

Abstract
Background
The widespread distribution of antimicrobial-resistant Shigella has become a recurrent challenge in many parts of the developing world. Previous studies indicate that the host of Shigella has expanded from humans to animals. This study aimed to investigate the prevalence of fluoroquinolone resistance and associated molecular characterization of S. dysenteriae 1 isolated from calves.
Results
All 38 unduplicated S. dysenteriae 1 isolates were collected from calves in Gansu Province from October 2014 to December 2016. According to MLST and PFGE analysis, these isolates were separated into 4 and 28 genotypes, respectively. The most common STs identified were ST228 (34.21%, 13/38) and ST229 (39.47%, 15/38), which were first found in the present study. All isolates harbored virulence genes, and the incidence of the seven virulence genes were ipaH (100%), ipaBCD (92.11%), stx (73.68%), ial (57.89%), sen (28.95%), set1A and set1B (0%). According to the results of antimicrobial susceptibilities, 76.32% (29/38) were resistant to fluoroquinolone and showed multidrug resistance. In a study on the polymorphism of quinolone resistance–determining region (QRDR) of gyrA/B and parC/E genes, we identified two mutations in gyrA (Ser83 → Leu and Asp87 → Asn) and parC (Ser80 → Ile and Ser83 → Leu), respectively. Among them, 55.17% (16/29) of resistant strains had the gyrA point mutations (Ser83 → Leu) and parC point mutation (Ser83 → Leu). Moreover, 41.38% (12/29) of isolates had all five point mutations of gyrA and parC. In addition, the prevalence of the plasmid-mediated quinolone resistance (PMQR) determinant genes was also investigated. All 29 fluoroquinolone-resistant isolates were positive for the aac (6′)-Ib-cr gene but negative for qepA, except for SD001. In addition, only 6 (20.69%, 6/29) isolates harbored the qnr gene, including two with qnrB (6.90%, 2/29) and four with qnrS (13.79%, 4/29).
Conclusion
Given the increased common emergence of multidrug resistant isolates, uninterrupted surveillance will be necessary to understand the actual epidemic burden and control this infection.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12866-020-02050-9.
Keywords: Shigella dysenteriae, Fluoroquinolone-resistant, QRDR, PMQR
Background
Shigella is one of the major pathogens that causes diarrheal diseases in humans and animals [1, 2]. Since the first highly toxigenic Shigella dysenteriae was isolated in 1898 [3], four species have been described in Shigella genus. Each subgroup can be divided into multiple subtypes according to biochemical and serological properties [4, 5]. Worldwide, there are approximately 164.7 million shigellosis infection cases in humans annually [5], while there are few data showing the prevalence in animals.
Antimicrobial therapy is the most effective way to fight shigellosis, so various kinds of drugs are widely used [6]. Fluoroquinolone, especially ciprofloxacin, was the first-line antibiotic for the treatment of shigellosis as recommended by the WHO [7]. However, antibiotic resistance usually occurs in the environment where antibiotics are frequently used [8, 9]. Furthermore, the accelerating accumulation and transmission of antimicrobial resistance genes (ARGs) among multidrug resistant (MDR) pathogens pose major difficulties in treating infections [10].
ARGs are widespread but cause problems only when present in pathogens [10]. Virulence genes have become significant marker of pathogenic bacteria. S. dysenteriae possesses diverse virulence genes, located on both the chromosome and/or the plasmid [11]. The common genetic loci are invasion plasmid antigen H (ipaH), invasion plasmid antigen genes (ipaBCD) and invasion associated locus (ial). In addition, the Shigella enterotoxin genes set1A, set1B (ShET-1), sen (SHET-2) and Shiga toxin gene stx were also reported to be responsible for initial watery diarrhea [12–14].
In this study, we aimed to investigate the epidemiology and molecular characteristics of S. dysenteriae isolates from calve farms in Gansu, China. Our analysis includes the antimicrobial resistance profiles, the molecular characterization of the mechanisms of resistance to fluoroquinolones, virulence gene profiles and the molecular characterization of S. dysenteriae isolates by pulsed-field gel electrophoresis (PFGE) and multilocus sequence typing (MLST).
Results
Bacterial isolation and identification
During our routine surveillance of bacillary dysentery, 38 S. dysenteriae were only found on six farms in Gansu Province. Detailed information on the study isolates is listed in Table S3. Among them, 20 isolates were isolated from 3 beef cattle farms, and 18 isolates were isolated from 3 dairy farms. There are 14 and 10 S. dysenteriae isolates from the same farms of Zhangye and Jinchang, respectively.
All 38 S. dysenteriae isolates were type 1 according to the results of the serotype reactions. In addition, based on the typical biochemical characteristics of Shigella spp., analysis of biochemical reactions indicated the presence of 3 biotypes (BTs) among these isolates (Tables 1 and S1). Among these BTs, BT2 (ability to ferment glucose, arabinose, and melibiose) was the predominant biotype, accounting for 86.84% (33/38). Furthermore, BT2 was widely isolated from each locus, with the exception of Baiyin.
Table 1.
Biochemical characteristics of S. dysentery 1 isolates
| Biotype | Total (n = 38) | ZY (n = 14) | BY (n = 3) | LX (n = 3) | LZ (n = 5) | WW (n = 3) | JC (n = 10) | |
|---|---|---|---|---|---|---|---|---|
| BT1 | glucose+, mannose+, arabinose+, melibiose- | 3 (7.89%) | 0 | 3 (100%) | 0 | 0 | 0 | 0 |
| BT2 | glucose+, mannose-, arabinose+, melibiose+ | 33 (86.84%) | 14 (100%) | 0 | 3 (100%) | 5 (100%) | 1 (33.33%) | 10 (100%) |
| BT3 | glucose+, mannose+, arabinose+, melibiose+ | 2 (5.26%) | 0 | 0 | 0 | 0 | 2 (66.67%) | 0 |
ZY Zhangye, BY Baiyin, LX Linxia, LZ Lanzhou, WW Wuwei, JC Jinchang
MLST-based genotype analysis
Thirty-eight S. dysenteriae isolates belonged to 4 MLST patterns (STs): ST57, ST191, ST228 and ST229. Among them, ST57 and ST191 were previously reported, while ST228 and ST229 were novel types. The allele number for each locus and the designation of the ST are listed in Fig. 1 and shared on the EcMLST website. The most common STs identified were ST228 (n = 13) and ST229 (n = 15), accounting for 73.68% (28/38). ST228 and ST229 were major ST types for dairy cows (Jinchang and Wuwei) and beef cattle (Zhangye and Baiyin) farms, respectively. In addition, these two ST types were different by 3 loci: arcA, clpX, mutS. In this study, each farm has a fixed ST type isolates, except Zhangye which contains ST229 and ST57 types (Figs. 1 and 2).
