

Significance Statement
Synaptopodin is a podocyte actin-binding protein that regulates the cytoskeleton. Despite the cytoskeleton's critical role in podocytes and its alteration in nephrotic syndrome and FSGS, mutations in the synaptopodin gene SYNPO have not been reported in patients. Studies in Synpo mutant mice suggest that a truncated form of synaptopodin partially compensates for loss of the full-length protein. Experiments using newly generated Synpo mutant mice demonstrated that complete absence of synaptopodin did not cause a phenotype, but it increased susceptibility to Adriamycin nephropathy, an FSGS model. In cultured podocytes from the mice, absence of synaptopodin altered the cytoskeleton. These findings indicate that synaptopodin is dispensable for normal podocyte homeostasis but is protective in injured podocytes, suggesting the cytoskeleton as a target for therapeutic intervention in podocytopathies.
Keywords: glomerulus podocyte, cytoskeleton, actin, glomerular filtration barrier
Visual Abstract

Abstract
Background
Synaptopodin (Synpo) is an actin-associated protein in podocytes and dendritic spines. Many functions in regulating the actin cytoskeleton via RhoA and other pathways have been ascribed to Synpo, yet no pathogenic mutations in the SYNPO gene have been discovered in patients. Naturally occurring Synpo isoforms are known (Synpo-short and -long), and a novel truncated version (Synpo-T) is upregulated in podocytes from Synpo mutant mice. Synpo-T maintains some Synpo functions, which may prevent a podocyte phenotype from emerging in unchallenged mutant mice.
Methods
Novel mouse models were generated to further investigate the functions of Synpo. In one, CRISPR/Cas9 deleted most of the Synpo gene, preventing production of any detectable Synpo protein. Two other mutant strains made truncated versions of the protein. Adriamycin injections were used to challenge the mice, and Synpo functions were investigated in primary cultured podocytes.
Results
Mice that could not make detectable Synpo (Synpo−/−) did not develop any kidney abnormalities up to 12 months of age. However, Synpo−/− mice were more susceptible to Adriamycin nephropathy. In cultured primary podocytes from mutant mice, the absence of Synpo caused loss of stress fibers, increased the number and size of focal adhesions, and impaired cell migration. Furthermore, loss of Synpo led to decreased RhoA activity and increased Rac1 activation.
Conclusions
In contrast to previous findings, podocytes can function normally in vivo in the absence of any Synpo isoform. Synpo plays a protective role in the context of podocyte injury through its involvement in actin reorganization and focal adhesion dynamics.
Podocytes have a complex cellular organization consisting of a cell body, major/primary processes, and foot processes (FPs) that form an interdigitating pattern with the FPs of neighboring podocytes. FPs are connected to each other by the glomerular slit diaphragm, a specialized cell-cell junction that when impaired, results in the loss of plasma proteins into the urine. The unique function of podocytes, which is to establish and maintain the glomerular capillaries and the filtration barrier to plasma proteins, is largely dependent on their specialized cellular architecture. The FPs contain an actin-based noncontractile cytoskeleton1 that is linked to the glomerular basement membrane (GBM) through integrin-based adhesion complexes.2 FPs are further characterized by a cortical network of short, branched actin filaments and the presence of highly ordered, parallel actin filament bundles, which are thought to modulate the permeability of the filtration barrier through changes in FP morphology.3 Therefore, proteins regulating the podocyte actin cytoskeleton should be critical for the maintenance of glomerular permselectivity. Mutations affecting podocyte proteins, such as nephrin, podocin, CD2AP, and α-actinin-4, lead to rearrangement of the actin cytoskeleton, disruption of the filtration barrier, and subsequent kidney disease.4–7 Proteins regulating the plasticity of the podocyte actin cytoskeleton, such as Rho guanine dissociation inhibitor-α, podocalyxin, FAT1, Nck1, and Nck2, are also of critical importance for sustained function of the glomerular filtration barrier.8–12
Synaptopodin (Synpo) is a proline-rich actin-binding protein that is present in a small number of actin-rich cellular compartments, including the FPs of podocytes and neuronal dendritic spines.13 Synpo exists in three isoforms14 (Figure 1B): neuronal Synaptopodin-short (Synpo-S; 690 amino acids [a.a.]), renal Synaptopodin-long (Synpo-L; 901 a.a.), and a truncated version (Synpo-T; 181 a.a.) that was shown to be upregulated in podocytes cultured from previously reported Synpo mutant mice in which lacZ and neo replace the Synpo-S coding sequence in exon 2 (herein referred to as SynpolacZ mice).15 Synpo-T overlaps with most of the C terminus of Synpo-L (Figure 1B).14 The brains of SynpolacZ mutant mice lack a dendritic spine apparatus.15 In contrast, these mice show an overtly normal kidney phenotype but display impaired recovery from protamine sulfate–induced podocyte FP effacement; the lack of a podocyte phenotype without a challenge was proposed to be due to the partial rescue of Synpo-L function by Synpo-T.14 In the absence of all three Synpo isoforms, cultured podocytes lack stress fibers and show impaired cell migration.14,16
Figure 1.
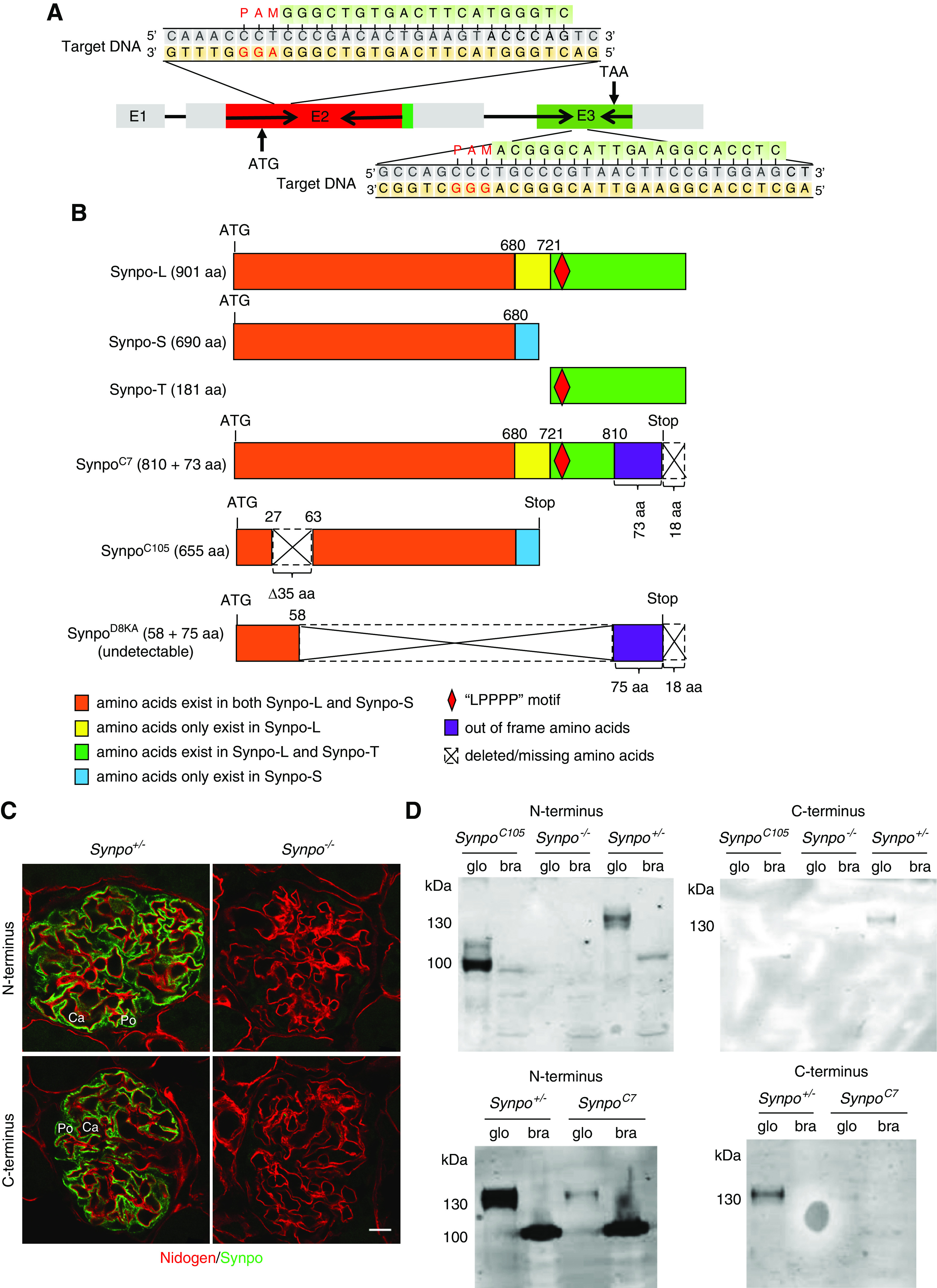
Novel mutations in mouse Synpo result in absence or truncations of the protein. (A) Schematic illustration of Synpo genomic structure. Exon 2 (E2) and E3, the only coding exons, were targeted. The clustered regulatory interspaced short palindromic repeats (CRISPR) target sites and protospacer adjacent motifs (PAMs) are indicated. (B) Schematics of the wild-type Synpo isoforms and mutant forms expressed in the mice utilized in this study. The orange boxes show the 680 a.a. shared by Synpo-S and Synpo-L. The yellow boxes show a.a. that exist in Synpo-L. The purple boxes show out-of-frame a.a. in SynpoC7 and SynpoD8KA. The red diamonds indicate the “LPPPP” motif. The blue boxes show the a.a. specific to Synpo-S. The dotted line frames show the a.a. stretches missing in the mutants. The SynpoD8KA allele produced no detectable protein and is considered null (Synpo−) in subsequent panels. (C) Synpo (green) was detected in podocytes of Synpo+/− but not Synpo−/− mice. Red indicates nidogen. Ca, capillary; Po, podocyte. Scale bar: 10 μm. (D) Expression of Synpo mutant proteins (or lack thereof) in glomeruli (glo) and brain (bra) was verified by western blotting with antibodies to the N and C termini of Synpo-L.
