

Abstract
We report a highly regio-, diastereo- and enantioselective vicinal dihalogenation of allyl amides. E-and Z-alkenes with both aryl and alkyl substituents were compatible with this chemistry. This is the result of exquisite catalyst controlled regioselectivity enabling use of electronically unbiased substrates. The reaction employs commercially available catalysts and halenium sources along with cheap inorganic halide salts to affect this transformation. A preliminary effort to extend this chemistry to heterodihalogenation is also presented.
Enantioselective alkene halogenation is a powerful transformation for rapidly increasing the molecular complexity of readily available and/or easily accessed motifs. With the advent of numerous methodologies for asymmetric halofunctionalization of alkenes,1 the challenging asymmetric vicinal dihalogenation reaction of alkenes has come into focus. Most well-established asymmetric halofunctionalizations reported have achieved enantioselective C–X (X = Cl, Br, I or F) bond formation along with concomitant formation of a C–O, C–N or even a C–C bond formation depending on the nucleophile employed in intercepting the putative intermediate. In contrast, halide nucleophiles that could lead to dihalogenated products have not been employed with the same levels of success. A timely and informative review of olefin dihalogenation by Denmark and co-workers provides a historical account of the field.2 Nonetheless, a few landmark achievements in this regard merit mention. Snyder’s group reported an enantioselective total synthesis of (–)-napyradiomycin A1 that featured an asymmetric dichlorination of an advanced precursor using chlorine gas and an excess of a chiral 1,1′-biphenanthryl promoter.3 The same group also reported the asymmetric dichlorination of unfunctionalized olefins with a chiral sulfide reagent.4 The Nicolaou group reported an asymmetric dichlorination of cinnamyl alcohols using (DHQD)2PHAL/ArICl2 reagent system.5 D enmark’s group reported the nonenantioselective syn-stereospecific dichlorination of alkenes.6 Burns and co-workers demonstrated the dibromination of cinnamyl alcohols using a chiral diol and dibromomalonate (as bromenium source) and a bromotitanium triisopropoxide (as bromide source).7 The highly regio- and enantioselctive vicinal asymmetric chlorobromination and dichlorination of aliphatic allyl alcohols using N-bromosuccinimide/tert-butylhypochlorite reagent system was also reported by the same group.8
From a mechanistic perspective, this transformation represents a unique challenge. First, facile olefin-to-olefin halenium transfer can rapidly erode the stereochemical fidelity of the putative chiral haliranium intermediate (Figure 1a).9 Second, a poor regioselectivity in the halide opening of the putative chiral haliranium ion intermediate can erode the enantioselectivity of the transformation; the two “constitutional isomers” resulting from the regioselectivity of the transformation are in fact the two enantiomers of the product (Figure 1a). Hence, in addition to exquisite face selectivity in alkene halogenation, excellent control of regioselectivity is also imperative. It is perhaps not surprising that many substrates that have succumbed to highly enantioselective dihalogenations are electronically biased: employing styryl systems leads to an inherent bias for the halide opening at the benzylic position. The development of catalyst-controlled regioselectivity as opposed to a substrate controlled process holds promise in significantly improving the scope of the transformation. It merits mention the second generation system reported by Burns et al.8a for dihalogenation of allyl alcohols represents the first example of a catalyst-controlled regioselection whereby a covalently tethered Ti-halide is postulated to impart high levels of regioselectivity for halide opening of the putative intermediate. The absence of the alcohol motif in other classes of substrates demands alternate means of achieving the required regioselectivity.8a
Figure 1.

(a) Mechanistic challenges for asymmetric dihalogenation. (b) Summary of catalytic asymmetric intermolecular halohydrin, haloetherification and haloesterification.
Our group has reported a highly enantioselective intermolecular haloetherifcation and haloesterification reaction of unsaturated amides (Figure 1b).10 One of the key features of the transformation was the excellent catalyst-controlled regioselectivity that renders a wide variety of alkyl-substituted alkenes as compatible substrates for the chemistry. We realized the potential to extend this chemistry to the enantioselective dihalogenation of related substrates by discovering an appropriate halide salt to intercept the same putative intermediate.
