

Abstract
Endothelial–to–mesenchymal transition (EndMT) is a cellular transdifferentiation program in which endothelial cells partially lose their endothelial identity and acquire mesenchymal-like features. Renal capillary endothelial cells can undergo EndMT in association with persistent damage of the renal parenchyma. The functional consequence(s) of EndMT in kidney fibrosis remains unexplored. Here, we studied the effect of Twist or Snail deficiency in endothelial cells on EndMT in kidney fibrosis. Conditional deletion of Twist1 (which encodes Twist) or Snai1 (which encodes Snail) in VE-cadherin+ or Tie1+ endothelial cells inhibited the emergence of EndMT and improved kidney fibrosis in two different kidney injury/fibrosis mouse models. Suppression of EndMT limited peritubular vascular leakage, reduced tissue hypoxia, and preserved tubular epithelial health and function. Hypoxia, which was exacerbated by EndMT, resulted in increased Myc abundance in tubular epithelial cells, enhanced glycolysis, and suppression of fatty acid oxidation. Pharmacological suppression or epithelial-specific genetic ablation of Myc in tubular epithelial cells ameliorated fibrosis and restored renal parenchymal function and metabolic homeostasis. Together, these findings demonstrate a functional role for EndMT in the response to kidney capillary endothelial injury and highlight the contribution of endothelial-epithelial crosstalk in the development of kidney fibrosis with a potential for therapeutic intervention.
Introduction
Endothelial cells exhibit the capacity to acquire mesenchymal features through a process known as endothelial to mesenchymal transition (EndMT) [1]. During this transition, endothelial cells partially lose endothelial identity and acquire mesenchymal characteristics, such as loss of cell-cell junctions, compromised cell polarity, acquisition of fibroblast-like morphology and gene expression profile. EndMT in embryonic heart development is a pre-requisite for the formation of cardiac cushions that are involved in adult heart valves and septa development [2]. EndMT was initially identified in the pathological settings of cardiac and kidney fibrosis [3, 4], and subsequently in cancer [5], atherosclerosis [6, 7], and tissue remodeling (reviewed in [8]). Pathological EndMT is associated with injury to the involved parenchyma of a given organ. In injured kidneys, renal capillary endothelial cells can undergo partial EndMT, as evidenced by the co-expression of endothelial (CD31) and mesenchymal markers (αSMA and FSP-1), as well as the use of endothelial lineage tracing studies. EndMT is reported in animal models of obstructive and diabetic nephropathies, ischemia-reperfusion injury, and Alport syndrome [4, 9–11]. EndMT has also been identified in kidney biopsies from patients with diabetic nephropathy, IgA nephropathy, lupus nephritis, and from transplant patients with allograft failure [12–16].
Endothelial cells exposed to the inflammatory cytokine TGFβ undergo mesenchymal transition, and silencing of Snail and Twist transcription factors is sufficient to limit the acquisition of the partial EndMT phenotype [17–21]. In vivo strategies to block TGFβ signaling pathway also lead to reduced EndMT and improvement of kidney fibrosis, supporting a central role of this signaling pathway in EndMT and its contribution to the pathogenesis of fibrosis [22, 23]. Similar to EndMT, the induction of epithelial to mesenchymal transition (EMT) also involves TGFβ signaling. In this regard, genetic deletion of Snail or Twist in mouse models of kidney fibrosis is effective at inhibiting the cognate process of EMT and suppresses fibrosis and restores parenchymal functionality [24, 25]. Although EndMT as such may not contribute to a substantial number of defined fibrosis-associated fibroblasts [26], it is associated with endothelial cell dysfunction. The specific, functional consequence(s) of EndMT in the emergence of kidney fibrosis remains to be evaluated. To this end, we engineered mouse models to define the rate-limiting roles of EndMT in kidney fibrosis. Specifically, we generated mice with conditional deletion of Twist1 or Snai1 in endothelial cells using multiple endothelial specific promoter-driven Cre-recombinase systems. Here, we showed that suppression of Snail- or Twist-induced EndMT ameliorated kidney fibrosis by preserving kidney interstitial vascular integrity, thereby limiting vascular leakage and peritubular tissue hypoxia. Hypoxia promotes kidney fibrosis by activating HIF1α, which mediates pro-fibrotic gene expression alterations and tissue damage [27, 28]. In this regard, capillary rarefaction and hypoxia are associated with human fibrosis [29, 30]. Our results support that EndMT-induced tissue hypoxia upregulates Myc in tubular epithelial cells, enhancing glycolysis [31–34]. Our findings mechanistically link EndMT with tubular epithelium metabolic dysfunction and offer new insights into potential anti-fibrosis therapies.
