

Abstract
Background
Trimethylamine-N-oxide (TMAO) is an independent risk factor for atherosclerosis. Consumption of hawthorn fruit is believed to be cardio-protective, yet whether it is able to suppress the TMAO-induced atherosclerosis remains unexplored. The present study was to investigate the effects of hawthorn fruit extract (HFE) on TMAO-exacerbated atherogenesis.
Methods
Five groups of male Apolipoprotein E knock-out (ApoE−/−) mice were fed a low-fat diet (LFD), a Western high-fat diet (WD), or one of the three WDs containing 0.2% TMAO (WD + TMAO), 0.2% TMAO plus 1% HFE (WD + TMAO + L-HFE), or 0.2% TMAO plus 2% HFE (WD + TMAO + H-HFE), respectively. After 12-weeks of intervention, plasma levels of TMAO, lipid profile, inflammatory biomarkers, and antioxidant enzyme activities were measured. Atherosclerotic lesions in the thoracic aorta and aortic sinus were evaluated. The sterols and fatty acids in the liver and feces were extracted and measured. Hepatic expressions of inflammatory biomarkers and antioxidant enzymes were analyzed.
Results
Dietary TMAO accelerated atherogenesis, exacerbated inflammation, and reduced antioxidant capacities in the plasma and the liver. TMAO promoted hepatic cholesterol accumulation by inhibiting fecal excretion of acidic sterols. HFE could dose-dependently reduce the TMAO-aggravated atherosclerosis and inflammation. HFE was also able to reverse the TMAO-induced reduction in antioxidant capacity by up-regulating the expression of antioxidant enzymes including superoxide dismutase 1 (SOD1), SOD2, glutathione peroxidase 3 (GSH-Px3), and catalase (CAT) in the liver. Moreover, the hepatic cholesterol content was lowered by HFE via enhanced fecal excretion of neutral and acidic sterols.
Conclusions
The present results indicated that HFE was able to reduce the TMAO-exacerbated atherogenesis by attenuating inflammation and improving antioxidant capacity at least in mice.
Graphic abstract

Keywords: Trimethylamine-N-oxide, Atherosclerosis, Hawthorn fruit extract, Inflammation, Antioxidant, Cholesterol
Introduction
Atherosclerosis is a chronic vascular disease and has become a major underlying cause of cardiovascular deaths worldwide due to its clinical manifestations including myocardial infarction, stroke, and peripheral vascular diseases. It has been well recognized that hypercholesterolemia, vascular inflammation, and oxidative stress are risk factors to initiate and accelerate atherogenesis [1–3]. In the recent decade, high plasma trimethylamine-N-oxide (TMAO) has been identified as an independent risk factor for cardiovascular diseases (CVDs) by inducing atherosclerosis [4–6]. In humans, most blood TMAO comes from dietary trimethylamine (TMA)-containing nutrients, for example, phosphatidylcholine, choline, and L-carnitine [4]. After ingestion, these dietary precursors liberate TMA molecule under the actions of bacterial enzymes in the intestine. TMA is then absorbed and oxidized to produce TMAO in the liver by flavin monooxygenases [7].
TMAO promotes atherogenesis mainly by inducing vascular inflammation. It has been reported that TMAO triggers inflammatory gene expression in the aorta of LDLR−/− mice by activating multiple inflammatory signaling pathways including mitogen-activated protein kinase (MAPK), extracellular signal-kinase (ERK), and nuclear factor κB (NF-κB) cascades [8]. The TMAO-triggered formation of NLRP3 inflammasome and subsequent production of interleukin (IL)-1β have been observed in both cultured vascular endothelial cells and mouse aorta [9, 10]. Moreover, TMAO has been shown to promote the formation of foam cells partially mediated by CD36/MAPK/JNK pathway [11]. Disturbance in redox balance by TMAO may also account for its atherogenic activity. It has been demonstrated that TMAO can inhibit the activity of antioxidant enzymes including superoxide dismutase (SOD) and catalase (CAT) in the aorta, leading to a weakened vascular antioxidant defense [10, 12, 13]. Meanwhile, TMAO exacerbates vascular oxidative stress by facilitating the generation of reactive oxygen species (ROS) [10, 12]. Apart from its detrimental effects on the arteries, TMAO has also been shown to disturb cholesterol metabolism and induce liver injury in mice [6, 14–18]. In addition, it has been reported that dietary TMAO aggravates glucose intolerance, obstructs hepatic insulin signaling pathway [19, 20], and cause renal fibrosis and dysfunction in murine models [21].
Hawthorn fruit is the bright red berry of Crataegus species. It has been popularly consumed as a fruit and utilized as an herbal medicine for some gastrointestinal disorders in China. Consumption of hawthorn fruit is believed to prevent the development of atherosclerosis with phenolic compounds such as (–)-epicatechin, hyperoside, and isoquercitrin being regarded as the active ingredients [22]. It has been reported that hawthorn ethanolic extract is able to improve vascular function by stimulating endothelium-dependent relaxation in rat isolated mesenteric arteries in vitro [23]. In in vivo studies, administration of hawthorn fruit or its extract can improve hyperlipidemia [24–28], increase the activity of endogenous antioxidant enzymes [29, 30], and reduce inflammation [31]. However, whether hawthorn fruit is able to suppress the TMAO-induced atherosclerosis remains unexplored. Herein, the present study was conducted to investigate if hawthorn fruit extract (HFE) could reduce TMAO-exacerbated atherogenesis in Apolipoprotein E knock-out (ApoE−/−) mice fed a Western high-fat diet.
Methods
Preparation of HFE
Dried seedless hawthorn fruit were purchased from HK JEBM Ltd. (Hong Kong, China). HFE was prepared according to the method described by Zhang et al. with minor modifications [25]. Briefly, hawthorn fruit was firstly ground into powder. 300 g of hawthorn fruit powder was extracted with one liter of 80% ethanol at room temperature for 24 h for three times. The liquid phase was filtered and pooled, followed by the removal of ethanol in a vacuum rotary evaporator. The aqueous phase was collected and then freeze-dried. The resultant HFE was stored at − 20 °C.
High performance liquid chromatography (HPLC) analysis of HFE
HPLC analysis was conducted to quantify the phenolic compounds present in HFE based on the method of Liu et al. with minor modifications [32]. Briefly, HFE dissolved in ethanol was filtered, and injected onto a VisionHT C18-HL column (250 × 4.6 mm, 5 μm; Grace, Columbia, MD, USA). The mobile phase consisted of water with 0.5% formic acid (solvent A), and acetonitrile/methanol = 80:20 (v/v) (solvent B), at a flow rate of 1 mL/min. Peaks were monitored at 280, 320, and 360 nm with a UV–visible detector. Phenolic compounds were identified by comparing the retention time with those of authentic standards and quantified according to the calibration curves. The HPLC chromatograms of HFE were shown in Fig. 1. HPLC results showed that 1 g of HFE contained 15.69 mg of total phenolic antioxidant compounds, including 5.93 mg of procyanidin B2, 5.36 mg of epicatechin, 2.45 mg of procyanidin C1, 1.31 mg of chlorogenic acid, 0.36 mg of hyperoside, and 0.28 mg of isoquercitrin.
Fig. 1.
