

Abstract
Background
Familial progressive hyper- and hypopigmentation (FPHH, MIM 145250) is a rare hereditary skin disorder that is predominantly characterized by progressive, diffuse, partly blotchy hyperpigmented lesions intermingled with scattered hypopigmented spots, lentigines and sometimes Cafe-au-lait spots (CALs). Heterozygous mutations of the KIT ligand (KITLG, MIM 184745) gene are responsible for FPHH. To date, only eight KITLG mutations have been reported to be associated with FPHH, and no clear genotype–phenotype correlations have been established. This study aimed to identify the causative mutations in the KITLG gene in two Chinese FPHH patients.
Methods
Direct sequencing of the coding regions of KITLG was performed. Pathogenicity prediction was performed using bioinformatics tools, including SIFT, Polyphen2, and SWISS-MODEL, and the results were further evaluated according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines.
Results
The novel mutation c.104A > T (p.Asn35Ile) and the recurrent mutation c.101C > T (p.Thr34Ile) in KITLG were identified. As shown using SIFT and Polyphen-2 software, both mutations identified in this study were predicted to be detrimental variations. Three-dimensional protein structure modeling indicated that the mutant KITLG proteins might affect the affinity of KITLG for its receptor, c-KIT. According to the 2015 ACMG guidelines, the novel mutation c.104A > T was ‘likely pathogenic’.
Conclusions
To date, most of the identified KITLG mutations have been clustered within the conserved VTNNV motif (amino acids 33–37) in exon 2. The known mutations are only involved in 33 V, 34 T, 36 N, and 37 V but not 35 N. We have now identified a novel mutation in KITLG, c.104A > T, that was first reported in FPHH within the conserved 35 N motif. These results strengthen our understanding of FPHH and expand the mutational spectrum of the KITLG gene.
Keywords: Familial progressive hyper- and hypopigmentation, KITLG gene, Mutation, VTNNV motif
Background
Familial progressive hyper- and hypopigmentation (FPHH, MIM 145250) is a rare genetic skin pigmentation anomaly disorder characterized by progressive, diffuse, partly blotchy hyperpigmented lesions that are intermixed with multiple café-au-lait spots, hypopigmented maculae and lentigines that are located on the face, neck, trunk and limbs and frequently on the palms, soles and oral mucosa. Dispigmentation patterns can range from well-isolated café-au-lait/hypopigmented patches on a background of normal-appearing skin to a confetti-like or mottled appearance [1, 2].
The FPHH locus was mapped at chromosome 12q21.31-q23.1 by genome-wide linkage analysis in a six-generation Chinese family. Positional candidate gene screening revealed that a heterozygous transversion (c.107A > G; p.Asn36Ser) in exon 2 of the KIT ligand (KITLG, MIM 184745) gene is responsible for this disorder [3].
The KITLG gene, also known as stem cell factor (SCF) and mast cell growth factor, encodes the ligand for the KIT receptor tyrosine kinase. By KITLG binding, KIT dimerizes and initiates diverse cellular responses and plays a crucial role in the development and maintenance of the melanocyte lineage in adult skin. Injection of the soluble form of sKITLG resulted in hyperpigmentation of the grafted skin tissue, while injection of the KIT- or KITLG-blocking antibodies into the explanted human skin led to a loss of melanocytes [4]. Functional analysis of the soluble form of KITLG (sKITLG) showed that mutant sKITLG (Asn36Ser) increased the content of melanin by 109% compared with wild-type sKITLG in human melanoma cells, and a gain-of-function effect of this missense mutation was indicated to possibly trigger hyperpigmentation of skin in patients [3].
Several mutations (HGMD, http://www.hgmd.cf.ac.uk) in the KITLG gene have been documented in a few FPHH families [2, 5–7]. Most of the FPHH-causing mutations in KITLG are clustered within the conserved VTNNV motif (amino acids 33–37) in exon 2, and a mutated VTNNV domain may increase the affinity of KITLG to the c-Kit receptor, suggesting that the mutation causes a downstream gain-of-function effect. However, many FPHH families without KITLG mutations have been identified, indicating additional locus heterogeneity for this disorder [5, 8, 9].