Results
Inducible and conditional genetic deletion of Twist1 or Snai1 in endothelial cells reduces fibrosis in mouse models of kidney fibrosis
CD31+ endothelial cells in human biopsies from patients with obstructive and chronic allograft nephropathy also expressed the mesenchymal marker αSMA (Fig. 1A, fig. S1A, and Data File S1). Similarly, EndMT was detected in fibrotic mouse kidneys from two distinct models of experimentally-induced nephropathies (unilateral ureteral obstruction (UUO) and folic acid-induced nephropathy) (Fig. 1A). To evaluate the functional role of EndMT, we bred mice with Twist1 and Snai1 conditional alleles (Twist1L/L and Snai1L/L) with mice expressing Cre-recombinase under the tamoxifen–inducible, endothelial–specific Cdh5 (VE-cadherin) promoter to generate conditional knock–out mice (Cdh5CreERT2+;TwistL/L – hereafter referred to as TwistEnd–cKO mice and Cdh5CreERT2+;Snai1L/L – hereafter referred to as SnailEnd–cKO mice) and control (Cdh5CreERT2–;Twist1L/L and Cdh5CreERT2–;Snai1L/L– hereafter referred to as wildtype (WT) mice) (fig. S1, B and C). TwistEnd–cKO mice and SnailEnd–cKO mice were born at the expected Mendelian ratio (fig. S1, D and E), were fertile, and did not exhibit any overt phenotype from 0 to 6 months of age.
Figure 1. Conditional deletion of Twist1 in endothelial cells ameliorates UUO-induced kidney fibrosis.
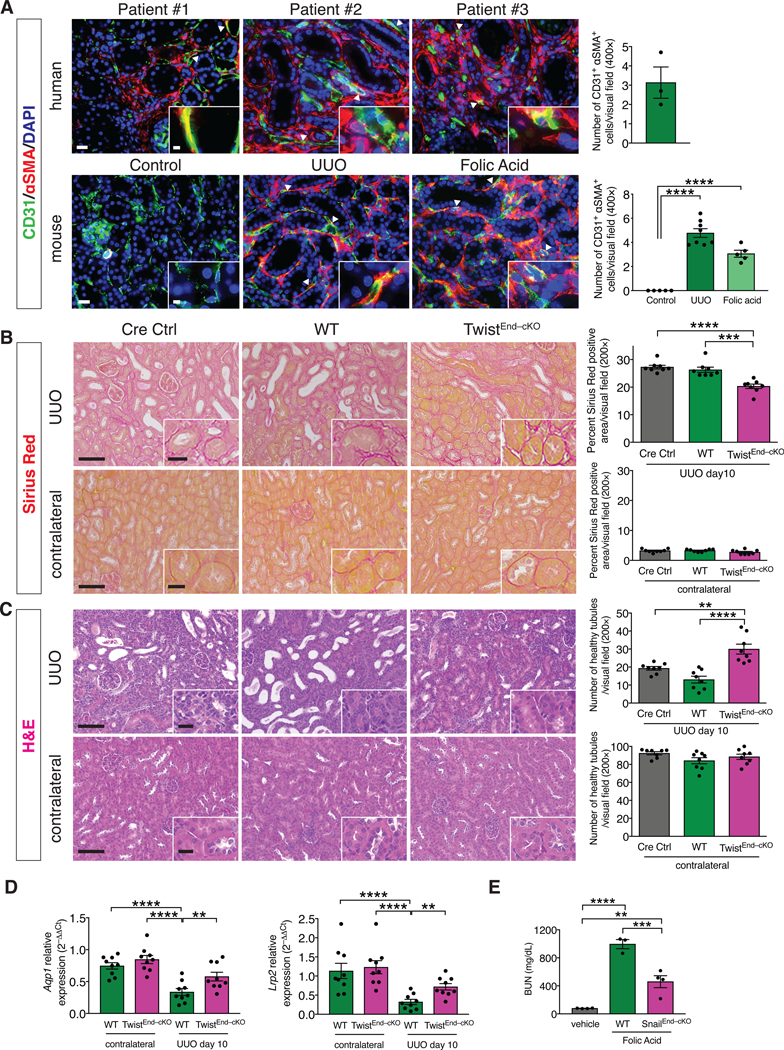
(A) Immunolabeling for CD31 and αSMA in kidneys from human biopsies of obstructive and allograft nephropathies, n = 3 patients (top), and from UUO and Folic acid-induced experimental models of kidney fibrosis, n = 8 and n = 5 mice for each group, respectively (bottom). Scale bars: 20 μm. Insets: 5 μm. DAPI: nuclei. (B) Representative images of Sirius Red staining and quantification of Sirius Red positive area in UUO (top) and contralateral (bottom) kidneys from the indicated experimental groups. Cre Ctrl n = 8 mice, WT n = 8 mice, TwistEnd–cKO n = 8 mice. (C) Representative H&E images and quantification of the number of healthy tubules in UUO (top) and contralateral (bottom) kidneys from the indicated experimental groups. Cre Ctrl n = 8 mice, WT n = 8 mice, TwistEnd–cKO n = 8 mice. Scale bars: 100 μm. Insets: 25 μm. (D) Relative transcript levels of Aqp1 (which encodes Aquaporin 1) and Lrp2 (which encodes Megalin) in the kidneys of the indicated experimental groups. WT and TwistEnd–cKO n = 9 mice for each group. (E) Blood urea nitrogen (BUN) levels. Vehicle n = 4 mice, WT n = 3 mice, SnailEnd–cKO n = 4 mice. Data are presented as mean ± s.e.m. One-way analysis of variance (ANOVA) with Dunnett’s post-hoc analysis (A) or Tukey post-hoc analysis (B–E). **P < 0.01, ***P < 0.001, ****P < 0.0001.
