

Abstract
Aberrant cell cycle activation is a hallmark of carcinogenesis. Recently three cell cycle targeting cyclin-dependent kinase 4/6 (CDK4/6) inhibitors have been approved for the treatment of metastatic breast cancer. CDK4/6 inhibitors suppress proliferation through inhibition of CDK4/6-dependent retinoblastoma-1 (Rb1) phosphorylation and inactivation, a key regulatory step in G1-to-S-phase transition. Importantly, aberrant cell cycle activation is also linked with several non-oncological diseases including acute kidney injury (AKI). AKI is a common disorder caused by toxic, inflammatory, and ischemic damage to renal tubular epithelial cells (RTECs). Interestingly, AKI triggered by the anti-cancer drug cisplatin can be mitigated by ribociclib, a CDK4/6 inhibitor, through mechanisms that remain unclear. Employing in vivo cell cycle analysis and functional Rb1 knock-down, here, we have examined the cellular and pharmacological basis of the renal protective effects of ribociclib during cisplatin nephrotoxicity. Remarkably, siRNA-mediated Rb1 silencing or RTEC-specific Rb1 gene ablation did not alter the severity of cisplatin-associated AKI; however, it completely abrogated the protective effects conferred by ribociclib administration. Furthermore, we find that cisplatin treatment evokes CDK4/6 activation and Rb1 phosphorylation in the normally quiescent RTECs, however, this is not followed by S-phase entry likely due to DNA-damage induced G1 arrest. The cytoprotective effects of ribociclib are thus not a result of suppression of S-phase entry but are likely dependent on the maintenance of Rb1 in a hypo-phosphorylated and functionally active form under stress conditions. These findings delineate the role of Rb1 in AKI and illustrate the pharmacological basis of the renal protective effects of CDK4/6 inhibitors.
Keywords: Acute kidney injury, Cisplatin nephrotoxicity, CDK4/6 inhibitors, Cell cycle, Retinoblastoma, Renal tubular epithelial cells
1. Introduction
In multicellular organisms, cell division and cell death must be coregulated to maintain tissue homeostasis [1]. Through finely balanced spatial and temporal regulation of cell division and cell death pathways proper embryonic development and adult tissue maintenance is achieved [2]. Cell division and cell death pathways are linked in part through the use of a common set of regulatory proteins. Strong evidence in favor of a link between cell cycle regulation and apoptosis is provided by several studies where it has been demonstrated that apoptosis is regulated by genes that are also involved in cell division [3]. This linkage has been recognized for p53, retinoblastoma susceptibility gene (Rb1), c-Myc, cyclin-dependent kinases (CDKs) and related genes [1]. For example, the tumor suppressor gene Rb1 is known to regulate both cell division and apoptosis through its regulation of the E2F family of transcription factors [4–7]. This Janus-faced activity is thought to provide dividing cells with the ability to escape inappropriate proliferative, genotoxic, or potentially oncogenic signals by activating cell death pathways [1].
Along with cancer, aberrant cellular proliferation is also a key contributor in the development and progression of various cardiovascular [8], neurological [9] and renal diseases [10], including acute kidney injury (AKI) [11]. AKI is associated with sudden disruption of renal tubular epithelial cell (RTEC) function due to direct and indirect cellular insults associated with sepsis [12], cardiac surgery [13], inflammation [14,15], ischemia [16], and cancer therapy [17]. In the case of cisplatin-associated kidney injury, transporter-mediated [18–20] accumulation of cisplatin in RTECs leads to activation of a number of signaling pathways [21–29] which finally culminate in RTEC dysfunction and cell death [30]. Interestingly, expression analysis of cell cycle regulatory proteins have shown that cell cycle activation is a key feature of AKI [31–35]. In a series of studies, it has been demonstrated that blocking cell cycle entry by modulating the CDK2-p21 pathway can provide protection from AKI [33,36,37]. Moreover, studies have shown that renal cell cycle entry is followed by G2/M cell cycle arrest, which contributes to development of fibrosis and chronic kidney disease [35].
The recent development of CDK4/6 inhibitors for anti-cancer treatment [38–40] has provided an opportunity to rigorously test the utility of modulating cell cycle pathways for the treatment or prevention of AKI. The emergence of these selective CDK4/6 inhibitors, ribociclib (Novartis), abemaciclib (Eli Lilly) and palbociclib (Pfizer) has enabled inhibition of G1-to-S-phase transition with improved effectiveness and fewer adverse effects [39,40]. These orally-administered compounds with similar structure can bind within the ATP binding pocket of CDK4 and CDK6 and have a high degree of selectivity for CDK4 and CDK6 as compared with other CDKs. Interestingly, recent studies have shown that CDK4/6 inhibitor palbociclib can mitigate nephrotoxic [41] and ischemia-associated [42] AKI. Our previous study [41] showed that the G1/S-regulating cyclin-dependent kinase 4/6-retinoblastoma-1 (CDK4/6-Rb1) pathway is activated early during the development of cisplatin-associated AKI. Targeted inhibition of CDK4/6 pathway by palbociclib and ribociclib, two FDA approved anti-cancer drugs, resulted in inhibition of Rb1 phosphorylation, amelioration of kidney injury, and improved overall survival [41].
Remarkably, we found that along with CDK4/6 inhibition, palbociclib and ribociclib also inhibit organic cation transporters [41], which are involved in cellular uptake of cisplatin [19] in RTECs. The relative contribution of inhibition of these distinct pathways on the renal protective effects of CDK4/6 inhibitors remains unknown. Notably, we found that along with wild type mice, ribociclib also affords renal protective effects in the Oct1/2 deficient mice, indicating an underlying cytoprotective role of an OCT2-independent pathway [41]. Whether this OCT2-independent pathway is Rb1 associated remains unknown. Interestingly, studies with cancer cell lines have shown that Rb1 is essential for cell cycle arrest caused by CDK4/6 inhibitors and Rb1 deficient cells fail to respond to CDK4/6 inhibition [43,44]. Here, we have carried out in vivo studies to elucidate the cell cycle-dependent renal protective mechanisms of CDK4/6 inhibitors by carrying out Rb1 knock-down as well as RTEC-specific Rb1 gene ablation. Our studies show that ribociclib mitigates cisplatin nephrotoxicity through Rb1-dependent mechanisms. These studies provide the pharmacological and cell biological basis for the kidney-specific protective effects of CDK4/6 inhibitors.
2. Materials and methods
2.1. Mice strains and genotyping
C57BL/6J mice, Rb1 floxed mice and Ggt1-Cre transgenic mice (stock numbers 000664, 008186 and 012841, respectively) were obtained from Jackson Laboratories (Bar Harbor, ME). Rb1 floxed mice were bred with Ggt1-Cre transgenic mice to generate renal tubular epithelial cell-specific Rb1 knockout mice. These transgenic mice express Cre recombinase in the renal tubular epithelial cells beginning at age 1–2 weeks [45]. Off-springs were genotyped by standard PCR-based methods. Primers used for amplification were Rb8302 (5′-CTC TAG ATC CTC TCA TTC TTC CC-3′) and Rb8303 (5′-CCT TGA CCA TAG CCC AGC AC-3′) and yield a 308-bp product for the Rb1 floxed allele, and a 258-bp product for the Rb1 wild-type allele. Primers for Ggt1-Cre are Cre5′ (AGG TGT AGA GAA GGC ACT TAG C), Cre3′ (CTA ATC GCC ATC TTC CAG CAG G) and produce a 405-bp product. PCR products were analyzed by electrophoresis using 1.5% agarose gels.
2.2. Animal care and treatment
Animals were housed in a temperature-controlled environment with a 12-hr light cycle and were given a standard diet and water ad libitum. All animal experiments were carried out in accordance with the animal use protocol approved by the Institutional Animal Care and User Committee of the Ohio State University. Mice were weighed weekly from 4 weeks to 8 weeks of age to monitor the effect of Rb1 deficiency.
2.3. Animal models of acute kidney injury
We carried out all the studies presented here in age-matched male mice at 8–12 weeks of age. In all the studies with conditional Rb1 knockout mice, we used male littermates from mice bred in-house. For experiments where only wild-type mice were used, 8-to12-wk-old male C57BL/6J mice were obtained from Jackson Laboratories.
For cisplatin nephrotoxicity experiments, single intraperitoneal injection of cisplatin (30 mg/kg; Sigma, St. Louis, MO) was carried out as described previously [25]. Subsequently, blood was collected on days 0–3 by submandibular vein bleed or on day 3 via cardiac puncture after carbon dioxide asphyxiation. Renal tissues were collected and processed for cell cycle, western blot, quantitative real-time PCR (qPCR) and histological analysis as described previously [41]. For CDK4/6 pharmacological inhibition studies, vehicle or ribociclib (Chemietek, Indianapolis, IN) were administered by oral gavage (150 mg/kg, dissolved in citrate buffer) four hours prior to cisplatin injection. For hydrodynamic injection, control (non-specific) or Rb1 targeting siRNAs from Ambion (25 μg in 0.5 ml of PBS; Austin, TX) or 0.5 ml of PBS was rapidly injected into the tail vein as described previously [46].
