

Significance Statement
The role of asymptomatic hyperuricemia in the progression of CKD has been unclear due to lack of animal models with clinically relevant uric acid (UA) levels. A new mouse model reveals that persistent asymptomatic hyperuricemia (approximately 15 mg/dl) does not cause CKD, nor accelerate progression, unless UA crystallizes in acidic tubular fluid. Crystallization initially causes tubular injury, inflammation, and interstitial fibrosis, and subsequently granulomatous interstitial nephritis with perilesional proinflammatory M1-like macrophage infiltrates. Modulating the M1-like macrophage phenotype, but not JAK/STAT inhibition, attenuates granulomatous nephritis.
Keywords: uric acid, granuloma, macrophages, inflammation, fibrosis, chronic kidney disease
Visual Abstract

Abstract
Background
The roles of asymptomatic hyperuricemia or uric acid (UA) crystals in CKD progression are unknown. Hypotheses to explain links between UA deposition and progression of CKD include that (1) asymptomatic hyperuricemia does not promote CKD progression unless UA crystallizes in the kidney; (2) UA crystal granulomas may form due to pre-existing CKD; and (3) proinflammatory granuloma-related M1-like macrophages may drive UA crystal-induced CKD progression.
Methods
MALDI-FTICR mass spectrometry, immunohistochemistry, 3D confocal microscopy, and flow cytometry were used to characterize a novel mouse model of hyperuricemia and chronic UA crystal nephropathy with granulomatous nephritis. Interventional studies probed the role of crystal-induced inflammation and macrophages in the pathology of progressive CKD.
Results
Asymptomatic hyperuricemia alone did not cause CKD or drive the progression of aristolochic acid I-induced CKD. Only hyperuricemia with UA crystalluria due to urinary acidification caused tubular obstruction, inflammation, and interstitial fibrosis. UA crystal granulomas surrounded by proinflammatory M1-like macrophages developed late in this process of chronic UA crystal nephropathy and contributed to the progression of pre-existing CKD. Suppressing M1-like macrophages with adenosine attenuated granulomatous nephritis and the progressive decline in GFR. In contrast, inhibiting the JAK/STAT inflammatory pathway with tofacitinib was not renoprotective.
Conclusions
Asymptomatic hyperuricemia does not affect CKD progression unless UA crystallizes in the kidney. UA crystal granulomas develop late in chronic UA crystal nephropathy and contribute to CKD progression because UA crystals trigger M1-like macrophage-related interstitial inflammation and fibrosis. Targeting proinflammatory macrophages, but not JAK/STAT signaling, can attenuate granulomatous interstitial nephritis.
CKD is a global public health problem with high morbidity and mortality.1 Because uric acid (UA) clearance declines as CKD progresses, CKD is usually accompanied with profound hyperuricemia (HU).2–5 HU is also associated with numerous comorbidities such as metabolic syndrome, hypertension, and cardiovascular disease, but causal relationships remain uncertain.6,7 Especially, whether asymptomatic HU contributes to the progression of CKD remains a subject of debate.8,9 So far, HU has a proven causative role only in diseases that involve UA in its crystalline form such as gouty arthritis, acute urate nephropathy, and urolithiasis.10–13 In the kidney, a low urine pH facilitates the precipitation of UA crystals, which occurs with certain diets or genetic syndromes, but not with CKD in general.14,15 In diagnostic kidney biopsy samples or autopsies medullary UA crystal deposits surrounded by granuloma-like macrophage infiltrates can occasionally be observed in CKD kidneys,10 which raises a chicken-and-egg dilemma on the causality among persistent HU, UA crystalluria, and CKD.16–20 Numerous small, single-center studies support the concept that asymptomatic HU contributes to CKD,21–25 and that UA crystal–induced kidney injury and –related infiltrates of proinflammatory M1-like macrophages are possible pathomechanisms in this context.26–28 However, several well-powered, multicenter, randomized, controlled trials did not find urate-lowering therapies to delay CKD progression29–33; hence the role of asymptomatic HU in CKD progression remains debated.34
Testing causality among asymptomatic HU, HU with UA crystalluria, and CKD progression requires a respective animal model with serum UA levels in a clinically relevant range, which so far does not exist. We hypothesized that (1) asymptomatic HU does not promote CKD progression unless UA crystallizes in the kidney; (2) UA crystal granulomas may form due to preexisting CKD; and (3) proinflammatory granuloma-related M1-like macrophages may drive UA crystal–induced CKD progression.
Methods
Animal Studies
All animal experiments were approved by the regional government authorities Regierung von Oberbayern (reference number: ROB-55.2–2532.Vet_02–15–189) or II LKE in Krakow (reference number: 70/2018, 257/2018) on the basis of the EU directive for the Protection of Animals Used for Scientific Purposes (2010/63/EU) and reported according to the ARRIVE guidelines.35 Mice were housed in groups of five in filter-top cages with enrichment and had free access to food and water ad libitum. Cages, nestlets, food, and water were sterilized by autoclaving before use.
Design of Animal Studies
Effect of HU on Aristolochic Acid I–Induced CKD Progression
Six-week-old Alb-creERT2;Glut9lox/lox mice and Glut9lox/lox control mice (kindly provided by Frédéric Preitner and Bernhard Thorens, University of Lausanne, Center for Integrative Genomics, Lausanne, Switzerland) were injected intraperitoneally with tamoxifen every alternate day for 1 week to deplete Glut9 expression in Alb-creERT2;Glut9lox/lox mice.36 Afterward, Alb-creERT2;Glut9lox/lox mice and Glut9lox/lox control mice were intraperitoneally injected with either aristolochic acid I (AAI; 5 mg/kg body wt in PBS) or vehicle (PBS) every second day for 2 weeks.37 All mice received a standard chow diet enriched with 25.6 g inosine per kg (Ssniff, Soest, Germany) for 42 days. Alb-creERT2;Glut9lox/lox mice developed HU and AAI-induced CKD (HU+AAI), Glut9lox/lox mice developed only AAI-induced CKD without HU (AAI), and Alb-creERT2;Glut9lox/lox mice with vehicle injection and chow diet with inosine developed HU without CKD (HU).
Model of Chronic UA Crystal Nephropathy
To induce chronic UA crystal nephropathy, Alb-creERT2;Glut9lox/lox mice and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen every alternate day for 1 week. Afterward, the mice were split into four groups: the first group of Alb-creERT2;Glut9lox/lox mice (HU+crystalluria [CU]) received an acidogenic diet with 58 kcal% fat and sucrose enriched with 25.6 g inosine per kg (Research Diets Inc., New Brunswick), the second group of Alb-creERT2;Glut9lox/lox mice received a standard chow diet with inosine (HU−CU), the third group of Glut9lox/lox mice was fed an acidogenic diet with inosine (healthy1), and the fourth group of Glut9lox/lox mice received a chow diet with inosine (healthy2) for 32 or 42 days. Tofacitinib (Selleckchem, Germany) was suspended in 0.5% methylcellulose/0.025% Tween 20 (Sigma-Aldrich, Germany) for in vivo studies. For intervention studies, HU+CU mice received daily vehicle (100 µl D-PBS) or tofacitinib (20 mg/kg in 100 µl D-PBS) for 2 weeks starting from day 14 by oral gavage. Mice with HU+CU were injected intraperitoneally with either adenosine (1 mg/ml in 200 µl D-PBS) (Sigma-Aldrich, Germany) or vehicle (200 µl D-PBS) every alternate day starting from day 14 until day 32. Group size calculation for all intervention studies was on the basis of GFR as a primary end point and quantitative assumptions obtained from our previous studies.38,39 We allocated mice by stratified randomization to the different treatments.
MALDI-FTICR Mass Spectrometry Imaging
Cryosections (10 µm) were prepared from frozen unfixed kidney tissue of healthy, HU−CU, and HU+CU mice using a cryostat (Leica Biosystems, Germany). The tissue sections were thaw mounted on Superfrost microscope glass slides (Thermo Scientific, Germany) and microscopically examined for birefringent crystalline deposits using polarized light. Before preparing for the MALDI–mass spectrometry (MS) measurement, we dried the slices in a vacuum desiccator at room temperature for at least 45 minutes.40 Then, for coating the tissue slides, a solution of α-cyano-4-hydroxycinnamic acid at 10 mg/ml in 1:1 acetonitrile/0.1% 2,2,2-trifluoroethanoic acid in water was applied using an iMatrixSpray sprayer.41 Four spraying cycles were applied at a flow rate of 5 ml/cm2. The tissue slides were analyzed on a 7 Tesla SolariX XR FTICR MS instrument with a MALDI source equipped with a Smartbeam-II Laser System (Bruker Bremen, Germany). Ions were detected in negative ionization mode over a mass range of 150–3000 m/z. Two hundred laser shots using sequential settings were applied per pixel at 2000 Hz, setting laser power to 20% at a lateral resolution of 30 μm. The signal for each pixel was normalized against the root mean square of all data points. Ion images were coregistered with an optical scan of the slice, which was generated before applying the matrix using an optical scanner (OpticLab H850; Plustek, Germany).
The MS images were generated and data were analyzed using FlexImaging 4.0, Data Analysis 4.3, and/or SCiLS Lab software (Bruker Bremen, Germany). The metabolite UA was identified by matching measured m/z values with the value calculated on the basis of its elemental formula (C5H4N4O3), within a mass window of 1 ppm (m/z 167.02103 D±1 ppm).40
Primary End Point of Interventional Studies
Transcutaneous Measurement of GFR
We measured GFR in conscious mice on days 0, 14, 32, and 42 (n=5–7 mice per group). Briefly, we anesthetized mice with isoflurane to mount a miniaturized imager device built from two light-emitting diodes, a photodiode, and a battery (MediBeacon Inc., Mannheim, Germany) onto the shaved neck of the animals.38,42 The background signal of the skin was recorded for 5 minutes, then mice received a single injection of FITC-sinistrin (iv, 150 mg/kg body wt) (MediBeacon Inc., Mannheim, Germany). Each mouse was kept in a single cage and the signal was recorded for 90 minutes. Data were analyzed using the imaging device MPD Studio software (MediBeacon Inc., Mannheim, Germany). GFR (µl/min per 100 g body wt) was calculated from the decrease of fluorescence intensity of FITC-sinistrin over time using the three-compartment model with linear correction (injection, plasma, interstitial compartment, and t1/2 of FITC-sinistrin), body weight of mouse, and an empiric conversion factor as per manufacturer’s protocol.42,43
Secondary End Points
Biochemical Parameters
Serum from mice was collected at various time points. For the assessment of HU and CKD, we measured serum BUN and creatinine (DiaSys, Holzheim, Germany), and serum UA (BioAssay Systems, Hayward), using commercially available kits and calculated the concentrations as per manufacturer’s protocol.
Assessment of Kidney Injury and Fibrosis
Upon euthanasia, we sampled the kidneys from mice. We used one kidney for flow cytometry analysis and the other was divided into three equal parts. One part was kept in RNA later solution at −80°C for RNA isolation and the second part fixed in 4% formaldehyde to be embedded in paraffin for histology analysis.39 The third part was fixed in 4% paraformaldehyde for 2 hours and transferred into 15% sucrose in D-PBS for an additional 2 hours at 4°C. Afterward, kidneys were kept in 30% sucrose in D-PBS overnight at 4°C and then stored at −80°C for histology. For assessment of kidney injury and fibrosis, we stained 2-μm-thick kidney sections with periodic acid–Schiff (PAS) reagent, as well as silver and Picro-sirius red stain, respectively. Renal immune cells and macrophages were identified on kidney sections by immunostaining using rat anti–mouse CD45 (BD Biosciences, Heidelberg, Germany) and rat anti–mouse F4/80 antibodies (Bio-Rad, Kidlington, UK).44 Quantification of immunostaining (% area) was done using ImageJ software. An observer blinded to the experimental condition performed all assessments.