Our studies commenced with identifying conditions that could transform 4a to 5a. Pilot studies indicated the best enantioselectivities were seen when MeCN or CF3CH2OH (TFE) was used as the solvent. It should be noted that competing intermolecular processes such as interception of the intermediate by the solvent leads to side products 6a (from TFE incorporation) or 8a (the Ritter product when CH3CN is employed).11 Also, the intramolecular halocyclization path yields the oxazoline 7a as a side product. Our initial screening of reactions conditions had to not only deliver the desired dihalogenated products in acceptable yields and enantioselectivity but also avoid the production of side products 6a–8a.
Numerous chloride sources were evaluated for this test reaction in the presence of 2.0 equiv of DCDMH, 10 mol % of (DHQD)2PHAL and acetonitrile (MeCN) as a solvent. Initially, soluble quaternary ammonium chloride salts were evaluated. Disappointingly, a mixture of products with a marginal preference for desired product 5a as a racemate were produced (5a:7a = 55:45, 50:50 er, Table 1, entry 1). Use of NaCl predominantly produced the Ritter product 8a (Table 1, entry 2). Encouragingly, LiCl fared much better despite its sparing solubility in organic solvents. Reactions run at ambient temperature with 15 equiv of LiCl gave significant amounts of the chlorocyclized byproduct 7a (5a:7a = 79:21, Table 1, entry 3). Lowering the temperature to −30 °C gave the dichlorinated product exclusively (5a:7a = 95:5 and 92:8 er, entry 4); although encouraging, this result gave significantly lower enantioselectivity for other substrates (Table S1).
Table 1.
Summary of Optimization Studies for Dichlorination
 | ||||||
| entry | solvent | Temp | XCI (equiv) | Yielda | 5a:6a:7a:8a | er (5a)b |
|---|---|---|---|---|---|---|
| 1 | MeCN | 23 | TEAC (15) | 82 | 55:0:45:0 | 50:50 |
| 2 | MeCN | 23 | NaCl (15) | 70 | 0:0:13:87 | nd |
| 3 | MeCN | 23 | LiCl (15) | 78 | 79:0:21:0 | 80:20 |
| 4 | MeCN | −30 | LiCl (15) | 84 | 95:0:5:0 | 92:8 |
| 5 | TFE | 23 | LiCl (15) | 82 | 45:56:0:0 | 97:3 |
| 6 | TFE | −30 | LiCl (50) | 86 | 86:14:0:0 | 98:2 |
| 7 | TFE | −30 | LiCl (100) | 95 | 95:5:0:0 | 98:2 |
| 8 | TFE | 23 | LiCl (100) | 90 | 95:5:0:0 | 92:8 |
| 9 | TFE | 23 | TEAC (100) | 84 | 66:34:0:0 | 93:7 |
| 10 | TFE | 23 | NaCl (100) | 82 | 0:89:11:0 | nd |
| 11 | TFE | 23 | CsCl (100) | 87 | 55:44:3:0 | 95:5 |
| 12C | TFE | 23 | Cl2 (gas) | 83 | 16:43:41:0 | 50:50 |
Combined yield, determined by NMR.
Determined by chiral HPLC.
Cl2 gas was generated in situ and bubbled into the reaction; DCDMH = dichlorodimethyl hydantoin; TFE = 2,2,2-trifluoroethanol; TEAC = tetraethylammonium chloride.
