

Significance Statement
The innate immune system is central to injury and repair in the kidney, but the heterogeneity of myeloid cell subsets behind these processes is unknown. Complementary technologies—including bulk tissue transcriptomics, integrated droplet– and plate-based single-cell RNA sequencing, and paired blood exchange—resolved myeloid cell heterogeneity in a murine model of reversible unilateral ureteric obstruction, creating a single-cell atlas. The identified novel myeloid subsets could be targeted to ameliorate injury or enhance repair, including an Arg1+ monocyte subset present during injury and Mmp12+ macrophages present during repair. Standard flow cytometry to detect cell surface markers would have missed these subsets. Complementary techniques capture the complexity and dynamics of monocyte, dendritic cell, and macrophage phenotypes in the injured and repairing kidney.
Keywords: fibrosis, kidney disease, scRNA sequencing, myeloid cells
Visual Abstract

Abstract
Background
Little is known about the roles of myeloid cell subsets in kidney injury and in the limited ability of the organ to repair itself. Characterizing these cells based only on surface markers using flow cytometry might not provide a full phenotypic picture. Defining these cells at the single-cell, transcriptomic level could reveal myeloid heterogeneity in the progression and regression of kidney disease.
Methods
Integrated droplet– and plate-based single-cell RNA sequencing were used in the murine, reversible, unilateral ureteric obstruction model to dissect the transcriptomic landscape at the single-cell level during renal injury and the resolution of fibrosis. Paired blood exchange tracked the fate of monocytes recruited to the injured kidney.
Results
A single-cell atlas of the kidney generated using transcriptomics revealed marked changes in the proportion and gene expression of renal cell types during injury and repair. Conventional flow cytometry markers would not have identified the 12 myeloid cell subsets. Monocytes recruited to the kidney early after injury rapidly adopt a proinflammatory, profibrotic phenotype that expresses Arg1, before transitioning to become Ccr2+ macrophages that accumulate in late injury. Conversely, a novel Mmp12+ macrophage subset acts during repair.
Conclusions
Complementary technologies identified novel myeloid subtypes, based on transcriptomics in single cells, that represent therapeutic targets to inhibit progression or promote regression of kidney disease.
CKD affects approximately 10% of the global population1 and is a major risk factor for ESKD and cardiovascular disease.2–4 It is now recognized that CKD is not always progressive, but that regression of albuminuria and improvement in renal function can occur if the injurious stimulus is removed.5–7 Furthermore, regression of established fibrosis, the best histologic predictor of outcome,8 has been observed after prolonged normalization of blood glucose levels after successful pancreas transplantation.9,10 However, the cellular and molecular pathways mediating injury regression are poorly understood, partly because renal biopsies are rarely performed in patients who are clinically improving.
The innate immune system has been implicated in both progression and regression of fibrosis in multiple organs, including the kidney.11–15 Recruitment of proinflammatory monocytes16,17 to the injured kidney via CCL1-CCR2 signaling18,19 may exacerbate tissue damage through the release of proinflammatory factors and by activating myofibroblasts. Tissue macrophages are heterogeneous and inherently plastic, and may adopt different phenotypes in response to environmental cues. Hence, they may be injurious, but, in addition, they may mediate repair by scavenging cell debris, degrading excess extracellular matrix (ECM), and by secreting factors that may promote regeneration of injured tissue.20–23
Most studies have used panels of cell surface markers to characterize myeloid cell subsets by flow cytometry; however, this approach is inherently biased and is unlikely to capture the full phenotypic spectrum. Recent advances in transcriptomics, including single-cell RNA sequencing (scRNA-seq), have facilitated detailed analysis of myeloid cells in the healthy kidney, and after AKI,24–26 and in other organs.27–31 However, macrophage heterogeneity during regression of fibrosis in the kidney remains uncertain. Hence, in this study, we employ scRNA-seq to characterize myeloid cell subsets in the reversible, unilateral-ureteric-obstruction (R-UUO) model, in which we32 and others33 have demonstrated regression of established tubulointerstitial fibrosis after reversal of obstruction. We identified myeloid cell subsets that were indistinguishable using standard flow cytometry markers, with the relative proportions of the subsets changing dynamically during injury and repair. Pseudotime analysis and paired blood exchange (PBE) support dynamic changes in monocyte and macrophage phenotype in response to induction and removal of injury. Similar myeloid cell phenotypes are observed in the human kidney, suggesting they may represent specific targets to slow progression of CKD or promote renal repair.
Methods
Animal Models
All protocols and surgical procedures were approved by the Animal Ethics Committee, University of Edinburgh. Animal experiments were conducted in accordance with the United Kingdom Animals Scientific Procedures Act 1986, under Home Office project licenses 70/8093 and 70/8867.
R-UUO Model
The R-UUO model was performed as previously described.32 Briefly, 8-week-old male C57BL/6JOlaHsd mice (Enviago) underwent laparotomy, and the left ureter was isolated and the distal portion was ligated twice with 6/0 black-braided silk sutures close to the bladder. In mice that required reversible ureteric obstruction, a silastic tube was placed around the ureter immediately proximal to the ligature to prevent excessive dilation. After 7 days of obstruction, the ureter was reanastomosed into the bladder, and the peritoneum and skin were sutured closed. Mice were euthanized by carbon dioxide narcosis and dislocation of the neck at day 2 (UUO-2) or day 7 (UUO-7) after UUO, or 7 (R-UUO 1 week), 14 (R-UUO 2 weeks), or 28 days (R-UUO 4 weeks) after ureteric reanastomosis after 7 days of obstruction. SMART-seq2 studies used MacGreen mice,34 in which the Csf1r promoter drives enhanced green fluorescent protein (EGFP) as a reporter specific to myeloid cells.
PBE
Male C57BL/6NCrl mice, which are homozygous for CD45.2, were paired with male Ly5.1 mice (Charles River), which are CD45.1/CD45.2 heterozygotes. Four pairs of mice were used. The PBE was performed as previously described.35 Briefly, all animals had a right jugular venous catheter inserted before the UUO surgery. At 1 day post-UUO, 15×150-µl aliquots of whole blood were exchanged between each animal in the pair over a 20-minute period. Two pairs were euthanized at both 2 and 7 days post-UUO, and whole blood and kidney tissue (UUO and contralateral kidney) was harvested.
Immunohistochemistry
Kidney tissue was fixed and formalin-fixed, paraffin-embedded, 4-μm tissue sections were prepared. Sections were rehydrated and staining was performed using the Sequenza system (Thermo Scientific, Waltham, MA). Sections were incubated with the avidin/biotin blocking kit (SP2001; Vector Laboratories) and blocked with serum-free protein block (X0909; Dako). Tissue sections were incubated with primary antibody (Supplemental Table 1) diluted in antibody diluent (S202230; Dako UK Ltd.), overnight at 4°C, before incubation with biotinylated secondary antibody (Supplemental Table 1) for 30 minutes at room temperature. Vectastain RTU ABC Reagent (PK7100; Vector Laboratories) was then applied, followed by incubation with the DAB+ Substrate Chromogen System (K3468; Dako), and then counterstaining with hematoxylin before dehydration and mounting with Pertex mounting medium (3808707E; Histolab Products AB). The stained section was scanned with a Zeiss Axio Scan.Z1 Slide Scanner (Carl Zeiss Microscopy). The percentage of DAB staining per section (n=6–8 per group) was determined by ImageJ.
Immunofluorescence
Slides were de-waxed in xylene (2×5 minutes), rehydrated, and antigen retrieved (7 minutes at 60% microwave power). Slides were allowed to cool at room temperature, mounted in Sequenza (Thermo Fisher) racks, rinsed twice in PBS, and blocked for 45 minutes in Gentex block (120 µl) at room temperature. Primary antibodies were incubated at the concentrations in Supplemental Table 1 in antibody diluent (Abcam) and incubated overnight at 4°C. Slides were then washed twice with PBS, and secondary antibodies were added at a concentration of 1:200, diluted in antibody diluent, and incubated at room temperature for 30 minutes. Slides were again washed twice with PBS and then blocked with Gentex block for 45 minutes at room temperature. The second primary antibody was added to the slides, and incubated at 4°C overnight. Slides were washed and secondary antibodies applied. For dual immunofluorescence, the slides were boiled in 10 mM citrate and blocked with serum-free protein block for 1 hour before incubation of the second primary antibody. The antibodies were visualized by incubation with tyramide red or green (Perkin Elmer) for 10 minutes. After washing, slides were mounted with 4′,6-diamidino-2-phenylindole (DAPI) Fluoromount-G (Southern Biotech) and a coverslip was applied before visualization.
Bone Marrow–Derived Macrophage Culture
The hind legs of C57BL/6JOlaHsd mice were removed before the skin and underlying muscle were excised with sterile scissors and forceps to isolate the femur. The bone marrow was then flushed out in DMEM (Gibco) containing 10% L929 conditioned medium, 10% FCS, and 1% penicillin-streptomycin. Cell suspensions were cultured for 1 week in 60-ml, sterile Teflon pots at 37°C with 5% carbon dioxide. Macrophages were then plated into six-well plates, incubated with 10 µg FITC-conjugated collagen (D12052), and left overnight. Cells were collected and run through flow cytometry to quantify the FITC signal.
RNA Extraction, Gene Expression, and Bulk RNA-seq
Total RNA from cortical kidney tissue was isolated using the RNeasy kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. For quantitative PCR analysis of targeted gene expression, cDNA was synthesized from 1 µg of template RNA, using the QuantiTect Reverse Transcription Kit (Qiagen, Venlo, Netherlands). Quantitative PCR was performed using the PerfeCTa FastMix II Probe Master (VWR, Lutterworth, United Kingdom) and TaqMan Gene Expression Assay–specific primers (Life Technologies; Supplemental Table 2) and normalized to hypoxanthine-guanine phosphoribosyltransferase.
Before RNA-seq, RNA integrity was checked using Agilent Nano Chips, and only samples with an RNA integrity number greater than seven were used in subsequent analysis. Four mice per group underwent RNA-seq, with the animals selected on the basis that their Havcr1 gene expression, as determined by quantitative RT-PCR, was closest to the mean of that group. A poly(A) library was constructed and run on a HiSeq2500, using 2×100-bp, paired-end (PE) sequencing. FastQC was used for initial quality control (QC), reads were mapped to the mm10 transcriptome using RSEM and Bowtie2, and DESeq2 was used for differential gene expression analysis. Data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE145053).
The shinyNGS R package was used to generate gene clusters in Figure 1C. Genes with an average fragments per kilobase of transcript per million mapped reads of less than one across all groups were excluded from analysis. Genes that were not significantly differentially expressed (DE; adjusted P<0.05, determined using DESeq2 R package) in any group (compared with sham) were excluded from analysis. Using the feature-based clustering module of the shinyNGS package, 7810 genes were assigned to one of six clusters, based on expression change between each of the groups.
Figure 1.
