

Abstract
Caveolin-1 (Cav-1) is a scaffolding protein and a major component of caveolae/lipid rafts. Previous reports have shown that endothelial dysfunction in Cav-1-deficient (Cav-1−/−) mice is mediated by elevated oxidative stress through endothelial nitric oxide synthase (eNOS) uncoupling and increased NADPH oxidase. Oxidant stress is the net balance of oxidant generation and scavenging, and the role of Cav-1 as a regulator of antioxidant enzymes in vascular tissue is poorly understood. Extracellular SOD (SOD3) is a copper (Cu)-containing enzyme that is secreted from vascular smooth muscle cells/fibroblasts and subsequently binds to the endothelial cells surface, where it scavenges extracellular and preserves endothelial function. SOD3 activity is dependent on Cu, supplied by the Cu transporter ATP7A, but whether Cav-1 regulates the ATP7A-SOD3 axis and its role in oxidative stress-mediated vascular dysfunction has not been studied. Here we show that the activity of SOD3, but not SOD1, was significantly decreased in Cav-1−/− vessels, which was rescued by re-expression of Cav-1 or Cu supplementation. Loss of Cav-1 reduced ATP7A protein, but not mRNA, and this was mediated by ubiquitination of ATP7A and proteasomal degradation. ATP7A bound to Cav-1 and was colocalized with SOD3 in caveolae/lipid rafts or perinucleus in vascular tissues or cells. Impaired endothelium-dependent vasorelaxation in Cav-1−/− mice was rescued by gene transfer of SOD3 or by ATP7A-overexpressing transgenic mice. These data reveal an unexpected role of Cav-1 in stabilizing ATP7A protein expression by preventing its ubiquitination and proteasomal degradation, thereby increasing SOD3 activity, which in turn protects against vascular oxidative stress-mediated endothelial dysfunction.
Keywords: ATP7A, caveolin-1, endothelial function, oxidative stress, SOD3
INTRODUCTION
Caveolin-1 (Cav-1) is a scaffolding protein and a major component of plasma membrane caveolae/lipid rafts (C/LR) and important for membrane trafficking, clustering of signaling molecules for efficient signal transduction, substrate transport, and endocytosis (8, 50). We and others have reported that Cav-1−/− mice have endothelial dysfunction (38, 57), aberrant angiogenesis (22, 33), and pulmonary hypertension (66) through eNOS hyperactivation and elevated oxidative stress. We have previously reported that Cav-1−/− mice exposed to hypoxia have increased NADPH oxidase (NOX2 and NOX4) expression and production and exacerbated pulmonary hypertension (6). Endothelial cell (EC)-specific Cav-1−/− mice exhibit impaired endothelium-dependent vasodilation and ischemia-induced angiogenesis via increased nitrosative stress (22). In disease states, elevated peroxynitrite oxidizes tetrahydrobiopterin (cofactor of eNOS) or disrupts endothelial caveolae, and loss of endothelial cell Cav-1 leads to eNOS uncoupling, which contributes to DOCA-salt hypertension (32), pulmonary hypertension (2, 45), and coronary microvascular dysfunction (5). Thus, increased NOX and eNOS uncoupling are considered to be major sources of elevated that induces endothelial dysfunction in blood vessels from Cav-1−/− mice (47). However, net elevation of is the result of both generation and scavenging, and the role of Cav-1 in regulating antioxidant enzymes that protect against oxidative stress-mediated endothelial dysfunction remains unknown.
The primary antioxidant defense system for are the family of superoxide dismutases (SODs), which includes three isoforms: the cytoplasmic copper (Cu)/Zn SOD (SOD1), the mitochondrial MnSOD (SOD2), and extracellular SOD (SOD3). SOD3 is highly expressed in the vasculature and is synthesized and secreted by vascular smooth muscle cells (VSMCs) and fibroblasts, but not ECs. Extracellular SOD3 then binds to the extracellular matrix and ECs surface (14), where it protects NO from inactivation by (14). Using SOD3−/− mice or overexpression strategies, we and others have shown that SOD3 plays an important role in preventing endothelial dysfunction in diabetes (63, 64), hypertension (7, 17, 25), and aging (4, 36). The R213G polymorphism in the SOD3 gene, which reduces its ability to bind to the endothelial surface and increases circulating SOD3 levels, is correlated with increased cardiovascular risk (26). Thus, in both mouse and man, SOD3 has an established function to protect against oxidative stress-induced vascular dysfunction and cardiovascular disease.
SOD3 is a Cu-containing enzyme, and its activity is dependent on the insertion of Cu, an essential micronutrient and catalytic cofactor (14, 15). The Cu-transporting ATPase (ATP7A) localizes to the trans-Golgi network (TGN), where it transports Cu for insertion into secretory Cu-containing enzymes, including SOD3 or lysyl oxidase (LOX), thereby increasing their specific activity (14, 15). Using ATP7A mutant mice, which have reduced Cu transporter activity (31, 40, 61), we reported that ATP7A functions to protect against endothelial dysfunction by increasing SOD3 activity in models of hypertension and diabetes (54, 63, 64). We also found that ATP7A protein was downregulated in vascular tissue from diabetic mice and patients, thereby decreasing SOD3 activity (63, 64). In addition, growth factor (PDGF) promoted the translocation of ATP7A from the TGN to the C/LR in cultured VSMC, enabling pro-LOX to obtain Cu to increase LOX activity, leading to VSMC migration (1). Overexpression of SOD3 in human ECs promoted VEGF receptor type2 (VEGFR2) signaling in C/LR, which enhanced angiogenic responses (48). Many of these processes are also regulated by Cav-1; however, a role of Cav-1 in regulating the ATP7A-SOD3 axis that protects against endothelial dysfunction has not been investigated.
Here, we demonstrate an unexpected role for Cav-1 as a positive regulator of SOD3 activity via stabilizing the Cu transporter ATP7A. Using Cav-1−/− mice and ATP7A overexpressing transgenic (Tg) mice, we found that SOD3 activity and ATP7A protein expression were significantly decreased in the vascular tissue of Cav-1−/− mice, resulting in impaired endothelium-dependent vasorelaxation, which was rescued by ATP7A overexpression. In vitro studies using Cav-1 siRNA, caveolae fractionation, coimmunoprecipitation, and colocalization analysis reveal that Cav-1 binds to and stabilizes ATP7A protein in the C/LR or Golgi, where it protects against proteasomal degradation of ATPA. Collectively, Cav-1 plays an important role in promoting ATP7A-mediated SOD3 activity in vascular tissues that protect endothelial function from excessive .
MATERIALS AND METHODS
Animals.
Control and Cav-1−/− mice on the C57bl/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME). ATP7A-transgenic (ATP7A Tg) mice overexpressing human ATP7A from a composite β-actin promoter (CAG) were generated as previously described (28). Cav-1−/−/ATP7A Tg mice were generated by intercross of Cav-1−/− mice with ATP7A Tg mice. Male mice were studied between 12 and 16 wk of age. Mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) before euthanasia. Then, aorta and mesenteric arteries were collected. The protocol for animal use was approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago and the Medical College of Georgia at Augusta University.
Cell culture.
Cav-1−/− and Cav-1 wild-type (WT) immortalized mouse embryonic fibroblast cells (mouse fibroblast cells) were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% bovine serum and 4.5 g/L glucose.
Superoxide dismutase activity assays.
Tissue harvesting and SOD activity assay were performed as described previously (54, 63). Briefly, tissues were homogenized in 10 vol of 50 mM potassium phosphate (pH 7.4) containing 0.3 M KBr, a cocktail of protease inhibitors (0.5 mM PMSF, 3 mM diethylene-triaminepentaacetic acid, 90 mg/L aprotinin, 10 mg/L pepstatin, 10 mg/L chymostatin, and 10 mg/L leupeptin), 1 mM sodium ascorbate, and 1 mM bathocuproinedisulfonate to limit the availability of copper during extraction, as previously described (12, 64). The homogenates were then sonicated and extracted at 4°C for 30 min. The extracts were then centrifuged at 3,000 g for 15 min. SOD activity was assayed by monitoring inhibition of the rate of xanthine/xanthine oxidase-mediated reduction of cytochrome c. Con A-Sepharose chromatography (Amersham Biosciences) was used to isolate SOD3 or SOD1 from vessels of Cav-1 WT or Cav-1−/− mice. For in vitro Cu treatment, ConA Sepharose-bound SOD3 or unbound SOD1/SOD2 fractions were treated with CuCl2 (10 μM for 60 min at room temperature), and their activity was measured, as we reported previously (54, 55).
Adenoviral vector and in vivo gene transfer.
Replication-deficient, adenovirus-expressing human SOD3 (Ad.SOD3) was purchased from the adenovirus core at University of Iowa (7). Ad.SOD3 and Lac Z (0.25 mL of 1 × 1012 particles/mL in 3% sucrose in PBS) were injected intravenously via retro-orbital injection. Three days after viral injection, mice were euthanized, and vascular tissue was collected for further experiments.
Vascular reactivity studies.
Isometric tension of mesenteric resistance arteries was measured using wire myograph (Model 610M; Danish Myo Technology, Denmark), as described previously (49). Endothelium-dependent vasorelaxation in response to acetylcholine and endothelium-independent relaxation to sodium nitroprusside (SNP) were evaluated in phenylephrine-preconstricted blood vessels. Results are expressed as percent relaxation of the phenylephrine-treated arteries, with 100% relaxation representing basal tension.
Immunoprecipitation and Immunoblotting.
