

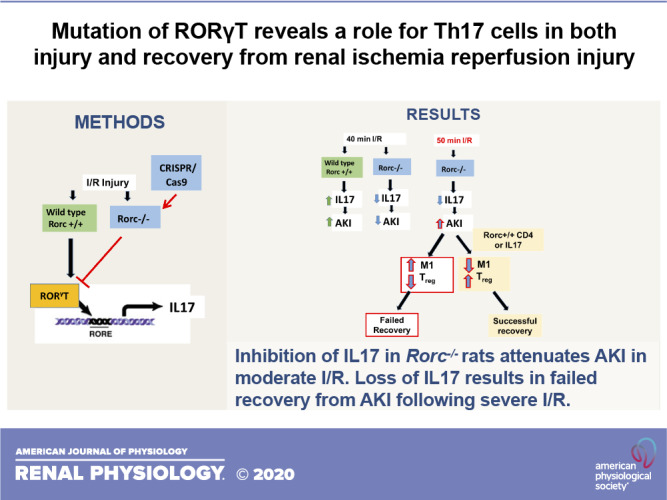
Keywords: acute kidney injury, cell proliferation, inflammation, lymphocytes
Abstract
To investigate T helper type 17 (Th17) cells in the setting of acute kidney injury, the gene encoding the master regulator of Th17 cell differentiation, that is, RAR-related orphan receptor-γ (RORγT), was mutated in Lewis rats using CRISPR/Cas9 technology. In response to 40 min of bilateral renal ischemia-reperfusion (I/R), RAR-related orphan receptor C (Rorc)−/− rats were resistant to injury relative to wild-type Rorc+/+ rats. This protection was associated with inhibition of IL-17 expression and reduced infiltration of CD4+ cells, CD8+ cells, B cells, and macrophages. To evaluate the effect of Th17 cells on repair, ischemia was increased to 50 min in Rorc−/− rats. This maneuver equalized the initial level of injury in Rorc−/− and Rorc+/+ rats 1 to 2 days post-I/R based on serum creatinine values. However, Rorc−/− rats, but not Rorc+/+ rats, failed to successfully recover renal function and had high mortality by 4 days post-I/R. Histological assessment of kidney tubules showed evidence of repair by day 4 post-I/R in Rorc+/+ rats but persistent necrosis and elevated cell proliferation in Rorc−/− rats. Adoptive transfer of CD4+ cells from the spleen of Rorc+/+ rats or supplementation of exogenous rIL-17 by an osmotic minipump improved renal function and survival of Rorc−/− rats following 50 min of I/R. This was associated with a relative decrease in the number of M1-type macrophages and a relative increase in the percentage of T regulatory cells. Taken together, these data suggest that Th17 cells have both a deleterious and a beneficial role in kidney injury and recovery, contributing to early postischemic injury and inflammation but also possibly being critical in the resolution of inflammation during kidney repair.
INTRODUCTION
Acute kidney injury (AKI) is defined by a rapid loss of renal function typically associated with ischemic or nephrotoxic injury, which is associated with high mortality rates. The disruption of renal blood flow and the presence of frank tubular damage represent important early features contributing to the pathogenesis of AKI. Studies in animal models have indicated that recovery from AKI involves resolution of renal blood flow and the initiation of an orchestrated renal repair response (5). However, incomplete repair of damaged tubules, rarefaction of renal peritubular capillaries, and activation of myofibroblasts represent maladaptive changes that may contribute to the development of chronic kidney disease (CKD) (5).
In addition, a variety of immune cells contributes to both the development of and recovery from AKI and the AKI-to-CKD transition (5, 18). For example, proinflammatory macrophages, often classified as “M1,” increase local oxidant stress and secrete cytokines such as TNF-α that contribute to tubular injury, whereas recovery from AKI may be hastened by a switch to a “M2” macrophage profile, characterized by secretion of cytokines and growth factors promoting kidney repair (16). In addition, effector T cells may differentiate into different T helper (Th) subtypes such as Th1, Th2, or Th17 under the influence of the renal injury milieu and influence the course of AKI. In contrast, a subset of CD4+ cells differentiate to T-regulatory cells, which have immunosuppressive activity and facilitate recovery from AKI, in part via secretion of IL-10 (22, 32).
Th17 cells are characterized by their secreted cytokines that include primarily IL-17A but also IL-17F, IL-21, IL-22, and IL-26 (10). Th17 cells are the most abundant Th population induced by renal ischemia-reperfusion (I/R) injury and are prominently reexpressed in rats challenged with high-salt diet following recovery from AKI (25). This treatment hastens the progression of CKD, which is significantly attenuated by treatment with the soluble receptor inhibitor IL-17Rc (24). However, the pathophysiological mechanisms that result in the loss of renal function in AKI or progression of the AKI-to-CKD transition remain incompletely defined.
The differentiation of Th17 cells is dependent on the activity of the transcription factor RAR-related orphan receptor-γ (RORγT) coded by the Rorc (RAR-related orphan receptor C) gene. RAR-related orphan receptor C (Rorc) (or RoRγT)-null mice display impaired differentiation of Th17 cells based on the expression of IL-17A and other Th17-associated cytokines (17). These mice have been previously used to study the role of Th17 cells in kidney disease models (7, 34). Although mouse models are routinely used to investigate mechanisms of kidney injury and the development of renal fibrosis, they have practical limitations. Progression of CKD and hypertension are associated with alterations in renal sodium handling as well as alterations in systemic and renal hemodynamic responses, which may be more easily investigated in rats. For these reasons, we sought to develop a Rorc mutant rat with the long-term goal of investigating the function of Th17 cells on AKI, CKD, blood pressure regulation, and hemodynamics. The current report describes initial findings using a CRISPR/Cas9 approach to mutate the Rorc gene in Lewis rats. As anticipated, the data suggest that Rorc mutation impairs Th17 activation and influences the severity of AKI in response to I/R. However, we also identified unanticipated effects of Rorc mutation on recovery following severe AKI, suggesting a potential beneficial role of Th17 cells in kidney repair.
MATERIALS AND METHODS
Animals.
Rats were maintained in accordance with the policies of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experimental protocols were approved by Institutional Animal Care and Use Committees of Indiana University or the Medical College of Wisconsin. A CRISPR/Cas9 system was used in Lewis rat (LEW/NCrl, Charles River Laboratories) embryos to induce a mutation of the rat Rorc gene (XM_032897831.1) using a previously described technique (1). The targeted sequence was GTGATCCCTTGCAAGATCTG. Genotyping included sequencing of the PCR products produced with the following primers: forward 5′-GCTCACTGAAGAAGTAGGGAGAGC-3′ and reverse 5′-CATGTGCACTGGTCTTAGGGAT-3′, and sequencing confirmed an 8-bp frame-shift deletion (GATCTGTG) in exon 3 (rn6-chr2:195,626,836–195,626,843).
F2 and further litters were used to establish a colony in the animal facilities initially at the Medical College of Wisconsin and then transferred to Indiana University for maintenance. Sequence information was used to predict the final protein product with ExPAsy-Translate (Swiss Institute of Bioinformatics) and illustrated using Protter (http://wlab.ethz.ch/protter). The resulting mutant is referred to as LEW-Rorcem3Mcwi (RGDID: 10054444) but is hereafter referred to as Rorc−/−, whereas wild-type Lewis rats derived from the same colony are referred to as Rorc+/+. Both strains are bred in house as homozygotes and are maintained on standard laboratory chow (Teklad). Western blot analysis of RORγT was conducted from the thymus homogenized with T-PER protein extraction reagent (Cat. No. 78510, ThermoFisher, Waltham, MA) in the presence of a protease inhibitor cocktail (Cat. No. P8340, Sigma, St. Louis, MO). After electrophoresis and transfer to 0.45-µm nitrocellulose membranes (GE Healthcare, Chicago, IL), blots were probed with mouse anti-RORγT antibody (0.2 µg/mL, Cat. No. 4G419, ThermoFisher) and bands visualized by chemiluminescence (SuperSignal ECL, ThermoFisher). Studies used both genotypes with targeted initial starting weight ∼225–300 g; for each study, efforts were made to ensure that body weights were similar between genotypes. The studies used only male rats because female rats were found to be intolerant to the anesthesia.