An important signaling system in all cells involves the Rho family of small GTPases that regulate the actin cytoskeleton.17 There are over 20 members of this family, but the best-studied members are the three most common and widely expressed: RhoA, Cdc42, and Rac1. In SynpolacZ podocytes, gene silencing of Synpo-T causes the loss of stress fibers and reduction of RhoA abundance and activity.14,16 Mechanistically, Synpo promotes stress fiber formation by blocking the c-Cbl–mediated ubiquitination and proteasomal degradation of Nck1,18 the Smurf1-mediated ubiquitination and proteasomal degradation of RhoA,16,19 and the Vav2-mediated activation of Rac1.20 In addition, Synpo can suppress filopodia by disrupting Cdc42:IRSp53:Mena signaling complexes.21
Although previous work, mostly performed in cultured immortalized podocytes, has demonstrated that Synpo regulates podocyte actin-cytoskeleton dynamics via Rho family GTPase signaling, the role of Synpo in vivo remains unclear. To further investigate the functions of Synpo in podocytes, we used a CRISPR/Cas9 approach to delete the majority of the Synpo gene and prevent production of any detectable Synpo-encoded protein. We found that the complete lack of Synpo caused no obvious phenotype under physiologic conditions, with no significant difference in the level of urinary protein between control and Synpo null mice. This is consistent with the lack of SYNPO mutations found in patients who are proteinuric, despite extensive exome sequencing in multiple cohorts. To determine whether Synpo has a role under pathologic conditions, we subjected mice to Adriamycin-induced nephropathy (AdrN) and found that Synpo deficiency accelerated the progression of AdrN and exacerbated the pathology. In vitro studies of primary podocytes revealed that the absence of Synpo caused loss of stress fibers, impaired cell migration, and changes in focal adhesion (FA) dynamics. Furthermore, loss of Synpo led to decreased RhoA activity and increased Rac1 activity. Collectively, our results indicate that Synpo is dispensable for the development and function of kidneys but is essential for protection of podocytes from an injury involving dramatic rearrangement and disruption of the actin cytoskeleton.
Methods
Mouse Models
Animal experiments conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Washington University Animal Studies Committee. To generate Synpo−/− total knockout mice via CRISPR/Cas9, two guide RNAs spaced approximately 8 kb apart (5′-CCCGACACTGAAGTACCCAG-3′; 5′-TGCCCGTAACTTCCGTGGAG-3′) targeting the only coding exons, exons 2 and 3 (Figure 1A), were microinjected into B6CBAF2/J zygotic pronuclei along with Cas9 mRNA. The resulting founders were analyzed by PCR using primers (Supplemental Figure 1) designed to amplify the 8-kb segment between the guide RNA target sites; these should only produce an amplicon if the 8 kb were deleted. In addition, next generation sequencing of amplicons generated using primer pairs flanking each target site (Supplemental Figure 1) was performed to identify smaller insertions and deletions. Dozens of different Synpo mutations were identified, including two independent deletions of the 8-kb segment. Three mutant alleles were analyzed in detail and are presented herein. F1 mice were generated by crossing founders to wild-type B6CBAF1/J mice, and these were intercrossed to generate F2 homozygotes and heterozygotes. The mice were maintained through subsequent generations by crossing homozygotes to heterozygotes, and heterozygotes were used as controls for comparisons with their homozygous littermates.
For AdrN studies, Synpo mutants were backcrossed to Balb/cJ for two generations. The resulting mice that were homozygous for the Prkdc mutation that confers susceptibility to AdrN22 were identified using a TaqMan assay designed by ThermoFisher. Prkdc−/− mice carrying Synpo alleles were used for AdrN studies. Mice weighing approximately 30 g at approximately 12 weeks of age were injected with Adriamycin (ADR) in 0.9% saline at 8 mg/kg into the retro-orbital sinus to induce nephropathy.
Antibodies
The following antibodies were used in immunofluorescence and western blot analyses. Guinea pig antibodies to the Synpo N terminus (03-GP94-N; recognizing Synpo-S and Synpo-L), Synpo C terminus (03-GP94-C; recognizing only Synpo-L and Synpo-T), and Synpo internal region (03-GP-94-IN; recognizing Synpo-S and Synpo-L) were from American Research Products. Guinea pig anti-mouse Synpo (163004; Synaptic Systems) recognizes Synpo-S and Synpo-L. Rat anti-Nidogen (clone ELM1) was from Millipore. Rabbit anti–α-actinin-44 was a gift from Alan Beggs (Children’s Hospital, Boston, MA). Mouse anti-human myosin IIA (clone 3C7) was from Abnova. Mouse antivinculin (clone VIN-11–5; V4505) was from Sigma. Rabbit anti–Wilms Tumor Protein [clone CAN-R9(IHC)-56–2; ab89901] was from Abcam.
Histology, Immunofluorescence Staining, and Quantification of Pathology
Mice were anesthetized by injection of ketamine/xylazine and then perfused with 20 ml of 4% paraformaldehyde. Paraffin sections of kidneys were stained with hematoxylin and eosin and periodic acid–Schiff (PAS). For transmission electron microscopy, kidneys were postfixed with 2% paraformaldehyde and 2% glutaraldehyde (buffered with 0.1 M sodium cacodylate buffer, pH 7.4). For scanning electron microscopy, isolated glomeruli were subjected to 2.5% glutaraldehyde and dehydration before mounting on stubs.
For immunofluorescence, fresh frozen kidney sections (4 μm) were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 for 3 minutes, and blocked for 30 minutes with 5% normal goat serum supplemented with 1% BSA in PBS. Sections were incubated with primary antibodies in blocking solution overnight at 4°C, washed with PBS, and incubated with secondary antibodies at 1:300 dilution in blocking buffer (AlexaFluor 488, 594 and AlexaFluor 647) for 1 hour at room temperature. Images were acquired with an Olympus FV1000 laser scanning confocal microscope and processed using National Institutes of Health ImageJ software.
For quantification of glomerulosclerosis, 50 full-sized glomeruli from each specimen were assessed after PAS staining by an observer blinded to genotypes. Glomeruli that exhibited adhesion of the capillary tuft to Bowman’s capsule, capillary obliteration, mesangial expansion, or fibrotic crescents were defined as sclerotic. Sclerotic glomeruli per section were assessed using a total number count as described previously.23 For quantitative ultrastructural analysis, FPs along at least 500 μm of total GBM length were quantified using ImageJ (http://rsbweb.nih.gov/ji/) on electron micrographs taken from at least four glomeruli of each animal. The total length of GBM in each micrograph was divided by the number of podocyte FPs adjacent to it in each image to determine the average length of podocyte FPs.
Isolation of Glomeruli and Culture of Primary Podocytes
Glomeruli were isolated as previously described.24 Briefly, anesthetized 2-month-old mice were perfused with 8×107 Dynabeads M-450 (Invitrogen, Carlsbad, CA). The kidneys were removed and minced on ice followed by digestion at 37°C with 1 mg/ml collagenase and 100 U/ml DNase I for 30 minutes and then, filtered twice with 100-μm Falcon cell strainers (BD Falcon, Bedford, MA). The tissue was pelleted by gentle centrifugation (200×g, 5 minutes), and glomeruli containing Dynabeads were collected using a magnetic particle concentrator (Dynal, Oslo, Norway) and washed in HBSS three times.
Isolated glomeruli were cultured on type I collagen–coated culture dishes (BD BIOCOAT, Bedford, MA) in 5% CO2 at 37°C in 47% DMEM/Ham’s F-12 (2:1), 46% 3T3-L1 cell-conditioned medium, 5% heat-inactivated FBS (Gibco), 1% Insulin-Transferrin-Selenium liquid media supplement (Gibco), and 100 U/ml penicillin-streptomycin (Gibco). After 4 days, colonies began sprouting around the glomeruli. These cells (passage 0 [P0]) showed an epithelial morphology with a polyhedral shape when confluence was reached. P1 and P2 podocytes were used for in vitro studies.
Immunofluorescence Staining of Primary Podocytes and Quantification of Actin Stress Fibers and Focal Adhesions
Primary podocytes seeded on collagen type I–coated coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in 1× PBS for 5 minutes, blocked with 2% BSA, and then incubated with the appropriate primary and secondary antibodies as described above. Images were acquired and processed as described above. Scoring of cellular structures was performed by an observer blinded to cell genotype.
For quantitative analysis of actin filament changes in primary podocytes, the various phalloidin staining patterns were grouped into four major classes and used for scoring as follows: type A: >90% of cell area filled with thick cables; type B: at least two thick cables running under the nucleus, with the rest of the cell area filled with fine cables; type C: no thick cables but some cables present; and type D: no cables visible in the central area of the cell.25 Stress fibers were also counted manually in each individual cell in independent images. A stress fiber was defined as a phalloidin-positive structure that was represented by a line spanning the length of the cell. “Broken” lines and “arcs” were not counted as stress fibers.
Quantification of FA numbers was performed by staining for vinculin. To obtain images with real size and number of FAs, a threshold was established. Quantification was performed with the “analyze particles” option. Results were expressed as FA sizes and total occupied area.
Urinalyses and BUN Assays
Urine samples were collected by manual restraint. Creatinine levels were quantified by a QuantiChrom Creatinine Assay Kit (DICT-500; BioAssay Systems, Hayward, CA), and albumin levels were determined with a QuantiChrom BCG Albumin Assay Kit (DIAG-250; BioAssay Systems) or an ELISA quantification kit (W90–13; Bethyl Laboratories, Montgomery, TX) according to the manufacturers’ specifications. Blood was collected by cheek puncture, and BUN was assayed with the QuantiChrom Urea Assay Kit (DIUR-100; BioAssay Systems).
Immunofluorescence Staining
Frozen kidney sections (4 μm) were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 for 3 minutes, and blocked for 30 minutes with 5% normal goat serum supplemented with 1% BSA in PBS. Sections were incubated overnight with primary antibodies in blocking solution at 4°C. Sections were then washed with PBS and incubated with secondary antibodies at 1:300 dilution in blocking buffer (AlexaFluor 488, 594 and AlexaFluor 647) for 1 hour.