Aortic tissue or cells were lysed with 500 μL of ice-cold lysis buffer, pH 7.4 (50 mM HEPES, 5 mM EDTA, 120 mM NaCl), 1% Triton X-100, 60 mM n-octyl-α-d-glucopyranoside, protease inhibitors (10 μg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin), and phosphatase inhibitors (50 mM sodium fluoride, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate). Immunoprecipitation was performed in lysates of aortic tissue (1,500 μg). Lysates were incubated with antibody overnight at 4°C and then precipitated with 20 μL of protein A/G-agarose beads for 2 h at 4°C. Total lysates (25 μg) or immunoprecipitates were size fractionated using SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes, blocked overnight in PBS containing 5% nonfat dry milk and 0.1% Tween 20, and incubated for overnight with primary antibodies. The following primary antibodies were used: anti-Atox1 (homemade) (23), anti-SOD3 (homemade) (13), anti-SOD1 (ab16831; Abcam), anti-ATP7A (B8162; LifeSpan Biosciences), anti-actin (SC-1616; Santa Cruz Biotechnology), anti-ubiquitin (SC-8017; Santa Cruz Biotechnology), anti-Cav-1 (610407 or 610060; BD Transduction), anti-paxillin (612405; BD Transduction), anti-flotillin-1 (610820; BD Transduction), anti-heparan sulfate (MAB2040; Millipore), anti-fibulin-5 (12188; Proteintech) or anti-collagen type-1 (Proteintech, 14695). After incubation with secondary antibodies [1706515, anti-rabbit IgG-horseradish peroxidase (HRP) conjugate, Bio-Rad; 1706516, anti-mouse IgG-HRP conjugate, Bio-Rad], protein expression was detected using ECL chemiluminescence.
Immunofluorescence analysis.
Human aortic smooth muscle cells on glass coverslips were rinsed quickly in ice-cold PBS, fixed in freshly prepared 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized in 0.05% Triton X-100 in PBS for 5 min, and rinsed sequentially in PBS, 50 µmol/L NH4Cl, and PBS for 10 min each. After incubation for 1 h in blocking buffer (PBS + 3% BSA), cells were incubated with anti-ATP7A antibody (GW21023; Sigma Aldrich) or anti-Cav-1 (610407; BD Transduction) for 18 h at 4°C, rinsed in PBS/BSA, and then incubated in Alexa Fluor conjugated goat anti-mouse (A11001; Invitrogen) or goat anti-chicken IgY FITC-conjugate (GAY-FITC; GenWay) for 1 h at room temperature, and cells were rinsed with PBS. Cells on coverslips were mounted onto glass slides using Vectashield (Vector Laboratories) and observed using confocal microscopy
Quantitative real-time PCR.
Total RNA from aortic tissue was isolated using the Tri Reagent (Molecular Research Center, Inc.). Reverse transcription was carried out using a high-capacity cDNA reverse transcription kit (Applied Biosystems) using 2 µg of total RNA. Quantitative PCR was performed using an ABI Prism 7000, a SYBR Green PCR kit (Qiagen), and QuantiTect Primers (Qiagen) for specific genes. Samples were all run in triplicates to reduce variability. Expression of genes was normalized and expressed as fold changes relative to 18S.
Detergent-free purification of C/LR membrane fractions.
C/LR fractions were separated using the sodium carbonate-based detergent-free method (1). Briefly, mouse aortae (400 mg) were homogenized in a solution containing 0.5 M sodium carbonate (pH 11), 1 mM sodium orthovanadate, and protease inhibitors. Homogenization was carried out sequentially in the following order using a loose-fitting Dounce homogenizer (10 strokes) and a sonicator (four 20-s bursts). The homogenates were adjusted to 45% sucrose by adding 90% sucrose in a buffer containing 25 mM Mes (pH 6.5) and 0.15 M NaCl and placed at the bottom of an ultracentrifugation tube. A 5–35% discontinuous sucrose gradient was formed above and centrifuged at 39,000 rpm at 4°C for 16–20 h in a Beckman SW-40Ti rotor. From the top of the tube, 13 fractions were collected, and an equal volume from each fraction was subjected to immunoblotting.
Statistical analysis.
Data are presented as means ± SE. Data were compared between groups of cells and animals by t test when one comparison was performed or by ANOVA for multiple comparisons. When significance was indicated by ANOVA, the Tukey-Kramer post hoc test was used to specify between-group differences. Values of P < 0.05 were considered statistically significant. Statistical tests were performed using Prism version 8 (GraphPad Software, San Diego, CA).
RESULTS
The specific activity of SOD3 is decreased in aorta from Cav-1−/− mice and rescued by Cu supplementation.
To determine whether Cav-1 is required for the activity of SOD3, we measured SOD3 activity and protein expression in Cav-1−/− aorta. We found that SOD3 activity was significantly decreased in Cav-1−/− mice compared with control wild-type (WT) mice, whereas SOD3 protein levels were not changed (Fig. 1 A and B). In contrast, SOD1 activity and protein level were not altered in Cav-1−/− vessels. Thus, the specific activity of SOD3, as determined by the ratio of activity to protein, was markedly decreased in Cav-1−/− vessels (i.e., increased levels of “inactive” SOD3 protein), whereas that of SOD1 was unchanged (Fig. 1C). To further investigate these findings, we next used cultured Cav-1−/− mouse fibroblasts. SOD3 protein was purified from the conditioned media of WT and Cav-1−/− cells using concanavalin A (Con A) sepharose chromatography, as described previously (54, 55). SOD3 activity in the conditioned media was also decreased in Cav-1−/− fibroblasts as compared with control cells, whereas SOD3 protein levels were not changed (Fig. 1, D and E). Thus, the specific activity of SOD3 was markedly decreased, whereas the activity and protein levels of SOD1 were not changed in Cav-1−/− fibroblasts (Fig. 1F). We next examined mechanisms by which SOD3 activity in vascular tissue is decreased in Cav-1−/− mice. Because SOD3 activity in tissue is dependent on the interaction with extracellular matrix of the cells (14), we first examined protein expression of extracellular matrix such as the heparan sulfate proteoglycan (7, 27), collagen type I (51), and fibulin-5 (41), which are required for binding of SOD3 at extracellular space and endothelial surface. However, we found that the expression of extracellular matrix proteins was not changed in aorta of Cav-1−/− mice (Fig. 2A), suggesting that decreased SOD3 activity in Cav-1−/− vessels was not due to decreased expression of extracellular matrix protein. Because the activity of SOD1 and SOD3 is dependent on the catalytic Cu cofactor (14), we next examined whether decreased SOD3-specific activity in Cav-1−/− vessels is due to deficiency of copper. We found that exogenous addition of Cu rescued specific activity of SOD3 purified from Cav-1−/− vessels. In contrast, the specific activity of SOD3 purified from control vessels or that of SOD1 from either Cav-1−/− or WT vessels was not affected by Cu addition (Fig. 2B). These results suggest that Cu loading to SOD3 is selectively impaired in Cav-1−/− vessels, whereas SOD3 enzyme from control vessels or SOD1 enzyme from either Cav-1−/− or control vessels is fully metallated.
Fig. 1.
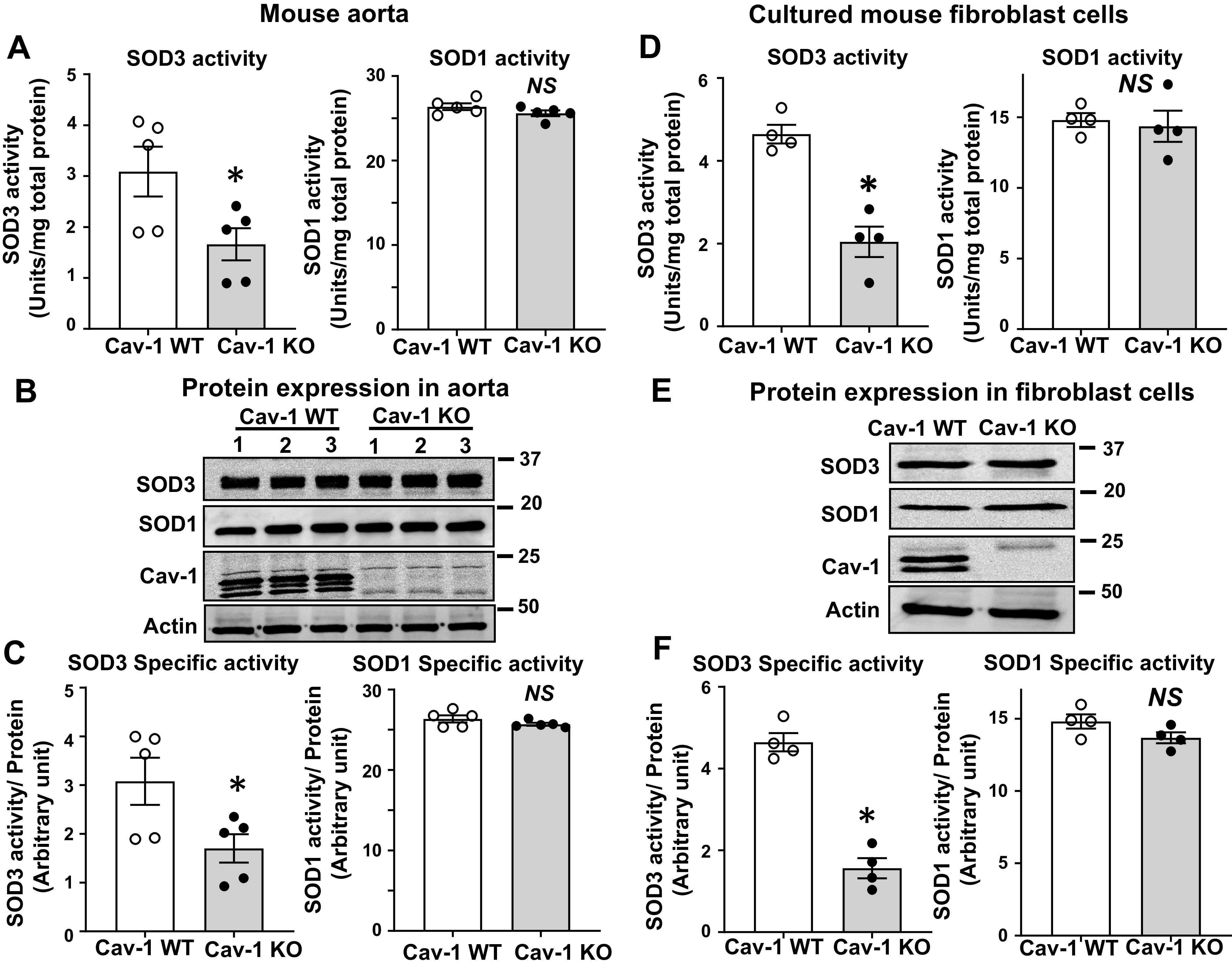
Specific activity of superoxide dismutase (SOD3) is decreased in vascular tissue and cultured fibroblast from caveolin-1 knockout (KO) mice (Cav-1−/−). A: activity of SOD3 and SOD1 in aortae from Cav-1 wild-type (WT) and Cav-1−/− mice as measured by inhibition of cytochrome c reduction in the presence of xanthine/xanthine oxidase. Concanavalin A (Con A)-Sepharose chromatography was used to isolate SOD3 from tissue homogenates. B: protein levels of SOD1, SOD3, Cav-1, and actin in aortic tissue. C: the specific activity of SOD1 and SOD3 was determined by the ratio of SOD activity relative to the amount of protein (n = 5). D–F: mouse fibroblast cells were cultured in 1% serum containing DMEM for 72 h. SOD3 secreted into the culture medium was collected and concentrated by concanavalin A-Sepharose chromatography. Activity of SOD3 in concentrated culture medium and SOD1 in cell lysates was assayed (D). Protein levels of SOD1 in cell lysate and SOD3 from conditional medium were determined by Western analysis (E). Specific activity of SOD1 and SOD3 was determined by the relative ratio of SOD activity to the amount of protein (n = 4) (F). Results are presented as means ± SE. *P < 0.05 vs. control. NS, not significant.