A series of studies was designed to address the effect of the RORγT mutation on the course of AKI. Study 1 was designed to evaluate the sensitivity of different rat strains to renal I/R at different levels of ischemia. Rorc−/−and Rorc+/+ rats were anesthetized with ketamine (100 mg/kg)/xylazine (5 mg/kg), and warm renal I/R was induced by bilateral clamping of the renal pedicles using a surgical approach that has been described previously (25). Ischemic times were 40 or 50 min, as indicated, and reestablishment of perfusion was verified by visual examination following removal of the clamps. Rats were provided postoperative analgesia using buprenorphine-SR (1 mg/kg) and allowed to recover for 2 or 7 days following surgery.
Study 2 used an adoptive transfer approach to provide either wild-type or mutant lymphocytes into Rorc−/− mutant rats following induction of renal I/R. Spleens from Rorc+/+ or Rorc−/− rats were harvested, and CD4+ cells were isolated following tissue digestion and antibody-mediated magnetic bead isolation (see later text). Freshly isolated CD4+ cells were administered to Rorc−/− rats within 15 min of reperfusion (following suturing of the incision) by an intraperitoneal injection at a dose of 1 × 106 cells per rat.
Study 3 sought to investigate the effect of IL-17 supplementation on recovery from renal injury in Rorc−/− rats. Recombinant IL-17 (Peprotech, Rock Hill, NJ) was loaded into osmotic minipumps (Alzet model 1007D) according to the manufacturer’s instructions at a concentration designed to achieve a dose of 50 or 100 ng·kg−1·day−1. Saline-loaded osmotic minipumps were used as controls. Pumps were placed intraperitoneally into rats immediately after reperfusion.
Measurements of renal function.
To measure creatinine, blood from rats was collected via tail clipping in heparin-containing tubes and spun to collect plasma. Plasma creatinine was measured using a Pointe Scientific analyzer and creatinine assay reagents using methods outlined by the manufacturer (Pointe Scientific, Canton, MI). In some experiments, aortic blood was obtained from anesthetized rats at the time of euthanasia and was used to determine plasma pH and serum analytes using a GEM Premier 4000 analyzer (Instrumentation Laboratories, Bedford, MA).
Renal histology and immunohistochemistry.
At the time of tissue harvest, kidneys were bisected, one half was fixed by immersion in 10% formalin and embedded in paraffin, and 5-µm sections stained with hematoxylin and eosin. Immunohistochemistry to identify proliferating cells used an antibody against Ki67 (rabbit anti-human/rat, Cat. No. PIPA516785, ThermoFisher) and staining was done using a Histostain-SP staining kit (Life Technologies, Carlsbad, CA). Images were obtained using Leica EC4 camera (Scientific Instruments, Columbus, OH) mounted on a Nikon Optimphot-2 microscope with a ×20 objective lens. Quantitative analysis of tissue damage was conducted based on approaches previously described (4). Briefly, five micrographs were obtained from the outer medulla of each kidney by an investigator blinded to the treatment groups, and scoring was conducted by another investigator also blinded to the treatment groups. Each tubule was categorized as severely damaged, mildly damaged, or undamaged, with severely damaged cells being classified as those with abundant cellular debris and a thinned epithelium, whereas moderately damaged cells showed evidence of cellular hypertrophy with minimal tubular debris. Data are expressed as the percentage of severely damaged tubules from ∼200–400 tubules scored per kidney.
FACS analysis of immune cells.
Harvested kidneys were minced and digested in liberase (2 μg/mL; Roche, Indianapolis, IN) for 15 min at 37°C with the help of Gentle MACs (Miltenyli, San Diego, CA). The digested tissue was filtered through a 100-μm filter mesh and washed with DMEM containing 10% fetal bovine serum (Sigma). The mononuclear cells were separated by Percoll (Sigma) and counted using a hemocytometer. To evaluate T-lymphocytes, the cells were stained with antibodies against rat CD4 (PE-Cy7; BD BioLegend, San Diego, CA) and CD8a (Alexa 647; BD BioLegend, San Diego, CA). To evaluate the cytokines secreted by T cells, the cells were stained for the CD4 surface marker, permeabilized using 0.1% saponin, and stained with antibodies against IL-17 (FITC; BD BioLegend) as described previously (25). To evaluate macrophages, cells were stained with macrophage/dendritic cell marker CD11b/c and subsequently analyzed for M1 (CD80 PE, BD BioLegend, and 86, BD PharMingen, San Diego, CA) or the M2 marker CD206 (PECy7, Bio-Rad, Hercules, CA). Cells were scanned using flow cytometry (FACSCalibur, BD Biosciences), and scans were analyzed using Flowjo software (Tree Star, Ashland, OR). Lymphocyte gating strategy was based on our previously described approach (25). Representative gating used for analysis of CD4+/CD8+ T cells is shown in Fig. 1, path A, and for dendritic cells and macrophages in Fig. 1, path B. For analysis of cell proliferation, Ki67 staining (PE; BD PharMingen) was conducted on total filtered kidney cells without Percoll separation. Cells were stained with antibodies against CD45 (FITC; BD PharMingen), and proliferating cells were defined as Ki67+ in either CD45+ or CD45− populations according to the strategy outlined in Fig. 1 (path C).
Fig. 1.

Representative gating strategy for flow cytometry measurements. Following digestion and filtration, cells were FACS-scanned, and forward scatter and side scatter were used to exclude cellular debris (left). A: lymphocytes were gated based on forward and side scatter plots. These were further gated based on the surface expression of CD4 and CD8. To analyze T-regulatory cells (T-regs), CD4+/FoxP3+ cells were identified using a contour plot. To evaluate T helper (Th) populations, such as Th1, Th2, or Th-17, cytokine-expressing cells were identified using histograms of either CD4+ or CD8+ cells following stimulation; an example of IL-17 expression for Th17 cells is shown. B: macrophages/dendritic cells were identified by expression of CD11b/c, and a population putatively identified as M1 was further identified using CD80 and CD86 staining. C: identification of Ki67 proliferating cells was conducted by separating live cells into CD45+ or CD45− populations and then evaluating each for Ki67 expression, as indicated.
T cell isolation and functional analysis.
For in vivo adoptive transfer studies, spleens were harvested from uninjured rats after euthanasia and subjected to tissue digestion, filtration, and Percoll separation as described previously. CD4+ T cells were then isolated using the MACS Pan-T cell microbead separation kit (Miltenyl, Glabach, Germany). In total, 1 × 106 CD4+T cells were suspended in sterile saline, counted, and administered to rats (study 2) at the time of reperfusion.
Functional analysis of IL-17 responses in T cells was based on previously described methods (25, 26). CD4+ T cells were stimulated with plate-bound anti-CD3 (precoated; 2 μg/mL) and soluble anti-CD28 (1 μg/mL). Cells (2.5 × 105 in 0.25 mL) were incubated for 12–14 h at 37°C in RPMI medium supplemented with 10% FBS (Invitrogen) in a 48-well plate. Cells were challenged with ANG II (Sigma, 10−7 M), and the extracellular Na+ was raised from 140 mM to 170 mM using a 1 M NaCl solution. The response of IL-17 mRNA was based on RT-PCR exactly as described previously (25, 26).
Statistical analysis.
All data are expressed as means ± SE. Differences in means were established by Student’s t test or one-way ANOVA with the Student–Newman–Keuls multiple-comparison test as indicated in the figures. Analysis was done with the aid of GraphPad Prism software (La Jolla, CA).
RESULTS
Generation of mutant Rorc rats to evaluate Th17 cells.
