

Abstract
The cellular communication network factor 1 (CCN1) is a matricellular protein that can modulate multiple tissue responses, including inflammation and repair. We have previously shown that adenoviral overexpression of Ccn1 is sufficient to cause acute lung injury in mice. We hypothesized that CCN1 is present in the airspaces of lungs during the acute phase of lung injury, and higher concentrations are associated with acute respiratory distress syndrome (ARDS) severity. We tested this hypothesis by measuring 1) CCN1 in bronchoalveolar lavage fluid (BALF) and lung homogenates from mice subjected to ventilation-induced lung injury (VILI), 2) Ccn1 gene expression and protein levels in MLE-12 cells (alveolar epithelial cell line) subjected to mechanical stretch, and 3) CCN1 in BALF from mechanically ventilated humans with and without ARDS. BALF CCN1 concentrations and whole lung CCN1 protein levels were significantly increased in mice with VILI (n = 6) versus noninjured controls (n = 6). Ccn1 gene expression and CCN1 protein levels were increased in MLE-12 cells cultured under stretch conditions. Subjects with ARDS (n = 77) had higher BALF CCN1 levels compared with mechanically ventilated subjects without ARDS (n = 45) (P < 0.05). In subjects with ARDS, BALF CCN1 concentrations were associated with higher total protein, sRAGE, and worse / ratios (all P < 0.05). CCN1 is present in the lungs of mice and humans during the acute inflammatory phase of lung injury, and concentrations are higher in patients with increased markers of severity. Alveolar epithelial cells may be an important source of CCN1 under mechanical stretch conditions.
Keywords: acute lung injury, acute respiratory distress syndrome, CCN1
INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a clinical syndrome characterized by the acute onset of significant hypoxemia and bilateral radiographic pulmonary opacities that are not fully explained by cardiac dysfunction (32). More than 23% of all patients supported on mechanical ventilation worldwide meet criteria for ARDS (3). Despite recent improvements in supportive care for ARDS, no specific therapies directed at a known underlying pathophysiology have been shown to improve patient outcomes. ARDS is associated with a 28-day mortality of ∼33%, and long-term physical and psychological morbidity in survivors is significant (3, 10). A better understanding of the pathogenic mechanisms regulating inflammation in ARDS could lead to novel therapeutic targets.
The cellular communication network factor 1 (CCN1), previously known as cysteine-rich protein 61 (CYR61), is a transcriptionally regulated matricellular protein that may play an important role in ARDS. Ccn1 is expressed in fibroblasts, smooth muscle, endothelial, alveolar macrophages (AMs), and epithelial cells (7, 9, 12, 26, 36). In the lung, CCN1 protein expression is markedly upregulated by hyperoxia in bronchiolar and alveolar cells in vivo and in pulmonary bronchiolar and primary pulmonary artery smooth muscle cells in vitro (13). Stimulation of bronchial epithelial cells with TNFα, IFNγ, IL-1β, and LPS also leads to significant increases in CCN1 protein expression (13). Secreted CCN1 can bind to multiple integrins that trigger various downstream signal transduction events that influence cell adhesion, migration, gene expression, proliferation, and survival (4). CCN1 can have multiple and even contradictory functions, depending on specific integrin interactions, and thus different models of lung injury can elicit distinct CCN1 expression patterns and associations with physiological end points.
We have previously shown that CCN1 expression is increased in two distinct murine models of acute lung injury (ALI) that heal with fibrosis, Fas-L and bleomycin instillation, and that adenoviral-mediated Ccn1 overexpression is sufficient to cause an ALI phenotype (8, 19). These data suggest that CCN1 plays a key role in various models of lung injury. However, its role in more clinically relevant models of ALI such as ventilator-induced lung injury (VILI) or in human ARDS is unknown. To determine whether our previous findings are relevant to ARDS, we asked whether CCN1 is increased in an experimental model of ALI, mechanically ventilated mice, as well as in the lungs of mechanically ventilated patients with and without ARDS. We hypothesized that bronchoalveolar lavage fluid (BALF) CCN1 levels are increased in mice subjected to mechanical ventilation as well as in humans with ARDS.
MATERIALS AND METHODS
Animal protocols.
All animal experiments were performed according to protocols approved by the Institutional Animal Research Committee of the University of Washington (UW). Mice were housed in a pathogen-free environment according to UW animal use guidelines. Male C57BL/6 mice 8 wk of age were anesthetized with 5% inhaled isoflurane and then endotracheally intubated with a 20-gauge angiocatheter. Mice were randomly assigned into either a mechanically ventilated group (MV) or a spontaneously breathing control group (no MV). In the MV group, BALF was obtained after 5 h of mechanical ventilation with a tidal volume = 20 mL/kg at an = 50%.
Cell stretch in vitro assays.
Mouse alveolar epithelial MLE-12 cells (ATCC; no. CRL-2210) were cultured in RPMI (supplemented with 10% FBS) at 37°C in a humidified atmosphere of 5% CO2-95% air. The Flexercell 2000 cell stretching system (Flexcell International) was used to stretch MLE-12 cells grown to a confluent monolayer, as determined by light microscopy. Amplitudes of 20% strain were used, and stretch was applied in square fashion to mimic mechanical ventilation stretch at a rate of 30 cycles/min (0.5 Hz).
CCN1 ELISA.
We developed an ELISA to measure animal BALF CCN1 concentrations, as previously described (8). Murine Ccn1 cDNA (NM 010516.2; Sino Biological) was cloned into a mammalian expression vector to generate a Ccn1 standard. The construct contained the entire Ccn1-coding cDNA, including the native start codon and a 10-histidine tag on the 3′ end of the gene. Human embryonic kidney (HEK)-293 cells were transfected with the plasmid using Lipofectamine (Life Technologies). Cell lysates were collected 5 days after transfection. The expressed mouse Ccn1-HIS fusion protein was purified with a Ni+-loaded affinity column. The specificity of purified mouse CCN1 was determined by immunoblotting and silver staining of SDS-PAGE. We used a goat anti-mouse CCN1 IgG (Santa Cruz Biotechnology) as a capture antibody and biotinylated sheep anti-mouse CCN1 IgG (R & D Systems) as a detection antibody. The lower limit of detection of the assay was 31.25 ng/mL.
Western blots.
