

Abstract
Electronic-cigarette, or vaping, product use–associated lung injury (EVALI) is a syndrome of acute respiratory failure characterized by monocytic and neutrophilic alveolar inflammation. Epidemiological and clinical evidence suggests a role of vitamin E acetate (VEA) in the development of EVALI, yet it remains unclear whether VEA has direct pulmonary toxicity. To test the hypotheses that aerosolized VEA causes lung injury in mice and directly injures human alveolar epithelial cells, we exposed adult mice and primary human alveolar epithelial type II (AT II) cells to an aerosol of VEA generated by a device designed for vaping oils. Outcome measures in mice included lung edema, BAL analysis, histology, and inflammatory cytokines; in vitro outcomes included cell death, cytokine release, cellular uptake of VEA, and gene-expression analysis. Comparison exposures in both models included the popular nicotine-containing JUUL aerosol. We discovered that VEA caused dose-dependent increases in lung water and BAL protein compared with control and JUUL-exposed mice in association with increased BAL neutrophils, oil-laden macrophages, multinucleated giant cells, and inflammatory cytokines. VEA aerosol was also toxic to AT II cells, causing increased cell death and the release of monocyte and neutrophil chemokines. VEA was directly absorbed by AT II cells, resulting in the differential gene expression of several inflammatory biological pathways. Given the epidemiological and clinical characteristics of the EVALI outbreak, these results suggest that VEA plays an important causal role.
Keywords: E-cigarette or vaping product use–associated lung injury, vitamin E acetate, acute respiratory distress syndrome, pulmonary edema
Clinical Relevance
We report that aerosolized vitamin E acetate (VEA) in mice causes dose-dependent pulmonary edema, an increase in alveolar-capillary barrier permeability to protein, and a bronchiolocentric pattern of inflammation with abundant lipid-laden and multinucleated macrophages. Furthermore, we found that VEA aerosol directly injures primary human type II alveolar epithelial cells, causing significant changes in gene expression and the secretion of inflammatory cytokines. In the context of prior evidence, this study strengthens the conclusion that VEA plays a causal role in electronic-cigarette, or vaping, product use–associated lung injury.
As of May 22, 2020, the outbreak of electronic-cigarette (e-cigarette), or vaping, product use–associated lung injury (EVALI) in the United States has affected more than 2,800 patients, resulting in at least 68 deaths. The clinical syndrome of EVALI is notable for the subacute onset of diffuse alveolar opacities after a prodome of constitutional and gastrointestinal symptoms (1). Analysis of pathology and BAL has yielded evidence of neutrophilic inflammation, lipid-containing macrophages, diffuse alveolar damage, bronchiolitis, and organizing pneumonia (1–3). The cause of EVALI remains unknown, and a recent National Institutes of Health workshop report emphasized the importance of conducting systematic and comprehensive laboratory-based studies into the toxicology and health effects of vaping products (4).
Most cases of EVALI have involved exposure to tetrahydrocannabinol (THC)-containing products obtained from the illicit market, although a minority of patients reported exposure only to nicotine-containing e-cigarettes (5). More recent work has demonstrated that nearly all patients with EVALI had detectable BAL concentrations of vitamin E acetate (VEA) (6), a viscous substance used in many of the devices to dilute THC oil or as a thickening ingredient in nicotine-containing vaping liquids (7).
When ingested orally, the α-tocopherol (α-TP) form of vitamin E is preferentially delivered to the tissues after gastrointestinal absorption, where it functions to scavenge free radicals and terminate lipid peroxidation (8, 9). Short-term exposure of aerosolized α-TP has been studied in rat and sheep models of inflammatory lung injury, with some evidence of benefit (10, 11). VEA, marketed for oral consumption because of its chemical stability and long shelf life, undergoes hydrolysis in the gastrointestinal tract, yielding α-TP (12). It remains unknown whether inhaled VEA is efficiently hydrolyzed by the lung.
The discovery of VEA in the airspaces of patients with EVALI suggests that it may play a causal role in its development. Alternatively, VEA may act synergistically with, or be a marker for, other harmful exposures. To date, there has been only one report of a toxic effect of aerosolized VEA in mice with limited characterization (13). No studies have yet been reported on the effects of aerosolized VEA in human lung tissue. We exposed mice to aerosolized VEA for up to 2 weeks and provide evidence that VEA causes dose-dependent neutrophilic and monocytic inflammation and lung injury. Furthermore, we report that primary human alveolar epithelial type II (AT II) cells grown in an air–liquid interface (ALI) culture readily absorb aerosolized VEA and undergo dose-dependent cell death in association with release of neutrophil and monocyte chemokines and large changes in gene expression. Taken together, the results suggest that VEA has direct lung toxicity, consistent with a causal role in EVALI.
Methods
For a detailed description of the methods used in this work, see the data supplement.
Aerosol Generation and Exposure Systems
VEA purchased from Sigma was aerosolized with an atomizer designed for vaping oils (1.5 Ω ceramic coil; ShenZhen Ocity Times Technology) using a Gram Universal Vaping Machine (Gram Research) at 9.4 W. VEA was aerosolized in its pure form (100%). Puff volume was 80 ml, drawn over 4 seconds into a syringe through an electronically controlled three-way valve and then injected into a vaping chamber over 2 seconds (see Figure E1 in the data supplement). JUUL (vegetable glycerin/propylene glycol [VG/PG] ratio = 30:70) (14) was aerosolized with the same volumes and puff times using cool mint pods (5% nicotine) for cell-culture experiments and Virginia Tobacco (5% nicotine) pods for animal experiments (substituted because of new purchase restrictions). The JUUL device was activated by airflow and was fully charged before each exposure. To fill the vaping chamber with aerosol, 10 puffs were initially injected over approximately 1 minute, followed by 110 puffs over 1 hour. In some experiments, VG/PG (50:50) was aerosolized using the same coils and vaping settings as were used for VEA aerosol generation. However, using a series of different mixtures of VG/PG and VEA was not feasible because these substances do not create a homogeneous mixture when combined together, and VEA was nearly exclusively used as an additive in nonpolar vaping liquids such as THC oils in the context of individuals who use these vaping liquids. The chamber was evacuated at a constant rate of 2.0 L/min during the exposure using a calibrated flowmeter (Dwyer) to draw in a mixture of fresh aerosol and room (or incubator) air. Upon completion of 110 puffs, the vacuum outflow speed was increased to clear the chamber over a period of 5 minutes, after which the mice or cells were removed from the chamber. JUUL and VEA aerosol exposures were well tolerated by mice, with no evidence of weight loss or hypothermia (data not shown).