Previous studies have shown that Th17 cells represent the predominant lymphocyte population following renal I/R injury as well as in response to subsequent exposure to elevated dietary sodium. To evaluate the role of Th17 cells in renal injury and repair following I/R, the Rorc gene, which codes for RORγT, the major transcription factor driving IL-17 expression and Th17 cell differentiation, was targeted for mutation. A CRISPR/Cas9 probe targeting the region between 163 and 184 of the rat Rorc gene was introduced into LEW/Crl embryos. Sequencing of founder DNA using a 373-bp PCR product indicated an 8-bp deletion at nt 177–184 (Fig. 2A). This PCR product contains a BglII site in wild-type rats that generates two fragments of 246 bp and 135 bp and is lost in the mutant rats (Fig. 2C) Based on sequence analysis, the wild-type Rorc gene encodes for 508-amino acid full-length protein, whereas the mutant gene is predicted to result in a missense mutation after amino acid 31 and termination after amino acid 45 (Fig. 2B). Western blot analysis of thymus tissue showed a ∼58-kDa band consistent with full-length RORγT in Rorc+/+ rats, which was absent in Rorc−/− rats (Fig. 2D). Figure 2E shows the results of a study using isolated CD4+ cells from the kidney 7 days following a 40-min I/R injury. Consistent with previous studies in Sprague-Dawley rats (25, 26), in vitro stimulation of the kidney CD4+ cells with 170 mM Na+ and ANG II resulted in a robust increase in IL-17 mRNA following injury in Rorc+/+ rats. In contrast, CD4+ cells from Rorc−/− rats did not manifest an increase in IL-17 expression, suggesting that Rorc−/− rats manifest a significant impairment in the Th17 differentiation pathway.
Fig. 2.

Generation and characterization of RAR-related orphan receptor C (Rorc) mutant rat. A: CRISPR/Cas9 was used to introduce a mutation in the rat Rorc gene of LEW/NCrl rat embryos by targeting the region shown in red of sequence XM_032897831.1. Gene sequencing identified an 8-bp deletion at nt 177–184. The resultant rat is referred to as LEW-Rorcem3Mcwi or hereafter in this report as Rorc −/−. B: the sequence predicted a full-length protein of 508 amino acids in Rorc+/+ rats, while the sequence in Rorc−/− rats predicted a missense mutation after amino acid 31, leading to premature truncation. The highlighted red box illustrates the area of missense relative to wild-type protein. C: genotyping of rats following PCR amplification and BglII digestion generated fragments in Rorc+/+ that are absent in Rorc −/−. D: Western blot using anti-RAR-related orphan receptor-γ (RORγT) identified a 55–60-kDa protein in the thymus from Rorc+/+ rats, which is not present in Rorc−/− rats. E: IL-17 mRNA expression in isolated CD4+ cells from the kidney following in vitro stimulation with elevated extracellular Na+ (170 mM) and ANG II (10−7 M). CD4+ cells were isolated from the kidney 7 days following ischemia-reperfusion (I/R) injury or sham from either Rorc+/+ or Rorc−/− rats. Data are normalized means ± SE of IL-17 mRNA, N = 3 rats per group, *P < 0.05 vs. sham, †P < 0.05 Rorc−/− vs. Rorc+/+ by one-way ANOVA and the Student–Newman–Keuls post hoc test.
Rorc−/− rats are bred as homozygotes and are slightly smaller than Rorc+/+ rats (at 12–14 wk, 316 ± 20 g body wt in Rorc+/+ and 248 ± 17 g body wt in Rorc−/−, P < 0.05). Otherwise, these rats do not manifest obvious developmental defects. In adult rats, alterations in thymic and splenic populations were evident. The percentage of monocytes identified as CD4+ was lower in the thymus and spleen, whereas the percentage of CD8+ cells was higher in Rorc−/− compared with Rorc+/+ rats. In addition, CD161+ cells, B cells, and CD11b/c+ cells were significantly lower in the thymus and spleen of Rorc−/− rats (Table 1).
Table 1.
Changes in percentages of lymphocytes in the thymus, spleen, and blood of Rorc+/+ vs. Rorc−/− LEW rats
| Thymus |
Spleen |
Blood |
||||
|---|---|---|---|---|---|---|
| Rorc+/+ | Rorc−/− | Rorc+/+ | Rorc−/− | Rorc+/+ | Rorc−/− | |
| CD3 | 52.1 ± 0.6 | 52.7 ± 0.7 | 54.6 ± 2.0 | 24.7 ± 1.7* | 52.1 ± 3.2 | 51.5 ± 1.2 |
| CD4 | 33.3 ± 1.2 | 15.6 ± 1.8* | 50.5 ± 2.5 | 24.2 ± 0.3* | 7.6 ± 1.2 | 5.2 ± 0.8* |
| CD8 | 22.1 ± 5.7 | 32.8 ± 1.8 | 12.2 ± 0.2 | 15.0 ± 1.4 | 9.8 ± 3.1 | 5.6 ± 0.6* |
| CD161 | 13.3 ± 1.0 | 4.5 ± 0.9* | 9.0 ± 0.4 | 4.1 ± 0.4* | 7.1 ± 1.7 | 14.7 ± 3.3* |
| B cells | 54.9 ± 0.9 | 22.0 ± 0.2* | 28.6 ± 3.6 | 13.9 ± 0.9* | 4.5 ± 2.2 | 1.3 ± 0.2* |
| CD11b/c | 12.1 ± 0.8 | 5.9 ± 0.3* | 15.2 ± 2.2 | 7.7 ± 1.0* | 2.5 ± 0.2 | 2.1 ± 0.2 |
Values are means ± SE and expressed as %monocytes. Rorc, RAR-related orphan receptor C.
P < 0.05 by Student’s t test.
Evaluation of renal injury in Rorc mutant rats.
To investigate the potential role of Th17 cells in the pathogenesis of AKI, both Rorc+/+ and Rorc−/− rats were subjected to 40 min of bilateral renal I/R and were allowed to recover for 48 h. The initial body weight of rats used in this study was not different between the two genotypes (Rorc+/+ = 320 ± 35 g; Rorc−/− = 317 ± 24 g; nonsignificant). Rorc−/− rats showed a significantly reduced serum creatinine 24 h following reperfusion relative to Rorc+/+ rats (Fig. 3A). The degree of renal inflammation present 48 h following I/R was also significantly reduced in Rorc−/− versus Rorc+/+ rats; specifically, the total numbers of mononuclear cells, total CD4+ cells, and total CD8+ cells were robustly induced by renal injury in Rorc+/+ rats, but these responses were strongly attenuated in Rorc−/− rats (Fig. 3, B–D). As expected, the numbers of IL-17 expressing cells (Fig.3E) and Th17 cells (CD4+/IL-17+; Fig. 3F) were almost completely abrogated in postischemic Rorc−/− rats. The numbers of B cells, CD11b/c+ cells, and Th1 cells (CD4+/interferon (IFN)-γ+) were also significantly attenuated in Rorc−/− versus Rorc+/+ rats, but there was no significant effect on T-regulatory cells 2 days following I/R (Table 2). When recovery was extended to 7 days, the infiltration of immune cells resolved relative to day 2, whereas the total number of infiltrating cells remained reduced in Rorc−/− rats relative to Rorc+/+ rats (Table 3). As an additional evaluation of renal injury, whole kidney Kim-1 mRNA was measured from similarly treated rats 24 h following I/R. kidney injury molecule-1 (Kim-1) mRNA was not detectable in sham control rats but was approximately twofold higher in Rorc+/+ rats versus Rorc−/− rats 24 h following I/R (2ΔΔCt = 4.8 ± 1.8 vs. 2.3 ± 0.8 SE, N = 4 per group), but these differences were not statistically significant.
Fig. 3.