Here, we reported two Chinese progressive hyper- and hypopigmentation cases, with one being familial and the other being sporadic. A novel mutation c.104A > T (p.Asn35Ile) and a recurrent mutation c.101C > T (p.Thr34Ile) in the KITLG gene were identified. Furthermore, we summarized the information on the mutations of the KITLG gene associated with the progressive hyper- and hypopigmentation previously reported.
Methods
Characteristics of the participants
Family 1 was a four-generation Chinese FPHH pedigree with seven affected individuals (three men and four women) (Fig. 1a; Additional file 1: Fig. S1). The pedigree presented an autosomal dominant inheritance pattern (Fig. 1a). The proband (III2) from family 1 was a 37-year-old woman. Generalized hyper- and hypopigmentation with irregular patches was found at birth, and the patches (0.2–0.8 cm) progressed successively over her face, neck, trunk and limbs with age. There were also a small number of larger pigmented lesions that were several centimeters in diameter on her trunk and limbs (Fig. 1b–e). All the affected individuals in this family had similar lesions, and none of them showed any other skin, nail, hair, teeth, mucosal or systemic diseases.
Fig. 1.
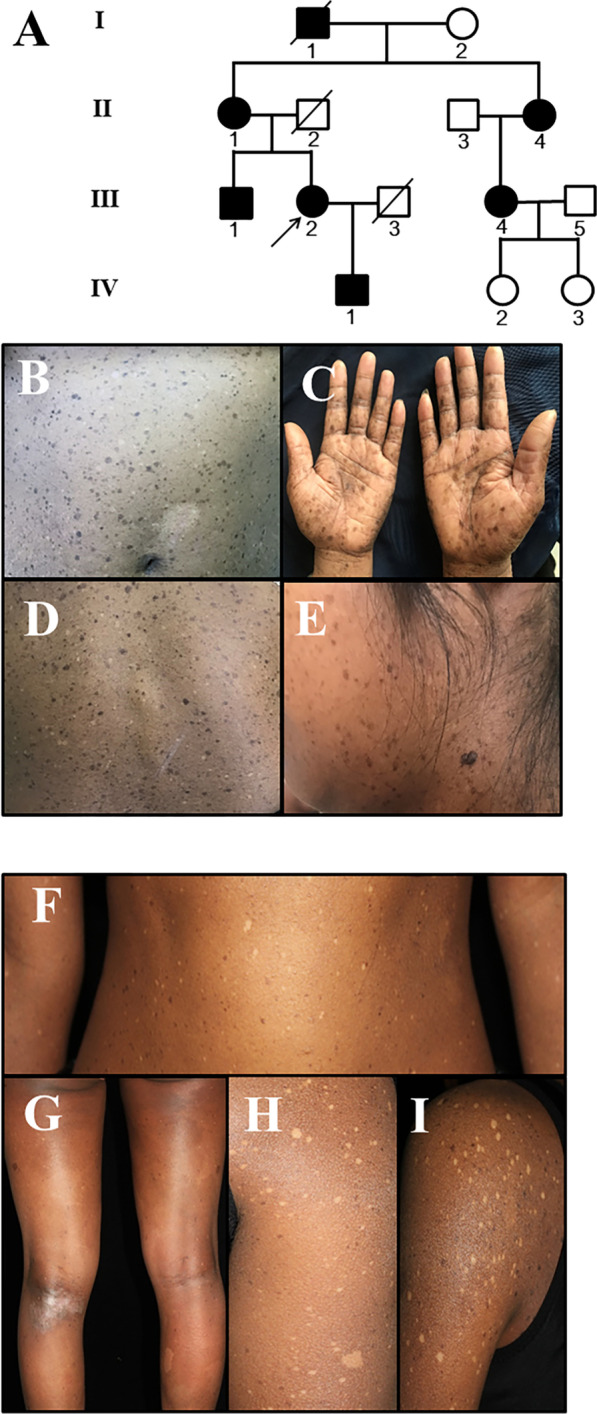
Pedigree and clinical features of the two cases with familial progressive hyper- and hypopigmentation. a Pedigree of family 1. b–e Generalized hyper- and hypopigmentation with irregular patches was found at birth, and the patches (0.2–0.8 cm) progressed successively over her trunk, limbs, face and neck with age. f–i With age, the lesions increased in both size and number and became more noticeable and appeared on the trunk and limbs
The sporadic case was a 26-year-old woman, and her parents were unaffected (Additional file 2: Fig. S2). The clinical appearances of this sporadic patient were mostly similar to those seen in the proband of family 1. However, diffuse hyperpigmentation was found on her entire body, and two vast café-au-lait (CAL)-like lesions presented on her legs at birth. One week after birth, it was shown that her diffuse hyperpigmented skin was intermixed with some small lentigines/CAL-like lesions, as well as hypopigmented macules and spots on her trunk. With increasing age, the lesions increased in both size and number and became more noticeable and appeared on her trunk and limbs (Fig. 1f–i). She was born to healthy, nonconsanguineous parents. Her nails, hair, teeth and mucosae were normal.