Adult (8 weeks old) mice were treated with daily injections of tamoxifen (Tmx) for five consecutive days (fig. S1F). One week after the last injection, the efficacy of the tamoxifen induced Cre-recombinase was evaluated by performing PCR for the recombined Twist1 and Snail alleles on genomic DNA extracted from TwistEnd–cKO, SnailEnd–cKO and WT kidneys. Tamoxifen-induced, Cre recombinase-mediated excision of exons 1–2 of Twist and exon 3 of Snai1 was confirmed with the detection of the expected ~300 bp PCR product in the TwistEnd–cKO [35] and SnailEndMT–cKO kidneys (fig. S1G–J). H&E, Sirius red and Masson Trichrome staining (MTS) showed no overt histopathological changes in the kidneys of TwistEnd–cKO and SnailEndMT–cKO mice following Tmx (day 7 after the last Tmx injection) when compared to WT control kidneys (fig. S2A–F). Next, Tmx-treated TwistEnd–cKO, SnailEnd–cKO and WT control mice were subjected to UUO to induce kidney fibrosis 7 days after the last Tmx injection, and the mice were euthanized 3 and 10 days after the UUO surgery (fig. S1F). Histopathological analyses of Sirius red and Masson trichrome stained kidneys showed a 25% reduction in cortical interstitial fibrosis in the obstructed (UUO) kidneys of TwistEnd–cKO mice when compared to WT control UUO kidneys (Fig. 1B and fig. S3A). Consistent with reduced fibrosis, immunolabeling for interstitial collagen I deposition and αSMA+ myofibroblasts were also significantly reduced in the UUO kidneys of TwistEnd–cKO mice compared to WT control (fig. S3B and S3C). Quantitative real-time PCR (qPCR) analysis showed decreased expression of Col1a1 (which encodes type I collagen alpha 1 chain) and Acta2 (which encodes αSMA) in TwistEnd–cKO kidneys compared to WT kidneys (fig. S3D). The kidneys of TwistEnd–cKO mice were significantly protected from tubular damage compared to WT mice, as determined by morphometric evaluation of the healthy tubules present in the fibrotic kidneys (Fig. 1C) and expression of the functional tubular epithelial-specific markers Aqp1 (which encodes Aquaporin 1) and Lrp2 (which encodes Megalin) (Fig. 1D).
Similar to TwistEnd–cKO mice, SnailEnd–cKO mice were also protected from UUO-induced kidney fibrosis compared to WT control mice, as indicated by reduced interstitial collagen I, improved tubular health, and decreased accumulation of αSMA+ myofibroblasts (fig. S4A–C and S5A–D). UUO and contralateral kidneys from mice carrying only the Cdh5CreERT2 transgene but lacking the conditional Twist1 or Snai1 alleles (Cdh5CreERT2+;Twist+/+ and Cdh5CreERT2+;Snail+/+ mice, respectively; hereafter referred as Cre Ctrl mice) were also analyzed to exclude Cre–dependent off target effects. Cre Ctrl UUO kidneys displayed a fibrotic phenotype comparable to that of the WT mice (Fig. 1B–C and fig. S3A–C, S4A–B, and S5A–C), and kidneys from control sham-operated mice did not present with fibrosis or compromised tubular health (fig. S6A–F). The amelioration of kidney fibrosis observed in the TwistEnd–cKO and SnailEnd–cKO mice was also evident at an early time point in disease progression (3 days after UUO), with TwistEnd–cKO and SnailEnd–cKO kidneys showing decreased interstitial fibrosis and improved tubular health when compared to WT kidneys (fig. S7A–F and S8A–F). Similarly, when given folic acid (FA) to induce nephropathy (fig. S9A), SnailEnd–cKO kidneys displayed reduced interstitial fibrosis, improved tubular health (fig. S9B–E), and improved renal function, when compared to FA–treated control WT mice (Fig. 1E). Together, these results indicate that conditional deletion of Twist1 and Snai1 in endothelial cells limits progression of interstitial fibrosis and preserves tubular epithelial health and function.