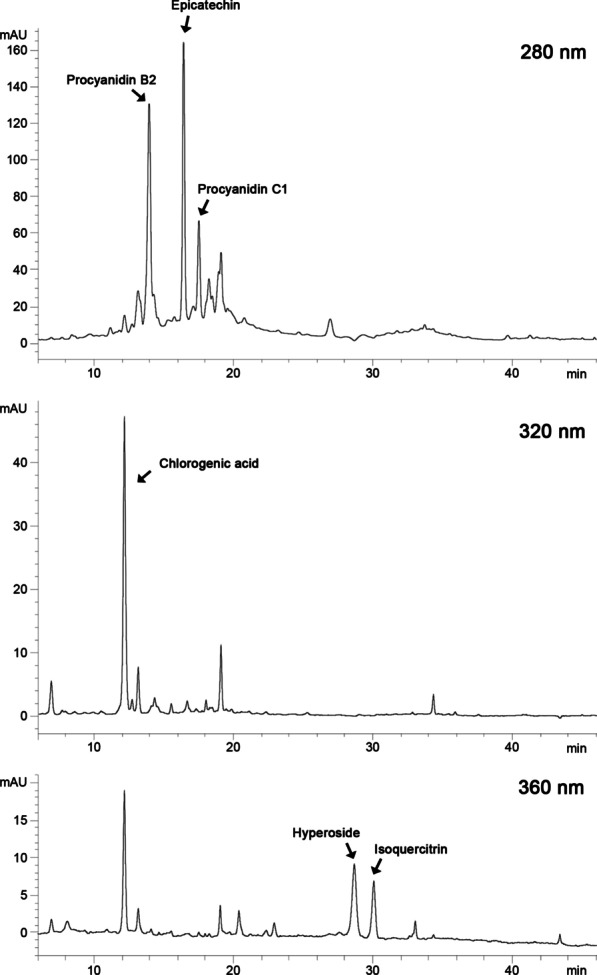
HPLC chromatograms of hawthorn fruit extract at 280, 320, and 360 nm
Preparation of diets
Five diets were formulated. Briefly, a low-fat diet (LFD) was prepared by mixing all the ingredients listed in Table 1. A Western diet (WD) with 40% energy from fat and 0.15% (w/w) cholesterol was similarly prepared. A WD plus 0.2% TMAO (WD + TMAO) was made by dissolving equimolar TMAO dihydrate (J&K Scientific Ltd., Beijing, China) in distilled water, which was then mixed with the other ingredients. Two experimental diets were prepared by adding 0.2% TMAO plus 1% HFE into WD as a low-dose HFE diet (WD + TMAO + L-HFE), and adding 0.2% TMAO plus 2% HFE into WD as a high-dose HFE diet (WD + TMAO + H-HFE). The diets were then cut into pieces of around 10–15 g cubes and stored at -20 °C until use.
Table 1.
Composition of five dietsa
| Ingredient (g) | LFD | WD | WD + TMAO | WD + TMAO + L-HFE | WD + TMAO + H-HFE |
|---|---|---|---|---|---|
| Corn starch | 600.2 | 414.6 | 414.6 | 414.6 | 414.6 |
| Casein | 140.0 | 167.1 | 167.1 | 167.1 | 167.1 |
| Sucrose | 100.0 | 69.1 | 69.1 | 69.1 | 69.1 |
| Powdered cellulose | 50.0 | 59.7 | 59.7 | 59.7 | 59.7 |
| Lard | 40.0 | 204.3 | 204.3 | 204.3 | 204.3 |
| AIN 76 mineral mix | 35.0 | 41.8 | 41.8 | 41.8 | 41.8 |
| Gelatin | 20.0 | 23.9 | 23.9 | 23.9 | 23.9 |
| AIN 93 vitamin mix | 10.0 | 11.9 | 11.9 | 11.9 | 11.9 |
| Choline bitartrate | 2.5 | 3.0 | 3.0 | 3.0 | 3.0 |
| DL-Methionine | 2.3 | 2.7 | 2.7 | 2.7 | 2.7 |
| Cholesterol | – | 1.5 | 1.5 | 1.5 | 1.5 |
| TMAOb | – | – | 2.0 | 2.0 | 2.0 |
| Hawthorn fruit extract | – | – | – | 10.0 | 20.0 |
| Total | 1000 | 1000 | 1002 | 1012 | 1022 |
| kcal/g | 3.9 | 4.6 | 4.6 | 4.6 | 4.6 |
| % kcal | |||||
| Carbohydrate | 73.8 | 43.1 | 43.1 | 43.1 | 43.1 |
| Protein | 16.9 | 16.9 | 16.9 | 16.9 | 16.9 |
| Fat | 9.4 | 40.0 | 40.0 | 40.0 | 40.0 |
aThe five diets include a low-fat diet (LFD), a Western diet (WD) containing 0.15% cholesterol and 40% energy from fat, a WD plus 0.2% TMAO (WD + TMAO), a WD with 0.2% TMAO and 1% hawthorn fruit extract (WD + TMAO + L-HFE), and a WD with 0.2% TMAO and 2% hawthorn fruit extract (WD + TMAO + H-HFE)
bTMAO dihydrate equivalent to 0.2% free TMAO was dissolved in distilled water and then mixed with the other ingredients
Mice
ApoE−/− mice are one of the most commonly used animal models for atherosclerosis studies because they can spontaneously develop hypercholesterolemia and produce atherosclerotic plaque, particularly when they were fed a typical Western high-fat diet [33, 34]. Most importantly, all the phases of atherosclerotic lesions developed in the arterial tree of ApoE−/− mice were very similar to humans [33]. In the present study, forty male ApoE−/− mice (8-week-old) were obtained from the Laboratory Animal Service Center, The Chinese University of Hong Kong. After one-week acclimation, mice were randomly divided into five groups (n = 8 each) and fed one of the five diets for 12 weeks. Food intake were recorded daily, and body weight were recorded weekly. The fecal samples were collected weekly. At the end of week 12, all the mice were sacrificed under isoflurane anesthesia after overnight fasting. Blood and organs were collected and stored at -80 °C until analyses. All the experimental protocols were approved by the Animal Experimental Ethical Committee, The Chinese University of Hong Kong (Ref No. 17/065/MIS).
Measurement of plasma TMAO
Plasma TMAO were determined as previously described [35]. In brief, 20 μL plasma was mixed with 80 uL methanol for protein precipitation. After centrifugation, the supernatant was collected and subjected to liquid chromatography with tandem mass spectrometry. TMAO was screened in multiple reaction monitoring mode based on the precursor-product ion transitions m/z 76 → 58. Different concentrations of TMAO standard were used to prepare the calibration curve.
Measurement of plasma lipids and inflammatory biomarkers
Total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and triacylglycerol (TG) in the plasma were measured by corresponding commercial enzymatic kits (Stanbio Laboratories, Boerne, TX, USA). Plasma non-HDL-C was determined by deducting HDL-C from TC. Plasma levels of tumor necrosis factor α (TNF-α), IL-1β, IL-6, monocyte chemoattractant protein 1 (MCP-1), and IL-10 were determined by ELISA kits according to the manufacturer’s instructions (CUSABIO Ltd., Wuhan, China).
Analyses of total antioxidant capacity (T-AOC), antioxidant enzyme activities, and malondialdehyde (MDA) in plasma
Plasma levels of T-AOC, SOD activity, GSH-Px activity, and MDA were determined using the corresponding commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China). Plasma T-AOC was determined by reducing Fe3+-TPTZ to Fe2+-TPTZ under acidic condition. The stable blue color of Fe2+-TPTZ was measured at 593 nm. T-AOC was expressed as mM, which was defined as 1 mM of T-AOC had equal antioxidant ability of 1 mM FeSO4 standard. The activities of antioxidant enzymes were expressed as a unit per milliliter of plasma (U/mL). The plasma MDA level was expressed as nmol MDA per milliliter of plasma (nmol/mL).