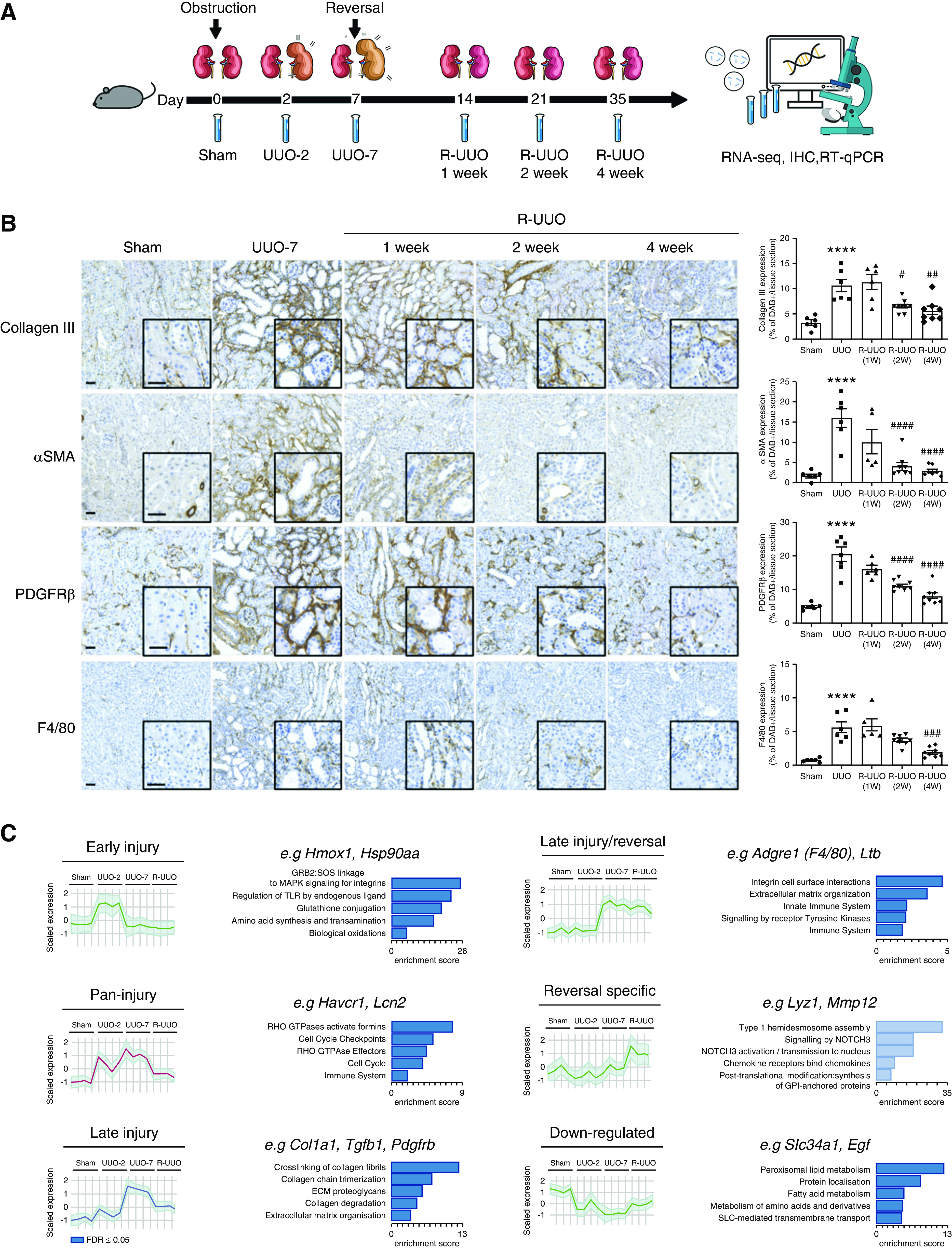
Phases of progression and regression of fibrosis in the R-UUO model are associated with dynamic changes in the renal transcriptome. (A) Male 6- to 8-week-old C57BL/6J mice underwent either UUO or sham surgery, and were either euthanized 2 days later, or left obstructed for 7 days and then euthanized, or had their ureter reimplanted to reverse obstruction before euthanasia at 1, 2, or 4 weeks post UUO (n=6–8 per group). (B) Representative images and quantification of fibrosis (collagen III) and fibroblast (PDGFR-β), myofibroblast (α-smooth muscle actin [α-SMA]), or macrophage (F4/80) accumulation during the R-UUO model. Scale bar, 50 µM. ****P<0.0001 versus sham, #P<0.05 versus UUO, ##P<0.01 versus UUO, ###P<0.001, ####P<0.0001 versus UUO. (C) Unbiased clustering analysis of bulk RNA sequencing data from the renal cortex of mice (n=4 per group) during the R-UUO time course identified six discrete temporal patterns of gene expression. Representative genes and enriched pathways are provided for each cluster. The number of genes included in each cluster is as follows: Down-regulated 1562; Early injury, 779; Pan-injury, 1479; Late injury, 1619; Late injury/reversal, 1708; and Reversal specific, 663. Shaded error range is the SD of the mean scaled gene expression for each animal. Dark and light blue pathways are those demonstrating gene enrichment at a false discovery rate of <0.05 and >0.05, respectively. FDR, false discovery rate; IHC, immunohistochemistry; RT-qPCR, quantitative RT-PCR.
Kidney Digestion for Flow Cytometry and Single-Cell Sequencing
Immediately after euthanasia, mice were perfused with 10 ml PBS. Kidneys were excised, decapsulated, and placed in ice-cold PBS. Equal portions of renal cortex from each mouse were finely minced in digest buffer (4.25 mg/ml Collagenase V [Sigma-Aldrich, St. Louis, MO], 6.25 mg/ml Collagenase D [Roche, Basel, Switzerland], 10 mg/ml Dispase [Thermo Scientific], and 300 µg/ml DNase [Roche] in RPMI 1640 [10% FCS, 1% penicillin/streptomycin/l-glutamine]) before homogenization in gentleMACS C Tubes, using the gentleMACS Dissociator (Miltenyi Biotec, Auburn, CA). Samples were incubated at 37°C with shaking to maximize digestion. The kidney suspension was then subjected to a second gentleMACS homogenization and digestion, neutralized with an equal volume of FACS buffer (PBS, 2mM EDTA, and 2% FCS). Kidney cell suspensions were then passed sequentially through 100-, 70-, and 40-µm sieves. Any residual red blood cells were lysed by Red Blood Cell Lysis Buffer (Sigma-Aldrich). Cells were resuspended in ice-cold FACS buffer, ready for use.
Flow Cytometry
Single-cell suspensions were incubated with Fc Block (BD Biosciences, San Jose, CA) and then incubated with preconjugated antibodies (Supplemental Table 3) in round-bottomed plates. Controls were set up, including unstained cells, beads with single stains of each antibody, and fluorophore minus one controls. DAPI was used to distinguish live and dead cells. For cellular composition analysis, the following antibodies were used: CD45, CD31, LTL, PDGFRβ, and F4/80. For SMART-seq2 experiments, cells were incubated with the antibodies CD45, CD11b, CD11c, F4/80, MHCII, CD206, CD64, and CD24; with a lineage dump gate including TCRβ (T cells), CD19 (B cells), Siglec-F (eosinophils), and Ly6G (neutrophils); all conjugated to BV421 and run on the BD FACS ARIA II. For PBE, blood and tissue were analyzed on the BD 6L LSRFortessa using the following antibody panels: for blood, CD45.1, CD45.2, CD3, F4/80, GR-1, CD11b, and CD19; for kidney tissue, CD45.1, CD45.2, F4/80, Ly6C, MHCII, CD11b, CD206, CD24, and CD64; with a dump gate including TCRβ, CD19, Siglec-F, and Ly6G. All files were exported in FCS format and analyzed with FlowJo software version 10.
Single-Cell Droplet Library Preparation
For scRNA-seq analysis on the 10× Genomics platform, single-cell suspensions from renal cortex were prepared from pools of three animals from each group, as outlined by the 10× Genomics Single Cell 3′ Reagent Kit User Guide version 2. A total of 50,000 live (DAPI−) cells were sorted on the BD FACS ARIA II. Samples were washed twice in PBS (Sigma), followed by centrifugation at 500 × g for 5 minutes at 4°C. Sample viability was assessed using trypan blue (Sigma) with an automated cell counter (Bio-Rad), and the appropriate volume for each sample was calculated. The chip was loaded with 10,700 cells per lane.
After droplet generation, samples were transferred onto a prechilled 96-well plate, heat sealed, and reverse transcription was performed using a C1000 Touch Thermal Cycler (Bio-Rad). After reverse transcription, cDNA was recovered using the 10× Genomics Recovery Agent, and a Silane DynaBead (Thermo Fisher) cleanup was performed. Purified cDNA was amplified and cleaned using SPRIselect beads (Beckman). Samples were diluted at 4:1 (elution buffer [Qiagen]/cDNA) and an initial concentration check was performed on a Qubit fluorometer (Invitrogen) to ensure adequate cDNA concentration. The final cDNA concentration was checked on a bioanalyzer (Invitrogen).
SMART-seq2 Library Preparation
For the SMART-seq2 experiment, we performed flow cytometry for n=3 animals per group, and sequenced one animal per group. The flow cytometry patterns within each group were broadly similar, mitigating against the selected animal being unrepresentative of the group. A single, live (DAPI+), EGFP+ (Csf1r+) cell was sorted into each well of a 96-well plate, and all fluorochrome information was recorded using the index-sort capability of the BD FACS ARIA II. Equal numbers of cells from each time point were sorted into each plate to reduce batch effect, with 192 cells per time point included in total. Single cells were processed as previously described.36 Briefly, cells were lysed immediately in lysis buffer containing 5% RNase inhibitor and 0.025% Triton X-100. Oligo(dT) primers were added and reverse transcription was performed. SMART-seq2 libraries were prepared according to the previously described protocol,36 with a few modifications37: at step 5, 0.1 µl of the External RNA Controls Consortium (ERCC) spike-in mix (10:5 diluted, 4456740; Life Technologies) was added with 0.1 µl of 100 µM oligo(dT) primer and 1 µl of dNTP mix and 0.8 µl of water, yielding the same concentrations of primer and oligo as originally reported. Fluidigm protocol (PN 100-7168 M1) was used for tagmentation library generation. The final cDNA concentration was checked on a bioanalyzer (Agilent).
Sequencing
The 10× libraries were pooled and normalized by molarity before being sequenced across four lanes on a single Illumina flow cell. Sequencing was performed on an Illumina HiSeq platform, with a target of approximately 350 million PE reads per lane, giving approximately 525 million PE reads per sample, comprising 2×150-bp PE configuration and 8-bp index reads. The SMART-seq2 libraries were sequenced as 8× 96-well plates, which were pooled and sequenced on an Illumina HiSeq 4000 (50-bp, single-end reads). Data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE140023).
scRNA-seq Analysis
For the droplet-based dataset, the cellranger mkfastq wrapper (Cell Ranger Single Cell Software suite 2.1.0, http://10xgenomics.com) de-multiplexed the Illumina output BCL files to library-specific FASTQ files. Subsequently, alignment was performed using the cellranger count function, using STAR aligner 2.5.1b38 against the Ensembl mouse reference genome version GRCm38.68. Correction and filtering of cell bar code and unique molecular identifiers followed, and the retained bar codes were quantified and used to generate a gene expression matrix. Summary sequencing statistics are provided in Supplemental Table 4. The SMART-seq2 raw reads were similarly mapped against the Ensembl mouse reference genome version GRCm38.68, using the STAR RNA-seq aligner,38 with the additional inclusion of the sequences for the ERCC spike-ins.
For our droplet-based dataset, a standard sequence of filtering, highly variable gene selection, dimensionality reduction, and clustering were performed using the scRNA-seq analysis R package Seurat (version 2.3.4).39 After alignment and initial preprocessing, we began our R workflow with 15,046 genes across 7073 cells in our sham group, 16,450 genes across 5088 cells in our UUO-2 group, 17,368 genes across 7124 cells in our UUO-7 group, and 17,227 genes across 6096 cells in our R-UUO group. To exclude low-quality cells in both single-cell experiments, we then filtered cells that expressed <300 genes and less than 500 unique molecular identifiers, and to exclude probable doublets, cells with >10,000 unique molecular identifiers and <3000 genes were removed. This would have removed the majority of injured and apoptotic cells.40 We used a mitochondrial filter to remove cells in which >50% of genes were mitochondrial, consistent with other renal-specific scRNA-seq projects.41,42 This is a higher filter than has been used in nonrenal single-cell analysis, but reflects the high mitochondrial content in renal tubular epithelial cells. Any gene not expressed in at least three cells was removed.
For the SMART-seq2 data, identical metrics were used as above, but the mitochondrial filter threshold was lowered to 25%, and cells with >25% of reads mapping to the ERCCs were additionally excluded. After filtering and QC, 15,046 genes across 4540 samples of sham mice, 16,450 genes across 3101 samples of the UUO-2 mice, 17,368 genes across 5563 samples of UUO-7 mice, 17,227 genes across 4308 samples of the R-UUO mice, and 13,517 genes across 362 samples in the SMART-seq2 data were taken forward for analysis, resulting in 92 cells in the sham group, 102 cells in UUO-2, 103 cells in UUO-7, and 65 cells from the R-UUO group.
Normalization was performed using the Seurat package to reduce biases introduced by technical variation, sequencing depth, and capture efficiency. We used the default global-scaling normalization method “logNormalize,” which normalized gene expression per cell by the total expression and multiplied the result by a scaling factor before log transformation. We then scaled the data and regressed out variation between cells due to the number of unique molecular identifiers and the percentage of mitochondrial genes.
The expression matrix subsequently underwent dimensionality reduction, using principal component analysis of the highly variable genes within the dataset. Using Seurat’s FindVariableGenes function (and computed using the LogVMR argument) we used log-mean expression values between 0.0125 and three, and a dispersion cutoff of 0.5 to select genes. Principal component analysis was performed using these selected genes, and 20 principal components were identified for subsequent analysis in each dataset, selected both visually using the elbow point on the elbow plot and via the jackstraw method.
Further cluster-based QC was performed in the droplet data using the density-based spatial clustering algorithm DBscan, which was used to identify cells on a t-distributed stochastic neighbor embedding (tSNE) map. We initially set an eps value of 0.5 and removed clusters with fewer than ten cells. The remaining cells were then clustered again with an eps value of one, followed by removing the clusters with <20 cells. Of note, this allowed identification of a cluster characterized by high expression of heat-shock genes, including Fos, Jun, and Atf3. This cluster was removed because it was considered to be an artifact of cell stress due to the experimental protocol, as recently described.43 This procedure removed 158 (3.4%) cells from a total of 4540 cells in sham mice, 137 (4.4%) of 3101 cells in UUO-2 mice, 167 (3%) of 5563 cells in UUO-7 mice, and 83 (1.9%) of 4308 cells in R-UUO mice.