For ischemia-reperfusion experiments, mice were anesthetized by isoflurane (Henry Schein Inc., Melville, NY) and placed on a surgical platform where the body temperature was monitored throughout the procedure. The skin was disinfected, kidneys were exposed and bilateral renal pedicles were clamped for 30 min. Consequently, the clamps were removed to initiate reperfusion followed by suturing to close the muscle and skin around the incision. To compensate for the fluid loss, 0.5 ml warm sterile saline was administered via intraperitoneal injection. Blood was collected on days 0–2 by submandibular vein bleed or on day 1 or 2 via cardiac puncture after carbon dioxide asphyxiation. Renal tissues were collected and processed for cell cycle, qPCR and western blot analysis. To induce rhabdomyolysis, 8–12 weeks old male C57BL/6J mice were injected with 7.5 ml/kg 50% glycerol (Fisher Scientific, Hampton, NH) intramuscularly to the two hind-legs or injected with saline as a control, followed by blood and tissue collection on day 0–2, followed by cell cycle, qPCR and western blot analysis of renal tissues.
2.4. Evaluation of renal damage
Renal damage was evaluated by biochemical analysis (blood urea nitrogen and creatinine), histological examination (H&E staining) and analysis of renal expression of injury biomarkers (western blot analysis of Ngal expression). Mouse serum samples collected at indicated time-points were utilized for blood urine nitrogen and creatinine measurements using QuantiChromTM Urea Assay Kit (DIUR-100; BioAssay Systems, Hayward, CA) and Creatinine Colorimetric Assay Kit (Cayman Chemical, Ann Harbor, MI). Histological analysis of renal tissues was carried out by harvesting the kidneys, followed by paraffin embedding, and tissue sectioning. Tissue sections (5 pm) were then stained with hematoxylin and eosin (H&E) by standard methods. H&E staining were conducted by the Comparative Pathology and Mouse Phenotyping Shared Resource at The Ohio State University. Histopathologic scoring was conducted in a blinded fashion by examining ten consecutive 100x fields per section from at least three mice per group as described previously [25,41]. Tubular damage was scored by calculating the percentage of tubules that depicted dilation, epithelial flattening, and cast formation, loss of brush border and nuclei, and denudation of the basement membrane. The degree of tissue damage was scored based on the percentage of damaged tubules: 0: no damage; 1: < 25%; 2: 25–50%; 3: 50–75%; 4: > 75%.
2.5. Quantitative real-time PCR
Total RNA was isolated from harvested renal cortical tissues using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD) according to the manufacture’s protocol. RNA concentrations were determined using a NanoDrop spectrophotometer (ThermoFisher Scientific, Waltham, MA). One microgram of total RNA was reversed transcribed using RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific) and qRT-PCR was run in QuantStudio 7 Flex Real-Time PCR System (ThermoFisher Scientific) using SYBR Green Master Mix (Alkali Scientific, Fort Lauderdale, FL) and gene-specific primers. The expression levels of samples were determined by the comparative CT (ΔΔCT) method, β-actin was used as the internal control. Primers for cyclin A, cyclin B, cyclin D, cyclin E, p21, and p27, β-actin were follows, cyclin A forward: 5′-CTG GCT TCG AAA TAT GAA GAG-3′, reverse: 5′-AGG ACT TTG AGT AGC AGA TG-3′; cyclin B forward: 5′-ATC TAA AGT CGG AGA GGT TG-3′, reverse: 5′-GTT GTC AAG AAT TTT CAG CG-3′; cyclin D forward: 5′-AAC ACT TCC TCT CCA AAA TG-3′, reverse: 5′-GAA CTT CAC ATC TGT GGC-3′; cyclin E forward: 5′-CAA AAC TTG AGG AAA TCT ACC C-3′, reverse: 5′-CCA CTT GGA CAT AGA CAT TC-3′; p21 forward: 5′-ACC TGA TGA TAC CCA ACT AC-3′, reverse: 5′-CTG TGG CAC CTT TTA TTC TG-3′; p27 forward: 5′-ATC TGC CTC TAA AAG CAT TG-3′, reverse: 5′-CCA TCC CTA ACG TTT ATG TG-3′; β-actin forward: 5′-GAT GTA TGA AGG CTT TGG TC-3′, reverse: 5′-TGT GCA CTT TTA TTG GTC TC-3′.
2.6. Immunoblot analysis
Renal cortical tissues were lysed in a modified RIPA buffer (Bead Mill 24 Homogenizer, Fisher Scientific) and lysates were prepared as previously described [41]. For each sample 75 pg protein was loaded on Bistris gradient mini or midi-gels (Invitrogen, Carlsbad, CA) followed by transfer to PVDF membrane (Bio-rad, Hercules, CA), incubation with primary and secondary antibodies, and development with ECL reagent (Cell Signaling, Danvers, MA) and subsequent signal detection with X-ray films (Amersham, Pittsburgh, PA). Membranes were incubated with primary (1:1000 dilution) and secondary antibodies (1:2000 dilution). Primary antibodies were from Cell Signaling Technology [CDK6 (3136), Rb1 (9313), and PCNA (2586)] or from Santa Cruz Biotechnology (Dallas, TX) [NGAL (50351), β-actin (47778), CDK4 (260), and E2F1 (193)] or from ProteinTech (Rosemont, IL) [Rb1 (10048), Rb1l (13354), and Rb12 (27251)]. Secondary HRP-labeled antibodies were from Jackson ImmunoResearch (West Grove, PA). Densitometric analysis was carried out using Image J software and the signals of indicated proteins were normalized by actin levels in the same samples.
2.7. Flow cytometry based analysis for cell cycle dynamics
Cell cycle analysis was carried out using procedures modified from our previous work [47]. Cells were isolated from frozen renal cortical tissues for 4′, 6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Invitrogen) staining as described recently [48]. This method is based on cell dispersion using EDTA instead of enzymatic treatment. Briefly, frozen kidneys were thawed in cold PBS/EDTA [PBS (pH 7.4), containing 0.1% (w/v) EDTA] (Fisher Scientific). Small pieces (about 3 × 3 × 3 mm) of thawed renal cortical tissues were mechanically dispersed using 100 μm and 35 μm nylon cell strainers. To achieve this, the 100 μm mesh was placed in a tissue culture dish containing PBS/EDTA, such that the bottom of the mesh remains in contact with the surface of the liquid. Thawed tissues were then pressed through the 100 μm mesh using the plunger of a 5 ml syringe which releases the cells immediately into the buffer. The mesh was then rinsed few times with 4 °C cold PBS/EDTA solution. The resulting cell suspension was then filtered through a 35 μm cell strainers and the total volume was adjusted with PBS/EDTA to 5 ml per sample. After centrifugation (310 × g; 4 °C; 6 min), the cell pellet was suspended in 0.5 ml 4 °C cold PBS/EDTA. To fix the cells, five ml −20 °C cold 80% ethanol was added drop-wise under constant, gentle vortexing. Samples were then incubated for 30 min on ice and subsequently stored overnight at −20 °C before being subjected to DAPI staining. Next day, the fixed cells were allowed to adapt to room temperature (RT) for 10 min. After centrifugation (310 × g, 4 °C, 6 min), pellets were re-suspended in 5 ml PBS/EDTA (RT) and incubated for 10 min at ambient temperature and pelleted again (310 × g, 4 °C, 6 min). Pellets were permeabilized by adding 1 ml of 0.2% (v/v) Triton X-100 (Fisher Scientific) in PBS for 15 min on ice. Permeabilized cells were then re-suspended in 1 ml DAPI solution (10 μg/ml) followed by incubation at room temperature for 30 min under dark conditions. Cellular DNA content was then determined with a LSRII (BD Biosciences, San Jose, CA) where gating was based on respective unstained cell populations. Flow cytometry was performed at the Analytical Cytometry Core facility the Ohio State University Comprehensive Cancer Center. The data were analyzed using FlowJo software (BD Bioscience).
2.8. BrdU staining
For analysis of S-phase entry in vivo, two BrdU injections, 2 and 24 h post cisplatin or ischemia surgery, were administered intraperitoneally (30 μl/g body weight, Invitrogen). After development of AKI, mice were euthanized, kidneys were harvested and fixed in 4% paraformaldehyde and cryosections were processed for immunostaining. After permeabilization, tissue sections were incubated in 2 M HC1 for 40 min at 37 °C, rinsed with PBS, followed by blocking in 3% BSA in PBS and BrdU antibody (Cell signaling, 5292) incubation. At least three animals for each condition were analyzed. Stained tissues were examined by confocal microscopy and total number of BrdU positive RTECs were estimated by counting a minimum of 1000 cells per group.