Samples containing equal amounts of protein were separated by SDS-PAGE under reducing conditions using NuPAGE Novex 4–12% Bis Tris polyacrylamide gradient gel with MES-SDS running buffer (Invitrogen). After electrophoresis, the gel contents were transferred onto a nitrocellulose membrane (Hybond-ECL; Amersham Biosciences) using NuPAGE transfer buffer (Invitrogen). Posttransfer, the membranes were incubated for 1 h with 5% nonfat milk, rinsed three times for 5 min in PBS with 0.1% Tween-20 (PBS-T; Sigma-Aldrich), and then incubated overnight at 4°C with primary antibody (rabbit anti-CCN1, cat no. ab24448; Abcam). After primary antibody labeling, the membranes were washed three times with PBS-T, and bound antibody was detected by incubation for 1 h with secondary antibody [goat anti-rabbit IgG-horseradish peroxidase (HRP)-conjugated, cat no. 1858415; Pierce]. The membranes were then washed five times with PBS-T and developed with ECL Prime Western Blotting detection reagent (Amersham). We used an Omega Ultra-Lum digital camera to visualize the membranes. After visualization of the signal for each respective antibody, the membranes were stripped of antibody by incubation for 15 min at room temperature in Restore Plus Western blot stripping buffer (Pierce). After being stripped, the membranes were washed three times in PBS-T and incubated for 30 min at room temperature in 5% nonfat milk. The blots were washed three times in PBS-T and then incubated overnight at 4°C with β-actin primary (rabbit anti-human β-actin, cat no. 4970; Cell Signaling Technology) and secondary antibody (goat anti-rabbit HRP conjugated; Invitrogen) for 1 h at room temperature, developed with the chemiluminescent substrate as described above, and visualized as described. Densitometry analysis of the immunoblots was performed using ImageJ software (National Institutes of Health).
Quantitative real-time PCR.
MLE-12 cells were lysed in TRIzol (Life Technologies). RNA was extracted in chloroform-isopropanol, washed in 75% ethanol, and resuspended in water. The samples were treated with DNAse for 30 min, and then cDNA was made with a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer’s instructions. The Ccn1 and Hprt (housekeeping gene) cDNA primer probes are listed in Supplemental Table S1 (all Supplemental Material for this article can be found online at https://doi.org/10.6084/m9.figshare.12733604). Quantitative PCR was performed using the SYBR green method. The ∆CT was the difference between the average CT for Ccn1 and Hprt. Data were interpreted using the ∆∆CT method.
Proliferation assay.
MLE-12 cells were seeded in 96-well microplates at a density of 3 × 104 cells/well and cultured in DMEM-F-12 (supplemented with 2% FCS) for 3 h at 37°C and 5% CO2. Growth media were then replaced with treatment media. Treatment conditions consisted of varying concentrations of recombinant CCN1 (0, 2, 20, 200, and 2,000 ng/mL) in serum-free 0.2% FCS and 2% FCS-supplemented media. Each treatment condition was performed in triplicate, and cells were incubated in treatment media for 24 h at 37°C and 5% CO2. Treatment media were aspirated after 24 h and plates stored in −80°C. Proliferation was measured using the CyQuantTM Cell Proliferation Assay kit (C7026; ThermoFisher Scientific). Frozen microplates were thawed, and assay reagent containing cell lysis buffer and fluorescent dye were added per the manufacturer’s instructions. Fluorescence was measured in a Biotek Synergy microplate reader (480 nm excitation and 520 nm emission). A standard fluorescence curve was generated with known MLE-12 cell numbers. The number of cells in each well was calculated using the measured fluorescence value referenced to the standard fluorescence curve. Three independent experiments were conducted, and proliferation data are presented as cell numbers per each treatment condition.
Chemokine (C-X-C motif) ligand 1 measurements.
MLE-12 cells were incubated on 96-well plates as described above until reaching 90% confluency. The media were replaced with media containing 10 µg/mL E. coli O11:B4 lipopolysaccharide (LPS; Sigma-Aldrich) or human recombinant CCN1 (Peprotech) at serial concentrations ranging from 20 to 2,000 ng/mL. The places were incubated at 37°C, 5% CO2, for 16 h. The conditioned media were retrieved, spun at 14,000 g, and stored at −80°C until measurement. Chemokine (C-X-C motif) ligand 1 (CXCL1) was measured by a commercial ELISA (R & D Systems).
Study population.
ARDS BALF samples (n = 77) were collected from subjects who were enrolled at five North American medical centers for a phase II, placebo-controlled trial of ω-3 fatty acids for the treatment of ARDS (31). Full inclusion and exclusion criteria have been described previously (23, 31). ARDS was adjudicated by the trial study team. Enrollment into the trial occurred within 48 h of ARDS onset. Samples were obtained within 48 h of enrollment and before subjects receiving treatment or placebo. Patients from both the placebo (n = 42) and treatment (n = 35) arms of the trial were included in our study. All studies were approved by the UW Human Subjects Division.
Non-ARDS BALF samples (n = 45) were collected from subjects who were enrolled in a prospective observational study conducted in the cardiac intensive care unit at the Hospital Universitario Central de Asturias in Oviedo, Spain, from February to June 2018. Patients were eligible if they remained on mechanical ventilation for >8 h due to a cardiac medical or surgical condition. Exclusion criteria included the presence of any chronic pulmonary condition, previous or current immunosuppression, previous or current cancer diagnosis, presence of a terminal condition, or inability to perform a research BAL (e.g., refractory hypoxemia). All samples were obtained within 24 h of onset of mechanical ventilation. The presence of cardiogenic pulmonary edema was adjudicated by two independent clinical researchers. Informed consent was obtained from patients’ next of kin, and the Clinical Research Ethics Committee of Principado de Asturias reviewed and approved the protocol.
Human BALF measurements.
CCN1 from the human BALF was measured using an ELISA (cat. no. DY4055) specific for human CCN1 per the manufacturer’s instructions (R & D Systems). BALF samples were diluted 1:2 before running the ELISA. All samples were assayed on the same day in a single batch. The lower limit of detection for the assay was 31.25 pg/mL.
Total protein concentration was determined using the bicinchoninic acid protein assay (ThermoFischer). BAL neutrophil percentages (%PMNs) were calculated from manual inspection of cytospin preparations made from each BALF cell pellet. / (P/F) ratio was extracted from the electronic medical record. BALF soluble form of receptor for advanced glycation end products (sRAGE) and IL-8 were measured using cytometric bead-based immunoassays per the manufacturer’s instructions (R & D Systems).
Alveolar macrophage isolation and gene expression.
In a subset of patients from the ω-3 fatty acid trial, AMs were purified from BALF by negative selection, as previously described (23–25). BALF was incubated with antibody-conjugated microbeads specific for the following markers: CD3 (T cells), CD15 (neutrophils), CD19 (B cells), CD235a (red blood cells), CD294 (eosinophils, basophils), and CD326 (epithelial cells). RNA extracted from isolated AMs was assessed for purity and then hybridized to Illumina HumanRef-8 Beadchips inclusive of 18,415 genes. We used the Bioconductor package lumi (6) to perform raw microarray data variance stabilization and quantile normalization. Gene expression is reported as log2 probe intensities.
Analyses.
Animal and in vitro data are expressed as individual data points, means, and standard deviations. Comparisons between two groups were done with a two-tailed t test. Comparisons between more than two groups were done with ANOVA with post hoc Tukey’s test. A P value of ≤0.05 was considered statistically significant.