Clusters were then identified using Seurat’s FindClusters function, built using the first ten principal components and a resolution parameter of 1.5. The original Louvain modularity optimization algorithm was used. tSNE (using the Rtsne package Barnes–Hut implementation) was then used for further dimensionality reduction and visualization, which was run on a reduced dimensional space of the first five to ten dimensions, using perplexity values of 15–50.
For all single-cell differential expression tests, we used the Wilcoxon rank-sum test to identify a unique expression profile for each cluster, with differential expression tested between each cluster and all other clusters combined. The FindAllMarkers test, as implemented in Seurat, returns an “adj_pval” (Bonferroni-adjusted P values) and an “avg_logFC” (average log fold change) for each gene. Genes were ranked in order of average log fold change and visualized using heatmaps.
The process of acquiring the myeloid subsets required a combination of cluster-based cell pruning and a gene-based cell filter. The clusters were annotated by cell type, using the generated DE marker genes, and recognized markers from known biology and from the single-cell literature to date. All myeloid clusters were then isolated, renormalized, and rescaled as described above, and were processed again through the pipeline described above, although without the DBscan QC step. Clustering resolution was lowered for each downstream implementation of the FindClusters function, and again clusters were identified based on DE genes. Any nonmyeloid cell clusters were removed, and the data were, once again, reprocessed and reclustered. This entire process was repeated three times in total, with successively higher resolutions used to generate greater numbers of clusters, to coerce any nonmyeloid cells into forming distinct clusters that could be removed as the data became cleaner. The expression of 52 key nonmyeloid genes was then assessed, and any cell expressing such genes was removed. Finally, tSNE graphs were visually inspected, and any unusual clusters were manually selected and DE genes inspected—any stray unwanted cluster was manually pruned and the data reprocessed as before.
Differential Proportion Analysis
Differential proportion analysis was developed to detect changes in population proportions across groups within single-cell experiments. The algorithm is described in detail, along with source code, by Farbehi et al.44 Briefly, this approach uses a novel permutation-based statistical test to analyze whether observed changes in proportions of cell populations were greater than expected by chance. This approach attempts to consider sources of technical variation within the experimental technique, such as differing absolute cell numbers within the experiment, cell-type capture bias (a known feature of current single-cell workflows45), and variation due to in silico analysis (cluster assignment accuracy, for example). A proportion table of clusters per phenotype/group is created from the count table, and the difference in cluster proportion is compared with a null distribution. This distribution is constructed by random permutations of random subsamples of cluster labels across a random proportion of total cells. A new proportion table is generated from these data, and the process is repeated multiple times, with the resulting difference in cluster proportions across the data forming the null distribution. The observed distribution is then compared with this null distribution, and final P values are calculated based on the minimum P values of any observed increases or decreases in proportion. As per the original paper,44 we used a w parameter of 0.1, where lower values will trend toward a stricter test (fewer significant hits), and higher values trend toward higher numbers of significant hits.
Assignment to Myeloid Cell Phenotype on the Immunological Genome Project Consortium
Cluster Identity Predictor (version 246) was used to generate Spearman correlation values for each cluster within our own data, as compared with cluster gene signatures with Immunological Genome Project (ImmGen) mouse immune cell datasets based on the entire gene expression dataset. The algorithm first subsets genes common to both datasets before using a one-to-many (cluster-to-references) calculation of correlation coefficients. A single correlation coefficient is calculated for each reference cell type and for each cluster, allowing each cluster in our experiment to be analyzed against each known cell type in the reference file and scored for its overall similarity. To further validate these assignments, we used SingleR27 to assign myeloid cell classification, using the murine ImmGen dataset, and create a consensus matrix with our classifications. Briefly, this pipeline is based on correlating reference bulk-transcriptomic datasets of pure cell types with single-cell gene expression. Similarly to the Cluster Identity Predictor, a Spearman coefficient was calculated for single-cell gene expression, with each of the samples in the reference dataset based only on the variable genes in the reference dataset. This is performed iteratively until a classification is reached. One myeloid cluster mapped to a mixture of cells and expressed cell cycle genes, such as Mki67 and Top2a, consistent with proliferation and was therefore assigned to the proliferation cluster.
Assignment to Myeloid Phenotype in Recovery from Ischemia-Reperfusion Injury
To compare the transcriptome from our myeloid cell clusters with that of myeloid cells in the kidney during recovery from renal ischemia-reperfusion injury (IRI), we used the datasets generated by Lever et al.47 and deposited in the Gene Expression Omnibus (GSE121410). We used SingleR software, as previously described, to align our macrophage clusters to embryonic and adult resident renal macrophages from healthy kidneys, and macrophages and infiltrating monocytes in kidneys from mice 6 days after IRI.
Ligand-Pair Interactions
Heatmaps and dotplots of number of ligand-pair interactions were generated using the CellphoneDB tool (https://www.cellphonedb.org/) developed by the Teichmann Lab (Wellcome Sanger Institute, Cambridge, United Kingdom).48 The lower cutoff for expression proportion of any ligand or receptor in a given cell type was set to 10%, and the number of permutations was set to 1000. The clusters were not subsampled.
Platform Integration
Because our SMART-seq2 library included an index sort, the transformed FACS data corresponding to each cell was then exported as .fcs files and analyzed using FlowJo software. The index-sort data were then extracted using the index FlowJo plugin available from FlowJo exchange. These data were then matched to the cell bar code and imported as both a separate “protein” assay and as metadata into the Seurat object to allow for visualization.
Our droplet-based and SMART-seq2 datasets were then integrated using the “anchoring” approach introduced in Seurat version 3.49 Here, we created an integrated reference dataset and transferred the cell type labels onto our SMART-seq2 data. Briefly, this approach requires identification of “anchors” between the datasets, which represent shared biologic states. This involves jointly reducing the dimensionality of both datasets using diagonalized canonic correlational analysis before searching for mutual nearest neighbors in the new shared space. The paired cells are treated as anchors that represented shared biology across the datasets. Anchors were identified using the default parameters of the FindIntegrationAnchors function, with the argument dims=1:20.
To map the monocyte, dendritic cell (DC), and macrophage clusters onto conventional flow cytometry myeloid cell gates, we used UUO-2, UUO-7, and R-UUO, respectively, because not all clusters were represented at all time points in the SMART-seq2 dataset.
Pseudotime Analysis
Lineage reconstruction and pseudotime inference was performed using the slingshot package.50 This method works by learning cluster relationships in an unsupervised manner and constructing smooth curves representing the lineage across two visualized pseudotime dimensions. Briefly, this involved first creating the raw expression matrix of the subsetted cells that were classified as “Arg1+ monocyte,” “Ly6c2+ monocyte,” and “Ccr2+ macrophage” by Seurat. This was followed by filtering genes not expressed in any cluster with less than ten cells, and having at least three reads within that cluster. Full quantile normalization was then performed before dimensionality reduction using diffusion maps via the destiny package.51 Next, cells were clustered to allow slingshot to infer a global structure of the lineage, using a Gaussian mixture modeling implemented in Mclust,52 revealing eight underling clusters in the data. Slingshot then constructed the cluster-based minimum spanning tree and fitted the principal curve.
Pathway Analysis
Gene set enrichment analysis (GSEA) and over-representation analysis (ORA) were used to identify enriched pathways based on the DE genes, using WebGestalt (http://www.webgestalt.org/). For GSEA, we generated a rank for each gene in the list of DE genes using the formula rank=(average log fold change)×(−log [adjusted P value]). To perform ORA of bulk RNA-seq data, significantly DE genes were selected for the algorithm based on a minimum of two-fold upregulation (or 50% of baseline for the downregulated genes) against the appropriate comparator (e.g., UUO-2 versus sham when considering early injury). Enrichment categories were discarded if they contained less than five or >2000 genes. These thresholds were calculated by WebGestalt based on the number of overlapping genes between the annotated genes in the category and the reference gene list for the ORA method. For the GSEA method, categories were discarded if they contained <15 genes or >500 genes. The Benjamini–Hochberg method was used to correct for multiple testing during ORA, and the top ten enriched categories—as ranked by false discovery rate—were selected. The reference gene list used was the Illumina MouseRef-8. We used pathway gene sets from the protein analysis through evolutionary relationships, PANTHER (http://www.pantherdb.org), Reactome, and Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/) as our reference gene lists.
Statistical Analyses
Animal group size was determined from previous pilot experiments. Comparisons between two unpaired, non-normally distributed data points were carried out via Mann–Whitney test. Comparisons between two unpaired, normally distributed data points were carried out via t test. Comparisons between multiple groups were performed with one-way ANOVA with the Tukey multiple comparison test. All statistical analysis was performed using GraphPad Prism version 10.
Results
Degradation of Excess ECM after Reversal of Ureteric Obstruction Is Associated with Persistence of Immune Cells
To determine the pathways that mediate renal injury and repair, we used the murine R-UUO model (Figure 1A). Within 7 days of ureteric obstruction (UUO-7), there was expansion of interstitial PDGF-β+ cells (Figure 1B), activation to α-smooth muscle actin+ myofibroblasts (Figure 1B, Supplemental Figure 1A), and collagen deposition (Figure 1B), with induction of tubular injury markers such as Havcr1 (encodes kidney injury molecule-1; Figure 1C). After reimplantation of the ureter, there was a decline in Havcr1 expression, a significant reduction in interstitial PDGFR-β+ cells with loss of myofibroblastic phenotype (α-smooth muscle actin−) (Supplemental Figure 1A), and a gradual regression of collagen deposition over 4 weeks (Figure 1B), as has been observed previously.33 Macrophages (F4/80+) accumulated in the kidney during UUO, and persisted through the early stages of R-UUO, before trending toward baseline levels by 4 weeks after reversal (Figure 1B), consistent with previous findings in this model.33
We first performed bulk RNA-seq of the renal cortex in the R-UUO model, which revealed six discrete temporal patterns of gene expression (Figure 1C, Supplemental Figure 1, B–D, Supplemental Table 5). Three of the clusters were characterized by gene upregulation predominantly during the injury phase: “early injury” genes (UUO-2 only) were enriched for damage-associated molecular pattern–Toll-like receptor (TLR) signaling, MAPK signaling, and oxidative stress pathways (Figure 1C); “pan-injury” genes included cell cycle genes and markers of kidney injury (Havcr1 and Lcn2); and “late injury” genes (UUO-7) were enriched for ECM components, ECM crosslinkers, and inhibitors of ECM degradation.
Two clusters were characterized by gene activation predominantly during R-UUO and were enriched for genes implicated in innate and adaptive immunity (Figure 1C). Remarkably, five of the top ten genes induced specifically after R-UUO (Lyz1, Mmp12, Gpnmb, Ccl8, and Retnla; Supplemental Figure 1, D and E) were also induced in macrophages in our model of resolution of liver fibrosis.23 Multiple podocyte-specific genes were also included in the cluster upregulated during reversal, consistent with the loss of tubular mass and relative glomerular preservation in this model of predominantly tubular injury.
The patterns of gene expression in the R-UUO model were consistent with those observed in a rat model of reversible diabetes and hypertension (Supplemental Figure 1, F and G),53 suggesting common mechanisms of injury and repair across multiple models and organs,23 with regression of fibrosis being characterized by the presence of a specific macrophage phenotype.
scRNA-seq Demonstrates Dynamic Changes in the Proportion of Intrinsic and Immune Cells during the R-UUO Model
To characterize the heterogeneity of cells during injury and repair and to ascribe the bulk-transcriptomic changes to specific cell types, we performed scRNA-seq on single-cell suspensions from the renal cortex of animals at four time points: baseline, UUO-2, UUO-7, and 2 weeks after R-UUO. Unsupervised clustering of the aggregated data from approximately 17,500 individual transcriptomes identified 15 discrete clusters (Figure 2, A and B, Supplemental Figure 2, A–D, Supplemental Table 6), which were classified using established cell-specific markers in murine kidneys (Figure 2C, Supplemental Figure 2B).41 Because this is the first scRNA-seq dataset during renal injury and reversal of fibrosis, we have created an interactive tool for data exploration at http://www.ruuo-kidney-gene-atlas.com/.
Figure 2.

scRNA-seq analysis identifies discrete renal cell types, with dynamic changes in the proportion and transcriptome of each cell type observed across the R-UUO model. tSNE plots of 17,136 cells from libraries pooled from mice that underwent sham, UUO-2, UUO-7, or R-UUO (2 weeks) (n=3 per time point) classified by (A) cell cluster and (B) time point. (C) Expression of selected marker genes for each cell classification projected onto tSNE plot. Color scale is log10 expression levels of genes. (D) Relative proportions of cells assigned to each cluster by time point. Statistical significance derived using differential proportional analysis, with a mean error of 0.1 over 100,000 iterations. *P<0.05. (E) Violin plots of Egf and Lcn2 (encodes neutrophil gelatinase-associated lipocalin) gene expression in the loop of Henle/distal convoluted cell cluster. The y axis shows the log-scale normalized read count. a–c, PCT subclusters colored by shared nearest neighbor; DCT, distal convoluted tubule; LoH, loop of Henle; Mac, macrophage; Mono, monocyte; NK, natural killer cell; PCT, proximal convoluted tubule; S1, S1 segment.