GTPase Activation Assay and Western Blots
Cells were seeded at 5×104 cells per ml and grown for 4 days. Cells were scraped into ice-cold cell lysis buffer supplemented with protease inhibitor cocktail (Cytoskeleton). Lysates were clarified by centrifugation at 10,000×g for 1 minute at 4°C, and total protein content was measured by BCA assay (Pierce). Twenty-five micrograms of protein was resolved in 12% SDS gels and electrotransferred to polyvinylidene difluoride membranes. Membranes were probed with antibodies against Rac1 or RhoA (Cytoskeleton) or β-actin. For Rho GTPase and Rac1 GTPase activation assays, 300 μg of total protein from primary podocyte lysates was incubated with Rhotekin-RBD or PAK-PBD protein beads (Cytoskeleton) at 4°C on a rotator for 1 hour. The beads were pelleted and washed with washing buffer (Cytoskeleton). The bound proteins were eluted with 2× Laemmli sample buffer by boiling. The samples were electrophoresed and analyzed by western blot with anti-RhoA and anti-Rac1 antibodies.
Statistical Analyses
A two-tailed t test was used to compare two groups. One-way ANOVA with a post hoc Tukey test was used to compare multiple groups. P=0.05 was considered significant. Values are reported as mean ± SEM.
Results
Generation of Synpo−/− Null and Truncation Mutant Mice
We used CRISPR/Cas9 with two guide RNAs targeting near the 5′ and 3′ ends of the Synpo gene’s coding region for Synpo-L in exons 2 and 3 to attempt to either delete most of the gene or mutate each end to prevent protein production (Figure 1A). Many mutations were discovered in 80 founder mice, mostly small deletions and insertions that shifted the reading frame. We focused our studies on three alleles (Figure 1B). The SynpoC7 allele is a deletion of 7 bp at the target site in exon 3, causing a frameshift and premature termination of translation (p.Arg811Serfs*74) such that the 91 COOH-terminal a.a. of Synpo-L and Synpo-T should be replaced by 73 out-of-frame a.a. The SynpoC105 allele is an in-frame deletion of 105 bp from exon 2 linked to a 3′ inversion of 5701 bp that prevents the splicing out of intron 2, such that the intronic stop codon of Synpo-S is used. The resulting protein (p.Val28_Val62del) is Synpo-S with a deletion of 35 a.a. near the NH2 terminus. The SynpoD8KA allele has a deletion of 7880 bp between the guide RNA target sites. This results in a frameshift and possible production of a protein (p.Asp59_Ala810delinsValfs*75) containing 58 a.a. from the NH2 terminus linked to 16 out-of-frame a.a. at the COOH terminus.
The putative mutant proteins were investigated by immunostaining and western blotting using domain-specific antibodies. In control mice, Synpo was detected with both anti–N terminus and anti–C terminus (Synpo-L) antibodies in podocyte cell bodies and FPs (Figure 1C), but no protein was detected in homozygous SynpoD8KA mice, which are designated Synpo−/− (null) mice (Figure 1C). By western blotting (Figure 1D), Synpo-L bands were detected at approximately 130 kD in control heterozygous Synpo+/− glomeruli with both anti–N terminus and anti–C terminus antibodies. The calculated molecular mass of Synpo-L is 96 kD. The difference between calculated mass and the mol wt determined by SDS-PAGE is consistent with other reports showing a higher than expected mass14 and may be the consequence of post-translational modifications or the high content of proline, leading to a mobility shift. As expected (Figure 1B), the Synpo C terminus was absent in homozygous SynpoC7 and SynpoC105 glomeruli (Figure 1D). A band near the expected relative mass was detected in SynpoC105 glomeruli with the anti–N terminus antibody, and a band of the same size as the heterozygous control (Synpo+/−) but with reduced intensity was detected in SynpoC7 glomeruli (Figure 1D). In brain lysates Synpo-S was detected with the anti–N terminus antibody (Figure 1D). These results are consistent with the predictions (Figure 1B) on the basis of DNA sequence analysis.
The Absence of Phenotype in Synpo−/− Mice
There were no significant differences in body weight between Synpo−/− and control mice (Figure 2A), and urinalysis revealed no significant differences in albumin:creatinine ratio (ACR) (Figure 2B). Histologic analyses of mice up to 1 year of age revealed no significant renal pathology by either hematoxylin and eosin or PAS staining (Figure 2C). By transmission electron microscopy, podocytes of Synpo−/− appeared structurally normal, and FP width was not different from controls (Figure 2E). Scanning electron microscopy showed the surface contours of FPs to be smooth and tightly apposed to each other in Synpo−/− glomeruli, similar to controls (Figure 2D).
Figure 2.
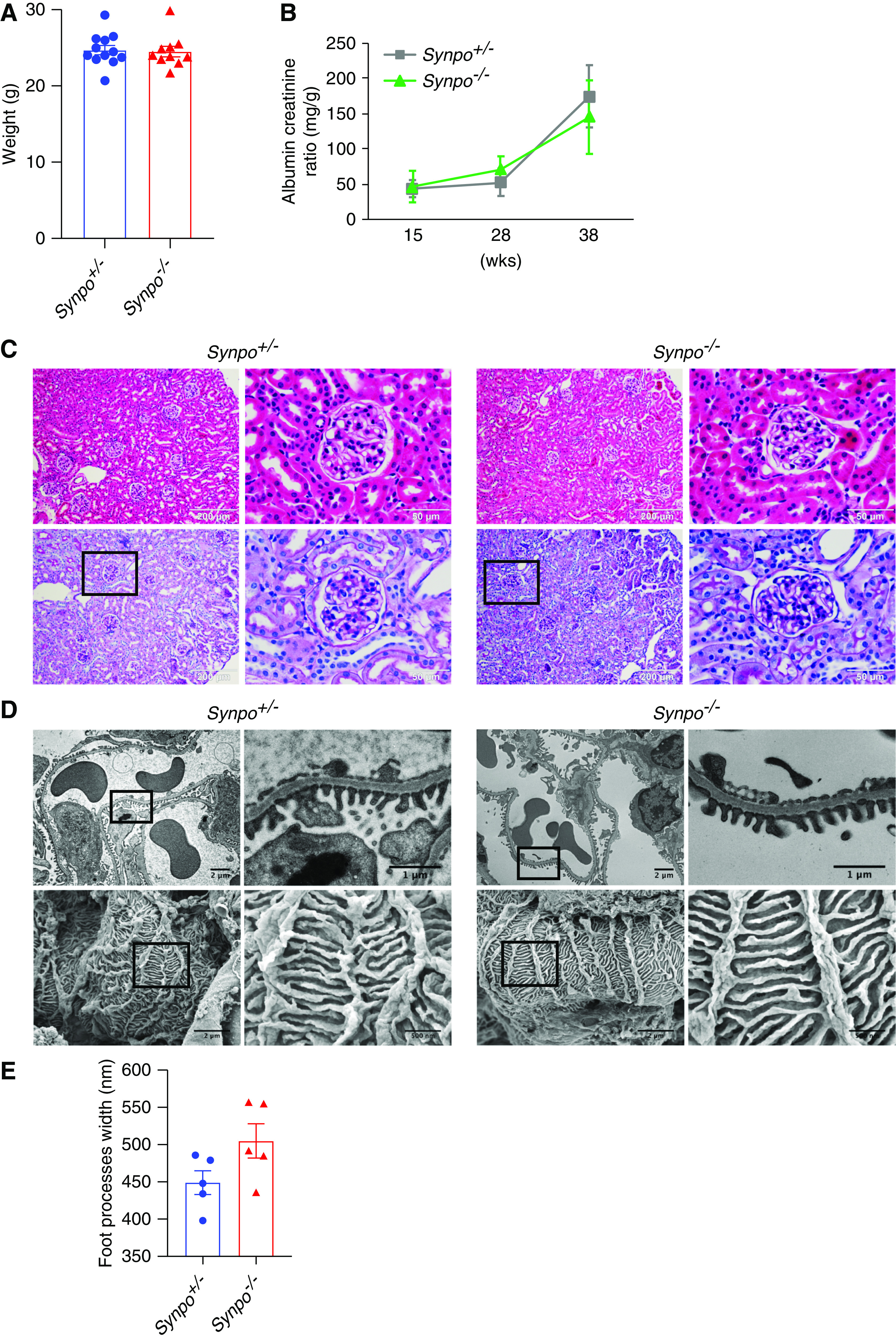
Synpo−/− mice exhbit no obvious defects in kidney or podocyte structure or function. (A) Quantification of body weights of Synpo+/− and Synpo−/− mice. n=10–12 mice. (B) Quantification of urinary ACRs at 15, 28, and 38 weeks of age. n=8 mice. (C) Hematoxylin and eosin and PAS staining of kidney sections shows normal glomeruli and tubules in both control and Synpo−/− mice at 36 weeks. (D) Electron microscopy (EM) reveals no FP effacement in control or Synpo−/− glomeruli. (Upper panel) Transmission EM. (Lower panel) Scanning EM. (E) Quantification of FP width in control and Synpo−/− mice at 36 weeks. n=5 mice. Data are presented as means ± SEM.
Previous in vitro studies demonstrated that Synpo-L localizes with and modulates the expression of α-actinin-4 in podocytes and elsewhere.26 To determine if the loss of Synpo affects the expression of α-actinin-4 in vivo, we immunostained mouse glomeruli for α-actinin-4. We found that α-actinin-4 localized in podocyte cell bodies and FPs in both Synpo+/− and Synpo−/− glomeruli (Supplemental Figure 2A). Western blot analysis showed no significant difference in α-actinin-4 expression level between Synpo−/− and control mice (Supplemental Figure 2, B and C).
Myosin IIA localizes to podocyte cell bodies and major processes and is responsible for the contractility of the cell, and podocyte injury in proteinuric diseases coincides with redistribution of myosin IIA toward the GBM.1 To study if the loss of Synpo affects the localization of myosin IIA, we immunostained for myosin IIA and observed that it localized in podocyte cell bodies and mesangial cells, and there were no differences in the localization and expression level between Synpo+/− and Synpo−/− glomeruli (Supplemental Figure 3). These data suggest that Synpo does not play a critical role in regulating glomerular structure or function in unchallenged mice.