2.9. Kinase assay
Renal tissues and cells were lysed with a buffer containing 150 mM NaCl (Fisher Scientific), 1 mM EDTA (Fisher Scientific), 1 mM EGTA (Fisher Scientific), 1% Triton X-100, 2.5 mM sodium pyrophosphate (Sigma), 1 mM β-glycerol phosphate (Sigma), 1 mM Na3VO4 (Sigma), 10 μg/ml leupeptin (Sigma), 10 μg/ml aprotinin (Sigma), 1 mM phenylmethylsulfonyl fluoride (Sigma), 50 mM NaF (Sigma), 0.2% dodecyl β-d-maltoside (ThermoFisher Scientific), and 20 mM Tris (pH 7.5, Fisher Scientific) as described previously [46]. The soluble extracts were then subjected to CDK4/6 immunoprecipitation. Briefly, 500 μg protein lysate was incubated with 4 μg IgG (Santa Cruz Biotech) or anti-CDK4/6 antibodies (Santa Cruz Biotech) at 4 °C overnight, followed by addition of 50 μl of agarose protein A/G beads (Santa Cruz Biotech). Bead-bound immunoprecipitates were washed and collected by centrifugation. Immunoprecipitates were added to a protein kinase reaction buffer containing 20 μM ATP (MP Biochemicals) and myelin basic protein (Millipore, Burlington, MA) as substrate and incubated at 30 °C for 30 min. The ADP-Glo™ Kinase Assay (promega, Madison, WI) kit was then used to measure kinase activity. Following termination of reaction, western blot analysis was performed to determine the level of inmmunoprecipitated proteins. Relative kinase activity was calculated by normalizing the kinase activity (luminescence) to the amount of immunoprecipitated protein (densitometry of CDK4/6 signal). While CDK4 and CDK6 kinase assays were performed separately for each sample, we combined the data to generate cumulative CDK4/6 activity. Percentage relative activity was calculated by setting the values obtained for the control (untreated) samples as baseline.
2.10. Cellular Thermal Shift Assay (CETSA)
Thermal shift assays were carried out using frozen kidney tissues (vehicle or ribociclib) using a modified version of a recently described method [49]. Renal cortical tissues were thawed and homogenized in cold PBS followed by 3 cycles of freeze-thawing using liquid nitrogen. Tissue lysates were separated from the cellular debris by centrifugation at 20,000 × g for 30 min at 4 °C. The tissue lysates were diluted with PBS containing protease inhibitors, divided into 50 μl aliquots and heated at different temperatures. This was followed by centrifugation at 20,000 × g for 30 min at 4 °C in order to separate the soluble fractions from precipitates. The soluble fraction was examined by western blot analysis using CDK4/6 antibodies followed by densitometric analysis and calculation of relative protein stability as compared to band intensities of the lowest temperature, β-actin was used as a negative control and we found no effect of ribociclib in the thermal shift assays for β-actin.
2.11. Statistical analysis
Data in all the graphs are presented as mean with s.e.m, unless stated otherwise. Statistical calculations were carried our using GraphPad Prism (San Diego, CA). p < 0.05 was considered as statistically significant. To calculate statistical significance between two groups, two-tailed unpaired Student’s t test or Mann-Whitney U test was performed. One-way ANOVA followed by Tukey’s or Dunnett’s multiple-comparisons test was used for comparisons among three or more groups.
3. Results
3.1. Ribociclib inhibits cisplatin-associated activation of CDK4/6 kinases.
To determine the pharmacological underpinnings of the protective effects of CDK4/6 inhibition during cisplatin-associated kidney injury, we initially sought to examine CDK4/6 kinase activity and inhibitor-target protein engagement in vivo. For these studies, we used a well-characterized mouse model of cisplatin-associated kidney injury [25], where a single intraperitoneal injection results in severe AKI after 72 h. As shown in Fig. 1a, we administered vehicle or ribociclib (150 mg/kg) by oral gavage, followed by intraperitoneal cisplatin injection (30 mg/kg) four hour later and subsequent examined renal function up to three days. We used ribociclib for these studies since it provided better renal protective and overall survival benefits than palbociclib at a similar dose of 150 mg/kg [41]. Consistent with studies [41] in FVB/NJ mice, ribociclib also provided significant protection from cisplatin associated kidney injury in C57BL/6J mice as seen with physiological (blood urea nitrogen and creatinine) and histological (H&E staining) analysis of kidney structure and function (Fig. 1b–d). Supporting our previous study [41] we also found a distinct increase in Rb1 phosphorylation (marker of CDK4/6 activation) in renal cortical tissues during the early phase of AKI (Fig. 1e). Importantly, ribociclib treatment significantly inhibited CDK4/6 kinase activity as shown by indirect (Rb1 phosphorylation) and direct (kinase assays) methods (Fig. 1e–f). We then used cellular thermal shift assays (CETSA) [49] to probe drug engagement (ribociclib) with target proteins (CDK4/6) in vivo. CETSA is based on the principle that drug binding can alter the thermal stability of target protein/s [49]. The observed changes in the thermal stability of a protein could be due to direct drug binding, drug-induced conformational changes, or drug-induced effects on post-translational modifications such as phosphorylation. CETSA assays using kidney lysates from vehicle and ribociclib treated mice showed that ribociclib increased the thermal stability (ATm describes the difference between the ribociclib treatment and control melting temperatures) of its main targets, namely CDK4 and CDK6 kinases (Fig. 1g–h). Altogether, these data support CDK4/6 target engagement and inhibition by ribociclib in vivo.
Fig. 1.
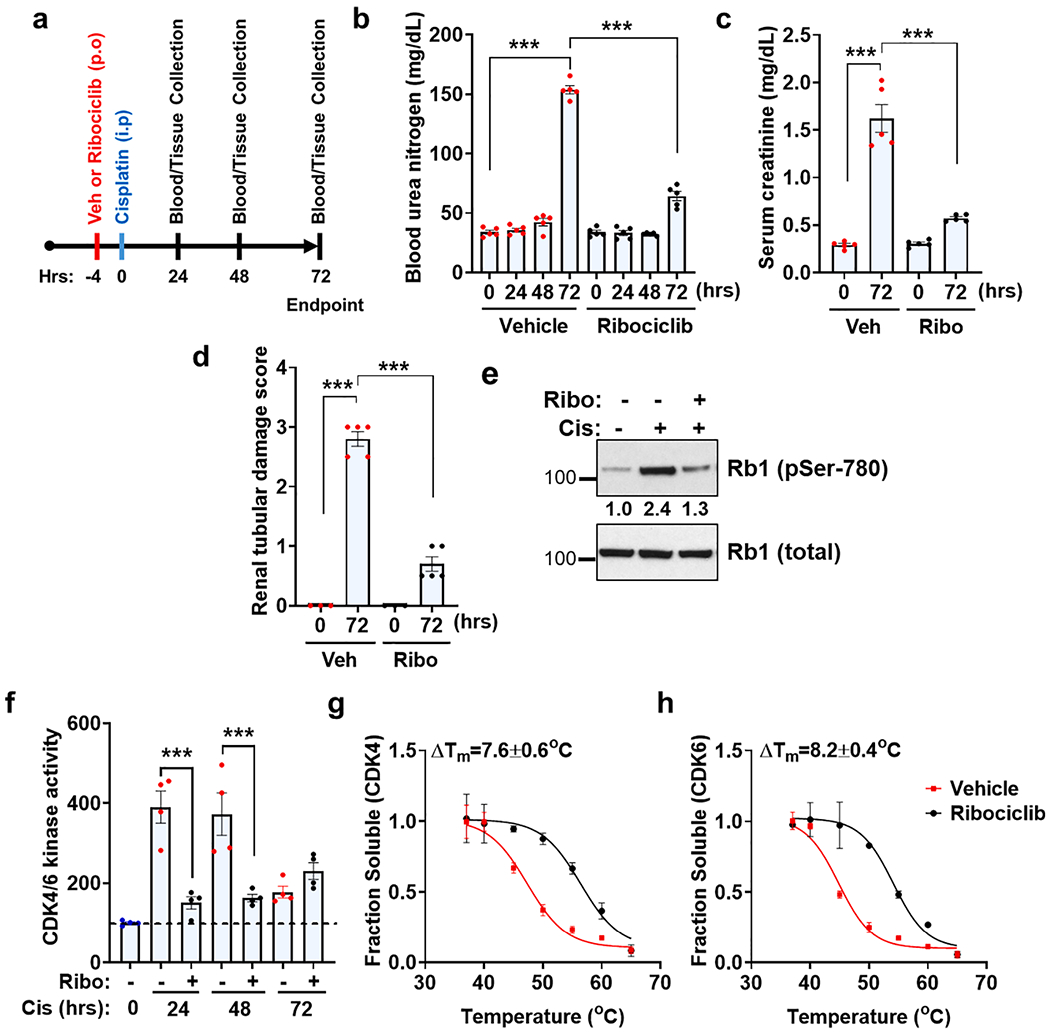
Ribociclib inhibits CDK4/6 activity and mitigates cisplatin-associated kidney injury. (a) Schematic representation of experimental treatment strategy. Age-matched male (8–12 weeks) C57BL/6 mice were administered a single oral dose of vehicle (citrate buffer) or ribociclib (150 mg/kg) followed by a single intraperitoneal injection of cisplatin (30 mg/kg) four hours later. (b) Blood urea nitrogen (c) Serum creatinine (d) Renal histological analysis (H&E) showed that ribociclib administration confers protection from cisplatin-associated AKI. Data (b-d) are presented as individual data points (n = 5 biologically independent samples), from one out of three independent experiments, all producing similar results. (e) Representative western blots showing ribociclib mediated suppression of cisplatin-associated Rb1 phosphorylation. Renal tissues were prepared 24 h post-cisplatin injection. (f) CDK4 and CDK6 proteins were immuno-precipitated from the kidneys of control and cisplatin treated mice, followed by in vitro kinase assays. The graphs represent data from a single experiment (n = 4 biologically independent samples), from one out of three independent experiments, all producing similar results. (g-h) Cellular thermal shift assay (CETSA) were carried out to identify drug engagement with target proteins in renal tissues 24 h post-cisplatin treatment. Thermal denaturation curves for CDK4 (g) and CDK6 (h) showed thermal stabilization upon ribociclib treatment in vivo. Data (g-h) are presented as mean (n = 3 biologically independent samples), from one out of three independent experiments, all producing similar results. In all the bar graphs, experimental values are presented as mean ± s.e.m. The height of error bar = 1 s.e. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparisons test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