Effects of RAR-related orphan receptor C (Rorc) mutation on renal ischemia-reperfusion (I/R) injury and lymphocyte expression. A: Lewis Rorc+/+ vs. Rorc−/− rats were subjected to 40 min of bilateral renal I/R injury and allowed to recover for 2 days. Serum creatinine values 24 h following recovery are shown. Results from two experimental cohorts were combined and pooled with values from individual animals shown. B–F: tissues from one cohort were analyzed for inflammatory cells. Shown are total mononuclear cells (B), total CD4+ cells (C), total CD8+ cells (D), total IL-17+ (E), and T helper (Th)17 cells (F) expressed as means ± SE positive cells per gram of kidney. *P < 0.05 vs. sham-operated control; †P < 0.05 Rorc+/+ vs. Rorc−/− rats, by one-way ANOVA and the Student–Newman–Keuls post hoc test.
Table 2.
Kidney lymphocyte and macrophage content in Rorc+/+ and Rorc−/− rats 2 days following 40 min of bilateral I/R
|
Rorc+/+ |
Rorc−/− |
|||
|---|---|---|---|---|
| Sham | I/R | Sham | I/R | |
| CD4/IFN-γ, cells/g | 95 ± 48 | 3,021 ± 829* | 20 ± 40 | 978 ± 274*† |
| FoxP3, +%CD4+ | 3.3 ± 0.36 | 0.66 ± 0.11* | 4.13 ± 0.7 | 1.35 ± 0.11* |
| Rt1B+, cells/g | 9,384 ± 294 | 31,773 ± 9,236* | 9,202 ± 436 | 10,905 ± 967*† |
| CD11b/c, cells/g | 10,366 ± 2,880 | 5,7216 ± 2,318 | 11,262 ± 1,191 | 10,522 ± 5,126*† |
Values are means ± SE. Rorc, RAR-related orphan receptor C; IFN, interferon.
P < 0.05 sham vs. ischemia-reperfusion (I/R);
P < 0.05 Rorc−/− vs. Rorc+/+ by one-way ANOVA and the Student–Newman–Keuls post hoc test.
Table 3.
Kidney lymphocyte and macrophage content in Rorc+/+ and Rorc−/− rats 7 days following 40 min of bilateral I/R
| Rorc+/+ | Rorc−/− | |
|---|---|---|
| CD4/IFN-γ, cells/g | 1,019 ± 300 | 400 ± 103 |
| FoxP3+, % of CD4+ | 1.57 ± 0.47 | 2.57 ± 0.47 |
| Rt1B+, cells/g | 5,464 ± 2,699 | 4,204 ± 898 |
| CD11b/c, cells/g | 15,523 ± 4,588 | 10,264 ± 967 |
Values are means ± SE; N = 5–6 per group. Rorc, RAR-related orphan receptor C; I/R, ischemia-reperfusion; IFN, interferon. No significant differences were observed.
As a less severe injury results in faster and more efficient recovery, the resistance to initial injury in Rorc−/− rats potentially confounds evaluating a role for Th17 cells in renal repair or progression to CKD. Therefore, experiments were conducted to equalize the initial level of injury in Rorc+/+ or Rorc−/− rats by increasing ischemic time in Rorc−/− rats. Figure 4A demonstrates that increasing ischemia to 50 min in Rorc−/− rats achieved a 24-h and 48-h post-I/R serum creatinine similar to 40 min of ischemia in Rorc+/+ rats, whereas 50 min of ischemia did not further increase serum creatinine values in Rorc+/+ rats. Histological assessment of kidneys in the renal outer medulla at 2 days postischemia revealed evidence of damage consistent with this stage of injury, including damaged tubules with sloughed or necrotic cell debris (black arrow). Such damage was readily observed in both Rorc+/+ and Rorc−/− rats following 40 min of I/R (Fig. 4B). The percentage of severely damaged tubules with necrotic or sloughed cells in the outer medulla tended to be slightly reduced in 40-min I/R Rorc−/− rats (32.1 ± 8.7%) versus 40-min I/R Rorc+/+ rats (37.2 ± 15.5%), whereas Rorc−/− rats subjected to 50 min of I/R tended to have moderately higher tubular damage (41.8 ± 5.6%). However, these differences were not statistically significant.
Fig. 4.

Effect of ischemic time on severity of renal injury and survival in RAR-related orphan receptor C (Rorc)+/+ vs. Rorc−/− rats. Rorc+/+ and Rorc−/− rats were subjected to either 40 or 50 min bilateral renal ischemia-reperfusion (I/R) and allowed to recover for up to 4 days. A: serum creatinine values are shown at 24 and 48 h following I/R in Rorc +/+ and Rorc−/− rats, respectively. *P < 0.05 Rorc−/− and Rorc+/+ rats following 40 min; Rorc+/+ and Rorc−/− rats were not different at 50 min of I/R. B: representative hematoxylin-eosin-stained cross sections through renal outer medulla are shown 2 days following renal I/R. Necrotic damage was evident in all samples (arrow) and no statistically significant differences were evident. Magnification is shown. C: Kaplan–Meier survival curve is shown indicating increased mortality beginning 2 days post-I/R in Rorc−/− rats following 50 min of injury vs. Rorc−/− rats following 40 min of injury or Rorc+/+ rats following 40 or 50 min of injury. N = 7–10 per group. Note that the lines defining +/+ 40 min I/R, +/+ 50 min I/R, and −/− 40 min I/R are overlapping at 100%.
The process of renal recovery was therefore examined in equivalently injured Rorc+/+ or Rorc−/− rats. Surprisingly, although Rorc−/− rats following 50 min of I/R manifest similar early levels of injury as Rorc+/+ rats after 40 or 50 min of I/R, the mutant rats showed a dramatic impairment in recovery. Figure 4C illustrates low survival (death or need for euthanasia for humane reasons) of Rorc−/− rats following 50 min of I/R, whereas Rorc+/+ rats after 40 or 50 min of I/R or Rorc−/− rats following 40 min of I/R survived. This experiment was initially intended to evaluate efficiency of repair for 7 days post-I/R but was terminated after 4 days due to the high mortality in the Rorc−/− 50-min I/R group. Subsequent studies on recovery from I/R were therefore conducted to a maximum of 4 days of recovery.
To investigate the lack of recovery in more detail and to determine if the lymphocyte RORγT activity underlies the failed recovery response, Rorc+/+ or Rorc−/− rats were injured for 40 or 50 min, and Rorc−/− rats were adoptively transferred with CD4+ splenocytes from either Rorc+/+ or Rorc−/− rats. Similar to the previous study, there was 100% survival in Rorc+/+ rats following 40-min I/R and recovery of serum creatinine toward normal values typically observed in sham controls by day 4 (Fig. 5, A and B). In contrast, 50-min I/R in Rorc−/− rats (+vehicle) resulted in increased mortality by day 4. Serum creatinine levels indicated sustained or worsening injury evident by day 3 post-I/R. Adoptive cell transfer of Rorc+/+ CD4+ cells resulted in a resolution of creatinine that was similar to Rorc+/+ rats at day 3 and day 4 and improved survival, whereas adoptive cell transfer of Rorc−/− CD4+ cells did not improve recovery of Rorc−/− rats (Fig. 5A). Interestingly, the level of inflammation in Rorc−/− rats based on total mononuclear cells, total CD4+ cells, and total CD11b/c+ cells (dendritic cells/macrophages) was increased relative to Rorc+/+ rats, whereas the percentage of Foxp3+/CD4+ T-regulatory cells was decreased. These differences were attenuated in Rorc−/− rats with adoptive cell transfer of Rorc+/+ CD4+ splenocytes (Fig. 5, C–F). Histological sections of renal outer medulla of Rorc−/− rats 4 days following injury had abundant areas with significant necrosis (Fig. 5G, thin black arrow) and dilated tubules with a thinned epithelial layer (thick black arrow). In contrast, both Rorc+/+ rats and Rorc−/− rats with Rorc+/+ adoptive cell transfer showed significantly reduced tubular damage relative to Rorc−/− vehicle-treated rats (Fig.5H); these kidneys displayed evidence of tubular repair, with less tubular debris and a thickened hyperplastic epithelium (Fig. 5G, red thick arrow).
Fig. 5.