Differential proportional analysis44 determined that, after UUO, there was a marked reduction in the proportion of cells derived from the proximal tubule, which partly reversed after R-UUO (Figure 2D). Additionally, there were dynamic changes in the tubular transcriptome after UUO, with induction of injury markers such as Lcn2 (adjusted P=1.45 × 10−293) and reduced expression of Egf, a biomarker of tubular cell health (adjusted P=5.67 × 10−05) (Figure 2E).54,55 There was early recruitment of neutrophils and natural killer cells to the obstructed kidney, followed by expansion of macrophages and T cells, which persisted beyond the reversal of obstruction, as observed previously in the repair phase in a model of IRI (Figure 2D).26 The changes in cell proportions were replicated on flow cytometry using markers of key cell types: proximal tubule cells (LTL), endothelial cells (CD31), fibroblasts/pericytes (PDGFR-β), immune cells (CD45), and F4/80Hi and F4/80Lo macrophages (Supplemental Figure 2E). Of note, although we detected a small number of cells expressing podocyte genes, these were too few to constitute a discrete cluster, as has been reported previously.42 This likely reflects the difficulty in isolating single glomerular cells from whole kidney cortex, whereas extraction of single nuclei is more efficacious, with small-nuclear RNA-seq detecting 20-fold more podocytes.42 Similarly, we may not have captured other glomerular cells, such as endothelial or mesangial cells and monocytes or macrophages, that are unique to the glomerulus.
scRNA-seq Reveals Myeloid Heterogeneity during Injury and Repair
Our previous data in the kidney and liver suggest a pivotal role for the plasticity of myeloid cells in injury and repair.23,53 To further characterize the myeloid cell heterogeneity and phenotype, we repeated the SNN clustering specifically on myeloid cells, partitioning these cells into 12 clusters (Figure 3A). We first assigned clusters as monocytes, macrophages, or DCs by generating Spearman correlation values for each cluster as compared with gene signatures of mouse immune cells obtained from ImmGen (Figure 3B, Supplemental Figure 3, A and C). We then refined this classification using cluster-defining DE genes (Supplemental Figure 3B, Supplemental Table 7) and the following genes encoding cell surface protein markers that define specific myeloid cell subsets on flow cytometry: Itgam (CD11b), Adgre1 (F4/80), Fcgr1 (CD64), Itgax (CD11c), and H2-Aa (MHCII) (Figure 3, C and D).
Figure 3.
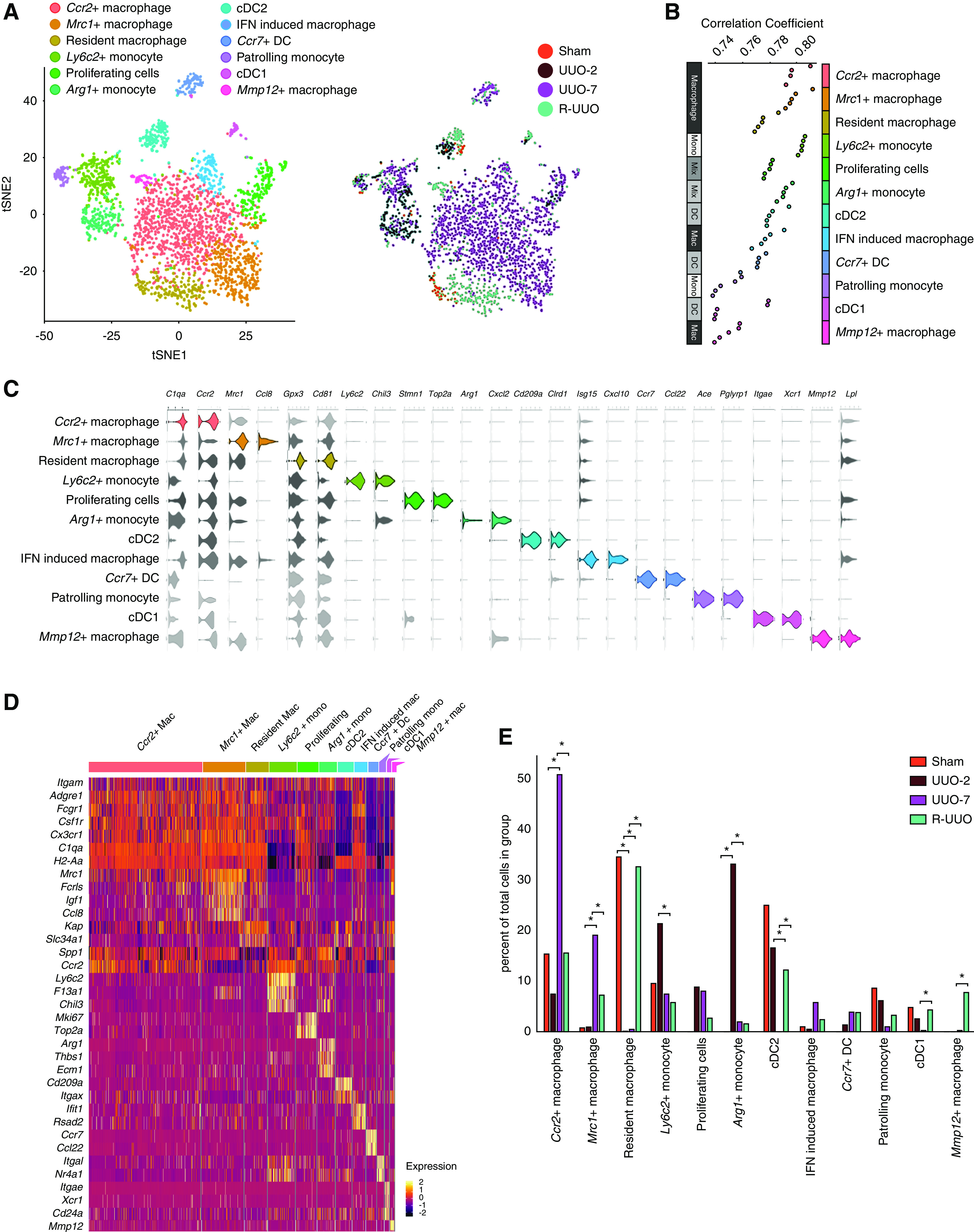
scRNA-seq analysis identifies 12 discrete myeloid cell clusters, with dynamic changes in the proportion of cells assigned to each cluster across the R-UUO model. (A) tSNE plot of 2956 pooled myeloid cells from each time point, annotated by cell type and time point. (B) Top five ImmGen reference immune cell types correlating with each cluster, ranked by Spearman correlation coefficients after a cluster-to-references analysis using Cluster Identify Predictor version 2. (C) Violin plots showing the expression levels of selected marker genes in each cluster. The x axis shows the log-scale normalized read count. (D) Heatmap of selected marker gene expression in each cluster, calculated using Wilcoxon signed-rank test. The color scheme is based on z-score distribution. (E) Relative proportions of each cell type at each time point. Statistical significance tested using differential proportional analysis with a mean error of 0.1 over 100,000 iterations. *P<0.05. Mac, macrophage; mono, monocyte; IFN, interferon.
Early Accumulation of Ly6c2+ and Arg1+ Monocytes after Ureteric Obstruction
We identified the first of three monocyte clusters as patrolling monocytes because they expressed Nr4a1 and Itgal, which are implicated in survival56,57 and adhesion56,58 of CX3CR1+/Ly6C− patrolling monocytes. We classified the second cluster as inflammatory Ly6C+ monocytes because they expressed Ly6c2, Ccr2, F13a1, and Chil3 genes.24 Cells in this cluster increased at UUO-2 (Figures 3, A and E and 4A), indicating early recruitment of Ly6C+ inflammatory monocytes to the kidney during injury.
Figure 4.
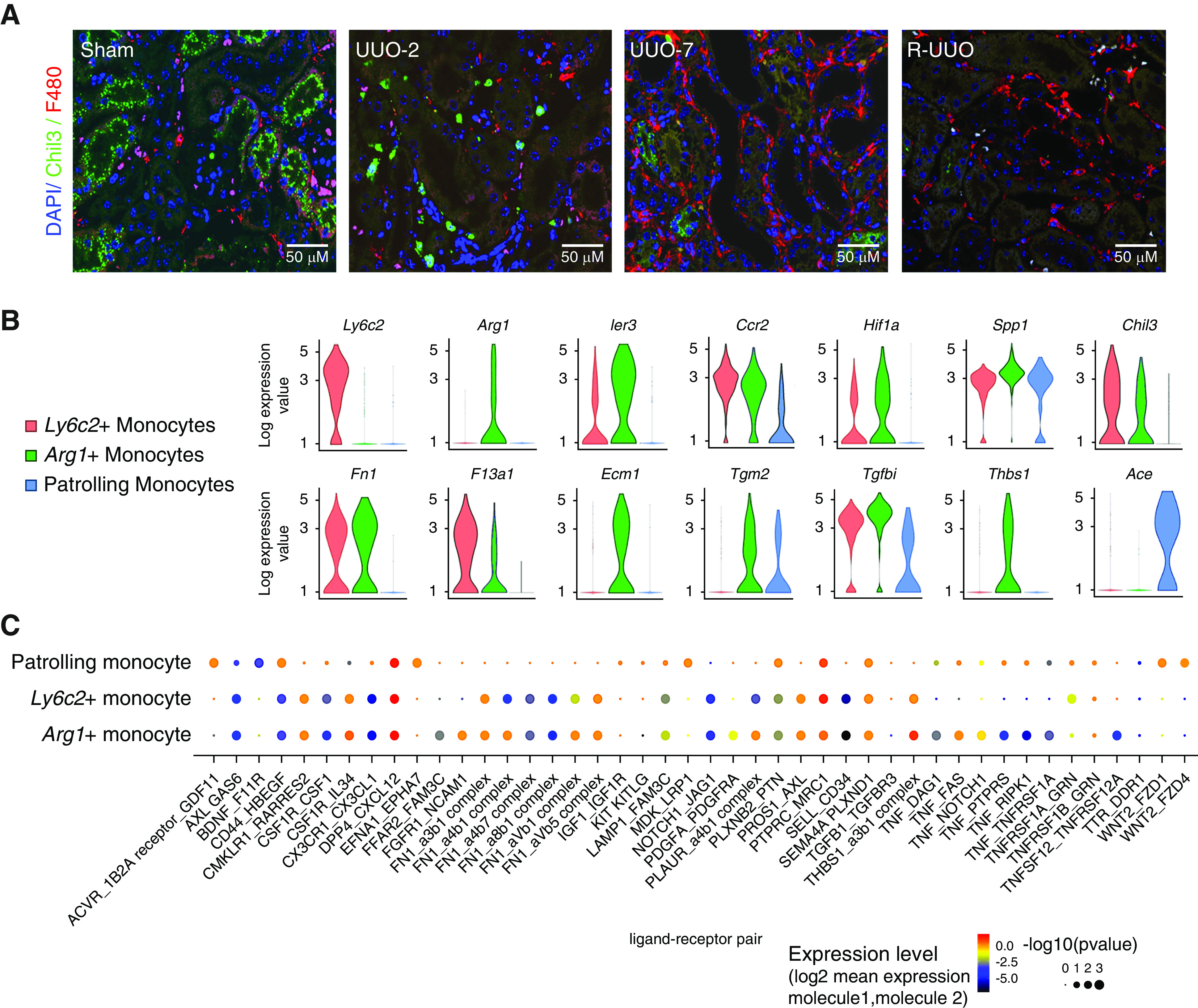
Monocytes recruited to the kidney at UUO-2 exhibit a pro-fibrotic phenotype that may activate mesenchymal cells. (A) Representative immunofluorescence images from each time point in the R-UUO model (sham, UUO-2, UUO-7, and R-UUO [2 weeks]) for Chil3 (marker of Ly6C+ monocytes, red) and the pan-macrophage marker F4/80 (green). (B) Violin plots showing log10 expression levels of selected genes in the three monocyte clusters. The y axis shows the log-scale normalized read count. (C) Significant monocyte ligand–mesenchymal receptor pairs across the three monocyte subclusters. Color of dot is proportional to mean expression values for all of the interacting partners, and size is inversely proportion to P value.