Importantly, a recent report highlights the possibility that thermal decomposition of VEA may produce the highly reactive compound, ketene (15). To generate ketene in that study, a metal coil was operated at 30 W or 50 W (11.0 or 15.5 A, respectively), which is considerably higher power and temperature than used in commercially available vape pens or in our experiments (9.4 W, 2.5 A).
Laboratory Animals
Adult C57BL6 mice were purchased from the National Cancer Institute, exposed to aerosol of either VEA (Sigma) or JUUL (Virginia tobacco or cool mint; 5% nicotine) for 1 hour twice daily for 6 or 15 days, and killed 12 hours from the last exposure. All experimental procedures were performed under protocols approved by the University of California, San Francisco, Institutional Animal Care and Use Committee. Separate animals underwent BAL by tracheotomy, histology (4% paraformaldehyde or optimal cutting temperature compound embedding), or lung harvest for measurement of excess extravascular lung water, as in our prior work (16). BAL total cell and differential cell counts were performed. BAL protein was measured with the bicinchoninic acid protein assay and cytokines were measured with a Luminex assay using a ProcartaPlex multiplex kit from Thermo Fisher Scientific.
Primary Cultures of Human AT II Cells
Human primary AT II cells were harvested (see data supplement) from adult human lungs from a single donor declined for lung transplantation (17). All experiments using cadaver human lung tissues were approved by the University of California, San Francisco, Biosafety Committee. AT II cells were cultured on Transwell membranes until they formed fully confluent monolayers and tight junctions, then cultured in ALI conditions. AT II cells were exposed to aerosol for between 20 minutes and 120 minutes daily for 3 days. During exposures, cells were cultured in serum-free conditions to optimize measurement of LDH (lactate dehydrogenase) and inflammatory cytokines in cell-culture media. LDH was measured by using the Pierce LDH cytotoxicity assay (Thermo Fisher Scientific), and chemokines were measured with a Luminex assay using a ProcartaPlex multiplex kit from Thermo Fisher Scientific.
Measurement of α-TP and α-TP Acetate
AT II cells on Transwell membranes (5–7.5 × 105 cells/membrane) were scraped, centrifuged, and frozen at −80°C. Lipids were extracted from cells using a one-phase solvent system as previously described (18). An aliquot of the supernatant was mixed with internal standards and Splash Lipidomix Mass Spec Standards (Avanti Polar Lipids) and underwent ultraperformance liquid chromatography–tandem mass spectrometry analysis (19). The α-TP acetate was identified using PeakView software (Sciex), and integration was performed using MultiQuant software (Sciex). Cholesterol quantities were identified by the software and quantitated by comparison with their internal standard quantities in the Splash mixture.
RNA Sequencing
AT II cells on Transwell membranes were lysed with RLT Plus buffer, and RNA was extracted using the RNeasy Plus Micro kit (Qiagen). RNA sequencing (RNA-Seq) libraries were built with the NEBNext Ultra II kit (New England Biolabs) and paired-end sequencing was performed on an Illumina NextSeq instrument. Gene counts were generated using STAR (Spliced Transcripts Alignment to a Reference) and human genome build 38 according to previously described methods (20). The DESeq 2 package (21) in the R computational environment (R Foundation for Statistical Computing) was used for normalization and differential expression analysis. Gene set enrichment analysis was performed on genes differentially expressed with a false discovery rate (FDR) < 0.1 using Web-Based Gene Set Analysis Toolkit overrepresentation analysis (22) and the Kyoto Encyclopedia of Genes and Genomes functional database. Pathways significantly enriched at an FDR < 0.1 were identified using a Benjamini-Hochberg multiple test correction.
Results
Aerosolized VEA Causes Lung Injury in Mice
Mice exposed for an hour twice daily to VEA for 6 or 15 days had a progressive increase in excess extravascular lung water compared with unexposed control mice (Figure 1A). Similarly, the concentration of protein in the BAL, an indicator of alveolar-capillary barrier integrity, was increased significantly by VEA at 6 and 15 days (Figure 1B). Plasma SP-D (surfactant protein-D), an indicator of alveolar epithelial injury and a prognostic biomarker in acute respiratory distress syndrome (23, 24), was increased by over 10-fold with 15 days of VEA exposure relative to control mice (Figure 1C). In contrast, short-term exposure to aerosol generated from a JUUL device caused no detectable lung injury by lung water, BAL protein, or plasma SP-D after 15 days of exposure. In a separate experiment, we tested whether VG/PG aerosolized by the same ceramic coils and using the same power settings and puffing protocol as VEA caused a similar degree of lung injury. As shown in Figure E2, 15 days of twice-daily exposure to aerosolized VEA was again demonstrated to cause lung injury compared with control exposure, with no detectable injury in mice exposed to an aerosol of VG/PG.
Figure 1.