Fig. 1.
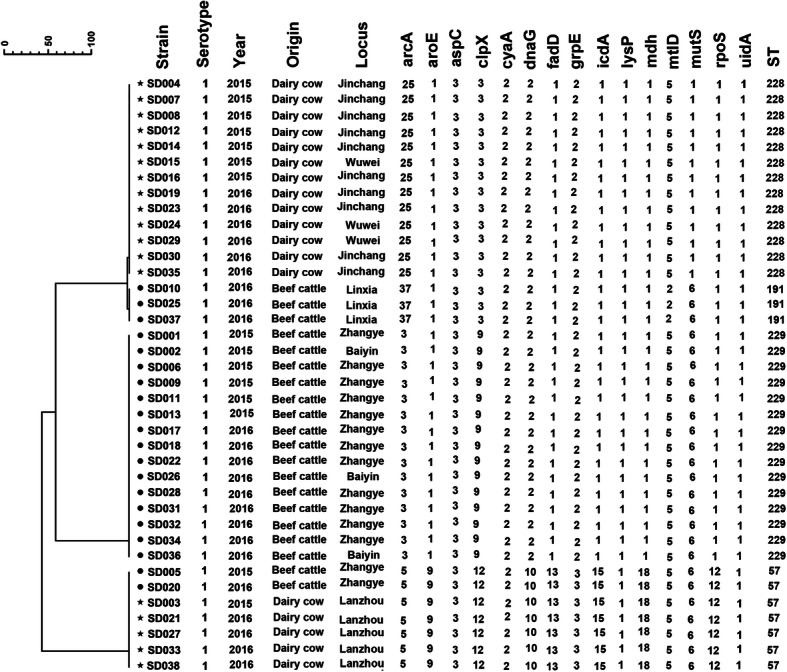
MLST clustering tree of S. dysenteriae isolates from 2014 to 2016 in diarrhea calves. The 38 isolates were analyzed using a 15 allele MLST as described in the Materials and Methods
Fig. 2.

Minimum spanning tree of the isolates of 38 S. dysenteriae isolates from diarrhea calves based on multilocus sequence typing (MLST). The minimum spanning tree was constructed using the 4 identified STs obtained from the 38 isolates using BioNumerics Software. Each circle corresponds to a single ST. The shadow zones of circles’ filling in green correspond to beef, and red to dairy cows. The size of the circle is proportional to the number of isolates and the numbers over the branches means the difference between the two ST types. The corresponding color, number of isolates and background information are shown to the right of the minimum spanning tree. The number on the lines represent the number of different genes between the two conjoint STs
PFGE-based genotype analysis
The genotypes and genetic relatedness of the 38 isolates were further determined by using PFGE. PFGE patterns of these S. dysenteriae isolates were heterogeneous; however, multiple PFGE patterns were present among these strains (Fig. 3). With approximately 80% similarity, XbaI-digested S. dysenteriae type 1 could be divided into 28 distinctive PFGE patterns (PT) and belonged to two major groups: A (A1-A4) and B (B1-B3), with 66% similarity. The cluster result of PFGE was similar with MLST, however, the same ST strain can be divided into several similar PT types. For example, the ST229 type is divided into 10 PT types, and the ST228 type is divided into 9 PT types. Interestingly, most strains in different geographical locations can be clustered individually, and strains in the farm can be divided into multiple PT types.
Fig. 3.

Dendrogram of 38 XbaI-digested S. dysenteriae isolates based on the cluster analysis of PFGE patterns. The dendrogram was constructed using the UPGMA clustering method. The PFGE pattern and background information for each strain are listed on the right side of the dendrogram
Prevalence of virulence genes
A total of five virulence genes were detected in those isolates involving ipaH, ipaBCD, ial, sen, and stx. The most frequently observed virulence genes are ipaH (100%), ipaBCD (92.11%), stx (73.68%), and ial (57.89%). The Shigella enterotoxin genes sen (28.95%) are occasionally present in Wuwei and Jinchang isolates. None of the studied strains possessed the set1A or set1B gene.
Regarding the differences in virulence gene distributions, the 38 S. dysenteriae isolates fell into 5 gene profile types (VT) (Table 2). Among these VTs, VT IV (n = 17) and VT V (n = 11) were the most common, accounting for 44.34 and 28.95%, respectively. One interesting finding was the presence of the same and/or similar VTs in the same locus. In addition, 92.11% of isolates carried two or more virulence genes. Three Linxia isolates (7.89%) belonged to VT I, which was only positive for ipaH.
Table 2.
Statistical the rate of each Virulence genes profile in S. dysentery serotype 1 isolates
| VT | Virulence genes profiles | No. (%) isolates | ||||||
|---|---|---|---|---|---|---|---|---|
| Total (n = 38) | ZY (n = 14) | BY (n = 3) | LX (n = 3) | LZ (n = 5) | WW (n = 3) | JC (n = 10) | ||
| I | ipaH + ipaBCD-ial-sen-stx- | 3 (7.89%) | 0 | 0 | 3 (100%) | 0 | 0 | 0 |
| II | ipaH + ipaBCD + ial-sen-stx- | 2 (5.26%) | 0 | 2 (66.67%) | 0 | 0 | 0 | 0 |
| III | ipaH + ipaBCD + ial + sen-stx- | 5 (13.16%) | 2 (14.29%) | 0 | 0 | 2 (40%) | 1 (33.33%) | 0 |
| IV | ipaH + ipaBCD + ial + sen-stx+ | 17 (44.34%) | 12 (85.71%) | 1 (33.33%) | 0 | 3 (60%) | 0 | 1 (10%) |
| V | ipaH + ipaBCD + ial-sen + stx+ | 11 (28.95%) | 0 | 0 | 0 | 0 | 2 (66.67%) | 9 (90%) |
VT Virulence genes profiles type, ZY Zhangye, BY Baiyin, LX Linxia, LZ Lanzhou, WW Wuwei, JC Jinchang
Antimicrobial resistance profiles
The antimicrobial resistance profiles of the 20 antimicrobials for 38 S. dysenteriae are shown in Table 3. All S. dysenteriae isolates were uniformly multidrug resistant to at least 3 types of antimicrobial agents. Among them, resistance to E was the most common (36,94.74%), followed by AMP (35, 92.11%), KZ (33, 86.84%), CRO (32, 84.21%), CTX (32, 84.21%), TE (31, 81.58%), CN (31, 81.58%), ENR (26, 68.42%), LEV (25, 65.79%), CIP (25, 65.79%), NOR (15, 39.47%), OFX (15, 39.47%), C (15, 39.47%), and S (15, 39.47%). Fortunately, all 38 isolates were sensitive to AMC, FOX, FEP, MEM, IPM and AK. However, the resistance rate of fluoroquinolone antibiotics has reached 39.47 to 68.42%, which will narrow the choice of antibiotics.