Synpo Deficiency Exacerbates ADR-Induced Podocyte Injury
The absence of an obvious phenotype, reminiscent of the previous study of SynpolacZ mice expressing Synpo-T,14 prompted us to investigate the role of Synpo in the context of a podocyte injury in which the cytoskeleton is disrupted. We used an AdrN mouse model. We collected urine from AdrN-susceptible Pkrdc mutant22 Synpo+/− and Synpo−/− mice at 1, 3, 5, and 7 days after ADR injection. Coomassie blue staining of urine samples showed increased levels of albumin in Synpo−/− mice versus controls (Figure 3A). We also detected higher urinary ACRs in Synpo−/− mice on day 3 and day 7 (Figure 3B). Renal function was assayed by measuring BUN, which was higher in Synpo−/− mice on day 7 compared with Synpo+/− littermates (Figure 3C). Hematuria was observed in both groups on day 14 after injection by dipstick, but it was more severe in Synpo−/− mice (Figure 3D). These results demonstrate that Synpo deficiency exacerbates AdrN.
Figure 3.
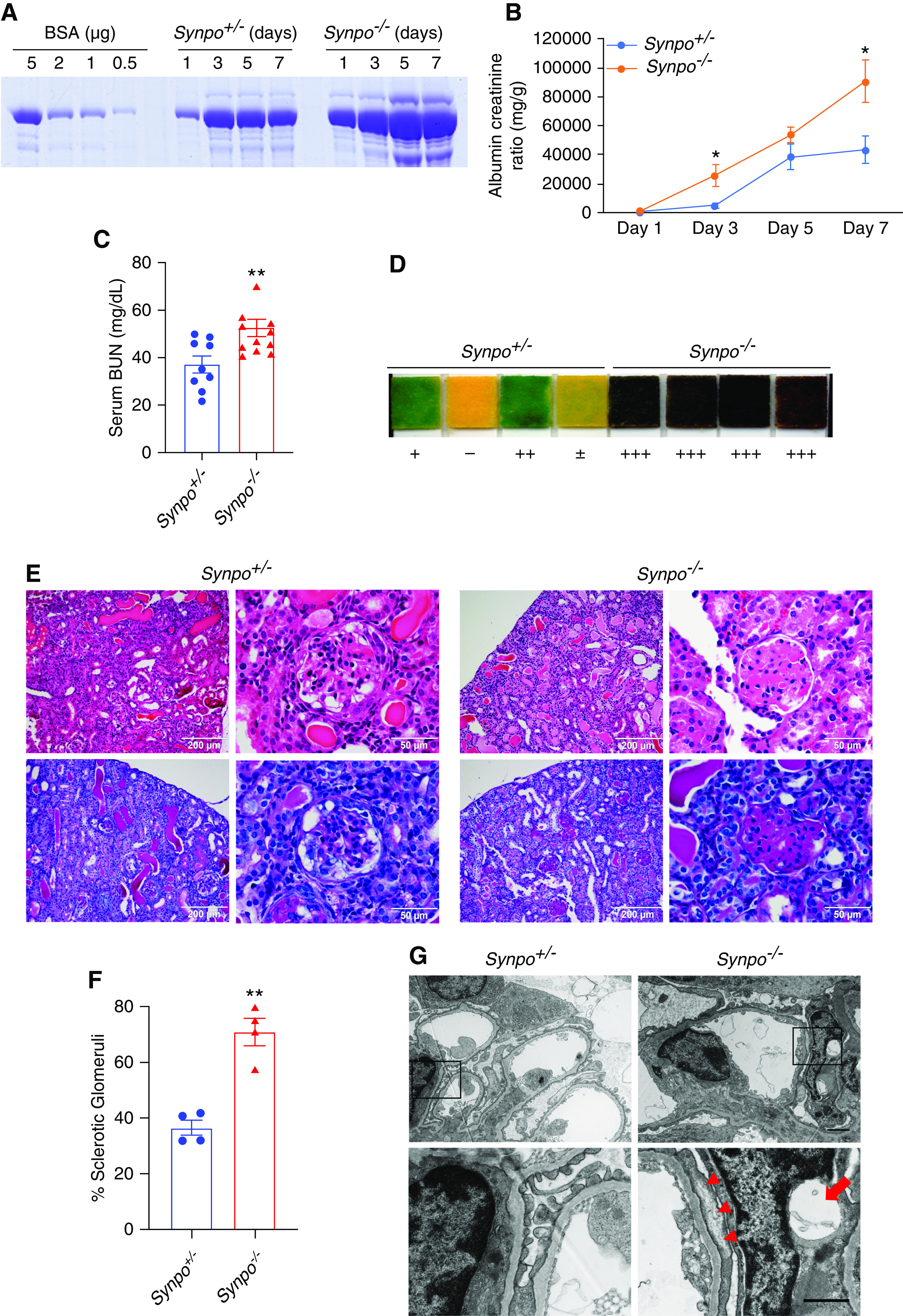
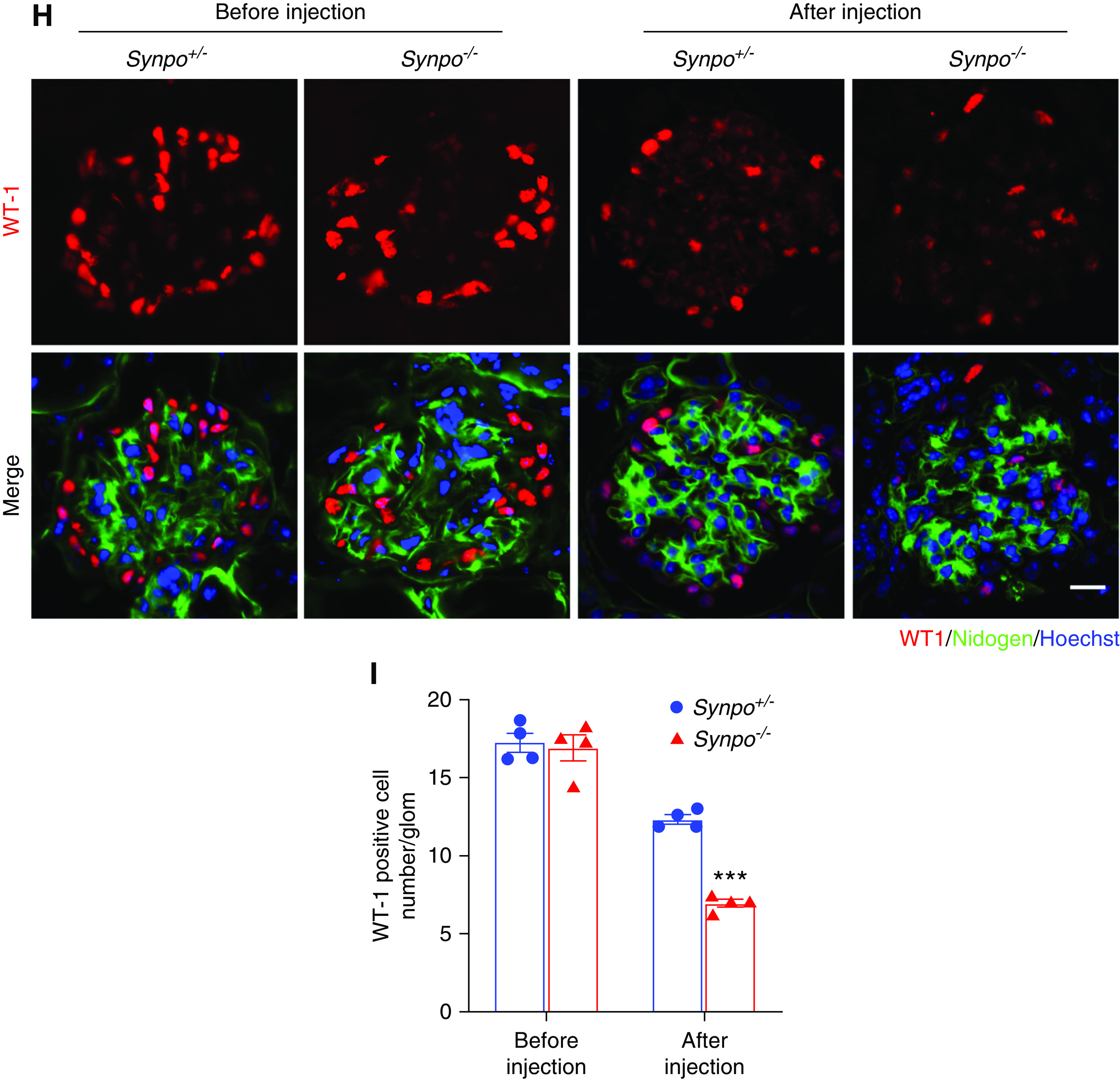
Synpo−/− mice treated with ADR exhibit increased proteinuria, hematuria, and glomerulosclerosis. (A) SDS-PAGE Coomassie blue staining of BSA standards and urine from Synpo+/− and Synpo−/− mice injected with ADR (8 mg/kg), demonstrating albuminuria at days 1, 3, 5, and 7 after injection; 1 μl of each urine was loaded. (B) Quantification of urinary ACRs at days 1, 3, 5, and 7 after ADR injection. n=7 mice per group. *P=0.05. (C) Elevated BUN in Synpo−/− mice at day 7 after ADR injection. n=9–11 mice per group. **P=0.01. (D) Hematuria dipstick assays 14 days after ADR injection. (E) Representative light microscopic images: (upper panel) hematoxylin and eosin and (lower panel) PAS of kidney sections. (F) Quantification of the percentage of sclerotic glomeruli. n=4 mice per group. **P=0.01. (G) Transmission electron micrographs illustrate more severe FP effacement in Synpo−/− mice 7 days after ADR injection. The arrowheads depict FP effacement, and the arrow depicts a podocyte vacuole. Scale bars: upper panel, 2 μm; lower panel, 1 μm. (H) Representative pictures of WT1-positive podocytes (red) before and 14 days after ADR injection. Sections were counterstained for nidogen (green) and DNA (blue). Scale bar: 10 μm. (I) Quantification of podocytes/glomerulus before and 14 days after ADR injection. The number of podocytes in Synpo−/− mice was significantly lower than that of Synpo+/− mice at day 14. Fifty glomeruli each in n=4 mice per group were analyzed. Data are presented as means ± SEM. ***P<0.001.