3.2. Cell cycle dynamics of renal tubular epithelial cells during cisplatin-associated AKI
We then investigated the effect of CDK4/6 inhibition on RTEC cell cycle dynamics. Instead of staining for cell cycle markers such as PCNA (proliferating cell nuclear antigen) [41], cell cycle profile was directly inspected by flow cytometry based analysis of cellular DNA content using methods modified from our previous study [47]. To this end, we utilized a recently described method [48] that is based on EDTA (Ethylene diamine tetra acetic acid) mediated dispersal of tissues instead of collagenase-dependent enzymatic methods [50]. Cells were isolated from the renal cortical tissues of the kidneys of control and cisplatin treated mice followed by examination of percentage of cells in various cell cycle phases (Fig. 2a). It is anticipated that approximately 80% cells that are being analyzed are renal tubular epithelial cells. For cell cycle analysis, we used HEK293 cells and murine liver cells as positive controls for actively cycling and polyploid cells respectively (Fig. 2b). As expected, most of the RTECs in normal adult mice (Fig. 2b and Table 1) were in a quiescent phase (G0/G1 with 2n DNA). Interestingly, we found that cisplatin treatment (alone or in combination with ribociclib) did not significantly alter the cell cycle dynamics of RTECs, with most cells still present in the G0/G1 phase (Fig. 2b and Table 1). The almost complete absence of cells in the S-phase was likely not due to issues with our cell isolation or staining method because in contrast to cisplatin treated mice, mice challenged with ischemia and rhabdomyolysis (Fig. 2c) showed a clear increase in S phase cells (Fig. 2d–e and Table 1). Along with S-phase cells, we also detected a distinct Sub-G1 (likely apoptotic) population in the kidneys of ischemic and rhabdomyolysis treated mice. The S-phase data (Fig. 2e) is not entirely unexpected because cisplatin-mediated DNA damage response is anticipated to activate cell cycle arrest, so that the cells with damage DNA do not undergo cell division. These results also show that PCNA induction observed in all the three mouse models of AKI (Fig. 2f), is more likely a marker of G1 activation rather than S-phase entry. Based on these results, we propose that cisplatin-associated CDK4/6 activation results in G1 activation, however most cells do not enter the S-phase, likely due to the activation of the DNA damage response and robust induction of cyclin-dependent kinase inhibitors (CKI) such as p21 (Fig. 2g) [33]. BrdU incorporation experiments also did not detect any S-phase entry in cisplatin treated tissues (Fig. 2h–i). These results suggest that RTECs are not undergoing active DNA replication during cisplatin-associated AKI and hence the renal protective effects of CDK4/6 inhibition are not likely due to block in S phase entry, but may be related to a block in G0-to-G1 activation.
Fig. 2.

In vivo cell cycle dynamics during cisplatin-associated kidney injury. (a) Schematic representation of procedure used for isolation, staining and cell cycle analysis of cells from renal tissues. (b) Age-matched male (8–12 weeks) C57BL/6 mice were administered a single oral dose of vehicle (citrate buffer) or ribociclib (150 mg/kg) followed by a single intraperitoneal injection of cisplatin (30 mg/kg) four hours later. Renal tissues were collected at indicated time-points followed by cell cycle analysis based on measurement of cellular DNA content (DAPI staining). These results indicated that RTEC cell cycle dynamics did not change during cisplatin treatment, with most cells in the G0/G1 phase with 2n DNA. The red arrow highlights the S phase population. (c) Bilateral renal ischemia was induced in male wild-type (C57BL/6) mice for 30 min followed by reperfusion, while rhabdomyolysis was induced in male wild-type (C57BL/6) mice by glycerol injection (7.5 ml/kg 50% glycerol) in the hind-leg muscles followed by measurement of renal function (blood urea nitrogen). (d) Renal tissues were collected at indicated time-points from mice that underwent bilateral renal ischemia or glycerol injection, followed by cell cycle analysis based on measurement of cellular DNA content (DAPI staining). The red arrow and blue asterisk highlights the S-phase and Sub-G1 population. Unlike cisplatin-associated kidney injury, kidneys from mice with ischemia and rhabdomyolysis showed a distinct injury induced population of S-phase (proliferating) and Sub-G1 phase (apoptotic) cells. (e) Graphical representation of total percentage of S phase cells in the normal and injured kidneys. (f) Representative blots showing similar CDK4/6 activation (phospho-Rb1), G1 activation (PCNA), and renal injury (NGAL) in the kidneys of mice undergoing cisplatin, ischemia and rhabdomyolysis-associated kidney injury. (g) qPCR analysis of mRNA expression indicated a robust p21 induction, while expression of cyclins were not changed during cisplatin nephrotoxicity (72 h). (h-i) Injury induced S-phase entry and DNA replication was measured in control, ischemic, and cisplatin treated mice by BrdU incorporation assay. BrdU positive RTECs were counted in control and injured kidneys by confocal microscopy. The graph represents cumulative data from two independent experiment (n = 5). Representative images show the presence of BrdU positive cells in the ischemic kidneys. The scale bar represents 100 μm and the images were taken at 60X magnification. The flow cytometry data (b & d) and graphs (c & e) represent data from one out of three independent experiments, all producing similar results. In all the bar graphs, experimental values are presented as mean ± s.e.m. The height of error bar = 1 s.e. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparisons test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
Table 1.
Cell cycle dynamics during acute kidney injury. Flow cytometry based cell cycle analysis (DAPI) of renal tissues was carried out in three distinct models of acute kidney injury, namely cisplatin, ischemia and rhabdomyolysis-associated AKI. A distinct increase in S-phase cell population was only observed in ischemic and rhabdomyolysis-associated kidney injury. Cisplatin treatment (in the presence or absence or ribociclib) did not alter the baseline cell cycle profile. Data (Mean ± SD, n = 3) presented in the table is from a single representative experiment. Similar results were obtained in three independent experiments.