For human association studies, BALF CCN1, IL-8, sRAGE, and total protein concentrations are expressed as individual values, medians, and interquartile ranges. sRAGE measurements were made on a random subset of subjects from the ARDS cohort (n = 29). Data were analyzed using nonparametric tests given their right-skewed distribution. Groups were divided by their median BALF CCN1, IL-8, sRAGE, and total protein concentrations. / (P/F) ratios were divided into mild/moderate (P/F > 150) versus moderate/severe (P/F < 150) based on a recent classification of ARDS severity (17). We used a Mann-Whitney test to identify significant differences between the medians of two groups and a Kruskal-Wallis test to identify significant variability in CCN1 concentrations between subjects with no pulmonary edema, cardiogenic pulmonary edema, and ARDS. For all analyses, P < 0.05 was considered statistically significant. All analyses were conducted in GraphPad Prism version 7.
RESULTS
Mechanical ventilation is associated with increased alveolar CCN1 concentrations.
We first tested whether CCN1 expression in the lungs increases in the setting of experimental lung injury. Mice were subjected to mechanical ventilation with a tidal volume of 20 mL/kg for 5 h (MV) and compared with spontaneously breathing mice (no MV). The MV mice developed ALI, as measured by an increase in the total number of alveolar neutrophils (PMNs) and in the total BALF protein concentrations (Fig. 1, A and B). The presence of soluble CCN1 in the alveolar compartment was assessed by measuring BALF CCN1 concentrations using a sandwich ELISA. We found a significant increase in soluble CCN1 in the BALF from the ventilated animals (means ± SD: MV = 1,290 pg/mL ± 129 vs. no MV = 322 pg/mL ± 49, P < 0.01; Fig. 1C). We next performed immunoblot analysis of total lung homogenates to assess the total CCN1 protein expression in the lungs. We found that total CCN1 protein expression was significantly increased in ventilated animals (Fig. 1, D and E).
Fig. 1.
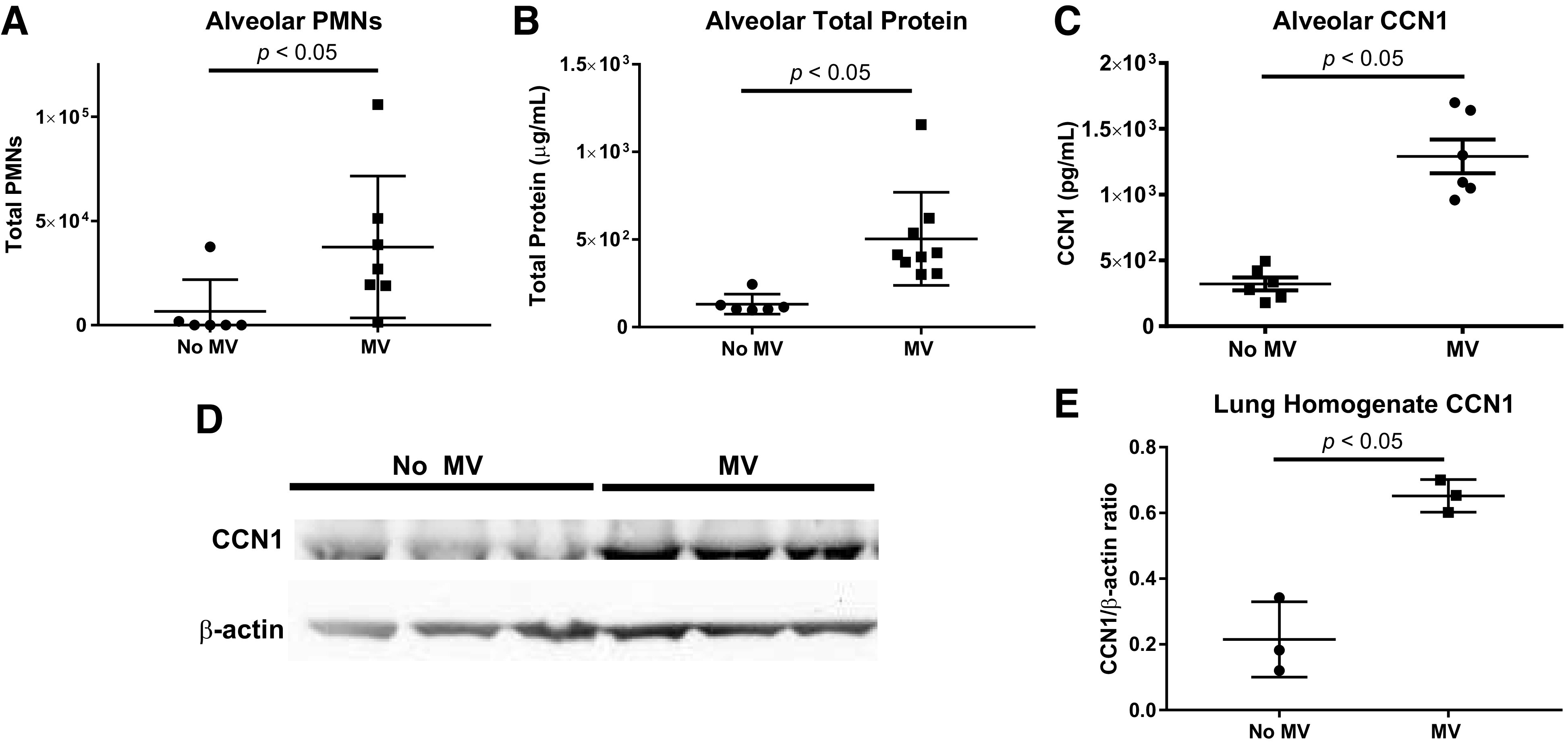
Soluble cellular communication network factor 1 (CCN1) bronchoalveolar lavage fluid (BALF) concentrations are increased in a ventilator-induced lung injury (VILI) model. We measured BALF total neutrophils (PMNs), total protein, and CCN1 concentration in mice subjected to mechanical ventilation at a tidal volume of 20 mL/kg for 5 h (MV) vs. spontaneously breathing control mice (no MV). Shown are the individual values, mean, and standard deviation. All comparisons were made using t tests. A: BALF total PMNs were increased in mice subjected to MV vs. no MV. B: BALF total protein concentrations were increased in mice subjected to MV vs. no MV. C: BALF CCN1 concentrations were increased in mice subjected to MV vs. no MV. D: Western blot probed for CCN1 and β-actin using protein lysates from lung homogenates collected from MV vs. no MV mice. The entire Western blot gel is shown in Supplemental Fig. S1. E: quantification by densitometry revealed a significant increase in CCN1/β-actin ratio in MV vs. no MV mice.
Mechanical stretch induces alveolar epithelial cell ccn1 expression in vitro.
CCN1 can be expressed in multiple cells in the lungs, including AMs and alveolar epithelial cells. To determine whether mechanical stretch of alveolar epithelial cells is sufficient to induce CCN1 expression, we next tested whether murine alveolar epithelial cell Ccn1 gene and protein expression are increased in response to mechanical stretch using an in vitro cell culture stretch model. MLE-12 cells are an immortalized murine cell line that exhibits characteristics of type II alveolar epithelial cells in vivo such as expression of surfactant proteins and secretion of phospholipids (20, 35). We measured Ccn1 gene expression in MLE-12 cells after exposure to cell culture stretch amplitudes of 20% at a rate of 30 cycles/min for 1 and 3 h and compared it with that of nonstretched controls (Fig. 2A). This level of epithelial cell stretching in vitro has previously been shown to approximate morphometric studies performed in ventilated animals (27). After 1 h of stretch, Ccn1 gene expression was significantly increased compared with nonstretched controls (P < 0.05). There was no difference in Ccn1 gene expression after 3 h of mechanical stretch. We then examined CCN1 protein expression in MLE-12 cells by Western blot analysis. CCN1 protein expression was induced by mechanical stretch in MLE-12 cells at 1 h compared with nonstretched controls (Fig. 2, B and C).