Fluorescence Staining and Immunohistochemistry
Kidneys of mice with HU+CU were fixed in 4% paraformaldehyde for 2 hours and transferred into 15% sucrose in D-PBS for an additional 2 hours at 4°C. Afterward, kidneys were kept in 30% sucrose in D-PBS overnight at 4°C and then stored at −80°C for histology. Fluorescent staining for Phalloidin (AlexaFluor-488–conjugated, green; Life Technologies, Milan, Italy) overnight and DAPI (nuclei; Life Technologies, Milan, Italy) for 30 minutes was performed on 40-μm-thick kidney sections to visualize UA crystal granulomas via z stack using a Leica SP5 AOBS confocal microscope (Leica, Wetzlar, Germany) equipped with a Chameleon Ultra-II two-photon laser (Coherent, Milan, Italy). Kidney sections (10 μm) from mice with HU+CU were stained with Phalloidin (AlexaFluor-546–conjugated, red; Life Technologies, Milan, Italy) for 30 minutes followed by staining with either dolichos biflorus agglutinin (DBA; FITC-conjugated, green; Vector Laboratories, Burlingame) or lotus tetragonolobus agglutinin (LTA; FITC-conjugated, green; Vector Laboratories, Burlingame) for 30 minutes, followed by DAPI stain for an additional 30 minutes.45 Kidney sections were also stained with anti-α smooth muscle actin (αSMA) antibody (Sigma-Aldrich, Germany) or with the anti–Tamm–Horsfall protein (THP) antibody (Cederlane, Burlington, Canada) for 30 minutes. Secondary antibodies were AlexaFluor-488 goat anti-mouse IgG2a or AlexaFluor-488 goat anti-mouse IgG2b (both from Life Technologies, Milan, Italy), with an incubation time of 30 minutes before a 30-minute DAPI staining. Representative images were taken on a Leica SP5 AOBS confocal microscope (Leica, Wetzlar, Germany) equipped with a Chameleon Ultra-II two-photon laser, and the number of granulomas per kidney section counted and the size of granulomas (μm) determined using the Leica LAS X software.
To identify undissolved crystal deposits, frozen (cryo) unfixed kidney sections from HU+CU mice on days 3, 14, and 32 were stained with hematoxylin and eosin (H&E) and UA crystal deposits were visualized under polarized light.
Human kidney biopsy samples from patients with UA crystal deposits, interstitial fibrosis, and cell infiltration were provided from the Arkana Laboratories (Little Rock, AR). Biopsy sections were fixed in Michel’s fixative or in 10% neutral buffered formalin and stained with trichrome, PAS, or H&E using standard protocols. We also performed immunostaining of nephrectomy tissue from a 65-year-old patient with urothelial carcinoma and CKD (adjacent tissue showed interstitial nephritis and hypertensive nephropathy). The surgical specimen was freshly collected and immediately fixed in 10% neutral buffered formalin at the Department of Pathology, University Hospital, CHRU in Lille, France. Biopsy sections were stained with the following antibodies: mouse anti-human CD68 antibody (1:250, M0814; DAKO, Denmark), mouse anti–human HLA-DR antibody (1:400, M0775; DAKO, Denmark), and rabbit anti-human CD163 antibody (1:400, DB 045–1; Biotech, Slovakia), using standard protocols (automated immunohistochemistry [IHC] on BenchMark ULTA IHC/ISH system; Roche), and viewed under light microscopy.
RNA Preparation and Real-Time Quantitative PCR
The RNA extraction kit from Qiagen (Düsseldorf, Germany) was used to isolate total RNA from mouse kidneys (n=5–7 mice per group) following the manufacturer’s instructions. RNA quality was assessed using 1% agarose gels before being transcribed into cDNA using reverse transcription (Superscript II; Invitrogen, Carlsbad, CA). Real-time RT-PCR was performed using SYBRGreen PCR master mix and analyzed with a Light Cycler 480 (Roche, Mannheim, Germany). All gene expression values were normalized using 18s rRNA as a housekeeping gene.46 All murine primers used for amplification were purchased from Metabion (Martinsried, Germany) and are listed in Supplemental Table 2.
Flow Cytometry Analysis of Murine Samples
We sampled kidneys from mice and digested them with collagenase/DNAse1 for 40 minutes at 37°C.38 Digested tissue was passed through a 70-μm filter and washed with cold D-PBS. The single-cell suspensions were then washed with wash buffer (0.1% BSA, 0.01% sodium azide in D-PBS) and FcR blocked with anti-mouse CD16/32 (2.4G2) for 5 minutes. After blocking, cells were stained with the surface antibodies PE/Cy5 anti-mouse CD45, V450 anti-mouse CD11b, BV510 anti-mouse MHCII, PE/Cy7 anti-mouse Gr1, APC/Cy7 anti-mouse CD11c (all from BioLegend, Fell, Germany), APC anti-mouse F4/80 (BioRad, München, Germany), FITC anti-mouse CD206 (BD Biosciences, Germany), and PE anti-mouse Cx3CR1 (BD Biosciences, Heidelberg, Germany) for 30 minutes at 4°C in the dark.47 After incubation, cells were washed with wash buffer and reconstituted in 1 ml of fresh wash buffer. We performed flow cytometry analysis using the BD FACSCanto II (Becton Dickinson, New Jersey) and analyzed data with FlowJo 8.7 (Tree Star Inc., Ashland, OR). We used Invitrogen AccuCheck counting beads (PCB100; Thermo Fisher Scientific, Langenselbold, Germany) to determine the absolute number of cells per micrometer.
ELISA
Concentrations of IL-6 in the serum were measured using the mouse ELISA kit for IL-6 (Ray Biotech, Norcross) according to the manufacturer’s protocol. The absorbance was measured on a Multiskan EX reader (Thermo Electron Corporation, Germany).
Single-Cell RNA Sequencing and Data Processing of Kidney Cells from a Mouse with UA Crystal Granulomas
The kidney of a mouse with UA crystal granulomas (HU+CU) was snap frozen in liquid nitrogen and at a later timepoint nuclei were isolated using a bounce dip homogenizer as described previously.48 Briefly, nuclei were isolated with Nuclei EZ Lysis buffer (NUC-101; Sigma-Aldrich, Germany) supplemented with RNAse inhibitor. Frozen tissues were thawed and cut into small pieces (<1 mm), and homogenized using a Dounce homogenizer. The homogenate was filtered through a 40-μm cell strainer (PluriSelect) and centrifuged at 500 × g for 5 minutes at 4°C. The pellet was resuspended and washed with 4 ml of resuspension buffer (1× PBS, 2% BSA).
The nuclei were run on the Chromium platform (10x Genomics) using v3.0 Chemistry according to the manufacturer instructions. Sequencing was performed on an Illumina Novaseq platform using S1 flow cells (Erasmus MC, Rotterdam, The Netherlands). The resulting RNA-sequencing reads were aligned to the Mus musculus Ensembl release 93 top-level assembly with Cell Ranger version 3.0.2 (10x Genomics). The estimated number of cells was 12,080, mean reads per cell was 7775, and median genes per cell was 1066. Sequencing performance was assessed for the total number of aligned reads, cells, and the total number of genes per cell detected. We used Seurat version 3.049 and the R package to identify non–immune and immune cell clusters. We filtered out cells, which had a low number of expressed genes (<200), high number of mitochondrial genes (>0.1%), and high number of total read counts (>50,000), and performed differential gene analysis, and generated a gene heatmap of different immune cells including macrophage subsets. Clusters were visualized as UMAP.
Statistical Analyses
Statistical analyses of data were performed using GraphPad Prism 7 software. Data were normally distributed and were compared by t test to calculate significance between two groups, by one-way ANOVA with Tukey’s post-hoc test for three or more groups, or by two-way ANOVA with Bonferroni’s comparison post-hoc test carried out when using two parameters with multiple groups. All data are presented as mean±SD and P<0.05 was considered statistically significant. Group sizes are indicated in each corresponding figure legend.
Results
Asymptomatic HU Does Not Affect the Progression of Aristolochic Acid–Induced CKD
First, we tested the concept that persistent asymptomatic HU may cause and contribute to CKD progression. We fed Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice a chow diet enriched with inosine and induced a progressive form of CKD by exposing the mice to AAI, a known nephrotoxin causing noncrystalline CKD in humans and mice37 (Figure 1A). At baseline, serum UA levels ranged between 4 and 6 mg/dl in all three groups of mice, which increased in Alb-creERT2;Glut9lox/lox mice to 12–20 mg/dl on day 42 (HU, HU+AAI), whereas Glut9lox/lox mice remained at baseline UA levels (AAI) (Figure 1B). HU per se was not associated with any crystalluria or signs of gouty arthritis (not shown), and can therefore be defined as asymptomatic HU. Of note, the presence of asymptomatic HU did not affect the progression of AAI-induced nephropathy (HU+AAI), as indicated by reduced BUN levels, GFR decline, tubular atrophy, and interstitial fibrosis, compared with AAI mice (Figure 1, C–F). Thus, robust and persistent asymptomatic HU does not affect the progression of AAI-induced CKD in mice.
Figure 1.

Asymptomatic HU does not induce kidney injury or contribute to the progression of AAI-induced CKD. (A) Schematic of experimental set-up. Alb-creERT2;Glut9lox/lox mice and Glut9lox/lox mice were injected intraperitoneally with tamoxifen followed by injections of either AAI to induce CKD or vehicle (control). All three groups were placed on a standard chow diet with inosine for 42 days. (B–D) Serum UA (B) and BUN levels (C), and GFR (D), of HU only, AAI only, and HU+AAI mice on days 0 and 42 (n=5 mice per group). (E–Eʺ) PAS (magnification, ×100) stain illustrates tubular injury in AAI only (E′) and HU+AAI (Eʺ) mice, but not in HU only (E) mice on day 42 (n=5 mice per group). (F–Fʺ) Interstitial fibrosis illustrated on Picro-sirius red–stained kidney sections in AAI only (F′) and HU+AAI (Fʺ) mice, but not in HU only (F) mice on day 42 (n=5 mice per group). Magnification, ×200. Data are mean±SD. *P<0.05; ** and §P<0.01; ***P<0.001; NS, not significant by two-way ANOVA.
HU with UA Crystalluria Causes CKD
In a model of acute urate nephropathy, the authors showed that low-to-mild HU with UA crystalluria induces kidney dysfunction associated with tubular injury, inflammation, infiltration of macrophages, and fibrosis in Alb-creERT2;Glut9lox/lox mice.36,50 To investigate whether HU with UA crystalluria drives CKD, we placed Alb-creERT2;Glut9lox/lox or Glut9lox/lox control mice on either an acidogenic diet enriched with the urate precursor inosine or a standard chow diet with inosine for 32 days (Figure 2A). In both groups of Alb-creERT2;Glut9lox/lox mice fed an inosine-rich diet, the serum UA levels significantly increased compared with Glut9lox/lox mice (healthy1 and 2), indicating that these mice developed HU (Figure 2B). However, only Alb-creERT2;Glut9lox/lox mice on an acidogenic diet with inosine developed CKD (HU+CU), as indicated by a significant increase in serum BUN and creatinine levels as well as a decline in GFR, compared with Alb-creERT2;Glut9lox/lox mice on chow diet with inosine (HU−CU) and Glut9lox/lox mice on both inosine diets (healthy1, healthy2) that remained healthy without kidney impairment (Figure 2, C–E).
Figure 2.