Further experimentation revealed employing trifluoroethanol (CF3CH2OH, TFE) as the reaction solvent gave reproducibly exquisite enantioselectivity (≥97:3 er) for the desired product (Table 1, entry 5), albeit at the expense of product yield (ca. 40%) due to formation of 6a. Formation of byproduct 6a could be greatly mitigated by increasing the stoichiometry of LiCl from 15 to 100 equiv (>20:1 5a:6a, Table 1, entry 7). This result was surprising given the low solubility of LiCl in TFE (ca. 20 mg/mL), i.e., only ~24 equiv of the 100 equiv added is actually solvated. Intrigued by the effect of solid LiCl, we studied the role of the counterion under optimized conditions with various chloride salts that have a wide range of solubilities in TFE. The fully soluble tetraethylammonium chloride produced a mixture of products with marginal preference for desired product 5a in high enantioselectivity (93:7 er, Table 1, entry 9). Treating compound 4a with sparingly soluble NaCl in TFE (0.03 M solubility) returned predominantly the TFE incorporated product 6a (Table 1, entry 10). These results are in complete contrast with LiCl (entry 8), which delivers the desired product in high chemo- and enantioselectivity. CsCl, exhibiting similar solubility as LiCl in TFE (0.53 M for CsCl vs 0.47 M for LiCl), also fails to deliver the product in high selectivity, yielding a nearly 1:1 ratio of 5a:6a. From these results, it is evident although solubility of the chloride source might be an important factor that dictates product distribution, the counterion is equally important. Additionally, the presence of undissolved LiCl is also essential for good selectivity. Finally, we ruled out the possibility that in situ generated Cl2 gas might be the active chlorenium and chloride source; in this instance, very low selectivity was observed for the desired product (Table 1, entry 12). Numerous control experiments suggest these reactions likely occur at the solid−liquid interface. These experiments are discussed later in the paper.
Mapping the generality of the dichlorination reaction, numerous cis-substituted allyl amides were examined under optimized condition (0.02 M substrate concentration in TFE, 100 equiv of LiCl and 2.0 equiv of DCDMH at −30 °C). Dichlorination of Z-aliphatic amides exhibit high diastereoselectivity (Table 2 see 5a to 5f, >99:1 dr). The identity of the benzamide motif had little influence on the enantioselectivity of this reaction; products 5a and 5b were both formed in >99:1 er (Table 2, entries 1 and 2). The other Z-alkyl substituted olefins afforded dichlorinated products in complete diastereo- and enantioselectivity (see 5c, 5d). The benzyloxy substituted alkene 4e gave lower enantioselectivity (89:11 er). Aryl-substituted Z-olefins gave corresponding products in high enantioselectivity and regioselectivity (>97:3 er and >99:1 rr, Table 2 entries 7–9). The diastereoselectivities and yields for these entries are varied (1.7:1 to >20:1 dr and 35% to 88% yield); as expected, reduced diastereoselectivity was seen with increasing benzylic cation stabilization. The poor yield for substrate 4g is attributed to formation of TFE incorporated product 6g and the six-member ring cyclized product,12 whereas the moderate yield for compound 4h is due to formation of TFE incorporated product 6h. Nonetheless, the trifluoromethyl substituted olefin 4i afforded the dichlorinated product with exquisite yield and stereoselectivity (88% yield, >99:1 dr, >99:1 er, Table 2, entry 9). The trans aliphatic substituted olefins showed high level of diastereoselectivity (>99:1 dr, see 5j to 5l). Changing 4-bromobenzamide to 4-nitrobenzamide gave identical results (~92:8 er, ~80% yield, see 5j, 5k). The benzyloxy protected substrate 4l formed dichlorinated product in 85% yield and 89:11 er (Table 2, entry 12). Compound 4m with aryl substituent on the alkene gave moderate yield (due to competing production of 6m and the corresponding six-member ring cyclized product12) and moderate enantioselectivity for product 5m (63% yield, 90:10 er, Table 2, entry 13). Trisubstituted alkene 4n was also compatible with this chemistry and returned the desired product in 73% yield and 92:8 er. It warrants emphasis for trisubstituted and aryl-substituted olefins, a higher substrate concentration (0.20 M) is required for mitigating formation of TFE incorporated byproduct (see SI, Table S2 for concentration studies). The quasienantiomeric catalyst, (DHQ)2PHAL, transformed two substrates (4a, 4j) to the corresponding enantiomeric products in comparable yield and selectivity (Table 2, entries 15 and 16).
Table 2.