Fig. 2.
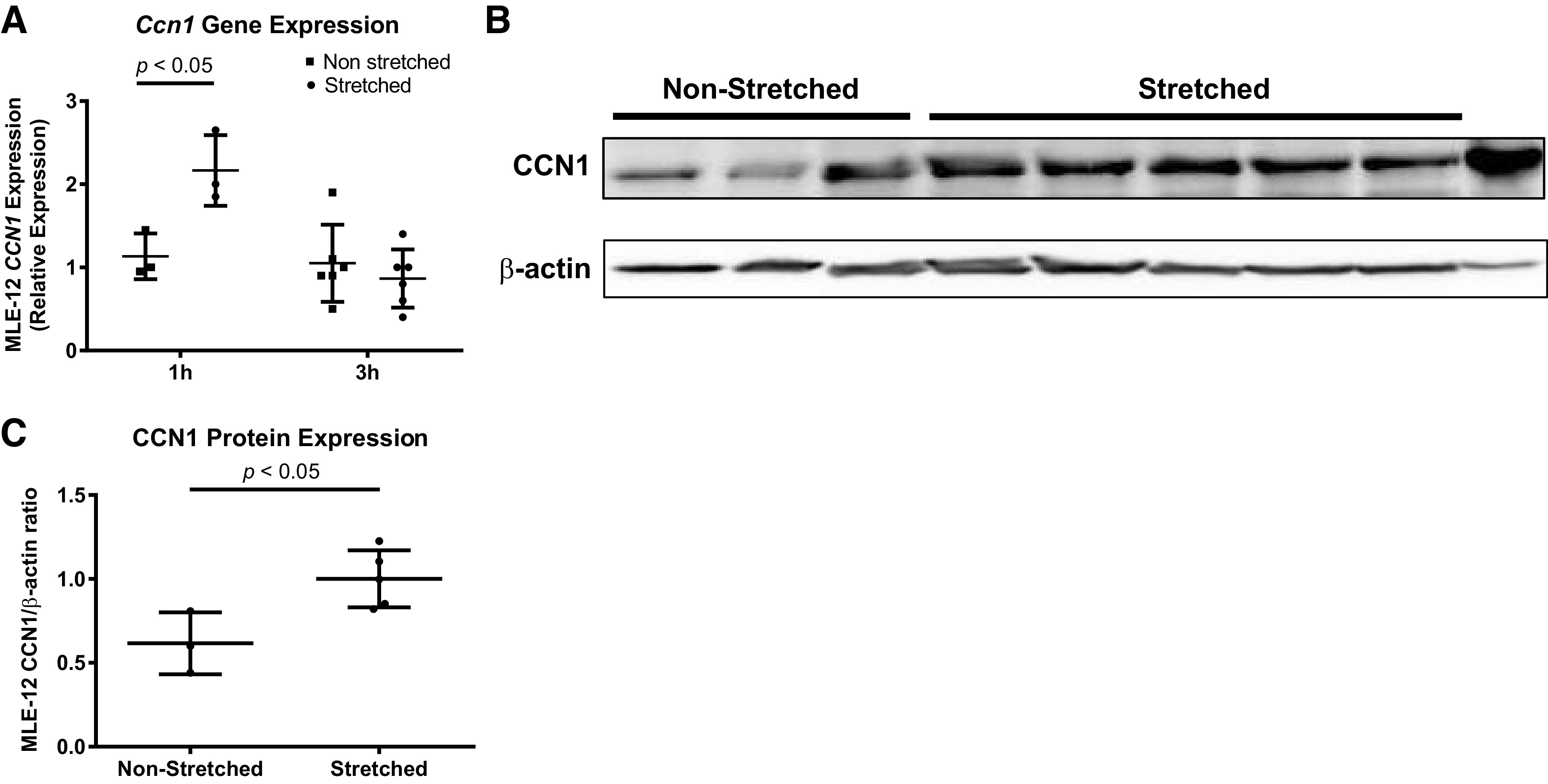
Mechanical stretch induces cellular communication network factor 1 (Ccn1) expression in alveolar epithelial cells. We exposed MLE-12 cells (a commonly used murine cell line expressing some features of normal type II alveolar epithelial cells) to 20% stretch at a cycling rate of 30/min. All comparisons were made using t tests. A: effect of 20% stretch on MLE-12 Ccn1 gene expression at 1 and 3 h. Shown are the individual values, mean, and standard deviation of Ccn1 relative gene expression. B: Western blot probed for CCN1 and β-actin using protein lysates from MLE-12 cells that were exposed to 20% stretch for 1 h. The entire Western blot gel is shown in Supplemental Fig. S2. C: quantification by band densitometry revealed a significant increase in CCN1/β-actin ratio in stretched vs. nonstretched controls. Shown are the individual values, mean, and standard deviation.
CCN1 induces alveolar epithelial cell proliferation and neutrophil chemokine release.
Given our findings that Ccn1 gene and protein expression were induced in both murine ALI and in vitro stretch models, we next investigated the direct effect of soluble CCN1 on alveolar epithelial cells. Previous research has shown that CCN1 can enhance bronchial epithelial cell IL-8 release (22) as well as accelerate repair of lung alveolar epithelium after inflammatory injury (37). We treated MLE-12 cells with increasing concentrations of soluble CCN1 and then measured neutrophil chemokine CXCL1/KC (murine functional IL-8 homologue) concentrations in the cell culture supernatants. Consistent with reports in bronchial epithelial cells, we found that high concentrations of CCN1 induced CXCL1 release from alveolar epithelial cells (Fig. 3A). We next sought to determine whether CCN1 might promote an alveolar epithelial cell proliferative response. We observed that high concentrations of CCN1 accelerated proliferation of alveolar epithelial cells (Fig. 3B). Overall, these findings suggest that CCN1 may induce alveolar epithelial cell injury responses, although the effects of CCN1 on alveolar epithelial cell proliferation and CXCL1 release were mainly observed at high doses of CCN1.
Fig. 3.

Cellular communication network factor 1 (CCN1) induces alveolar epithelial cell proliferation and neutrophil chemokine release. We treated MLE-12 cells with increasing concentrations of CCN1 and measured cell proliferation and chemokine (C-X-C motif) ligand 1 (CXCL1) supernatant concentrations. A: increasing doses of CCN1 were associated with increasing CXCL1 supernatant concentrations. Variance was tested with 1-way ANOVA. Post hoc Tukey’s test demonstrated a significant difference in the mean CXCL1 concentration between the 2,000 ng/mL CCN1 treatment group and all other treatment groups (P < 0.05). B: increasing doses of CCN1 were associated with increasing cell proliferation at a 0.2% FCS media condition. Variance was tested with 1-way ANOVA for each media condition. NS, not significant.