Table 3.
Statistical and analysis the result of antimicrobial susceptibility to 20 antibiotics for 38 S. dysentery
| Antimicrobial | Antimicrobial resistance rate No. (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Total (n = 38) | Beef farm(n = 20) | Dairy farm(n = 18) | |||||||
| Zhangye(n = 14) | Baiyin(n = 3) | Linxia(n = 3) | Jinchang(n = 10) | Lanzhou(n = 5) | Wuwei(n = 3) | ||||
| Fluoroquinolone | Norfloxacin | NOR | 15(39.47%) | 12(85.71%) | 3(100%) | 0 | 0 | 0 | 0 |
| Enrofloxacin | ENR | 26(68.42%) | 13(92.86%) | 0 | 0 | 10(100%) | 0 | 3(100%) | |
| Levofloxacin | LEV | 25(65.79%) | 12(85.71%) | 0 | 0 | 10(100%) | 0 | 3(100%) | |
| Ciprofloxacin | CIP | 25(65.79%) | 12(85.71%) | 0 | 0 | 10(100%) | 0 | 3(100%) | |
| Ofloxacin | OFX | 15(39.47%) | 12(85.71%) | 3(100%) | 0 | 0 | 0 | 0 | |
| β-lactam | Ampicillin | AMP | 35(92.11%) | 14(100%) | 3(100%) | 0 | 10(100%) | 5(100%) | 3(100%) |
| Amoxycillin/Clavulanic acid | AMC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Cephazolin | KZ | 33(86.84%) | 14(100%) | 3(100%) | 0 | 9(90%) | 5(100%) | 2(66.67%) | |
| Cefoxitin | FOX | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Ceftriaxone | CRO | 32(84.21%) | 14(100%) | 0 | 0 | 10(100%) | 5(100%) | 3(100%) | |
| Cefotaxime | CTX | 32(84.21%) | 14(100%) | 0 | 0 | 10(100%) | 5(100%) | 3(100%) | |
| Cefepime | FEP | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Meropenem | MEM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Imipenem | IPM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Macrolides | Erythromycin | E | 36(94.74%) | 12(85.71%) | 3(100%) | 3(100%) | 10(100%) | 5(100%) | 3(100%) |
| Chloramphenicol | Chloramphenicol | C | 15(39.47%) | 12(85.71%) | 3(100%) | 0 | 0 | 0 | 0 |
| Tetracyclines | Tetracycline | TE | 31(81.58%) | 12(85.71%) | 3(100%) | 3(100%) | 10(100%) | 0 | 3(100%) |
| Aminoglycosides | Streptomycin | S | 15(39.47%) | 12(85.71%) | 3(100%) | 0 | 0 | 0 | 0 |
| Gentamicin | CN | 31(81.58%) | 12(85.71%) | 3(100%) | 3(100%) | 10(100%) | 0 | 3(100%) | |
| Amikacin | AK | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
Moreover, most of the isolates (29/38, 76.32%) were resistant to fluoroquinolone antibiotics. And 4 isolates were resistant to other fluoroquinolones other than CIP, including the resistance of SD020 to ENR and the resistance of SD002, SD026, SD036 to NOR and OFX. These resistant isolates were divided into 5 antimicrobial-resistance profile types (RT) (Table 4). Among these RTs, RT 4 (n = 11) and RT 5 (n = 12) were detected easily, accounting for 37.93 and 41.38%, respectively. All of the fluoroquinolone-resistant isolates were multidrug resistant (MDR). To be specific, 41.8% (12/29) of fluoroquinolone-resistant isolates were resistant to each class of antibiotics. Regarding the test for the virulence genes of ipaBCD, sen, stx, there are statistically significant difference between RT type and fluoroquinolone sensitive strains (P<0.05) (Table 5).
Table 4.
Antimicrobial profile and amino acid types in QRDRs and PMQRs analysis of S. dysentery with resistance to fluoroquinolones
| Strain name | Antimicrobial profile | QRDRs | PMQR | ST | PT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| gyrA | parC | aac(6′)-Ib-cr | qnr | qepA | |||||||
| 83 (S) | 87 (D) | 80 (S) | 83 (S) | ||||||||
| SD020 | RT1 | ENR/KZ/CRO/CTX | L | D | I | L | + | – | – | 57 | 15 |
| SD002 | RT2 | NOR/OFX/KZ/E/C/TE/S/CN | L | D | S | L | + | – | – | 229 | 6 |
| SD026 | RT2 | NOR/OFX/KZ/E/C/TE/S/CN | L | D | S | L | + | – | – | 229 | 6 |
| SD036 | RT2 | NOR/OFX/KZ/E/C/TE/S/CN | L | D | S | L | + | – | – | 229 | 7 |
| SD015 | RT3 | ENR/LEV/CIP/ CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 23 |
| SD016 | RT3 | ENR/LEV/CIP/ CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 25 |
| SD004 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 28 |
| SD007 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 26 |
| SD008 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | qnrB | – | 228 | 26 |
| SD012 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 26 |
| SD014 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 27 |
| SD019 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 22 |
| SD023 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | qnrB | – | 228 | 22 |
| SD024 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 21 |
| SD029 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 20 |
| SD030 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 24 |
| SD035 | RT4 | ENR/LEV/CIP/ KZ/CRO/CTX/E/TE/CN | L | D | S | L | + | – | – | 228 | 23 |
| SD001 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | + | 229 | 8 |
| SD006 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | – | 229 | 8 |
| SD009 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | – | 229 | 8 |
| SD011 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | – | 229 | 9 |
| SD013 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | – | 229 | 8 |
| SD017 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | – | 229 | 8 |
| SD018 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | – | – | 229 | 10 |
| SD022 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | qnrS | – | 229 | 6 |
| SD028 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | qnrS | – | 229 | 4 |
| SD031 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | qnrS | – | 229 | 3 |
| SD032 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | qnrS | – | 229 | 1 |
| SD034 | RT5 | NOR/ENR/LEV/CIP/OFX/ KZ/CRO/CTX/E/C/TE/S/CN | L | N | I | L | + | qnrS | – | 229 | 2 |
Table 5.