| Cisplatin |
|||||||
|---|---|---|---|---|---|---|---|
| Vehicle |
Ribociclib |
||||||
| Hours | 0 | 24 | 48 | 72 | 0 | 24 | 48 |
| Sub G1 | – | – | – | – | – | – | – |
| G0/G1 | 84.7 ± 0.61 | 85.67 ± 0.20 | 86.57 ± 0.15 | 83 ± 0.62 | 84.7 ± 0.61 | 85.47 ± 1.55 | 85.1 ± 1.23 |
| S | 1.58 ± 0.10 | 1.53 ± 0.22 | 1.72 ± 0.12 | 1.86 ± 0.09 | 1.58 ± 0.10 | 1.43 ± 0.05 | 1.7 ± 0.06 |
| G2/M | 13.40 ± 0.67 | 12.97 ± 0.62 | 12.33 ± 0.46 | 14.70 ± 0.80 | 13.40 ± 0.67 | 13.23 ± 1.53 | 13.50 ± 0.90 |
| Cisplatin |
Ischemia | Rhabdomyolysis | |||||
| Ribociclib |
|||||||
| Hours | 72 | 0 | 24 | 48 | 0 | 24 | 48 |
| Sub G1 | – | – | 6.4 ± 0.25 | 5.72 ± 0.22 | – | 7.06 ± 0.99 | 12.37 ± 1.31 |
| G0/G1 | 83.03 ± 0.44 | 85.6 ± 0.15 | 75.53 ± 0.49 | 74.03 ± 0.15 | 85.67 ± 0.15 | 75.17 ± 0.75 | 66.47 ± 1.56 |
| S | 1.78 ± 0.29 | 1.66 ± 0.04 | 5.18 ± 0.19 | 7.39 ± 0.13 | 1.77 ± 0.12 | 7.19 ± 0.38 | 8.95 ± 0.26 |
| G2/M | 14.47 ± 0.69 | 12.6 ± 0.15 | 11.73 ± 0.41 | 13.53 ± 0.22 | 12.27 ± 0.48 | 11.2 ± 0.42 | 11.23 ± 0.20 |
3.3. In vivo siRNA-mediated Rb1 knock-down does not affect the severity of cisplatin nephrotoxicity.
Once we had determined the cell cycle dynamics during cisplatin-associated AKI, next we sought to examine the functional role of Rb1 pathway in the renal protective effects of CDK4/6 inhibitors. To this end, we initially sought to knock-down Rb1 in RTECs in vivo using a previously established hydrodynamic siRNA injection approach [46,51]. As shown in the Fig. 3a–b, two distinct siRNAs targeting the Rb1 gene were able to knock-down Rb1 levels by approximately 75% as compared to the vehicle and non-specific siRNA group. Subsequent experiments (Fig. 3c–f) showed that Rb1 knock-down did not affect the extent of cisplatin-associated AKI as seen with blood urea nitrogen (BUN), serum creatinine, and histological analysis (H&E staining and renal damage score). These results suggest that Rb1 is possibly not crucial for normal renal function in adult mice. Moreover, since CDK4/6 activation during cisplatin treatment leads to Rb1 phosphorylation and inactivation, it is likely that most of the Rb1 protein is present in an inactivated (phosphorylated) state under stress conditions and hence its knock-down does not influence the severity of AKI.
Fig. 3.
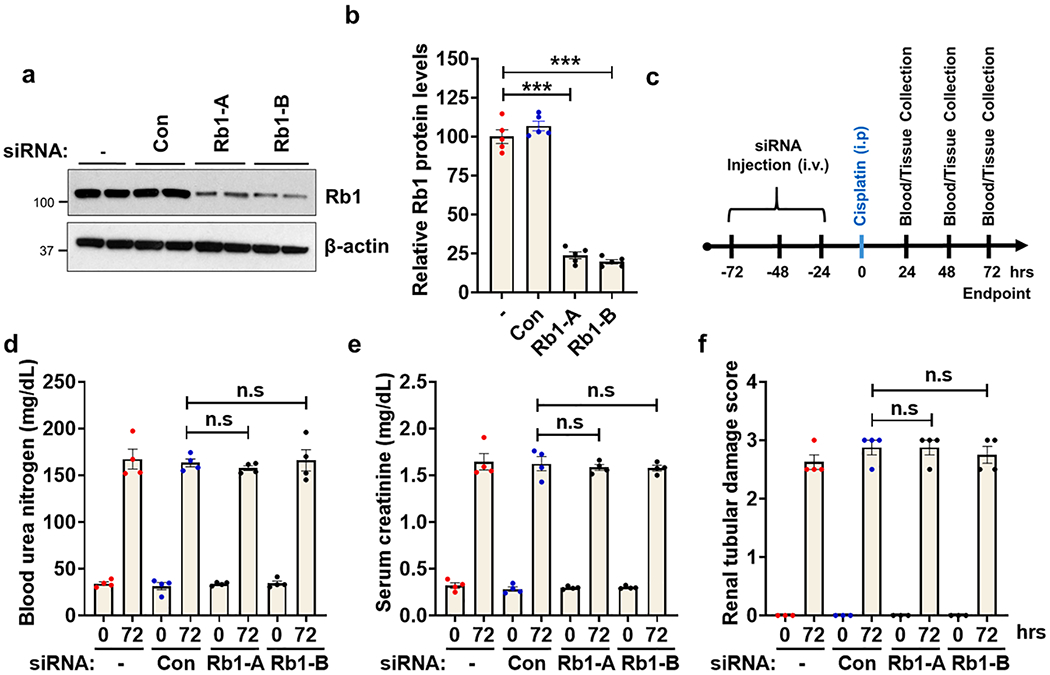
In vivo siRNA-mediated Rb1 knock-down does not influence cisplatin-associated kidney injury. Age-matched male (8–12 weeks) C57BL/6 mice were administered with three once-daily intravenous injections (hydrodynamic) of control (non-specific) or Rb1 (Rb1-A and Rb1-B) targeting siRNAs (25 μg in 0.5 ml of PBS). In one group (-) 0.5 ml of PBS was injected. Two days after the last injection, renal tissues were collected for western blot analysis of Rb1 protein. (a) Representative blots and (b) densitometric analysis show that both the Rb1 targeting siRNAs were able to knock-down Rb1 proteins levels by approximately 75%. Blots are representative of three independent experiments, all producing similar results. (c) Schematic representation of experimental treatment strategy. Briefly, mice were injected with three doses of siRNA to knock-down Rb1 gene followed by cisplatin injection and examination of renal injury. (d) Blood urea nitrogen (e) Serum creatinine and (f) Renal histological analysis show that Rb1 knock-down did not influence cisplatin-associated kidney injury. Data presented in d-f are from one (n = 4 biological samples) out of three experiments, all producing similar results. In all the bar graphs, experimental values are presented as mean ± s.e.m. The height of error bar = 1 s.e. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparisons test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. Rb1 is essential for renal protective effects of CDK4/6 inhibition.
Subsequently we carried out experiments to determine if ribociclib mediated renal protection is influenced by Rb1 knock-down. Strikingly, we found that the control siRNA mice were protected from cisplatin-associated AKI by ribociclib administration, however this effect was completely absent in the mice with Rb1 knock-down (Fig. 4a–d). Protein kinase assays indicated similar CDK4/6 inhibition in control and Rb1 siRNA groups (Fig. 4e). Furthermore, western blot analysis showed that CDK4/6 inhibition prevented the increase in both PCNA (G1 activation) and neutrophil gelatinase-associated lipocalin (NGAL, kidney injury marker) induction only in the control mice (Fig. 4f). These results suggested that inhibition of the CDK4/6 mediated Rb1 phosphorylation is critical for the renal protective effects of CDK4/6 inhibitors. Therefore, we hypothesized that ribociclib-mediated CDK4/6 inhibition results in the maintenance of hypo-phosphorylated Rb1, which might play a protective role during cisplatin-associated AKI.
Fig. 4.