To further investigate the effect of Synpo loss on AdrN, we examined renal histology. On day 7, partial glomerulosclerosis and expansion of the mesangial area appeared in the ADR-injected Synpo−/− mice (data not shown). Synpo−/− mice had developed even more glomerulosclerosis by day 14 (Figure 3E), and the sclerotic glomeruli:total glomeruli ratio was significantly higher in Synpo−/− mice versus controls (Figure 3F). Electron microscopy showed that many podocytes in Synpo−/− kidneys were abnormal, and vacuole-like structures were observed in the cell body and major processes on day 7. In contrast, few podocytes in Synpo+/− kidneys showed vacuoles. On day 14, the ADR-treated Synpo−/− mice had developed a more severe podocyte injury as shown by GBM thickening and global FP broadening and effacement (Figure 3G). Podocytes are the target of ADR-induced injury, and reduced podocyte number contributes to the development of glomerulosclerosis and leads to progressive renal failure.27 To determine the number of resident podocytes, WT1-positive nuclei (Figure 3H) were counted. Before ADR injection, there were no significant differences in the number between Synpo+/− and Synpo−/− mice, but on day 14 after injection, the number of WT1-positive cells per glomerulus was significantly decreased in Synpo−/− mice versus Synpo+/− mice (Figure 3I).
The Loss of a Domain Containing Synpo’s “LPPPP” Motif Causes Severe ADR-Induced Podocyte Injury
In previous studies, it was shown that Synpo-L but not Synpo-S contains an LPPPP motif,14 which is a consensus motif for Ena/VASP binding (Figure 1B). This motif typically appears in proteins involved in actin cytoskeleton assembly and disassembly,28 processes that involve the Ena/VASP family of proteins.29 To investigate the functions of the truncated proteins in podocytes, we induced AdrN in susceptible (i.e., Pkrdc−/−) Synpo+/− (control), SynpoC7, SynpoC105, and Synpo−/− mice. The LPPPP motif is preserved in SynpoC7 mice but lost in SynpoC105 mice. After one single ADR injection, we collected urine at different time points. By day 8, Synpo−/− mice showed significantly elevated ACR compared with Synpo+/− mice. On days 12 and 15, both Synpo−/− and SynpoC105 mice showed significantly increased albuminuria compared with the control littermates (Figure 4A). However, SynpoC7 mice showed no significant differences compared with controls at any time point (Figure 4A). Histologic examination revealed more severe pathologies in SynpoC105 and Synpo−/− mice, including more tubular protein casts and sclerotic glomeruli (Figure 4, B and C). These results suggest that the LPPPP motif present in SynpoC7 mice but not in the other two mutants plays an essential role in actin dynamics that helps protect podocytes from the effects of AdrN.
Figure 4.

ADR causes increased proteinuria and glomerulosclerosis in SynpoC105 mice but not in SynpoC7 mice. (A) Bar graphs show urinary ACRs for Synpo+/−, SynpoC7, SynpoC105, and Synpo−/− mice at the indicated days after ADR injection. n=4–10 mice per group. *P=0.05. (B) Representative images of renal histology by PAS staining in Synpo+/−, SynpoC7, SynpoC105, and Synpo−/− mice 15 days after ADR injection. Scale bars: upper panel, 100 μm; lower panel, 10 μm. (C) Quantification as percentage of sclerotic glomeruli. n=4–5 mice per group. Data are presented as means ± SEM. **P=0.01; ***P<0.001.
Loss of Synpo Affects the Actin Cytoskeleton and Focal Adhesions in Primary Podocytes
To explore further the functions of Synpo, we investigated the effect of its loss on primary podocytes. Synpo interacts with and regulates the actin cytoskeleton,14,16 so we visualized the architecture of the actin cytoskeleton in primary podocytes isolated from Synpo+/− and Synpo−/− mice by staining with phalloidin. Synpo−/− podocytes showed a remarkable reduction in stress fiber formation (Figure 5, A and B; quantified in Figure 5C). To investigate whether Synpo plays an important role in the actin cytoskeleton in podocytes under pathologic conditions, we visualized stress fibers in ADR-treated podocytes. Before ADR treatment, the percentage of stress fiber–containing cells was lower but NS in Synpo−/− versus Synpo+/− podocytes (66.9±9.7 versus 44.2±5.7, respectively; P=0.07). After ADR treatment (0.2 μg/ml for 24 hours), the percentage of stress fiber–containing cells decreased by 38% in Synpo+/− podocytes and by 47% in Synpo−/− podocytes. ADR treatment aggravated the reduction of stress fiber formation in Synpo−/− podocytes (Figure 5, B–D).
Figure 5.
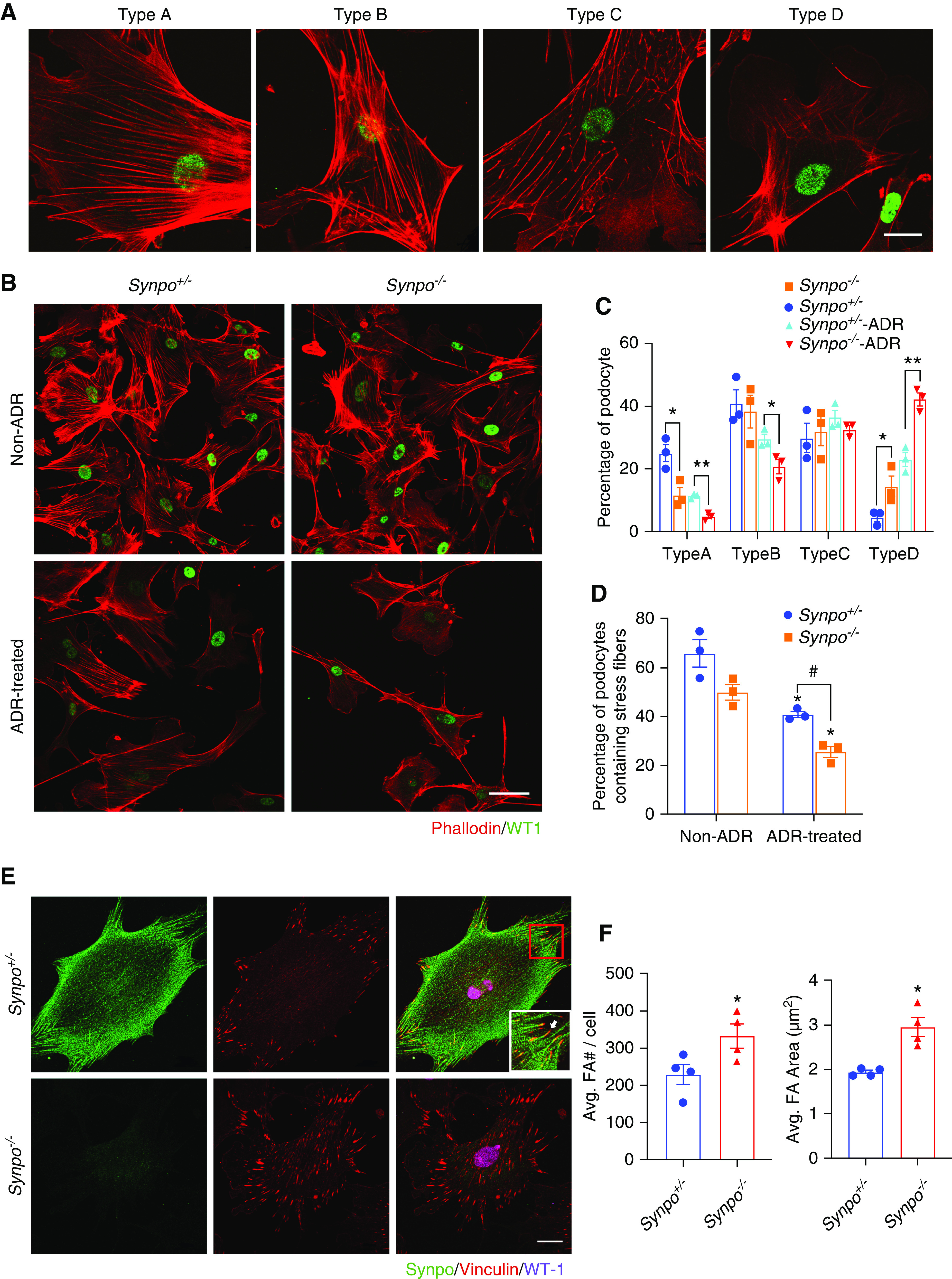
Synpo deficiency affects stress fibers and FA number and size. (A) Representative confocal images of the different phalloidin staining patterns observed in isolated Synpo+/− (control) podocytes. Type A: >90% of cell area filled with thick cables; type B: at least two thick cables running under the nucleus, with the rest of the cell area filled with fine cables; type C: no thick cables but some cables present; and type D: no cables visible in the central area of the cell. Scale bar: 20 μm. (B) Representative confocal images of podocytes stained with phalloidin and WT1 before and after ADR treatment. Scale bar: 50 μm. (C) Quantification of phalloidin staining without and with ADR treatment. Synpo−/− podocytes had fewer cells with the type A pattern and more with the type D pattern when compared with Synpo+/− podocytes at baseline. ADR-treated Synpo−/− podocytes had fewer cells with type A and type B staining patterns and more with the type D pattern. n=3 independent primary podocyte isolations per genotype; at least 60 cells from each group were analyzed. *P=0.05; **P=0.01. (D) Quantification of stress fiber–containing podocytes without and with ADR treatment for 24 hours. n=3 independent primary podocyte isolations per genotype; at least 60 cells from each group were analyzed. *P=0.05 versus non-ADR; #P=0.01 versus the relevant Synpo+/− group. (E) Representative images of podocytes stained for Synpo, vinculin, and WT1. Scale bar: 20 μm. Inset shows that the FA contains Synpo, but Synpo is absent from the very end (arrow). (F) Quantification of number and area of FAs in podocytes. Synpo−/− podocytes had more and larger FAs. n=4 independent primary podocyte isolations per genotype; 40 cells from each isolation were analyzed. Data are presented as means ± SEM. *P=0.05.