Prevalence of virulence genes in the different antimicrobial-resistance profile types strains
| Gene | Total (n = 38) | RT1 (n = 1) | RT2 (n = 3) | RT3 (n = 2) | RT4 (n = 11) | RT5 (n = 12) | Other (n = 9) | P-value |
|---|---|---|---|---|---|---|---|---|
| ipaH | 38 (100%) | 1 (100%) | 3 (100%) | 2 (100%) | 11 (100%) | 12 (100%) | 9 (100%) | – |
| ipaBCD | 35 (92.11%) | 1 (100%) | 3 (100%) | 2 (100%) | 11 (100%) | 12 (100%) | 6 (66.67%) | 0.001 |
| ial | 22 (57.89%) | 1 (100%) | 1 (33.33%) | 0 (0%) | 2 (18.18%) | 12 (100%) | 6 (66.67%) | 0.542 |
| sen | 11 (28.95%) | 0 (0%) | 0 (0%) | 2 (100%) | 9 (81.82%) | 0 (0%) | 0 (0%) | 0.028 |
| stx | 28 (73.68%) | 0 (0%) | 1 (33.33%) | 2 (100%) | 10 (90.91%) | 12 (100%) | 3 (33.33%) | 0.002 |
Other: fluoroquinolone sensitive strains
In addition, the resistance profiles of strains which isolated from the same farms were very similar. And the isolates belonging to ST229 (A1 and A2 groups) and ST228 (B group) were resistant to fluoroquinolone antibiotics, while the isolates belonging to ST 57 (A4) and ST191 (A3) were sensitive to those antibiotics, except for SD020 isolated from Zhangye area with ST57 (PT15).
ARG analysis for fluoroquinolone-resistant S. dysenteriae isolates
To determine the molecular characterization in fluoroquinolone-resistant S. dysenteriae, both SNPs in QRDR of gyrA/B and parC/E genes and PMQR genes were analyzed. In 29 fluoroquinolone-resistant isolates, there was no strain that displayed mutations in the gyrB and parE genes, although some mutations in gyrA and parC were identified in each resistant isolate (Table 4). All resistant isolates in the present study carried common mutations in gyrA codon 83 (S → L) and parC codon 83 (S → L). Furthermore, the isolates with ST228 only carried the mutations in gyrA codon 83 (S → L) and parC codon 83 (S → L). While most of the isolates with ST229 carried all of the four mutated locuses, except isolates from Baiyin.
The PMQR genes qnr, aac (6′)-Ib-cr, and qepA occur worldwide and are increasingly detected in clinical isolates of Enterobacteriaceae [15, 16]. In our study, aac (6′)-Ib-cr was the most common PMQR gene and harbored by each isolate; however, all isolates were negative for qepA, except SD001. Only seven (7/29, 24.14%) isolates harbored the qnr gene, and no isolate harbored qnr, aac (6′)-Ib-cr, and qepA simultaneously. Interestingly, the qnrB (n = 2) gene and qnrS (n = 5) were belonged into ST228 and ST229, respectively.
Discussion
Invasive diarrhea due to Shigella species remains an important public health problem in developing countries [17]. All four species of Shigella can cause shigellosis, but S. flexneri and S. sonnei are the most prevalent [18]. Present study confirmed the existence of diverse Shigella isolates in calves in local epidemiological studies. A total of 136 Shigella isolates were collected in this study, including 54 S. flexneri (1.63%, 54/3321), 44 S. sonnei (1.32%, 44/3321), and 38 S. dysenteriae (1.14%, 38/3321), and no S. boydii was found.
Strain molecular characterization is important for epidemiological studies. MLST, PFGE and MLVA techniques were widely used in laboratory for Shigella study, unfortunately, we don’t have a widely recognized MLVA methods to study S. dysentery. The next generation sequencing is the reference technique today, however, it requires professional analytical methods and tedious operations, making it unable to be widely carried out in all laboratories compared with MLST and PFGE.
In our study, the majority of isolates (n = 13 and n = 15) belonged to new ST types ST228 and ST229. However, ST148, ST252, and ST1739 were previously reported in human isolates [19]. PFGE is the most common typing procedure currently used with Shigella spp. because of its high discriminatory power [20]. Taking PFGE as a reference epidemiological tool, the clustering of these diverse PTs allows us to learn more about the epidemiological characteristics of S. dysenteriae in specific geographical regions.
The ability of Shigella spp. to cause shigellosis is attributed to the expression of arrays of virulence genes associated with colonization, invasion/penetration and toxin-mediated disease [21]. Virulence genes responsible for the pathogenesis of shigellosis are often multifactorial and coordinately regulated [22]. Estimating the existence of virulence determinants in Shigella would help us better understand its pathogenicity [21]. IpaH takes responsibility for the strain spread from cell to cell and the modification of host response to infection [23]. All Shigella spp. were positive for ipaH as a typical marker because this gene exists in multiple copies on both the chromosome and the invasion plasmid. Additionally, in this study, most isolates simultaneously harbored the stx, ial and sen genes. The diversity of the observed virulence genes results in dysentery in hosts. Notably, these virulence genes are indistinguishable from human Shigella.
For shigellosis, antibiotic therapy can reduce the duration and severity of the illness [24]. Based on previous reports, fluoroquinolones and third-generation cephalosporins are the best choice for empiric treatment for severe bacterial diarrheas caused by S. dysenteriae [25]. However, resistance will emerge to any antimicrobial agent used intensively, and the differential selection pressures of antimicrobials lead to differences in drug resistance [26]. These factors have contributed to Shigella strains acquiring resistance to the latest drug of choice, together with other effective drugs after therapy shigellosis. In addition, the resistance rate of S. dysenteriae 1 isolates to fluoroquinolones has achieved at an alarming rate, and the isolates have demonstrated multidrug resistance. Our current study indicated that continuous surveillance of the resistance pattern in pathogens would be essential for the choice of appropriate antimicrobial therapy and control the spread of Shigella.