Suppression of EndMT in renal fibrosis using inducible, conditional genetic deletion of Twist1 or Snai1 in endothelial cells reduces inflammation
We performed RNA-Seq to comprehensively define the transcriptomic changes in renal fibrosis with and without the conditional deletion of Twist1 and Snai1 in endothelial cells. Whole kidneys from WT, TwistEnd–cKO and SnailEnd–cKO mice undergoing UUO (day 10) were compared to contralateral as well as healthy kidney controls. Gene set enrichment analysis (GSEA) revealed a significant reduction of the inflammatory response (Fig. 2A), as well as in all of the major inflammatory pathways (IL6/JAK/STAT3, TNFα and IL2/STAT5 signaling, IFNα and IFNγ response, fig. S10, A and C) in TwistEnd–cKO and SnailEnd–cKO UUO kidneys compared to WT. Real-time qPCR analysis of transcriptional levels of proinflammatory genes such as Il1b, Il6, Tnfa, Tgfb, Ccl2, and Ccl5 confirmed a significant transcriptional upregulation of these inflammatory cytokines in the fibrotic kidneys compared to contrataleral controls, and showed a significant reduction in transcript levels in the TwistEnd–cKO and SnailEnd–cKO kidneys compared to the WT UUO kidneys (Fig. 2B and fig. S10, B and D).
Figure 2. Endothelial deletion of Twist1 or Snai1 reduces inflammation and CD8+ T cells infiltration in the fibrotic kidney.
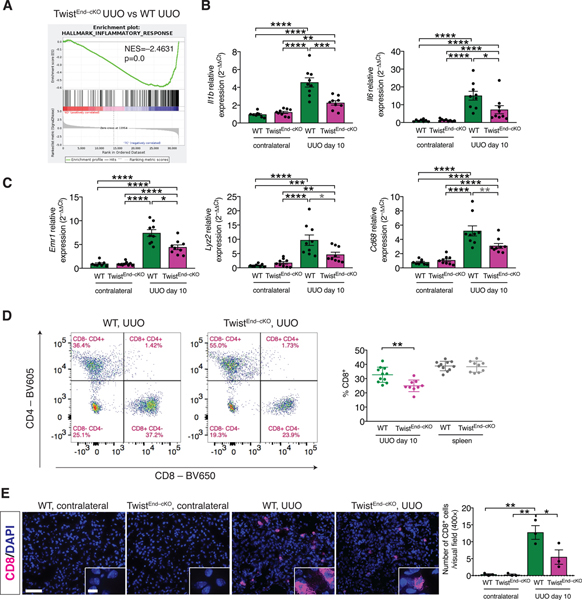
(A) GSEA enrichment plots of Hallmark gene dataset associated with the inflammatory response in the kidneys of TwistEnd–cKO UUO compared to WT UUO mice. All groups, n = 3 mice for each group. (B) Relative transcript levels of Il1b (which encodes interleukin-1β) and Il6 (which encodes interleukin-6) in the kidneys of the indicated experimental groups. WT contr. n = 8 mice, WT UUO n = 9, TwistEnd–cKO n = 9 mice. (C) Relative transcript levels of Emr1 (which encodes F4/80), Lyz2 (which encodes lysozyme) and Cd68 in the kidneys of the indicated experimental groups. WT contr. n = 8 mice, WT UUO n = 9, TwistEnd–cKO n = 9 mice. (D) Flow cytometry analysis of the percentage of CD8+ cells in kidneys and spleens of the indicated experimental groups. Data are presented as mean ± s.d. WT n = 11 mice, TwistEnd–cKO n = 9 mice. (E) Immunolabeling for CD8 in UUO and contralateral kidneys of the indicated experimental groups and respective quantification. All groups, n = 3 mice for each group. Scale bars: 50 μm. Insets: 12.5 μm. DAPI: nuclei. Data are presented as mean ± s.e.m in A, B, C, E. One-way analysis of variance (ANOVA) with Tukey post-hoc analysis, with gray stars indicating the use of unpaired two-tailed t-test. * P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Because immune infiltration substantially contributes to fibrosis [36] and endothelial cells exert a prominent role in the recruitment of immune cells at the site of the damage, we profiled the immune cell infiltrate of UUO and contralateral kidneys and spleens by flow cytometry (fig. S11A). UUO kidneys showed a significant increase in immune infiltration compared to the contralateral kidneys (fig. S11B and fig. S12A), including monocytes/macrophages (CD11b+GR–), myeloid-derived suppressor cells (CD11b+Ly6C+ and CD11b+Ly6G+), and lymphocytes (CD4+ effector T cells, CD4+FoxP3+ regulatory T cells and cytotoxic CD8+ T cells) (fig. S11B and fig. S12A), of of which have been implicated in the pathogenesis of renal fibrosis [37, 38]. Although the frequency of CD11b+GR– monocytes/macrophages was not changed in EndMT–cKO fibrotic kidneys compared to WT fibrotic kidneys, real-time qPCR analysis of differentiation markers such as Lyz2 (which encodes lysozyme), Emr1 (which encodes F4/80) and Cd68 (which encodes Cd68) indicated a decrease in these transcripts in the UUO kidneys of TwistEnd–cKO and SnailEnd–cKO mice compared to the WT (Fig. 2C). TwistEnd–cKO and SnailEnd–cKO fibrotic kidneys also presented with a reduced frequency of CD8+ T cells compared to WT fibrotic kidneys (Fig. 2D and fig. S12B). CD8 immunolabeling of kidney frozen sections showed a significant decrease in CD8+ T cell infiltration in the fibrotic kidneys of TwistEnd–cKO and SnailEnd–cKO compared to WT fibrotic kidneys (Fig. 2E and fig. S12C). The decrease in the frequency of CD8+ T cells was evident at the early phase of the injury response (day 3 after UUO), albeit only in the TwistEnd–cKO UUO kidneys (fig. S13A and B and fig. S14A–C). The frequencies of CD8+Ki67+ cells present in TwistEnd–cKO UUO kidneys was also significantly lower than in WT UUO day 10 kidneys (fig. S13C and D). We evaluated whether a differential expression of endothelial adhesion molecules in the EndMT–cKO kidneys could be associated with the decreased CD8+ T cell infiltration. Real-time qPCR analysis indicated that, although the expression of the adhesion genes Vcam1 and Icam1 was markedly increased in the fibrotic kidneys compared to the contralateral controls, the transcriptional levels were similar between the TwistEnd–cKO and SnailEnd–cKO UUO kidneys and WT kidneys (fig. S14D). Collectively these results support that endothelial cell deletion of Twist and Snail limits interstitial fibrosis by dampening inflammation and inflammatory signaling, resulting in a reduced infiltration of cytotoxic CD8+ T cells.