To gain further insight into the function of Synpo in podocytes, we investigated the localization of Synpo and the localization and morphology of FAs. We immunostained primary mouse podocytes for Synpo, the podocyte marker WT1, and the FA marker vinculin. Synpo was found in a punctate pattern on actin filaments in Synpo+/− podocytes but was absent in Synpo−/− cells (Figure 5E). Synpo was also detected in FAs (Figure 5E). The distribution of FAs was different in Synpo+/− versus Synpo−/− cells. In control, FAs were mainly distributed near the edges of the cells, but in Synpo−/− cells, FAs were located throughout the cells. FAs were also significantly larger and more abundant in Synpo−/− cells than in controls (Figure 5, E and F). To study how the changes in the size and localization of FAs relate to the reorganization of actin stress fibers, we stained primary podocytes with phalloidin and antivinculin. Synpo−/− podocytes showed less organized actin stress fibers, which were associated with increased number and size of FAs (Supplemental Figure 4).
Absence of Synpo’s “LPPPP” Motif and Mislocalization of α-Actinin-4
In previous studies, it was shown that Synpo-L interacts with α-actinin-4, colocalizes with it along stress fibers, and bundles and elongates α-actinin-4–induced actin filaments.14
To investigate the effects of Synpo mutations on the actin cytoskeleton bundling and the expression of α-actinin-4, we stained primary podocytes derived from Synpo+/−, SynpoC7, SynpoC105, and Synpo−/− mice with antibodies to the Synpo N terminus and to α-actinin-4. As expected, full-length Synpo in Synpo+/− cells was robustly associated with stress fibers (Figure 6A), similar to the previously described differentiated immortalized podocytes.14 The mutant protein encoded by SynpoC7 (Figure 1B) was also associated with stress fibers (Figure 6A). In contrast, the mutant Synpo encoded by SynpoC105 that lacks the LPPPP motif did not localize along most stress fibers (Figure 6A). Synpo colocalized with α-actinin-4 in SynpoC105 podocytes as well as in Synpo+/− and SynpoC7 cells, but the proteins localized diffusely and/or discontinuously along stress fibers or were absent from some thick and fine cables (Figure 6A). Podocytes isolated from SynpoC105 and Synpo−/− mice revealed a significant reduction in actin stress fiber formation (Figure 6B). Together, these observations suggest that the LPPPP motif missing in SynpoC105 is normally required for Synpo’s robust association with stress fibers and regulates α-actinin-4 bundling of stress fibers. Previous work has demonstrated that Synpo induces stress fibers by increasing the levels of active RhoA. Therefore, an alternative explanation could be that Synpo is required for stress fiber assembly via the regulation of RhoA activity and that the reduction in the number of stress fibers then leads to the reduced level of α-actinin-4 associated with stress fibers.
Figure 6.
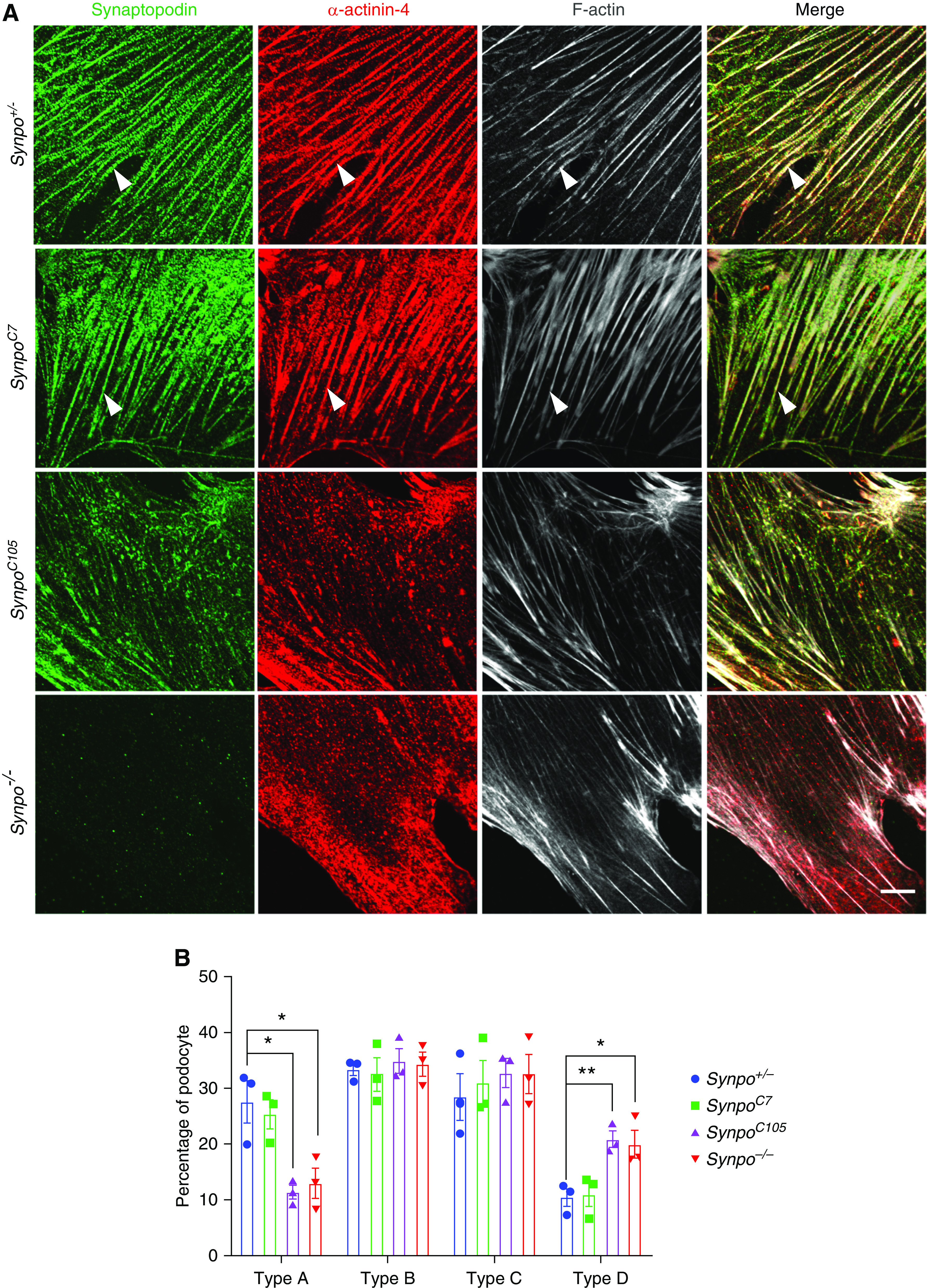
Synpo mutations induce mislocalization and decreased expression of α-actinin-4. (A) Representative confocal images of podocytes isolated from the indicated mice. Cells were stained for Synpo (green), α-actinin-4 (red), and phalloidin (gray). Arrowheads point to Synpo and α-actinin-4 colocalized in stress fibers in both Synpo+/− and SynpoC7 podocytes. In SynpoC105 podocytes, Synpo and α-actinin-4 were colocalized but appeared shortened, discontinuous, or absent in some thick or fine cables. Scale bar: 10 μm. (B) Quantification of phalloidin staining in podocytes isolated from Synpo+/−, SynpoC7, SynpoC105, and Synpo−/− mice. Synpo−/− and SynpoC105 podocytes had fewer cells with the type A pattern and more with the type D pattern when compared with Synpo+/− podocytes at baseline. n=3 independent primary podocyte isolations per genotype; at least 60 cells from each group were analyzed. *P=0.05; **P=0.01.
The Absence of Synpo Impairs Cell Motility
Our studies of Synpo-deficient podocytes revealed reduced actin stress fiber formation but increased number and size of FAs (Figure 5). These data suggest that the absence of Synpo impairs cell motility. To test this hypothesis, we analyzed migration of Synpo+/− and Synpo−/− podocytes. First, using a Transwell system in which a type I collagen–coated membrane filter separated the upper and lower compartments, we found Synpo−/− podocytes to be less motile than Synpo+/− podocytes (Figure 7, A and B). To investigate whether Synpo deficiency also impairs directed podocyte migration, we performed wound-healing assays. We created scratch wounds in cultures of subconfluent primary podocytes and examined the effect of Synpo on directional movement 8 and 24 hours postwounding. Synpo deficiency attenuated directional podocyte migration (Figure 7C).
Figure 7.