Fig. 2.

A: extracellular matrix protein expression in aorta of caveolin-1 knockout (KO) mice (Cav-1−/−) and Cav-1 wild-type (WT) mice. Protein levels of extracellular matrix proteins (heparin sulfate, fibulin-5,collagen type 1) and actin in aortic tissue. Densitometric analysis is shown (n = 3). B: effect of in vitro copper (Cu) treatment on superoxide dismutase (SOD3)-specific activity purified from aortas of Cav-1-WT and Cav-1−/− mice. Concanavalin A (Con A)-Sepharose-bound SOD3 or -unbound SOD1 proteins from aortas of Cav-1 WT and Cav-1−/− mice were treated with or without CuCl2 (10 mmol/L, 1 h at room temperature), and the specific activity of SOD3 and SOD1 was measured (n = 3) as described in Fig. 1. Results are presented as means ± SE. *P < 0.05 vs control. NS, not significant.
Endothelium-dependent relaxation is impaired in mesenteric arteries from Cav-1−/− mice and rescued by gene transfer of SOD3.
Previous studies have reported that Cav-1 deficiency induces endothelial dysfunction via increased oxidative stress (6, 38, 57, 66). To examine whether impaired endothelium-dependent vasorelaxation in Cav-1−/− mice (38) is due to decreased SOD3 activity, we next assessed blood vessel function by using mesenteric arteries (∼200 mm in diameter) with the wire myograph, which contributes to blood pressure and tissue perfusion (21). Figure 3A shows that acetylcholine (ACh)-induced endothelium-dependent vasorelaxation was significantly impaired in resistance arteries of Cav-1−/− mice compared with WT mice, consistent with previous reports (37). Furthermore, the impaired vasorelaxation response of Cav-1−/− vessels was significantly improved by gene transfer of adenovirus expressing SOD3 (Ad-SOD3) as compared with a control virus (Ad-Null). In contrast, sodium nitroprusside (SNP)-induced endothelium-independent vasorelaxation was not different among WT mice treated with Ad-Null, Cav-1−/− mice treated with Ad-Null, or Ad-SOD3 (Fig. 3B). These results suggest that impaired endothelium-dependent vasorelaxation in Cav-1−/− mice is due at least in part to decreased SOD3 activity.
Fig. 3.
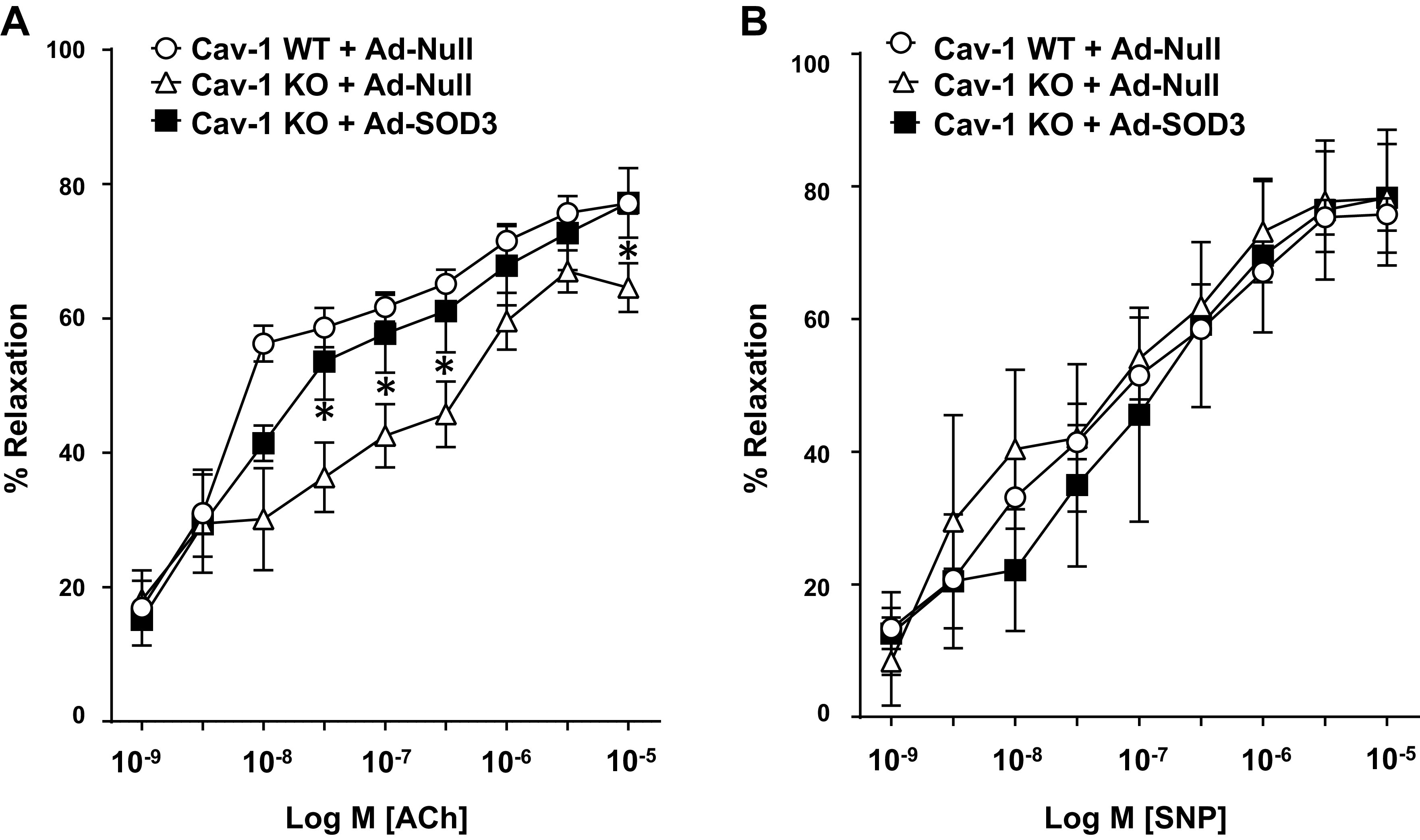
Decreased superoxide dismutase (SOD3) activity in blood vessels contributes to impaired endothelium-dependent relaxation in caveolin-1 knockout (KO) mice (Cav-1−/−). A and B: isometric tension in mesenteric resistance arteries from Cav-1 wild-type (WT) and Cav-1−/− mice was measured in isolated organ chambers using wire myography. Vasodilation was evoked by ACh (A) and sodium nitroprusside (SNP) (B) after preconstriction with phenylephrine (1–5 µM) in the presence of adeno-SOD3 (1 × 1012 particles per mice) 3 days after intravenous injection in the tail vein of mice. Results are presented as means ± SE (n = 4–8). *P < 0.05 vs control.
Expression of the Cu transporter ATP7A is decreased in vascular tissues from Cav-1−/− mice and rescued by re-expression of Cav-1.
Based on data that the selectively reduced activity of SOD3 in Cav-1−/− mice was rescued by addition of Cu, we next examined whether the expression of Cu transporter proteins, which transport Cu to the SOD3, was altered in Cav-1−/− mice. Unexpectedly, we found that the protein but not mRNA expression of ATP7A was significantly decreased in aortae from Cav-1−/− mice as compared with WT mice (Fig. 4, A and B). ATP7A protein expression was also significantly downregulated in fibroblasts isolated from Cav-1−/− mice (Fig. 4C). Reduced ATP7A expression and its downstream SOD3-specific activity were rescued by re-expression of Cav-1 using a Myc-Cav-1 plasmid (Fig. 4, D and E). Of note, protein expression of the Cu chaperone Atox1, which delivers Cu to ATP7A, was not altered by loss of Cav-1−/− in aorta or fibroblasts (Fig. 4, A and C). These results suggest that loss of Cav-1 reduces ATP7A protein expression without altering its mRNA levels in vascular cells, which contributes to the decreased activity of SOD3.
Fig. 4.