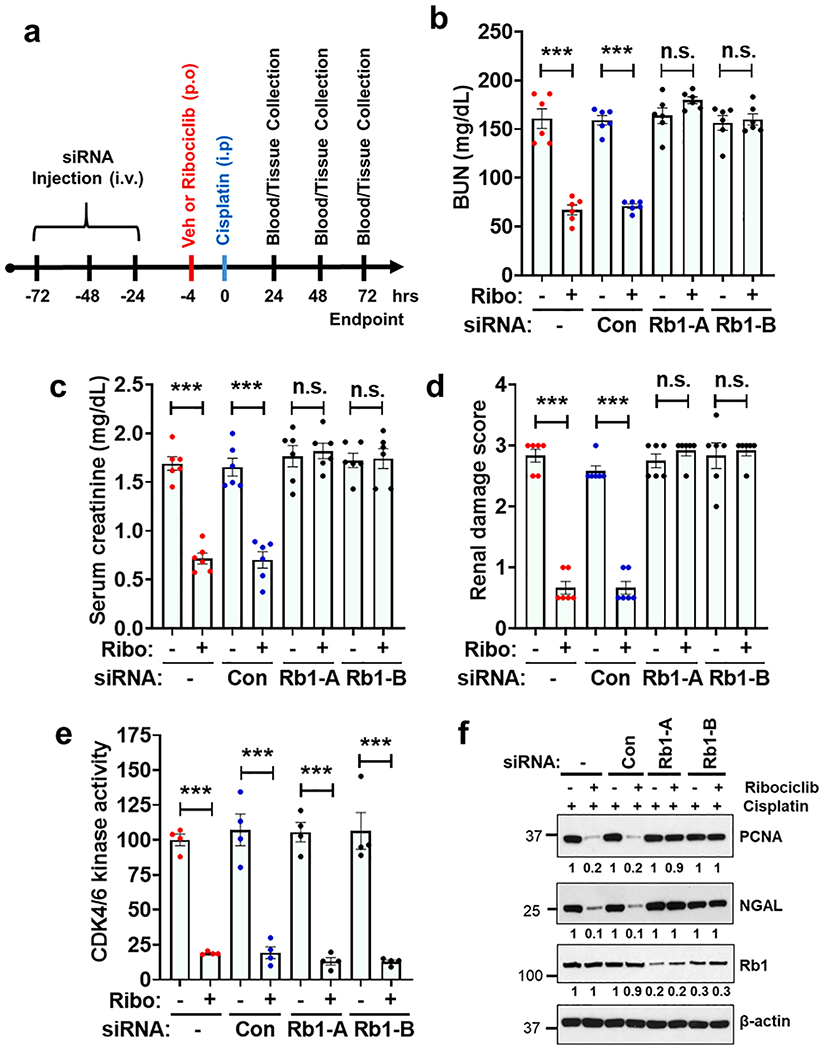
Ribociclib mediated renal protective effects are Rb1 dependent. (a) Age-matched male (8–12 weeks) C57BL/6 mice were administered with three once-daily intravenous injections (hydrodynamic) of control (non-specific) or Rb1 (Rb1-A and Rb1-B) targeting siRNAs (25 μg in 0.5 ml of PBS). In one group (-) 0.5 ml of PBS was injected. One day later oral administration of either vehicle (citrate buffer) or ribociclib (150 mg/kg) was carried out, followed by a single intraperitoneal injection of cisplatin (30 mg/kg) four hours later. Renal function and histology was examined from 1 to 3 days post-cisplatin injection. (b) Blood urea nitrogen (c) Serum creatinine and (d) Renal histological analysis show that while Rb1 knock-down did not influence cisplatin-associated kidney injury, it however abolished the renal protective effect of ribociclib. The graphs (b-d) represent cumulative data from two out of four independent experiments (n = 6 biologically independent samples), all producing similar results. (e) CDK4 and CDK6 proteins were immuno-precipitated from renal tissues followed by in vitro kinase assays. The graph represents data from a single experiment (n = 4 biologically independent samples), from one out of three independent experiments, all producing similar results. (f) Western blot analysis of renal tissues (72 h post-cisplatin injection) showed that ribociclib treatment can ameliorate cisplatin associated PCNA and NGAL induction in the control siRNA injected mice but not the Rb1 siRNA injected mice. Blots are representative of four independent experiments. In all the bar graphs, experimental values are presented as mean ± s.e.m. The height of error bar = 1 s.e. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Dunnett’s multiple-comparisons test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
3.5. Characterization of renal tubular epithelial cell-specific Rb1 knockout mice
To further confirm the results obtained with in vivo siRNA-mediated Rb1 knock-down, we next sought to generate a renal tubule specific Rb1 knockout mice. To this end, we crossed the Rb1 floxed mice with the Ggt1-Cre mice. In Ggt1-Cre mice, Cre recombinase is expressed in renal tubular epithelial cells 7–10 days after birth, such that Cre induction occurs after the completion of renal development [45]. Mutant mice were identified by PCR analysis of genomic DNA and Rb1 gene ablation was confirmed by immunoblot analysis (Fig. 5a–b). The Rb1PT−/− (RTEC-specific Rb1 knockout) mice developed normally, were fertile, and exhibited no obvious phenotypes. Specifically, body weight, renal function and histology were not different in the Rb1PT−/− mice as compared with the control littermates (Fig. 5c–d). Rb1PT−/− mice were also monitored for a year and we did not observe any obvious phenotypic differences. These results suggest that renal function was not affected by Rb1 deficiency in the RTECs under normal conditions. Littermate control and Rb1PT−/− mice were then challenged with a single dose of intra-peritoneal cisplatin injection, followed by assessment of renal function and histology. Concordant with the in vivo siRNA results, we found that RTEC-specific Rb1 knockout did not influence cisplatin-associated AKI (Fig. 5e–f). Expression analysis of cell cycle regulatory proteins including Rb-related proteins pi 07 (RBL1) and pl30 (RBL2) also showed no significant difference in the kidneys of WT and Rb1PT−/− mice (Fig. 5g). The protein expression of PCNA, NGAL, RBL2, and E2F1 was significantly and similarly increased in both WT and Rb1PT−/− mice during cisplatin-associated kidney injury.
Fig. 5.

Rb1 gene deletion in tubular epithelial cells does not influence cisplatin-associated kidney injury. To generate mice with RTEC specific Rb1 knockout, Ggt1-Cre mice were crossed with Rb1 floxed mice. (a) Representative genotyping data. (b) Renal cortical tissues from 8 week old littermate control and Rb1 conditional knockout mice (indicated by Rb1PT−/−) were examined by western blot analysis. Densitometric analysis showed a 80% reduction in Rb1 protein expression in renal tissues. Blots are representative of at least three independent experiment. (c-d) Under baseline conditions, the control and If RB1PT−/− mice did not show any differences in body weight or renal function up to 8 weeks of age. (e-f) Control and Rb1-deifient littermate male mice were treated with cisplatin (30 mg/kg) followed by examination of renal function at 72 h. Measurement of blood urea nitrogen and examination of renal histology showed that cisplatin-associated kidney damage was similar in both the control and RB1PT−/− mice. Data in graph (e) is cumulative of three independent experiments (n = 5–10 biologically independent samples). Histological pictures (40× magnification and black scale bar = 100 μm) are representative of three independent samples. Asterisks show damaged renal tubules. (g) Western blot analysis of renal tissues (72 h post-cisplatin injection) showed that cisplatin associated PCNA and NGAL induction is not influenced by Rb1 gene ablation in tubular epithelial cells. Blots are representative of two independent experiments. In all the bar graphs, experimental values are presented as mean ± s.e.m. The height of error bar = 1 s.e. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparisons test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, p < 0.001.
3.6. Rb1 is crucial for ribociclib mediated renal protection during cisplatin-associated AKI
The preceding data suggested that Rb1PT−/− mice are a suitable model to examine the pharmacological basis of CDK4/6 inhibitors during AKI progression. So, next we administered a single oral dose of ribociclib (150 mg/kg) followed by cisplatin injection in littermate control and Rb1PT−/− mice. Similar to our previous results, ribociclib treatment remarkably suppressed cisplatin-induced AKI as seen with measurement of blood urea nitrogen, serum creatinine, and histological analysis in the control (Rb1 proficient) mice (Fig. 6a–c). Tissue histological examination further supported that ribociclib prevents cisplatin-induced renal injury in the control mice (Fig. 6d). However, ribociclib treatment had no protective effects in the Rb1PT−/− mice. Interestingly, ribociclib treatment seemed to slightly increase blood urea nitrogen and serum creatinine in the Rb1PT−/− mice as compared to the vehicle group. Furthermore, as compared to suppression in control mice, protein expression of PCNA and E2F1 remained elevated in ribociclib-treated Rb1PT−/− mice (Fig. 6e–f). These findings indicate that the renal protective effect of ribociclib is dependent on a functional CDK4/6-Rb1 pathway.
Fig. 6.

Ribociclib mediated renal protective effects are abolished in RTEC-specific Rb1 deficient mice. Control and Rb1-deifient littermate male mice (8–12 weeks old) were administered with either vehicle (-) or ribociclib (150 mg/kg) followed by a single intraperitoneal injection of cisplatin (30 mg/kg) four hours later. Renal function and histology was examined from 1 to 3 days post-cisplatin injection. (a) Blood urea nitrogen (b) Serum creatinine and (c-d) Renal histological analysis show that while Rb1 deficiency did not influence cisplatin-associated kidney injury, it however abolished the renal protective effect of ribociclib. The graphs (b-d) represent data from three independent experiments (n = 4 biologically independent samples), all producing similar results. (e-f) Western blot analysis of renal tissues (72 h post-cisplatin injection) showed that ribociclib treatment can ameliorate cisplatin associated PCNA, NGAL, and E2F1 induction in the control mice but not the Rb1 deficient mice. Blots are representative of three independent experiments, data from which was pooled for densitometric analysis shown in the graph (** indicates comparison with cisplatin treated control group). In all the bar graphs, experimental values are presented as mean ± s.e.m. The height of error bar = 1 s.e. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparisons test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
4. Discussion
Cyclin-dependent kinases (CDKs) and their cyclin partners are critical drivers of cell cycle progression in embryonic and adult tissues [40]. The CDK4/6/cyclin D complex is essential for G1/S phase transition and is responsible for phosphorylation of the retinoblastoma-1 (Rb1) protein, the activation of the E2F family of transcription factors, and the subsequent initiation of DNA replication [7,52]. However, aberrant G1/S activation can result in cell death activation through E2F1 dependent and independent mechanisms [1], Here we report that during cisplatin-associated acute kidney injury, CDK4/6 kinase activation and Rb1 phosphorylation are observed, however, the renal tubular epithelial cells do not enter the S phase. Importantly, ribociclib-mediated CDK4/6 inhibition mitigated AKI in an Rb1-dependent manner.
The mammalian cell cycle is a tightly controlled and regulated process that ensures faithful DNA replication and cell division. Cell cycle entry is coordinated by multiple pathways including growth factors and signaling networks that monitor genomic integrity to ascertain the absence of DNA damage [40,53]. Subsequent progression through G0/G1, S, G2 and M phases is regulated by several evolutionarily conserved CDKs. In most adult tissues, cells are in a cell cycle arrested state termed GO phase, which can be either transient (quiescence) or permanent (terminal differentiation or senescence) [2]. As part of normal cell turnover or in response to injury, quiescent cells can be triggered to re-enter the cell cycle through stimulation by mitogenic factors. These mitogenic factors activate the CDK4/6 pathway to drive cell cycle progression from GO or G1 phase into S phase, in which DNA replication occurs. Interestingly, a majority of renal tubular epithelial cells are quiescent under normal conditions and are thought to enter the cell cycle in response to ischemic and other injuries [16]. Through mechanisms that are not completely understood, injury induced cell cycle entry contributes to regeneration in the kidney under appropriate conditions [16] and fibrosis when this process is dysregulated [35].