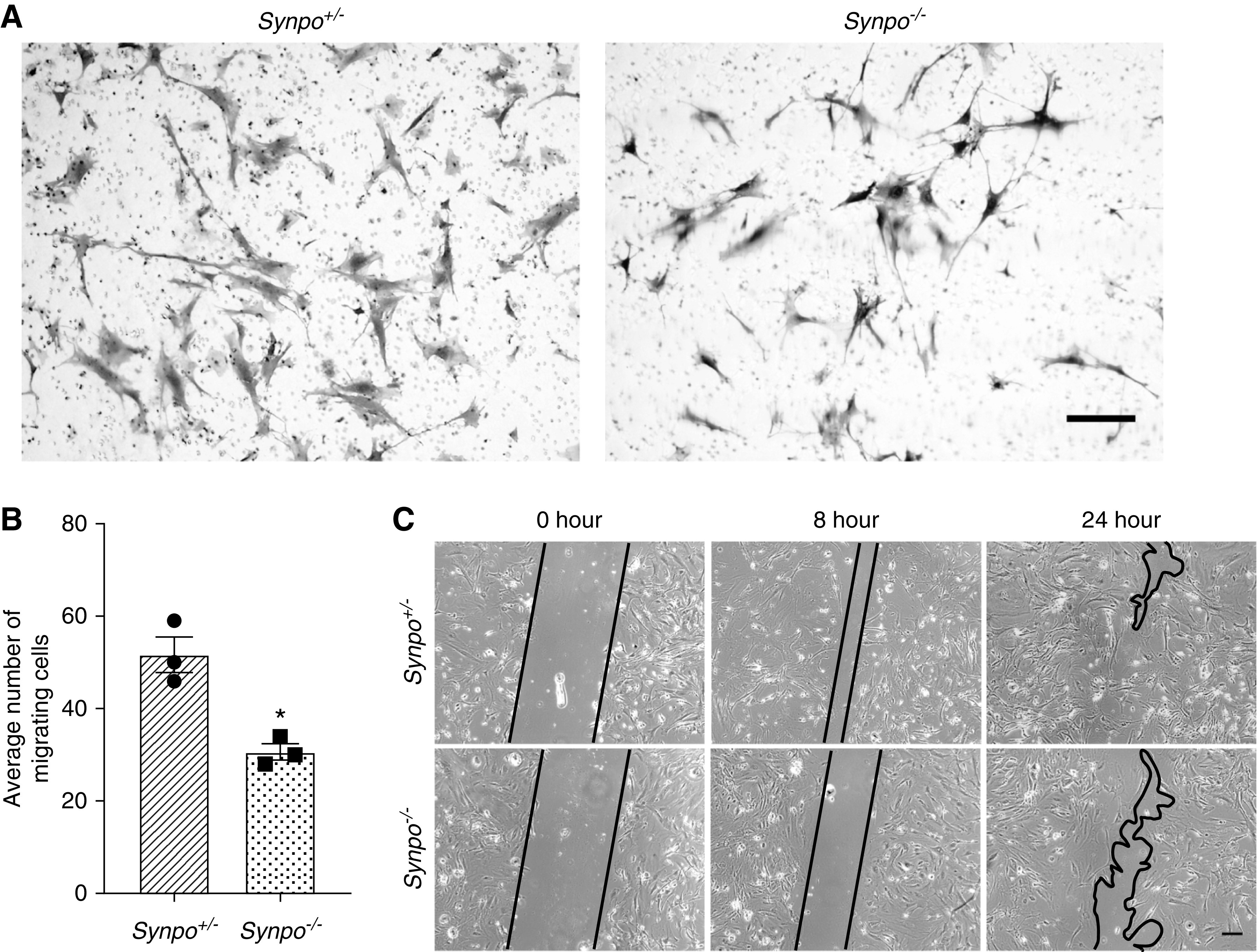
Synpo deficiency impairs the migration of primary podocytes. (A) Representative images of transwell migration assays. Scale bar: 10 μm. (B) Quantification of transwell assays shows Synpo−/− podocytes displayed reduced migration. n=3 independent primary podocyte isolations per group. Data are presented as means ± SEM. *P=0.01. (C) Scrape wound assays were performed to study the migration of podocytes over 24 hours. Images show different time points (0, 8, and 24 hours). The gap between the migrating areas is marked by black lines and revealed that Synpo−/− cells migrated more slowly into the gap than Synpo+/− cells. Scale bar: 50 μm.
Synpo Deficiency Decreases RhoA Activation and Increases Rac1 Activation
Previous studies have shown that gene silencing of Synpo causes the loss of stress fibers and reduction of RhoA abundance and activity.14,16 Mechanistically, Synpo promotes stress fiber formation by blocking the c-Cbl–mediated ubiquitination and proteasomal degradation of Nck118 and the Smurf1-mediated ubiquitination and proteasomal degradation of RhoA.16,19 It also has been shown that degradation of Synpo reduces its capacity to block the Vav2-mediated activation of Rac1, leading to increased reactive oxygen species (ROS) production and decreased RhoA activity.20 To investigate if Synpo deficiency affects the abundance and activity of GTPase family proteins in primary podocytes, we performed a pull-down assay for GTP-bound (active) RhoA and Rac1. We found that the absence of Synpo did not change the levels of total RhoA and Rac1 (Figure 8), but it decreased the levels of GTP-bound active RhoA (Figure 8A) and increased the levels of GTP-bound active Rac1 (Figure 8B). The level of Nck1 did not change in the absence of Synpo (Figure 8B). These results suggest that the absence of Synpo shifts the activation of Rho protein balance from RhoA toward Rac1, leading to the loss of stress fibers.
Figure 8.

Synpo−/− podocytes have reduced active RhoA and increased active Rac1. GTP-bound (active) and total RhoA and Rac1 levels were quantified and are presented as the ratio of RhoA-GTP to Rac1-GTP levels divided by the corresponding total RhoA to Rac1 levels relative to that of control cells. (A) RhoA pull downs from Synpo+/− and Synpo−/− podocytes show similar levels of total RhoA but decreased active RhoA in the mutant. (B) Rac1 pull downs and assay of Nck1 levels in Synpo+/− and Synpo−/− podocytes. Total Rac1 levels were similar in control and mutant podocytes, but active Rac1 was increased in the mutant. The level of Nck1 was unchanged. n=3–4 independent primary podocyte isolations per genotype. Data are presented as means ± SEM. *P=0.05; **P=0.01.
Discussion
In the previously reported studies of SynpolacZ mice, it was concluded that ectopic expression of Synpo-T (Figure 1B) prevented an overtly abnormal kidney phenotype.14 Here, we directly tested this by generating Synpo−/− null mice. We found that even the complete lack of detectable Synpo due to an approximately 8-kb gene deletion encompassing most of both coding exons caused no obvious phenotype in unchallenged mice, indicating that Synpo-T expression is likely irrelevant to the normal phenotype reported in SynpolacZ mice.14 This is consistent with the lack of any reports of SYNPO mutations causing kidney disease in patients who are proteinuric. Myopodin (Synpo2), a Synpo homolog, is expressed in skeletal muscle and cardiac muscle.30 The lack of distinct phenotypes in Synpo−/− mice is unlikely due to functional redundancy with myopodin because we did not observe expression of myopodin in Synpo−/− kidneys (Supplemental Figure 5).
It has been demonstrated that podocytes are relatively stationary under normal conditions.31 Yet, following podocyte injury, as in AdrN, there is podocyte movement and reorganization of the actin cytoskeleton.32 Synpo has been shown to regulate RhoA, Rac1, and Cdc42 signaling to stabilize the kidney filter by blocking the reorganization of the actin cytoskeleton into a promigratory configuration.33 Although our results show that Synpo is dispensable in normal stationary podocytes, it is likely involved in (1) attenuating the reorganization of the podocyte actin cytoskeleton when podocytes are injured and become migratory; or (2) promoting normalization of the cytoskeleton after injury to facilitate recovery.
Consistent with the finding that recovery from LPS-induced proteinuria was slowed in SynpolacZ mice,14 here we found that the lack of Synpo exacerbated AdrN, as significantly higher levels of proteinuria and hematuria and higher numbers of sclerotic glomeruli were detected in Synpo−/− mice versus controls. Moreover, cultured Synpo−/− primary podocytes showed decreased stress fiber formation, increased number and size of FAs, and impaired cell migration. In addition, ADR treatment in vitro aggravated the reduction of stress fiber formation in Synpo−/− podocytes. Taken together, Synpo is dispensable for development and maintenance of podocytes and for function of the glomerular filtration barrier; however, in injured podocytes and for those in culture, Synpo is protective via the regulation of actin cytoskeleton dynamics. These results raise the question of why Synpo deficiency results in obvious phenotypes in vitro but not in vivo.
Podocytes in culture behave very differently from those in vivo in many ways. One likely reason (of many) is that type I collagen–coated substrates (glass or plastic) upon which podocytes are typically grown are much stiffer than the GBM that podocytes adhere to in vivo. The consequences of stiffness differences have been studied in glomerular disease models in vivo34 as well as in vitro using Tg26 podocytes, which express much of the HIV genome and have a disordered cytoskeleton. These cells spread normally and develop normal stress fibers and FAs on 5-kPa gels, but they do not spread and show abnormal stress fibers and FAs on gels of reduced stiffness (1.5 kPa). This suggests that Tg26 podocytes exhibit abnormal responses to their mechanical environment.35 Of relevance here, Synpo−/− podocytes may have impaired responsiveness to substrate stiffness both in vivo and in vitro, and the high stiffness in vitro likely provides an injury-like state that allows manifestations of cytoskeletal defects (Figures 5 and 6).
Cell culture also imposes a state of oxidative stress on cells. Cell culture medium can catalyze the oxidation of added compounds, resulting in apparent cellular effects that are in fact due to oxidation products, such as ROS.36 It has been reported that loss of Synpo increases Vav2-mediated activation of Rac1, leading to increased ROS production and thereby, promoting RhoA inactivation.20 Thus, the absence of Synpo may make podocytes more sensitive to oxidative stress.
We demonstrated that the loss of Synpo results in mislocalization, increased number, and increased size of FAs in podocytes (Figure 5, E and F). FA mislocalization was associated with short disorganized stress fibers, suggesting a defect in FA turnover. FAs, by which cells are anchored to the matrix, are a major determinant of actin cytoskeleton dynamics and cell motility in vitro.37 We speculate that the FA defects we observed in vitro relate to the increased severity of AdrN in Synpo−/− mice. A mature FA contains hundreds of proteins that can be grouped on the basis of their contribution to four basic processes: receptor/matrix binding, linkage to the actin cytoskeleton, intracellular signal transduction, and polymerization information.38 Exactly how Synpo modulates FA composition and turnover remains to be characterized.
How might Synpo modulate the actin cytoskeleton reorganization? In previous studies, it was demonstrated that Synpo not only functions by physical linkage to actin filaments, but it also regulates actin dynamics via the Rho signaling pathway.14,16 Synpo alters the affinity or avidity of α-actinin-4 for actin, thereby inducing the formation of long, unbranched parallel bundles.14 Our results are consistent with these observations, as we found that Synpo deficiency induces decreased expression and mislocalization of α-actinin-4 in primary podocytes (Figure 6), resulting in more short branched filament–containing cells (Figure 5, A–D). Moreover, the absence of Synpo led to increased Rac1 activity and inactivation of RhoA (Figure 8). Degradation of Synpo was reported to abrogate its capacity to block the c-Cbl–induced proteasomal degradation of the RhoA activator Nck118 and the Smurf1-induced proteasomal degradation of RhoA.16 Our findings are inconsistent with these observations, as we observed no significant differences in the expression levels of Nck1 and total RhoA between Synpo+/− and Synpo−/− podocytes. Our data reveal that Synpo regulates podocyte actin dynamics by promoting α-actinin-4 binding to actin and regulating the balance of active RhoA and Rac1.