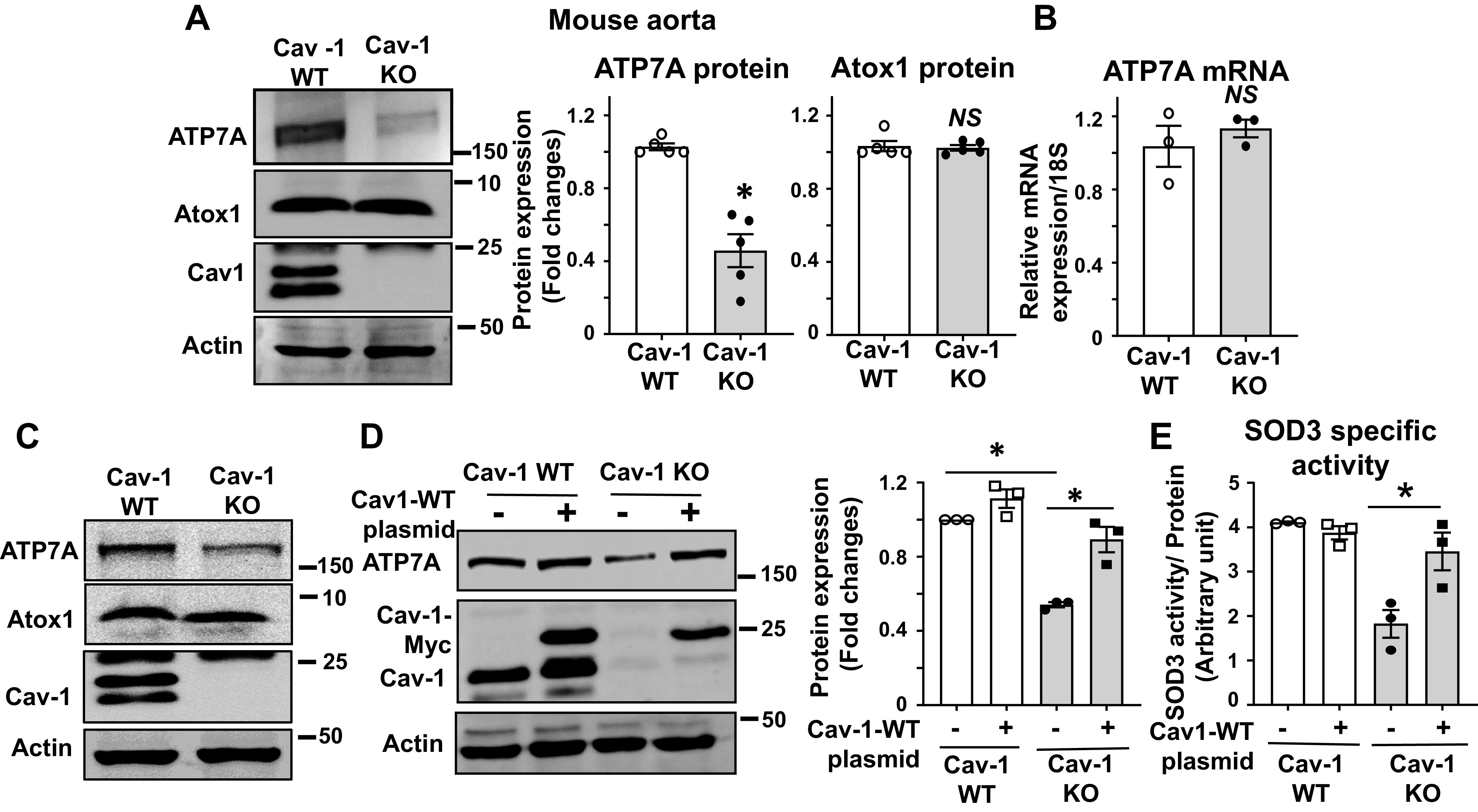
Protein expression of the copper transporter ATP7A is decreased in blood vessels from caveolin-1 knockout (KO) mice (Cav-1−/−). A: relative protein expression of ATP7A and Atox1 in aortas from Cav-1 wild-type (WT) and Cav-1−/− mice was determined by Western blotting with antibodies specific to respective proteins. Densitometric analysis is shown (right, n = 5). B: ATP7A mRNA expression in aortas from Cav-1 WT and Cav-1−/− mice was determined by real-time quantitative RT-PCR (n = 3). C: protein expression for ATP7A and Atox1 in mouse fibroblasts isolated from Cav-1 WT and Cav-1−/− mice (n = 3). D: Cav-1 WT and Cav-1−/− mouse fibroblasts mice were transfected with Cav-1 WT plasmid. Lysates were used to measure ATP7A, Cav-1, and actin protein expression. (n = 3). E: superoxide dismutase (SOD3)-specific activity in conditional medium of Cav-1 WT plasmid transfected Cav-1−/− mouse fibroblast cells. Cav-1 WT and Cav-1−/− mouse fibroblast cells were transfected with Cav-1-WT plasmid. The specific activity of SOD3 was determined by the ratio of activity to relative amount of protein in culture conditional medium. Results are presented as means ± SE. *P < 0.05, NS, not significant. ATP7a, Menkes ATPase, copper-transporting P-type ATPase.
Cav-1 stabilizes ATP7A protein expression by preventing ubiquitination and proteasomal degradation.
We next examined the posttranslational mechanisms that Cav-1 might regulate to alter ATP7A protein stability. Ubiquitination is a major pathway of cellular protein degradation (59). To investigate whether Cav-1 regulates the ubiquitination of ATP7A, we immunoprecipitated (IP) ATP7A from WT and Cav-1−/− fibroblast lysates, performed SDS PAGE, and then immunoblotted (IB) using an anti-ubiquitin antibody. The levels of ubiquitinated ATP7A were increased in Cav-1−/− fibroblasts (Fig. 5A) and Cav-1−/− aorta (Supplemental Fig. S1B; all Supplemental Material is available at http://doi.org/10.6084/m9.figshare.12921710). Of note, ATP7A ubiquitination and its protein downregulation in Cav-1−/− fibroblast were prevented by re-expression of Cav-1 (Supplemental Fig. S1A). Because ubiquitination can tag proteins for degradation via the proteasome or lysosome pathway (30), we next examined the effects of proteasomal or lysosomal inhibitors on the expression levels of ATP7A. Figure 5B shows that the proteasome inhibitor MG132, but not the lysosomal inhibitor chloroquine, significantly rescued the decreased protein expression of ATP7A in Cav-1−/− fibroblasts. These results suggest that Cav-1 stabilizes ATP7A protein expression by preventing its ubiquitination and proteasomal degradation.
Fig. 5.
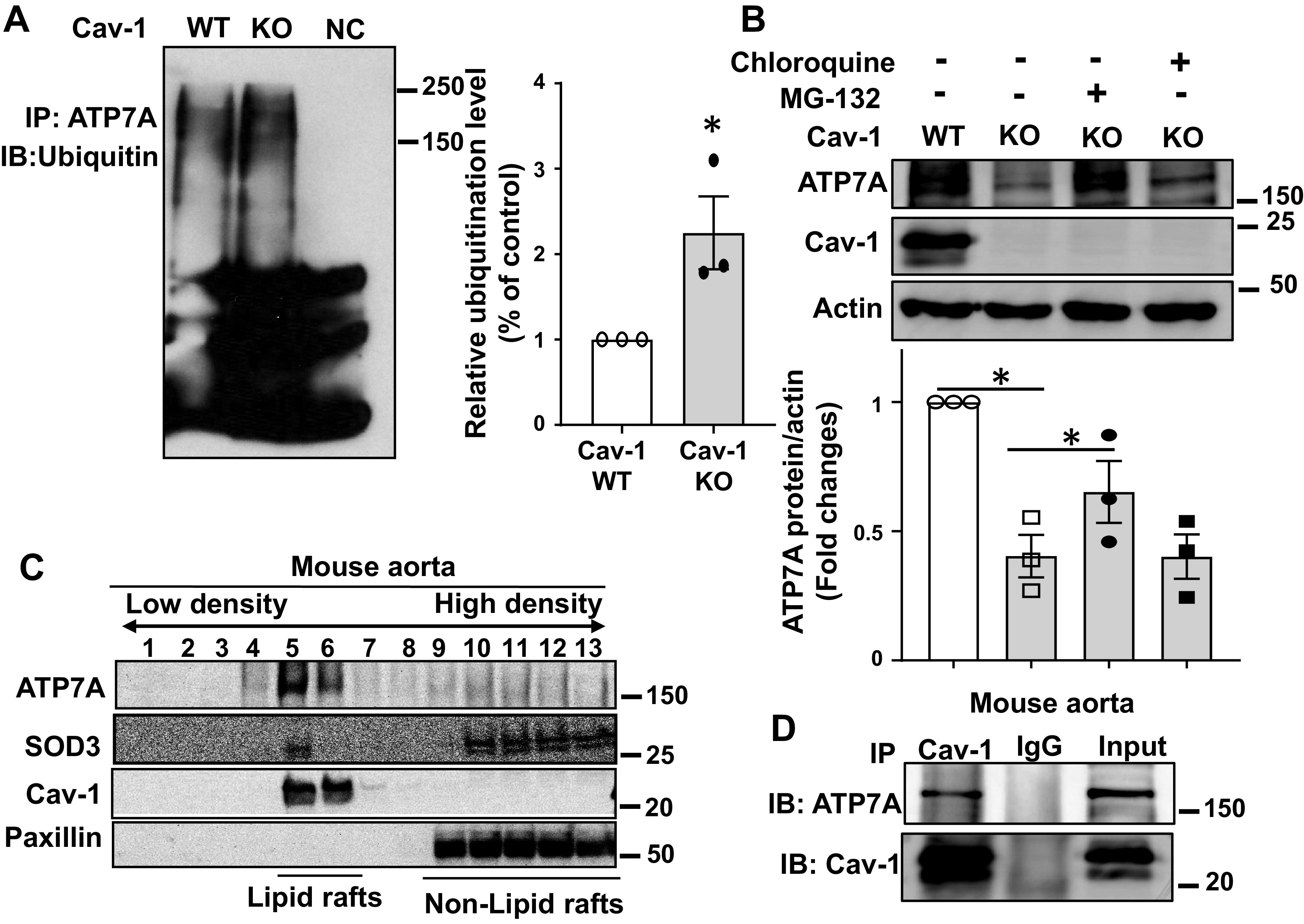
Caveolin-1 (Cav-1) is required for ATP7A protein stabilization and prevents proteosomal degradation. A: Cav-1 wild-type (WT) and Cav-1−/− [knockout (KO)] mouse fibroblasts were lysed and immunoprecipitated (IP) with anti-ATP7A, followed by immunoblotting (IB) with an anti-ubiquitin antibody. Right: averaged data for ATP7A ubiquitination (n = 3). B: mouse fibroblast cells were incubated with an inhibitor of the proteasome MG132 (20 μmol/L) for 24 h or an inhibitor of the lysosome chloroquine (100 μmol/L) for 24 h, and the protein expression of ATP7A was determined by Western blot (n = 3). C: membrane fractionation of mouse aorta. Equivolume fractions isolated from the top (fraction 1) to the bottom (fraction 13) were immunoblotted with antibodies as indicated. D: IP of Cav-1 using an anti-Cav-1 antibody in aortic lysates, followed by IB with an ATP7A antibody (n = 3). Results are presented as means ± SE. *P < 0.05. ATP7a, Menkes ATPase, copper-transporting P-type ATPase.
Cav-1 colocalizes with ATP7A in the C/LR and the perinucleus in vascular tissues and cells.