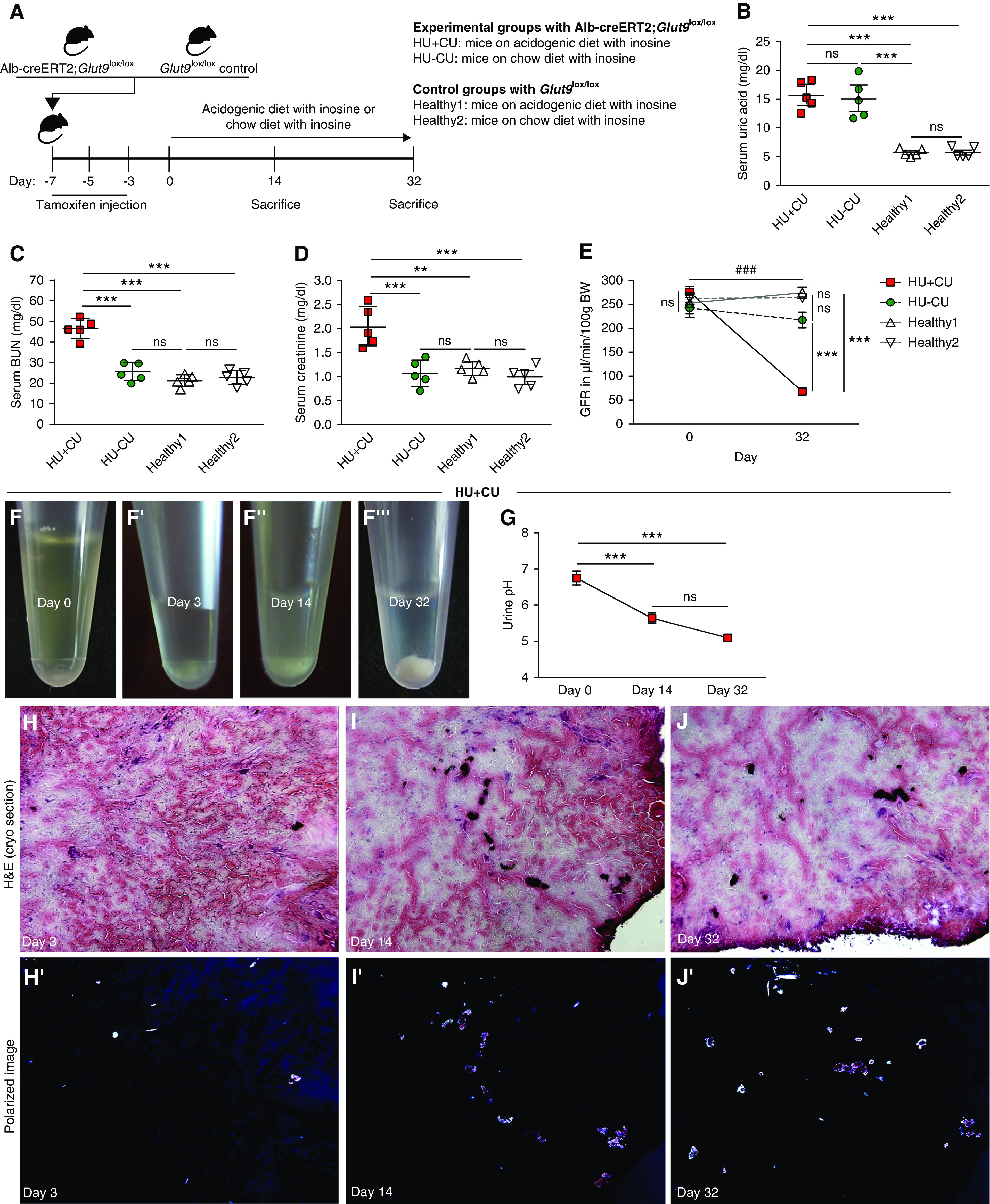
Renal crystal deposits lead to CKD in mice. (A) Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine up to 32 days. (B–D) Serum UA (B), BUN (C), and creatinine (D) levels of Alb-creERT2;Glut9lox/lox mice with acidogenic diet and inosine (HU+CU) or chow diet with inosine (HU−CU), and Glut9lox/lox mice with acidogenic diet with inosine (healthy2) or chow diet with inosine (healthy1) on day 32 (n=5 mice per group, using one-way ANOVA). (E) GFR of all four groups from day 0 to day 32 (n=5 mice per group, two-way ANOVA). (F) Urine was collected from HU+CU mice that were fed an acidogenic diet with inosine on days 0, 3, 14, and 32. Images illustrate UA crystals in urine on days 3, 14, and 32 (F’–F’’’). (G) Urinary pH was measured from HU+CU mice using a pH meter (n=5 mice per group, one-way ANOVA). (H–J) H&E staining of frozen unfixed kidney sections (H–J) and crystal deposits visualized under a polarizable light microscope from Alb-creERT2;Glut9lox/lox mice with acidogenic diet and inosine (HU+CU) on days 3, 14, and 32 (H’–J’). Data are mean±SD. **P<0.01; *** or ###P<0.001; NS, not significant.
Reports have shown that a low urine pH promotes the crystallization of soluble UA and the development of UA crystal nephropathy or UA urolithiasis.14,51,52 To investigate whether urinary crystallization is responsible for the observed kidney dysfunction, we checked the urine of Alb-creERT2;Glut9lox/lox mice that were fed an acidogenic diet with inosine (HU+CU) and found significant crystalluria (Figure 1F) as well as a drop in urinary pH (Figure 1G) from day 3 onwards. Crystal deposits were also observed on frozen unfixed kidney sections stained with H&E that showed birefringence under polarized light (Figure 2, H–J). This indicates that both inosine and the acidogenic diet were required to induce HU with crystalluria in Alb-creERT2;Glut9lox/lox mice, whereas an acidogenic diet or chow diet alone was not sufficient to induce HU or crystal-induced CKD (Supplemental Figure 1, A–D).
To identify the composition and location of such crystal deposits, we performed polarized light microscopy and identified birefringent crystal deposits in frozen unfixed kidney sections from HU+CU mice (Figure 3A), whereas crystals were absent in HU−CU and healthy mice (Figure 3, A’ and A’’). Crystal deposits predominantly localized to the inner medulla, but also in focal areas of the cortex. MALDI-MS imaging matched the molecular distribution of the analyte UA (blue) with the polarized image of the tissue section and UA colocalized with the detected crystal deposits (Figure 3, A and B). MALDI-MS imaging revealed that the main analyte was UA, as illustrated in blue, with a signal of m/z 167.02103 and a mass accuracy of ±1 ppm (Figure 3B). This corresponds to the deprotonated form of UA. To confirm that the urinary crystals from HU+CU mice were UA crystals, we added recombinant uricase and found that UA crystals completely dissolved after 5 minutes (Figure 3C), whereas calcium oxalate crystals remained unaffected (Supplemental Figure 1E). Body weight in HU+CU mice was unaffected after 32 days, whereas HU and healthy mice gained weight after starting the special diets (Figure 3D). Kidneys from HU+CU mice on day 32 were smaller in size and had a rough and pale cortex with white crystal deposits, and weighed more on day 32 (Figure 3, E’ and H) compared with kidneys on day 3 (Figure 3E) and from HU and healthy mice on day 32 (Figure 3, F–H).
Figure 3.
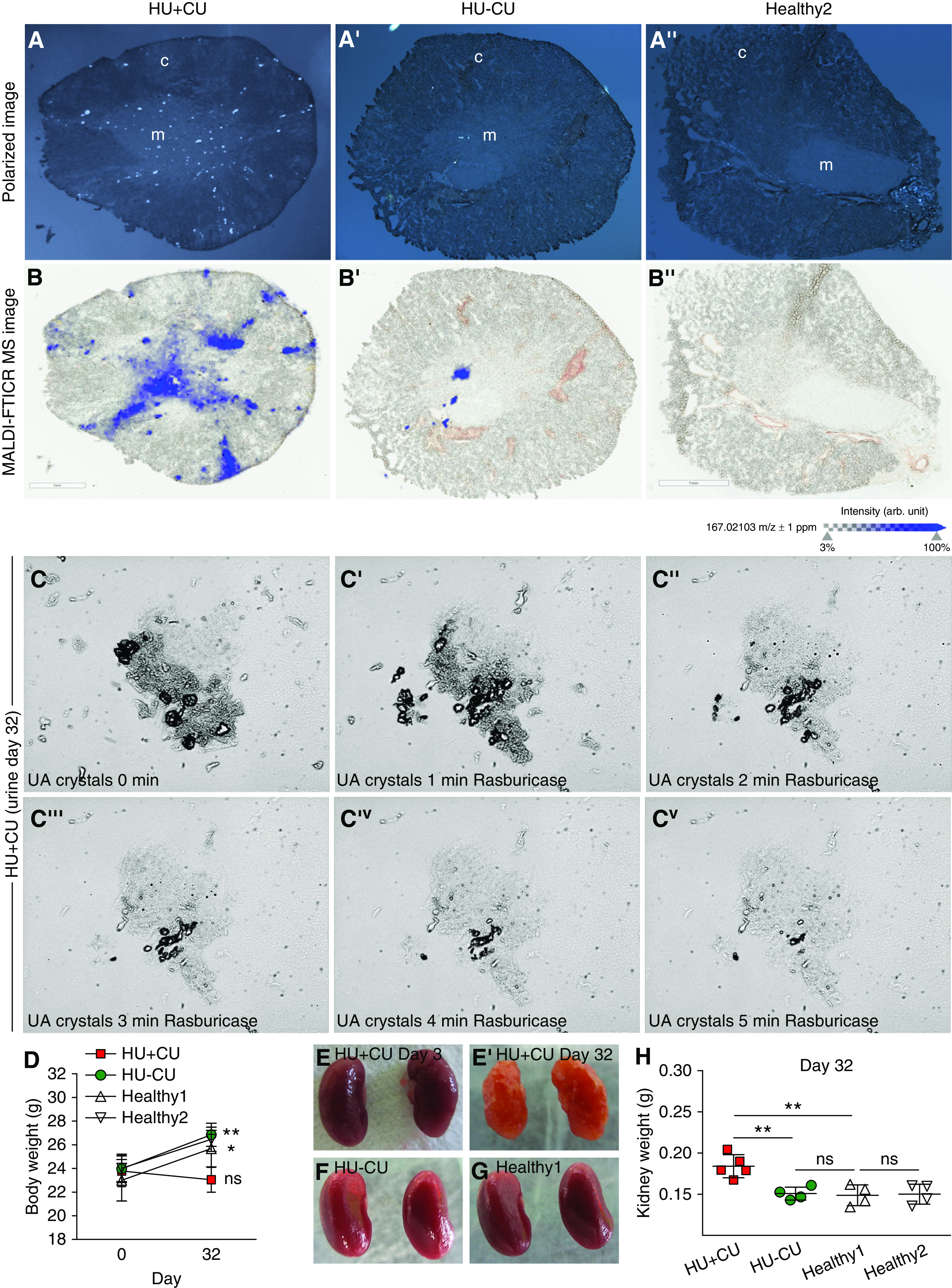
Medullary crystal deposits are composed of UA. Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine up to 32 days. (A) Polarized light microscopy images (before MALDI-MS imaging) visualizing birefringent crystal deposits in frozen unfixed kidney tissue sections from mice with HU+CU (A) and HU−CU (A’) and healthy mice (A’’). (B) Overlay of MALDI-MS images with an optical scan to visualize spatial distribution of UA (m/z 167.02103 D) in the same tissue sections of HU+CU mice as in (A) (B), but very much less in HU−CU mice (B’) and absent in healthy mice (B’’). c, cortex; m, medulla. (C) Urinary crystals were observed under the light microscope from HU+CU mice on day 32 (C). Rasburicase was added to dissolve UA crystals within 5 minutes (C’–Cv). Magnification, ×400. (D) Body weight from all four groups of mice on days 0 and 32 (n=5 mice per group, two-way ANOVA). (E–G) Macroscopic images of kidneys from HU+CU mice on day 3 (E) and day 32 (E’), and from HU−CU (F) or healthy mice (G). (H) Kidney weight of mice from all four groups on day 32 (n=4–5 mice per group, one-way ANOVA). Data are mean±SD. *P<0.05; **P<0.01; ***P<0.001; NS, not significant.
PAS stain revealed diffuse tubular injury and UA crystal granulomas only in mice with HU+CU (Figure 4, A–A’’’, black arrows) as well as an increase in the tubular injury marker KIM-1 (Figure 4B), but not in HU−CU and healthy controls, on day 32. Between five and 21 granulomas per kidney section were present (Figure 4C). All granulomas appeared predominantly in the medulla, with a central clear space of dissolved UA crystal clefts surrounded by multinucleated giant cells (Figure 4, D and E, Supplemental Movies 1 and 2). The size of these granulomas ranged from 50 to 150 µm; most granulomas measured between 70 and 110 µm (Figure 4F), with a positive correlation between total granuloma size and size of the crystal clefts (slope of 0.35, correlation coefficient of r2=0.55; Figure 4G). We did not find granulomas in the cortex so the crystal deposits noted on frozen unfixed kidney sections most likely relate to UA precipitations inside tubules being washed out during the fixation process (Figures 2, H–J and 3, A and B).
Figure 4.