Rescue of RAR-related orphan receptor C (Rorc)−/− rats following bilateral ischemia-reperfusion (I/R) by adoptive transfer of Rorc+/+ splenocytes. Rorc +/+ rats were subjected to 40 min of bilateral I/R and Rorc−/− rats were subjected to 50 min bilateral renal I/R. At the time of reperfusion, rats were provided with vehicle or adoptive transfer (AdT) of 1 × 106 splenocytes from either Rorc+/+ or Rorc−/− by intraperitoneal injection. A: daily serum creatinine values are shown for up to 4 days following I/R. B: Kaplan–Meier curve showing percent survival of rats following I/R injury and the effect of adoptive transfer. C–F: quantitative analysis of infiltrating cells 4 days following recovery from I/R injury expressed as positive cells per g kidney. Shown are total CD45+ cells (C), CD4+ cells (D), CD11/b/c+ (E), and % T-regulatory cells (F). G: representative renal histology 4 days following renal I/R of Rorc+/+ or Rorc−/− with adoptive transfer as labeled. Note the presence of necrotic cellular debris (thin black arrow) and thinned epithelial layer (thick black arrow) in outer medulla of Rorc−/− rats (arrows), while evidence of tubular repair containing a thicker epithelium (red arrows) was evident in Rorc+/+ kidneys or Rorc−/− kidneys following Adt of splenocytes from Rorc+/+ rats. H: percentage of severely damaged tubules from renal outer medulla of kidneys shown in G) Magnification is shown. For A, C–F, and H, values are means ± SE. *P < 0.05 in Rorc+/+vs. Rorc−/−rats; †P < 0.05 Rorc+/+adoptive transfer vs. Rorc−/−adoptive transfer in Rorc−/−rats, by one-way ANOVA and the Student–Newman–Keuls post hoc test.
To evaluate whether impaired recovery of Rorc−/− rats was due to loss of secreted IL-17, Rorc−/− rats were subjected to 50 min of I/R and administered different doses of recombinant IL-17 (50 or 100 ng·kg−1·day−1) or vehicle by an osmotic minipump. In this study, Rorc+/+ rats subjected to 40 or 50 min were also included for comparison. All groups showed equivalent increases in serum creatinine 24 h following I/R, and only the Rorc+/+ group subjected to 40 min of I/R had lower levels of creatinine on day 2 relative to Rorc+/+ or Rorc−/− 50-min I/R groups. Importantly, serum creatinine levels of Rorc−/− 50-min I/R rats treated with IL-17 improved relative to vehicle, by day 3 and day 4, and the recovery was more efficient with the higher IL-17 dose (Fig. 6A). Similar to data in Figs. 4 and 5, a significant percentage of Rorc−/− vehicle-treated rats died or required euthanasia by day 4, but there was no mortality in rats treated with IL-17, and only 1 of 10 Rorc+/+ rats subjected to 50 min of I/R died before day 4 (Fig. 6B). While there was an apparent improvement in serum creatinine in the Rorc−/− vehicle-treated group between day 3 and day 4, the reduction in creatinine was due to death of rats with higher creatinine levels and not due to improvement of creatinine in any surviving animals (data not shown). Although differences in serum creatinine were not evident between Rorc−/− vehicle-treated rats versus either Rorc+/+ or Rorc−/− IL-17-treated rats by day 2, Rorc−/− vehicle-treated rats exhibited hyperkalemia and acidosis by day 2 versus Rorc+/+ rats or Rorc−/− rats treated with a high dose of IL-17 (Table 4). It is noteworthy that the degree of inflammation indicated by total mononuclear cells, CD4+ cells, and CD11b/c cells was significantly attenuated in Rorc−/− rats following administration of IL-17, whereas the percentage of T regulatory cells was increased by IL-17 (Fig. 6, C–F).
Fig. 6.

Rescue of RAR-related orphan receptor C (Rorc)−/− rats following bilateral ischemia-reperfusion (I/R) by exogenous IL-17. Rorc +/+ rats were subjected to 40 or 50 min of ischemia and Rorc−/− rats were subjected to 50 min bilateral renal I/R (as indicated) and provided rIL-17 (50 or 100 ng·kg−1·day−1) or vehicle by osmotic minipump. A: daily serum creatinine values are shown for up to 4 days following I/R. B: Kaplan–Meier curve showing percent survival of rats following I/R injury and the effect of IL-17; Rorc−/− vehicle-treated rats showed mortality by day 4, whereas curves for Rorc−/− rats treated with IL-17-treated overlap at 100% survival. C–F: quantitative analyses of infiltrating cells 4 days following recovery from I/R injury expressed as positive cells per gram kidney. Shown are total CD45+ cells (C), total CD4+ cells (D), total CD11b/c+ cells (E), and % T-regulatory cells (F). For A and C–F, data are means ± SE. †P < 0.05 in Rorc−/− IL-17 treated vs. Rorc−/− vehicle-treated rats; ‡P < 0.05 in 50 min Rorc−/−vs. 40 min I/R Rorc+/+rats at day 2 by one-way ANOVA and the Student–Newman–Keuls post hoc test.
Table 4.
Blood chemistry 48 h following ischemia/reperfusion in Rorc+/+ and Rorc −/− rats
|
Rorc+/+ Sham |
Rorc−/− Sham |
Rorc+/+ I/R (50 min) |
Rorc−/− I/R (50 min) |
Rorc−/− I/R (50 min) IL-17 |
|
|---|---|---|---|---|---|
| n | 6 | 5 | 6 | 6 | 5 |
| pH | 7.38 ± 0.02 | 7.37 ± 0.02 | 7.37 ± 0.02 | 7.24 ± 0.04* | 7.37 ± 0.03† |
| Na+, mmol/L | 139.2 ± 0.5 | 140.4 ± 1.1 | 143.3 ± 1.1 | 137.0 ± 2.3 | 141.0 ± 0.6 |
| K+, mmol/L | 3.7 ± 0.1 | 3.5 ± 0.1 | 4.1 ± 0.2 | 5.0 ± 0.5* | 4.3 ± 0.3† |
| Cl−, mmol/L | 108.5 ± 0.8 | 110.2 ± 1.9 | 110 ± 0.7 | 101.5 ± 2.0* | 106.0 ± 1.5† |
| , mmol/L | 27.8 ± 1.0 | 27.5 ± 1.0 | 29.5 ± 0.7 | 21.6 ± 2.7* | 24.2 ± 2.8 |
| Base deficit, mmol/L | 2.0 ± 1.0 | 1.4 ± 0.7 | 3.3 ± 0.7 | −6.0 ± 2.9* | 2.0 ± 1.3† |
| Anion gap | 6.6 ± 0.5 | 6.2 ± 0.6 | 8.9 ± 0.6 | 18.9 ± 2.5* | 15.1 ± 3.7 |
Values are means ± SE. IL-17 was administered at a dose of 100 ng·kg−1·day−1 by an osmotic minipump. I/R, ischemia-reperfusion. Rorc, RAR-related orphan receptor C.
P < 0.05 Rorc−/− vs. Rorc+/+;
P < 0.05 Rorc−/− vehicle vs. Rorc−/− IL-17 treated, by one-way ANOVA and the Student–Newman–Keuls test.
These data suggested the possibility that the renal repair response was inhibited in Rorc−/− rats. As cell proliferation is an important component of renal repair, Ki67 staining was conducted on tissues from postischemic Rorc−/− or Rorc+/+ rats. Abundant Ki67 staining was localized primarily in the tubular epithelium primarily in the outer medulla of Rorc+/+ rat kidneys (Fig. 7A, thick white arrow). In addition, there was also abundant Ki67 staining in the kidneys of Rorc−/− rats, indicating that proliferation is not impaired in Rorc−/− rats. However, due to the high level of necrosis and relatively thin epithelial layer, it was difficult to determine whether Ki67+ was expressed in proliferating tubular epithelial cells or infiltrating inflammatory cells in the surrounding interstitium (inset, thin black arrow).
Fig. 7.