While cell cycle activation in RTECs is a common phenomenon in response to injury, cisplatin-associated AKI might differ from ischemic and other forms of renal damage [16,35]. Cisplatin is known to preferentially accumulate in RTECs, in part through transporter mediated mechanisms [19] leading to activation of DNA damage response [26]. One of the major consequences of the activation of DNA damage response is induction of cell cycle arrest [53]. Our previous studies [41] and current work suggests that during cisplatin-associated AKI, CDK4/6 activation and Rb1 phosphorylation occurs within 24 h post-treatment; however, we could not detect any cells in the S phase up to 72 h post cisplatin-injection. Interestingly, PCNA induction occurs within 24 h, however we believe that this is most likely a result of G1-activation and not S phase entry per se. Based on these results we propose that CDK4/6 activation during the acute phase of cisplatin-associated AKI is marked by Rb1 phosphorylation, and G1 activation, but not S phase entry. While we do not observe a marked increase in S, G2/M or polyploid [54] cells during the 72 h period, it is likely that these cells may enter the cell cycle at later time points, as shown in a recent study [55], where cell cycle dynamics were examined at 96 h post cisplatin-injection.
These results raise the question of the mechanisms underlying the renal protective effects of CDK4/6 inhibitors. Interestingly, administration of CDK4/6 inhibitors has been shown to protect normal bone marrow and skin stem cells from the effects of chemo- and radiotherapy, by reducing the proliferation of progenitor cells [56–58]. However, as compared to the cycling progenitor cells in the bone marrow and skin, most of the RTECs in the kidney are quiescent under normal conditions and we did not detect any increase in the number of cells in the S phase during cisplatin-associated AKI. One possible explanation is that CDK4/6 activation and Rb1 phosphorylation during cisplatin-associated AKI leads to activation of E2F1, which might contribute to cell-death activation instead of S-phase initiation. Indeed, previous studies have clearly shown that E2F1 plays a pathogenic role during cisplatin-associated AKI [36]. We speculate that irrespective of the underlying etiology, CDK4/6 pathway is activated in RTECs as part of a regenerative process. However during cisplatin-associated AKI, G1 activation (CDK4/6 activation and Rb1 phosphorylation) and DNA damage induced cell cycle arrest occurs concurrently, which might tilt the balance from S-phase entry towards pro-cell-death signaling.
While future studies are required to understand how CDK4/6 activation leads to renal dysfunction and AKI, our study provides strong evidence that the renal protective effects of CDK4/6 inhibitors are dependent on the presence of functional Rb1 in RTECs. Both siRNA-mediated Rb1 knock-down and RTEC-specific Rb1 knockout abrogated the renal protective effects of ribociclib. Importantly, RTEC-specific Rb1 knockout did not lead to an obvious phenotype under normal conditions. Although Rb1 is a tumor suppressor, its loss-of-function did not cause any obvious cell cycle defects or malignancy in the conditional knockout mice. The possibility of functional compensation by other retinoblastoma family members remains a possibility. However, multiple gene mutations might be required to observe a cell cycle specific defect as observed with renal epithelium-specific deletion of Vhl, Trp53 and Rb1, which causes renal cell carcinoma [59]. RTEC-specific Rb1 deletion also did not influence the severity of cisplatin-associated AKI. One possible reason could be that in the wild type mice, during cisplatin-associated AKI, Rb1 is inactivated through phosphorylation by CDK4/6 kinases and hence, the influence of Rb1 knockout on cisplatin-associated AKI is minimal. However, when wild type mice are treated with CDK4/6 inhibitors, Rb1 phosphorylation and inactivation is inhibited and under these conditions, hypo-phosphorylated Rb1 plays a protective role. As a result, the phenotype in Rb1 deficient mice is only observed when CDK4/6 kinases are inhibited (Fig. 7).
Fig. 7.

Proposed Model. We propose that cisplatin-associated cellular injury induces CDK4/6 activation in renal tubular epithelial cells which is manifested as Rb1 phosphorylation. Rb1 phosphorylation and inactivation under stress conditions causes RTEC dysfunction, cell death and kidney injury likely through E2F1 dependent and independent manner. When the mice are treated with ribociclib, CDK4/6 activation is reduced, resulting in higher levels of hypo-phosphorylated Rb1, which results in epithelial cell protection and mitigation of kidney injury. In the Rb1 deficient mice, CDK4/6 is inactivated, but hypo-phosphorylated Rb1 is absent and hence no renal protective effects are observed.
Our study raises several questions that merit future investigations. Firstly, are the mechanisms underlying the renal protective effects of CDK4/6 inhibition distinct between cisplatin and ischemia-associated AKI? Secondly, while CDK4/6 inhibitors mitigate AKI, do they influence the regenerative processes important for recovery of renal function post-injury? Further in-depth studies in various short (single-dose) and long-term (multiple-dose) models of cisplatin-associated AKI could reveal RTEC cell cycle dynamics during AKI and provide better understanding of how to potentially use CDK4/6 inhibitors as renal therapeutics. Collectively, our findings provide insights into the cell cycle dynamics during cisplatin-associated acute kidney injury, illuminates the role of Rb1 in renal injury, and uncovers the pharmacological basis of the renal protective effects of CDK4/6 inhibitors.
Acknowledgements
This study was supported by the American Heart Association Scientist Development Grant (17SDG33440070) and funds from the Ohio State University Comprehensive Cancer Center, and the Pelotonia foundation. Y.B. was supported by a postdoctoral fellowship from American Heart Association. We thank the Analytical Cytometry core facility (P30CA016058) at The Ohio State University Comprehensive Cancer Center and the Comparative Pathology and Mouse Phenotyping Shared Resource at the College of Veterinary Medicine for assistance with flow cytometry and histological analysis.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Pucci B, Kasten M, Giordano A, Cell cycle and apoptosis, Neoplasia 2 (4) (2000) 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nabel EG, CDKs and CKIs: molecular targets for tissue remodelling, Nat. Rev. Drug Discov 1 (8) (2002) 587–598. [DOI] [PubMed] [Google Scholar]
- [3].King KL, Cidlowski JA, Cell cycle regulation and apoptosis, Annu. Rev. Physiol 60 (1998) 601–617. [DOI] [PubMed] [Google Scholar]
- [4].Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, Greenberg ME, Orkin S, Nevins JR, Robinson ML, Leone G, The E2F1-3 transcription factors are essential for cellular proliferation, Nature 414 (6862) (2001) 457–462. [DOI] [PubMed] [Google Scholar]
- [5].Zebell SG, Dong X, Cell-cycle regulators and cell death in immunity, Cell Host Microbe 18 (4) (2015) 402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A, RBI dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy, Oncotarget 6 (20) (2015) 17873–17890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dyson NJ, RBI: a prototype tumor suppressor and an enigma, Genes Dev. 30 (13) (2016) 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, Srivastava D, Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration, Cell 173 (1) (2018) 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Herrup K, Yang Y, Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci 8 (5) (2007) 368–378. [DOI] [PubMed] [Google Scholar]
- [10].Ferenbach DA, Bonventre JV, Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD, Nat. Rev. Nephrol 11 (5) (2015) 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zuk A, Bonventre JV, Acute kidney injury, Annu. Rev. Med 67 (2016) 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Peerapornratana S, Manrique-Caballero CL, Gomez H, Kellum JA, Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment, Kidney Int. 96 (5) (2019) 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang Y, Bellomo R, Cardiac surgery-associated acute kidney injury: risk factors, pathophysiology and treatment, Nat. Rev. Nephrol 13 (11) (2017) 697–711. [DOI] [PubMed] [Google Scholar]
- [14].Li L, Huang L, Vergis AL, Ye H, Bajwa A, Narayan V, Stricter RM, Rosin DL, Okusa MD, IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury, J. Clin. Invest 120 (1) (2010) 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ramesh G, Reeves WB, TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity, J. Clin. Invest 110 (6) (2002) 835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chang-Panesso M, Kadyrov FF, Lalli M, Wu H, Ikeda S, Kefaloyianni E, Abdelmageed MM, Herrlich A, Kobayashi A, Humphreys BD, FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury, J. Clin. Invest 129 (12) (2019) 5501–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rosner MH, Perazella MA, Acute kidney injury in patients with cancer, N. Engl. J. Med 376 (18) (2017) 1770–1781. [DOI] [PubMed] [Google Scholar]