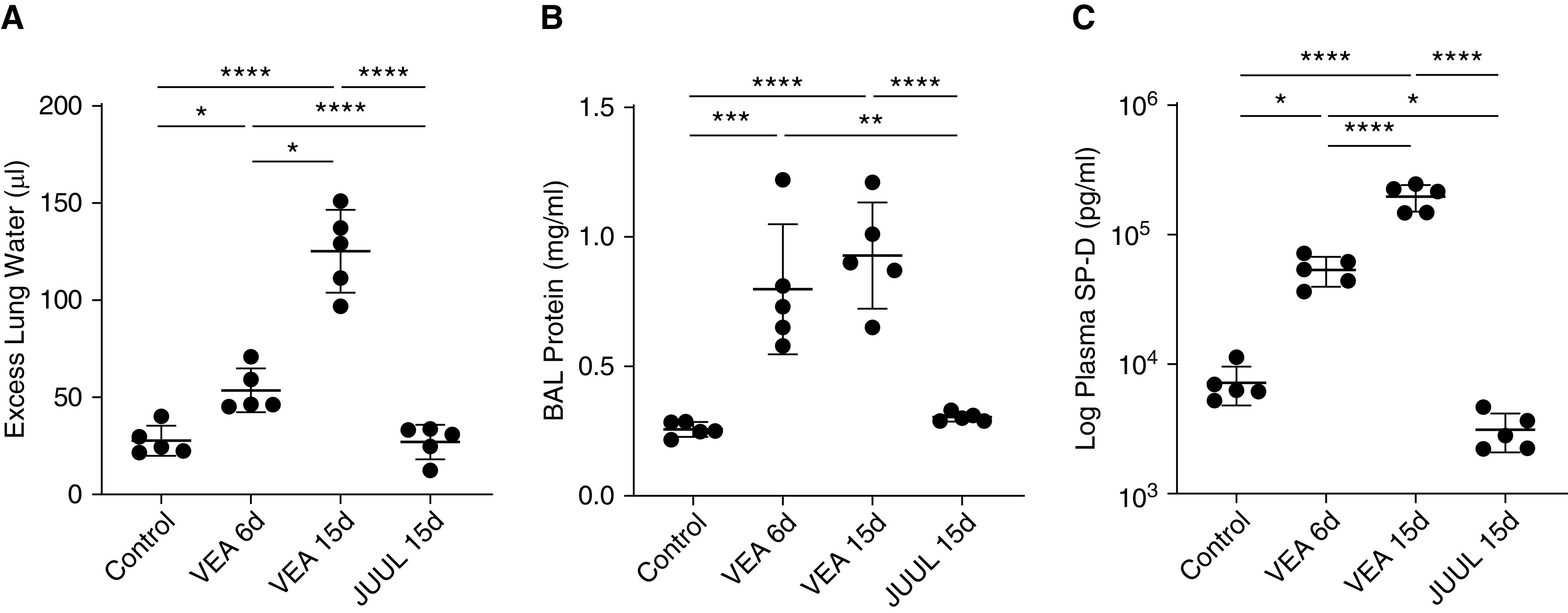
Vitamin E acetate (VEA) aerosol exposure increases lung water, alveolar-capillary barrier permeability, and plasma SP-D (surfactant protein-D), a marker of alveolar epithelial injury. (A) Excess extravascular lung water was significantly increased after 6 and 15 days of twice-daily VEA aerosol exposure compared with control and JUUL-exposed mice. P < 0.0001 by ANOVA. (B) VEA aerosol increased the concentration of protein in the BAL. P < 0.0001 by ANOVA. (C) Plasma SP-D mirrored the lung-water data, consistent with alveolar epithelial injury. P < 0.0001 by ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Tukey multiple-comparison test (MCT).
Lung Injury Is Associated with Neutrophilic Inflammation and Lipid-Laden Macrophages
VEA aerosol exposure for 15 days resulted in an influx of inflammatory cells into the airspaces (Figure 2). The total BAL cell count was increased relative to control and JUUL aerosol exposure (Figure 2A). Airspace neutrophils, which are present at very low levels in health, were increased at both 6 and 15 days of VEA exposure (Figure 2C). Furthermore, VEA-exposed mice had many large vacuolated macrophages, frequently containing multiple nuclei (Figure 2D), a sign of macrophage activation and foreign-body reaction previously reported in exogenous lipoid pneumonia (25, 26). Some EVALI clinical reports (3) have emphasized the presence of lipid droplets in phagocytic cells, identified by staining with the dye Oil Red O. The heavily vacuolated macrophages in VEA-exposed mice stained positive for Oil Red O (Figure E3). The concentration of the potent neutrophil chemoattractant KC (murine homolog of IL-8) and monocyte chemokine MCP-3 were both increased significantly in the airspaces with VEA exposure, consistent with the increase in BAL neutrophils and monocytes and macrophages (Figure 2E).
Figure 2.
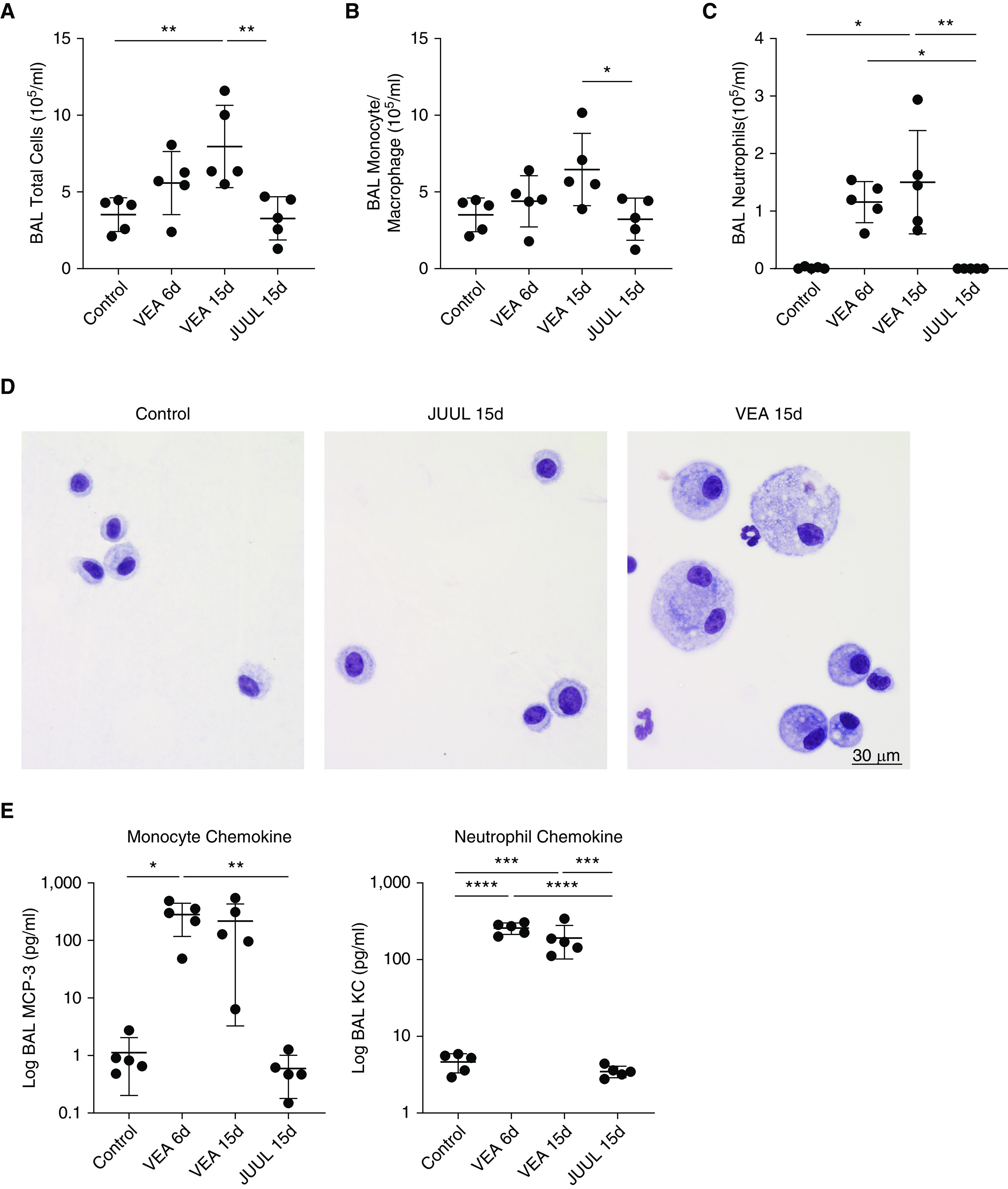
VEA causes mixed monocytic and neutrophilic airspace inflammation in association with monocyte and neutrophil chemokines. (A) BAL total cells were increased following 15 days of VEA exposure. P < 0.01 by ANOVA. (B) BAL monocytes are increased with 15 days of VEA exposure. P < 0.05 by ANOVA. (C) BAL neutrophils were also increased with VEA exposure. P < 0.01 by Kruskal-Wallis test; *P < 0.01 and **P < 0.05 by Dunn MCT. (D) Representative photomicrographs of BAL cytospins demonstrated the presence of neutrophils and large multinucleated foamy macrophages after VEA exposure. Scale bar, 30 μm. (E) The monocyte chemokine MCP-3 is increased in the BAL after 6 days of VEA exposure. P < 0.01 by ANOVA. Similarly, BAL KC (the murine homolog of IL-8) was increased by VEA. P < 0.0001 by ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Tukey MCT in A, B, and E. KC = murine homolog of IL-8; MCP-3 = monocyte chemotactic protein 3.