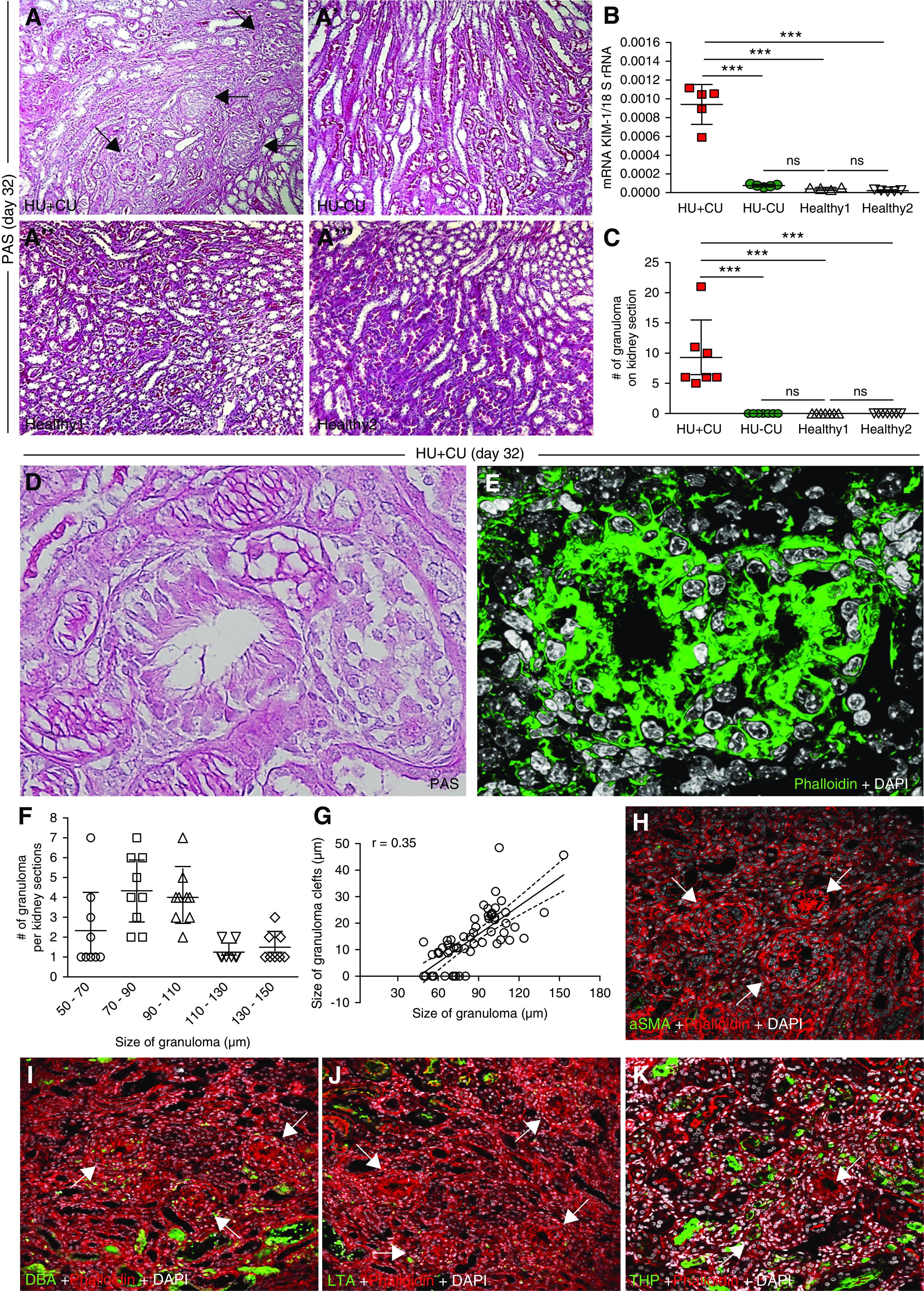
Renal UA crystal deposits associate with tubular injury, inflammation, and interstitial fibrosis in mice with chronic UA crystal nephropathy. Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine for 32 days. (A) PAS stain illustrates tubular injury and UA crystal granulomas (black arrows) in HU+CU mice (A), but not in HU−CU (A’) and healthy mice (A’’ and A’’’). Magnification, ×200. (B) Intrarenal mRNA expression levels of the tubular injury marker KIM-1 from all four groups of mice on day 32. (C) The number of granulomas counted on PAS-stained kidney sections from HU+CU mice. No granulomas were observed in HU−CU and healthy mice (n=7 mice per group, one-way ANOVA). (D) PAS stain illustrates UA crystal granulomas in HU+CU mice. Magnification, ×400. (E) Fluorescence microscopy of frozen unfixed kidney sections shows UA granulomas with centered clefts of dissolved UA crystals from mice with HU+CU as illustrated by Phalloidin (actin filaments, green) and DAPI (nuclei, white). Magnification, ×630. (F and G) Quantification of the size of UA granuloma (µm) per UA granuloma counted on kidney sections (F), and the linear correlation between the size of clefts (µm) versus the size of UA granuloma (µm) with r2=0.35 (G). (H) Fluorescence microscopy of UA granulomas (white arrows) and fibrosis from mice with HU+CU, as illustrated by αSMA (green), phalloidin (actin filaments, red), and DAPI (nuclei, white). Magnification, ×400. (I–K) Immunostaining of UA granulomas (white arrows) from mice with HU+CU as illustrated by DBA (I, distal tubules, green), LTA (J, proximal tubules, green), and THP (K, thick ascending limbs, green). Phalloidin was used as membrane stain (actin filaments, red) and DAPI to visualize nuclei (white). Magnification, ×400. Data are mean±SD. ***P<0.001; NS, not significant.
Next, we asked whether UA crystal granulomas were composed of myofibroblasts or tubular epithelial cells. We performed immunostaining of frozen fixed kidney sections from HU+CU mice and found that UA crystal granulomas were negative for αSMA, DBA, LTA, and THP (Figure 4, H–K, white arrows), ruling out a tubular epithelial cell composition.
Like UA crystal granulomas in kidney sections from HU+CU mice, medullary UA crystal deposits in human kidney biopsy samples appeared as empty spaces in the form of needle-shaped or feathery empty clefts representing dissolved UA crystals after formalin fixation and tissue processing (Supplemental Figure 2A, black arrows). UA crystal granulomas showed moderate-to-severe cellular inflammation, epithelioid mononuclear cell layers (Supplemental Figure 2A), foreign–body type giant cells (Supplemental Figure 2B, black arrow), and interstitial fibrosis (Supplemental Figure 2C). UA crystals that survived the fixation process showed birefringence under polarized light (Supplemental Figure 2D). Taken together, lowering urine pH turns our mouse model of robust and persistent asymptomatic HU into a model mimicking chronic UA crystal nephropathy characterized by granulomatous interstitial nephritis.
Crystalline Granulomas Form Late in the Course of Chronic UA Crystal Nephropathy
Whether crystalline UA granulomas develop before or after CKD and interstitial fibrosis is currently unknown. To investigate this, we studied the evolution of UA granulomas during a time course of CKD interstitial fibrosis. The intrarenal mRNA expression levels of Fibronectin 1, Collagen1α1, and fibroblast specific protein (Fsp)–1 significantly increased in mice with HU+CU compared with the other three groups on day 32 (Figure 5A). Picro-sirius red stain revealed that interstitial fibrosis was already present on day 14 and further increased until the end of the study in mice with HU+CU, as indicated by the percentage of Picro-sirius red area (Figure 5, B and C). This is consistent with a GFR decline over time (Figure 5D). When determining the association between GFR decline and extent of interstitial fibrosis, we found a linear relationship between the two variables with a slope of −2.26 and a correlation coefficient of r2=0.67 (Figure 5E). Despite a GFR decline of 50% (day 0 versus 14; Figure 5D) and a significant increase in fibrotic lesions (Figure 5C), we noticed that the number of UA crystal granulomas in HU+CU mice was very low on day 14 compared with day 32 (Figure 5F). Statistical analysis between the number of granulomas and the extent of fibrosis revealed no linear regression, but rather a plateau from day 14 to 32 (r2=0.54; Figure 5G). Thus, in this model of chronic UA crystal nephropathy, granulomas form late in the process of the disease, not before CKD and interstitial fibrosis are already established.
Figure 5.
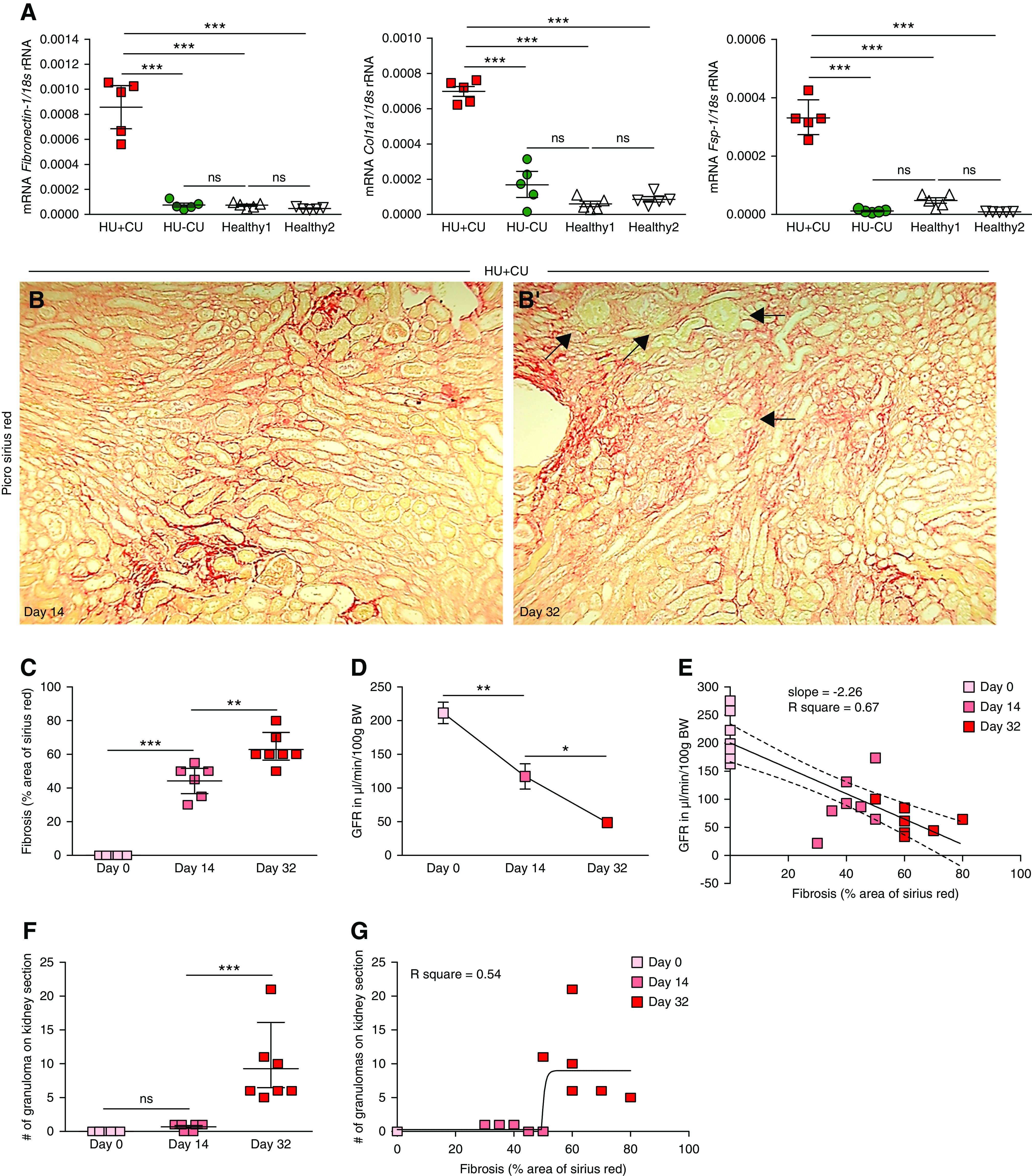
Renal UA crystal granulomas formed after interstitial fibrosis was established. Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine for 32 days. (A) Intrarenal mRNA expression levels of the fibrosis marker Fibronectin 1, collagen (Col)1α1, and Fsp-1 of all four groups of mice (n=5 mice per group, one-way ANOVA). (B) Picro-sirius red staining illustrating interstitial fibrosis in kidney sections of HU−CU mice on days 14 (B) and 32 (B’). Black arrows point at UA granulomas. Magnification, ×200. (C) Quantification of Picro-sirius red–stained kidney sections from HU+CU mice (n=5–7 mice per group, one-way ANOVA). (D) GFR of HU+CU mice on days 0, 14, and 32 (n=5–7 mice per group, one-way ANOVA). (E) Linear regression analysis between GFR and fibrosis of mice with HU+CU over time (r2=0.67). (F) Number of UA granulomas counted on PAS-stained kidney sections from HU+CU mice on days 0, 14, and 32 (n=5–7 mice per group, one-way ANOVA). (G) Illustration of a nonlinear regression with curve fit between the number of granulomas per kidney section and the extent of interstitial fibrosis (r2=0.54). Data are mean±SD. *P<0.05; **P<0.01; ***P<0.001; NS, not significant.