Evaluation of atherosclerotic lesions
Oil red O staining was used to quantify the atherosclerotic lesions in the thoracic aorta. In brief, the thoracic aorta was carefully cleaned and cut open vertically under microscope, followed by staining in 0.5% oil red O and scanning with a table scanner (Epson 1220 perfection, Epson Co., Tokyo, Japan). The plaque area in the thoracic aorta was quantified by Image J (National Institutes of Health, USA).
For evaluation of atherosclerotic lesions in the aortic sinus, the upper part of heart was fixed, dehydrated, and embedded in paraffin. Serial sections at 5 μm was obtained at aortic sinus using a microtome. The sections were then subjected to routine H&E staining to quantify the area of plaque and necrotic core, and to Masson’s Trichome staining to determine the collagen content in the plaque. Digital micrographs were taken by a Carl Zeiss PALM Inverted microscope (Carl Zeiss, Jena, Germany). Image J was used to quantify the areas of plaque, necrotic core, and collagen.
Analysis of hepatic cholesterol
Liver cholesterol content was determined using a gas chromatography (GC) method as previously described [36]. Basically, 5α-cholestane was added into liver as an internal standard, followed by extraction with chloroform/methanol (2:1, v/v). After saponification in 1 mol/L NaOH in 90% ethanol, cholesterol was extracted with cyclohexane, converted to its trimethylsilyl (TMS) derivative, and injected onto a SAC-5 capillary column (30 m × 0.25 mm, i.d.; Supelco, Inc., Bellefonte, PA, USA) in a Shimadzu GC-14B equipped with a flame ionization detector for GC analysis.
Analysis of fecal neutral and acidic sterols
Fecal contents of neutral and acidic sterols were determined using a GC method [35]. In brief, the whole week feces were lyophilized and ground into powder. 5α-Cholestane and hyodeoxycholic acid were added as internal standards for neutral and acidic sterols, respectively. Weighted fecal powder were saponified. The unsaponified fraction was extracted with cyclohexane for neutral sterol analysis, while the saponified aqueous phase was collected for acidic sterol extraction. All the sterols were converted to their corresponding TMS derivatives for GC analysis.
Measurement of hepatic and fecal lipid contents
Hepatic and fecal lipid contents were quantified by a GC method as previously described [37]. In brief, 100 mg of liver or 300 mg of fecal powder with 0.6 mg of heptadecanoic acid as an internal standard was used for analysis. Total lipid was extracted with chloroform/methanol (2:1, v/v). Fatty acids (FAs) were released and converted to their corresponding fatty acid methyl esters (FAMEs) with 14% boron trifluoride in methanol. The resultant FAMEs were injected onto a HP Innowax capillary column (30 m × 0.32 mm, 0.50 µm film thickness; Agilent Technologies, Santa Clara, CA, USA) for GC analysis.
Quantitative real-time PCR
Quantitative real-time PCR analyses were conducted as previously described [35]. Basically, RNA was extracted from liver tissue with Trizol (Invitrogen, Carlsbad, CA, USA) and converted to its complementary DNA. Real-time PCR was then performed in a StepOnePlus Real-Time System (Thermo Fisher Scientific, Waltham, MA, USA) using primers shown in Table 2 and SYBR green as a fluorophore. The abundance of mRNA was normalized with that of β-actin.
Table 2.
Quantitative real-time PCR primer sequences
| Gene | Forward primer, 5′ to 3′ | Reverse primer, 5′ to 3′ |
|---|---|---|
| SOD1 | AACCAGTTGTGTTGTCAGGAC | CCACCATGTTTCTTAGAGTGAGG |
| SOD2 | CAGACCTGCCTTACGACTATGG | CTCGGTGGCGTTGAGATTGTT |
| GSH-Px3 | CCTTTTAAGCAGTATGCAGGCA | CAAGCCAAATGGCCCAAGTT |
| CAT | TGGCACACTTTGACAGAGAGC | CCTTTGCCTTGGAGTATCTGG |
| MCP-1 | CCACAACCACCTCAAGCACT | TAAGGCATCACAGTCCGAGTC |
| IL-6 | GAGTGGCTAAGGACCAAGACC | AACGCACTAGGTTTGCCGA |
| IL-10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| IL-1β | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| TNF-α | CCTGTAGCCCACGTCGTAG | GGGAGTAGACAAGGTACAACCC |
| β-actin | TGTCCACCTTCCAGCAGATGT | AGCTCAGTAACAGTCCGCCTAGA |
Determination of hepatic levels of inflammatory cytokines, T-AOC, antioxidant enzyme activities, and MDA
1.8 mL of phosphate-buffered saline was added into 200 mg of liver tissue, followed by homogenization. After centrifuge, the supernatant was collected and subjected to protein concentration measurement with a Bicinchoninic Acid Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Hepatic levels of inflammatory cytokines, T-AOC, activities of antioxidant enzyme, and MDA were measured based on the methods described in 2.6 and 2.7.
Statistical analysis
All the data were expressed as mean ± SD. One-way ANOVA with post hoc LSD was performed in SPSS software (version 21.0, Chicago, IL, USA) to determine any differences among five groups. Ordinary linear regression was carried out across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups to examine the dose-dependent effect of HFE. Statistical difference was set at p < 0.05.
Results
Food intake, body weight, and organ weight
After 12-weeks of intervention, results showed that WD feeding raised body weight of mice compared with LFD feeding (Table 3). Addition of TMAO into WD did not affect the body weight. Mice in WD + TMAO + H-HFE group had slightly higher body weight gain compared with those in WD + TMAO group. No difference in daily energy intake or organ weight was found among the five groups of mice (Table 3).
Table 3.
Food intake, body weight, and organ weight in ApoE−/− mice
| LFD | WD | WD + TMAO | WD + TMAO + L-HFE | WD + TMAO + H-HFE | p value | |
|---|---|---|---|---|---|---|
| Food intake | ||||||
| Daily food intake (g/d) | 3.96 ± 0.23a | 3.32 ± 0.10b | 3.16 ± 0.10b | 3.20 ± 0.15b | 3.29 ± 0.29b | 0.002 |
| Daily energy intake (kcal/d) | 15.25 ± 0.89 | 15.28 ± 0.46 | 14.51 ± 0.47 | 14.54 ± 0.67 | 14.81 ± 1.31 | 0.662 |
| Body weight (g) | ||||||
| Initial | 24.8 ± 1.4 | 24.8 ± 1.1 | 25.0 ± 1.0 | 25.2 ± 0.8 | 24.8 ± 0.8 | 0.950 |
| Final | 28.1 ± 1.4c | 30.0 ± 1.1ab | 29.1 ± 1.2bc | 30.2 ± 1.4ab | 30.7 ± 1.6a | 0.006 |
| Organ weight (% body weight) | ||||||
| Liver | 4.00 ± 0.22 | 3.84 ± 0.06 | 3.86 ± 0.14 | 3.89 ± 0.08 | 3.85 ± 0.11 | 0.133 |
| Testis | 0.61 ± 0.10 | 0.62 ± 0.05 | 0.69 ± 0.06 | 0.63 ± 0.12 | 0.57 ± 0.12 | 0.202 |
| Kidney | 1.05 ± 0.08 | 1.00 ± 0.07 | 1.02 ± 0.05 | 1.05 ± 0.09 | 1.03 ± 0.05 | 0.544 |
| Epididymal adipose tissue | 2.21 ± 0.37 | 1.77 ± 0.61 | 1.91 ± 0.54 | 2.03 ± 0.33 | 2.15 ± 0.74 | 0.513 |
| Perirenal adipose tissue | 0.77 ± 0.26 | 0.59 ± 0.28 | 0.66 ± 0.25 | 0.67 ± 0.23 | 0.87 ± 0.31 | 0.317 |
| Heart | 0.64 ± 0.05 | 0.63 ± 0.09 | 0.70 ± 0.07 | 0.67 ± 0.06 | 0.69 ± 0.06 | 0.231 |
Mice were fed a low-fat diet (LFD), a Western diet (WD), or one of the three WDs containing 0.2% TMAO (WD + TMAO), 0.2% TMAO plus 1% hawthorn fruit extract (WD + TMAO + L-HFE), or 0.2% TMAO plus 2% hawthorn fruit extract (WD + TMAO + H-HFE) for 12 weeks. Data are expressed as mean ± SD, n = 8. a,b,cmeans in the same row with different letters differ significantly at p < 0.05
Plasma lipid profiles
After 12-week intervention, WD feeding disturbed plasma lipid profiles in mice by raising plasma levels of TC, TG, and non-HDL-C, and reducing plasma HDL-C concentration compared with LFD feeding (Table 4). Neither TMAO nor HFE significantly altered plasma lipids in WD-fed mice. Compared to feeding WD + TMAO diet, high-dose HFE supplementation tended to decrease plasma TC (p = 0.101) but this decrement did not reach statistical significance.