Mutation screening of the KITLG gene by direct sequencing
Genomic DNA was extracted from blood samples of family 1 and a sporadic case using the QIA-amp® DNA Blood Mini Kit (Qiagen, Shanghai, China). All exons and their flanking intronic sequences of the KITLG gene were amplified by polymerase chain reaction (PCR) as described previously [8]. Purified PCR products were sequenced directly using an ABI Prism® 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). DNA sequences were analyzed by comparison to the human KITLG reference sequence (NM_000899.5, https://www.ncbi.nlm.nih.gov/nuccore/NM_000899). The mutations were checked with HGMD, Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/) and the 1000 Genomes project (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). Furthermore, samples from 100 unrelated normal Chinese Han individuals were also sequenced to exclude polymorphic variants.
KITLG protein function prediction and molecular modeling
Online in silico programs were applied to predict the potential impact of an amino acid substitution on the structure and function of the KITLG protein with Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/), respectively. Furthermore, we performed analysis using an online server, SWISS-MODEL (http://swissmodel.expasy.org/), to construct the three-dimensional structure of KITLG. The pathogenicity prediction was evaluated following the American College of Medical Genetics and Genomics (ACMG) guidelines of 2015.
Results
Identification of KITLG gene mutations
All subsequently detected variants were then filtered on the basis of population. After filtering all variants, a novel heterozygous missense mutation c.104A > T (p.Asn35Ile) and a recurrent mutation c.101C > T (p.Thr34Ile) in the KITLG gene were revealed in family 1 (Fig. 2a) and in the sporadic case (Fig. 2b). These two mutations were not detected in the unaffected family members or 100 unrelated population-matched controls (Fig. 2c). The variation c.102 T > A (p.Thr34Thr) in family 1 is a synonymous mutation (Fig. 2a).
Fig. 2.
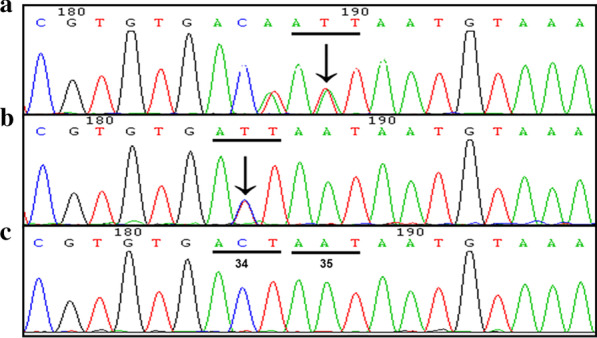
KITLG gene mutation analysis in two Chinese FPHH cases. a A novel mutation c.104A > T (p.Asn35Ile) was identified in family 1. The variation c.102 T > A (p.Thr34Thr) is a synonymous mutation. b A recurrent mutation c.101C > T (p.Thr34Ile) was identified in the sporadic case. c Sequence of the wild-type allele showing translation of the threonine residue at codon 34 (ACT) and asparagine at codon 35 (AAT)
According to the 2015 ACMG guidelines, the prevalence of the variant in affected individuals was significantly increased compared with the prevalence in controls, meeting the criterion of pathogenic strong 4 (PS4). These two mutations match the criterion of pathogenic moderate 1 (PM1) since all of them are in hot spot regions. A novel heterozygous missense mutation c.104A > T (p.Asn35Ile) was cosegregated in each affected family member, indicating that they matched the criterion of pathogenic supporting 2 (PP2).