Substrate Scope for Asymmetric Dichlorination
 | ||||||
| entry | R1 | R2 | R3 | Prod | %yielda/dr | erb |
|---|---|---|---|---|---|---|
| 1c | H | C3H7 | NO2 | 5a | 91/>99:1 | >99:1 |
| 2 | H | C3H7 | Br | 5b | 75/>99:1 | >99:1 |
| 3 | H | C2H5 | NO2 | 5c | 85/>99:1 | >99:1 |
| 4 | H | C5H11 | NO2 | 5d | 90/>99:1 | >99:1 |
| 5 | H | BnOCH2 | NO2 | 5e | 79/>99:1 | 89:11 |
| 6 | H | TBDPSOC2H4 | NO2 | 5f | 82/>99:1 | >99:1 |
| 7f,g | H | pMe-Ph | NO2 | 5g | 35c/1.7:1 | 97:3e |
| 8d,g | H | Ph | NO2 | 5h | 62c/11:1 | >99:1 |
| 9g | H | pCF3-Ph | NO2 | 5i | 88c/>99:1 | >99:1 |
| 10 | C3H7 | H | NO2 | 5j | 76/>99:1 | 93:7 |
| 11 | C3H7 | H | Br | 5k | 82/>99:1 | 92:8 |
| 12 | BnOCH2 | H | NO2 | 51 | 85>99:1 | 89:11 |
| 13d,g | Ph | H | NO2 | 5m | 63d/53:1 | 90:10 |
| 14d,g | Me | Me | NO2 | 5n | 73c/na | 92:8 |
| 15h | H | C3H7 | NO2 | ent-5a | 94/>99:1 | 98:2 |
| 16h | C3H7 | H | NO2 | ent-5j | 80/>99:1 | 96:4 |
Isolated yield on a 0.1 mmol scale.
Enantioselectivity determined by chiral HPLC.
Performed on 1 g scale with 1% cat.
Mass balance is TFE incorporated products.
The minor diastereomer shows 91:9 er.
Mass balance is cyclized and TFE incorporated products.
Substrate concentration was 0.2 M.
Reactions were performed with quasienantiomeric (DHQ)2PHAL catalyst.
This chemistry also delivers vicinal dibrominated and chlorobrominated products with high stereoselectivity. Treating 4a in TFE (0.2 M) with 100 equiv of LiCl as chloride source and 2.0 equiv of NBS as bromenium source gave 9a in 97% yield and >99:1 er (Table 3, entry 1). Using LiBr with NBS gave the dibrominated product 9a′ in 90% yield and 83:17 er (Table 3, entry 2). The Z-aromatic olefin 4g returned chlorobrominated product 9g in 96% yield with high stereoselectivity (>99:1 er and >99:1 dr, see entry 3, Table 3). Chlorobromination of E-amides 4j and 4m formed desired products 9j and 9m in high diastereoselectivity and good enantioselectivity. Yield for aromatic substrate 4m suffers due to the formation of the corresponding six-member ring cyclized product12 (58% yield, see entry 5, Table 3). Noteworthy, chlorobrominated products shed light on the regiochemical course of the reaction. This information is not easily obtained from either the dichlorination or dibromination reactions, because the nucleophile and elecrophile are not distinguished in the final products of the latter two transformations. The catalyst’s exquisite control in the dihalogenation reaction was demonstrated via chlorobromination of 4b in presence and absence of (DHQD)2PHAL (Table S7).
Table 3.
Regio- and Enantioselective Hetero-Dihalogenation
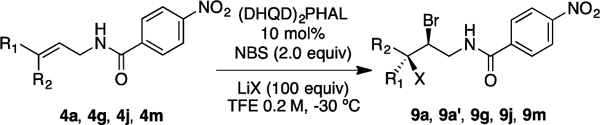 | |||||||
| entry | R1 | R2 | LiX | X | prod | %yieldadr | erb |
|---|---|---|---|---|---|---|---|
| 1 | H | c3h7 | LiCl | Cl | 9a | 97/>99:1 | >99:1 |
| 2 | H | c3h7 | LiBr | Br | 9a’ | 90/>99:1 | 83:17 |
| 3 | H | 4-CF3Ph | LiCl | Cl | 9g | 96/>99:1 | >99:1 |
| 4 | C3H7 | H | LiCl | Cl | 9j | 85/>99:1 | 92:8 |
| 5 | Ph | H | LiCl | Cl | 9m | 58/>99:1 | 89:11 |
Isolated yield.
Determined by chiral HPLC.