VE-cadherin–Cre and Tie1–Cre driven Twist1 and Snai1 conditional deletion generates viable adult mice with reduced propensity for kidney fibrosis
Tie2-Cre–driven endothelial deletion of Snail results in embryonic lethality due to vascular defects [39]. However, mice with Tie2-Cre–driven deletion of Twist1 are viable [40–44], although they display lung abnormalities [40]. These results raise the possibility of a distinct contribution of endothelial-derived Twist or Snail in embryonic development. To further address the functional contribution of Twist and Snail associated endothelial cells in embryonic development and kidney fibrosis, we bred Twist1L/L and Snai1L/L mice with mice expressing Cre under the constitutive Cdh5 (VE-cadherin) promoter (fig. S15 A–B). Cdh5Cre+;TwistL/L and Cdh5Cre+;Snai1L/L mice were viable and genotypes matched the expected Mendelian ratio (fig. S15C–D and fig. S16A–H).
Cdh5Cre+;TwistL/L and Cdh5Cre+;Snai1L/L mice were challenged with UUO to induce kidney fibrosis. Kidneys from Cdh5Cre+;TwistL/L mice subjected to UUO displayed improved renal outcome, compared to those from Cdh5Cre–;TwistL/L and Cdh5Cre+;Twist+/+ (WT) mice, as indicated by the reduction of interstitial fibrosis and improved tubular health (fig. S18A–C). Similarly, we observed reduced fibrosis and improved tubular health in UUO kidneys of the Cdh5Cre+;Snai1L/L mice compared to those of Cdh5Cre–;Snai1L/L or Cdh5Cre+;Snai1+/+ (WT) mice (fig. S19A–C). Similarly, fibrosis was decreased, although to a milder extent, in Tie1Cre+;TwistL/L and Tie1Cre+;Snai1L/L UUO kidneys compared to WT UUO kidneys (fig. S15E–H, fig. S17A–H, fig. S20A–C and fig. S21A–C). Collectively, the results, obtained using three distinct endothelial-driven Cre-LoxP mediated conditional targeting of Twist1 and Snai1, indicate that the deletion of these EndMT transcription factors limits kidney fibrosis.
Endothelial conditional deletion of Twist1 and Snai1 inhibits EndMT
The mesenchymal-driving transcription factors Twist and Snail have been implicated in the activation of EndMT [17, 44]. Twist overexpression induces EndMT in human pulmonary arterial endothelial cells [44] and its siRNA-mediated silencing blocks hypoxia-induced EndMT in pulmonary microvascular endothelial cells [21]. In vitro silencing of Snail prevents TGFβ– and shear stress–induced EndMT in human cutaneous microvascular endothelial cells, embryonic stem cell-derived endothelial cells, and HUVECs [17–19]. To measure EndMT suppression in vivo during kidney fibrosis in our genetic mouse models, we first confirmed the fidelity of our Cre/Lox systems by evaluating the DNA of kidneys for recombined Twist1 and Snai1 alleles (fig. S22A–D). Real-time qPCR analysis confirmed reduced expression of Twist1 and Snai1 in UUO kidneys (fig. S22E–F). Confocal imaging of TwistEnd–cKO and WT kidney tissues co-immunolabeled for the endothelial marker CD31 and the mesenchymal marker αSMA showed endothelial cells undergoing partial EndMT (as indicated by co-localization of CD31 and αSMA) in WT fibrotic kidneys 10 days after UUO, and significantly reduced partial EndMT in TwistEnd–cKO and SnailEnd–cKO fibrotic kidney compared to WT (Fig. 3A). In line with previous studies reporting the activation of EndMT in the early phases of the injury response [10, 45, 46], we detected partial EndMT in the WT UUO kidneys at day 3 after UUO (Fig. 3A). GSEA analysis of RNA sequencing of fibrotic kidneys confirmed the reduction of the EndMT signature in the UUO kidneys from TwistEnd–cKO and SnailEnd–cKO mice compared to WT (Fig. 3B). A heat map representation of the expression levels of known Twist and Snail transcriptional targets during EndMT, as identified by published studies [17–21], also indicate a decrease in the expression of Twist and Snail transcriptional targets in TwistEnd–cKO and SnailEnd–cKO UUO kidneys compared to the WT UUO (Fig. 3C).