Effect of IL-17 supplementation on proliferation and macrophage infiltration following ischemia-reperfusion (I/R) injury in RAR-related orphan receptor C (Rorc)−/− rats. Rorc−/−or Rorc+/+ rats were subjected to 40 or 50 min bilateral renal I/R respectively or sham surgery. A: Ki67 staining is shown through renal outer medulla 4 days following I/R surgery. Abundant Ki67+ cells were observed in tubules of Rorc+/+ and Rorc−/− kidneys (white arrow). Tubules with necrotic debris were frequently observed in Rorc−/− rats at 4 days following I/R that were surrounded by Ki67+ cells, which could not be clearly localized within tubules (inset, black arrow). Magnification is shown in A. B: quantitative analysis of cell proliferation of Ki67+/CD45− cells based on FACS at 2 or 4 days following sham or I/R. C: quantitative analysis of M1 macrophages defined as CD11/bc+/CD80+/CD86+ based on FACS at 2 or 4 days after sham or I/R. B and C: rats were implanted with minipumps containing either IL-17 or vehicle (V). Note the lines for representing sham Rorc+/+ and Rorc−/− rats overlap in B and C. N = 5–6 rats per group at 2 days and 8–9 per group at 4 days. Data are means ± SE. *P < 0.05 vs. sham-operated control, †P < 0.05 Rorc−/−IL-17 vs. Rorc−/− vehicle, ‡P < 0.05 in Rorc−/− vs. Rorc+/+ rats, by one-way ANOVA and the Student–Newman–Keuls post hoc test.
Therefore, to determine if cell proliferation was present in infiltrating cells or resident kidney cells, additional studies were conducted using FACS analysis to differentiate Ki67 signal as either resident kidney cells (CD45−) or cells of hematopoietic origin (CD45+). FACS analysis demonstrated that >90% of all Ki67+ cells were resident kidney cells (i.e., CD45−), suggesting that the vast majority of proliferating cells were nonhematopoietic and likely represent regenerating tubules (data not shown). Moreover, the percentage of CD45+ or CD45− Ki67+ cells did not differ between Rorc+/+ versus Rorc−/− rats (data not shown). Figure 7B illustrates quantitative analysis of Ki67+/CD45− (presumptive tubular) cells in Rorc+/+ versus Rorc−/− rats at 2 or 4 days post-I/R. As expected, abundant Ki67+ cells were observed in Rorc+/+ rats, which was significantly elevated at 2 days post-I/R versus sham, but declined by day 4. In contrast, Rorc −/− rats showed a similar level of proliferation as Rorc+/+ rats at day 2 but remained persistently elevated at day 4. Rorc−/− rats given IL-17 showed reduced proliferation relative to vehicle at both 2 and 4 days post-I/R.
It is possible that sustained proliferation in Rorc−/− rats is secondary to persistent ongoing injury up to 4 days postischemia, whereas proliferation in Rorc+/+ rats declines with ongoing repair. As M1-type macrophages represent a potential injurious signal in the early post-I/R period, M1 responses (defined as CD11b/c+/CD80+/86+) were evaluated at 2 and 4 days post-I/R (Fig. 7C). M1 macrophages were similar in both genotypes at 2 days post-I/R but diverged significantly between day 2 and day 4, resolving mildly in Rorc+/+ rats but increasing significantly in Rorc−/− rats. Interestingly, IL-17 treatment of Rorc−/− rats decreased M1 macrophages between day 2 and day 4. IL-17 treatment did not have an effect on M2 macrophages (data not shown).
DISCUSSION
The pathogenesis of AKI comprises an interplay between hemodynamics, cellular energetics, cell death pathways, and an elaborate tissue repair response. AKI is also a significant inflammatory condition involving the influx of a variety of leukocytes, including T lymphocytes. Several lines of evidence have established a role for lymphocytes in the pathogenesis of AKI. For example, pioneering studies by Rabb et al. (31) used CD4+- or CD8+-deficient mice, which were protected from ischemic AKI, whereas other studies demonstrated that blockade of the CD28-B7 costimulatory pathway, considered important in T cell activation, attenuates renal injury in response to I/R (12). A potential role for T cells in the AKI-to-CKD transition was suggested in studies that demonstrated a sustained influx of both activated and effector memory T cells for up to 6 wk following I/R in mice (2). Pechman et al. (30) demonstrated that lymphocyte inhibitor mycophenolate mofetil (MMF) given to rats between 5 and 9 wk post-I/R attenuated the development of renal fibrosis, inflammation, and hypertension following exposure to a high-salt diet.
Using the same rat model, Mehrotra et al. (26) investigated the cytokine profile of kidney-derived T-lymphocytes and demonstrated that Th17 cells (CD4+/IL-17+) were the most abundant Th population, with Th1 (IFN-γ+) or Th2 (IL-4+) being far less abundant. Interestingly, protection against AKI in a rat model of renal I/R by treatment with either human adipocyte-derived stromal cells or conditioned media from human cord blood-derived endothelial colony-forming cells was associated with a dramatic decrease in Th17 cell induction (8, 9). In contrast, vitamin D deficiency, which exacerbates kidney injury to I/R, was associated with a significant increase in Th17 cell induction (11). Moreover, following an initial peak of activity between 1 and 3 days following I/R, Th17 cells are also robustly restimulated following exposure to a high-salt diet that hastens the development of hypertension and CKD (25). Recent clinical studies reported elevated levels of plasma IL-17 or IL-17+ peripheral blood mononuclear cells in patients with AKI (23, 26). Taken together, these reports suggest that Th17 cells represent a potentially important target in AKI and the AKI-to-CKD transition.
An initial approach to investigate the role of activated T cells in the AKI-to-CKD transition used athymic rats (Foxn1rnu−/rnu). These rats are characterized by impaired thymus development resulting in lack of CD4+ cell activation. Surprisingly, whereas conventional Th17 cell (CD4+/IL-17+) differentiation was impaired in athymic rats following I/R injury, IL-17 expression was not. Rather, IL-17 expression in athymic rats was shifted to a population of MMF-insensitive natural killer cells (NK cell/CD161+). However, in both athymic and euthymic rats, IL-17 blockade with a soluble receptor inhibitor attenuated fibrosis and inflammation in response to elevated dietary salt following initial recovery from AKI, suggesting that IL-17 activity contributed to the pathogenesis of the AKI-to-CKD transition (24).
The current experiment was designed to overcome the compensatory activity of athymic rats to induce IL-17 expression. RORγT is a transcription factor purported to drive the expression of IL-17 and is a master regulator of Th17 cell differentiation. We hypothesized that mutation of Rorc, the gene coding for RORγT, would manifest impaired Th17 activation, as well as potential IL-17 production from NK cells, and thus permit the investigation of the Th17 cells in AKI and subsequent CKD progression. Initial studies used a standard 40-min bilateral I/R that showed a modest but significant reduction in the degree of renal injury in Rorc−/− rats versus wild-type rats. Importantly, Th17 cell induction, as well as expression of IL-17 from other cell types, was blocked in Rorc−/− rats. Moreover, we did not observe any compensation by Th1 cells following I/R. Th1 cells were far less abundant than Th17 cells following I/R in wild-type rats, and there was a further reduction in these cells in mutant rats.
These observations are consistent with other studies suggesting that IL-17 contributes to the development of AKI. For example, IL-17−/− mice or Rorγt−/− mice were shown to be resistant to AKI in a model using cisplatin (7). Recently, Wang et al. (35) demonstrated that the isoform IL-17C is produced by injured proximal tubules and suggested that tubular IL-17C may activate Th17 cell differentiation. Blockade of IL-17C with a specific neutralizing antibody or knockout of the IL-17C receptor, IL-17RE, attenuated Th17 cell activation and the severity of injury in response to renal I/R (35). In addition, our group recently demonstrated that blockade with soluble IL-17Rc significantly attenuated AKI in response to I/R in rats (26). Moreover, we also demonstrated that the store-operated Ca2+ channel, Orai1, was critical to the activation of Th17 cells and that an Orai1 inhibitor protected rats against both I/R and glycerol-induced AKI (26). The mechanism of protection due to the lack of Th17 cells is not yet clear. However, these cells secrete many factors such as IL-17A, IL-17F, IL-21, IL-22, IL-23, and TNF-α. IL-17 receptors are found in epithelial cells and could influence the production of neutrophil-attracting CXC chemokines such as IL-8 chemokines or could directly mobilize neutrophils (27). The cumulative activity of Th17 cells could have direct consequences on tubular injury or could secondarily influence the development of vascular congestion contributing to renal hypoxia.