Table 4.
Plasma lipids in ApoE−/− mice
| LFD | WD | WD + TMAO | WD + TMAO + L-HFE | WD + TMAO + H-HFE | p value | p for dose effect | |
|---|---|---|---|---|---|---|---|
| TC (mg/dL) | 673 ± 73b | 1163 ± 156a | 1250 ± 134a | 1177 ± 153a | 1138 ± 133a | < 0.001 | 0.117 |
| HDL-C (mg/dL) | 74 ± 20a | 46 ± 15b | 46 ± 20b | 41 ± 19b | 43 ± 13b | 0.016 | 0.745 |
| TG (mg/dL) | 131 ± 22b | 216 ± 43a | 226 ± 28a | 255 ± 48a | 224 ± 42a | < 0.001 | 0.143 |
| Non-HDL-C (mg/dL) | 600 ± 67b | 1117 ± 154a | 1204 ± 152a | 1136 ± 161a | 1095 ± 130a | < 0.001 | 0.919 |
| Non-HDL-C/HDL-C | 8.64 ± 2.27b | 27.06 ± 11.32a | 31.99 ± 15.98a | 31.92 ± 12.74a | 27.30 ± 8.81a | 0.006 | 0.464 |
| HDL-C/TC | 0.11 ± 0.03a | 0.04 ± 0.01b | 0.04 ± 0.02b | 0.04 ± 0.02b | 0.04 ± 0.01b | < 0.001 | 0.982 |
Mice were fed a low-fat diet (LFD), a Western diet (WD), or one of the three WDs containing 0.2% TMAO (WD + TMAO), 0.2% TMAO plus 1% hawthorn fruit extract (WD + TMAO + L-HFE), or 0.2% TMAO plus 2% hawthorn fruit extract (WD + TMAO + H-HFE) for 12 weeks. Data are expressed as mean ± SD, n = 8
TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; TG, triacylglycerol; Non-HDL-C, non-HDL cholesterol
a,bMeans in the same row with different letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
Plasma TMAO level
Similar plasma TMAO level was observed in mice maintained on LFD and WD (Fig. 2a). Addition of 0.2% TMAO into WD increased plasma TMAO level in mice by four folds compared with feeding of diets without TMAO. HFE supplementation did not affect plasma TMAO concentration in mice (Fig. 2a), suggesting that the absorption of dietary TMAO was not altered by HFE administration.
Fig. 2.

Plasma levels of TMAO, inflammatory cytokines, and oxidative stress-related parameters in ApoE−/− mice. a Plasma level of TMAO. The inflammatory cytokines include b tumor necrosis factor α (TNF-α), c interleukin (IL)-1β, d IL-6, e monocyte chemoattractant protein 1 (MCP-1), and f IL-10. Plasma levels of g total antioxidant capacity (T-AOC), h superoxide dismutase (SOD) activity, i glutathione peroxidase (GSH-Px) activity, and j malondialdehyde (MDA) in ApoE−/− mice. Data are expressed as mean with SD, n = 8. a,b,cmeans with different superscript letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
Plasma inflammatory biomarkers
Results on plasma inflammatory biomarkers revealed that WD feeding raised plasma levels of pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and MCP-1 compared with LFD feeding (Fig. 2b–e). Addition of TMAO into WD aggravated inflammation by further elevating TNF-α, IL-1β, and MCP-1 levels in the plasma (Fig. 2b, c, e). Compared with WD + TMAO diet, addition of HFE reduced plasma concentrations of TNF-α and MCP-1 in a dose-dependent manner (Fig. 2b, e). No significant difference in plasma IL-10 level was observed among the five groups of mice (Fig. 2f).
Plasma T-AOC, antioxidant enzyme activities, and MDA
Mice maintained on WD had lower plasma T-AOC and higher plasma level of MDA, a marker of lipid peroxidation [38], compared with those on LFD (Fig. 2g, j). Addition of TMAO into WD exacerbated oxidative stress by reducing plasma T-AOC, decreasing the activity of antioxidant enzymes SOD and GSH-Px, and elevating plasma MDA concentration (Fig. 2g–j). The TMAO-induced reductions in plasma T-AOC and GSH-Px activity were restored by HFE administration (Fig. 2g, i). Moreover, it was observed that HFE dose-dependently reduced plasma MDA level against TMAO in mice (Fig. 2j).
Atherosclerotic lesions
WD feeding promoted the formation of plaque in mouse aorta (Fig. 3a–g). Compared with those in WD group, mice in WD + TMAO group had plaque area increased by 87% in thoracic aorta and by 32% in aortic sinus, respectively (Fig. 3a–d). Addition of HFE was able to diminish plaque development by both longitudinal and cross-sectional evaluations, and inhibit the formation of necrotic core in aortic sinus in a dose-dependent fashion (Fig. 3a–d, f). However, no significant difference in collagen content or the ratio of collagen to necrotic core, an indicator for plaque stability, was found among the four groups of mice fed high-fat WDs.
Fig. 3.

Atherosclerotic lesions in ApoE−/− mice. a Representative images and b quantification of en face oil-red-O stained plaque area in the thoracic aorta. c Representative images of cross-sections at the aortic sinus stained with H&E and Masson’s Trichrome. Images were taken at 40 × and 100 × magnification. Quantification of d plaque area, e collagen content, f necrotic core area, and g the ratio of collagen to necrotic core in the aortic sinus of ApoE−/− mice. Data are expressed as mean with SD, n = 8. a,b,c,dmeans with different superscript letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
Hepatic cholesterol and lipid contents
Compared with LFD feeding, WD feeding increased hepatic cholesterol content and the accumulation of lipids in the liver (Fig. 4a–e). Addition of TMAO promoted the liver cholesterol accumulation but it did not alter the hepatic levels of FAs (Fig. 4a–e). Compared with WD + TMAO diet, HFE supplementation decreased hepatic cholesterol level in a dose-dependent manner (Fig. 4a). In addition, the hepatic accumulation of total FAs, saturated fatty acids (SFAs), and polyunsaturated fatty acids (PUFAs) was significantly reduced in WD + TMAO + H-HFE group (Fig. 4b, c, e).
Fig. 4.