Macrophage Phenotypes in Chronic UA Crystal Nephropathy with Granulomatous Interstitial Nephritis
Next, we performed single-cell RNA sequencing of kidneys from HU+CU mice on day 32. Known cell type–specific markers,53–56 identified cell clusters of usual parenchymal kidney cells (Figure 6, A and B), as well as leukocyte clusters were all positive for Ptprc (Cd45), as expected (Figure 6C). In addition, we used common genes from the literature to identify inflammatory signaling pathways such as JAK/STAT (Figure 6D), and different immune cell subsets57,58 including proinflammatory M1-like (Figure 6E) and alternatively activated M2-like macrophages (Figure 6F), neutrophils (Figure 6G), and conventional dendritic cell (cDC) subsets (Figure 6, H and I). These data imply a chronic inflammatory response during chronic UA crystal nephropathy associated with the infiltration of immune cells.
Figure 6.
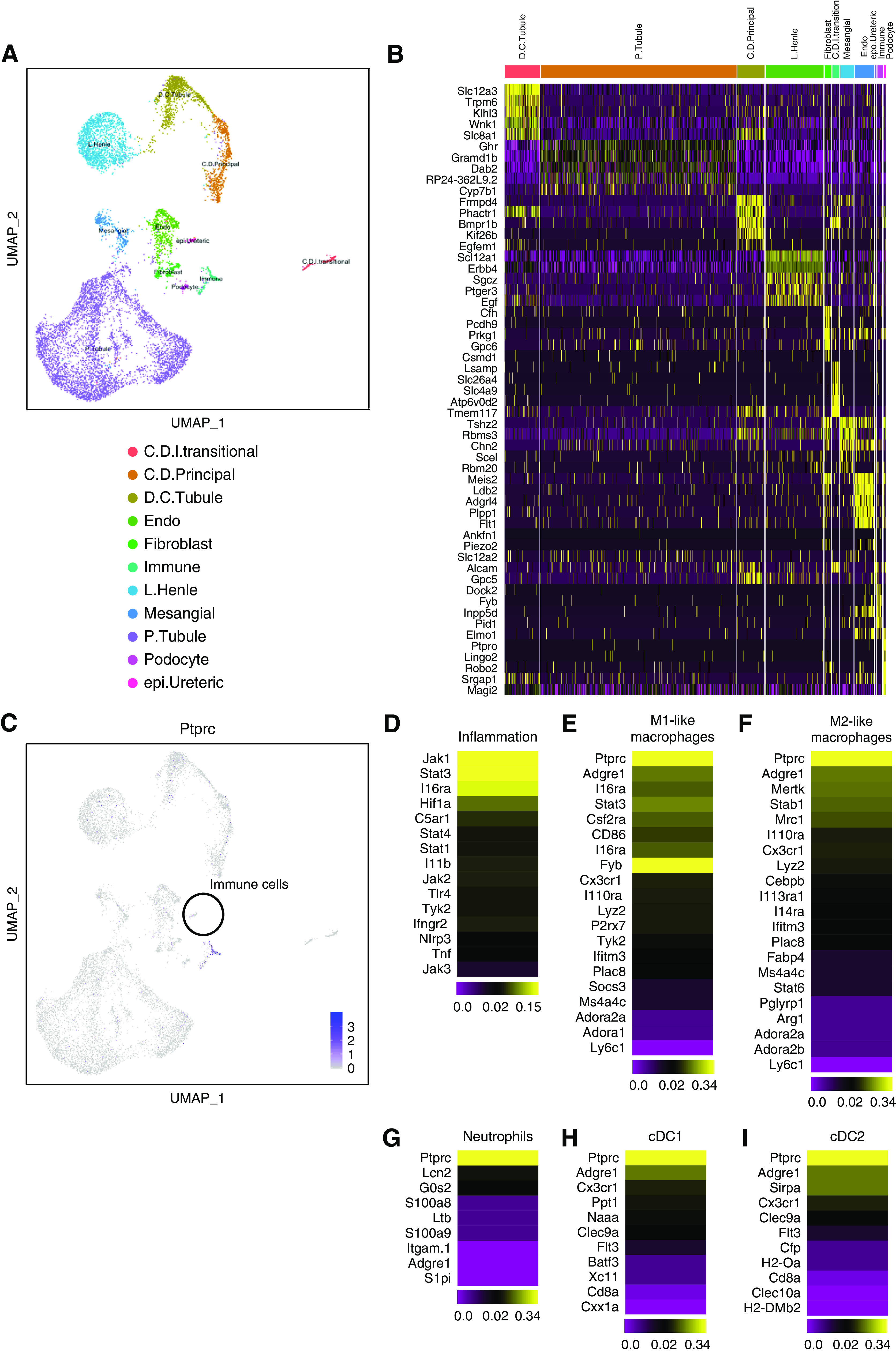
Identification of inflammatory signaling pathways and immune cell subsets. Single-cell RNA sequencing. (A and B) Single-cell RNA sequencing of kidneys from mice with chronic UA crystal nephropathy and granulomatous nephritis (HU+CU) on day 32. A total of 12 clusters of nonimmune and immune cells were identified. Cluster analysis is illustrated as UMAP (A) and as heat map (B). C.D.I.transitional, collecting duct intercalated and transitional cells; C.D.Principal, collecting duct principal cells; D.C.Tubule, distal convoluted tubule; L.Henle, loop of Henle cells; Mesangial, mesangial cells; P.Tubule, proximal tubule; epi.Ureteric, ureteric epithelial cells. (C–I) Single-cell RNA sequencing of kidneys from HU+CU mice identifies immune cell clusters according to Ptprc gene expression demonstrated as UMAP (C). Heat maps of specific genes within the immune cell clusters characteristic for inflammatory signaling pathways (D), M1-like (E) and M2-like macrophages (F), neutrophils (G), and cDCs cDC1 (H) and cDC2 (I).
Macrophages act as amplifiers of local microenvironments during homeostasis as well as in all different phases of acute and chronic disease processes.58,59 To assess which infiltrating leukocytes and, in particular, macrophage phenotypes may contribute to the formation of granulomas in chronic UA crystal nephropathy, we first performed flow cytometric analysis of kidney cell suspensions. The data revealed that the majority of leukocytes were CD45+MHCII+F4/80hi macrophages. These were further distinguished into M1-like proinflammatory macrophages (CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206−) and alternatively activated M2-like macrophages (CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206+) (Figure 7, A and B). cDCs (CD45+MHCII+F4/80+CD11bhiCD11chi) as well as neutrophils and monocytes (CD45+MHCII−CD11bhiGr1hi) were also present (Figure 7, A and B). Immunostaining of kidney sections confirmed a high number of infiltrating F4/80+ macrophages in HU+CU mice compared with the other three groups on day 32 (Figure 7D). Interestingly, the number of M1-like macrophages was higher compared with M2-like macrophages in HU+CU mice on day 14, an observation which reversed on day 32 (Figure 7C). UA crystal granulomas were CD45+ and F4/80+ positive (Figure 7, E and F), indicative of M1-like macrophages. The M2-like macrophage marker CD163 was negative (Figure 7G).
Figure 7.
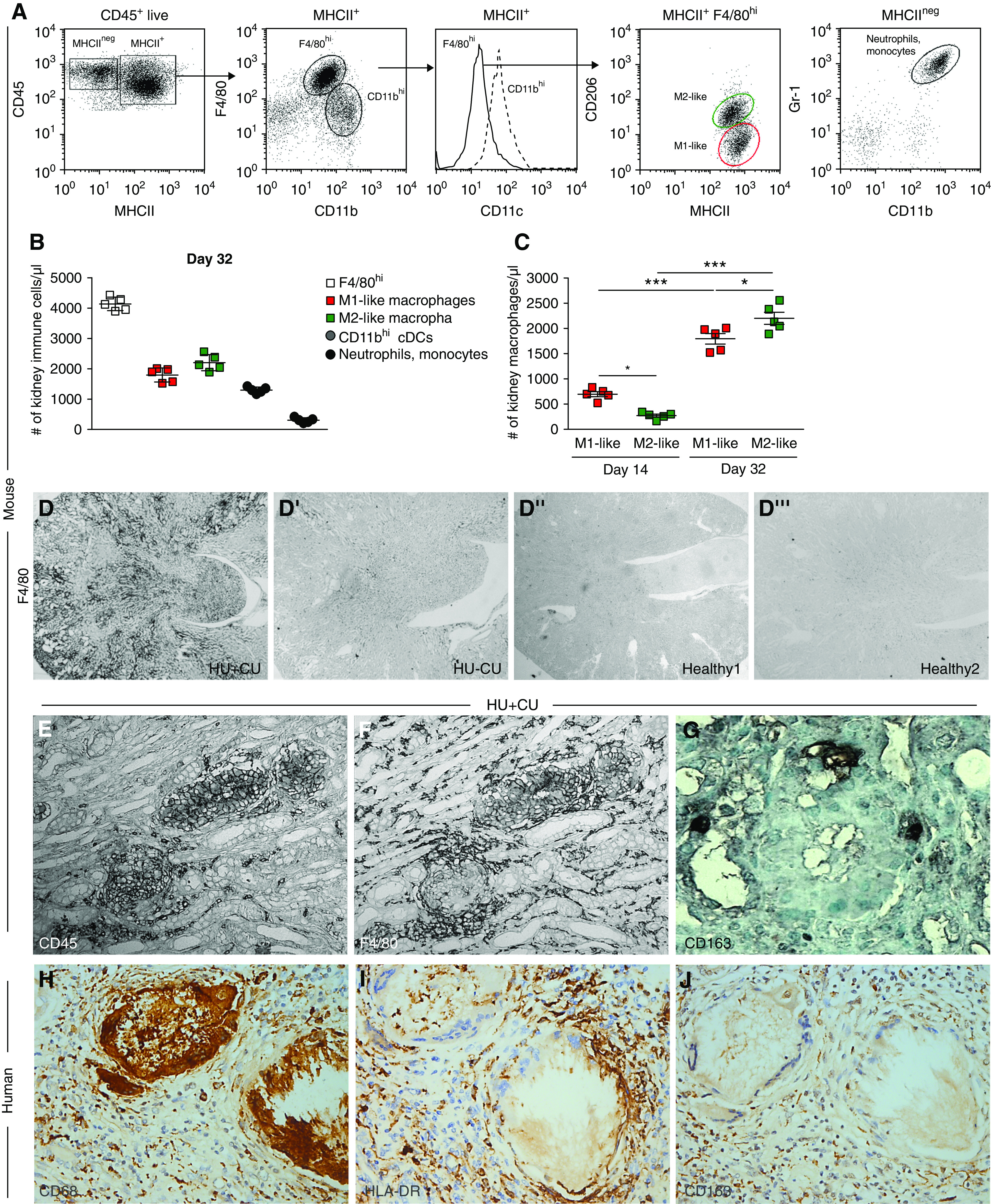
Macrophages contribute to UA crystal granuloma formation. Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine for 14 and 32 days. (A and B) Flow cytometric analysis of whole kidneys from HU+CU mice. Gating strategy to identify macrophages (CD45+MHCII+CD11b+F4/80hiCD11clo), proinflammatory M1-like macrophages (CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206−), alternatively activated M2-like macrophages (CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206+), neutrophils (CD45+MHCII−CD11b+Gr-1+), and cDCs (CD45+MHCII+F4/80loCD11bhiCD11chi) (A). Absolute numbers of leukocyte subsets from kidneys of HU+CU mice on day 32 (n=5 mice per group) (B). (C) Absolute numbers of M1- and M2-like macrophages from HU+CU mice on days 14 and 32 determined by flow cytometry (n=5 mice per group, two-way ANOVA). Data are mean±SD. *P<0.05; ***P<0.001. (D) F4/80 immunostaining of kidney sections illustrating the infiltration of F4/80+ cells in HU+CU mice (D), but not in HU−CU (D’) and healthy mice (D’’ and D’’’) on day 32. Magnification, ×25. (E–G) Immunostaining of kidneys from mice with HU+CU shows that UA crystal granulomas were positive for the immune cell marker CD45 (E) and F4/80 (F), but not for the M2-like macrophage marker CD163 (G) on day 32. Magnification, ×200. (H–J) Immunostaining of a nephrectomy specimen reveals numerous UA crystal granulomas positive for CD68+ macrophages (H) and HLA-DR+ proinflammatory M1-like macrophages (I), but not for CD163+ alternatively activated M2-like macrophages (J) (magnification, ×400). The patient was a 65-year-old man with a history of urothelial carcinoma, interstitial nephritis and vascular nephropathy, type 2 diabetes, ischemic cardiopathy and high BP, and serum creatinine 2 mg/dl.