The fact these reactions required up to 100 equiv of LiCl for optimal results was counterintuitive, given the sparing solubility of LiCl in organic solvents. Additionally, a significant amount of the added LiCl remained undissolved during the reaction and could be recovered at the end. To determine whether suspended LiCl plays a role in this reaction and if indeed the reaction is occurring on a solid–liquid interface, two sets of control experiments were executed. In the first set, a saturated solution of LiCl in TFE (0.47 M concentration) was prepared and employed in dichlorination reactions with different substrate concentrations (Table 4, entries 1–4). Two key observations were made. First, all reactions gave similar product ratios regardless of substrate concentration or substrate:LiCl ratio (5.8–6.6:1 ratio of 5a:6a). Second, the ratio of 5a:6a was significantly worse than that observed under optimized reaction conditions that employed a large excess of LiCl (>20:1 5a:6a), i.e., reactions in the presence of suspended/undissolved LiCl were significantly more selective.
Table 4.
Effect of LiCl Stoichiometry, Agitation, and Concentration
 | |||||
| entry | TFE (mL) | conc. (4a) | conc. (LiCl) | LiClc (equiv) | 5a:6aa,b |
|---|---|---|---|---|---|
| 1 | 0.5 | 0.20 | 0.47 | 2 | 5.8:1 |
| 2 | 0.5 | 0.08 | 0.47 | 5 | 6.6:1 |
| 3 | 2 | 0.02 | 0.47 | 20 | 6.4:1 |
| 4 | 7 | 0.006 | 0.47 | 67 | 6.2:1 |
| conc | RPM | equiv of LiCl | yield%a | ||
| 5 | 0.02 | 0 | 15 | 95 | 1.0:1.0 |
| 6 | 0.02 | 100 | 15 | 88 | 3.5:1.0 |
| 7 | 0.02 | 300 | 15 | 90 | 3.5:1.0 |
| 8 | 0.02 | 0 | 100 | 90 | 1.5:1.0 |
| 9 | 0.02 | 300 | 100 | 95 | >20:1 |
| 10 | 0.20 | 0 | 100 | 82 | >20:1 |
| 11 | 0.20 | 300 | 100 | 87 | >20:1 |
Ratios and yields determined by NMR.
1% to 3% of cyclized product 7a was seen by NMR.
0.40 M solution of LiCl in TFE was prepared by saturating TFE with LiCl, filtering undissolved LiCl and determining molarity of the dissolved salt from the difference in mass of recovered LiCl.
A second set of control experiments was performed to probe mixing and mass-transfer effects. The stirring speed was altered, first in the soluble regime (15 equiv of LiCl, 0.3 M in LiCl) and then in the insoluble regime. The stirring speed had a remarkable effect on product distribution. In the absence of stirring (0 rpm), a significant amount of byproduct 6a was formed (5a:6a = 1:1, Table 4, entry 5). At 100 and 300 rpm, this ratio improved to 3.5:1 (entries 6 and 7, Table 4). In the insoluble regime (100 equiv of LiCl, 0.02 M substrate concentration, entries 8–11, Table 4), this effect was more pronounced. At 0 rpm, the ratio of 5a:6a was 1.5:1. Increasing the rate of stirring to 300 rpm gave the desired product almost exclusively (95% yield, 5a:6a = >20:1, Table 4, entry 9). With further increase in substrate concentration to 0.20 M, the effects of mass transfer become less pronounced (5a:6a = >20:1 at 0 rpm as well as at 300 rpm, see entries 10 and 11 in Table 4). The combination of results from Tables 1 and 4, highlighting the requirement for a Li cation, and also the dependence on the heterogeneous nature of the reaction, strongly suggests success in greatly limiting the TFE incorporated side product 6 is due to the reaction proceeding at the liquid–solid interface. We find a small but reproducible effect on product ratios as a function of LiCl particle size. Smaller mesh salt yields the highest selectivity for the desired dichlorinated product by minimizing the TFE incorporated side product (see Table S5). Finally, dichlorination of 4a was compared in the presence and absence of 12-crown-4 ether (Table S6). Promoting the increased solubility of LiCl in the presence of 12-C-4 leads to diminshed selectivity for the dichlorinated product, suggesting that soluble LiCl is not effective to circumvent the production of the TFE incorporated product.