The most common mechanism of the highly level resistance to quinolone has been mostly attributed to accumulation of sequential mutations in DNA gyrase and DNA topoisomerase IV, typically of gyrA at codon 83 and/or 87, and of parC at codon 80 [27, 28]. In addition, different mutation loci and different mutations at the same locus may result in different quinolone susceptibility levels [29, 30]. To date, limited studies have examined quinolone resistance mechanisms among Shigella isolates in animals. In the current study, we successfully investigated fluoroquinolone resistance-conferring mutations by sequencing QRDR of S. dysenteriae 1 isolated in different geographical locations. According to our results, only two main combinations of mutations were observed: gyrA codons 83 (Ser → Leu), and parC codons 83 (Ser → Leu) (55.17%, 16/29); gyrA codons 83 (Ser → Leu), 87 (Asp→Asn) and parC codons 80 (Ser → Ile), 83 (Ser → Leu) (41.38%, 12/29). This phenomenon of high resistance rate and single mutation pattern is likely related to the large-scale use of single quinolone antibiotics by veterinarians.
Generally, the PMQR determinants are located on mobile genetic elements, which may be associated with mobile or transposable elements among members of the Enterobacteriaceae family [31]. Over the past few years, the dissemination of PMQR genes among quinolone-resistant Shigella isolates has been surveyed in some studies and has emerged as an important issue across the world [32–35]. It has been reported that aac (6′)-Ib-cr significantly increases the frequency of selection of chromosomal mutants to reduce ciprofloxacin activity by N-acetylation at the amino nitrogen on its piperazinyl substituent [36]. The aac (6′)-Ib-cr–positive Shigella was first isolated in 1998 [37]; however, the most striking finding of our previous and present study was the aac (6′)-Ib-cr widely present in all quinolone-resistant isolates [38, 39]. The qnr family could protect DNA gyrase against quinolones and confer low-level resistance to nalidixic acid and reduced susceptibility to fluoroquinolone, especially to ciprofloxacin [40]. The qnr gene has occurred worldwide and contains a variety of subtypes, but it was not a common epidemic genotype according to our study. Otherwise, the plasmid-mediated qepA gene can encode a 14-transmembrane segment efflux pump and extrude several quinolones from bacteria, thereby conferring low levels of resistance to these antimicrobial agents and favoring the selection of new alterations that are able to induce full quinolone resistance [41, 42]. Compared with other PMQR determinants, qepA is a new resistance gene, first described in 2007 [43], so it is not difficult to understand the lowest frequencies.
The QRDR mutation and PMQR determinants are both important to quinolone resistance. Compared with mutations in the QRDR, PMQR could not be the direct reason for quinolone and fluoroquinolone resistance [44]. However, the presence of PMQR genes may facilitate the selection of QRDR mutations that result in higher levels of quinolone resistance.
Conclusion
In this study described fluoroquinolone resistance and virulence genes among S. dysenteriae strains isolated from calves and the molecular characterization involved. To systematically understand S. dysenteriae, PFGE and MLST methods were applied to genetically characterize the 38 isolates. PFGE based on XbaI digestion divided the 38 isolates into 28 PTs, while MLST based on 15 housekeeping genes differentiated the 38 isolates into 4 STs. Although MLST provided suitable discrimination in S. dysenteriae subtyping, PFGE might exhibit a higher discriminatory ability. Overall, the data from this study will provide a useful typing resource, which will provide a scientific basis for addressing clinical and epidemiological issues regarding S. dysenteriae. Given this knowledge, continuous and extensive surveillance will be essential to explore and prevent the spread of the epidemic.
Methods
Bacterial strains and serotyping
From October 2014 to December 2016, we tracked the calf diarrhea caused by Shigella from 26 farms in northwestern regions of China. A total of 3321 fresh stool samples from calves with diarrhea (3 to 20 days) were collected for detecting Shigella. All of the isolates were collected directly from fresh stool samples following plating on Salmonella-Shigella (SS) selective agar and confirming on MacConkey (MAC) agar with 37 °C for 24 h. The colorless, semitransparent, smooth, and moist circular plaques were considered presumptive Shigella for further 16S rDNA (TaKaRa, Japan) and biochemical confirmation.
The biochemical reactions of the presumptive positive Shigella isolates were tested by API20E kits (bioMerieux, France), and the serotype was tested by a commercially available agglutinating antibody kit (Denka Seiken, Tokyo, Japan) according to the manufacturers’ recommendations.
Multilocus sequence typing (MLST)
All isolates were subjected to MLST according to the protocols described in the EcMLST database (http://www.shigatox.net/ecmlst). PCR amplification conditions of 15 housekeeping genes were as follows: 95 °C for 5 min; 30 cycles of 94 °C for 30 s, 55 °C for 90 s, and 72 °C for 1 min; and 72 °C for 5 min with ExTaq DNA polymerase (Takara, Dalian, China). The PCR products were sequenced bidirectionally, and sequences of the 15 housekeeping genes were edited by using SeqMan 7.0. Then, these sequences were uploaded to the EcMLST website for comparison, which allowed us to determine the gene and ST type [45]. Clustering and minimum spanning tree (MST) analysis were used to infer relationships among the isolates using the fingerprint analysis software BioNumerics (version 7.1).
Pulsed-field gel electrophoresis (PFGE)
DNA fingerprinting was performed by PFGE. Intact agarose-embedded chromosomal DNA of S. dysenteriae isolates was digested with restriction enzyme XbaI (TaKaRa, Japan), and restriction fragments were separated on the CHEF Mapper XA system (Bio-Rad) in 1% SeaKem Gold agarose gel (Lonza, USA) in 0.5x TBE buffer with a size range of 30–700 kb. Electrophoretic parameters were determined by multiple screening for the most suitable value, which included switching times from 2.16 to 54.17 s, voltage 6 v/cm, angle 120° and run time 21 h. Salmonella enterica serotype Braenderup strain H9812 was used as a molecular size marker for this analysis.
The gel image was captured by Universal Hood II (Bio-RAD, USA), and the PFGE profiles were analyzed by BioNumerics (version 7.1). Degrees of homology were determined by comparing the bands, and the clustering tree indicating relative genetic similarity was constructed by the unweighted pair group method with arithmetic averages (UPGMA).