To confirm this also in humans, IHC of a kidney biopsy sample from a patient with HU revealed a high infiltration of CD68+ macrophages within the granulomas and interstitium (Figure 7H). Medullary UA crystal granulomas were composed predominantly of HLA-DR+ proinflammatory M1-like macrophages (Figure 7I), whereas CD163+ alternatively activated M2-like macrophages were distributed diffusely in the wider surrounding interstitium (Figure 7J). The data imply that in patients with HU-related crystalluria, medullary UA crystal granulomas are associated with structural interstitial remodeling characteristic of progressive CKD; such lesions involve proinflammatory M1-like macrophages.
JAK/STAT Inhibition with Tofacitinib Does Not Affect Crystal Granuloma Formation and CKD Progression in Chronic UA Crystal Nephropathy
Human microarray gene expression data60 showed that the majority of inflammatory genes including STAT1, IL4R, IL13RA1, IL6ST, IL6R, IL13, and INFG, all of which are involved in JAK/STAT signaling, were expressed in most CKD entities, albeit being most prominent in highly inflammatory kidney diseases such as lupus nephritis (Figure 3, Supplemental Methods) or FSGS.61 However, a role for JAK/STAT signaling in crystal nephropathies is currently unknown. This raised the question of whether targeting crystal-induced inflammation could modulate the progression of chronic UA crystal nephropathy and specifically granulomatous interstitial nephritis.
The JAK/STAT inhibitor tofacitinib is approved for the treatment of chronic forms of inflammation such as rheumatoid arthritis62 and psoriatic arthritis,63 which served as an additional rationale to test tofacitinib in our model of chronic UA crystal nephropathy with granulomatous nephritis. HU+CU mice received 20 mg/kg tofacitinib from day 14 when interstitial fibrosis, but not yet crystal granuloma, had established (Supplemental Figure 4A). Tofacitinib significantly reduced serum IL-6 levels compared with vehicle-treated mice on day 32 (Supplemental Figure 4B). However, no difference between tofacitinib and vehicle treatment was observed in serum UA levels, BUN levels, GFR, tubular injury, and intrarenal mRNA expression levels of KIM-1, Il6, Tnfα, iNos, Arg1, Fibronectin 1, and Col1α1 (Supplemental Figure 4, B–I). Furthermore, tofacitinib did not alter the numbers of M1-like and M2-like macrophages (Supplemental Figure 4, J and K), as well as the number of UA crystal granulomas (Supplemental Figure 4L), compared with vehicle-treated HU+CU mice. Thus, JAK/STAT inhibition with tofacitinib did not affect UA crystal–induced inflammation, macrophage-mediated granuloma formation, and CKD progression in our mouse model of chronic UA crystal nephropathy.
Suppressing M1 Macrophages with Adenosine Reduces Granuloma Formation and CKD Progression of Chronic UA Crystal Nephropathy
If JAK/STAT inhibition was a too unspecific anti-inflammatory strategy, we speculated on a more specific intervention such as suppressing the M1-like phenotype of interstitial macrophages. Activation of adenosine A2 receptors on macrophages can suppress M1-like activation of macrophages, a concept previously shown to prevent progressive interstitial fibrosis in other models of CKD.64–66 Therefore, we examined the effect of the receptor agonist adenosine in mice with HU+CU from day 14 until euthanasia on day 32 (Figure 8A). Adenosine had no effect on serum UA levels (Figure 8B), but significantly reduced the levels of serum BUN and increased GFR on day 32 (Figure 8, C and D). Immunostaining of kidney sections revealed less tubular injury (Figure 8, E and F), a reduced number of UA crystal granulomas (Figure 8G), and less interstitial fibrosis (Figure 8H) as well as a decrease in the number of intrarenal CD45+ cells (Figure 8I) upon adenosine treatment. As expected, adenosine treatment induced a macrophage switch from an M1- into M2-like macrophage phenotype as indicated by a decrease in the number of CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206− M1-like macrophages (Figure 8J), but an increase in the number of CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206+ M2-like macrophages (Figure 8K) on day 32. Taken together, asymptomatic HU does not affect CKD unless UA crystallizes inside the kidney and causes UA crystal nephropathy (Figure 8L, left and middle panel). UA crystal granulomas develop late in the course of chronic UA crystal nephropathy (Figure 8L, right panel). Granulomatous nephritis is associated with diverse macrophage phenotypes, but M1-like macrophages are enriched in UA crystal granulomas. Suppressing M1-like macrophage activation with adenosine attenuates crystal granuloma formation and subsequent CKD progression. Thus, M1-like macrophages play an essential role in the progression of chronic UA crystal nephropathy with granulomatous interstitial nephritis.
Figure 8.
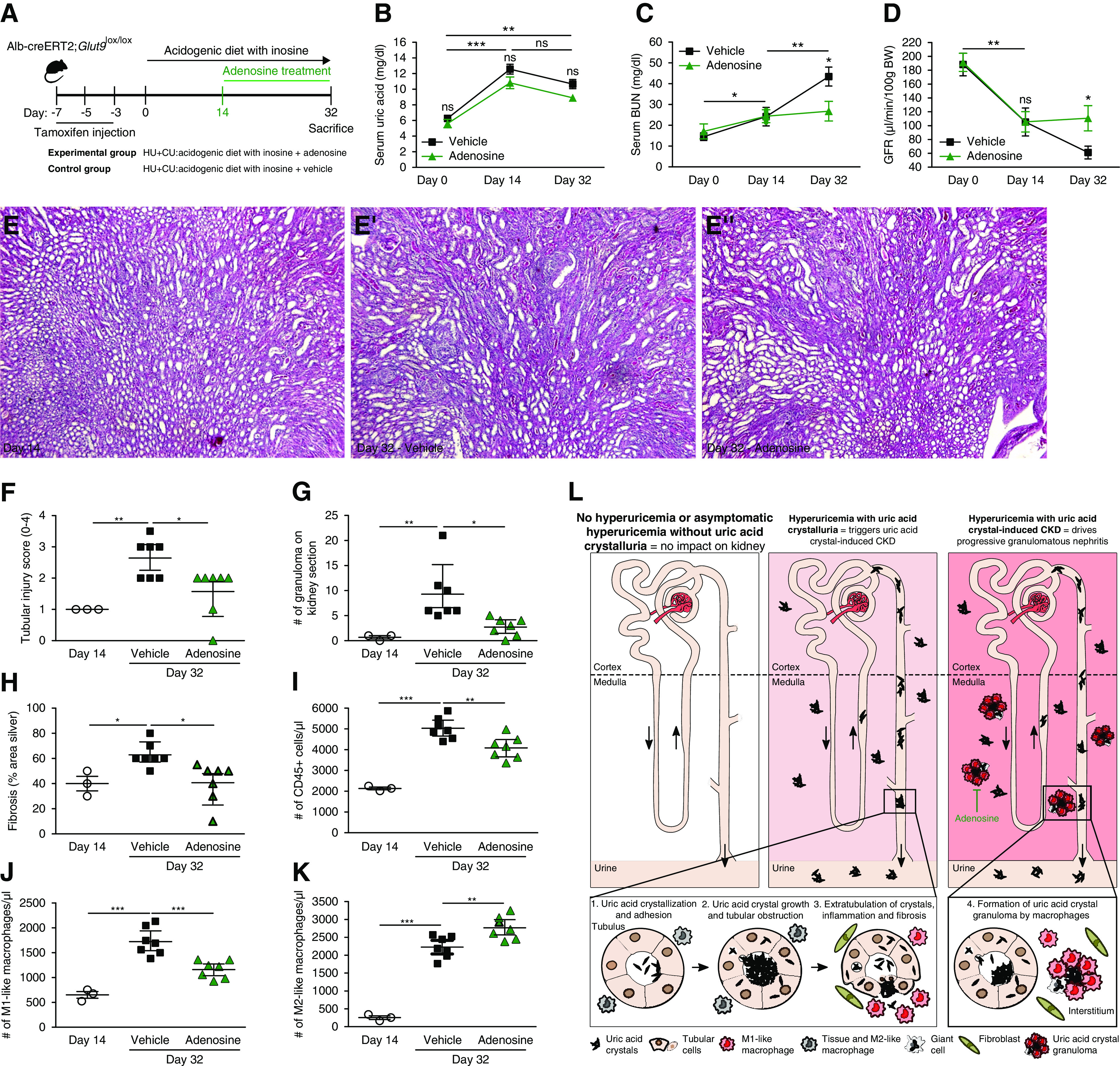
Adenosine treatment reduces granuloma-associated CKD progression by activating adenosine receptor signaling in macrophages. (A) Schematic of experimental set-up. Alb-creERT2;Glut9lox/lox mice were injected intraperitoneally with tamoxifen and then fed an acidogenic diet with inosine for 32 days. On day 14, HU+CU mice were injected with adenosine or vehicle every alternate day. (B–D) Serum UA (B) and BUN levels (C), and GFR (D), of adenosine- and vehicle-treated HU+CU mice on days 0–32 (n=5 per group). (E and F) PAS stain (E) illustrates tubular injury and tubular injury score (F) in HU+CU mice on day 14 and in adenosine- and vehicle-treated HU+CU mice on day 32 (n=3–7 mice per group). Magnification, ×100. (G) Number of granulomas on PAS-stained kidney sections (n=3–7 mice per group). (H) Percentage (%) of interstitial fibrosis on silver-stained kidney sections (n=3–7 mice per group). (I–K) Absolute numbers of renal CD45+ cells (I), proinflammatory M1-like macrophages (J, CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206−), and alternatively activated M2-like macrophages (K, CD45+MHCII+F4/80hiCD11b+CD11cloCX3CR1+CD206+) (n=3–7 mice per group). Data are mean±SD. *P<0.05; **P<0.01; ***P<0.001; NS, not significant by two-way ANOVA. (L) Proposed mechanisms of asymptomatic HU and UA crystalluria. The absence or presence of asymptomatic HU without UA crystalluria does not cause CKD (left panel). Only when UA precipitates in the urine due to a low urinary pH do UA crystals trigger tubular obstruction, inflammation, and interstitial fibrosis, as in chronic UA crystal nephropathy (middle panel). Eventually the crystal plugs translocate into the interstitial compartment. In the interstitium, frustrated phagocytosis of large crystal masses causes the formation of UA crystal granulomas that are composed of M1-like macrophages. These UA crystal granulomas contribute to CKD progression (right panel), which can be inhibited by adenosine.
Discussion
We hypothesized (1) that asymptomatic HU does not promote CKD progression unless UA crystallizes in the kidney; (2) that UA crystal granulomas may not precede CKD, but develop upon preexisting CKD; and (3) that the phenotype of the granuloma-related macrophages drives CKD progression. Our experimental data verify that asymptomatic HU does not cause CKD or promote the progressive decline of kidney function. Only HU with crystalluria due to urinary acidification induces tubular injury, inflammation, and interstitial fibrosis, characteristics of CKD. Subsequently, UA crystal granulomas form and contribute to CKD progression (Figure 8L). Targeting M1-like macrophages using adenosine could prevent the formation of UA crystal granulomas and progressive interstitial fibrosis, whereas the JAK/STAT inhibitor tofacitinib had no such effects. These findings disprove a role for asymptomatic HU in CKD progression and imply UA crystalluria and UA stone formation as potential indicators.
Whether asymptomatic HU contributes to CKD progression has remained a controversial issue for years.8,67 Although a meta-analysis of all single-center studies supported a benefit from urate-lowering drugs on the progression of unselected forms of CKD at an odds ratio of 0.9, a meta-analysis of all well-powered, multicenter, randomized, controlled trials on this topic showed an odds ratio of 1, and hence disproved a causal link between asymptomatic HU and the progression of unselected forms of CKD.9 This conclusion is not only consistent with the results of two Mendelian randomization studies on this issue,68,69 but also with our in vivo data showing that even robust and persistent asymptomatic HU alone does not cause CKD or accelerate AAI-induced CKD. In addition, we could recently show that asymptomatic HU acts as an intrinsic negative regulator of innate immunity with the capacity to inhibit monocyte activation via the urate transporter SLC2A9-mediated intracellular uptake of UA in acute tissue inflammation,70,71 highlighting an immunomodulatory role for asymptomatic HU.
However, the story is different when UA precipitates in the kidney, e.g., under the influence of an acidogenic diet36 or other factors that reduce urine pH.14,15 Such UA crystal deposits cause tubular obstruction, inflammation, infiltration of immune cells including macrophages, and interstitial fibrosis, similar to that observed in calcium oxalate crystal–induced CKD.38,71 Impaired kidney function, urine acidification, excessive dietary intake of purine-rich foods, endogenous UA overproduction, and UA precipitation are all known risk factors for chronic UA crystal nephropathy.72–77 Indeed, lowering urinary pH was sufficient to turn asymptomatic HU into chronic UA crystalline nephropathy.