Figure 3. EndMT occurs in UUO-induced kidney fibrosis and is inhibited by the conditional deletion of Twist1 or Snai1.
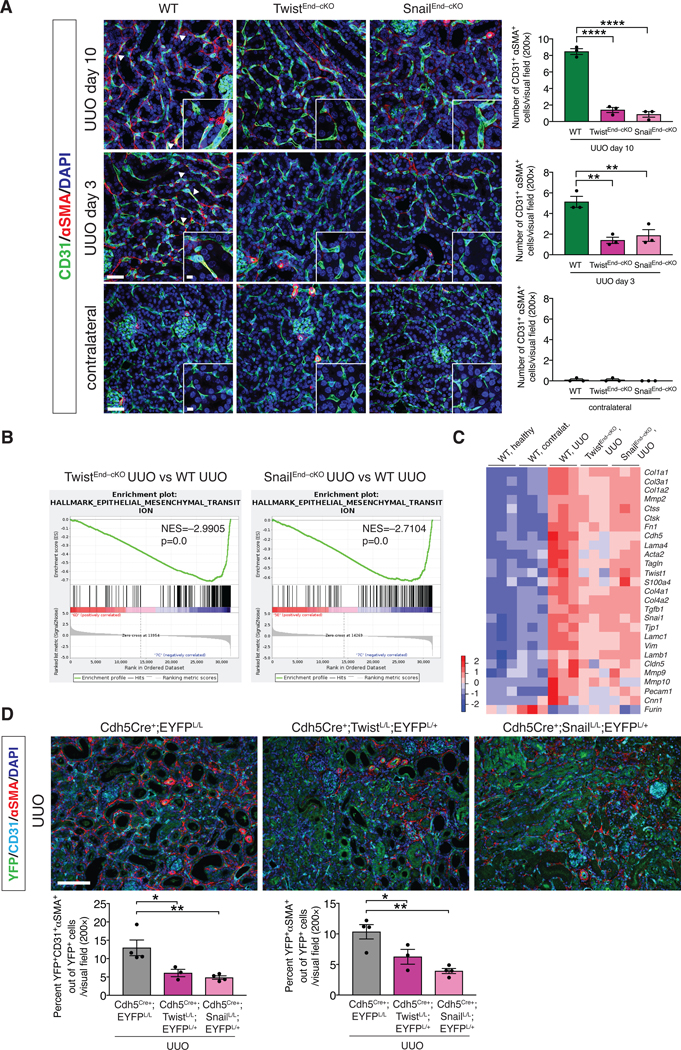
(A) Maximum intensity orthogonal projections of CD31 and αSMA co-immunolabeling of UUO kidneys from the indicated experimental groups and respective quantification of the number of CD31+αSMA+ double positive cells per visual field (200×). Cre Ctrl, WT and TwistEnd–cKO, n = 3 mice; Cre Ctrl, WT and SnailEnd–cKO n = 3 mice. Scale bars: 40 μm. Insets: 10 μm. DAPI: nuclei. (B) GSEA enrichment plots of Hallmark gene dataset associated with epithelial-to-mesenchymal transition in the kidneys of TwistEnd–cKO UUO or SnailEnd–cKO UUO compared to WT UUO mice. All groups, n = 3 mice for each group. (C) Heatmap representing the intensity of expression of EndMT-associated Twist and Snail transcriptional target genes in the kidneys of the indicated experimental groups. Columns represent individual mouse kidney samples. All groups, n = 3 mice for each group. (D) Immunolabeling for YFP, CD31 and αSMA of UUO kidneys from the indicated experimental groups. Cdh5Cre+;EYFPL/L n = 4 mice, Cdh5Cre+;TwistL/L;EYFPL/+ n = 3 mice, Cdh5Cre+;Snai1L/L;EYFPL/+ n = 4 mice. Scale bars: 100 μm. Data is presented as mean ± s.e.m. One-way analysis of variance (ANOVA) with Tukey post-hoc analysis. *P < 0.05, **P < 0.01, ***P < 0.001.