Prediction of the potential impacts of the mutations
The mutations c.104A > T (p.Asn35Ile) and c.101C > T (p.Thr34Ile) were predicted to be ‘possible damaging’ and ‘deleterious’ with Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/), respectively. These predictions indicated that the two variants may have an effect on protein function, meeting the criterion of pathogenic supporting 3 (PP3) according to the ACMG guidelines of 2015.
The substitutions 34Thr and 35Asn were identified in KITLG (Fig. 3), in which polar, neutral, hydrophilic R-based amino acids were changed to Ile, a nonpolar, hydrophobic R-based amino acid, in family 1 and the sporadic case with SWISS-MODEL (http://swissmodel.expasy.org/ ).
Fig. 3.

Three-dimensional structure of KITLG. a–c Protein three-dimensional structure overall picture. d–f Partial map of protein three-dimensional structure. e Thr and Asn, as polar, neutral R-based amino acids, became Ile, nonpolar, hydrophobic R-based amino acids. When Thr changed to Ile at position 34, the side chain of Ile 34 changed. f When Asn changed to Ile at position 35, the side chain of Ile 35 changed
Finally, a novel heterozygous missense mutation c.104A > T (p.Asn35Ile) and a recurrent mutation c.101C > T (p.Thr34Ile) in the KITLG gene were revealed in family 1 (Fig. 2a) and in the sporadic case and were classified as ‘likely pathogenic’ in accordance with the 2015 ACMG guidelines. The details of each mutation are shown in Additional file 3: Table S1, and the genotype information is provided in Additional file 4: Table S2.
Discussion
FPHH is a rare autosomal dominant disorder with variable penetrance caused by mutations in the KITLG gene, which encodes the C-Kit ligand [5]. Because FPHH is very rare with reduced penetrance, no clear incidence rate of this disease has been documented. Patients with FPHH often do not have systemic symptoms. Growth retardation and intellectual disability in some affected members were reported by Westerhof et al. [10]. A 2-year-old Chinese female FPHH patient with frequent seizures, impaired temperature regulation and susceptibility to infection also had mild intellectual disability [11].
Nine publications in PubMed (https://www.ncbi.nlm.nih.gov/pubmed) were related to pathogenic mutations of the KITLG gene. Mutations in the KITLG gene are associated with autosomal dominant nonsyndromic deafness-69 (DFNA69, MIM 616697), Waardenburg syndrome-2 (WS2, MIM 193510), and FPHH. Seco Z et al. [12] reported that certain mutations of KITLG, including c.286_303delinsT (p.Ser96Ter), c.200_202del (p.His67_Cys68delinsArg), and c.310C > G (p.Leu104Val), cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2 (WS2). Ogawa Y et al. [13] reported a patient with WS2 who had the unusual complication of large, pigmented macules caused by a homozygous KITLG mutation (c.94G > A, p.Arg32Cys). It was speculated that the mechanism of the mutation underlying WS2 leading to membrane incorporation and reducing secretion of KITLG occurs via a gain-of-function or dominant-negative effect. A de novo mosaic KITLG variant (NM_000899.3:c.329A > G; p.Asp110Gly) was found in a 6-year-old boy who had congenital linear and mottled hyperpigmentation [14]. However, all the phenotypes presented in these three publications with KITLG are not defined as clearly associated with FPHH; therefore, we only summarize all the other KITLG mutations associated with FPHH here in this study.
Including the novel mutation (c.104A > T, p.Asn35Ile) we reported in this study, eight different missense mutations in the KITLG gene responsible for FPHH have been identified (Additional file 5: Table S3, Fig. 4). Seven out of eight mutations were clustered in a short amino acid sequence (VTNNV, amino acids 33–37) in exon 2 (Fig. 4), except for c.337G > A, which was in exon 4 of the KITLG gene. Most pathogenic mutations in FPHH occur within the VTNNV domain of the KITLG protein (amino acids 33–37) and lie within the third b-strand of the protein. Only the p.Val37 change represents the first amino acid of the second a-helix (amino acids 37–46). To date, the reported mutations are only involved in 33 V, 34 T, 36 N, and 37 V but not 35 N. We are the first to report the c.104A > T (p.Asn35Ile) mutation at 35 N (Fig. 4) with FPHH patients.
Fig. 4.