Alveolar CCN1 levels are increased in subjects with ARDS and are associated with measures of severity.
To determine the human relevance of our animal and in vitro data, we asked whether CCN1 alveolar concentrations are increased in humans with ARDS. We measured CCN1 concentrations in the BALF from a subset of patients previously enrolled in a phase II trial to study ω-3 fatty acids for the treatment of ARDS (31). As controls, we used cardiac patients supported on invasive mechanical ventilation for ≥8 h. Subject characteristics for both cohorts are shown in Table 1. Subjects from both cohorts were predominantly male. The P/F ratios, VFDs, and 28-day mortality were all worse in the ARDS cohort.
Table 1.
Subject characteristics
| Characteristic | ARDS Cohort (n = 77) | Non-ARDS Cohort (n = 45) |
|---|---|---|
| Demographic | ||
| Age (means ± SD) | 50 ± 16 | 45 ± 11 |
| Sex (%male) | 60% | 76% |
| Clinical diagnosis | ||
| ARDS | 77 (100%) | 0 (0%) |
| Cardiogenic pulmonary edema | 0 (0%) | 26 (58%) |
| No pulmonary edema | 0 (0%) | 19 (42%) |
| ARDS risk factor, n (%)* | ||
| Sepsis | 49 (64%) | NA |
| Pneumonia | 31 (42%) | NA |
| Trauma | 28 (36%) | NA |
| Other | 8 (11%) | NA |
| Physiological | ||
| P/F ratio (median, IQR) | 157 (121–208) | 214 (140–310) |
| Outcome | ||
| VFDs (median, IQR) | 14 (0–21) | 25 (17–27) |
| Mortality (28 days), n, % | 11 (20%) | 8 (18%) |
ARDS, acute respiratory distress syndrome; IQR, interquartile range; NA, not applicable; P/F: /; VFDs, ventilator-free days.
ARDS risk factors are not mutually exclusive.
BALF CCN1 concentrations were significantly higher in mechanically ventilated subjects with ARDS versus without ARDS (Fig. 4A). We next divided the non-ARDS subjects into those with cardiogenic pulmonary edema versus no pulmonary edema. BALF CCN1 concentrations were significantly different between these three groups and were highest in subjects with ARDS (Fig. 4B). We next analyzed only subjects with ARDS and tested whether there was a relationship between the BALF CCN1 concentrations and ARDS severity. Subjects with higher total protein concentrations had higher BALF CCN1 concentrations (Fig. 5A). There was a trend toward an association between higher percent PMNs and higher BALF CCN1 concentrations that did not achieve statistical significance (Fig. 5B).
Fig. 4.

Cellular communication network factor 1 (CCN1) bronchoalveolar lavage fluid (BALF) concentrations are higher in subjects with acute respiratory distress syndrome (ARDS) compared with non-ARDS controls. CCN1 concentrations in BALF collected from mechanically ventilated subjects without ARDS (non-ARDS) and from subjects with ARDS (ARDS). Non-ARDS subjects were comprised of patients with cardiogenic pulmonary edema (CPE; n = 26) and patients without any pulmonary edema (no CPE; n = 19). A: CCN1 concentrations were significantly higher in BALF from ARDS vs. non-ARDS subjects. Shown are the individual values, median, and interquartile range (Mann-Whitney U test). B: CCN1 concentrations were different in BALF from subjects with no cardiogenic pulmonary edema (no CPE), cardiogenic pulmonary edema (CPE), and ARDS. Shown are the individual values, median, and interquartile range (Kruskal-Wallis test). CCN1 was measured by ELISA.
Fig. 5.
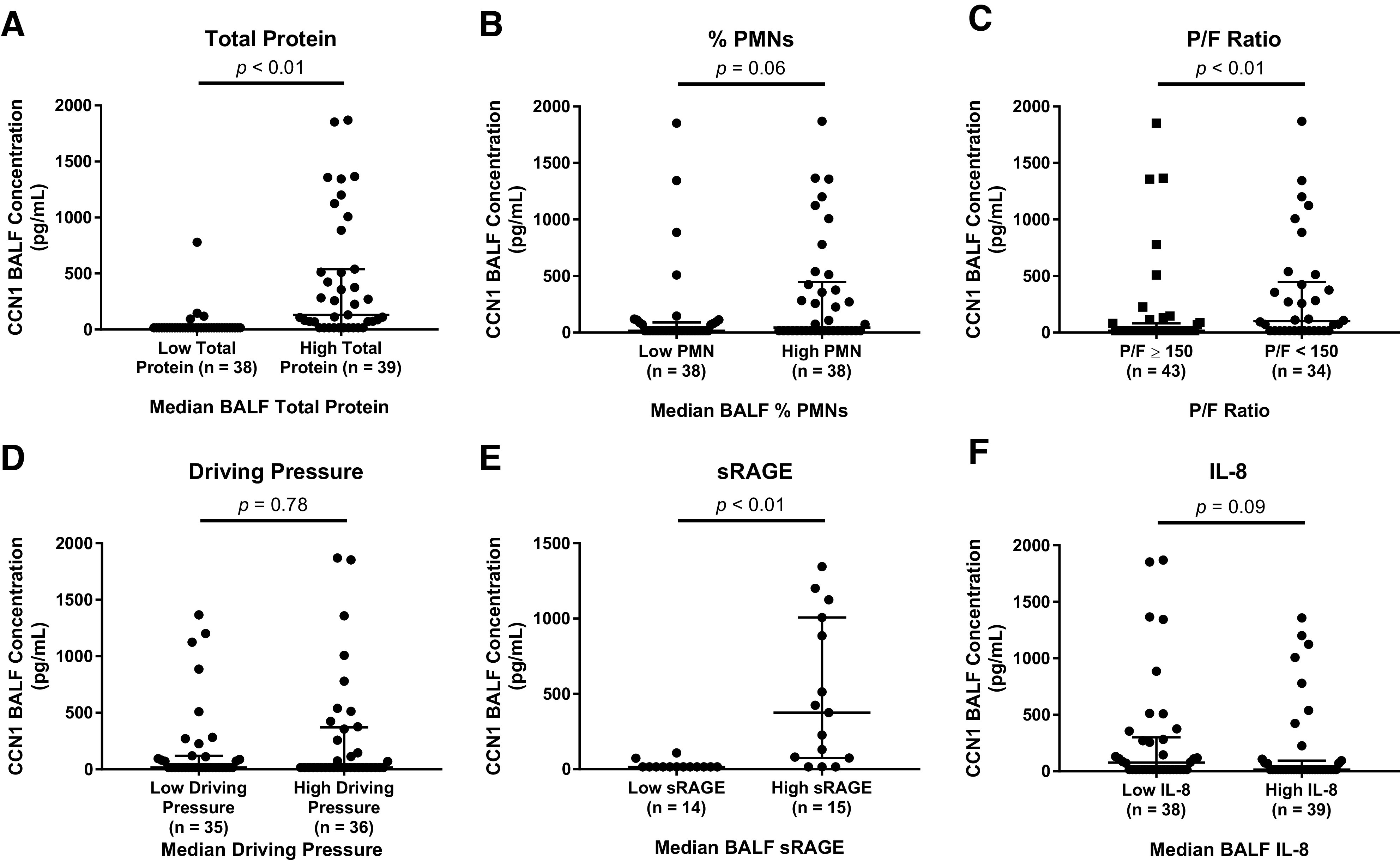
Cellular communication network factor 1 (CCN1) concentrations in bronchoalveolar lavage fluid (BALF) are associated with total protein and acute respiratory distress syndrome (ARDS) severity. A: CCN1 concentrations were higher in subjects with high BALF total protein as compared with low BALF total protein. Subjects were divided by the median alveolar total protein concentration. B: CCN1 concentrations were higher in BALF from ARDS subjects with high %PMN (total number of alveolar neutrophils) vs. low %PMNs. Subjects were divided by the median %PMNs. C: BALF CCN1 concentrations were higher in ARDS subjects with a / (P/F) ratio of <150 vs. subjects with a P/F ratio of ≥150. D: BALF CCN1 concentrations were not different between subjects with low vs. high driving pressures. Subjects were stratified by the median driving pressure. E: BALF CCN1 concentrations were higher in ARDS subjects with high BALF sRAGE vs. low BALF sRAGE concentrations. Subjects were stratified by the median alveolar soluble form of receptor for advanced glycation end products (sRAGE) concentration. F: BALF CCN1 concentrations were not associated with BALF IL-8 concentrations in subjects with ARDS. Subjects were stratified by the median alveolar IL-8 concentration. We used an ELISA to measure CCN1 concentrations from BALF collected from subjects with ARDS. Shown are the individual values, median, and interquartile range. All comparisons were made using a Mann-Whitney test.