Changes in hepatic lipid contents in ApoE−/− mice. Hepatic contents of a cholesterol, b total fatty acids (FAs), c saturated fatty acids (SFAs), d monounsaturated fatty acids (MUFAs), and e polyunsaturated fatty acids (PUFAs) in ApoE−/− mice. Data are expressed as mean with SD, n = 8. a,b,cmeans with different superscript letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
Fecal sterols and lipids
Results showed that coprostanol, coprostanone, cholesterol, and dihydrocholsterol were the predominant neutral sterols in mouse feces (Table 5). Mouse fecal acidic sterols mainly consisted of lithocholic acid, deoxycholic acid, chenodeoxycholic acid, and cholic acid. WD feeding strikingly promoted the excretion of total neutral and acidic sterols in feces (Table 5). Addition of TMAO into WD did not influence the fecal neutral sterol contents, but it significantly decreased the fecal output of total acidic sterols by 41%. Compared with those on WD + TMAO diet, mice on WD + TMAO + L-HFE and WD + TMAO + H-HFE diets had fecal contents of total neutral sterols increased by 68% and 100%, respectively. Moreover, the TMAO-induced reduction in fecal output of acidic sterols was reversed by HFE supplementation in a dose-dependent manner (Table 5).
Table 5.
Fecal contents of sterols and lipids at week 12 in ApoE−/− mice
| LFD | WD | WD + TMAO | WD + TMAO + L-HFE | WD + TMAO + H-HFE | p value | p for dose effect | |
|---|---|---|---|---|---|---|---|
| Week 12 fecal neutral sterols (mg/d) | |||||||
| Coprostanol | 0.01 ± 0.00c | 0.39 ± 0.07b | 0.44 ± 0.15b | 0.70 ± 0.21a | 0.61 ± 0.12a | < 0.001 | 0.123 |
| Coprostanone | 0.01 ± 0.01 | 0.02 ± 0.02 | 0.09 ± 0.10 | 0.16 ± 0.16 | 0.10 ± 0.13 | 0.105 | 0.921 |
| Cholesterol | 0.09 ± 0.03c | 0.53 ± 0.25b | 0.38 ± 0.32bc | 0.69 ± 0.35b | 1.13 ± 0.56a | < 0.001 | 0.006 |
| Dihydrocholesterol | 0.02 ± 0.01c | 0.05 ± 0.01ab | 0.04 ± 0.02b | 0.05 ± 0.02ab | 0.06 ± 0.02a | 0.001 | 0.064 |
| Total neutral sterols | 0.12 ± 0.03c | 0.99 ± 0.19b | 0.95 ± 0.28b | 1.60 ± 0.41a | 1.90 ± 0.41a | < 0.001 | < 0.001 |
| Week 12 fecal acidic sterols (mg/d) | |||||||
| Lithocholic acid | 0.24 ± 0.02c | 0.74 ± 0.24bc | 0.66 ± 0.21bc | 1.25 ± 0.34ab | 1.61 ± 0.55a | 0.003 | 0.016 |
| Deoxycholic acid | 0.40 ± 0.06b | 0.80 ± 0.35a | 0.46 ± 0.12b | 0.59 ± 0.05ab | 0.57 ± 0.01ab | 0.101 | 0.119 |
| Chenodeoxycholic acid | 0.17 ± 0.04c | 0.54 ± 0.16a | 0.30 ± 0.07bc | 0.40 ± 0.03ab | 0.31 ± 0.03bc | 0.003 | 0.807 |
| Cholic acid | 0.31 ± 0.12b | 0.97 ± 0.21a | 0.39 ± 0.15b | 0.51 ± 0.28b | 0.45 ± 0.05b | 0.008 | 0.693 |
| Total acidic sterols | 1.11 ± 0.22c | 3.05 ± 0.84a | 1.81 ± 0.47bc | 2.75 ± 0.36ab | 2.94 ± 0.62a | 0.006 | 0.028 |
| Week 12 fecal lipids (mg/d) | |||||||
| Total SFAs | 0.59 ± 0.08b | 7.04 ± 1.02a | 7.08 ± 0.46a | 6.76 ± 1.76a | 8.20 ± 2.55a | < 0.001 | 0.471 |
| Total MUFAs | 0.21 ± 0.03b | 0.78 ± 0.11a | 0.76 ± 0.05a | 0.84 ± 0.24a | 1.04 ± 0.49a | < 0.001 | 0.162 |
| Total PUFAs | 0.10 ± 0.01c | 0.20 ± 0.03b | 0.19 ± 0.01b | 0.21 ± 0.04b | 0.28 ± 0.10a | < 0.001 | 0.176 |
| Total FAs | 0.90 ± 0.12b | 8.02 ± 1.15a | 8.04 ± 0.51a | 7.81 ± 2.03a | 9.52 ± 3.13a | < 0.001 | 0.411 |
Mice were fed a low-fat diet (LFD), a Western diet (WD), or one of the three WDs containing 0.2% TMAO (WD + TMAO), 0.2% TMAO plus 1% hawthorn fruit extract (WD + TMAO + L-HFE), or 0.2% TMAO plus 2% hawthorn fruit extract (WD + TMAO + H-HFE) for 12 weeks. Data are expressed as mean ± SD, n = 8
SFA, saturated fatty acid; MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acid; FA, fatty acid
a,b,cMeans in the same row with different letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
For fecal FA analysis, a ninefold increment in fecal total FA content was observed in WD-fed mice compared with those in LFD-fed mice (Table 5). Addition of TMAO into WD did not alter the fecal output of FAs. Compared with those on WD + TMAO diet, mice on WD + TMAO + H-HFE diet had higher fecal content of PUFAs. However, HFE administration did not significantly affect the fecal excretion of SFAs, MUFAs, or total FAs (Table 5).
Hepatic gene expression of inflammatory cytokines
WD feeding stimulated hepatic inflammation by facilitating the expression of MCP-1 at both transcriptional and translational levels, and increasing the mRNA level of TNF-α and IL-1β (Fig. 5a–c). Addition of TMAO into WD further exacerbated inflammation in the liver by up-regulating the mRNA expression of MCP-1, TNF-α, and IL-1β (Fig. 5a–c). Compared with feeding WD + TMAO diet, feeding WD + TMAO + L-HFE and WD + TMAO + H-HFE diets significantly lowered the mRNA and protein abundance of MCP-1 in the liver (Fig. 5a). Besides, the mRNA expression of IL-1β was dose-dependently down-regulated by dietary HFE (Fig. 5c). Neither TMAO nor HFE significantly affected the mRNA or protein levels of IL-6 and IL-10 in the liver (Fig. 5d, e).
Fig. 5.

Relative expression of inflammatory cytokines in the liver of ApoE−/− mice. The mRNA and protein levels of a monocyte chemoattractant protein 1 (MCP-1), b tumor necrosis factor α (TNF-α), c interleukin (IL)-1β, d IL-6, and e IL-10 in the liver of ApoE−/− mice. Data are expressed as mean with SD, n = 8. a,b,cmeans with different superscript letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
Hepatic gene expression of antioxidant enzymes and their activities
The hepatic expression of several antioxidant enzymes was then examined. SOD 1 and SOD 2 genes encode the cytoplasmic Cu/Zn-SOD and mitochondrial Mn-SOD, respectively [39]. GSH-Px3 is responsible for the major GSH-Px activity in plasma [40]. CAT catalyzes the decomposition of hydrogen peroxide [41]. After 12-week intervention, WD feeding lowered the antioxidant defense capacity in the liver by down-regulating the mRNA expression of SOD1, SOD2, and CAT, and reducing the activity of SOD (Fig. 6a, b, d, f). Compared with those on WD diet, a significant decrement in the mRNA level of CAT, and lowered activity of SOD and CAT were observed in mice maintained on WD + TMAO diet (Fig. 6d, f, h). The mRNA abundance of SOD1, SOD2, GSH-Px3, and CAT was dose-dependently raised by dietary HFE (Fig. 6a–d). Furthermore, the TMAO-induced reduction in SOD activity was reversed by both low-dose and high-dose HFE, while the TMAO-reduced CAT activity was improved in WD + TMAO + H-HFE group (Fig. 6f, h). However, no obvious difference in hepatic T-AOC, GSH-Px activity, or MDA level were observed among the five groups of mice (Fig. 6e, g, i).