A third cluster uniquely expressed Arg1 (Figure 3, C and D) and additionally markers of Ly6C+ inflammatory monocytes, including Ccr2, Chil3, and F13a1, although not Ly6c2 (Figures 3D and 4B). DE genes in this cluster included early response genes (Ier3, Fos, Jun), hypoxia genes (Hif1a, Vegfa), proinflammatory genes (Thbs1, Spp1), profibrotic genes (Tgfb1, Tgfbi), and genes encoding ECM components (Fn1, Ecm1) or ECM crosslinkers (Tgm2; Figure 4B). These Arg1+ cells were exclusively present at UUO-2 (Figure 3, A and E), and the profibrotic gene expression suggested they may initiate fibrosis by interacting with mesenchymal cells. Accordingly, we determined expression of ligand-receptor pairs between each monocyte subset and mesenchymal cells (Figure 4C). More monocyte ligand–mesenchymal receptor pairs were expressed in Ly6c2+ and Arg1+ monocytes than patrolling monocytes. In addition, compared with the Ly6c2+ cells, the Arg1+ cells demonstrated greater potential for Fn1-integrin, Pdgfa-Pdgfrß, and Tnf-Tnfsfr1 signaling to mesenchymal cells. Taken together, these data suggest Arg1+ cells may be derived from recruited Ly6C+ monocytes that become activated acutely in the hypoxic and inflammatory milieu of the injured kidney toward a profibrotic phenotype.
Macrophages Adopt Differing Phenotypes during Injury and Resolution Phases
We identified five clusters (Figure 3B) that expressed genes consistent with macrophage identity, including those encoding CSF1 receptor (Csf1r), MHCII (H2-Aa), and Cd81, a conserved marker of renal macrophages (Figure 3, C and D).24 We define a first cluster as quiescent resident macrophages, detected exclusively in kidneys that underwent sham surgery or were de-obstructed (Figure 3, A and E). In contrast to other macrophage clusters, these cells did not express Spp1, which encodes osteopontin, a marker of activated macrophages59 that promotes renal injury after UUO.60 Although they were tagged by proximal tubular genes, the expression was much lower than in tubular cells (Figure 2C); therefore, this likely represents ambient tubular RNA, which is most prevalent in sham or R-UUO mice due to the higher proportion of proximal tubular cells (Figure 2D).
A second macrophage cluster expressed high levels of Mrc1 (encodes mannose receptor), and cells from this cluster were most commonly observed at UUO-7 and, to a lesser extent, at R-UUO (Figures 3, A and E and 5A). The Mrc1+ cells expressed multiple scavenger receptors (Mrc1, Fcrls, Stab1), suggesting a role in scavenging debris/excess ECM (Figure 3D). In addition, they expressed Igf1, which is upregulated in reparative macrophages in the liver23 and promotes regression of cirrhosis and liver regeneration,61 and Apoe, which dampens inflammation62 and promotes regeneration.63 The Mrc1+ cells exhibited lower H2-Aa expression, indicating downregulation of MHCII. This is of interest because resident renal macrophages downregulate MHCII and adopt a phenotype consistent with embryonic macrophages during repair from acute ischaemia-reperfusion injury (IRI).47 To assess this further, we compared the transcriptome of our macrophage clusters with those of embryonic kidney macrophages, resident renal macrophages before and after IRI, and infiltrative monocytes.47 The transcriptome of the Mrc1+ cells most closely aligned to embryonic macrophages, consistent with reprogramming toward a developmental phenotype to facilitate renal repair (Figure 5B).
Figure 5.
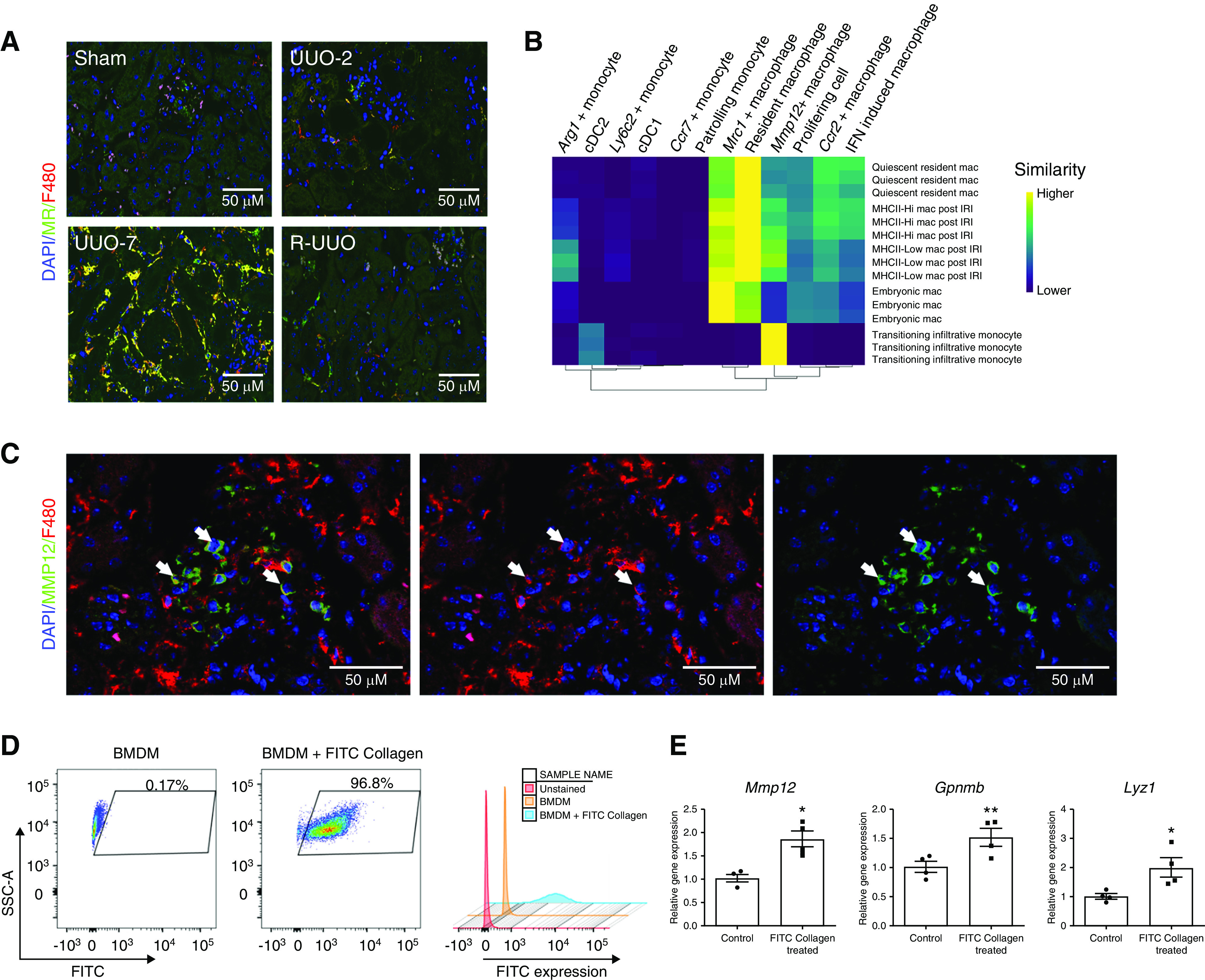
Mannose receptor (MR)+ F4/80Hi macrophages and MMP12+F4/80Lo cells are observed in late injury or specifically following reversal of obstruction, respectively. (A) Representative immunofluorescence images across the R-UUO time course (sham, UUO-2, UUO-7, and R-UUO [2 weeks]) for mannose receptor (MR, marker of Mrc1+ macrophages, green) and F4/80 (red). (B) Single R analysis comparing the transcriptome of the myeloid clusters with those of embryonic macrophages, adult macrophages before and after renal IRI, and infiltrating monocytes during repair of renal IRI. (C) Immunofluorescence images showing colocalisation of MMP12 (green) and F4/80 (red) in renal macrophages. (D) Flow cytometry plots of bone marrow–derived macrophages (BMDMs) demonstrating fluorescence after phagocytosis of FITC-collagen. (E) Expression of reparative macrophage genes measured by quantitative RT-PCR in BMDMs after phagocytosis of FITC-collagen versus BMDMs cultured in medium alone (control). n=4 replicates. *P<0.05, **P<0.01. Mac, macrophage.
A third macrophage cluster predominantly comprised cells from UUO-7 kidneys. Cells in this cluster expressed Ccr2 (Figure 3D), raising the possibility they may be derived from Ly6C+/CCR2+ monocytes that are recruited to the injured kidney before transitioning to adopt a transcriptome highly similar to resident macrophages (Figures 3, C and D and 5B).
A fourth cluster was uniquely characterized by expression of IFN-stimulated genes (Figure 3, C and D). Although their function remains unknown, similar cells have been observed after injury in other organs, including the heart.28
Intriguingly, a final macrophage cluster was observed solely in kidneys that had undergone R-UUO. These cells were characterized by expression of Mmp12, a macrophage-specific metalloproteinase, and scavenger receptors (Mrc1, Fcrls), suggesting they may be involved in matrix remodeling (Figures 3, C and D and 5C). In addition, the Mmp12+ cluster–expressed genes implicated in efferocytosis and lipid transport, suggesting they may be involved in clearance of apoptotic cells (Supplemental Figure 3D). This is in keeping with our previous work, where Mmp12+ expression defines a reparative macrophage phenotype that mediates regression of liver fibrosis.23 In response to phagocytosis of collagen (Figure 5D), bone marrow–derived macrophages upregulated the degradative enzymes expressed in reparative macrophages including Mmp12, Gpnmb, and Lyz123 (Figure 5E), suggesting they may switch to a matrix-degrading phenotype on encountering scarred matrix. Although the Mmp12+ cells mapped to macrophages on the ImmGen databases (Figure 3B), they morphologically resembled monocytes and expressed low levels of F4/80 (Figure 5C) but high levels of Ccr2 (Figure 3C). Furthermore, their transcriptome most closely aligned to monocytes infiltrating the kidney during recovery from IRI (Figure 5B). Taken together, these data suggest they may be derived from monocytes that adopt a unique Mmp12+ phenotype on encountering a kidney during the reparative phase after injury.
DCs Adopt a Migratory Phenotype during Late-Stage Injury and Resolution
We assigned three clusters as DCs, expressing MHC genes (H2-Aa) but not macrophage markers (Cd81, C1q; Figure 3D). One cluster expressed Itgae (encodes CD103), whereas another expressed Cd209a, consistent with type 1 and type 2 conventional DCs (cDC1, cDC2), respectively.24 cDC1s and cDC2s were proportionally fewer in UUO-7 before returning during R-UUO (Figure 3, A and E). In contrast, the third DC cluster, which expressed Ccr7, was not detected in sham animals, but appeared by UUO-7 and persisted through R-UUO (Figure 3, A and E). This cluster mapped specifically to lymph node DCs in the ImmGen database (Supplemental Figure 3A). Taken together, the data suggest that, after kidney injury, resident DCs upregulate Ccr7 and this may promote migration to draining lymph nodes by binding to CCR19/CCR21.64
Conventional Flow Cytometry Does Not Capture the Full Heterogeneity of Myeloid Cells
To assess how our scRNA-seq–derived clusters corresponded to conventional cytometry, we performed scRNA-seq using plate-based SMART-seq2 technology, which enabled linkage of each transcriptome to an abundance of cell surface markers (FACS intensity) using index sorting.65 We repeated the R-UUO model using MacGreen mice (express EGFP under the Csf1r promoter) and performed flow cytometry, gating on CD45+MacGreen+TCRβ−CD19−Ly6G−Siglec-F− myeloid cells. There was expansion of the CD11b+F4/80Lo population at UUO-2, consistent with early recruitment of monocytes in response to injury (Figure 6A). By UUO-7, monocyte recruitment had diminished, but there was an increase in CD11b+F4/80Hi macrophages, which persisted through 2 weeks after R-UUO (Figure 6A).
Figure 6.