Therefore, we suggest that this model could be useful in evaluating the effect of Th17 cell induction and the development of renal fibrosis, renal sodium handling, and vascular responses associated with the progression of CKD following AKI. However, as Rorc−/− rats have a reduced level of initial injury relative to wild-type rats, it may be difficult to interpret data on progression as being attributable to impaired Th17 activation rather than being a consequence of a reduced level of initial kidney damage (6). For that reason, we conducted studies to equalize the level of injury by increasing ischemic time to 50 min in Rorc−/− rats. This approach resulted in similar levels of initial renal damage as observed in Rorc+/+ rats based on plasma creatinine values and renal histological assessment at 1–2 days post-I/R, potentially allowing a comparison of recovery in the two genotypes. Nevertheless, in consideration of the relative insensitivity of serum creatinine as a marker of renal injury and the multiple different pathophysiological processes induced by I/R, it is possible that injury is not truly equalized by this procedure, and additional evaluation of renal injury should be considered.
However, as initial serum creatinine levels were equalized by increasing ischemic time to 50 min in Rorc−/− rats, this injury evoked an unexpected effect leading to high mortality and failure to recover kidney function. That this effect is due to the loss of Th17 cells is suggested by the fact that adoptive transfer of CD4+ splenocytes from Rorc+/+ rats, but not Rorc−/− rats, and supplementation with recombinant IL-17 improved survival and resulted in recovery of kidney function similar to that observed in Rorc+/+ rats. Thus, Th17 cells have a paradoxical effect of not only contributing to renal injury but also having a positive effect on recovery following more severe injury. Our suggestion that Th17 cells contribute to recovery in Rorc−/− rats is based on the fact that adoptive transfer of Rorc+/+ splenocytes or rIL-17 facilitates recovery of serum creatinine after peaking at day 2 postischemia. It is important to note that we cannot rule out that these treatments did not attenuate the acute manifestation of injury in a way that was not detectable by measuring serum creatinine. Indeed, Rorc−/− rats manifest hyperkalemia and acidosis detectable by day 2, relative to IL-17-treated rats, suggesting that rats may suffer more acute or more prolonged injury.
Kidney repair is a process by which damaged epithelial cells recover from sublethal injury and/or invoke a regenerative response characterized by cell proliferation and redifferentiation (5, 13, 20, 29). The suppression of inflammation, reestablishment of perfusion, and activation of growth factors to support kidney repair are essential elements of recovery from AKI. Histological assessment indicated that Rorc−/− rats showed persistent cell necrosis up to 4 days following renal I/R, whereas Rorc+/+ rats displayed little necrosis and thickening tubular epithelium in the outer medulla at this time point, consistent with the time course of repair in this model. This suggests that renal recovery is impaired in Rorc−/− rats. However, Rorc−/− rats showed abundant cell proliferation in the aftermath of injury. In rat models of renal I/R, cell proliferation is reported to peak between days 2 and 3 and then subsequently resolves as new cells repopulate the tubular basement membrane (5, 33, 36, 37). Quantitative FACS analysis of Ki67 staining indicated that cell proliferation decreases in Rorc+/+ rats between days 2 and 4 but continues to rise between days 2 and 4 in Rorc−/− rats. We suggest that persistent elevation in tubular proliferation represents a compensatory response to sustained tissue injury in Rorc−/− rats.
Several reports using mouse models of AKI have described that polarized macrophages have differing effects on tissue injury. M1-type macrophages are present abundantly in the early postischemic period, and they are thought to contribute to tubular damage and loss of renal function by producing reactive oxygen species and proinflammatory cytokines such as IL-1β and TNF-α (15, 21). However, macrophages shift their polarization from an inflammatory M1 phenotype to a prorepair and anti-inflammatory M2 phenotype. Lee et al. demonstrated that adoptive transfer of macrophages differentiated in vitro toward M2 phenotype-enhanced tubular cell proliferation following renal I/R (15, 21).
To investigate whether loss of Th17 cells influences the expression or polarization of macrophages, we used a FACS-based approach to characterize these subtypes by staining for either CD80/CD86 or CD206 for M1 and M2, respectively. Although these markers provide an imperfect assessment of macrophage polarization, they help to provide an initial assessment of population responses during recovery. Interestingly, M1 macrophages showed a downward trend between day 2 and day 4 post-I/R in Rorc+/+ rats, whereas Rorc−/− rats showed abundant enhancement of M1 macrophage expression at 4 days of recovery. Surprisingly, we were unable to identify any effect on M2 macrophage expression by 4 days. We suggest that the persistent abundance of M1 macrophages may represent a source of sustained tissue injury, which was unexpectedly inhibited in rats treated with IL-17.
These data suggest that IL-17 may be part of a complex feedback system to limit inflammation. Indeed, while Th17 activation is generally considered proinflammatory, other studies have suggested that IL-17 may attenuate inflammation under some conditions. For example, Mohamed et al. (28) using a model of diabetic nephropathy in mice demonstrated that inflammation was exacerbated in kidneys of IL-17−/− mice versus wild-type mice and that a low dose of rIL-17 could reverse renal damage and inflammation.
There are now considerable data suggesting that T-regulatory cells represent an important anti-inflammatory lymphocyte promoting recovery of AKI, possibly mediated by the secretion of IL-10 (19, 32). The percentage of T-regulatory cells (defined as CD25+/FoxP3+) was significantly reduced in Rorc−/− rats relative to Rorc+/+ rats. Moreover, adoptive transfer of Rorc+/+ splenocytes, as well as infusion of rIL-17, increased the percentage of T-regulatory cells to levels in Rorc+/+ control rats. Thus, protection by IL-17 following severe injury may be due in part to T-regulatory cells. The balance between Th17 cells and T-regulatory cells is complex, and an imbalance of Th17 cells versus T-regulatory cells, with elevations in Th17 cells and reductions in T-regulatory cells, has been observed in models of autoimmune disease (3). In a study of allergic rhinitis, IL-17 inhibition resulted in the stable formation of T-regulatory cells and improved outcome (14). Therefore, our observation that IL-17 supplementation was associated with increased T-regulatory cell expression following I/R in Rorc−/− rats is initially surprising. The mechanism of this response is unclear and will require further investigation.
Taken together, these data suggest a complex role of Th17 cell induction in the setting of ischemic AKI. The impairment of Th17 cells plays a critical role in the initiation of injury, as Th17 cells may exacerbate the renal inflammatory response to I/R. However, Th17 cells following a more severe insult may be an essential feature to resolve inflammation. Future studies will be required to examine effects of Th17 induction on the cellular mechanisms mediating inflammation and altered renal vascular responses contributing to tissue injury as well as the long-term effects of these cells on progression.
GRANTS
This work is supported by National Institutes of Health (NIH) Grant DK063114 (to D.P.B.) and the Gene Editing Rat Resource Center at the Medical College of Wisconsin (NIH Grant HL114474).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.M., M.R.D., A.M.G., and D.P.B. conceived and designed research; P.M., M.U., J.A.C., S.L.M., and D.P.B. performed experiments; P.M. and D.P.B. analyzed data; P.M. and D.P.B. interpreted results of experiments; P.M., J.A.C., and D.P.B. prepared figures; P.M. and D.P.B. drafted manuscript; P.M., M.R.D., A.M.G., and D.P.B. edited and revised manuscript; P.M., M.U., J.A.C., S.L.M., M.R.D., A.M.G., and D.P.B. approved final version of manuscript.
