et al. PDGFRα signaling in cardiac fibroblasts modulates quiescence, metabolism and self-renewal, and promotes anatomical and functional repair. bioRxiv 2018, 225979. [Google Scholar]
- 144.Vainio LE; Szabó Z; Lin R; Ulvila J; Yrjölä R; Alakoski T; Piuhola J; Koch WJ; Ruskoaho H; Fouse SD; et al. Connective Tissue Growth Factor Inhibition Enhances Cardiac Repair and Limits Fibrosis After Myocardial Infarction. JACC Basic to Transl. Sci 2019, 4, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang P; Su J; Mende U Cross talk between cardiac myocytes and fibroblasts: from multiscale investigative approaches to mechanisms and functional consequences. Am. J. Physiol. Circ. Physiol 2012, 303, H1385–H1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kohl P; Gourdie RG Fibroblast-myocyte electrotonic coupling: Does it occur in native cardiac tissue? J. Mol. Cell. Cardiol 2014, 70, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Quinn TA; Camelliti P; Rog-Zielinska EA; Siedlecka U; Poggioli T; O’Toole ET; Knöpfel T; Kohl P Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 14852–14857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hynes RO The extracellular matrix: Not just pretty fibrils. Science (80-. ) 2009, 326, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Myllyharju J; Kivirikko KI Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [DOI] [PubMed] [Google Scholar]
- 150.Aszódi A; Pfeifer A; Wendel M; Hiripi L; Fässler R Mouse models for extracellular matrix diseases. J. Mol. Med 1998, 76, 238–252. [DOI] [PubMed] [Google Scholar]
- 151.Wang JH-C; Thampatty BP; Lin J-S; Im H-J Mechanoregulation of gene expression in fibroblasts. Gene 2007, 391, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cardoso-Moreira M; Velten B; Mort M; Cooper DN; Huber W; Kaessmann H Developmental gene expression differences between humans and mammalian models. bioRxiv 2019, 747782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Luther DJ; Thodeti CK; Shamhart PE; Adapala RK; Hodnichak C; Weihrauch D; Bonaldo P; Chilian WM; Meszaros JG Absence of type vi collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ. Res 2012, 110, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.C. LDW; K.P. Q; G. I; Black Young developmental age cardiac extracellular matrix promotes the expansion of neonatal cardiomyocytes in vitro. 2014, 76, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.De Castro Brás LE; Ramirez TA; DeLeon-Pennell KY; Chiao YA; Ma Y; Dai Q; Halade GV; Hakala K; Weintraub ST; Lindsey ML Texas 3-Step decellularization protocol: Looking at the cardiac extracellular matrix. J. Proteomics 2013, 86, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lindsey ML; Hall ME; Harmancey R; Ma Y Adapting extracellular matrix proteomics for clinical studies on cardiac remodeling post-myocardial infarction. Clin. Proteomics 2016, 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Padmanabhan Iyer R; Chiao YA; Flynn ER; Hakala K; Cates CA; Weintraub ST; de Castro Brás LE Matrix metalloproteinase-9-dependent mechanisms of reduced contractility and increased stiffness in the aging heart. Proteomics - Clin. Appl 2016, 10, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Byron A; Humphries JD; Humphries MJ Defining the extracellular matrix using proteomics. Int. J. Exp. Pathol 2013, 94, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Didangelos A; Yin X; Mandal K; Saje A; Smith A; Xu Q; Jahangiri M; Mayr M Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: A proteomics approach. Mol. Cell. Proteomics 2011, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Johnson TD; Dequach JA; Gaetani R; Ungerleider J; Elhag D; Nigam V; Behfar A; Christman KL Human versus porcine tissue sourcing for an injectable myocardial matrix hydrogel. Biomater. Sci 2014, 2, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Johnson TD; Hill RC; Dzieciatkowska M; Nigam V; Behfar A; Christman KL; Hansen KC Quantification of Decellularized Human Myocardial Matrix: A Comparison of Six Patients HHS Public Access. Proteomics Clin Appl 2016, 10, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]