We next employed a lineage tracing (fate–mapping) approach to label endothelial cells by breeding both the inducible and constitutive Cdh5-driven Cre mice with mice carrying the Rosa26-LSL-EYFP reporter allele (fig. S23A–D). The number of YFP+ endothelial cells co-expressing αSMA was significantly reduced in both TwistEnd–cKO and SnailEnd–cKO UUO kidneys compared to WT UUO kidneys (fig. S24A–B). Finally, we utilized tyramide signal amplification (TSA) staining to capture EndMT by immunolabeling the endothelial marker CD31, lineage traced (EYFP) endothelial cells, and the mesenchymal marker αSMA (Fig. 3D). Among the YFP+ endothelial lineage traced cells in the UUO kidneys, 13% co-expressed CD31 and αSMA, representing endothelial cells with partial EndMT program. Approximately 10% of YFP+ cells expressed αSMA without detectable expression of CD31, possibly capturing endothelial cells with a more advanced EndMT program (Fig. 3D). Both partial and more advanced states of EndMT were reduced in TwistEnd–cKO and SnailEnd–cKO UUO kidneys compared to WT UUO kidneys (Fig. 3D). Together, multiple techniques support the specific decrease in EndMT in fibrotic kidneys upon deletion of Twist or Snail in endothelial cells.
Suppression of EndMT reduces vascular leakage in kidney fibrosis
Injury to the renal vasculature is associated with activation of a fibrogenic response, leading to damage of the parenchyma [47]. We investigated whether the activation of the EndMT program contributes to the injury-induced endothelial dysfunction in kidney fibrosis. We first ascertained whether EndMT associated with kidney fibrosis impaired endothelial cell-cell junctions, possibly impacting vascular permeability. Systemic injection of FITC-conjugated dextran (70 kDa) revealed that TwistEnd–cKO and SnailEnd–cKO UUO kidneys displayed a significantly lower FITC-dextran leakage compared to WT UUO kidneys (Fig. 4A-B and fig. S25A–B). These data suggest that inhibition of EndMT preserves the integrity of the peritubular capillaries. Contralateral kidneys did not display vascular leakage, as demonstrated a lack of FITC-dextran in the kidney interstitium (fig. S25A–B). These findings were recapitulated when using intersitial albumin as a measure of vascular leakage instead of FITC-dextran. Immunohistochemistry analysis revealed that albumin was confined to blood vessels in contralateral control kidneys, whereas it was extravasated in the interstitial area in UUO kidneys in WT mice (Fig. 4C–D). TwistEnd–cKO and SnailEnd–cKO UUO kidneys displayed significantly less albumin in the interstitial space (Fig. 4C–D). Additionally, vascular density, measured by counting the number of patented albumin+ capillaries per tubular segment, indicated a substantial decrease in the capillaries/tubule ratio in the WT UUO kidneys compared to contralateral, suggestive of vascular rarefaction, which was significantly reversed in the UUO kidneys of TwistEnd–cKO and SnailEnd–cKO mice (Fig. 4E).
Figure 4. Blocking EndMT limits vascular leakage.
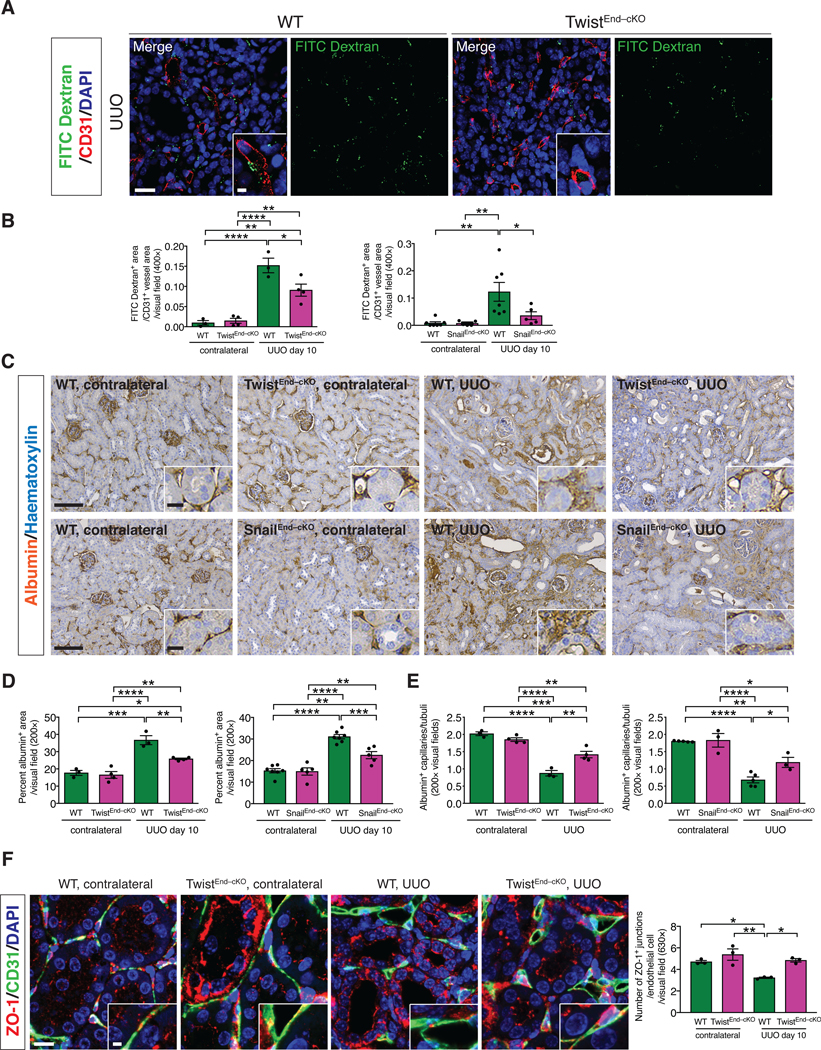
(A) Immunolabeling for CD31 and visualization of FITC-conjugated dextran in kidneys from the indicated experimental groups. Scale bars: 20 μm. DAPI: nuclei. (B) Quantification of the ratio of FITC-dextran+ area per CD31+ vessel area in UUO and contralateral kidneys of the indicated experimental groups. WT n = 3 mice, TwistEnd–cKO n = 4 mice; WT n = 7 mice and SnailEnd-cKO n = 5 mice. (C–D) Immunohistochemistry analysis of albumin in kidneys from the indicated experimental groups (C) and respective quantification (D). WT n = 3 mice, TwistEnd–cKO n = 4 mice; WT n = 7 and SnailEnd-cKO