Detection of virulence genes
All S. dysenteriae isolates were examined for the presence of seven virulence genes, invasion plasmid antigen H (ipaH), invasion plasmid antigen genes (ipaBCD), invasion associated locus (ial), Shiga toxin gene (stx), and Shigella enterotoxin genes (set1A, set1B, and sen) by PCR according to published procedures [13, 27]. The primers for these virulence genes are listed in Table S2. Amplification products were separated by 1% agarose gel electrophoresis and stained with ethidium bromide.
Determination of antimicrobial susceptibility
All of the isolated strains were tested for their antimicrobial susceptibility against a variety of 20 antibiotic discs (Oxoid, UK) on Mueller–Hinton agar following the guidelines of the Clinical and Laboratory Standards Institute (CLSI) [46]. The antimicrobial discs used in this study were norfloxacin (NOR, 10 μg), enrofloxacin (ENR, 5 μg), levofloxacin (LEV, 5 μg), ciprofloxacin (CIP, 5 μg), ofloxacin (OFX, 5 μg), ampicillin (AMP, 10 μg), amoxycillin/clavulanic acid (AMC, 30 μg), cephazolin (KZ, 30 μg), cefoxitin (FOX, 30 μg), ceftriaxone (CRO, 30 μg), cefotaxime (CTX, 30 μg), cefepime (FEP, 30 μg), imipenem (IPM, 10 μg), meropenem (MEM, 10 μg), erythromycin (E, 15 μg), chloramphenicol (C, 30 μg), tetracycline (TE, 30 μg), streptomycin (S, 10 μg), gentamicin (CN, 10 μg), and amikacin (AK, 30 μg). E. coli ATCC 25922 was used for quality control organisms according to the CLSI standard.
Detection of fluoroquinolone resistance genes
To understand the underlying mechanism of fluoroquinolone resistance, four QRDR genes, DNA gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE), and six PMQR determinant genes, qnrA, qnrB, qnrD, qnrS, aac (6′)-Ib-cr and qepA, were amplified for fluoroquinolone-resistant isolates in our study [4, 47, 48]. The obtained PCR fragments were sequenced after purification and analyzed through comparison with sequences in GenBank. The primer information is listed in Table S3.
Statistical analysis
The P-value in each gene of prevalence rates across RT and Other were calculated using Chi-square test (SPSS 19.0 version, IBM, New York, USA) to find any relationship. A P<0.05 was statistically significant.
Supplementary Information
Additional file 1: Table S1. Strain information of S. dysenteriae isolates from diarrheal calves in this study.
Additional file 2: Table S2. Primers for the detection of virulence genes.
Additional file 3: Table S3. Primers for the detection of fluoroquinolone resistance–determining genes.
Acknowledgements
Not Applicable.
Abbreviations
- PFGE
Pulsed-field gel electrophoresis
- MLST
Multilocus sequence typing
- ARGs
Antimicrobial resistance genes
- MDR
Multidrug resistant
- BTs
Biotypes
- VT
Virulence profile types
- NOR
Norfloxacin
- ENR
Enrofloxacin
- LEV
Levofloxacin
- CIP
Ciprofloxacin
- OFX
Ofloxacin
- AMP
Ampicillin
- AMC
Amoxycillin/Clavulanic
- KZ
Cephazolin
- FOX
Cefoxitin
- CRO
Ceftriaxone
- CTX
Cefotaxime
- FEP
Cefepime
- MEM
Meropenem
- IPM
Imipenem
- E
Erythromycin
- C
Chloramphenicol
- TE
Tetracycline
- S
Streptomycin
- CN
Gentamicin
- AK
Amikacin
Authors’ contributions
Z.Z. and J.Y.Z. designed the study; Z.Z., M.Z.C., Y.X.S., X.Z.Z., G.H.L and B.L. generated and provided the dataset; Z.Z., L.W.Z and W.W.W. performed the experiments, analyzed the data, and wrote the manuscript. All authors have read and approved the final manuscript.
Funding
The design of the study including collection, analysis, and interpretation of data was supported by the grants from the National Natural Science Foundation of China (No:31872520) and Drug Development and Clinical Drug Use Posts of National Beef Yak Industry Technical System (No: CARS-37); And the writing and submission the manuscript was supported by the Natural Science Foundation of Hebei Province (No: C2019402114) and a grant (2018SKLID308) from the State Key Laboratory for Infectious Disease Prevention and Control (China CDC).
Availability of data and materials
The data supporting the findings of this study are contained within the manuscript.
Ethics approval and consent to participate
This study has gained ethical approval for this study. Our study was conducted according to the Animal Administration and Ethics Committee of Lanzhou Institute of Husbandry and Pharmaceutical Sciences of CAAS (Permit No. SYXK-2014-009). And we gained consent from the owners of the animals for them to be used in the study.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mingze Cao and Weiwei Wang contributed equally to this work.
Contributor Information
Zhen Zhu, Email: zhuzhen234@yeah.net.
Jiyu Zhang, Email: infzjy@sina.com.