Fig. 6.

The expression and activity of antioxidant enzyme in the liver of ApoE−/− mice. The mRNA levels of a superoxide dismutase 1 (SOD1), b SOD2, c glutathione peroxidase 3 (GSH-Px3), and d catalase (CAT) in mice. e Hepatic total antioxidant capacity (T-AOC) in mice. Activity of f SOD, g GSH-Px, and h CAT in the liver of mice. i Hepatic malondialdehyde (MDA) level in mice. Data are expressed as mean with SD, n = 8. a,b,c,dmeans with different superscript letters differ significantly at p < 0.05 by one-way ANOVA. p for dose effect was analyzed using ordinary linear regression across WD + TMAO, WD + TMAO + L-HFE, and WD + TMAO + H-HFE groups
Discussion
Recent studies have shown that TMAO is positively correlated with the risk of adverse CVDs in humans [4, 5]. Hawthorn fruit consumption is considered beneficial to cardiovascular health. To the best of our knowledge, the present study was the first of its kinds to investigate the effect of HFE on TMAO-exacerbated atherogenesis in mice. The present results clearly demonstrated that TMAO accelerated atherogenesis, exacerbated inflammation, and aggravated oxidative stress in ApoE−/− mice fed a high-fat WD. Consistent with the previous report [29], the present results demonstrated that HFE was able to inhibit the formation of atherosclerotic plaque in mice fed a TMAO-containing diet.
Atherosclerosis is a progressive inflammatory vascular disease [2]. Activated vascular inflammation can trigger endothelial dysfunction, promote the formation of foam cells, and accelerate atherosclerosis development [42]. Amongst various inflammatory cytokines and chemokines, the production of MCP-1 by aortic endothelial cells is markedly enhanced at the initiation stage of atherosclerosis [43, 44]. MCP-1 functions to recruit monocytes and facilitate their infiltration into the vascular intima [44]. Overexpression of MCP-1 could accelerate atherogenesis in ApoE−/− mice by promoting the accumulation of macrophages and oxidized lipids in the aorta [43]. TNF-α exerts a potent pro-atherogenic activity by up-regulating the expression of adhesion molecules and MCP-1, increasing the permeability of endothelium, and stimulating the activation of macrophages [45]. IL-1β and IL-6 are the predominant pro-inflammatory ILs in atherosclerosis mainly by aggravating subsequent inflammatory cascades, mediating the acute-phase response, and promoting the fatty streak formation in atherogenesis [46]. IL-10 is generally considered as an anti-atherogenic cytokine due to its anti-inflammatory properties [47]. Our results clearly showed that dietary TMAO increased plasma levels of TNF-α, IL-1β, and MCP-1 in mice fed a high-fat WD (Fig. 2b, c, e). Consistently, the hepatic mRNA expression of these three proinflammatory cytokines were up-regulated by TMAO (Fig. 5a–c). Together with the findings that dietary TMAO elevated serum concentrations of MCP-1 and IL-1β, and induced inflammation in adipose tissue of high-fat-fed mice [19, 20], our results demonstrated that TMAO was pro-atherogenic and aggravated extensive inflammation. In the present study, HFE supplement significantly lowered plasma levels of TNF-α and MCP-1 in a dose-dependent manner (Fig. 2b, e) and down-regulated the hepatic expression of MCP-1 and IL-1β (Fig. 5a, c). Our data were in agreement with the study of Zhang et al., who reported that the sugar-free aqueous extract of hawthorn fruit lowered serum levels of inflammatory biomarkers including IL-1β, IL-8, IL-18, and C-reactive protein (CRP) in rats fed a high-fat diet [31]. The present results suggested that HFE supplement could alleviate TMAO-exacerbated inflammation.
Oxidative stress is a redox imbalance with increased production of ROS and overwhelmed antioxidant defense capacity [48]. Endogenous antioxidant enzymes including SOD, GSH-Px, and CAT are critical for the maintenance of redox balance by scavenging excessive ROS [41]. Severe oxidative stress can lead to endothelial dysfunction, LDL oxidative modification, and atherogenesis [48]. Previous studies showed that TMAO stimulated ROS production and inhibited the SOD activation both in human umbilical vein endothelial cells (HUVEC) and rodent animals [10, 12, 13]. Similarly, the present results demonstrated that dietary TMAO lowered plasma T-AOC, reduced the activities of SOD and GSH-Px in the plasma, and increased the plasma level of MDA (Fig. 2g–j). The hepatic activity of SOD and CAT were also decreased in mice exposed to TMAO-containing diet (Fig. 6f, h). The present data suggested that dietary TMAO exacerbated oxidative stress and weakened the antioxidant defenses in mice.
The cardio-protective effect of HFE was partially attributed to its potent antioxidant capacity. Previous study showed that seven antioxidant phenolic compounds (epicatechin, chlorogenic acid, hyperoside, isoquecitrin, protocatechuic acid, rutin, and quercetin) isolated from the ethyl acetate fraction of HFE were able to prevent the Cu2+-mediated oxidation of human low-density lipoprotein (LDL) and the peroxy free radical-induced oxidation of α-tocopherol in human LDL in vitro [22]. In the present study, HFE containing 15.69 mg/g total phenolic antioxidants conspicuously enhanced the antioxidant defenses and relieved the TMAO-exacerbated oxidative stress both in the plasma and the liver (Figs. 2g–j, 6f, h). At a molecular level, the hepatic mRNA expression of SOD1, SOD2, GSH-Px3, and CAT was up-regulated by HFE in a dose-dependent manner (Fig. 6a–d). Together with the finding that HFE could improve the expression of antioxidant enzymes in the serum, liver, and brain of senescence-accelerated mice [30], our results clearly demonstrated that HFE possessed antioxidant capacity in both the plasma and the liver, thereby contributing to its anti-atherogenic activity against TMAO in mice.
Disturbed cholesterol metabolism may lead to cholesterol accumulation and the development of atherosclerosis [49, 50]. In the present study, TMAO administration elevated hepatic cholesterol concentration without affecting FA content in the liver (Fig. 4a–e). Fecal excretions of neutral and acidic sterols are the major routes for cholesterol elimination [50]. Consistent with the report of Chen et al. [14] and our previous studies [35, 51], the present results showed that the TMAO-induced hepatic cholesterol accumulation was probably mediated by decreasing the fecal output of acidic sterols (Table 5). However, no significant alterations in plasma TC, HDL-C, or non-HDL-C was observed in mice fed dietary TMAO (Table 4), which was in line with the findings that 0.12% dietary TMAO induced atherogenesis but it did not change the plasma TC in ApoE−/− mice [4, 6]. In contrast, some researchers observed an increment in plasma TC in mice given diets containing high content of TMAO or choline, a dietary precursor of TMAO [14, 16, 17, 35]. Contradictorily, it was reported that 0.2% dietary TMAO led to decreased plasma TC and TG levels in high-fat-fed C57BL/6 J mice [19]. The inconclusive effect of TMAO on plasma cholesterol may be affected by other dietary components, as well as the rodent models adopted.