Previous studies have established that Cav-1 directly interacts with proteins present in the C/LR via its scaffolding domain to facilitate complex formation and changes in protein expression or activity (44). To address the mechanism by which Cav-1 stabilizes protein expression of ATP7A, we next examined whether Cav-1 colocalizes with and binds to ATP7A and its downstream substrate SOD3 in the C/LR. Of note, ATP7A has three Cav-1-binding consensus motifs but has not yet been shown to interact with Cav-1. Gradient centrifugation was used to isolate C/LR membranes, and both ATP7A and SOD3 were colocalized within the C/LR fraction in mouse aorta (Fig. 5C). Consistently, Cav-1 was found together in immune complexes with ATP7A in aortic lysates (Fig. 5D). Together, these results suggest that Cav-1 binding to ATP7A in C/LR may be required for preventing the proteasomal degradation of ATP7A.
Since Cav-1 is also localized in other organelles, such as Golgi (35), we next examined the subcellular location of Cav-1 and ATP7A using immunofluorescence (IF) colocalization analysis in cultured VSMCs. We found that Cav-1 was localized at the plasma membranes (presumably C/LR) and the perinuclear region, where it colocalized with ATP7A in the basal state (Supplemental Fig. S2). Note that we could not detect SOD3 in cultured VSMCs because it was secreted to cultured media after synthesis. Because it has been shown that ATP7A is localized at TGN at the basal state in various cells (52, 65), these results suggest that Cav-1-ATP7A association not only in the C/LR but also in the perinucleus (Golgi or TGN) seems to be important for stabilizing ATP7A protein by preventing its ubiquitination and targeting for proteasomal degradation.
Decreased specific activity of SOD3 and endothelial dysfunction in Cav-1−/− mice are rescued by overexpressing ATP7A.
Because ATP7A functions as a Cu transporter for SOD3 and is important for full activity, we next examined whether the specific downregulation of ATP7A protein in Cav-1−/− mice is responsible for the impaired endothelium-dependent vasorelaxation and SOD3 activity. To rescue ATP7A expression, Cav-1−/− mice were intercrossed with ATP7A-overexpressing transgenic (ATP7A Tg) mice. Figure 6, A and B, shows that the reduction in ATP7A protein expression and SOD3-specific activity in Cav-1−/− vessels could be rescued by restoring ATP7A expression in double-transgenic (ATP7A-Tg/Cav-1−/−) mice. Furthermore, impaired endothelium-dependent relaxation to Ach was significantly improved by ATP7A overexpression in Cav-1−/− mice as compared with Cav-1−/− mice, whereas endothelial-independent relaxation to SNP was not different between the two groups (Fig. 6C). These findings suggest that decreased ATP7A expression in Cav-1−/− vessels contributes to decreased SOD3-specific activity and endothelial dysfunction. We also examined the localization of ATP7A and SOD3 in the C/LR and the non-C/LR membrane fractions in WT, Cav-1−/−, and ATP7A-Tg/Cav-1−/− mouse aorta using gradient centrifugation. Figure 6D shows that expression of ATP7A and SOD3 protein, but not flotillin-1 (lipid raft marker), was abolished in Cav-1−/− mice, which was not altered in ATP7A-Tg/Cav-1−/− mice that lack caveolae (8). Of note, ATP7A protein expression was increased only in the non-C/LR fraction by ATP7A overexpression in Cav-1−/− mice (Fig. 6D). Taken together, these results suggest that localization of ATP7A and SOD3 in the C/LR is dependent on Cav-1 but is not required for ATP7A-mediated Cu delivery to SOD3 to increase its specific activity.
Fig. 6.
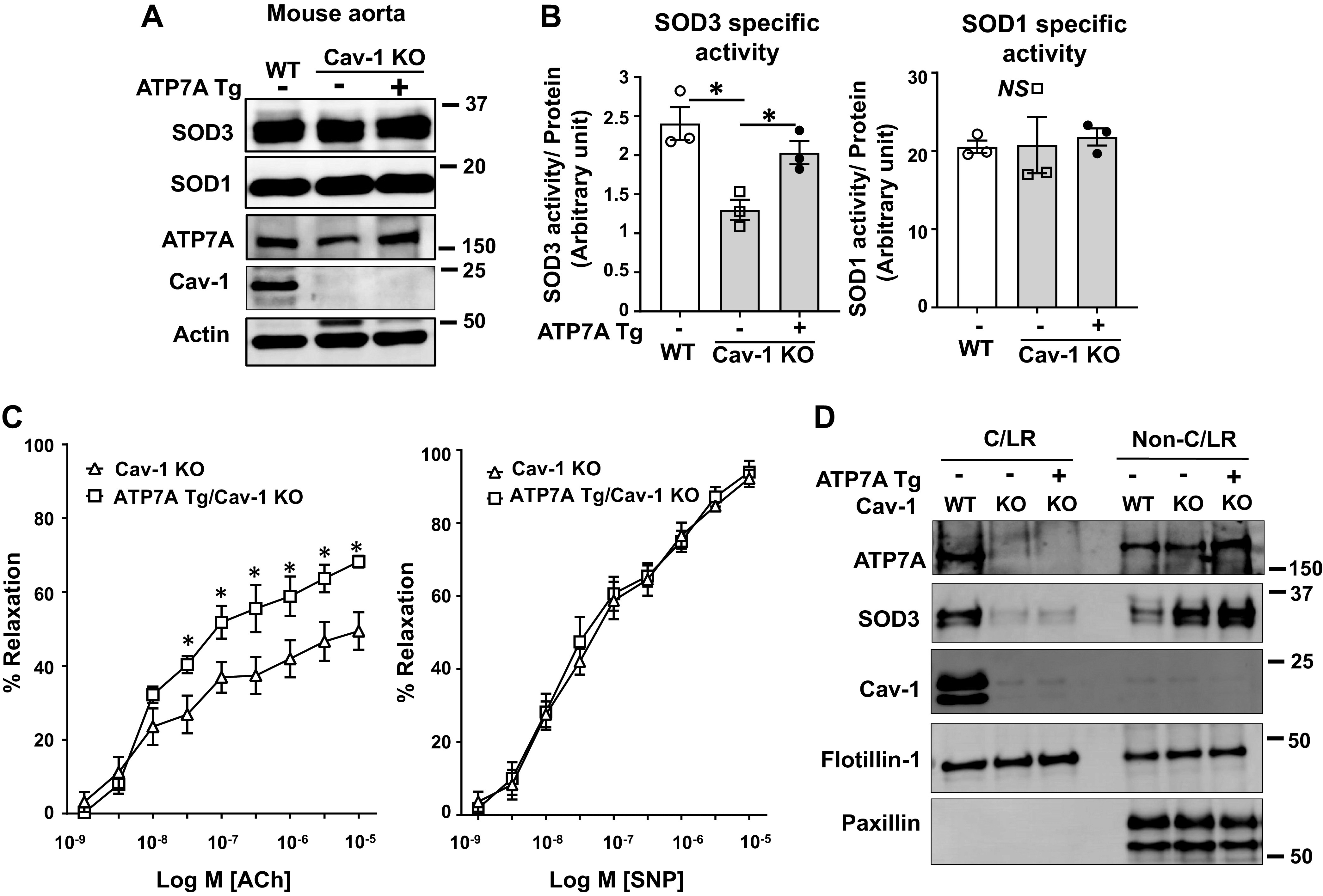
Transgenic mice overexpressing Menkes ATPase, copper-transporting P-type ATPase (ATP7A; ATP7A Tg) rescued superoxide dismutase (SOD3) activity and endothelium-dependent relaxation in caveolin-1 knockout (KO) (Cav-1−/−) mice. A: protein levels of ATP7A, SOD3, SOD1, Cav-1, and actin in aortas from Cav-1 wild-type (WT) or Cav-1−/− double Tg mice overexpressing ATP7A were measured (n = 3). B: specific activity of SOD3 and SOD1 in tissue homogenates were assayed as described in Fig. 1. C: endothelium-dependent or -independent relaxation of mesenteric resistance arteries from Cav-1 WT or Cav-1−/− double Tg mice overexpressing ATP7A. Vasorelaxation was evoked by acetylcholine (ACh) and sodium nitroprusside (SNP) after preconstriction with phenylephrine (n = 6). D: membrane fractionation of mouse aorta of Cav-1 WT, Cav-1−/−, and ATP7A-Tg/Cav-1−/− mice. Equal amounts of caveolin-enriched lipid raft fractions (caveolae/lipid rafts; fraction 4 to 5) and non-caveolae/lipid rafts; fractions 10 to 13) from a pool of 4 mouse aortas were immunoblotted with antibodies as indicated. Results are presented as means ± SE. *P < 0.05. ATP7a, Menkes ATPase, copper-transporting P-type ATPase.
In summary, impaired endothelium-dependent vasorelaxation in Cav-1−/− mice lacking caveolae was rescued by gene transfer of SOD3 or by crossing with ATP7A-overexpressing transgenic mice. These data reveal an unexpected role of Cav-1 in stabilizing ATP7A protein expression by preventing its ubiquitination and proteasomal degradation, thereby increasing SOD3 activity, which in turn protects against vascular oxidative stress-mediated endothelial dysfunction.
DISCUSSION
Endothelial dysfunction precedes cardiovascular disease, but the mechanisms involved and specific therapeutics to treat it remain elusive. Cav-1 has a well-established role in regulating endothelial function and dysfunction and, more recently, the ability to regulate the production of oxidants. Our study identifies a novel role of Cav-1 in the regulation of antioxidant systems, and more specifically, SOD3. Blood vessels from Cav-1−/− mice have impaired endothelial function and reduced activity of SOD3 but not SOD1. In ex vivo experiments, specific activity of SOD3 purified from Cav1−/− vessels was rescued with the addition of exogenous Cu. Expression of the Cu transporter ATP7A, but not the Cu chaperone Atox1, was decreased in aorta or cultured fibroblasts from Cav-1−/− mice. We found that Cav-1 was necessary for the protein stabilization of the ATP7A, which supplies Cu to SOD3 to enable its catalytic activity. ATP7A was found together with Cav-1 and SOD3 in the C/LR and Cav-1 bound to ATP7A. Loss of Cav-1 expression promoted the proteasomal degradation of ATP7A, decreasing Cu incorporation into SOD3 and reducing its activity in blood vessels, elevating extracellular and impairing endothelial function (Fig. 7). Rescue of ATP7A expression in ATP7A-Tg/Cav1−/− mice lacking caveolae was enough to reverse SOD3 activity and endothelial dysfunction in Cav-1−/− mice.