References
- 1.Mather AE, Baker KS, McGregor H, Coupland P, Mather PL, Deheer-Graham A, et al. Bacillary dysentery from world war 1 and NCTC1, the first bacterial isolate in the National Collection. Lancet. 2014;384:1720. doi: 10.1016/S0140-6736(14)61790-6. [DOI] [PubMed] [Google Scholar]
- 2.Priamukhina NS, Kilesso VA, Tikhomirov ED, et al. Animal carriers of Shigella and their possible epidemiological importance. Zh Mikrobiol Epidemiol Immunobiol. 1984;11:20–24. [PubMed] [Google Scholar]
- 3.Baker KS, Mather AE, McGregor H, Coupland P, Langridge GC, Day M, et al. The extant world war 1 dysentery bacillus NCTC1: a genomic analysis. Lancet. 2014;384:1691–1697. doi: 10.1016/S0140-6736(14)61789-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui X, Wang J, Yang C, Liang B, Ma Q, Yi S, et al. Prevalence and antimicrobial resistance of Shigella flexneri serotype 2 variant in China. Front Microbiol. 2015;6:435. doi: 10.3389/fmicb.2015.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perepelov AV, Shekht ME, Liu B, Shevelev SD, Ledov VA, Senchenkova SN, et al. Shigella flexneri O-antigens revisited: final elucidation of the O-acetylation profiles and a survey of the O-antigen structure diversity. FEMS Immunol Med Microbiol. 2012;66:201–210. doi: 10.1111/j.1574-695X.2012.01000.x. [DOI] [PubMed] [Google Scholar]
- 6.Tajbakhsh M, Garcia Migura L, Rahbar M, Svendsen CA, Mohammadzadeh M, Zali MR, et al. Antimicrobial-resistant Shigella infections from Iran: an overlooked problem? J Antimicrob Chemoth. 2012;67:1128–1133. doi: 10.1093/jac/dks023. [DOI] [PubMed] [Google Scholar]
- 7.WHO International note on antibiotics in the management of shigellsosis. Wkly Epidemiol Rec. 2004;79:202–203. [Google Scholar]
- 8.Poole K, Srikumar R. Multidrug efflux in Pseudomonas aeruginosa: components, mechanisms and clinical significance. Curr Top Med Chem. 2001;1:59. doi: 10.2174/1568026013395605. [DOI] [PubMed] [Google Scholar]
- 9.McEwen SA, Fedorka-Cray PJ. Antimicrobial use and resistance in animals. Clin Infect Dis. 2002;34:93–106. doi: 10.1086/340246. [DOI] [PubMed] [Google Scholar]
- 10.Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DGJ. The structure and diversity of human, animal and environmental resistomes. Microbiome. 2016;4:54. doi: 10.1186/s40168-016-0199-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutta S, Nandy S, Jain P, Yoshida S, Matsushita S. Molecular characterization of serologically atypical provisional serovars of Shigella isolates from Kolkata, India. J Med Microbiol. 2014;63:1696–1703. doi: 10.1099/jmm.0.081307-0. [DOI] [PubMed] [Google Scholar]
- 12.Vargas M, Gascon J, Maria TJ, Vila J. Prevalence of Shigella enterotoxins 1 and 2 among Shigella strains isolated from patients with Traveler's diarrhea. J Clin Microbiol. 1999;37:3608–3611. doi: 10.1128/JCM.37.11.3608-3611.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das A, Natarajan M, Mandal J. The emergence of quinolone resistant Shigella sonnei, Pondicherry, India. PLoS One. 2016;11:e160290. doi: 10.1371/journal.pone.0160290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faruque SM, Khan R, Kamruzzaman M, Yamasaki S, Ahmad QS, Azim T, et al. Isolation of Shigella dysenteriae type 1 and S. flexneri strains from surface waters in Bangladesh: comparative molecular analysis of environmental Shigella isolates versus clinical strains. Appl Environ Microbiol. 2002;68:3908–3913. doi: 10.1128/AEM.68.8.3908-3913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robicsek A, Jacoby GA, Hooper DC. The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect Dis. 2006;6:629–640. doi: 10.1016/S1473-3099(06)70599-0. [DOI] [PubMed] [Google Scholar]
- 16.Pu X, Pan J, Gu Y, Zheng W, Li J, Yu H. Complete sequences and characterization of two novel plasmids Carryingaac (6′)-Ib-cr andqnrS gene inShigella flexneri. Microb Drug Resist. 2016;22:115–122. doi: 10.1089/mdr.2015.0082. [DOI] [PubMed] [Google Scholar]
- 17.Ansaruzzaman M. Isolation and characterization of provisional serovar Shigella boydii E16553 from diarrhoeal patients in Bangladesh. J Med Microbiol. 2005;54:477–480. doi: 10.1099/jmm.0.45889-0. [DOI] [PubMed] [Google Scholar]
- 18.Livio S, Strockbine NA, Panchalingam S, Tennant SM, Barry EM, Marohn ME, et al. Shigella isolates from the global enteric multicenter study inform vaccine development. Clin Infect Dis. 2014;59:933–941. doi: 10.1093/cid/ciu468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chattaway MA, Schaefer U, Tewolde R, Dallman TJ, Jenkins C. Identification of Escherichia coli and Shigella species from whole-genome sequences. J Clin Microbiol. 2017;55(2):616–623. doi: 10.1128/JCM.01790-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navia MM, Capitano L, Ruiz J, Vargas M, Urassa H, Schellemberg D, et al. Typing and characterization of mechanisms of resistance of Shigella spp. isolated from feces of children under 5 years of age from Ifakara, Tanzania. J Clin Microbiol. 1999;37:3113–3117. doi: 10.1128/JCM.37.10.3113-3117.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang CL, Liu QZ, Wang J, Chu X, Shen LM, Guo YY. Epidemic and virulence characteristic of Shigella spp. with extended-spectrum cephalosporin resistance in Xiaoshan District, Hangzhou, China. BMC Infect Dis. 2014;14:260. doi: 10.1186/1471-2334-14-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu M, Zhang X, Liu G, Huang Y, Jia L, Liang W, et al. An eight-year study of Shigella species in Beijing, China: serodiversity, virulence genes, and antimicrobial resistance. J Infect Dev Ctries. 2014;8:904–908. doi: 10.3855/jidc.3692. [DOI] [PubMed] [Google Scholar]
- 23.Parsot C. Shigella spp. and enteroinvasiveEscherichia coli pathogenicity factors. FEMS Microbiol Lett. 2005;252:11–18. doi: 10.1016/j.femsle.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 24.Pazhani GP, Niyogi SK, Singh AK, Sen B, Yamasaki S, Taneja N, et al. Molecular characterization of multidrug-resistant Shigella species isolated from epidemic and endemic cases of shigellosis in India. J Med Microbiol. 2008;57:856–863. doi: 10.1099/jmm.0.2008/000521-0. [DOI] [PubMed] [Google Scholar]
- 25.Gendrel D, Cohen R. Diarrhées bactériennes et antibiotiques : les recommandations européennes. Arch Pediatr. 2008;15:S93–S96. doi: 10.1016/S0929-693X(08)74223-4. [DOI] [PubMed] [Google Scholar]
- 26.Heddini A, Cars O, Qiang S, Tomson G. Antibiotic resistance in China—a major future challenge. Lancet. 2009;373:30. doi: 10.1016/S0140-6736(08)61956-X. [DOI] [PubMed] [Google Scholar]