Based on the finding that subjects with increased alveolar capillary leak (total protein) and PMN recruitment had higher BALF CCN1 concentrations, we next tested whether BALF CCN1 concentrations were associated with more severe ARDS based on P/F ratios and driving pressures. Subjects with more severe ARDS based on P/F ratios had significantly higher BALF concentrations compared with subjects with less severe ARDS (Fig. 5C). Surprisingly, BALF CCN1 concentrations were not different between subjects with low versus high driving pressures (Fig. 5D), perhaps reflecting significant patient heterogeneity in our cohort.
We next tested whether BALF CCN1 concentrations were associated with a biomarker linked with alveolar epithelial function, sRAGE, in subjects with ARDS. sRAGE is an isoform of membrane-bound RAGE, a transmembrane receptor constitutively expressed on alveolar epithelial cells (34). Plasma and BALF sRAGE levels are associated with impaired alveolar fluid clearance and have been used as a functional readout for decreased alveolar epithelial cell function (1, 11, 14). Subjects with high BALF sRAGE concentrations had higher BALF CCN1 concentrations (Fig. 5E). Notably, there was no difference in BALF CCN1 concentrations between subjects with high and low BALF IL-8 levels (Fig. 5F). AMs are the major source of the chemokine IL-8 in the airspaces (18).
Alveolar macrophage CCN1 expression and ARDS inflammation.
In exploratory analysis, we analyzed AM-specific CCN1 gene expression in a subset of n = 30 ARDS subjects from the fish oil trial who had AMs isolated by negative selection (25). Subjects with higher percent PMNs had higher AM-specific CCN1 gene expression (Supplemental Fig. S3A). AM-specific CCN1 gene expression was not associated with total protein concentration or P/F ratios (Supplemental Fig. S3, B and C).
DISCUSSION
The main findings from this study are that alveolar epithelial cells are an important source of CCN1 under mechanical stretch conditions and that alveolar CCN1 concentrations are increased in the acute inflammatory stage of lung injury in both animals and humans with ARDS. Mechanical stretch of murine alveolar epithelial cells is sufficient to induce Ccn1 gene and protein expression. High doses of CCN1 can induce proliferative and chemokine responses in murine alveolar epithelial cells. In subjects with ARDS, BALF CCN1 concentrations are associated with ARDS severity. Taken together, these findings support an overall biological model whereby mechanical stress increases alveolar epithelial cell CCN1 gene and protein expression, which in turn may contribute to ARDS pathogenesis and related adverse outcomes.
Our data clarify the role that CCN1 might play in clinically relevant models of ALI, such as mechanical ventilation. We have previously shown that increasing expression of Ccn1 in the lung using adenoviral vectors is sufficient to elicit lung injury in mice (8); however, mechanistic questions such as which cell types are producing and responding to Ccn1 are unknown. Herein, we have demonstrated that mechanical ventilation in mice is sufficient to increase Ccn1 gene and protein expression (Fig. 1) and that alveolar epithelial cells release CCN1 in response to mechanical stretch (Fig. 2). Our data also suggest that alveolar epithelial cells might respond to CCN1 in an autocrine fashion. High doses of CCN1 stimulated alveolar epithelial cell proliferation and neutrophil chemokine release (Fig. 3).
The pathogenesis of ALI is complex and poorly understood; however, it is tempting to speculate that these alveolar epithelial cell responses to CCN1 are mediated through integrins. There are at least eight integrins (αVβ3, α2β1, α5β1, α6β1, αVβ5, αIIbβ3, αMβ2, and αDβ2) that have been identified as key receptors of CCN1, and specific integrin binding by CCN1 can induce distinct cellular responses (4). For example, CCN1 must interact with both α6β1 and αVβ5 to elicit TNFα-mediated apoptosis (5). Mechanical strain leading to conformational changes in alveolar epithelial cell integrins is clearly implicated in transmitting stretch signals to downstream cellular functions (27, 29). CCN1 has been shown to influence cell survival, apoptosis, senescence, migration, and proliferation through integrin activation (4). Our data demonstrate that alveolar epithelial cell Ccn1 gene and protein expression are induced by mechanical stretch (Fig. 2) and that soluble alveolar concentrations of CCN1 are increased in mechanically ventilated mice compared with spontaneously breathing mice (Fig. 1). CCN1 has been shown to have distinct effects on cells, depending on specific integrin interactions. Future studies are needed to determine which integrin receptors mediate alveolar epithelial cell proliferation and chemokine responses to CCN1.
Our findings also extend the established role CCN1 plays in various other lung injury responses to ALI and ARDS. Moon and colleagues (21, 22) have shown that CCN1 secretion induced by cigarette smoking extracts augments IL-8 release from bronchial epithelial cells. Our data suggest that high concentrations of CCN1 may augment IL-8 release from alveolar epithelial cells (Fig. 3). ARDS is associated with a neutrophilic alveolitis, and alveolar IL-8 has been robustly implicated in the pathogenesis of ARDS (18, 33). It is possible that CCN1 may play an important role in how alveolar epithelial cells interact with neutrophils in ARDS. Interestingly, BALF IL-8 concentrations were not associated with CCN1 in our ARDS cohort (Fig. 5F). There are many other important sources of alveolar IL-8, including AMs, which may explain the discordance between our clinical and in vitro data.