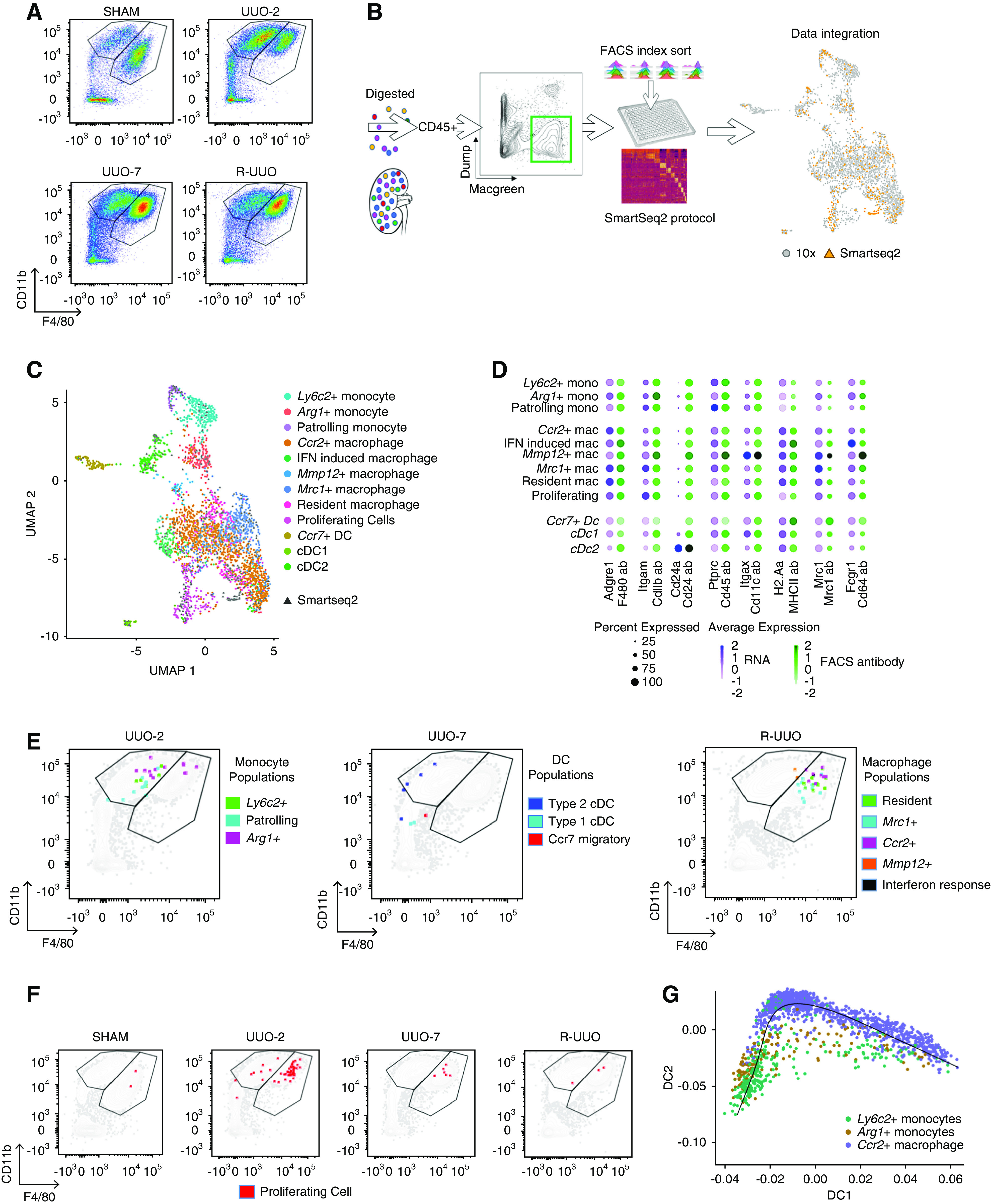
Integration of droplet– and plate-based scRNA-seq datasets determines that clusters identified by scRNA-seq would not be readily discriminated on conventional flow cytometry. (A) Representative flow cytometry plots from kidney cell suspensions from each time point after gating on CD45+MacGreen+TCRβ−CD19−Ly6G−Siglec-F− myeloid cells. Cells segregated into CD11b+F4/80Lo monocyte and CD11b+F4/80Hi macrophage gates. (B) Strategy to integrate gene and cell surface protein expression at the single-cell level. Kidneys were digested into single-cell suspensions and single CD45+MacGreen+TCR1β−CD19−Ly6G−Siglec-F− myeloid cells were sorted into individual wells after index linkage to cell surface marker expression. They underwent scRNA-seq using the SMART-seq2 protocol before integration with the 10× dataset. (C) Uniform Manifold Approximation and Projection (UMAP) of the combined 10× and SMART-seq2 dataset. (D) Dotplot of cell surface protein and corresponding gene expression in each cluster. The size of the dot denotes the percentage of cells in each cluster expressing the relevant gene/protein; the intensity of color represents mean gene/protein expression. (E) Representative flow cytometry plots from UUO-2, UUO-7, and R-UUO (2 weeks) illustrate mapping of cells from each myeloid cluster onto the CD11b+F4/80Lo monocyte and CD11b+F4/80Hi macrophage gates. (F) Mapping of proliferating cells (red) at each time point onto the flow cytometry plots. (G) Pseudotime analysis of the transcriptomes of the Ly6c2+, Arg1+, and Ccr2+ clusters. Mac, macrophage; mono, monocyte.
We captured 192 individual CD45+MacGreen+TCR1β−CD19−Ly6G−Siglec-F− myeloid cells from each time point for scRNA-seq on the SMART-seq2 platform (Figure 6B). This dataset was then integrated with the original droplet dataset, with SMART-seq2 cells distributed through every cluster (Figure 6, B and C). Index linkage demonstrated moderate correlation between the gene and corresponding surface protein expression in each cluster (Figure 6D). Next, we mapped the cells from each myeloid cluster onto the monocyte and macrophage gates on flow cytometry (Figure 6E). Cells from the Ly6c2+ and patrolling monocyte clusters mapped to the CD11b+F4/80Lo monocyte gate, as expected. Conversely, resident, Mrc1+, Ccr2+, and IFN-response macrophages all mapped appropriately to the CD11b+F4/80Hi macrophage gate. Cells from the Arg1+ cluster straddled the monocyte and macrophage gates (Figure 6E), with a proportion of the Arg1+ cells colocating with Ccr2+ macrophages in the CD11bHiF4/80Hi region, suggesting they may be transitioning to Ccr2+ macrophages. Furthermore, trajectory analysis supported transition of Ly6c2+ monocytes to Ccr2+ macrophages, with Arg1+ monocytes representing an intermediate transitional state (Figure 6G). Cells from the Mmp12+ cluster also straddled the monocyte and macrophage gates (Figure 6E), consistent with their intermediate F4/80 expression on immunofluorescence (Figure 5B). Cells from the cDC1 and Ccr7+ clusters were CD11b−F4/80−, whereas the cDC2s mapped to the CD11b+F4/80Lo monocyte gate (Figure 6E). Cells from the proliferating myeloid cluster were observed predominantly at UUO-2 and mapped to the monocyte and, more particularly, the macrophage gates (Figure 6F). Consistent with this, the scRNA-seq cell cluster designated as proliferating cells (express Mki67) predominantly expressed macrophage markers, with representation of all macrophage clusters.
Monocytes Recruited Early after UUO Transition to a Macrophage Phenotype
Pseudotime trajectory analysis and flow cytometry data suggested that Ly6c+ monocytes recruited to the obstructed kidney transitioned to Ccr2+ macrophages by UUO-7. To test this hypothesis, we performed PBE between Ly5.1 mice (CD45.1/CD45.2 heterozygous) and C57BL6/J mice (CD45.2 homozygous) (Figure 7A). Immediately after the PBE, approximately 40% of total CD45+ circulating cells were derived from the donor (Figure 7B). The proportion of donor-derived circulating monocytes/neutrophils fell rapidly to approximately 1% within 24 hours, with negligible numbers persisting through to UUO-7 (Figure 7C, full gating strategy in Supplemental Figure 4A). There was a similarly rapid reduction in the proportion of donor-derived circulating T and B lymphocytes; however, a small number of donor lymphocytes persisted in the circulation through UUO-7 (Figure 7C).
Figure 7.
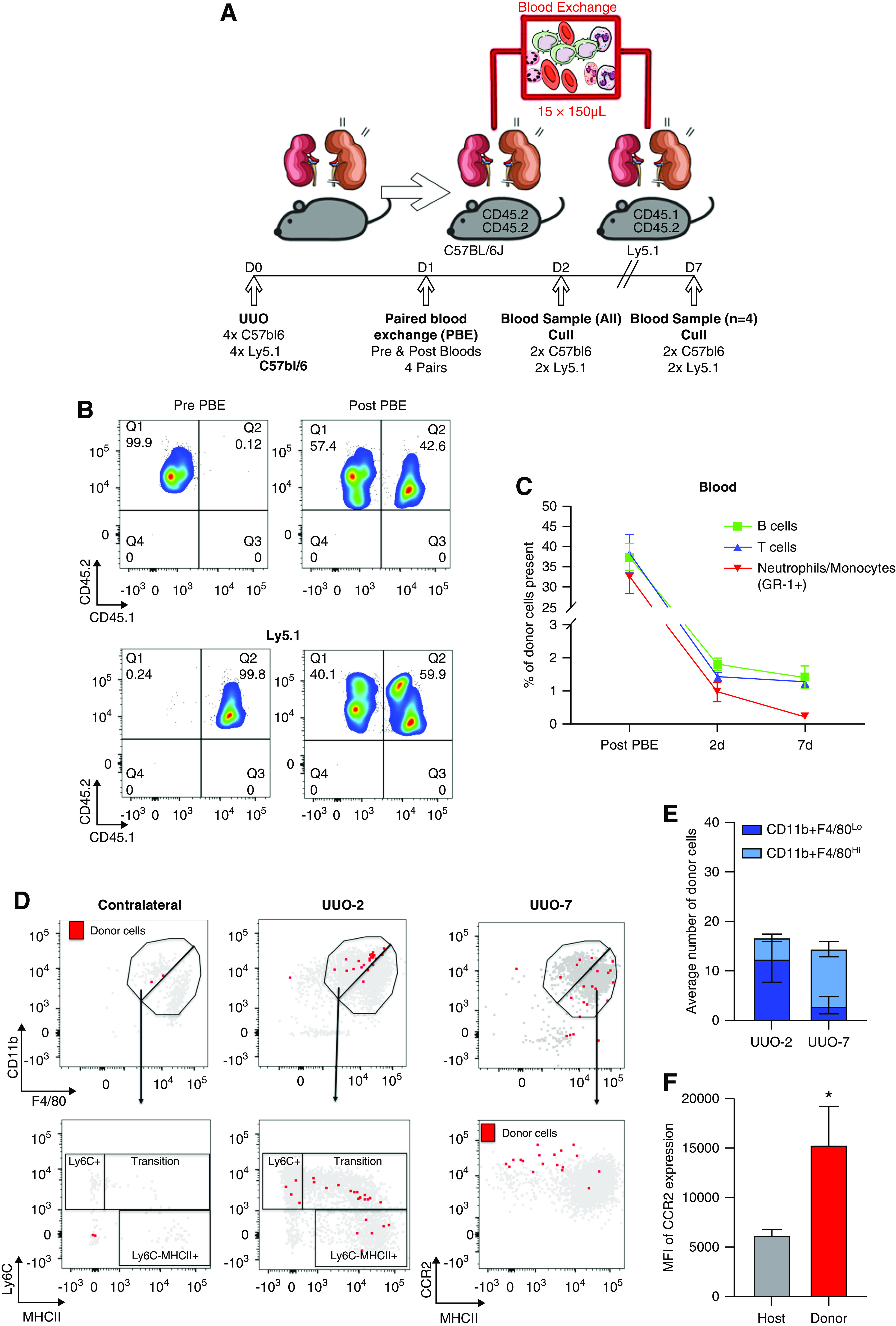
Paired blood exchange (PBE) demonstrates that CCR2+ macrophages observed at UUO-7 are derived from donor monocytes. (A) Schemata of experimental strategy for PBE to track fate of immune cells recruited to the kidney. One day after UUO, whole blood exchange was performed between pairs of C57BL/6 and Ly5.1 mice (n=4) and pairs were euthanized either 2 or 7 days after UUO. (B) Representative flow cytometry plots of circulating CD45+ cells from pairs of mice pre- and immediately post-blood exchange, illustrating approximately 40% of circulating cells were derived from donors after the exchange (CD45.1+CD45.2+ cells in C57BL/6; CD45.2+/+ cells in Ly5.1 mice). (C) Percentage of circulating immune cells derived from paired donor over the experimental time course (n=4 pairs immediately post-PBE and at UUO-2; n=2 pairs at UUO-7). (D) Illustrative flow cytometry plots mapping donor cells (red) and recipient cells (gray) to the CD11b+F4/80Lo monocyte and CD11b+F4/80Hi macrophage gates, the monocyte waterfall in obstructed and contralateral kidneys at 2 days post-UUO, and the expression of CCR2 and MHCII 7 days post-UUO. (E) Average number of donor cells mapping to the CD11b+F4/80Lo monocyte and CD11b+F4/80Hi macrophage gates in obstructed kidneys at 2 and 7 days after UUO. (F) The expression of CCR2 (mean fluorescent intensity, MFI) on the donor cells compared with the host cells from the CD11b+F4/80Hi macrophage gate in obstructed kidneys 7 days after UUO. n=4 per group. *P<0.05 by Mann–Whitney test.