We report an experimentally expedient dihalogenation reaction catalyzed with (DHQD)2PHAL, yielding products in high yield and enantioselectivity. Exquisite catalyst controlled regioselectivity has allowed for a broad substrate scope that includes alkyl and aryl substituted allyl amides. The stereochemistry of the double bond is of little consequence, as good results are obtained with both E- and Z-olefins. Of particular interest is the role of LiCl, the chloride source for the reaction. Our screening demonstrated TFE as the optimal choice for solvent, although its incorporation as the nucleophile in the reaction was initially a problem. Use of excess LiCl drastically reduces the TFE incorporated side product. Our preliminary work suggests a role not only for the solid salt in solution but also for the presence of Li salt, for the success of this transformation. Mechanistic investigations are underway to elaborate on the nature of interactions, presumably at the solid/liquid interface, that lead to the observed effects.
Supplementary Material
Acknowledgments
Funding
Generous support was provided by the NIH (GM110525).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACSPublications website at DOI: 10.1021/jacs.6b09203.
Experimental details (PDF), and data for C13H16Cl2N2O3 (5a) (CIF) and C13H16BrClN2O3 (9a) (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Denmark SE; Kuester WE; Burk MT Angew. Chem., Int. Ed 2012, 51, 10938. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whitehead DC; Yousefi R; Jaganathan A; Borhan BJ Am. Chem. Soc 2010, 132, 3298. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yousefi R; Ashtekar KD; Whitehead DC; Jackson JE; Borhan BJ Am. Chem. Soc 2013, 135, 14524. [DOI] [PubMed] [Google Scholar]; (d) Jaganathan A; Garzan A; Whitehead DC; Staples RJ; Borhan B Angew. Chem., Int. Ed 2011, 50, 2593. [DOI] [PubMed] [Google Scholar]; (e) Jaganathan A; Borhan B Org. Lett 2014, 16, 3616. [DOI] [PubMed] [Google Scholar]; (f) Cheng YA; Yu WZ; Yeung YY Org. Biomol. Chem 2014, 12, 2333. [DOI] [PubMed] [Google Scholar]; (g) Chen J; Zhou L Synthesis 2014, 46, 586. [Google Scholar]; (h) Hennecke U Chem. - Asian J 2012, 7, 456. [DOI] [PubMed] [Google Scholar]; (i) Castellanos A; Fletcher SP Chem. - Eur. J 2011, 17, 5766. [DOI] [PubMed] [Google Scholar]; (j) Tan CK; Zhou L; Yeung YY Synlett 2011, 2011, 1335. [Google Scholar]; (k) Hennecke U; Wilking M Synlett 2014, 25, 1633. [Google Scholar]; (l) Chemler SR; Bovino MT ACS Catal. 2013, 3, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Murai K; Fujioka H Heterocycles 2013, 87, 763. [Google Scholar]; (n) Tan CK; Yeung YY Chem. Commun 2013, 49, 7985. [DOI] [PubMed] [Google Scholar]; (o) Pan H; Huang H; Liu W; Tian H; Shi Y Org. Lett 2016, 18, 896. [DOI] [PubMed] [Google Scholar]; (p) Nakatsuji H; Sawamura Y; Sakakura A; Ishihara K Angew. Chem., Int. Ed 2014, 53, 6974. [DOI] [PubMed] [Google Scholar]; (q) Cheng YA; Yu WZ; Yeung YY Angew. Chem., Int. Ed 2015, 54, 12102. [DOI] [PubMed] [Google Scholar]; (r) Cai Y; Zhou P; Liu X; Zhao J; Lin L; Feng X Chem. - Eur. J 2015, 21, 6386. [DOI] [PubMed] [Google Scholar]; (s) Chen G; Ma S Angew. Chem., Int. Ed 2010, 49, 8306. [DOI] [PubMed] [Google Scholar]; (t) Zheng SQ; Schienebeck CM; Zhang W; Wang HY; Tang WP Asian