ACKNOWLEDGMENTS
Portions of this work were presented at the 2019 meeting of the American Society of Nephrology. We thank Dr. Robert Bacallao and Weimin Xu for assistance with Western blot analysis.
REFERENCES
- 1.Arkhipov SN, Potter DL, Geurts AM, Pavlov TS. Knockout of P2rx7 purinergic receptor attenuates cyst growth in a rat model of ARPKD. Am J Physiol Renal Physiol 317: F1649–F1655, 2019. doi: 10.1152/ajprenal.00395.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, Hassoun HT, Rabb H. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int 75: 526–535, 2009. doi: 10.1038/ki.2008.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Astry B, Venkatesha SH, Moudgil KD. Involvement of the IL-23/IL-17 axis and the Th17/Treg balance in the pathogenesis and control of autoimmune arthritis. Cytokine 74: 54–61, 2015. doi: 10.1016/j.cyto.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile DP, Dwinell MR, Wang S-J, Shames BD, Donohoe DL, Chen S, Sreedharan R, Van Why SK. Chromosome substitution modulates resistance to ischemia reperfusion injury in Brown Norway rats. Kidney Int 83: 242–250, 2013. doi: 10.1038/ki.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C; ADQI XIII Work Group . Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 27: 687–697, 2016. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan AJ, Alikhan MA, Odobasic D, Gan PY, Khouri MB, Steinmetz OM, Mansell AS, Kitching AR, Holdsworth SR, Summers SA. Innate IL-17A-producing leukocytes promote acute kidney injury via inflammasome and toll-like receptor activation. Am J Pathol 184: 1411–1418, 2014. doi: 10.1016/j.ajpath.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 8.Collett JA, Mehrotra P, Crone A, Shelley WC, Yoder MC, Basile DP. Endothelial colony-forming cells ameliorate endothelial dysfunction via secreted factors following ischemia-reperfusion injury. Am J Physiol Renal Physiol 312: F897–F907, 2017. doi: 10.1152/ajprenal.00643.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collett JA, Traktuev DO, Mehrotra P, Crone A, Merfeld-Clauss S, March KL, Basile DP. Human adipose stromal cell therapy improves survival and reduces renal inflammation and capillary rarefaction in acute kidney injury. J Cell Mol Med 21: 1420–1430, 2017. doi: 10.1111/jcmm.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crome SQ, Wang AY, Levings MK. Translational mini-review series on Th17 cells: function and regulation of human T helper 17 cells in health and disease. Clin Exp Immunol 159: 109–119, 2010. doi: 10.1111/j.1365-2249.2009.04037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Bragança AC, Volpini RA, Mehrotra P, Andrade L, Basile DP. Vitamin D deficiency contributes to vascular damage in sustained ischemic acute kidney injury. Physiol Rep 4: e12829, 2016. doi: 10.14814/phy2.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Greef KE, Ysebaert DK, Dauwe S, Persy V, Vercauteren SR, Mey D, De Broe ME. Anti-B7-1 blocks mononuclear cell adherence in vasa recta after ischemia. Kidney Int 60: 1415–1427, 2001. doi: 10.1046/j.1523-1755.2001.00944.x. [DOI] [PubMed] [Google Scholar]
- 13.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu ZW, Wang YX, Cao ZW. Neutralization of interleukin-17 suppresses allergic rhinitis symptoms by downregulating Th2 and Th17 responses and upregulating the Treg response. Oncotarget 8: 22361–22369, 2017. doi: 10.18632/oncotarget.15652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huen SC, Cantley LG. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol 30: 199–209, 2015. doi: 10.1007/s00467-013-2726-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huen SC, Cantley LG. Macrophages in renal injury and repair. Annu Rev Physiol 79: 449–469, 2017. doi: 10.1146/annurev-physiol-022516-034219. [DOI] [PubMed] [Google Scholar]
- 17.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133, 2006. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 18.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol 11: 88–101, 2015. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 19.Kinsey GR. Macrophage dynamics in AKI to CKD progression. J Am Soc Nephrol 25: 209–211, 2014. doi: 10.1681/ASN.2013101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kusaba T, Lalli M, Kramann R, Kobayashi A, Humphreys BD. Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci USA 111: 1527–1532, 2014. [Erratum in Proc Natl Acad Sci USA 111: 5754, 2014.] doi: 10.1073/pnas.1310653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi B-S, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SA, Noel S, Sadasivam M, Hamad ARA, Rabb H. Role of immune cells in acute kidney injury and repair. Nephron 137: 282–286, 2017. doi: 10.1159/000477181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maravitsa P, Adamopoulou M, Pistiki A, Netea MG, Louis K, Giamarellos-Bourboulis EJ. Systemic over-release of interleukin-17 in acute kidney injury after septic shock: Clinical and experimental evidence. Immunol Lett 178: 68–76, 2016. doi: 10.1016/j.imlet.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Mehrotra P, Collett JA, McKinney SD, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: compensatory role of natural killer cells in athymic rats. Am J Physiol Renal Physiol 312: F385–F397, 2017. doi: 10.1152/ajprenal.00462.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int 88: 776–784, 2015. doi: 10.1038/ki.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehrotra P, Sturek M, Neyra JA, Basile DP. Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J Clin Invest 129: 4951–4961, 2019. doi: 10.1172/JCI126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mi S, Li Z, Yang H-Z, Liu H, Wang J-P, Ma Y-G, Wang X-X, Liu H-Z, Sun W, Hu Z-W. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-β1-dependent and -independent mechanisms. J Immunol 187: 3003–3014, 2011. doi: 10.4049/jimmunol.1004081. [DOI] [PubMed] [Google Scholar]
- 28.Mohamed R, Jayakumar C, Chen F, Fulton D, Stepp D, Gansevoort RT, Ramesh G. Low-dose IL-17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J Am Soc Nephrol 27: 745–765, 2016. doi: 10.1681/ASN.2014111136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nony PA, Schnellmann RG. Mechanisms of renal cell repair and regeneration after acute renal failure. J Pharmacol Exp Ther 304: 905–912, 2003. doi: 10.1124/jpet.102.035022. [DOI] [PubMed] [Google Scholar]
- 30.Pechman KR, Basile DP, Lund H, Mattson DL. Immune suppression blocks sodium-sensitive hypertension following recovery from ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol 294: R1234–R1239, 2008. doi: 10.1152/ajpregu.00821.2007. [DOI] [PubMed] [Google Scholar]
- 31.Rabb H, Daniels F, O’Donnell M, Haq M, Saba SR, Keane W, Tang WW. Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 279: F525–F531, 2000. doi: 10.1152/ajprenal.2000.279.3.F525. [DOI] [PubMed] [Google Scholar]
- 32.Sharma R, Kinsey GR. Regulatory T cells in acute and chronic kidney diseases. Am J Physiol Renal Physiol 314: F679–F698, 2018. doi: 10.1152/ajprenal.00236.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spurgeon KR, Donohoe DL, Basile DP. Transforming growth factor-β in acute renal failure: receptor expression, effects on proliferation, cellularity, and vascularization after recovery from injury. Am J Physiol Renal Physiol 288: F568–F577, 2005. doi: 10.1152/ajprenal.00330.2004. [DOI] [PubMed] [Google Scholar]
- 34.Steinmetz OM, Summers SA, Gan P-Y, Semple T, Holdsworth SR, Kitching AR. The Th17-defining transcription factor RORγt promotes glomerulonephritis. J Am Soc Nephrol 22: 472–483, 2011. doi: 10.1681/ASN.2010040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang F, Yin J, Lin Y, Zhang F, Liu X, Zhang G, Kong Y, Lu Z, Wu R, Wang N, Xing T, Qian Y. IL-17C has a pathogenic role in kidney ischemia/reperfusion injury. Kidney Int 97: 1219–1229, 2020. doi: 10.1016/j.kint.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 36.Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 93: 2175–2188, 1994. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol Dial Transplant 15: 1562–1574, 2000. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]