Histological analysis after 6 days of exposure to VEA indicated monocytic and neutrophilic alveolar and interstitial inflammation in a bronchiolocentric pattern with increased foamy macrophages in the airspaces, similar to what is seen in EVALI patients (2) (Figures 3A and 3B). Mice exposed to JUUL for 15 days had no obvious histological injury (data not shown). As shown in Figure 3C, mice exposed to VEA-containing aerosol for 15 days had a significant accumulation of large, lipid-containing phagocytic cells in the alveolar space in association with a dense inflammatory infiltrate affecting the alveolar septa (Figure 3C). Pulmonary blood vessels were frequently surrounded by lymphocyte-rich cell aggregates, with progression between 6 and 15 days of VEA exposure (Figure 4).
Figure 3.

Histological characterization of VEA-induced pulmonary toxicity. (A) Representative photomicrographs from mice exposed to VEA aerosol for 6 days demonstrated alveolar and interstitial mixed predominantly monocytic and neutrophilic (arrow) inflammation in a bronchiolocentric pattern. Scale bar, 100 μm. (B) Higher magnification revealed clusters of foamy alveolar macrophages (arrowheads) and neutrophils (arrow). Scale bar, 30 μm. (C) Oil Red O staining with modified Mayer hematoxylin counterstain after 15 days of VEA exposure demonstrates multiple alveolar macrophages filled with oil on a background of interstitial inflammation. Scale bar, 20 μm.
Figure 4.

VEA causes lymphocyte-predominant perivascular inflammation. Representative photomicrographs from the lungs of mice exposed to VEA for 6 or 15 days revealed lymphocyte-rich inflammation (arrows) that progressed over time. Scale bars, 100 μm. BV = blood vessel.
Aerosolized VEA Is Directly Toxic to the Human Alveolar Epithelium
To test whether aerosolized VEA is directly toxic to surfactant-producing AT II cells, we isolated primary human AT II cells from lungs declined for transplantation and grew them on collagen-coated Transwells in ALI culture (Figure 5A). Aerosolized VEA for between 20 and 120 minutes daily for 3 days caused dose-dependent cell death of AT II cells, as demonstrated by an increase in supernatant LDH (Figure 5B) and TUNEL staining (Figure E4). We confirmed this dose-dependent cytotoxicity in a subsequent experiment using AT II cells derived from a separate donor lung (Figure E5). In contrast, JUUL aerosol was well tolerated for up to 60 minutes daily for 3 days in comparison with just 20 minutes of VEA (Figure 5C). As in mouse BAL, VEA exposure increased the release of monocyte and neutrophil chemokines relative to control conditions or JUUL aerosol (Figures 5D and 5E) and did so in a dose-dependent fashion (Figure E6).
Figure 5.
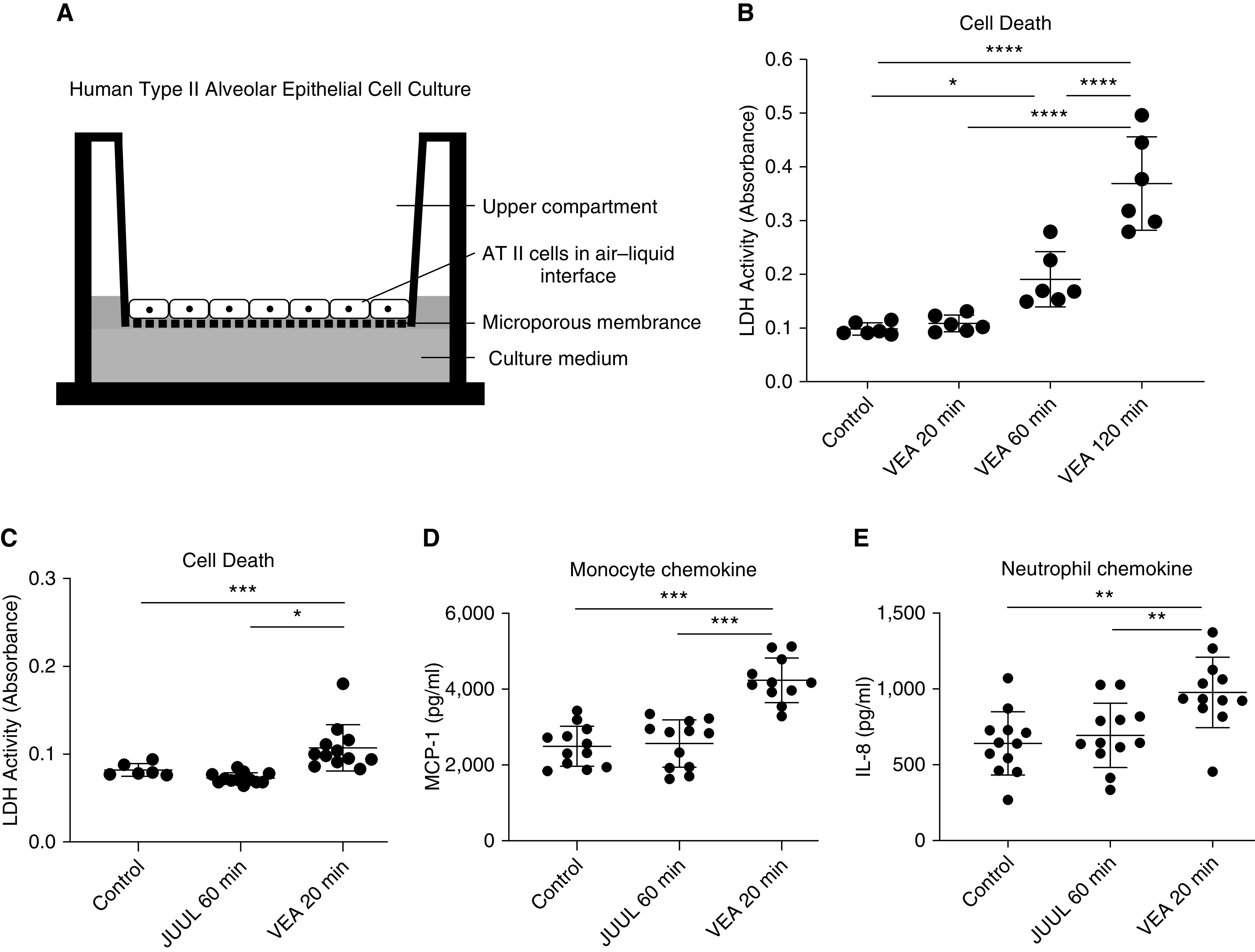