Summary of KITLG gene mutations associated with FPHH. All the mutations associated with FPHH are located within the VTNNV region in exon 2, except c.337G > A in exon 4 of the KITLG gene. The mutations reported previously are in BLUE, while the novel mutation (c.104A > T) at 35 N reported in this study is in RED
Except for diffuse hyper- and hypopigmentation, vast CAL-like lesions have been detected as the most common skin problems present in FPHH patients. Vitiligo was found in one family. Sparse lateral eyebrows and malignancy (pharyngeal cancer, papillary thyroid cancer and melanoma) were found in two families. Short sutures were found in only one family, and mental retardation was not present in these FPHH patients (Additional file 5: Table S3).
KITLG, as KIT LIGAND, is produced locally in human skin by epidermal keratinocytes and endothelial cells, where it induces the migration, development and survival of melanocytes. The signaling of KITLG and its receptor KIT plays an important role in melanocyte proliferation and pigment production [15, 16]. The role of the KIT/KITLG system in melanogenesis has been experimentally confirmed using animal models [3, 17]. After KITLG binds the c-KIT receptor, dimerization is triggered. It initiates signal transduction via the RAS/MAPK pathway to upregulate melanoblast proliferation [16, 18]. The KITLG/C-KIT/RAS/MAPK signaling pathways play important roles in the regulation of hematopoiesis, stem cell survival, gametogenesis, mast cell development, migration and function, and skin color [19, 20]. Mutant alleles of the KITLG gene are lethal in homozygous mice and produce a variable level of coat-color dilution in heterozygous mice [20]. It has been reported in human studies that variations in the KITLG gene are also associated with skin, hair, and eye pigmentation (MIM 611664), autosomal dominant nonsyndromic deafness-69, WS2 and FPHH.
Here, we reported a novel c.104A > T (p.Asn35Ile) mutation of KITLG in a Chinese FPHH family. According to the 2015 ACMG guidelines, the mutation was initially identified as a ‘likely pathogenic’ mutation [21]. To the best of our knowledge, only eight different missense KITLG mutations have been reported to cause FPHH (Additional file 5: Table S3). Notably, seven known mutations were clustered in a highly conserved short amino acid sequence VTNNV (amino acids 33–37) (Fig. 4). The VTNNV domain of the KITLG protein (amino acids 33–37) lies within the third b-strand of the protein and is responsible for its binding functions. Both mutations c.104A > T (p.Asn35Ile) and c.101C > T (p.Thr34Ile) found in this study were located in the VTNNV domain and were predicted to be detrimental variations by the SIFT and Polyphen-2 tools. Using the Swiss-Model servers [22, 23], three-dimensional structures of mutant KITLG proteins were found to change compared with the wild type (Fig. 3). Both 35Asn and 34Thr are polar, hydrophilic amino acids, and the mutant became nonpolar, hydrophobic isoleucine; therefore, it might change the features of the protein and affect the ligand affinity to its receptor c-Kit, thus affecting the migration of melanoblasts, melanosome transfer and melanin synthesis, conferring a phenotype with hyper- and hypopigmentation. The precise mechanisms need to be elucidated by further experiments. Our findings revealed a novel KITLG mutation associated with FPHH and reinforced the evidence that VTNNV was a hot spot for mutation. However, definitive functional analyses of this mutation are needed to determine the structure–function relationship in patients with FPHH.
Conclusions
In summary, a novel mutation c.104A > T (p.Asn35Ile) in the KITLG gene was reported in a Chinese FPHH patient. The correlations between genotypes and phenotypes of FPHH were summarized. These results strengthen our understanding of FPHH and expand the mutational spectrum of the KITLG gene.
Supplementary Information
Additional file 1. Fig. S1: Sequencing results of seven individuals from family 1. A: II1, B: II4, C: III1, D: III4, E: III5, F: IV1, G: IV3
Additional file 2. Fig. S2: Sequencing results of the proband’s parents from the sporadic case. A: Proband’s father, B: Proband’s mother.
Additional file 3. Table S1: Classification of KITLG mutations in this study according to the 2015 ACMG guidelines.
Additional file 4. Table S2: Family 1 (A) and the sporadic case (B) genotype information details.
Additional file 5. Table S3: The reported cases and KITLG mutations of familial progressive hyper- and hypopigmentation to date.
Acknowledgements
We would like to thank all the individuals and their families who participated in this project.