In the present study, we observed that HFE administration could dose-dependently decrease the hepatic cholesterol content via enhancement on the fecal excretion of neutral and acidic sterols (Fig. 4a and Table 5), but it did not significantly modulate plasma lipid profiles in ApoE−/− mice exposed to dietary TMAO (Table 4). The inconsistent effect of TMAO or HFE on plasma cholesterol and liver cholesterol might be explained by the complete deletion of ApoE gene in ApoE−/− mice. Wild-type mice are generally resistant to atherosclerosis, because most of their cholesterol is transported in HDL particles, but not in non-HDL particles as in humans [34]. ApoE, a 34 kDa glycoprotein, is a structural component of lipoprotein particles, and functions as a recognition factor for the clearance of chylomicrons and very low-density lipoprotein (VLDL) remnants from circulation by the liver [52]. Genetic manipulation by knocking out ApoE gene in mice results in spontaneous development of hypercholesterolemia (dominated by non-HDL-C) and atherosclerosis [34]. Moreover, similar lesion distribution and progression as those in humans could be observed in ApoE−/− mice [33, 53], making them a particularly popular animal model in atherosclerosis studies. Study showed that compared to their wild-type counterparts, ApoE−/− mice had 10 times higher plasma cholesterol level, whereas their liver cholesterol content was less affected and only increased by 56% [54]. Therefore, the absence of ApoE impedes the exchange between plasma cholesterol and hepatic cholesterol, and might alter the impact of TMAO or HFE on plasma cholesterol regulation.
Conclusion
In summary, the present study demonstrated that TMAO accelerated atherosclerosis development by inducing extensive inflammation and oxidative stress in high-fat-fed ApoE−/− mice. Moreover, TMAO disturbed cholesterol metabolism by inhibiting fecal acidic sterol excretion, resulting in elevated hepatic cholesterol accumulation. Phenolic antioxidants-enriched HFE could reduce atherogenesis, alleviate inflammation, improve antioxidant defense capacity, and partially reverse the TMAO-induced elevation in hepatic cholesterol content in mice given dietary TMAO. It was therefore concluded that HFE was protective against the TMAO-exacerbated atherogenesis in ApoE−/− mice mainly via its anti-inflammatory and antioxidant activities.
Acknowledgements
We thank Hong Kong Research Grants Council General Research Fund for supporting this research and Mr. Hing Man Ho (School of Chinese Medicine, Hong Kong Baptist University) for his kind technical assistance.
Abbreviations
- ApoE
Apolipoprotein E
- CAT
Catalase
- CVD
Cardiovascular disease
- ERK
Extracellular signal-kinase
- FA
Fatty acid
- FAME
Fatty acid methyl ester
- GC
Gas chromatography
- GSH-Px3
Glutathione peroxidase 3
- HDL-C
High-density lipoprotein cholesterol
- HFE
Hawthorn fruit extract
- HPLC
High performance liquid chromatography
- IL
Interleukin
- LFD
Low-fat diet
- MAPK
Mitogen-activated protein kinase
- MCP-1
Monocyte chemoattractant protein 1
- MDA
Malondialdehyde
- MUFA
Monounsaturated fatty acid
- NF-κB
Nuclear factor κB
- PUFA
Polyunsaturated fatty acid
- ROS
Reactive oxygen species
- SFA
Saturated fatty acid
- SOD1
Superoxide dismutase 1
- SOD2
Superoxide dismutase 2
- T-AOC
Total antioxidant capacity
- TC
Total cholesterol
- TG
Triacylglycerol
- TMA
Trimethylamine
- TMAO
Trimethylamine-N-oxide
- TMS
Trimethylsilyl
- TNF-α
Tumor necrosis factor α
- WD
Western high-fat diet
Authors’ contributions
ZH and ZYC designed the research. ZYC supervised the research and provide research funding. ZH, EK, WH, HZ, JL, and KYM participated in data acquisition and analysis. ZH and ZYC wrote and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by Hong Kong Research Grants Council General Research Fund (Project Number CUHK 14105820).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All the experimental protocols were approved by the Animal Experimental Ethical Committee, The Chinese University of Hong Kong (Ref No. 17/065/MIS).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular Risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown JM, Hazen SL. Microbial modulation of cardiovascular disease. Nat Rev Microbiol. 2018;16:171–181. doi: 10.1038/nrmicro.2017.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. 2016;5:e002767. doi: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boini KM, Hussain T, Li PL, Koka S. Trimethylamine-N-oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell Physiol Biochem. 2017;44:152–162. doi: 10.1159/000484623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017;6:e006347. doi: 10.1161/JAHA.117.006347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geng J, Yang C, Wang B, Zhang X, Hu T, Gu Y, et al. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed Pharmacother. 2018;97:941–947. doi: 10.1016/j.biopha.2017.11.016. [DOI] [PubMed] [Google Scholar]
- 12.Ke Y, Li D, Zhao M, Liu C, Liu J, Zeng A, et al. Gut flora-dependent metabolite trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic Biol Med. 2018;116:88–100. doi: 10.1016/j.freeradbiomed.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Yang G, Lin CC, Yang Y, Yuan L, Wang P, Wen X, et al. Nobiletin prevents trimethylamine oxide-induced vascular inflammation via inhibition of the NF-κB/MAPK pathways. J Agric Food Chem. 2019;67:6169–6176. doi: 10.1021/acs.jafc.9b01270. [DOI] [PubMed] [Google Scholar]
- 14.Chen ML, Yi L, Zhang Y, Zhou X, Ran L, Yang J, et al. Resveratrol attenuates trimethylamine-N-oxide (TMAO)-induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. mBio. 2016;7:e02210–e02215. doi: 10.1128/mBio.02210-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y, Zhao Y, Yuan L, Yang X. Protective effects of tartary buckwheat flavonoids on high TMAO diet-induced vascular dysfunction and liver injury in mice. Food Funct. 2015;6:3359–3372. doi: 10.1039/C5FO00581G. [DOI] [PubMed] [Google Scholar]
- 16.Guo J, Meng Y, Zhao Y, Hu Y, Ren D, Yang X. Myricetin derived from Hovenia dulcis Thunb. ameliorates vascular endothelial dysfunction and liver injury in high choline-fed mice. Food Funct. 2015;6:1620–1634. doi: 10.1039/C4FO01073F. [DOI] [PubMed] [Google Scholar]
- 17.Lu Y, Li W, Yang X. Soluble soybean polysaccharides enhance the protective effects of genistein against hepatic injury in high l-carnitine-fed mice. Food Funct. 2017;8:4364–4373. doi: 10.1039/C7FO00907K. [DOI] [PubMed] [Google Scholar]
- 18.Jia M, Ren D, Nie Y, Yang X. Beneficial effects of apple peel polyphenols on vascular endothelial dysfunction and liver injury in high choline-fed mice. Food Funct. 2017;8:1282–1292. doi: 10.1039/C7FO00147A. [DOI] [PubMed] [Google Scholar]