To determine the fate of donor monocytes recruited to the obstructed kidney, we performed flow cytometry on kidney cell suspensions, gating on CD45+CD64+TCRβ−CD19−Ly6G−Siglec-F− myeloid cells (Supplemental Figure 4B). At UUO-2, donor cells were recruited preferentially to the obstructed kidney (Figure 7D) and mapped almost exclusively to the CD11b+F4/80Lo monocyte gate (Figure 7E). Indeed, they spanned the monocyte “waterfall,” suggesting transition from Ly6CHi/MHCLo monocytes toward a Ly6CLoMHCIIHi macrophage-like phenotype Figure 7D). By UUO-7, the majority of donor cells were located in the CD11b+F4/80Hi macrophage gate, and they expressed high levels of CCR2 compared with the global macrophage population (Figure 7F). In combination with the trajectory analysis and flow cytometry data, these data suggest donor monocytes are recruited selectively to the obstructed kidney by UUO-2, transition to a CCR2Hi macrophage by UUO-7, and, hence, are the likely source of the cells in the Ccr2+ macrophage cluster observed at UUO-7 in the scRNA-seq dataset (Figure 3A).
Myeloid Cell Subsets Correlate with Fibrosis in Human Kidney Disease
We next assessed whether similar myeloid cell phenotypes were observed in the human kidney using the Human Protein Atlas (Figure 8A). Cells that stained with F13A1 (marker of Ly6c2+ monocytes) and DOK2 (Arg1+ monocytes) were located specifically in focal areas of injury/inflammation, whereas ITGAL (patrolling monocytes) was largely restricted to cells within the circulation. CD68, a pan-macrophage marker (including a marker of resident macrophages), was widely distributed in the healthy kidney, whereas mannose receptor (Mrc1+ macrophages) and CCR2 (Ccr2+ macrophages) localized to areas of tissue injury. IRF8 and CD209, markers of types 1 and 2 conventional DCs, respectively, localized to areas of renal injury, with CCR7 (Ccr7+ migratory DCs) staining a cluster of cells that resembled a tertiary lymphoid follicle. Furthermore, in biopsy specimens of healthy donors and patients with diabetic nephropathy or FSGS (https://www.nephroseq.org/resource/login.html), expression of each myeloid marker correlated with expression of Col1a1 (encodes collagen I; Figure 8B). Expression of Mmp12 was not detected in the healthy human kidney or in patients with CKD, which is consistent with the fact that cells in the Mmp12+ cluster were specific to the resolution phase of kidney injury.
Figure 8.
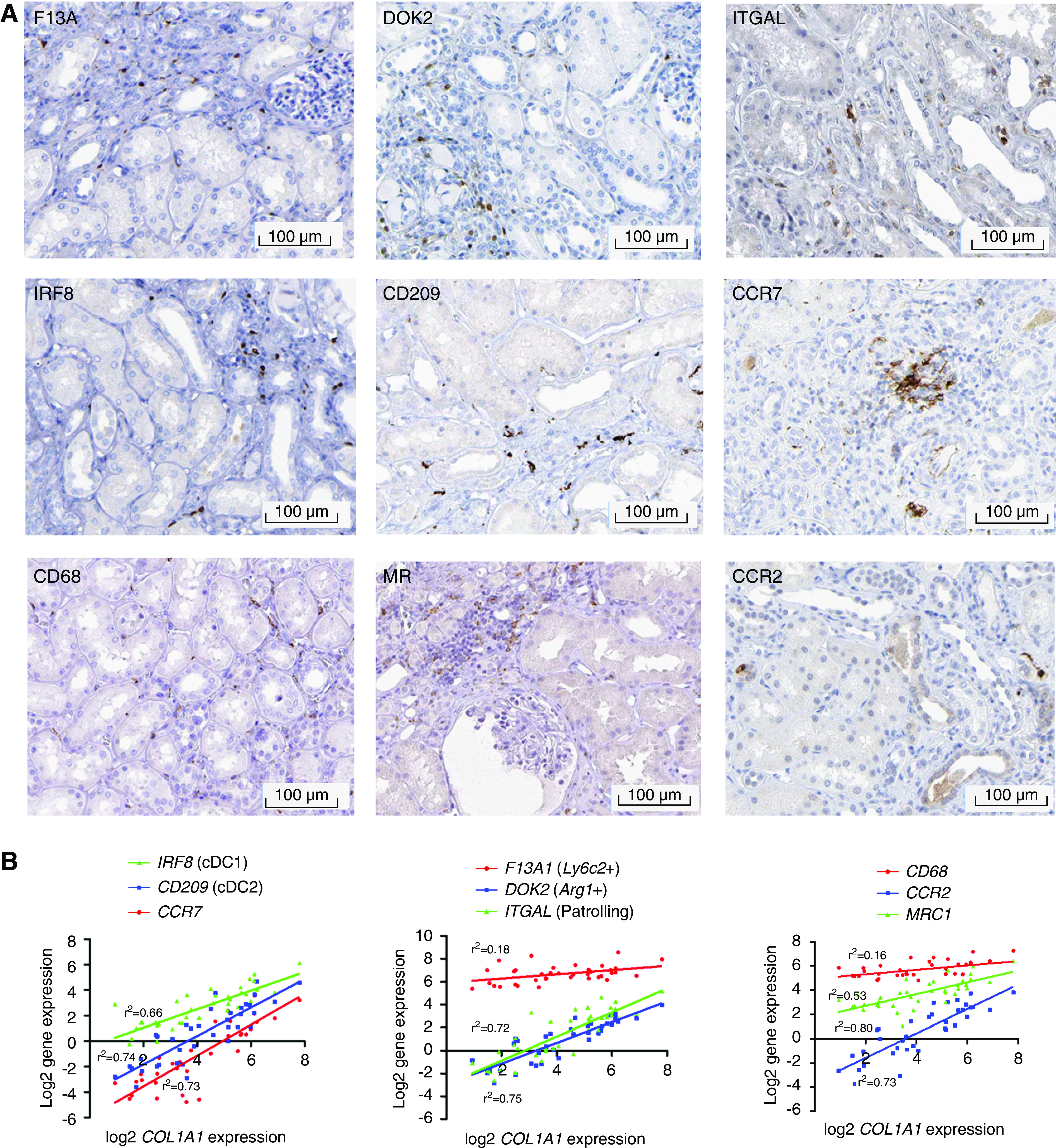
Markers of murine myeloid subsets are also observed in human kidney disease. (A) Immunostaining for myeloid cluster–defining markers in human kidney tissue obtained from the Human Protein Atlas.66 (B) Gene expression of cluster-defining markers for DCs (left), monocytes (center), and macrophages (right) against COL1A1 gene expression in the tubulointerstitium of kidneys from healthy controls (n=9), patients with diabetic nephropathy (n=10), and patients with FSGS (n=18). Data obtained from www.Nephroseq.org.
Discussion
Our scRNA-seq studies, the first detailed characterization of myeloid cell heterogeneity in the kidney during progression and regression of fibrosis, have identified novel monocyte and macrophage subsets not previously observed in the kidney.
Acute injury induces a novel population of cells that are transcriptomically aligned to monocytes but that uniquely express Arg1. Although Arg1 has traditionally been thought of as a marker of alternative macrophage activation,67 the Arg1+ cells do not express other markers of alternative activation, such as Mrc1 or MHCII-encoding genes, suggesting that in vitro immune activation assays do not reflect the complex in vivo milieu. Indeed, the Arg1+ cells express proinflammatory and profibrotic genes, and future work should determine whether specific depletion of these cells could reduce disease severity. Intriguingly, a novel Mmp12+ macrophage subset emerged specifically during the resolution phase. We have previously reported a similar macrophage phenotype during the resolution of liver disease, suggesting common reparative mechanisms across organs.23 Our in vitro studies suggest that ingestion of excess ECM or cell debris may be a stimulus for induction of this phenotype; however, their cellular origin and role in resolution requires further study because strategies to induce this phenotype may enhance scar degradation in the diseased kidney and other organs. To definitively conclude that the Mmp12+ macrophages we identified are essential to repair, as our studies infer, functional studies in MMP12−/− mice demonstrating a lack of repair would be required.
Uniquely, we have integrated plate-based and droplet scRNA-seq with index linkage to map our myeloid subsets onto monocyte and macrophage gates on flow cytometry. Importantly, the novel myeloid subsets would not have been identified by conventional markers on flow cytometry, highlighting the utility of scRNA-seq to characterize myeloid heterogeneity in an unbiased and more detailed way. Based on the DE genes in each subset, we suggest a panel of cell surface markers could be used in combination to differentiate the cells from each myeloid cluster on flow cytometry (Table 1), with the caveat that there is only a modest correlation between gene expression and cell surface marker expression of the corresponding protein.
Table 1.
Cell surface markers for assignment of myeloid clusters by flow cytometry
| Cluster | F4/80 (Adgre1) | CD11b (Itgam) | CD64 (Fcgr1) | MR (Mrc1) | CD40 (Cd40) | CCR2 (Ccr2) | Ly6C (Ly6c2) | CCR1 (Ccr1) | CD11a (Itgal) | CD203 (Itgae) | CD209 (Cd209a) | CCR7 (Ccr7) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Resident macs | +++ | ++ | ++ | + | − | − | − | − | − | − | − | |
| Mrc1+ macs | +++ | +++ | ++ | +++ | − | − | + | − | − | − | − | |
| IFN-response macs | +++ | ++ | +++ | + | ++ | − | − | − | − | − | − | − |
| Ccr2+ macs | ++ | ++ | ++ | + | ++ | − | − | − | − | − | − | |
| Mmp12+ macs | + | + | − | ++ | + | − | − | − | − | − | − | |
| Ly6c2+ monos | − | ++ | − | − | +++ | +++ | + | + | − | − | − | |
| Arg1+ monos | − | +++ | + | − | + | − | +++ | − | − | − | − | |
| Patrolling monos | − | + | − | − | − | − | − | +++ | − | − | − | |
| cDC1 | − | − | − | − | − | − | − | − | +++ | − | − | |
| cDC2 | − | ++ | − | − | − | − | + | − | − | +++ | − | |
| Ccr7+ DCs | − | − | − | − | − | − | − | − | − | − | +++ |
Gene expression (italicised in brackets) of corresponding cell surface protein markers that could be used to assign cells to myeloid clusters on flow cytometry. Macs, macrophages; monos, monocytes; IFN, interferon.
To track the fate of circulating immune cells recruited to the kidney, we used PBE, which has previously been used to assess the effects of donor serum35 but not, to our knowledge, to track immune cells. By combining PBE, flow cytometry, and pseudotime analyses, we demonstrate early recruitment of monocytes specifically to the obstructed kidney and that these subsequently adopt a macrophage phenotype, but continue to express CCR2. These results suggest circulating monocytes are the source of the large Ccr2+ macrophage cluster observed at UUO-7 in the scRNA-seq dataset, which is consistent with lineage tracing and parabiosis studies after myocardial infarction.28 Remarkably, although they continue to express Ccr2, their transcriptome is otherwise almost identical to resident macrophages. Genetic or pharmacologic inactivation of CCR2 after renal IRI reduces the expansion of F4/80+ macrophages in the kidney and the severity of renal fibrosis, suggesting CCR2+ cells may be detrimental,19 although it is unclear whether they remain detrimental once they transition to a macrophage phenotype. One advantage of PBE over parabiosis or bone marrow transfer is that the donor cells persist at large numbers in the circulation for a relatively short time, and, therefore, enable the tracking of cells at multiple discrete time points after injury or during resolution of disease. Future studies using PBE may help determine whether cells in the Mmp12+ cluster are indeed derived from infiltrating monocytes entering a repairing kidney. The short circulating time of donor cells may also be a limitation of the technique, in that only a small proportion of recruited cells are derived from the donor, therefore, a limited number of cells are available for downstream analysis. Further refinements including performing several consecutive PBEs may increase the yield.
In summary, by combining multiple complementary technologies, our studies have identified novel subsets of myeloid cells, which may also be present in human kidney disease and, hence, may represent therapeutic targets to inhibit progression and enhance resolution of kidney disease.
Disclosures
All authors have nothing to disclose.
Funding
This work was funded by Kidney Research UK project grants RP30/2015 and RP_046_20170303, and a Medical Research Council Discovery Award MC_PC_15075. E. O’Sullivan is funded by Kidney Research UK Clinical Training Fellowship grant TF_006_20161125. L. Denby is supported by a Kidney Research UK Senior Fellowship grant SF_001_20181122 and a previous Intermediate Fellowship grant PD6/2012. T. Chandra was supported by a University of Edinburgh Chancellor’s Fellowship. P. Ramachandran was supported by a Medical Research Council Clinician Scientist Fellowship grant MR/N008340/1. N. Henderson was supported by a Wellcome Trust Senior Research Fellowship in Clinical Science (reference 103749). D.J. Simpson is funded by the Medical Research Council studentship (Doctoral Training Programme in Precision Medicine).
Supplementary Material
Acknowledgments
We acknowledge Mr. Gary Borthwick (Centre for Cardiovascular Science, University of Edinburgh) for his assistance in performing the R-UUO surgery. Flow cytometry data were generated with support from the Queen’s Medical Research Institute Flow Cytometry and Cell Sorting Facility, University of Edinburgh. Thanks to Drs. Mike and Irina Conboy (University of California, Berkley) for assistance with establishing the PBE model.