Abbreviations
- FPHH
Familial progressive hyper- and hypopigmentation
- CALS
Café-au-lait spots
- sKITLG
The soluble form of KITLG
- MIM
Mendelian Inheritance in Man
Authors’ contributions
NHZ, SMZ, XLL, ZLL, ZLY, and ML collected clinical information and performed genetic analysis. JBW, JLL, WSL and MS performed molecular genetics experiments and analyzed the data. JBW and WSL wrote the manuscript. MS and ML designed the study and edited the final manuscript. All authors revised the paper critically for intellectual content and approved the final version. All authors agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigated and properly resolved. All authors read and approved the final manuscript.
Funding
This work was supported by Henan medical science and technology project jointly built by the ministry and the province (SB201904011) and the National Natural Science Foundation of China (31401071).
Availability of data and materials
The datasets generated during the current study are available in the Mendeley repository (10.17632/yftvffrkyt.1).
Ethics approval and consent to participate
This study was performed in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for medical research involving human subjects and was approved by the ethics committee of Henan Provincial People’s Hospital. Written informed consent was obtained from all individuals who participated in this study and from parents on behalf of minors.
Consent for publication
Written informed consent for publication of identifying images or other personal or clinical details was obtained from all of the participants or the parents of any participant under the age of 18.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jianbo Wang and Weisheng Li: Co-first authors
Contributor Information
Miao Sun, Email: miaosunsuda@163.com.
Min Li, Email: lmpfdoctor@163.com.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-020-00851-5.
References
- 1.Chernosky ME, Anderson DE, Chang JP, Shaw MW, Romsdahl MM. Familial progressive hyperpigmentation. Arch Dermatol. 1971;103:581–591. doi: 10.1001/archderm.1971.04000180007002. [DOI] [PubMed] [Google Scholar]
- 2.Gulseren D, Guleray N, Akgun-Dogan O, Simsek-Kiper PO, Utine EG, Alikasifoglu M, et al. Cafe noir spots: a feature of familial progressive hyper- and hypopigmentation. J Eur Acad Dermatol Venereol. 2020;34:e76–e77. doi: 10.1111/jdv.15968. [DOI] [PubMed] [Google Scholar]
- 3.Wang ZQ, Si L, Tang Q, Lin D, Fu ZJ, Zhang J, et al. Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. Am J Hum Genet. 2009;84:672–677. doi: 10.1016/j.ajhg.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grichnik JM, Burch JA, Burchette J, Shea CR. The SCF/KIT pathway plays a critical role in the control of normal human melanocyte homeostasis. J Invest Dermatol. 1998;111:233–238. doi: 10.1046/j.1523-1747.1998.00272.x. [DOI] [PubMed] [Google Scholar]
- 5.Amyere M, Vogt T, Hoo J, Brandrup F, Bygum A, Boon L, et al. KITLG mutations cause familial progressive hyper- and hypopigmentation. J Invest Dermatol. 2011;131:1234–1239. doi: 10.1038/jid.2011.29. [DOI] [PubMed] [Google Scholar]
- 6.Cuell A, Bansal N, Cole T, Kaur MR, Lee J, Loffeld A, et al. Familial progressive hyper- and hypopigmentation and malignancy in two families with new mutations in KITLG. Clin Exp Dermatol. 2015;40:860–864. doi: 10.1111/ced.12702. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Cheng R, Liang J, Ni C, Li M, Yao Z. Report of a child with sporadic familial progressive hyper- and hypopigmentation caused by a novel KITLG mutation. Br J Dermatol. 2016;175:1369–1371. doi: 10.1111/bjd.14694. [DOI] [PubMed] [Google Scholar]
- 8.Fang XK, He YX, Li YJ, Chen LR, Wang HP, Sun Q. Familial progressive hyper- and hypopigmentation: a report on a Chinese family and evidence for genetic heterogeneity. An Bras Dermatol. 2017;92:329–333. doi: 10.1590/abd1806-4841.20175567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng L, Zheng XD, Liu LH, Fu LY, Zuo XB, Chen G, et al. Familial progressive hyperpigmentation and hypopigmentation without KITLG mutation. Clin Exp Dermatol. 2016;41:927–929. doi: 10.1111/ced.12923. [DOI] [PubMed] [Google Scholar]