Fig. 7.
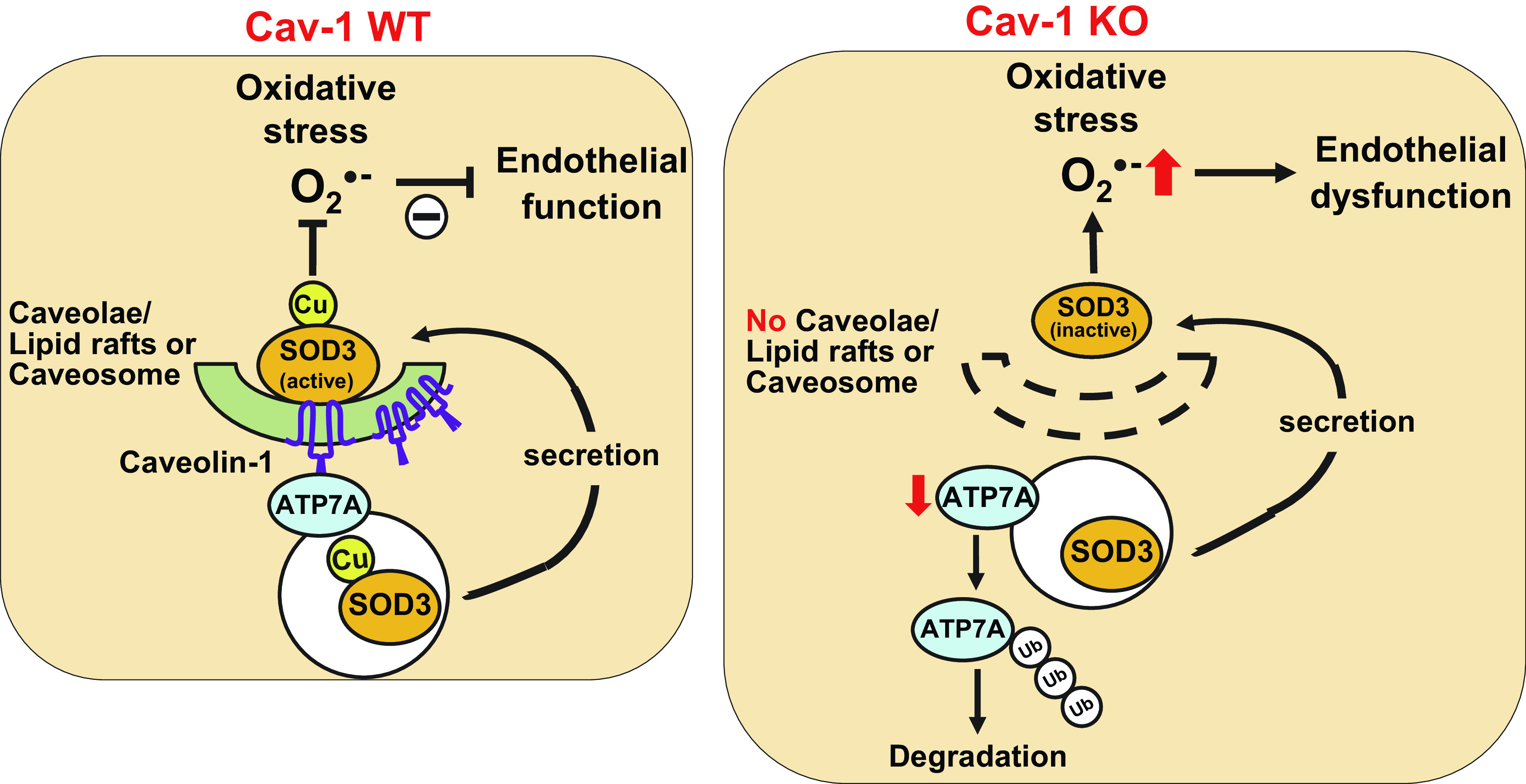
Proposed working model showing how caveolin-1 (Cav-1) is required for stabilizing protein expression of the copper transporter ATP7A and enabling copper delivery to extracellular superoxide dismutase 3 (SOD3) in vascular tissue to enable its full activity and protection of endothelial function against oxidative stress. ATP7A, Menkes ATPase, copper (Cu)-transporting P-type ATPase; Ub, ubiquitin.
Unexpectedly, we found that mice lacking Cav-1 exhibit an impairment of the antioxidant defense system that was specific to SOD3 but not another Cu-containing enzyme, SOD1. Loss of activity occurred without affecting the expression of these proteins in vascular tissues. Impaired activity of SOD3 will promote oxidative stress-mediated inactivation of NO, which normally traverses from ECs to VSMCs in the extracellular space, and thereby promote endothelial dysfunction (14). Indeed, the functional consequences of the compromised SOD3 activity induced by Cav-1 deficiency showed that the impaired endothelium-dependent relaxation in mesenteric-resistant arteries in Cav-1−/− mice (38) can be rescued by gene transfer of SOD3. Of note, previous reports have shown increased and decreased vascular function in Cav-1−/− mice, which may be due to differences in vessel size, such as conduit arteries (11, 56) versus resistance arteries (57), respectively. Furthermore, endothelium-dependent relaxation in mesenteric-resistant arteries depends on not only NO but also on H2O2, which has been proposed to function as an endothelium-dependent hyperpolarizing factor (EDHF) (60). Recent studies have shown that endothelial Cav-1 protects EDHF-mediated relaxation from NO-mediated nitrative stress in the microcirculation in mice (22). Given that SOD3 functions to scavenge to generate the vasodilator H2O2 in the extracellular space (14, 48), it is possible that the loss of SOD3 activity in Cav-1−/− mice may reduce the amount of H2O2 produced, which contributes to the impaired vasorelaxation in Cav-1−/− blood vessels. These findings support a critical role of Cav-1 in regulating vascular function via increasing SOD3 activity.
Full activity of SOD3 is dependent on the catalytic cofactor Cu, which is delivered via the Cu chaperone Atox1, and the Cu transporter ATP7A (14, 15). We found that addition of Cu restored the reduced activity of SOD3 in aortic lysates of the ConA Sepharose-purified SOD3, suggesting that impaired access to Cu and Cu loading into the SOD3 may be regulated by Cav-1. Expression of ATP7A protein, but not its mRNA or that of Atox1, was decreased in vascular tissue of Cav-1−/− mice. Re-expression of Cav-1 in Cav-1−/− cells increased ATP7A protein expression, indicating that Cav-1 may regulate ATP7A protein stability. Furthermore, reduction of ATP7A protein in Cav-1−/− fibroblast was associated with an increased ubiquitination and proteosomal degradation. Consistent with these results, previous studies have demonstrated that Cav-1 provides protection against the degradation of the insulin receptor (IR), GLUT4, Pma1 protein (16, 18, 19, 20, 34, 58), pannexib-1 (10), and BMPR2 (46), among others. We recently reported that insulin signaling to Akt2 stabilizes ATP7A protein in VSMCs, which is required for full SOD3 activation (63). Our initial hypothesis was that Cav-1-mediated stabilization of ATP7A is mediated through increasing insulin signaling. However, in preliminary experiments, Cav-1 knockdown in VSMCs had no effects on insulin signaling (data not shown), suggesting that the Cav-1-insulin signaling axis may be cell type specific. Importantly, the decrease in both SOD3 activity and endothelium-dependent vasodilation in Cav-1−/− vessels was rescued in Cav-1−/− mice crossed with ATP7A-Tg mice or supplemental Cu. Thus, these results suggest that decreased SOD3 activity in Cav-1−/− mice is due to decreased ability of ATP7A to insert Cu.
In the present study, fractionation of membrane components of isolated aorta revealed that both ATP7A and SOD3 colocalized with Cav-1 in the C/LR fractions in intact vessels that have been exposed to various growth factors. Consistent with this, using cultured VSMCs, we previously reported that ATP7A translocates from the TGN to plasma membrane C/LR, which was associated with activation of the Cu-containing enzyme lysyl oxidase (likely via Cu delivery) in response to the growth factor PDGF (1). Thus, we thought that SOD3 might be activated similarly by ATP7A in the C/LR. However, the present study revealed that ATP7A and SOD3 protein, but not flotillin-1, was abolished in Cav-1−/− aorta in C/LR, which was not altered in ATP7A Tg/Cav-1−/− aorta (Fig. 6D). Of note, ATP7A protein expression was increased only in non-C/LR fraction by ATP7A overexpression in Cav-1−/− compared with Cav-1−/− aorta, which may contribute to restoring SOD3 activity. These results suggest that localization of ATP7A and SOD3 in C/LR is dependent on Cav-1 but seems not to be required for transferring Cu from ATP7A to SOD3 to increase its specific activity in blood vessels. Furthermore, we cannot exclude the possibility that colocalization of ATP7A with Cav-1 in C/LR is important for Cav-1-mediated ATP7A protein stabilization.
Cav-1 has also been shown to bind to various signaling proteins and enzymes via its scaffolding domain to enhance or inhibit their function (9). In this study, we used coimmunoprecipitation to determine whether ATP7A interacts with Cav-1. In lysates from mouse aorta, we found that ATP7A bound to Cav-1. In support of this finding, sequence analysis showed that ATP7A protein identified three Cav-1-binding consensus motifs. Because Cav-1 binding has been shown to stabilize Cav-2 (56), GLUT4, and IR (18) protein expression, we hypothesize that Cav-1 binding to ATP7A via its scaffolding domains is required to prevent the ubiquitination and proteosomal degradation of ATP7A in vascular tissue. The mechanism by which this occurs by displacing ubiquitin ligases or changes in folding of ATP7A remains to be determined. It has been shown that ATP7A is localized at TGN at the basal state in various cells (52, 65) including VSMCs (1), whereas Cav-1 is localized at the Golgi (35) and has been proposed to cycle between intracellular compartments (caveosomes) and the cell surface. Interestingly, in serum-starved, cultured VSMCs, we found that Cav-1 was localized at the perinuclear region and plasma membranes (C/LR), whereas it was colocalized with ATP7A at perinuclear region (Supplemental Fig. S2). Thus, it is likely that Cav-1 binding to ATP7A at the Golgi or TGN is required for ATP7A stability and loss of Cav-1 may lead to ATP7A accumulation in the Golgi, thereby leading to ATP7A ubiquitination and degradation, resulting in reduced SOD3 activity and EC dysfunction. In addition, although we could not detect SOD3 by IF in cultured VSMCs due to its secretion, it is possible that Cav-1 binding to ATP7A at TGN or Golgi may be required for transferring Cu to the secretory Cu enzyme SOD3 to increase its activity.