L. Denby and B. Conway were responsible for funding acquisition, supervision, conceptualization, project administration, methodology, investigation, analysis, and writing and editing the manuscript; E. O’Sullivan was responsible for methodology, investigation, analysis, and writing and editing the manuscript; J. Hughes was responsible for funding acquisition, conceptualization, methodology, editing the manuscript, and analysis; T. Chandra was responsible for hardware provision, funding acquisition, supervision, conceptualization, methodology, analysis, and editing the manuscript; C. Cairns was responsible for investigation, analysis, and writing the manuscript; J. O’Sullivan, O. Teenan, P. Ding, D. Humphries, and K. Stewart were responsible for investigation and analysis; A. Salzano was responsible for investigation; N. Henderson was responsible for hardware and experimental methodology; P. Ramachandran was responsible for hardware, experimental methodology, and analysis; D. Simpson was responsible for data analysis; K. Connor and R. Pius were responsible for database resources; and D. Ferenbach and C. Benezech were responsible for experimental methodology and analysis.References
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Contributor Information
Collaborators: Brad Astor, Lawence J Appel, Tom Greene, Teresa Chen, John Chalmers, Min Jun, Toshiaki Ohkuma, Mark Woodward, Adeera Levin, Ognjenka Djurdjev, Jesse Schold, Joseph Nally, Sankar Navaneethan, Susana Arrigain, Alex R. Chang, H. Lester Kirchner, Jamie Green, Kevin Ho, Brian J Lee, Varda Shalev, Cheli Melzer Cohen, Inbal Goldshtein, Gabriel Chodick, Jack Wetzels, Peter J Blankestijn, Arjan van Zuilen, Jan van de Brand, Mark Sarnak, Lesley Inker, Timothy Kenealy, C Raina Elley, Simon Moyes, John F Collins, Csaba Kovesdy, Praveen K Potukuchi, Keiichi Sumida, Miklos Z Molnar, Kamyar Kalantar-Zadeh, Dick de Zeeuw, Michelle J Pena, Hiddo JL Heerspink, Juan Jesus Carrero, Hong Xu, Marco Trevisan, David Naimark, Navdeep Tangri, Josef Coresh, Ron T Gansevoort, Morgan E. Grams, Stein Hallan, Csaba P Kovesdy, Andrew S Levey, Kunihiro Matsushita, Mark Woodward, Shoshana H Ballew, Jingsha Chen, Lucia Kwak, Yingying Sang, Aditya Surapeneni, Ron Gansevoort, Kai-Uwe Eckardt, Hrefna Gudmundsdottir, Adeera Levin, Romaldas Maciulaitis, Tom Manley, Vlado Perkovic, Kimberly Smith, Norman Stockbridge, Aliza Thompson, Thorsten Vetter, Kerry Willis, and Luxia Zhang
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020060806/-/DCSupplemental.
Supplemental Figure 1. Immunofluorescence and bulk RNA-seq analysis.
Supplemental Figure 2. Whole library analysis of single cell RNA-seq and cell proportions determined by flow cytometry.
Supplemental Figure 3. Analysis of myeloid transcriptomes.
Supplemental Figure 4. Flow cytometry gating strategy.
Supplemental Table 1. Antibodies utilized in immunohistochemistry/immunofluorescence studies.
Supplemental Table 2. Taqman probes used for qRT-PCR in studies.
Supplemental Table 3. Antibodies utilized in flow cytometry.
Supplemental Table 4. Summary of 10× sequencing statistics
Supplemental Table 5. Bulk RNA-seq matrix of differentially expressed genes in each gene cluster, expressed in FKPM.
Supplemental Table 6. Single cell RNA-seq table of differentially expressed genes in the global data as described in Figure 2, expressed per cluster.
Supplemental Table 7. Single cell RNA-seq table of differentially expressed genes in the myeloid only data as described in Figure 3, expressed per cluster.
References
- 1.Mills KT, Xu Y, Zhang W, Bundy JD, Chen CS, Kelly TN, et al.: A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int 88: 950–957, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Astor BC, Matsushita K, Gansevoort RT, van der Velde M, Woodward M, Levey AS, et al.: Chronic Kidney Disease Prognosis Consortium : Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int 79: 1331–1340, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen TK, Knicely DH, Grams ME: Chronic kidney disease diagnosis and management: A review. JAMA 322: 1294–1304, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY: Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 351: 1296–1305, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Hu B, Gadegbeku C, Lipkowitz MS, Rostand S, Lewis J, Wright JT, et al.: African-American Study of Kidney Disease and Hypertension Group : Kidney function can improve in patients with hypertensive CKD. J Am Soc Nephrol 23: 706–713, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS: Regression of microalbuminuria in type 1 diabetes. N Engl J Med 348: 2285–2293, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Ruggenenti P, Perticucci E, Cravedi P, Gambara V, Costantini M, Sharma SK, et al.: Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 19: 1213–1224, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariani LH, Martini S, Barisoni L, Canetta PA, Troost JP, Hodgin JB, et al.: Interstitial fibrosis scored on whole-slide digital imaging of kidney biopsies is a predictor of outcome in proteinuric glomerulopathies. Nephrol Dial Transplant 33: 310–318, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M: Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 339: 69–75, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Fioretto P, Sutherland DE, Najafian B, Mauer M: Remodeling of renal interstitial and tubular lesions in pancreas transplant recipients. Kidney Int 69: 907–912, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Duffield JS: Macrophages in kidney repair and regeneration. J Am Soc Nephrol 22: 199–201, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, et al.: Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol 167: 1207–1219, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al.: Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J: The renal mononuclear phagocytic system. J Am Soc Nephrol 23: 194–203, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ricardo SD, van Goor H, Eddy AA: Macrophage diversity in renal injury and repair. J Clin Invest 118: 3522–3530, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin SL, Castaño AP, Nowlin BT, Lupher ML Jr., Duffield JS: Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol 183: 6733–6743, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Montgomery TA, Xu L, Mason S, Chinnadurai A, Lee CG, Elias JA, et al.: Breast regression protein-39/chitinase 3-like 1 promotes renal fibrosis after kidney injury via activation of myofibroblasts. J Am Soc Nephrol 28: 3218–3226, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH: Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int 69: 73–80, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Xu L, Sharkey D, Cantley LG: Tubular GM-CSF promotes late MCP-1/CCR2-mediated fibrosis and inflammation after ischemia/reperfusion injury. J Am Soc Nephrol 30: 1825–1840, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin SL, Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, et al.: Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci U S A 107: 4194–4199, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Chang J, Yao B, Niu A, Kelly E, Breeggemann MC, et al.: Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells, and recovery in acute kidney injury. Kidney Int 88: 1274–1282, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, et al.: CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest 122: 4519–4532, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al.: Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A 109: E3186–E3195, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, et al.: Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol 30: 767–781, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, et al. : Spatiotemporal immune zonation of the human kidney. Science 365: 1461–1466, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.do Valle Duraes F, Lafont A, Beibel M, Martin K, Darribat K, Cuttat R, et al.: Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight 5: e130651, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al.: Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, et al.: Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20: 29–39, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al.: Single-cell immune landscape of human atherosclerotic plaques. Nat Med 25: 1576–1588, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al.: Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566: 388–392, 2019. [DOI] [PubMed] [Google Scholar]
- 31.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al.: Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575: 512–518, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hesketh EE, Vernon MA, Ding P, Clay S, Borthwick G, Conway B, et al. : A murine model of irreversible and reversible unilateral ureteric obstruction. J Vis Exp [94]: 52559, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cochrane AL, Kett MM, Samuel CS, Campanale NV, Anderson WP, Hume DA, et al.: Renal structural and functional repair in a mouse model of reversal of ureteral obstruction. J Am Soc Nephrol 16: 3623–3630, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Sasmono RT, Williams E: Generation and characterization of MacGreen mice, the Cfs1r-EGFP transgenic mice. Methods Mol Biol 844: 157–176, 2012. [DOI] [PubMed] [Google Scholar]
- 35.Rebo J, Mehdipour M, Gathwala R, Causey K, Liu Y, Conboy MJ, et al.: A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat Commun 7: 13363, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R: Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9: 171–181, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Kirschner K, Chandra T, Kiselev V, Flores-Santa Cruz D, Macaulay IC, Park HJ, et al.: Proliferation drives aging-related functional decline in a subpopulation of the hematopoietic stem cell compartment. Cell Rep 19: 1503–1511, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al.: STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, Marioni JC, et al.: Classification of low quality cells from single-cell RNA-seq data. Genome Biol 17: 29, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al. : Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu H, Kirita Y, Donnelly EL, Humphreys BD: Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30: 23–32, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Flanagan CH, Campbell KR, Zhang AW, Kabeer F, Lim JLP, Biele J, et al.: CRUK IMAXT Grand Challenge Team : Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol 20: 210, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al.: Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 8: e43882, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Sullivan ED, Mylonas KJ, Hughes J, Ferenbach DA: Complementary roles for single-nucleus and single-cell RNA sequencing in kidney disease research. J Am Soc Nephrol 30: 712–713, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ekiz HA, Conley CJ, Stephens WZ, O’Connell RM: CIPR: a web-based R/shiny app and R package to annotate cell clusters in single cell RNA sequencing experiments. BMC Bioinformatics 21: 191, 2020. 10.1186/s12859-020-3538-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lever JM, Hull TD, Boddu R, Pepin ME, Black LM, Adedoyin OO, et al.: Resident macrophages reprogram toward a developmental state after acute kidney injury. JCI Insight 4: 125503, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R: CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15: 1484–1506, 2020. [DOI] [PubMed] [Google Scholar]
- 49.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. : Comprehensive integration of single-cell data. Cell 177: 1888–1902.e21, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al.: Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19: 477, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, Buettner F: Destiny: Diffusion maps for large-scale single-cell data in R. Bioinformatics 32: 1241–1243, 2016. [DOI] [PubMed] [Google Scholar]
- 52.Scrucca L, Fop M, Murphy TB, Raftery AE: Mclust 5: Clustering, classification and density estimation using Gaussian finite mixture models. R J 8: 289–317, 2016. [PMC free article] [PubMed] [Google Scholar]
- 53.Conway BR, Betz B, Sheldrake TA, Manning JR, Dunbar DR, Dobyns A, et al. : Tight blood glycaemic and blood pressure control in experimental diabetic nephropathy reduces extracellular matrix production without regression of fibrosis. Nephrology (Carlton) 19: 802–813, 2014 [DOI] [PubMed] [Google Scholar]
- 54.Betz BB, Jenks SJ, Cronshaw AD, Lamont DJ, Cairns C, Manning JR, et al.: Urinary peptidomics in a rodent model of diabetic nephropathy highlights epidermal growth factor as a biomarker for renal deterioration in patients with type 2 diabetes. Kidney Int 89: 1125–1135, 2016. [DOI] [PubMed] [Google Scholar]
- 55.Ju W, Nair V, Smith S, Zhu L, Shedden K, Song PXK, et al.: ERCB, C-PROBE, NEPTUNE, and PKU-IgAN Consortium : Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med 7: 316ra193, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al.: Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 153: 362–375, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al.: The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol 12: 778–785, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. : Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317: 666–670, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Guo H, Cai CQ, Schroeder RA, Kuo PC: Osteopontin is a negative feedback regulator of nitric oxide synthesis in murine macrophages. J Immunol 166: 1079–1086, 2001. [DOI] [PubMed] [Google Scholar]
- 60.Ophascharoensuk V, Giachelli CM, Gordon K, Hughes J, Pichler R, Brown P, et al.: Obstructive uropathy in the mouse: Role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int 56: 571–580, 1999. [DOI] [PubMed] [Google Scholar]
- 61.Sobrevals L, Rodriguez C, Romero-Trevejo JL, Gondi G, Monreal I, Pañeda A, et al.: Insulin-like growth factor I gene transfer to cirrhotic liver induces fibrolysis and reduces fibrogenesis leading to cirrhosis reversion in rats. Hepatology 51: 912–921, 2010. [DOI] [PubMed] [Google Scholar]
- 62.Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al.: ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med 25: 496–506, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnold L, Perrin H, de Chanville CB, Saclier M, Hermand P, Poupel L, et al.: CX3CR1 deficiency promotes muscle repair and regeneration by enhancing macrophage ApoE production. Nat Commun 6: 8972, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, et al.: Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: Role of interleukin-6 and macrophage colony-stimulating factor. Blood 92: 4778–4791, 1998. [PubMed] [Google Scholar]
- 65.See P, Lum J, Chen J, Ginhoux F: A single-cell sequencing guide for immunologists. Front Immunol 9: 2425, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al.: Proteomics. Tissue-based map of the human proteome. Science 347: 1260419, 2015. [DOI] [PubMed] [Google Scholar]
- 67.Gordon S: Alternative activation of macrophages. Nat Rev Immunol 3: 23–35, 2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.