Although CCN1 has been implicated in lung inflammatory responses, it has also been linked with acute reparative and fibroproliferative processes. Zemans et al. (37) have previously demonstrated that CCN1 is critical for epithelial wound repair in an in vitro neutrophil migration injury model. Our data expand upon these previous findings by showing that CCN1 may independently promote alveolar epithelial monolayer repair by increasing cell proliferation (Fig. 3). These data suggest that CCN1 is associated with acute repair, although other studies suggest that repeated cycles of injury may dysregulate the CCN1 pathway and lead to a profibrotic response (15, 16).
Our present study has confirmed that CCN1 levels are detectable in the BALF of mechanically ventilated subjects and are increased in subjects with ARDS. ARDS is a syndrome where mechanical stretch and VILI are presumed to play a causal role in the disease pathogenesis (2, 28, 30). This may explain why BALF CCN1 levels were higher in mechanically ventilated subjects with ARDS versus without ARDS (Fig. 4). Interestingly, increased BALF CCN1 levels were associated with increased BALF sRAGE concentrations (Fig. 5E). sRAGE is a biomarker that has been used as a functional readout for alveolar epithelial cell dysfunction. It is possible that alveolar epithelial cell damage from mechanical stretch may be linked to both increased BALF CCN1 and sRAGE levels; however, this hypothesis will need to be tested in future in vivo mechanistic studies.
There are several limitations in the present study. First, the cellular sources of BALF CCN1 in humans remains unclear. Our in vitro data (Fig. 2) and our prior findings suggest that alveolar epithelial cells may be an important source of CCN1 after acute injury, but we are unable to test this in humans with ARDS (8). However, associations between BALF CCN1 and sRAGE indicate a possible link between alveolar epithelial cell dysfunction and CCN1 secretion in ARDS. Additionally, we have shown that AMs express CCN1, and this might be associated with increased percent PMNs (Supplemental Fig. S3). Second, the BAL method (in mice and humans) only samples soluble CCN1 in the alveolar space and does not measure membrane-bound or extracellular-matrix bound CCN1, which could be biologically active. Finally, our findings from ARDS patients were derived from a single cohort and will need to be replicated in larger ARDS studies that employ alveolar sampling.
In conclusion, we have demonstrated that alveolar CCN1 concentrations are increased in a mechanical ventilation model of ALI and in patients with ARDS. In patients with ARDS, alveolar CCN1 levels are associated with increased total protein in the alveolar space as well as reduced P/F ratio. Alveolar epithelial cells cultured under mechanical stretch conditions have increased Ccn1 gene and protein expression. To our knowledge, this is the first study that has evaluated the role of CCN1 in the alveolar fluid of subjects with ARDS. Our report highlights the need for future mechanistic studies to determine the cellular origins of CCN1 in the lung as well as its mechanistic targets.
GRANTS
This work was supported by NIH Grant K23-HL-144916 and a Francis Family Foundation/Parker B. Francis Fellowship (E. D. Morrell), NIH Grant P50-HL-073996 (M. M. Wurfel), Swiss National Science Foundation Grant PBGEP3-142293 (S. Grazioli), and Merit Award No. I01BX002914 from the US Department of Veterans Affairs Biomedical Laboratory R & D (BLRD) Service (G. Matute-Bello).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.D.M., S.G. and G.M.B conceived and designed research; E.D.M., S.G., C.F.H., O.K., L.A. and S.K. performed experiments; E.D.M., M.W., S.G., O.K., S.K., R.S., S.A.G., L.A., and G.A., and G.M.B. analyzed data; E.D.M., M.W., S.G., C.F.H., S.K., R.S., S.A.G., L.A., G.A. and G.M.B. interpreted results of experiments; E.D.M., S.G. and G.M.B. prepared figures; E.D.M., S.G. and G.M.B drafted manuscript; E.D.M., M.W., G.A., S.G., C.F.H., O.K., S.K., R.S., S.G., L.A., and G.M.B. edited and revised manuscript; E.D.M., M.W., G.A., S.G., C.F.H., O.K., S.K., R.S., S.A.G., L.A., and G.M.B. approved final version of manuscript.
REFERENCES
- 1.Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 297: L1035–L1041, 2009. doi: 10.1152/ajplung.00214.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beitler JR, Malhotra A, Thompson BT. Ventilator-induced Lung Injury. Clin Chest Med 37: 633–646, 2016. doi: 10.1016/j.ccm.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, Ranieri M, Rubenfeld G, Thompson BT, Wrigge H, Slutsky AS, Pesenti A; LUNG SAFE Investigators; ESICM Trials Group . Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315: 788–800, 2016. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 4.Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 41: 771–783, 2009. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen CC, Young JL, Monzon RI, Chen N, Todorović V, Lau LF. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J 26: 1257–1267, 2007. doi: 10.1038/sj.emboj.7601596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24: 1547–1548, 2008. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 7.Gashaw I, Stiller S, Böing C, Kimmig R, Winterhager E. Premenstrual regulation of the pro-angiogenic factor CYR61 in human endometrium. Endocrinology 149: 2261–2269, 2008. doi: 10.1210/en.2007-1568. [DOI] [PubMed] [Google Scholar]
- 8.Grazioli S, Gil S, An D, Kajikawa O, Farnand AW, Hanson JF, Birkland T, Chen P, Duffield J, Schnapp LM, Altemeier WA, Matute-Bello G. CYR61 (CCN1) overexpression induces lung injury in mice. Am J Physiol Lung Cell Mol Physiol 308: L759–L765, 2015. doi: 10.1152/ajplung.00190.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanna M, Liu H, Amir J, Sun Y, Morris SW, Siddiqui MA, Lau LF, Chaqour B. Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J Biol Chem 284: 23125–23136, 2009. doi: 10.1074/jbc.M109.019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herridge MS, Tansey CM, Matté A, Tomlinson G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart TE, Kudlow P, Cook D, Slutsky AS, Cheung AM; Canadian Critical Care Trials Group . Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 364: 1293–1304, 2011. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 11.Jabaudon M, Blondonnet R, Roszyk L, Bouvier D, Audard J, Clairefond G, Fournier M, Marceau G, Déchelotte P, Pereira B, Sapin V, Constantin J-M. Soluble receptor for advanced glycation end-products predicts impaired alveolar fluid clearance in acute respiratory distress syndrome. Am J Respir Crit Care Med 192: 191–199, 2015. doi: 10.1164/rccm.201501-0020OC. [DOI] [PubMed] [Google Scholar]