Our current findings may have implications for pathophysiological situations where endothelial function is compromised. We recently found that ATP7A is decreased in diabetic blood vessels (64) and aortic aneurysms (62) in which ATP7A plays an important role in maintaining vascular function. Cav-1 protein expression is also decreased in crude membrane fraction of renal cortex in diabetic rats (29), in the brain of diabetic mice (3), in aged mice (42, 43), in hypertensive pulmonary arteries from mice and humans (6) and the membrane fraction from diabetic EC (5) which results in endothelial dysfunction. Thus, these findings suggest that pathophysiological conditions associated with a loss of Cav-1 may lead to decreased expression of ATP7A and a subsequent loss of SOD3 activity, which together elevate oxidative stress and compromise endothelial function in blood vessels. In the current study, the molecular mechanism by which Cav-1 stabilizes ATP7A protein expression remains to be identified. Previous studies have demonstrated that COMMD (Cu metabolism Murr1 domain) 1 facilitates the degradation of the ATP7A via proteasomal degradation (39) and also regulates the endosomal trafficking of ATP7A (53). Thus, Cav-1 binding to ATP7A may limit the recruitment of COMMD1 to ATP7A to prevent its degradation. This possibility should be clarified in future studies.
In conclusion, our study provides compelling evidence that Cav-1 plays an important role in maintaining endothelial and vascular function by stabilizing ATP7A protein expression and increasing SOD3 activity in blood vessels. Our findings provide novel insights into a Cav-1-ATP7A-SOD3 signaling axis that may be useful in the development of potential therapeutics for preventing oxidative stress-dependent endothelial dysfunction and various cardiovascular diseases.
GRANTS
This research was supported by 5NIH R01 HL070187-14 (to T.F.), Department of Veterans Affairs Merit Review Grant 2I01BX001232-05 (to T.F.), R01HL133613, R01HL116976 (to T.F. and M.U.-F.), NIHR01 HL077524, HL077524-S1, R21HL112293 (to M.U.-F.), NIH R01 HL083298, HL125356, HL142636 (to R.D.M.), and American Heart Association Grant 15SDG25700406 (to V.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.S., M.U. and T.F. conceived and designed research; V.S. and M.N.O. performed experiments; V.S. analyzed data; V.S., M.U. and T.F. interpreted results of experiments; V.S. and T.F. prepared figures; V.S. and T.F. drafted manuscript; V.S., M.N.O., J.P.O., R.D.M., D.F., M.U. and T.F. edited and revised manuscript; V.S., M.N.O., J.P.O., R.D.M., D.F., M.U. and T.F. approved final version of manuscript.
REFERENCES
- 1.Ashino T, Sudhahar V, Urao N, Oshikawa J, Chen GF, Wang H, Huo Y, Finney L, Vogt S, McKinney RD, Maryon EB, Kaplan JH, Ushio-Fukai M, Fukai T. Unexpected role of the copper transporter ATP7A in PDGF-induced vascular smooth muscle cell migration. Circ Res 107: 787–799, 2010. doi: 10.1161/CIRCRESAHA.110.225334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakhshi FR, Mao M, Shajahan AN, Piegeler T, Chen Z, Chernaya O, Sharma T, Elliott WM, Szulcek R, Bogaard HJ, Comhair S, Erzurum S, van Nieuw Amerongen GP, Bonini MG, Minshall RD. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ 3: 816–830, 2013. doi: 10.1086/674753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonds JA, Shetti A, Bheri A, Chen Z, Disouky A, Tai L, Mao M, Head BP, Bonini MG, Haus JM, Minshall RD, Lazarov O. Depletion of caveolin-1 in type 2 diabetes model induces Alzheimer’s disease pathology precursors. J Neurosci 39: 8576–8583, 2019. doi: 10.1523/JNEUROSCI.0730-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown KA, Chu Y, Lund DD, Heistad DD, Faraci FM. Gene transfer of extracellular superoxide dismutase protects against vascular dysfunction with aging. Am J Physiol Heart Circ Physiol 290: H2600–H2605, 2006. doi: 10.1152/ajpheart.00676.2005. [DOI] [PubMed] [Google Scholar]
- 5.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, Belin de Chantemele E, Feher A, Romero MJ, Bagi Z. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes 63: 1381–1393, 2014. doi: 10.2337/db13-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen F, Barman S, Yu Y, Haigh S, Wang Y, Black SM, Rafikov R, Dou H, Bagi Z, Han W, Su Y, Fulton DJ. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med 73: 201–213, 2014. [Erratum in Free Radic Biol Med 81: 184, 2015]. doi: 10.1016/j.freeradbiomed.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu Y, Iida S, Lund DD, Weiss RM, DiBona GF, Watanabe Y, Faraci FM, Heistad DD. Gene transfer of extracellular superoxide dismutase reduces arterial pressure in spontaneously hypertensive rats: role of heparin-binding domain. Circ Res 92: 461–468, 2003. doi: 10.1161/01.RES.0000057755.02845.F9. [DOI] [PubMed] [Google Scholar]
- 8.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev 84: 1341–1379, 2004. doi: 10.1152/physrev.00046.2003. . [DOI] [PubMed] [Google Scholar]
- 9.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 272: 6525–6533, 1997. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 10.DeLalio LJ, Keller AS, Chen J, Boyce AKJ, Artamonov MV, Askew-Page HR, Keller TCS IV, Johnstone SR, Weaver RB, Good ME, Murphy SA, Best AK, Mintz EL, Penuela S, Greenwood IA, Machado RF, Somlyo AV, Swayne LA, Minshall RD, Isakson BE. Interaction between pannexin 1 and caveolin-1 in smooth muscle can regulate blood pressure. Arterioscler Thromb Vasc Biol 38: 2065–2078, 2018. doi: 10.1161/ATVBAHA.118.311290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293: 2449–2452, 2001. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 12.El Meskini R, Culotta VC, Mains RE, Eipper BA. Supplying copper to the cuproenzyme peptidylglycine alpha-amidating monooxygenase. J Biol Chem 278: 12278–12284, 2003. doi: 10.1074/jbc.M211413200. [DOI] [PubMed] [Google Scholar]
- 13.Fukai T, Galis ZS, Meng XP, Parthasarathy S, Harrison DG. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J Clin Invest 101: 2101–2111, 1998. doi: 10.1172/JCI2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 15: 1583–1606, 2011. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukai T, Ushio-Fukai M, Kaplan JH. Copper transporters and copper chaperones: roles in cardiovascular physiology and disease. Am J Physiol Cell Physiol 315: C186–C201, 2018. doi: 10.1152/ajpcell.00132.2018. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong X, Chang A. A mutant plasma membrane ATPase, Pma1-10, is defective in stability at the yeast cell surface. Proc Natl Acad Sci USA 98: 9104–9109, 2001. doi: 10.1073/pnas.161282998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension 48: 473–481, 2006. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 18.González-Muñoz E, López-Iglesias C, Calvo M, Palacín M, Zorzano A, Camps M. Caveolin-1 loss of function accelerates glucose transporter 4 and insulin receptor degradation in 3T3-L1 adipocytes. Endocrinology 150: 3493–3502, 2009. doi: 10.1210/en.2008-1520. [DOI] [PubMed] [Google Scholar]
- 19.Imamura T, Haruta T, Takata Y, Usui I, Iwata M, Ishihara H, Ishiki M, Ishibashi O, Ueno E, Sasaoka T, Kobayashi M. Involvement of heat shock protein 90 in the degradation of mutant insulin receptors by the proteasome. J Biol Chem 273: 11183–11188, 1998. doi: 10.1074/jbc.273.18.11183. [DOI] [PubMed] [Google Scholar]
- 20.Imamura T, Takata Y, Sasaoka T, Takada Y, Morioka H, Haruta T, Sawa T, Iwanishi M, Hu YG, Suzuki Y, , et al. Two naturally occurring mutations in the kinase domain of insulin receptor accelerate degradation of the insulin receptor and impair the kinase activity. J Biol Chem 269: 31019–31027, 1994. [PubMed] [Google Scholar]
- 21.Intengan HD, Schiffrin EL. Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants. Hypertension 36: 312–318, 2000. doi: 10.1161/01.HYP.36.3.312. [DOI] [PubMed] [Google Scholar]
- 22.Ito A, Shiroto T, Godo S, Saito H, Tanaka S, Ikumi Y, Kajitani S, Satoh K, Shimokawa H. Important roles of endothelial caveolin-1 in endothelium-dependent hyperpolarization and ischemic angiogenesis in mice. Am J Physiol Heart Circ Physiol 316: H900–H910, 2019. doi: 10.1152/ajpheart.00589.2018. [DOI] [PubMed] [Google Scholar]
- 23.Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, Fukai T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem 283: 9157–9167, 2008. doi: 10.1074/jbc.M709463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwanishi M, Haruta T, Takata Y, Ishibashi O, Sasaoka T, Egawa K, Imamura T, Naitou K, Itazu T, Kobayashi M. A mutation (Trp1193-->Leu1193) in the tyrosine kinase domain of the insulin receptor associated with type A syndrome of insulin resistance. Diabetologia 36: 414–422, 1993. doi: 10.1007/BF00402277. [DOI] [PubMed] [Google Scholar]
- 25.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res 93: 622–629, 2003. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 26.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation 109: 59–65, 2004. doi: 10.1161/01.CIR.0000105720.28086.6C. [DOI] [PubMed] [Google Scholar]
- 27.Karlsson K, Lindahl U, Marklund SL. Binding of human extracellular superoxide dismutase C to sulphated glycosaminoglycans. Biochem J 256: 29–33, 1988. doi: 10.1042/bj2560029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ke BX, Llanos RM, Wright M, Deal Y, Mercer JF. Alteration of copper physiology in mice overexpressing the human Menkes protein ATP7A. Am J Physiol Regul Integr Comp Physiol 290: R1460–R1467, 2006. doi: 10.1152/ajpregu.00806.2005. [DOI] [PubMed] [Google Scholar]