- 10.Vélez A, Salido R, Amorrich-Campos V, Garnacho-Saucedo G, Alvarez-López MA, Galán M, et al. Hereditary congenital hypopigmented and hyperpigmented macules (Westerhof syndrome) in two siblings. Br J Dermatol. 2009;161:1399–1400. doi: 10.1111/j.1365-2133.2009.09428.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhang RZ, Zhu WY. Familial progressive hypo- and hyperpigmentation: a variant case. Indian J Dermatol Venereol Leprol. 2012;78:350–353. doi: 10.4103/0378-6323.95453. [DOI] [PubMed] [Google Scholar]
- 12.Zazo Seco C, Serrao de Castro L, van Nierop JW, Morin M, Jhangiani S, Verver EJ, et al. Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and waardenburg syndrome type 2. Am J Hum Genet. 2015;97:647–60. [DOI] [PMC free article] [PubMed]
- 13.Ogawa Y, Kono M, Akiyama M. Pigmented macules in Waardenburg syndrome type 2 due to KITLG mutation. Pigment Cell Melanoma Res. 2017;30:501–504. doi: 10.1111/pcmr.12597. [DOI] [PubMed] [Google Scholar]
- 14.Sorlin A, Maruani A, Aubriot-Lorton MH, Kuentz P, Duffourd Y, Teysseire S, et al. Mosaicism for a KITLG mutation in linear and whorled nevoid hypermelanosis. J Invest Dermatol. 2017;137:1575–1578. doi: 10.1016/j.jid.2017.01.035. [DOI] [PubMed] [Google Scholar]
- 15.Kato M, Yagami A, Tsukamoto T, Shinkai Y, Kato T, Kurahashi H. Novel mutation in the KITLG gene in familial progressive hyperpigmentation with or without hypopigmentation. J Dermatol. 2020;47:669–672. doi: 10.1111/1346-8138.15313. [DOI] [PubMed] [Google Scholar]
- 16.Steel KP, Davidson DR, Jackson IJ. Trp-2/Dt, a new early melanoblast marker, shows that steel growth-factor (C-Kit Ligand) is a survival factor. Development. 1992;115:1111–1119. doi: 10.1242/dev.115.4.1111. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida H, Kunisada T, Grimm T, Nishimura EK, Nishioka E, Nishikawa SI. Review: melanocyte migration and survival controlled by SCF/c-kit expression. J Investig Dermatol Symp Proc. 2001;6:1–5. doi: 10.1046/j.0022-202x.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- 18.Spritz RA. Molecular basis of human piebaldism. J Invest Dermatol. 1994;103:137S–S140. doi: 10.1038/jid.1994.25. [DOI] [PubMed] [Google Scholar]
- 19.Mahakali Zama A, Hudson FP, 3rd, Bedell MA. Analysis of hypomorphic KitlSl mutants suggests different requirements for KITL in proliferation and migration of mouse primordial germ cells. Biol Reprod. 2005;73:639–647. doi: 10.1095/biolreprod.105.042846. [DOI] [PubMed] [Google Scholar]
- 20.Rajaraman S, Davis W, Mahakali-Zama A, Evans H, Russell L, Bedell M. An allelic series of mutations in the Kit ligand gene of mice. I. Identification of point mutations in seven ethylnitrosourea-induced kitlsteel alleles. Genetics. 2002;162:331–40. doi: 10.1093/genetics/162.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL repository and associated resources. Nucl Acids Res. 2009;37(Database issue):D387–92. [DOI] [PMC free article] [PubMed]
- 23.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucl Acids Res. 2014;42(W1):W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Fig. S1: Sequencing results of seven individuals from family 1. A: II1, B: II4, C: III1, D: III4, E: III5, F: IV1, G: IV3
Additional file 2. Fig. S2: Sequencing results of the proband’s parents from the sporadic case. A: Proband’s father, B: Proband’s mother.
Additional file 3. Table S1: Classification of KITLG mutations in this study according to the 2015 ACMG guidelines.
Additional file 4. Table S2: Family 1 (A) and the sporadic case (B) genotype information details.
Additional file 5. Table S3: The reported cases and KITLG mutations of familial progressive hyper- and hypopigmentation to date.
Data Availability Statement
The datasets generated during the current study are available in the Mendeley repository (10.17632/yftvffrkyt.1).