Medullary UA crystal granulomas develop secondary to chronic UA crystal nephropathy and interstitial fibrosis, probably related to tubular crystal plugs and extratubulation that remained invisible with standard pathology and were only detectable on frozen unfixed kidney sections, consistent as observed in human kidney biopsy sampes.17,26 In this process, macrophages surround and translocate UA crystals into the interstitium followed by granuloma formation,78 which in turn contributes to CKD progression by driving interstitial inflammation via M1-like macrophage–related kidney inflammation. Direct interaction or even uptake of UA crystals strongly stimulates macrophage activation toward this proinflammatory phenotype.79–81 Granulomas consist of different macrophage subsets. Such activated proinflammatory macrophages directly interact with infectious organisms or microparticles at the center of the granuloma surrounded by alternatively activated macrophages.28,78,82–84 M1-like macrophages were present in mice with chronic UA crystal nephropathy and suppressing the M1-like phenotype with adenosine significantly attenuated granulomatous interstitial nephritis and the progression to CKD. This finding is consistent with previous in vitro and in vivo reports that demonstrated beneficial effects of targeting adenosine receptor signaling in macrophages in various animal models of CKD64–66,85–89 including other chronic crystalline nephropathies.47,90 An effect of adenosine on other immune cell subsets inside and outside of the kidney may also contribute to the functional and structural outcomes.
Although approved only for the treatment of rheumatoid and psoriatic arthritis, numerous experimental studies document that inhibition of the JAK/STAT signaling pathway can suppress inflammation and organ damage in a wide spectrum of autoimmune and inflammatory disease models including asthma,91 giant cell arteritis,92 ulcerative colitis,93 chronic pancreatitis,94 and lupus nephritis.95,96 Human microarray gene expression data60 reveal overexpression of many JAK/STAT signaling pathway–related transcripts in highly inflammatory disorders such as lupus nephritis. A transcriptome analysis of chronic UA crystal nephropathy kidneys gave consistent results. However, the JAK/STAT inhibitor tofacitinib did not show renoprotective effects in our model. We assume that JAK/STAT-related cytokines are not critically involved in granulomatous interstitial nephritis that follows a more chronic cause. However, tofacitinib acts specifically on Jak1 and Jak3, and we cannot exclude that JAK/STAT inhibitors with other specificities may elicit different effects.
Our experiment was designed to reach levels of HU that mimic profound HU in humans. One may argue that UA is handled completely differently in mice and humans, that mice are not programmed to such high UA acid levels, and that actually a milder form of HU would better mimic the relative elevation in humans. However, together with the results of a previous study, we can conclude that even high serum UA levels do not accelerate CKD progression unless they cause crystalluria and intrarenal UA deposition in mice.50
In conclusion, our data show that asymptomatic HU does not affect CKD progression unless UA crystallizes in the kidney, as indicated by UA crystalluria leading to chronic UA crystal nephropathy (Figure 8L). UA crystal granulomas develop late in this process, but contribute to CKD progression because UA crystals trigger M1-like macrophage–related interstitial inflammation and fibrosis. Specifically targeting these proinflammatory M1-like macrophages, but not JAK/STAT inhibition, can attenuate granulomatous interstitial nephritis. Our current scientific evidence shows that only HU with UA crystalluria drives CKD, and that identifying urinary UA crystals can serve as an additional diagnostic parameter as to whether or not patients with asymptomatic HU need to be treated with urate-lowering therapy.
Disclosures
R. Kramann reports grants from Chugai Pharma, outside the submitted work. H.-J. Anders reports personal fees from Secarna, personal fees from Previpharma, personal fees from Noxxon, personal fees from Inositec, and personal fees from GlaxoSmithKline, outside the submitted work. All remaining authors have nothing to disclose.
Funding
This work was funded by Deutsche Forschungsgemeinschaft grants STE2437/2-1 and STE2437/2-2 (to S. Steiger), LE2621/6-1 (to M. Lech), and AN372/16-2, AN372/24-1 (to H. Anders). S. Steiger received support from the Ludwig-Maximilians-Universität München Excellence Initiative and M. Lech from the Narodowe Centrum Nauki (Polish National Science Centre) (2016/23/B/NZ6/00086). The European Research Council under the European Union’s Horizon 2020 Research and Innovation programme (H2020 European Research Council) supported P. Romagnani (grant agreement No. 648274). The Ministero dell’Istruzione, dell’Università e della Ricerca PRIN 2017 supported P. Romagnani.
Supplementary Material
Acknowledgments
We thank Jana Mandelbaum and Dan Draganovici for expert technical support. Götz Schlipf and Bernd Steinhuber are greatly acknowledged for the practical assistance in cryosectioning and matrix assisted laser desorption ionization-mass spectrometry imaging. We thank the Joint Research Centre for Computational Biomedicine (JRC-COMBINE) for support, which is partially funded by Bayer.
This work was previously presented in part in the thesis project of Mr. Markus Sellmayr and Mr. Moritz Roman Hernandez Petzsche to the Medical Faculty of the Ludwig-Maximilian's-University, Munich. Dr. Stefanie Steiger and Prof. Hans-Joachim Anders designed the study concept and experiments. Mr. Markus Sellmayr, Mr. Moritz Roman Hernandez Petzsche, Dr. Qiuyue Ma, Mr. Nils Krüger, Dr. Maciej Lech, and Dr. Stefanie Steiger conducted experiments, and acquired and analyzed data. Prof. Helen Liapis provided the diagnostic kidney biopsy data. Dr. Andreas Brink and Dr. Barbara Lenz performed polarized light microscopy and matrix assisted laser desorption ionization-Fourier transform ion cyclotron resonance mass spectrometry imaging. Dr. Maria Lucia Angelotti and Prof. Paola Romagnani performed fluorescence imaging, and Gnemmi immunohistochemistry of human biopsy samples. Dr. Christoph Kuppe, Mrs. Hyojin Kim, Dr. Ferenc Tajti, Dr. Eric Moniqué Johannes Bindels, Dr. Julio Saez-Rodriguez, and Prof. Rafael Kramann carried out single-cell RNA sequencing, processed the data, and provided the human microarray gene expression data. Dr. Stefanie Steiger and Prof. Hans-Joachim Anders wrote the manuscript. All contributing authors reviewed and revised the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020040523/-/DCSupplemental.
Supplemental Methods. Human microarray gene expression data of different chronic kidney disease entities.
Supplemental Figure 1. Only an acidogenic diet with inosine causes impairment in kidney function but not acidogenic and chow diet without inosine.
Supplemental Figure 2. Histopathological abnormalities on kidney biopsies from patients with uric acid crystal deposits.
Supplemental Figure 3. Human Genome arrays from different human CKD disease entities.
Supplemental Figure 4. Tofacitinib therapy does not prevent crystalluria, granuloma formation and CKD progression.
Supplemental Movie 1. Uric acid crystal granuloma in 3D.
Supplemental Movie 2. Uric acid crystal granuloma in 3D moves along the z axis.
Supplemental Table 1. Murine primer sequences.
References
- 1.GBD Chronic Kidney Disease Collaboration : Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 395: 709–733, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards NL: The role of hyperuricemia and gout in kidney and cardiovascular disease. Cleve Clin J Med 75: S13–S16, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Choi HK, Mount DB, Reginato AM; American College of Physicians; American Physiological Society : Pathogenesis of gout. Ann Intern Med 143: 499–516, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Reginato AM, Mount DB, Yang I, Choi HK: The genetics of hyperuricaemia and gout. Nat Rev Rheumatol 8: 610–621, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roch-Ramel F, Guisan B: Renal transport of urate in humans. News Physiol Sci 14: 80–84, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Sattui SE, Singh JA, Gaffo AL: Comorbidities in patients with crystal diseases and hyperuricemia. Rheum Dis Clin North Am 40: 251–278, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feig DI, Kang DH, Johnson RJ: Uric acid and cardiovascular risk. N Engl J Med 359: 1811–1821, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato Y, Feig DI, Stack AG, Kang DH, Lanaspa MA, Ejaz AA, et al. : The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat Rev Nephrol 15: 767–775, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Steiger S, Ma Q, Anders HJ: The case for evidence-based medicine for the association between hyperuricaemia and CKD. Nat Rev Nephrol 16: 422, 2020. [DOI] [PubMed] [Google Scholar]
- 10.Gustafsson D, Unwin R: The pathophysiology of hyperuricaemia and its possible relationship to cardiovascular disease, morbidity and mortality. BMC Nephrol 14: 164, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chonchol M, Shlipak MG, Katz R, Sarnak MJ, Newman AB, Siscovick DS, et al. : Relationship of uric acid with progression of kidney disease. Am J Kidney Dis 50: 239–247, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Desai J, Steiger S, Anders HJ: Molecular pathophysiology of gout. Trends Mol Med 23: 756–768, 2017. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Chang Y, Yun KE, Jung HS, Lee SJ, Shin H, et al. : Development of nephrolithiasis in asymptomatic hyperuricemia: A cohort study. Am J Kidney Dis 70: 173–181, 2017. [DOI] [PubMed] [Google Scholar]
- 14.Pazos Pérez F: Uric acid renal lithiasis: New concepts. Contrib Nephrol 192: 116–124, 2018. [DOI] [PubMed] [Google Scholar]
- 15.Bell DS: Beware the low urine pH--the major cause of the increased prevalence of nephrolithiasis in the patient with type 2 diabetes. Diabetes Obes Metab 14: 299–303, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Beck LH: Requiem for gouty nephropathy. Kidney Int 30: 280–287, 1986. [DOI] [PubMed] [Google Scholar]
- 17.Nickeleit V, Mihatsch MJ: Uric acid nephropathy and end-stage renal disease--review of a non-disease. Nephrol Dial Transplant 12: 1832–1838, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Modern FW, Meister L: The kidney of gout, a clinical entity. Med Clin North Am 21: 941–951, 1952. [PubMed] [Google Scholar]
- 19.Linnane JW, Burry AF, Emmerson BT: Urate deposits in the renal medulla. Prevalence and associations. Nephron 29: 216–222, 1981. [DOI] [PubMed] [Google Scholar]
- 20.Verger D, Leroux-Robert C, Ganter P, Richet G: [Gouty tophi in the renal medulla in chronic uremia. Study of 17 cases discovered from among 62 autopsies]. Nephron 4: 356–370, 1967. [DOI] [PubMed] [Google Scholar]
- 21.Bayram D, Tuğrul Sezer M, İnal S, Altuntaş A, Kıdır V, Orhan H: The effects of allopurinol on metabolic acidosis and endothelial functions in chronic kidney disease patients. Clin Exp Nephrol 19: 443–449, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Goicoechea M, de Vinuesa SG, Verdalles U, Ruiz-Caro C, Ampuero J, Rincón A, et al. : Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol 5: 1388–1393, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goicoechea M, Garcia de Vinuesa S, Verdalles U, Verde E, Macias N, Santos A, et al. : Allopurinol and progression of CKD and cardiovascular events: Long-term follow-up of a randomized clinical trial. Am J Kidney Dis 65: 543–549, 2015. [DOI] [PubMed] [Google Scholar]
- 24.Goldfarb DS, MacDonald PA, Gunawardhana L, Chefo S, McLean L: Randomized controlled trial of febuxostat versus allopurinol or placebo in individuals with higher urinary uric acid excretion and calcium stones. Clin J Am Soc Nephrol 8: 1960–1967, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wada T, Hosoya T, Honda D, Sakamoto R, Narita K, Sasaki T, et al. : Uric acid-lowering and renoprotective effects of topiroxostat, a selective xanthine oxidoreductase inhibitor, in patients with diabetic nephropathy and hyperuricemia: A randomized, double-blind, placebo-controlled, parallel-group study (UPWARD study). Clin Exp Nephrol 22: 860–870, 2018. [DOI] [PubMed] [Google Scholar]
- 26.Ayoub I, Almaani S, Brodsky S, Nadasdy T, Prosek J, Hebert L, et al. : Revisiting medullary tophi: A link between uric acid and progressive chronic kidney disease? Clin Nephrol 85: 109–113, 2016. [DOI] [PubMed] [Google Scholar]
- 27.Mulay SR, Anders HJ: Crystal nephropathies: Mechanisms of crystal-induced kidney injury. Nat Rev Nephrol 13: 226–240, 2017. [DOI] [PubMed] [Google Scholar]
- 28.Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. : Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med 22: 531–538, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yood RA, Ottery FD, Irish W, Wolfson M: Effect of pegloticase on renal function in patients with chronic kidney disease: A post hoc subgroup analysis of 2 randomized, placebo-controlled, phase 3 clinical trials. BMC Res Notes 7: 54, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosoya T, Ohno I, Nomura S, Hisatome I, Uchida S, Fujimori S, et al. : Effects of topiroxostat on the serum urate levels and urinary albumin excretion in hyperuricemic stage 3 chronic kidney disease patients with or without gout. Clin Exp Nephrol 18: 876–884, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saag KG, Whelton A, Becker MA, MacDonald P, Hunt B, Gunawardhana L: Impact of febuxostat on renal function in gout patients with moderate-to-severe renal impairment. Arthritis Rheumatol 68: 2035–2043, 2016. [DOI] [PubMed] [Google Scholar]