VEA aerosol causes dose-dependent cell death of human alveolar type II cells. (A) Schematic depicting air–liquid interface culture of primary human alveolar epithelial cells isolated from healthy areas of lungs declined for transplantation. The alveolar epithelial type II (AT II) cells were cultured in serum-free media during aerosol exposure for an hour daily for 3 consecutive days. (B) The concentration of LDH (lactate dehydrogenase) in the media increased in a dose-dependent manner. P < 0.0001 by ANOVA. (C) In a subsequent experiment, 20 minutes of VEA aerosol was more toxic to AT II cells than control and 60 minutes of JUUL aerosol. P < 0.001 by ANOVA (note the scale difference in the y-axis between B and C). (D) MCP-1, a monocyte chemokine, was increased by acute exposure to VEA but was not increased by exposure to JUUL aerosol. P < 0.0001 by ANOVA. (E) Acute exposure to VEA increased alveolar epithelial cell release of the potent neutrophil chemokine IL-8. P < 0.01 by ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Tukey MCT.
VEA Is Absorbed by Alveolar Epithelial Cells and Causes Changes in Gene Expression
We next tested whether AT II cells might absorb VEA in ALI culture. As shown in Figure 6A, numerous AT II cells exposed to VEA aerosol develop punctate cytoplasmic staining with Oil Red O. Analysis by mass spectrometry of AT II cells after a 3-day exposure for between 20 and 120 minutes reveals a dose-dependent increase in VEA, consistent with cellular uptake (Figure 6B). α-TP increased with VEA, suggesting that deacetylation occurs slowly in the cells or spontaneously in the serum-free culture media employed in these experiments.
Figure 6.
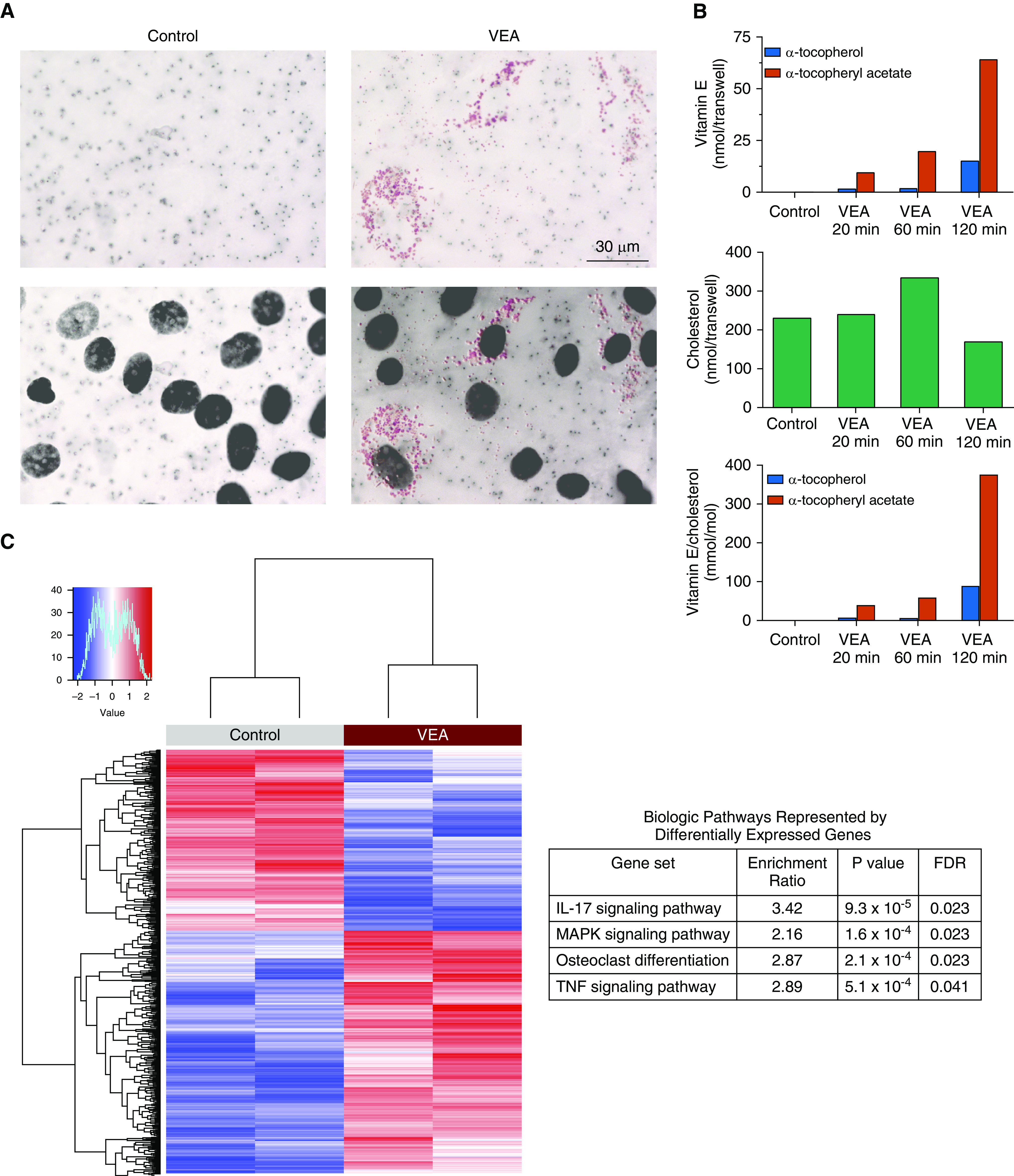
VEA was absorbed by alveolar epithelial cells and induced inflammatory gene expression. (A) Oil Red O staining (upper panels) of Transwell membranes revealed punctate staining of alveolar epithelial cells exposed to VEA aerosol. Superimposition of nuclear counterstain (DAPI, lower panels) demonstrated that the oil staining is cytoplasmic. Scale bar, 30 μm. (B) Mass spectrometry of alveolar cell pellets after 3 days of aerosol exposure (for 20, 60, or 120 min daily) demonstrated dose-dependent accumulation of VEA, quantified in nanomoles per Transwell (upper panel) and in millimoles per mole of cholesterol (bottom panel, corrects for intact cells). A fraction of VEA (α-tocopheryl acetate [red bars]) was deacetylated into α-tocopherol (blue bars). (C) RNA sequencing performed on two VEA-exposed and two unexposed control AT II cells harvested from a single human lung donor identified 752 differentially expressed genes at an FDR < 0.1, with enrichment in inflammatory gene pathways and genes related to osteoclast differentiation. FDR = false discovery rate; MAPK = mitogen-activated protein kinase.