- 12.Jin Y, Kim HP, Cao J, Zhang M, Ifedigbo E, Choi AMK. Caveolin-1 regulates the secretion and cytoprotection of Cyr61 in hyperoxic cell death. FASEB J 23: 341–350, 2009. doi: 10.1096/fj.08-108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y, Kim HP, Ifedigbo E, Lau LF, Choi AMK. Cyr61 protects against hyperoxia-induced cell death via Akt pathway in pulmonary epithelial cells. Am J Respir Cell Mol Biol 33: 297–302, 2005. doi: 10.1165/rcmb.2005-0144OC. [DOI] [PubMed] [Google Scholar]
- 14.Jones TK, Feng R, Kerchberger VE, Reilly JP, Anderson BJ, Shashaty MGS, Wang F, Dunn TG, Riley TR, Abbott J, Ittner CAG, Christiani DC, Mikacenic C, Wurfel MM, Ware LB, Calfee CS, Matthay MA, Christie JD, Meyer NJ. Plasma sRAGE acts as a genetically regulated causal intermediate in sepsis-associated acute respiratory distress syndrome. Am J Respir Crit Care Med 201: 47–56, 2020. doi: 10.1164/rccm.201810-2033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kulkarni T, Kurundkar AR, Kim Y-I, de Andrade J, Luckhardt T, Thannickal VJ. The senescence-associated matricellular protein CCN1 in plasma of human subjects with idiopathic pulmonary fibrosis. Respir Med 161: 105821, 2020. doi: 10.1016/j.rmed.2019.105821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurundkar AR, Kurundkar D, Rangarajan S, Locy ML, Zhou Y, Liu R-M, Zmijewski J, Thannickal VJ. The matricellular protein CCN1 enhances TGF-β1/SMAD3-dependent profibrotic signaling in fibroblasts and contributes to fibrogenic responses to lung injury. FASEB J 30: 2135–2150, 2016. doi: 10.1096/fj.201500173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maiolo G, Collino F, Vasques F, Rapetti F, Tonetti T, Romitti F, Cressoni M, Chiumello D, Moerer O, Herrmann P, Friede T, Quintel M, Gattinoni L. Reclassifying Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 197: 1586–1595, 2018. doi: 10.1164/rccm.201709-1804OC. [DOI] [PubMed] [Google Scholar]
- 18.Martin TR. Lung cytokines and ARDS: Roger S. Mitchell lecture. Chest 116, Suppl: 2S–8S, 1999. doi: 10.1378/chest.116.suppl_1.2S. [DOI] [PubMed] [Google Scholar]
- 19.Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, Madtes DK, Shapiro SD, Martin TR. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol 37: 210–221, 2007. doi: 10.1165/rcmb.2006-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mir-Kasimov M, Sturrock A, McManus M, Paine R III. Effect of alveolar epithelial cell plasticity on the regulation of GM-CSF expression. Am J Physiol Lung Cell Mol Physiol 302: L504–L511, 2012. doi: 10.1152/ajplung.00303.2010. [DOI] [PubMed] [Google Scholar]
- 21.Moon HG, Kim SH, Gao J, Quan T, Qin Z, Osorio JC, Rosas IO, Wu M, Tesfaigzi Y, Jin Y. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am J Physiol Lung Cell Mol Physiol 307: L326–L337, 2014. doi: 10.1152/ajplung.00102.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon HG, Zheng Y, An CH, Kim YK, Jin Y. CCN1 secretion induced by cigarette smoking extracts augments IL-8 release from bronchial epithelial cells. PLoS One 8: e68199, 2013. doi: 10.1371/journal.pone.0068199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrell ED, Bhatraju PK, Mikacenic CR, Radella F II, Manicone AM, Stapleton RD, Wurfel MM, Gharib SA. Alveolar macrophage transcriptional programs are associated with outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 200: 732–741, 2019. doi: 10.1164/rccm.201807-1381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrell ED, Mikacenic C, Gong K-Q, Kosamo S, Wurfel MM, Manicone AM. Alveolar MMP28 is associated with clinical outcomes and measures of lung injury in acute respiratory distress syndrome. Crit Care 24: 141, 2020. doi: 10.1186/s13054-020-02847-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrell ED, Radella F II, Manicone AM, Mikacenic C, Stapleton RD, Gharib SA, Wurfel MM. Peripheral and alveolar cell transcriptional programs are distinct in acute respiratory distress syndrome. Am J Respir Crit Care Med 197: 528–532, 2018. doi: 10.1164/rccm.201703-0614LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pendurthi UR, Allen KE, Ezban M, Rao LV. Factor VIIa and thrombin induce the expression of Cyr61 and connective tissue growth factor, extracellular matrix signaling proteins that could act as possible downstream mediators in factor VIIa x tissue factor-induced signal transduction. J Biol Chem 275: 14632–14641, 2000. doi: 10.1074/jbc.275.19.14632. [DOI] [PubMed] [Google Scholar]
- 27.Pugin J. Molecular mechanisms of lung cell activation induced by cyclic stretch. Crit Care Med 31, Suppl: S200–S206, 2003. doi: 10.1097/01.CCM.0000057844.31307.ED. [DOI] [PubMed] [Google Scholar]
- 28.Sahetya SK, Mancebo J, Brower RG. Fifty Years of Research in ARDS. Vt Selection in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 196: 1519–1525, 2017. doi: 10.1164/rccm.201708-1629CI. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheppard D. Functions of pulmonary epithelial integrins: from development to disease. Physiol Rev 83: 673–686, 2003. doi: 10.1152/physrev.00033.2002. [DOI] [PubMed] [Google Scholar]
- 30.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 369: 2126–2136, 2013. doi: 10.1056/NEJMra1208707. [DOI] [PubMed] [Google Scholar]
- 31.Stapleton RD, Martin TR, Weiss NS, Crowley JJ, Gundel SJ, Nathens AB, Akhtar SR, Ruzinski JT, Caldwell E, Curtis JR, Heyland DK, Watkins TR, Parsons PE, Martin JM, Wurfel MM, Hallstrand TS, Sims KA, Neff MJ. A phase II randomized placebo-controlled trial of omega-3 fatty acids for the treatment of acute lung injury. Crit Care Med 39: 1655–1662, 2011. doi: 10.1097/CCM.0b013e318218669d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS; ARDS Definition Task Force . Acute respiratory distress syndrome: the Berlin Definition. JAMA 307: 2526–2533, 2012. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 33.Thompson BT, Chambers RC, Liu KD. Acute Respiratory Distress Syndrome. N Engl J Med 377: 562–572, 2017. doi: 10.1056/NEJMra1608077. [DOI] [PubMed] [Google Scholar]
- 34.Uchida T, Shirasawa M, Ware LB, Kojima K, Hata Y, Makita K, Mednick G, Matthay ZA, Matthay MA. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med 173: 1008–1015, 2006. doi: 10.1164/rccm.200509-1477OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, Whitsett JA. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci USA 90: 11029–11033, 1993. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.You JJ, Yang CM, Chen MS, Yang CH. Regulation of Cyr61/CCN1 expression by hypoxia through cooperation of c-Jun/AP-1 and HIF-1α in retinal vascular endothelial cells. Exp Eye Res 91: 825–836, 2010. doi: 10.1016/j.exer.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Zemans RL, McClendon J, Aschner Y, Briones N, Young SK, Lau LF, Kahn M, Downey GP. Role of β-catenin-regulated CCN matricellular proteins in epithelial repair after inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol 304: L415–L427, 2013. doi: 10.1152/ajplung.00180.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]