- [18].Pabla N, Murphy RF, Liu K, Dong Z, The copper transporter Ctrl contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity, Am. J. Physiol. Renal. Physiol 296 (3) (2009) F505–F511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A, Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity, Clin. Pharmacol. Ther 86 (4) (2009) 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ciarimboli G, Deuster D, Knief A, Sperling M, Holtkamp M, Edemir B, Pavenstadt H, Lanvers-Kaminsky C, Zehnhoff-Dinnesen A, Schinkel AH, Koepsell H, Jurgens H, Schlatter E, Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions, Am. J. Pathol 176 (3) (2010) 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pabla N, Dong Z, Cisplatin nephrotoxicity: mechanisms and renoprotective strategies, Kidney Int. 73 (9) (2008) 994–1007. [DOI] [PubMed] [Google Scholar]
- [22].Ramesh G, Reeves WB, p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice, Am. J. Physiol. Renal. Physiol 289 (1) (2005) F166–F174. [DOI] [PubMed] [Google Scholar]
- [23].Bolisetty S, Traylor A, Joseph R, Zarjou A, Agarwal A, Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury, Am. J. Physiol. Renal. Physiol 310 (5) (2016) F385–F394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Arany I, Megyesi JK, Kaneto H, Price PM, Safirstein RL, Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells, Am. J. Physiol. Renal. Physiol 287 (3) (2004) F543–F549. [DOI] [PubMed] [Google Scholar]
- [25].Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z, Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer, J. Clin. Invest 121 (7) (2011) 2709–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pabla N, Huang S, Mi QS, Daniel R, Dong Z, ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis, J. Biol. Chem 283 (10) (2008) 6572–6583. [DOI] [PubMed] [Google Scholar]
- [27].Cummings BS, Schnellmann RG, Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways, J. Pharmacol. Exp. Ther 302 (1) (2002) 8–17. [DOI] [PubMed] [Google Scholar]
- [28].Nowak G, Price PM, Schnellmann RG, Lack of a functional p21WAFl/CIPl gene accelerates caspase-independent apoptosis induced by cisplatin in renal cells, Am. J. Physiol. Renal. Physiol 285 (3) (2003) F440–F450. [DOI] [PubMed] [Google Scholar]
- [29].Funk JA, Schnellmann RG, Persistent disruption of mitochondrial homeostasis after acute kidney injury, Am. J. Physiol. Renal. Physiol 302 (7) (2012) F853–F864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z, Regulated cell death in AKI, J. Am. Soc. Nephrol 25 (12) (2014) 2689–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Megyesi J, Di Mari J, Udvarhelyi N, Price PM, Safirstein R, DNA synthesis is dissociated from the immediate-early gene response in the post-ischemic kidney, Kidney Int. 48 (5) (1995) 1451–1458. [DOI] [PubMed] [Google Scholar]
- [32].Megyesi J, Udvarhelyi N, Safirstein RL, Price PM, The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure, Am. J. Physiol 271 (6 Pt 2) (1996) F1211–F1216. [DOI] [PubMed] [Google Scholar]
- [33].Megyesi J, Safirstein RL, Price PM, Induction of p21WAFl/CIPl/SDIl in kidney tubule cells affects the course of cisplatin-induced acute renal failure, J. Clin. Invest 101 (4) (1998) 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Price PM, Megyesi J, Saf Irstein RL, Cell cycle regulation: repair and regeneration in acute renal failure, Kidney Int. 66 (2) (2004) 509–514. [DOI] [PubMed] [Google Scholar]
- [35].Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV, Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury, Nat. Med 16 (5) (2010) 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yu F, Megyesi J, Safirstein RL, Price PM, Involvement of the CDK2-E2F1 pathway in cisplatin cytotoxicity in vitro and in vivo, Am. J. Physiol. Renal. Physiol 293 (1) (2007) F52–F59. [DOI] [PubMed] [Google Scholar]
- [37].Price PM, Safirstein RL, Megyesi J, The cell cycle and acute kidney injury, Kidney Int. 76 (6) (2009) 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Killock D, CDK4/6 inhibitors prolong OS, Nat. Rev. Clin. Oncol 16 (12) (2019) 722. [DOI] [PubMed] [Google Scholar]
- [39].Asghar U, Witkiewicz AK, Turner NC, Knudsen ES, The history and future of targeting cyclin-dependent kinases in cancer therapy, Nat Rev Drug Discov 14 (2) (2015) 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sherr CJ, Beach D, Shapiro GI, Targeting CDK4 and CDK6: from discovery to therapy, Cancer Discov 6 (4) (2016) 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pabla N, Gibson AA, Buege M, Ong SS, Li L, Hu S, Du G, Sprowl JA, Vasilyeva A, Janke LJ, Schlatter E, Chen T, Ciarimboli G, Sparreboom A, Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions, Proc. Natl. Acad. Sci. USA 112 (16) (2015) 5231–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].DiRocco DP, Bisi J, Roberts P, Strum J, Wong KK, Sharpless N, Humphreys BD, CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury, Am. J. Physiol. Renal. Physiol 306 (4) (2014) F379–F388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES, Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure, Oncogene 29 (28) (2010) 4018–4032. [DOI] [PubMed] [Google Scholar]
- [44].Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL, Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts, Mol. Cancer Ther 3 (11) (2004) 1427–1438. [PubMed] [Google Scholar]
- [45].Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG, Evidence that fibroblasts derive from epithelium during tissue fibrosis, J. Clin. Invest 110 (3) (2002) 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sprowl JA, Ong SS, Gibson AA, Hu S, Du G, Lin W, Li L, Bharill S, Ness RA, Stecula A, Offer SM, Diasio RB, Nies AT, Schwab M, Cavaletti G, Schlatter E, Ciarimboli G, Schellens JH, Isacoff EY, Sali A, Chen T, Baker SD, Sparreboom A, Pabla N, A phosphotyrosine switch regulates organic cation transporters, Nat. Commun 7 (2016) 10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pabla N, Bhatt K, Dong Z, Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of Chkl that regulates cell cycle and DNA damage checkpoints, Proc. Natl. Acad. Sci. USA 109 (1) (2012) 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Heinlein C, Deppert W, Braithwaite AW, Speidel D, A rapid and optimization-free procedure allows the in vivo detection of subtle cell cycle and ploidy alterations in tissues by flow cytometry, Cell Cycle 9 (17) (2010) 3584–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P, Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay, Science 341 (6141) (2013) 84–87. [DOI] [PubMed] [Google Scholar]
- [50].Manolopoulou M, Matlock BK, Nlandu-Khodo S, Simmons AJ, Lau KS, Phillips-Mignemi M, Ivanova A, Alford CE, Flaherty DK, Gewin LS, Novel kidney dissociation protocol and image-based flow cytometry facilitate improved analysis of injured proximal tubules, Am. J. Physiol. Renal. Physiol 316 (5) (2019) F847–F855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hamar P, Song E, Kokeny G, Chen A, Ouyang N, Lieberman J, Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury, Proc. Natl. Acad. Sci. USA 101 (41) (2004) 14883–14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bertoli C, Skotheim JM, de Bruin RA, Control of cell cycle transcription during G1 and S phases, Nat. Rev. Mol. Cell Biol 14 (8) (2013) 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ciccia A, Elledge SJ, The DNA damage response: making it safe to play with knives, Mol. Cell 40 (2) (2010) 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lazzeri E, Angelotti ML, Peired A, Conte C, Marschner JA, Maggi L, Mazzinghi B, Lombardi D, Melica ME, Nardi S, Ronconi E, Sisti A, Antonelli G, Becherucci F, De Chiara L, Guevara RR, Burger A, Schaefer B, Annunziato F, Anders HJ, Lasagni L, Romagnani P, Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury, Nat. Commun 9 (1) (2018) 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kishi S, Brooks CR, Taguchi K, Ichimura T, Mori Y, Akinfolarin A, Gupta N, Galichon P, Elias BC, Suzuki T, Wang Q, Gewin L, Morizane R, Bonventre JV, Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses, J. Clin. Invest 129 (11) (2019) 4797–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, Ramsey MR, Jin J, Wong KK, Su L, Zhou D, Sharpless NE, Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition, J. Clin. Invest 120 (7) (2010) 2528–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].He S, Roberts PJ, Sorrentino JA, Bisi JE, Storrie-White H, Tiessen RG, Makhuli KM, Wargin WA, Tadema H, van Hoogdalem EJ, Strum JC, Malik R, Sharpless NE, Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion, Sci. Transl. Med 9 (387) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Purba TS, Ng’andu K, Brunken L, Smart E, Mitchell E, Hassan N, O’Brien A, Mellor C, Jackson J, Shahmalak A, Paus R, CDK4/6 inhibition mitigates stem cell damage in a novel model for taxane-induced alopecia, EMBO Mol. Med 11 (10) (2019) e11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Harlander S, Schonenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, Moch H, Wild PJ, Frew IJ, Combined mutation in Vh1, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice, Nat. Med 23 (7) (2017) 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]