RNA-Seq was performed after 3 days of hourly exposures to control conditions or the aerosol of VEA or JUUL. A principal component analysis revealed clear separation between VEA-exposed and control AT II cells (Figure E7). Differential gene expression between VEA-exposed and control (unexposed) cells identified 752 genes at an FDR < 0.1 (Figure 6C). Pathway analysis revealed enrichment in the IL-17, MAPK, and TNF signaling pathways, together with genes related to osteoclast differentiation (Table E1 and Figure E8). Although we found a dose–response effect (data not shown), as this analysis was performed on the AT II cells from only a single human lung donor, the results must be considered as hypothesis-generating, warranting future validation.
Discussion
The recent outbreak of EVALI has highlighted how little is understood about the health effects of these rapidly evolving products (1). Over the last several months, attention has focused on VEA, a viscous diluent recently introduced into vaping products that has been found in many of the devices used by EVALI patients (27) and has been nearly uniformly recoverable in their BAL (6). However, the mere presence of this chemically stable molecule in the airspaces of patients several days into their hospital course is not proof of causation. To date, only a single study of inhaled VEA in mice has been published, with a limited characterization of the nature of the lung injury (13). Furthermore, no studies have yet tested the effect of VEA on the human alveolar epithelium.
We here provide evidence that VEA aerosolized by ceramic coils of the type used by EVALI patients has direct toxicity to the lung. Similar to patients exposed to VEA (1–3), mice exposed to VEA develop diffuse lung injury with an increase in lung water and BAL protein in association with mixed neutrophilic and monocytic alveolar and interstitial inflammation in a bronchiolocentric pattern, including the presence of numerous lipid-filled and large multinucleated macrophages. Furthermore, we found that surfactant-producing human AT II cells are directly injured by VEA aerosol, which incorporates into the cells and causes the release of monocyte and neutrophil chemokines in the context of major inflammatory changes in gene expression. Strengths of this study include the demonstration of 1) a dose–response relationships with VEA as aerosolized by real-world devices, 2) similar inflammatory signaling changes in the mouse and human AT II cell experiments, and 3) recapitulation of several key pathological features of EVALI.
It has long been known that the lung has difficulty removing long-chain hydrocarbons from the airspaces. Exogenous lipoid pneumonia is most frequently caused by the aspiration of animal or mineral oils and is characterized by macrophage oil uptake and subacute-to-chronic inflammation, resulting in fibrotic or granulomatous lung lesions, radiographic opacities, and gas-exchange abnormalities (28, 29). In experimental models, paraffin oil has been shown to cause AT II cell injury, with loss of microvilli and cytoplasmic vacuolization (30). The type of inflammation induced by oils in the airspaces depends both on the chemical nature of the oil and the droplet size (31). Unlike the aspiration of large oil globules into focal lung areas, the vaping of drugs dissolved in oils by atomizers produces very small droplets capable of reaching the distal airways and the alveoli uniformly across all areas of the lung. This novel biological exposure differs fundamentally from that evaluated in prior reports of lipoid pneumonia and would be expected to result in new patterns of lung injury. For example, evidence of fat attenuation on computed tomographic images caused by large globules of fat in aspiration-induced lipoid pneumonia (29) would be unlikely to occur after exposure to an oil aerosol, and this has not been reported in EVALI (32).
What might be the mechanisms by which VEA causes lung injury? Injury to the alveolar epithelium appears to be central to the pathogenesis, consistent with prior experimental and clinical studies of acute respiratory distress syndrome (33). Identification of VEA-induced transcriptional changes provides some insight regarding the underlying mechanisms of inflammatory lung injury in EVALI. The marked proinflammatory effect of VEA is reflected in the TNF- and MAPK-signaling pathway enrichment. IL-17 signaling in the lung is important for immune defense against bacterial respiratory pathogens (34), and enrichment in IL-17–related genes suggests that aerosolized VEA may induce a host response similar to that elicited by bacterial pneumonia. Notably, an aerosol of very small oil droplets may form a hydrophobic layer on the apical surface of the cells that impairs oxygen diffusion, although the differential-gene-expression analysis did not reveal pathways induced by hypoxia.
The demonstration of the toxicity of VEA does not exclude the possibility of other interacting insults in patients with EVALI, which may include subclinical toxicity from nicotine-containing e-cigarettes (35), microbial or toxin contamination of the devices (36), and the ongoing use of combustible marijuana or tobacco products (37). Moreover, the lack of direct lung injury observed with brief exposures to aerosol from nicotine pod devices (JUUL) should not be interpreted as evidence of their safety. Importantly, recent studies in mice demonstrate that several months of exposure to VG/PG in e-cigarettes results in profound alterations in surfactant metabolism and the accumulation of lipids in macrophages, highlighting the significant potential for synergistic toxicity with preceding or concomitant exposure to oil aerosols (38). Prolonged exposure to inhaled nicotine in animal models has resulted in obstructive lung physiology and pathological changes consistent with emphysema (39).