- 27.Talukder KA, Khajanchi BK, Islam MA, Islam Z, Dutta DK, Rahman M, et al. Fluoroquinolone resistance linked to both gyrA and parC mutations in the quinolone resistance–determining region of Shigella dysenteriae type 1. Curr Microbiol. 2006;52:108–111. doi: 10.1007/s00284-005-0140-9. [DOI] [PubMed] [Google Scholar]
- 28.Dutta S, Kawamura Y, Ezaki T, Nair GB, Iida K, Yoshida S. Alteration in the GyrA subunit of DNA gyrase and the ParC subunit of topoisomerase IV in quinolone-resistant Shigella dysenteriae serotype 1 clinical isolates from Kolkata, India. Antimicrob Agents Chemother. 2005;49:1660–1661. doi: 10.1128/AAC.49.4.1660-1661.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JY, Kim SH, Jeon SM, Park MS, Rhie HG, Lee BK. Resistance to fluoroquinolones by the combination of target site mutations and enhanced expression of genes for efflux pumps in Shigella flexneri and Shigella sonnei strains isolated in Korea. Clin Microbiol Infec. 2008;14:760–765. doi: 10.1111/j.1469-0691.2008.02033.x. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz J. Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J Antimicrob Chemoth. 2003;51:1109–1117. doi: 10.1093/jac/dkg222. [DOI] [PubMed] [Google Scholar]
- 31.Folster JP, Pecic G, Bowen A, Rickert R, Carattoli A, Whichard JM. Decreased susceptibility to ciprofloxacin amongShigella isolates in the United States, 2006 to 2009. Antimicrob Agents Chemoth. 2011;55:1758–1760. doi: 10.1128/AAC.01463-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pu XY, Pan JC, Zhang W, Zheng W, Wang HQ, Gu YM. Quinolone resistance-determining region mutations and the plasmid-mediated quinolone resistance gene qnrS played important roles in decreased susceptibility to fluoroquinolones among Shigella isolates in Southeast China between 1998 and 2013. Int J Antimicrob Agents. 2015;45:438–439. doi: 10.1016/j.ijantimicag.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Xiong W, Sun Y, Ding X, Wang M, Zeng Z. Selective pressure of antibiotics on ARGs and bacterial communities in manure-polluted freshwater-sediment microcosms. Front Microbiol. 2015;6:194. doi: 10.3389/fmicb.2015.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian L, Zhu X, Chen Z, Liu W, Li S, Yu W, et al. Characteristics of bacterial pathogens associated with acute diarrhea in children under 5 years of age: a hospital-based cross-sectional study. BMC Infect Dis. 2016;16:253. doi: 10.1186/s12879-016-1603-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu B, Qin T, Fan W, Bi R, Chen Y, Li Y, et al. Novel mutations in gyrA and parC among Shigella sonnei strains from Jiangsu Province of China, 2002-2011. Int J Infect Dis. 2017;59:44–49. doi: 10.1016/j.ijid.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 36.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Hye Park C, et al. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med. 2006;12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 37.Pu XY, Pan JC, Wang HQ, Zhang W, Huang ZC, Gu YM. Characterization of fluoroquinolone-resistant Shigella flexneri in Hangzhou area of China. J Antimicrob Chemoth. 2009;63:917–920. doi: 10.1093/jac/dkp087. [DOI] [PubMed] [Google Scholar]
- 38.Zhu Z, Cao M, Zhou X, Li B, Zhang J. Epidemic characterization and molecular genotyping of Shigella flexneri isolated from calves with diarrhea in Northwest China. Antimicrob Resist Infect Control. 2017;6:92. doi: 10.1186/s13756-017-0252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Z, Shi Y, Zhou X, Li B, Zhang J. Molecular characterization of fluoroquinolone and/or cephalosporin resistance in Shigella sonnei isolates from yaks. BMC Vet Res. 2018;14:177. doi: 10.1186/s12917-018-1500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taneja N, Kumar A, Appannanavar S, Verma G, Sharma M. Plasmid-mediated quinolone resistance in Shigella isolates over a decade in India. J Glob Antimicrob Res. 2014;2:59–60. doi: 10.1016/j.jgar.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Yamane K, Wachino JI, Suzuki S, Kimura K, Shibata N, Kato H, et al. New plasmid-mediated Fluoroquinolone efflux pump, QepA, found in an Escherichia coli clinical isolate. Antimicrob Agents Chemoth. 2007;51:3354–3360. doi: 10.1128/AAC.00339-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruiz J. Transferable Mechanisms of Quinolone Resistance from 1998 Onward. Clin Microbiol Rev. 2019;32(4):e00007-19. [DOI] [PMC free article] [PubMed]
- 43.Perichon B, Courvalin P, Galimand M. Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic Fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob Agents Chemoth. 2007;51:2464–2469. doi: 10.1128/AAC.00143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang H, Duan G, Zhu J, Zhang W, Xi Y, Fan Q. Prevalence and characterisation of plasmid-mediated quinolone resistance and mutations in the gyrase and topoisomerase IV genes among Shigella isolates from Henan, China, between 2001 and 2008. Int J Antimicrob Ag. 2013;42:173–177. doi: 10.1016/j.ijantimicag.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 45.Schaumburg F, Alabi AS, Kaba H, Lell B, Becker K, Grobusch MP, et al. Molecular characterization of Shigella spp. from patients in Gabon 2011-2013. T Roy Soc Trop Med H. 2015;109:275–279. doi: 10.1093/trstmh/tru175. [DOI] [PubMed] [Google Scholar]
- 46.Clinical Laboratory Standards Institute . Performance standards for antimicrobial disk susceptibility testing; Twenty-second informational supplement. m100 –s23. Wayne: Clinical and Laboratory Standards Institute; 2013. [Google Scholar]
- 47.Colobatiu L, Tabaran A, Flonta M, Oniga O, Mirel S, Mihaiu M. First description of plasmid-mediated quinolone resistance determinants and β-lactamase encoding genes in non-typhoidal Salmonella isolated from humans, one companion animal and food in Romania. Gut Pathog. 2015;7:16. doi: 10.1186/s13099-015-0063-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu LF, Li JB, Ye Y, Li X. Mutations in the GyrA subunit of DNA gyrase and the ParC subunit of topoisomerase IV in clinical strains of fluoroquinolone-resistant Shigella in Anhui, China. J Microbiol. 2007;45:168–170. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Strain information of S. dysenteriae isolates from diarrheal calves in this study.
Additional file 2: Table S2. Primers for the detection of virulence genes.
Additional file 3: Table S3. Primers for the detection of fluoroquinolone resistance–determining genes.
Data Availability Statement
The data supporting the findings of this study are contained within the manuscript.