Synpo exists in three isoforms. In mouse kidneys, the C terminus of Synpo-L is identical to Synpo-T, which was reported to maintain some Synpo functions, thus preventing a major podocyte phenotype from emerging.14 However, which specific domains of Synpo-T are associated with Synpo’s functions in podocytes, such as binding to actin and regulating actin dynamics, is unclear. Here, we observed that the SynpoC105 mutant, which only makes Synpo-S with a 35-a.a. deletion near the NH2 terminus (Figure 1B), was more sensitive to ADR-induced injury. In contrast, the mutant encoded by SynpoC7, which is Synpo-L with an abnormal COOH terminus (Figure 1B), maintained some of Synpo’s protective functions during AdrN (Figure 4). Consistent with these observations, in vitro studies revealed that Synpo functions were maintained in SynpoC7 podocytes but not in SynpoC105 podocytes (Figure 6). Together, these results suggest that the critical a.a. reside either in the N terminus of Synpo-T and/or in the 35 a.a. deleted from near the NH2 terminus of SynpoC105. In support of the importance of the N terminus of Synpo-T, this region contains the LPPPP motif (Figure 1B), which is the binding site for the EVH1 domain of Ena/VASP proteins. The Ena/VASP family of proteins has a universal role in control of cell motility and actin dynamics. These proteins consist of an N-terminal EVH1 domain and a central proline-rich region, which acts as a ligand for the actin monomer binding protein profilin, as well as several SH3 domain-containing proteins, such as ABL, and a C-terminal EVH2 domain involved in oligomerization and F-actin binding.39 Therefore, we speculate that the LPPPP motif may play a critical role in the actin bundling activity of Synpo. To further elucidate if the LPPPP motif is important for Synpo functions, more finely targeted domain-specific mutations will be necessary.
In conclusion, our studies suggest that Synpo has a renoprotective effect that is dependent on its ability to maintain (or restore) the podocyte’s actin cytoskeleton after injury. However, why no SYNPO mutations have been discovered in patients with kidney disease remains a mystery. It is possible that, in contrast to the mouse, Synpo has a critical role in the human brain such that a mutation that increases susceptibility to kidney disease is not tolerated in neurons. If true, mutations that affect exon 3 (Synpo-L), which should not affect neurons, may not affect podocytes because the two α-actinin-4 binding domains in Synpo-S14 could suffice to allow Synpo to perform its role as a regulator of actin cytoskeleton dynamics.
Disclosures
J. Miner acknowledges consulting income from Janssen. All remaining authors have nothing to disclose.
Funding
This work was funded by National Institute of Diabetes and Digestive and Kidney Diseases grants R01DK058366 and R01DK078314. Production of mutant mice was supported in part by Diabetes Research Center Transgenic and Embryonic Stem Cell Core grant P30. J. Miner and L. Ning report grants from National Institute of Diabetes and Digestive and Kidney Diseases during the conduct of the study.
Supplementary Material
Acknowledgments
We thank Ms. Jennifer Richardson for genotyping mice and for preparing sections for electron microscopy and Dr. Ying Maggie Chen for advice on growing primary podocytes and for providing 3T3-L1 cells. We also thank the Transgenic Vectors Core for design of guide RNAs, the Mouse Genetics Core for producing mutant mice and for mouse husbandry, the Genome Engineering and iPSC Core for performing next generation sequencing, the Pulmonary Morphology Core for paraffin histology, and the Washington University Center for Cellular Imaging for scanning electron microscopy.
Jeffrey H. Miner and Liang Ning were responsible for the overall study design; Liang Ning and Hani Suleiman performed all experiments; Jeffrey H. Miner, Liang Ning, and Hani Suleiman interpreted the data; and Jeffrey H. Miner and Liang Ning wrote and edited the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020050572/-/DCSupplemental.
Supplemental Figure 1. Schematic illustration of Synpo genomic structure and indications of guide RNAs and primers used for genotyping.
Supplemental Figure 2. The absence of Synpo did not change the localization and expression level of α-actinin-4 in glomeruli.
Supplemental Figure 3. The absence of Synpo did not change the expression of myosin IIA in glomeruli.
Supplemental Figure 4. Increased number and size of FAs are associated with less organized, shortened, and discontinuous stress fibers.
Supplemental Figure 5. Synpo2/myopodin is not found in Synpo+/− or Synpo−/− kidneys.
References
- 1.Suleiman HY, Roth R, Jain S, Heuser JE, Shaw AS, Miner JH: Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. JCI Insight 2: e94137, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suh JH, Miner JH: The glomerular basement membrane as a barrier to albumin. Nat Rev Nephrol 9: 470–477, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greka A, Mundel P: Cell biology and pathology of podocytes. Annu Rev Physiol 74: 299–323, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al.: Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24: 251–256, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al.: Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al.: NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Shih N-Y, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, et al.: Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286: 312–315, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, et al.: Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene 18: 5373–5380, 1999. [DOI] [PubMed] [Google Scholar]
- 9.Schmieder S, Nagai M, Orlando RA, Takeda T, Farquhar MG: Podocalyxin activates RhoA and induces actin reorganization through NHERF1 and Ezrin in MDCK cells. J Am Soc Nephrol 15: 2289–2298, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Moeller MJ, Soofi A, Braun GS, Li X, Watzl C, Kriz W, et al.: Protocadherin FAT1 binds Ena/VASP proteins and is necessary for actin dynamics and cell polarization. EMBO J 23: 3769–3779, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al.: Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440: 818–823, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB: Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest 116: 1346–1359, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mundel P, Heid HW, Mundel TM, Krüger M, Reiser J, Kriz W: Synaptopodin: An actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol 139: 193–204, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, et al.: Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. J Clin Invest 115: 1188–1198, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deller T, Korte M, Chabanis S, Drakew A, Schwegler H, Stefani GG, et al.: Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc Natl Acad Sci U S A 100: 10494–10499, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P: Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 8: 485–491, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Ridley AJ, Hall A: The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70: 389–399, 1992. [DOI] [PubMed] [Google Scholar]
- 18.Buvall L, Rashmi P, Lopez-Rivera E, Andreeva S, Weins A, Wallentin H, et al.: Proteasomal degradation of Nck1 but not Nck2 regulates RhoA activation and actin dynamics. Nat Commun 4: 2863, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong JS, Iorns E, Rheault MN, Ward TM, Rashmi P, Weber U, et al.: Rescue of tropomyosin deficiency in Drosophila and human cancer cells by synaptopodin reveals a role of tropomyosin α in RhoA stabilization. EMBO J 31: 1028–1040, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buvall L, Wallentin H, Sieber J, Andreeva S, Choi HY, Mundel P, et al.: Synaptopodin is a coincidence detector of tyrosine versus serine/threonine phosphorylation for the modulation of Rho protein crosstalk in podocytes. J Am Soc Nephrol 28: 837–851, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagida-Asanuma E, Asanuma K, Kim K, Donnelly M, Young Choi H, Hyung Chang J, et al.: Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol 171: 415–427, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papeta N, Zheng Z, Schon EA, Brosel S, Altintas MM, Nasr SH, et al.: Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J Clin Invest 120: 4055–4064, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asanuma K, Akiba-Takagi M, Kodama F, Asao R, Nagai Y, Lydia A, et al.: Dendrin location in podocytes is associated with disease progression in animal and human glomerulopathy. Am J Nephrol 33: 537–549, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y, et al.: A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol 161: 799–805, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian X, Kim JJ, Monkley SM, Gotoh N, Nandez R, Soda K, et al.: Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J Clin Invest 124: 1098–1113, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kannan N, Tang VW: Synaptopodin couples epithelial contractility to α-actinin-4-dependent junction maturation. J Cell Biol 211: 407–434, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mundel P, Shankland SJ: Podocyte biology and response to injury. J Am Soc Nephrol 13: 3005–3015, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Renfranz PJ, Beckerle MC: Doing (F/L)PPPPs: EVH1 domains and their proline-rich partners in cell polarity and migration. Curr Opin Cell Biol 14: 88–103, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Lambrechts A, Van Troys M, Ampe C: The actin cytoskeleton in normal and pathological cell motility. Int J Biochem Cell Biol 36: 1890–1909, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Chalovich JM, Schroeter MM: Synaptopodin family of natively unfolded, actin binding proteins: Physical properties and potential biological functions. Biophys Rev 2: 181–189, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brähler S, Yu H, Suleiman H, Krishnan GM, Saunders BT, Kopp JB, et al.: Intravital and kidney slice imaging of podocyte membrane dynamics. J Am Soc Nephrol 27: 3285–3290, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian X, Ishibe S: Targeting the podocyte cytoskeleton: From pathogenesis to therapy in proteinuric kidney disease. Nephrol Dial Transplant 31: 1577–1583, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu SM, Nissaisorakarn P, Husain I, Jim B: Proteinuric kidney diseases: A podocyte’s slit diaphragm and cytoskeleton approach. Front Med (Lausanne) 5: 221, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wyss HM, Henderson JM, Byfield FJ, Bruggeman LA, Ding Y, Huang C, et al.: Biophysical properties of normal and diseased renal glomeruli. Am J Physiol Cell Physiol 300: C397–C405, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Embry AE, Liu Z, Henderson JM, Byfield FJ, Liu L, Yoon J, et al.: Similar biophysical abnormalities in glomeruli and podocytes from two distinct models. J Am Soc Nephrol 29: 1501–1512, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halliwell B: Oxidative stress in cell culture: An under-appreciated problem? FEBS Lett 540: 3–6, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Parsons JT, Horwitz AR, Schwartz MA: Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11: 633–643, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bershadsky AD, Balaban NQ, Geiger B: Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol 19: 677–695, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS: Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell 101: 703–715, 2000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.