Furthermore, studies of nicotine-containing e-cigarette aerosol in primary human bronchial epithelial cells grown in ALI have revealed impaired barrier function (39), increased production of IL-8 (40), reactive oxygen species (41), and proteins involved in inflammation (42). To our knowledge, however, no studies of the effects of e-cigarette aerosol on primary human alveolar epithelial cells in ALI have yet been published.
Limitations of this study include the lack of THC experiments due to ongoing strict federal research restrictions and a lack of an assessment of potential synergistic toxicity between VEA and nicotine-containing aerosols. Furthermore, over these shorter exposure durations, we have not observed obvious signs of systemic inflammation, which have been reported in some patients with EVALI, although we did observe increasing perivascular lymphocyte-rich inflammation after 15 days of exposure, which is frequently seen in patients with EVALI (43). There was a limited number of samples for RNA-Seq because of the pandemic-related laboratory shutdown; thus, further studies will be needed in the future for validation. In addition, measuring the breakdown products of VEA aerosol by mass spectrometry would be of future interest.
In conclusion, we present evidence from adult mice and cultures of human alveolar epithelial cells that aerosolized VEA has direct pulmonary toxicity, consistent with the observed pattern in humans with EVALI. In the context of recent studies showing the temporal emergence of this chemical in devices in 2019 and its presence in the airspaces of nearly all EVALI patients, our data strengthen the conclusion that VEA plays a causal role in this syndrome of acute respiratory failure clinically recognized as vaping-associated lung injury.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Scott Leonard at the Linus Pauling Institute, College of Public Health and Human Sciences, Oregon State University, for outstanding technical assistance with the mass spectrometric work.
Footnotes
Supported by U.S. National Heart, Lung, and Blood Institute grants U54 HL147127, R01 HL134828, and R35 HL140026.
Author Contributions: S.M.: conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing and editing. X.F., M.G.T., K.D.J., C.L., and P.H.S.: collection and assembly of data, data analysis, and data interpretation. C.S.C. and M.A.M.: data analysis, data interpretation, and financial support. J.E.G.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, editing, and approval of the final version to be published.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0209OC on August 21, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Layden JE, Ghinai I, Pray I, Kimball A, Layer M, Tenforde MW, et al. Pulmonary illness related to E-cigarette use in Illinois and Wisconsin - final report. N Engl J Med. 2020;382:903–916. doi: 10.1056/NEJMoa1911614. [DOI] [PubMed] [Google Scholar]
- 2.Butt YM, Smith ML, Tazelaar HD, Vaszar LT, Swanson KL, Cecchini MJ, et al. Pathology of vaping-associated lung injury. N Engl J Med. 2019;381:1780–1781. doi: 10.1056/NEJMc1913069. [DOI] [PubMed] [Google Scholar]
- 3.Maddock SD, Cirulis MM, Callahan SJ, Keenan LM, Pirozzi CS, Raman SM, et al. Pulmonary lipid-laden macrophages and vaping. N Engl J Med. 2019;381:1488–1489. doi: 10.1056/NEJMc1912038. [DOI] [PubMed] [Google Scholar]
- 4.Alexander LEC, Ware LB, Calfee CS, Callahan SJ, Eissenberg T, Farver C, et al. NIH workshop report. E-cigarette or vaping product use associated lung injury (EVALI): developing a research agenda. Am J Respir Crit Care Med. doi: 10.1164/rccm.201912-2332WS. [online ahead of print] 3 Apr 2020; DOI: 10.1164/rccm.201912-2332WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatham-Stephens K, Roguski K, Jang Y, Cho P, Jatlaoui TC, Kabbani S, et al. Lung Injury Response Epidemiology/Surveillance Task Force; Lung Injury Response Clinical Task Force. Characteristics of hospitalized and nonhospitalized patients in a nationwide outbreak of e-cigarette, or vaping, product use-associated lung injury: United States, November 2019. MMWR Morb Mortal Wkly Rep. 2019;68:1076–1080. doi: 10.15585/mmwr.mm6846e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blount BC, Karwowski MP, Shields PG, Morel-Espinosa M, Valentin-Blasini L, Gardner M, et al. Lung Injury Response Laboratory Working Group. Vitamin E acetate in bronchoalveolar-lavage fluid associated with EVALI. N Engl J Med. 2020;382:697–705. doi: 10.1056/NEJMoa1916433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blount BC, Karwowski MP, Morel-Espinosa M, Rees J, Sosnoff C, Cowan E, et al. Evaluation of bronchoalveolar lavage fluid from patients in an outbreak of e-cigarette, or vaping, product use-associated lung injury: 10 States, August-October 2019. MMWR Morb Mortal Wkly Rep. 2019;68:1040–1041. doi: 10.15585/mmwr.mm6845e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burton GW, Ingold KU. Autoxidation of biological molecules: 1. antioxidant activity of vitamin E and related chain-breaking phenolic antioxidants in vitro. J Am Chem Soc. 1981;103:6472–6477. [Google Scholar]
- 9.Traber MG. Vitamin E. In: Erdman JW, MacDonald IA, Zeisel SH, editors. Present knowledge in nutrition. 10th ed. Hoboken, NJ: Wiley-Blackwell; 2012. pp. 214–229. [Google Scholar]
- 10.Morita N, Traber MG, Enkhbaatar P, Westphal M, Murakami K, Leonard SW, et al. Aerosolized alpha-tocopherol ameliorates acute lung injury following combined burn and smoke inhalation injury in sheep. Shock. 2006;25:277–282. doi: 10.1097/01.shk.0000208805.23182.a7. [DOI] [PubMed] [Google Scholar]
- 11.Hybertson BM, Chung JH, Fini MA, Lee YM, Allard JD, Hansen BN, et al. Aerosol-administered alpha-tocopherol attenuates lung inflammation in rats given lipopolysaccharide intratracheally. Exp Lung Res. 2005;31:283–294. doi: 10.1080/01902140590918560. [DOI] [PubMed] [Google Scholar]