- 19.Gao X, Liu X, Xu J, Xue C, Xue Y, Wang Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J Biosci Bioeng. 2014;118:476–481. doi: 10.1016/j.jbiosc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Gao X, Xu J, Jiang C, Zhang Y, Xue Y, Li Z, et al. Fish oil ameliorates trimethylamine N-oxide-exacerbated glucose intolerance in high-fat diet-fed mice. Food Funct. 2015;6:1117–1125. doi: 10.1039/C5FO00007F. [DOI] [PubMed] [Google Scholar]
- 21.Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448–455. doi: 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Chang Q, Zhu M, Huang Y, Ho WKK, Chen ZY. Characterization of antioxidants present in hawthorn fruits. J Nutr Biochem. 2001;12:144–152. doi: 10.1016/S0955-2863(00)00137-6. [DOI] [PubMed] [Google Scholar]
- 23.Chen ZY, Zhang ZS, Kwan KY, Zhu M, Ho WKK, Huang Y. Endothelium-dependent relaxation induced by hawthorn extract in rat mesenteric artery. Life Sci. 1998;63:1983–1991. doi: 10.1016/S0024-3205(98)00476-7. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Ho WKK, Huang Y, James AE, Lam LW, Chen ZY. Hawthorn fruit is hypolipidemic in rabbits fed a high cholesterol diet. J Nutr. 2002;132:5–10. doi: 10.1093/jn/132.1.5. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Ho WKK, Huang Y, Chen ZY. Hypocholesterolemic activity of hawthorn fruit is mediated by regulation of cholesterol-7alpha-hydroxylase and acyl CoA: cholesterol acyltransferase. Food Res Int. 2002;35:885–891. doi: 10.1016/S0963-9969(02)00099-6. [DOI] [Google Scholar]
- 26.Niu CS, Chen CT, Chen LJ, Cheng KC, Yeh CH, Cheng JT. Decrease of blood lipids induced by Shan-Zha (fruit of Crataegus pinnatifida) is mainly related to an increase of PPARα in liver of mice fed high-fat diet. Horm Metab Res. 2011;43:625–630. doi: 10.1055/s-0031-1275325. [DOI] [PubMed] [Google Scholar]
- 27.Shih CC, Lin CH, Lin YJ, Wu JB. Validation of the antidiabetic and hypolipidemic effects of hawthorn by assessment of gluconeogenesis and lipogenesis related genes and AMP-activated protein kinase phosphorylation. Evid Based Complement Alternat Med. 2013;2013:597067. doi: 10.1155/2013/597067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu HJ, Luo XG, Dong QQ, Mu A, Shi GL, Wang QT, et al. Ethanol extract of Zhongtian hawthorn lowers serum cholesterol in mice by inhibiting transcription of 3-hydroxy-3-methylglutaryl-coa reductase via nuclear factor-kappa B signal pathway. Exp Biol Med. 2016;241:667–674. doi: 10.1177/1535370215627032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Zhang L, Geng Y, Geng Y. Hawthorn fruit attenuates atherosclerosis by improving the hypolipidemic and antioxidant activities in apolipoprotein E-deficient mice. J Atheroscler Thromb. 2014;21:119–128. doi: 10.5551/jat.19174. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Zhang Z, Guo Y, Sun P, Lv X, Zuo Y. Hawthorn fruit increases the antioxidant capacity and reduces lipid peroxidation in senescence-accelerated mice. Eur Food Res Technol. 2011;232:743–751. doi: 10.1007/s00217-011-1435-7. [DOI] [Google Scholar]
- 31.Zhang J, Liang R, Wang L, Yan R, Hou R, Gao S, et al. Effects of an aqueous extract of Crataegus pinnatifida Bge. var. major N.E.Br. fruit on experimental atherosclerosis in rats. J Ethnopharmacol. 2013;148:563–569. doi: 10.1016/j.jep.2013.04.053. [DOI] [PubMed] [Google Scholar]
- 32.Liu P, Kallio H, Yang B. Phenolic compounds in hawthorn (Crataegus grayana) fruits and leaves and changes during fruit ripening. J Agric Food Chem. 2011;59:11141–11149. doi: 10.1021/jf202465u. [DOI] [PubMed] [Google Scholar]
- 33.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.ATV.14.1.133. [DOI] [PubMed] [Google Scholar]
- 34.Oppi S, Lüscher TF, Stein S. Mouse models for atherosclerosis research—which is my line? Front Cardiovasc Med. 2019;6:46. doi: 10.3389/fcvm.2019.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He Z, Lei L, Kwek E, Zhao Y, Liu J, Hao W, et al. Ginger attenuates trimethylamine-N-oxide (TMAO)-exacerbated disturbance in cholesterol metabolism and vascular inflammation. J Funct Foods. 2019;52:25–33. doi: 10.1016/j.jff.2018.10.022. [DOI] [Google Scholar]
- 36.Liu J, Hao W, He Z, Kwek E, Zhao Y, Zhu H, et al. Beneficial effects of tea water extracts on the body weight and gut microbiota in C57BL/6J mice fed with a high-fat diet. Food Funct. 2019;10:2847–2860. doi: 10.1039/C8FO02051E. [DOI] [PubMed] [Google Scholar]
- 37.Zhu H, Chen J, He Z, Hao W, Liu J, Kwek E, et al. Soybean germ oil reduces blood cholesterol by inhibiting cholesterol absorption and enhancing bile acid excretion. Food Funct. 2019;10:1836–1845. doi: 10.1039/C8FO02585A. [DOI] [PubMed] [Google Scholar]
- 38.Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis. 2005;15:316–328. doi: 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. doi: 10.1016/S0891-5849(02)00905-X. [DOI] [PubMed] [Google Scholar]
- 40.Lee YS, Kim AY, Choi JW, Kim M, Yasue S, Son HJ, et al. Dysregulation of adipose glutathione peroxidase 3 in obesity contributes to local and systemic oxidative stress. Mol Endocrinol. 2008;22:2176–2189. doi: 10.1210/me.2008-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 43.Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, Rollins BJ, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–1525. doi: 10.1161/01.ATV.19.6.1518. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez-Quesada C, Frangogiannis NG. Monocyte chemoattractant protein-1/CCL2 as a biomarker in acute coronary syndromes. Curr Atheroscler Rep. 2009;11:131–138. doi: 10.1007/s11883-009-0021-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fatkhullina AR, Peshkova IO, Koltsova EK. The role of cytokines in the development of atherosclerosis. Biochemistry (Mosc.) 2016;81:1358–1370. doi: 10.1134/S0006297916110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017;19:42. doi: 10.1007/s11883-017-0678-6. [DOI] [PubMed] [Google Scholar]
- 49.Berenson GS, Srinivasan SR, Bao W, Newman WP, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338:1650–1656. doi: 10.1056/NEJM199806043382302. [DOI] [PubMed] [Google Scholar]
- 50.Chen ZY, Jiao R, Ma KY. Cholesterol-lowering nutraceuticals and functional foods. J Agric Food Chem. 2008;56:8761–8773. doi: 10.1021/jf801566r. [DOI] [PubMed] [Google Scholar]
- 51.He Z, Hao W, Kwek E, Lei L, Liu J, Zhu H, et al. Fish oil is more potent than flaxseed oil in modulating gut microbiota and reducing trimethylamine-N-oxide-exacerbated atherogenesis. J Agric Food Chem. 2019;67:13635–13647. doi: 10.1021/acs.jafc.9b06753. [DOI] [PubMed] [Google Scholar]
- 52.Getz GS, Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50(Suppl):S156–S161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24:1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- 54.Kuipers F, Van Ree JM, Hofker MH, Wolters H, In’t Veld G, Havinga R, et al. Altered lipid metabolism in apolipoprotein E-deficient mice does not affect cholesterol balance across the liver. Hepatology. 1996;24:241–247. doi: 10.1002/hep.510240138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.