- 29.Komers R, Schutzer WE, Reed JF, Lindsley JN, Oyama TT, Buck DC, Mader SL, Anderson S. Altered endothelial nitric oxide synthase targeting and conformation and caveolin-1 expression in the diabetic kidney. Diabetes 55: 1651–1659, 2006. doi: 10.2337/db05-1595. [DOI] [PubMed] [Google Scholar]
- 30.Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci 42: 873–886, 2017. doi: 10.1016/j.tibs.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 31.La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Camakaris J, Mercer JF. Intracellular localization and loss of copper responsiveness of Mnk, the murine homologue of the Menkes protein, in cells from blotchy (Mo blo) and brindled (Mo br) mouse mutants. Hum Mol Genet 8: 1069–1075, 1999. doi: 10.1093/hmg/8.6.1069. [DOI] [PubMed] [Google Scholar]
- 32.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003. doi: 10.1172/JCI200314172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin MI, Yu J, Murata T, Sessa WC. Caveolin-1-deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res 67: 2849–2856, 2007. doi: 10.1158/0008-5472.CAN-06-4082. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab 8: 310–317, 2008. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luetterforst R, Stang E, Zorzi N, Carozzi A, Way M, Parton RG. Molecular characterization of caveolin association with the Golgi complex: identification of a cis-Golgi targeting domain in the caveolin molecule. J Cell Biol 145: 1443–1459, 1999. doi: 10.1083/jcb.145.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lund DD, Chu Y, Miller JD, Heistad DD. Protective effect of extracellular superoxide dismutase on endothelial function during aging. Am J Physiol Heart Circ Physiol 296: H1920–H1925, 2009. doi: 10.1152/ajpheart.01342.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mao M, Sudhahar V, Ansenberger-Fricano K, Fernandes DC, Tanaka LY, Fukai T, Laurindo FR, Mason RP, Vasquez-Vivar J, Minshall RD, Stadler K, Bonini MG. Nitroglycerin drives endothelial nitric oxide synthase activation via the phosphatidylinositol 3-kinase/protein kinase B pathway. Free Radic Biol Med 52: 427–435, 2012. doi: 10.1016/j.freeradbiomed.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao M, Varadarajan S, Fukai T, Bakhshi FR, Chernaya O, Dudley SC Jr, Minshall RD, Bonini MG. Nitroglycerin tolerance in caveolin-1 deficient mice. PLoS One 9: e104101, 2014. doi: 10.1371/journal.pone.0104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Materia S, Cater MA, Klomp LW, Mercer JF, La Fontaine S. Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J Biol Chem 287: 2485–2499, 2012. doi: 10.1074/jbc.M111.302216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mercer JF. Menkes syndrome and animal models. Am J Clin Nutr 67, Suppl: 1022S–1028S, 1998. doi: 10.1093/ajcn/67.5.1022S. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen AD, Itoh S, Jeney V, Yanagisawa H, Fujimoto M, Ushio-Fukai M, Fukai T. Fibulin-5 is a novel binding protein for extracellular superoxide dismutase. Circ Res 95: 1067–1074, 2004. doi: 10.1161/01.RES.0000149568.85071.FB. [DOI] [PubMed] [Google Scholar]
- 42.Oh YS, Khil LY, Cho KA, Ryu SJ, Ha MK, Cheon GJ, Lee TS, Yoon JW, Jun HS, Park SC. A potential role for skeletal muscle caveolin-1 as an insulin sensitivity modulator in ageing-dependent non-obese type 2 diabetes: studies in a new mouse model. Diabetologia 51: 1025–1034, 2008. doi: 10.1007/s00125-008-0993-0. [DOI] [PubMed] [Google Scholar]
- 43.Oh YS, Lee TS, Cheon GJ, Jang IS, Jun HS, Park SC. Modulation of insulin sensitivity and caveolin-1 expression by orchidectomy in a nonobese type 2 diabetes animal model. Mol Med 17: 4–11, 2011. doi: 10.2119/molmed.2009.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem 273: 5419–5422, 1998. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 45.Oliveira SDS, Castellon M, Chen J, Bonini MG, Gu X, Elliott MH, Machado RF, Minshall RD. Inflammation-induced caveolin-1 and BMPRII depletion promotes endothelial dysfunction and TGF-β-driven pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 312: L760–L771, 2017. doi: 10.1152/ajplung.00484.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliveira SDS, Chen J, Castellon M, Mao M, Raj JU, Comhair S, Erzurum S, Silva CLM, Machado RF, Bonini MG, Minshall RD. Injury-induced shedding of extracellular vesicles depletes endothelial cells of Cav-1 (caveolin-1) and enables TGF-β (transforming growth factor-β)-dependent pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 39: 1191–1202, 2019. doi: 10.1161/ATVBAHA.118.312038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliveira SDS, Minshall RD. Caveolin and endothelial NO signaling. Curr Top Membr 82: 257–279, 2018. doi: 10.1016/bs.ctm.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 48.Oshikawa J, Urao N, Kim HW, Kaplan N, Razvi M, McKinney R, Poole LB, Fukai T, Ushio-Fukai M. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS One 5: e10189, 2010. doi: 10.1371/journal.pone.0010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozumi K, Sudhahar V, Kim HW, Chen GF, Kohno T, Finney L, Vogt S, McKinney RD, Ushio-Fukai M, Fukai T. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: a key regulator of extracellular superoxide dismutase. Hypertension 60: 476–486, 2012. doi: 10.1161/HYPERTENSIONAHA.111.189571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14: 98–112, 2013. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 51.Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, Thøgersen IB, Jacobsen C, Bowler RP, Fattman CL, Crapo JD, Enghild JJ. Extracellular superoxide dismutase (EC-SOD) binds to type i collagen and protects against oxidative fragmentation. J Biol Chem 279: 13705–13710, 2004. doi: 10.1074/jbc.M310217200. [DOI] [PubMed] [Google Scholar]
- 52.Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15: 6084–6095, 1996. doi: 10.1002/j.1460-2075.1996.tb00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phillips-Krawczak CA, Singla A, Starokadomskyy P, Deng Z, Osborne DG, Li H, Dick CJ, Gomez TS, Koenecke M, Zhang JS, Dai H, Sifuentes-Dominguez LF, Geng LN, Kaufmann SH, Hein MY, Wallis M, McGaughran J, Gecz J, Sluis B, Billadeau DD, Burstein E. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol Biol Cell 26: 91–103, 2015. doi: 10.1091/mbc.e14-06-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin Z, Gongora MC, Ozumi K, Itoh S, Akram K, Ushio-Fukai M, Harrison DG, Fukai T. Role of Menkes ATPase in angiotensin II-induced hypertension: a key modulator for extracellular superoxide dismutase function. Hypertension 52: 945–951, 2008. doi: 10.1161/HYPERTENSIONAHA.108.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J 20: 334–336, 2006. doi: 10.1096/fj.05-4564fje. . [DOI] [PubMed] [Google Scholar]
- 56.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138, 2001. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 57.Saito H, Godo S, Sato S, Ito A, Ikumi Y, Tanaka S, Ida T, Fujii S, Akaike T, Shimokawa H. Important role of endothelial caveolin-1 in the protective role of endothelium-dependent hyperpolarization against nitric oxide-mediated nitrative stress in microcirculation in mice. J Cardiovasc Pharmacol 71: 113–126, 2018. doi: 10.1097/FJC.0000000000000552. [DOI] [PubMed] [Google Scholar]
- 58.Sawa T, Imamura T, Haruta T, Sasaoka T, Ishiki M, Takata Y, Takada Y, Morioka H, Ishihara H, Usui I, Kobayashi M. Hsp70 family molecular chaperones and mutant insulin receptor: differential binding specificities of BiP and Hsp70/Hsc70 determines accumulation or degradation of insulin receptor. Biochem Biophys Res Commun 218: 449–453, 1996. doi: 10.1006/bbrc.1996.0080. [DOI] [PubMed] [Google Scholar]
- 59.Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 18: 949–963, 2019. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- 60.Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol 39: 725–732, 2005. doi: 10.1016/j.yjmcc.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Starcher B, Madaras JA, Fisk D, Perry EF, Hill CH. Abnormal cellular copper metabolism in the blotchy mouse. J Nutr 108: 1229–1233, 1978. doi: 10.1093/jn/108.8.1229. [DOI] [PubMed] [Google Scholar]
- 62.Sudhahar V, Das A, Horimatsu T, Ash D, Leanhart S, Antipova O, Vogt S, Singla B, Csanyi G, White J, Kaplan JH, Fulton D, Weintraub NL, Kim HW, Ushio-Fukai M, Fukai T. Copper transporter ATP7A (copper-transporting P-type ATPase/menkes ATPase) limits vascular inflammation and aortic aneurysm development: role of microRNA-125b. Arterioscler Thromb Vasc Biol 39: 2320–2337, 2019. doi: 10.1161/ATVBAHA.119.313374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sudhahar V, Okur MN, Bagi Z, O’Bryan JP, Hay N, Makino A, Patel VS, Phillips SA, Stepp D, Ushio-Fukai M, Fukai T. Akt2 (protein kinase B beta) stabilizes ATP7A, a copper transporter for extracellular superoxide dismutase, in vascular smooth muscle: novel mechanism to limit endothelial dysfunction in type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol 38: 529–541, 2018. doi: 10.1161/ATVBAHA.117.309819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sudhahar V, Urao N, Oshikawa J, McKinney RD, Llanos RM, Mercer JF, Ushio-Fukai M, Fukai T. Copper transporter ATP7A protects against endothelial dysfunction in type 1 diabetic mice by regulating extracellular superoxide dismutase. Diabetes 62: 3839–3850, 2013. doi: 10.2337/db12-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamaguchi Y, Heiny ME, Suzuki M, Gitlin JD. Biochemical characterization and intracellular localization of the Menkes disease protein. Proc Natl Acad Sci USA 93: 14030–14035, 1996. doi: 10.1073/pnas.93.24.14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest 119: 2009–2018, 2009. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]