- 32.Badve SV, Pascoe EM, Tiku A, Boudville N, Brown FG, Cass A, et al. ; CKD-FIX Study Investigators : Effects of allopurinol on the progression of chronic kidney disease. N Engl J Med 382: 2504–2513, 2020. [DOI] [PubMed] [Google Scholar]
- 33.Doria A, Galecki AT, Spino C, Pop-Busui R, Cherney DZ, Lingvay I, et al. ; PERL Study Group : Serum urate lowering with allopurinol and kidney function in type 1 diabetes. N Engl J Med 382: 2493–2503, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moe OW: Posing the question again: Does chronic uric acid nephropathy exist? J Am Soc Nephrol 21: 395–397, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG: Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preitner F, Laverriere-Loss A, Metref S, Da Costa A, Moret C, Rotman S, et al. : Urate-induced acute renal failure and chronic inflammation in liver-specific Glut9 knockout mice. Am J Physiol Renal Physiol 305: F786–F795, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Honarpisheh M, Foresto-Neto O, Steiger S, Kraft F, Koehler P, von Rauchhaupt E, et al. : Aristolochic acid I determine the phenotype and activation of macrophages in acute and chronic kidney disease. Sci Rep 8: 12169, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steiger S, Grill JF, Ma Q, Bäuerle T, Jordan J, Smolle M, et al. : Anti-transforming growth factor β IgG elicits a dual effect on calcium oxalate crystallization and progressive nephrocalcinosis-related chronic kidney disease. Front Immunol 9: 619, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Q, Steiger S, Anders HJ: Sodium glucose transporter-2 inhibition has no renoprotective effects on non-diabetic chronic kidney disease. Physiol Rep 5: e13228, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lenz B, Brink A, Siam M, De Paepe A, Bassett S, Eichinger-Chapelon A, et al. : Application of imaging techniques to cases of drug-induced crystal nephropathy in preclinical studies. Toxicol Sci 163: 409–419, 2018. [DOI] [PubMed] [Google Scholar]
- 41.Stoeckli M, Staab D, Wetzel M, Brechbuehl M: iMatrixSpray: A free and open source sample preparation device for mass spectrometric imaging. Chimia (Aarau) 68: 146–149, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, et al. : Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Friedemann J, Heinrich R, Shulhevich Y, Raedle M, William-Olsson L, Pill J, et al. : Improved kinetic model for the transcutaneous measurement of glomerular filtration rate in experimental animals. Kidney Int 90: 1377–1385, 2016. [DOI] [PubMed] [Google Scholar]
- 44.Ninichuk V, Gross O, Reichel C, Khandoga A, Pawar RD, Ciubar R, et al. : Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol 16: 977–985, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Peired AJ, Antonelli G, Angelotti ML, Allinovi M, Guzzi F, Sisti A, et al. : Acute kidney injury promotes development of papillary renal cell adenoma and carcinoma from renal progenitor cells [published correction appears in Sci Transl Med 12: eabd2685, 2020]. Sci Transl Med 12: eaaw6003, 2020. [DOI] [PubMed] [Google Scholar]
- 46.Lech M, Anders HJ: Expression profiling by real-time quantitative polymerase chain reaction (RT-qPCR). Methods Mol Biol 1169: 133–142, 2014. [DOI] [PubMed] [Google Scholar]
- 47.Anders HJ, Suarez-Alvarez B, Grigorescu M, Foresto-Neto O, Steiger S, Desai J, et al. : The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int 93: 656–669, 2018. [DOI] [PubMed] [Google Scholar]
- 48.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, et al. : The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A 116: 19619–19625, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Preitner F, Pimentel A, Metref S, Berthonneche C, Sarre A, Moret C, et al. : No development of hypertension in the hyperuricemic liver-Glut9 knockout mouse. Kidney Int 87: 940–947, 2015. [DOI] [PubMed] [Google Scholar]
- 51.Chhana A, Lee G, Dalbeth N: Factors influencing the crystallization of monosodium urate: A systematic literature review. BMC Musculoskelet Disord 16: 296, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mulay SR, Evan A, Anders HJ: Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant 29: 507–514, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Karaiskos N, Rahmatollahi M, Boltengagen A, Liu H, Hoehne M, Rinschen M, et al. : A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 29: 2060–2068, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kramann R, Machado F, Wu H, Kusaba T, Hoeft K, Schneider RK, et al. : Parabiosis and single-cell RNA sequencing reveal a limited contribution of monocytes to myofibroblasts in kidney fibrosis. JCI Insight 3: e99561, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al. : Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Combes AN, et al. : Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk [published correction appears in Development 146: dev182162, 2019]. Development 146: dev178673, 2019 [DOI] [PubMed] [Google Scholar]
- 57.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, et al. : Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol 30: 767–781, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salei N, Rambichler S, Salvermoser J, Papaioannou NE, Schuchert R, Pakalniškytė D, et al. : The kidney contains ontogenetically distinct dendritic cell and macrophage subtypes throughout development that differ in their inflammatory properties. J Am Soc Nephrol 31: 257–278, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weidenbusch M, Anders HJ: Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair and fibrosis. J Innate Immun 4: 463–477, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tajti F, Kuppe C, Antoranz A, Ibrahim MM, Kim H, Ceccarelli F, et al. : A functional landscape of CKD entities from public transcriptomic data. Kidney Int Rep 5: 211–224, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tao J, Mariani L, Eddy S, Maecker H, Kambham N, Mehta K, et al. : JAK-STAT signaling is activated in the kidney and peripheral blood cells of patients with focal segmental glomerulosclerosis. Kidney Int 94: 795–808, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. : EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis 79: 685–699, 2020. [DOI] [PubMed] [Google Scholar]
- 63.Mease P, Hall S, FitzGerald O, van der Heijde D, Merola JF, Avila-Zapata F, et al. : Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med 377: 1537–1550, 2017. [DOI] [PubMed] [Google Scholar]
- 64.Garcia GE, Truong LD, Chen JF, Johnson RJ, Feng L: Adenosine A(2A) receptor activation prevents progressive kidney fibrosis in a model of immune-associated chronic inflammation. Kidney Int 80: 378–388, 2011. [DOI] [PubMed] [Google Scholar]
- 65.Awad AS, Huang L, Ye H, Duong ET, Bolton WK, Linden J, et al. : Adenosine A2A receptor activation attenuates inflammation and injury in diabetic nephropathy. Am J Physiol Renal Physiol 290: F828–F837, 2006. [DOI] [PubMed] [Google Scholar]
- 66.Xiao H, Shen HY, Liu W, Xiong RP, Li P, Meng G, et al. : Adenosine A2A receptor: A target for regulating renal interstitial fibrosis in obstructive nephropathy. PLoS One 8: e60173, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumagai T, Ota T, Tamura Y, Chang WX, Shibata S, Uchida S: Time to target uric acid to retard CKD progression. Clin Exp Nephrol 21: 182–192, 2017. [DOI] [PubMed] [Google Scholar]
- 68.Jordan DM, Choi HK, Verbanck M, Topless R, Won HH, Nadkarni G, et al. : No causal effects of serum urate levels on the risk of chronic kidney disease: A mendelian randomization study. PLoS Med 16: e1002725, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li X, Meng X, Timofeeva M, Tzoulaki I, Tsilidis KK, Ioannidis JP, et al. : Serum uric acid levels and multiple health outcomes: Umbrella review of evidence from observational studies, randomised controlled trials, and mendelian randomisation studies. BMJ 357: j2376, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma Q, Honarpisheh M, Li C, Sellmayr M, Lindenmeyer M, Böhland C, et al. : Soluble uric acid is an intrinsic negative regulator of monocyte activation in monosodium urate crystal-induced tissue inflammation. J Immunol 205: 789–800, 2020. [DOI] [PubMed] [Google Scholar]
- 71.Mulay SR, Eberhard JN, Pfann V, Marschner JA, Darisipudi MN, Daniel C, et al. : Oxalate-induced chronic kidney disease with its uremic and cardiovascular complications in C57BL/6 mice. Am J Physiol Renal Physiol 310: F785–F795, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cameron MA, Maalouf NM, Adams-Huet B, Moe OW, Sakhaee K: Urine composition in type 2 diabetes: Predisposition to uric acid nephrolithiasis. J Am Soc Nephrol 17: 1422–1428, 2006. [DOI] [PubMed] [Google Scholar]
- 73.Daudon M, Lacour B, Jungers P: High prevalence of uric acid calculi in diabetic stone formers. Nephrol Dial Transplant 20: 468–469, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Johnson RJ, Nakagawa T, Jalal D, Sánchez-Lozada LG, Kang DH, Ritz E: Uric acid and chronic kidney disease: Which is chasing which? Nephrol Dial Transplant 28: 2221–2228, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maalouf NM, Sakhaee K, Parks JH, Coe FL, Adams-Huet B, Pak CY: Association of urinary pH with body weight in nephrolithiasis. Kidney Int 65: 1422–1425, 2004. [DOI] [PubMed] [Google Scholar]
- 76.Hediger MA, Johnson RJ, Miyazaki H, Endou H: Molecular physiology of urate transport. Physiology (Bethesda) 20: 125–133, 2005. [DOI] [PubMed] [Google Scholar]
- 77.Maalouf NM: Metabolic syndrome and the genesis of uric acid stones. J Ren Nutr 21: 128–131, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klinkhammer BM, Djudjaj S, Kunter U, Palsson R, Edvardsson VO, Wiech T, et al. : Cellular and molecular mechanisms of kidney injury in 2,8-dihydroxyadenine nephropathy. J Am Soc Nephrol 31: 799–816, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. : MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 116: 2262–2271, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241, 2006. [DOI] [PubMed] [Google Scholar]
- 81.Martin WJ, Shaw O, Liu X, Steiger S, Harper JL: Monosodium urate monohydrate crystal-recruited noninflammatory monocytes differentiate into M1-like proinflammatory macrophages in a peritoneal murine model of gout. Arthritis Rheum 63: 1322–1332, 2011. [DOI] [PubMed] [Google Scholar]
- 82.Ramakrishnan L: Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 12: 352–366, 2012. [DOI] [PubMed] [Google Scholar]
- 83.Williams GT, Williams WJ: Granulomatous inflammation--a review. J Clin Pathol 36: 723–733, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pagán AJ, Ramakrishnan L: The Formation and function of granulomas. Annu Rev Immunol 36: 639–665, 2018. [DOI] [PubMed] [Google Scholar]
- 85.Ibrahim O, Oteh M, Syukur AA, Hassan CH, Fadilah SW, Rahman MM: Evaluation of aspirin and clopidogrel resistance in patients with Acute Coronary Syndrome by using Adenosine Diposphate Test and Aspirin Test. Pak J Med Sci 29: 97–102, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Truong LD, Trostel J, McMahan R, Chen JF, Garcia GE: Macrophage A2A adenosine receptors are essential to protect from progressive kidney injury. Am J Pathol 186: 2601–2613, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang YM, McRae JL, Robson SC, Cowan PJ, Zhang GY, Hu M, et al. : Regulatory T cells participate in CD39-mediated protection from renal injury. Eur J Immunol 42: 2441–2451, 2012. [DOI] [PubMed] [Google Scholar]
- 88.Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, et al. : Elevated ecto-5′-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res 112: 1466–1478, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Csóka B, Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. : Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J 26: 376–386, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lech M, Gröbmayr R, Ryu M, Lorenz G, Hartter I, Mulay SR, et al. : Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy. J Am Soc Nephrol 25: 292–304, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P: The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol 582: 154–161, 2008. [DOI] [PubMed] [Google Scholar]
- 92.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM: Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation 137: 1934–1948, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, et al. ; Study A3921063 Investigators : Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med 367: 616–624, 2012. [DOI] [PubMed] [Google Scholar]
- 94.Komar HM, Serpa G, Kerscher C, Schwoegl E, Mace TA, Jin M, et al. : Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci Rep 7: 1787, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. : Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol 69: 148–160, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. : Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J Immunol 187: 3840–3853, 2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.