- 12.Schmölz L, Birringer M, Lorkowski S, Wallert M. Complexity of vitamin E metabolism. World J Biol Chem. 2016;7:14–43. doi: 10.4331/wjbc.v7.i1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhat TA, Kalathil SG, Bogner PN, Blount BC, Goniewicz ML, Thanavala YM. An animal model of inhaled vitamin E acetate and EVALI-like lung injury. N Engl J Med. 2020;382:1175–1177. doi: 10.1056/NEJMc2000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvanko AM, Havel CM, Jacob P, Benowitz NL. Characterization of nicotine salts in 23 electronic cigarette refill liquids. Nicotine Tob Res. 2020;22:1239–1243. doi: 10.1093/ntr/ntz232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu D, O’Shea DF. Potential for release of pulmonary toxic ketene from vaping pyrolysis of vitamin E acetate. Proc Natl Acad Sci U S A. 2020;117:6349–6355. doi: 10.1073/pnas.1920925117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su X, Lee JW, Matthay ZA, Mednick G, Uchida T, Fang X, et al. Activation of the alpha7 nAChR reduces acid-induced acute lung injury in mice and rats. Am J Respir Cell Mol Biol. 2007;37:186–192. doi: 10.1165/rcmb.2006-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross JT, Nesseler N, Lee J-W, Ware LB, Matthay MA. The ex vivo human lung: research value for translational science. JCI Insight. 2019;4:e128833. doi: 10.1172/jci.insight.128833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDougall MQ, Choi J, Stevens JF, Truong L, Tanguay RL, Traber MG. Lipidomics and H2(18)O labeling techniques reveal increased remodeling of DHA-containing membrane phospholipids associated with abnormal locomotor responses in α-tocopherol deficient zebrafish (Danio rerio) embryos. Redox Biol. 2016;8:165–174. doi: 10.1016/j.redox.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi J, Leonard SW, Kasper K, McDougall M, Stevens JF, Tanguay RL, et al. Novel function of vitamin E in regulation of zebrafish (Danio rerio) brain lysophospholipids discovered using lipidomics. J Lipid Res. 2015;56:1182–1190. doi: 10.1194/jlr.M058941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langelier C, Kalantar KL, Moazed F, Wilson MR, Crawford ED, Deiss T, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci U S A. 2018;115:E12353–E12362. doi: 10.1073/pnas.1809700115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ware LB, Koyama T, Billheimer DD, Wu W, Bernard GR, Thompson BT, et al. NHLBI ARDS Clinical Trials Network. Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest. 2010;137:288–296. doi: 10.1378/chest.09-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisner MD, Parsons P, Matthay MA, Ware L, Greene K Acute Respiratory Distress Syndrome Network. Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax. 2003;58:983–988. doi: 10.1136/thorax.58.11.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sung S, Tazelaar HD, Crapanzano JP, Nassar A, Saqi A. Adult exogenous lipoid pneumonia: a rare and underrecognized entity in cytology: a case series. Cytojournal. 2018;15:17. doi: 10.4103/cytojournal.cytojournal_29_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumarguru BN, Natarajan M, Biligi DS, Raghupathi AR. Giant cell lesions of lungs: a histopathological and morphometric study of seven autopsy cases. J Clin Diagn Res. 2015;9:EC12–EC16. doi: 10.7860/JCDR/2015/15035.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor J, Wiens T, Peterson J, Saravia S, Lunda M, Hanson K, et al. Lung Injury Response Task Force. Characteristics of e-cigarette, or vaping, products used by patients with associated lung injury and products seized by law enforcement: Minnesota, 2018 and 2019. MMWR Morb Mortal Wkly Rep. 2019;68:1096–1100. doi: 10.15585/mmwr.mm6847e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hadda V, Khilnani GC. Lipoid pneumonia: an overview. Expert Rev Respir Med. 2010;4:799–807. doi: 10.1586/ers.10.74. [DOI] [PubMed] [Google Scholar]
- 29.Marchiori E, Zanetti G, Mano CM, Hochhegger B. Exogenous lipoid pneumonia: clinical and radiological manifestations. Respir Med. 2011;105:659–666. doi: 10.1016/j.rmed.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Baskerville A. Ultrastructural changes in experimental lipid pneumonia. Res Vet Sci. 1969;10:4–6. [PubMed] [Google Scholar]
- 31.Paterson JLH. An experimental study of pneumonia following the aspiration of oily substances: lipoid cell pneumonia. J Pathol Bacteriol. 1938;46:151–164. [Google Scholar]
- 32.Henry TS, Kanne JP, Kligerman SJ. Imaging of vaping-associated lung disease. N Engl J Med. 2019;381:1486–1487. doi: 10.1056/NEJMc1911995. [DOI] [PubMed] [Google Scholar]
- 33.Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwanaga N, Kolls JK. Updates on T helper type 17 immunity in respiratory disease. Immunology. 2019;156:3–8. doi: 10.1111/imm.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gotts JE, Jordt S-E, McConnell R, Tarran R. What are the respiratory effects of e-cigarettes? BMJ. 2019;366:l5275. doi: 10.1136/bmj.l5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee M-S, Christiani DC. Microbial toxins in nicotine vaping liquids. Am J Respir Crit Care Med. 2020;201:741–743. doi: 10.1164/rccm.201911-2178LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellington S, Salvatore PP, Ko J, Danielson M, Kim L, Cyrus A, et al. Lung Injury Response Epidemiology/Surveillance Task Force. Update: product, substance-use, and demographic characteristics of hospitalized patients in a nationwide outbreak of e-cigarette, or vaping, product use-associated lung injury: United States, August 2019-January 2020. MMWR Morb Mortal Wkly Rep. 2020;69:44–49. doi: 10.15585/mmwr.mm6902e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madison MC, Landers CT, Gu B-H, Chang C-Y, Tung H-Y, You R, et al. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J Clin Invest. 2019;129:4290–4304. doi: 10.1172/JCI128531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Arcos I, Geraghty P, Baumlin N, Campos M, Dabo AJ, Jundi B, et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax. 2016;71:1119–1129. doi: 10.1136/thoraxjnl-2015-208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crotty Alexander LE, Drummond CA, Hepokoski M, Mathew D, Moshensky A, Willeford A, et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am J Physiol Regul Integr Comp Physiol. 2018;314:R834–R847. doi: 10.1152/ajpregu.00270.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moses E, Wang T, Corbett S, Jackson GR, Drizik E, Perdomo C, et al. Molecular impact of electronic cigarette aerosol exposure in human bronchial epithelium. Toxicol Sci. 2017;155:248–257. doi: 10.1093/toxsci/kfw198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herr C, Tsitouras K, Niederstraßer J, Backes C, Beisswenger C, Dong L, et al. Cigarette smoke and electronic cigarettes differentially activate bronchial epithelial cells. Respir Res. 2020;21:67. doi: 10.1186/s12931-020-1317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukhopadhyay S, Mehrad M, Dammert P, Arrossi AV, Sarda R, Brenner DS, et al. Lung biopsy findings in severe pulmonary illness associated with e-cigarette use (vaping) Am J Clin Pathol. 2020;153:30–39. doi: 10.1093/ajcp/aqz182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.