J. Org. Chem 2014, 3, 366. [Google Scholar]; (u) Dobish MC; Johnston JN J. Am. Chem. Soc 2012, 134, 6068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Fang C; Paull DH; Hethcox JC; Shugrue CR; Martin SF Org. Lett 2012, 14, 6290. [DOI] [PMC free article] [PubMed] [Google Scholar]; (w) Veitch GE; Jacobsen EN Angew. Chem., Int. Ed 2010, 49, 7332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (x) Wang Y-M; Wu J; Hoong C; Rauniyar V; Toste FD J. Am. Chem. Soc 2012, 134, 12928. [DOI] [PubMed] [Google Scholar]; (y) Arai T; Watanabe O; Yabe S; Yamanaka M Angew. Chem., Int. Ed 2015, 54, 12767. [DOI] [PubMed] [Google Scholar]; (z) Mizar P; Burrelli A; Gunther E; Softje M; Farooq U; Wirth T Chem. - Eur. J 2014, 20, 13113. [DOI] [PubMed] [Google Scholar]; (aa) Xie W; Jiang G; Liu H; Hu J; Pan X; Zhang H; Wan X; Lai Y; Ma D Angew. Chem., Int. Ed 2013, 52, 12924. [DOI] [PubMed] [Google Scholar]; (ab) Ke Z; Tan CK; Chen F; Yeung YY J. Am. Chem. Soc 2014, 136, 5627. [DOI] [PubMed] [Google Scholar]; (ac) Wilking M; Muck-Lichtenfeld C; Daniliuc CG; Hennecke UJ Am. Chem. Soc 2013, 135, 8133. [DOI] [PubMed] [Google Scholar]; (ad) Zhang W; Liu N; Schienebeck CM; Zhou X; Izhar II; Guzei IA; Tang WP Chem. Sci 2013, 4, 2652. [Google Scholar]; (ae) Yin Q; You SL Org. Lett 2014, 16, 2426. [DOI] [PubMed] [Google Scholar]; (af) Toda Y; Pink M; Johnston JN J. Am. Chem. Soc 2014, 136, 14734. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ag) Tripathi CB; Mukherjee S Org. Lett 2015, 17, 4424. [DOI] [PubMed] [Google Scholar]; (ah) Cai YF; Liu XH; Hui YH; Jiang J; Wang WT; Chen WL; Lin LL; Feng XM Angew. Chem., Int. Ed 2010, 49, 6160. [DOI] [PubMed] [Google Scholar]
- (2).Cresswell AJ; Eey ST; Denmark SE Angew. Chem., Int. Ed 2015, 54, 15642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Snyder SA; Tang ZY; Gupta RJ Am. Chem. Soc 2009, 131, 5744. [DOI] [PubMed] [Google Scholar]
- (4).Snyder SA; Treitler DS; Brucks AP J. Am. Chem. Soc 2010, 132, 14303. [DOI] [PubMed] [Google Scholar]
- (5).Nicolaou KC; Simmons NL; Ying YC; Heretsch PM; Chen JS J. Am. Chem. Soc 2011, 133, 8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cresswell AJ; Eey ST; Denmark SE Nat. Chem 2015, 7, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hu DX; Shibuya GM; Burns NZ J. Am. Chem. Soc 2013, 135, 12960. [DOI] [PubMed] [Google Scholar]
- (8) (a). Landry ML; Hu DX; McKenna GM; Burns NZ J. Am. Chem. Soc 2016, 138, 5150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu DX; Seidl FJ; Bucher C; Burns NZ J. Am. Chem. Soc 2015, 137, 3795. [DOI] [PubMed] [Google Scholar]; (c) Bucher C; Deans RM; Burns NZ J. Am. Chem. Soc 2015, 137, 12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Denmark SE; Burk MT; Hoover AJ J. Am. Chem. Soc 2010, 132, 1232. [DOI] [PubMed] [Google Scholar]
- (10).Soltanzadeh B; Jaganathan A; Staples RJ; Borhan B Angew. Chem., Int. Ed 2015, 54, 9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Relative stereochemistry for 6a, 7a and 8a is shown; however, absolute stereochemistry is unknown.
- (12).Aryl substituted olefins can lead to production of the six-member ring dihydro-4H-1,3-oxazine side product, which is the result of intramolecular attack at the benzylic position as reported in ref 1d.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.