n = 5 mice. Scale bars: 100 μm. Insets: 25 μm. (E) Quantification of peritubular patented capillary density performed on the albumin immunohistochemistry analysis presented in (C). WT n = 3 mice, TwistEnd–cKO n = 4 mice; WT n = 5 and SnailEnd-cKO n = 3 mice. (F) Maximum intensity orthogonal projections of ZO-1 and CD31 co-immunolabeling and respective quantification of kidneys from the indicated experimental groups. All groups, n = 3 mice for each group. Scale bars: 30 μm. Insets: 7.5 μm. Data are presented as mean ± s.e.m. One-way analysis of variance (ANOVA) with Tukey post-hoc analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We tested whether the rescue in the increased vascular permeability of EndMT–cKO fibrotic kidneys could be associated with a compromised VEGF receptor 2 (VEGFR2) expression on endothelial cells. Endothelial cells significantly upregulated the expression of VEGFR2 under fibrotic conditions, as demonstrated by the increase in the fraction of CD31+VEGFR2+ cells in UUO kidneys compared to contralateral kidneys (fig. S25C). The increase in VEGFR2 was similar between WT and TwistEnd–cKO or SnailEnd–cKO UUO kidneys, suggesting that the modulation of vascular permeability by EndMT was not due to alterations in VEGFR2 signaling (fig. S25C). Twist may act as a negative regulator of Tie2 expression in a mouse model of lung fibrosis [40]; however, the percentage of CD31+Tie2+ cells in UUO kidneys of EndMT–cKO mice did not differ from those in control mice (fig. S25D).
We next investigated whether EndMT could alter vascular permeability by modulating the stability of the endothelial intercellular junctions. Contralateral and UUO kidneys were co-immunolabed with CD31 and the tight junction-associated protein ZO-1. The number of ZO-1+ junctions per CD31+ endothelial cell was decreased in WT UUO kidneys compared to the contralateral kidneys. Conversely, the number of ZO-1+ tight junctions was preserved in the UUO kidneys of TwistEnd–cKO or SnailEnd–cKO mice compared to the WT UUO kidneys (Fig. 4F and fig. S25E). Together, these results support that EndMT facilitates fibrosis in the kidney by inducing vascular rarefaction, disrupting vascular integrity and promoting leakage through altering the endothelial cell-cell junctions.
EndMT induces tissue hypoxia and reprograms tubular epithelium metabolism
Insufficient blood perfusion and increased interstitial pressure from leaky vessels may result in inadequate oxygen tension in the affected tissues, generating hypoxia in the injured parenchyma [48]. We tested whether the decreased vascular leakage observed in the TwistEnd–cKO and SnailEnd–cKO fibrotic kidneys limited tissue hypoxia associated with fibrosis. Immunofluorescence analysis revealed that tubular epithelial cells in WT UUO kidneys displayed high levels transmembrane expression of CAIX, a marker for hypoxia, both at 3 and 10 days after surgery (Fig. 5A and fig. S26A and S27A), and that CAIX was barely detected in healthy kidneys (fig. S26A). CAIX+ tubular epithelial cells were decreased in TwistEnd–cKO and SnailEnd–cKO UUO kidneys compared to the WT kidneys, both at 3 and 10 days after UUO surgery (Fig. 5A and fig. S26C,G and fig. S27A,E) supporting a positive correlation between decreased vascular leakage and reduced number of hypoxic tubular epithelial cells in the kidneys of EndMT–cKO mice during fibrosis. GSEA analysis confirmed the reduced transcriptional hypoxic signature in the fibrotic kidneys of TwistEnd–cKO and SnailEnd–cKO mice compared to the WT (Fig. 5B and fig. S26B).
Figure 5. Blocking EndMT protects against tubular hypoxia and preserves metabolic homeostasis.
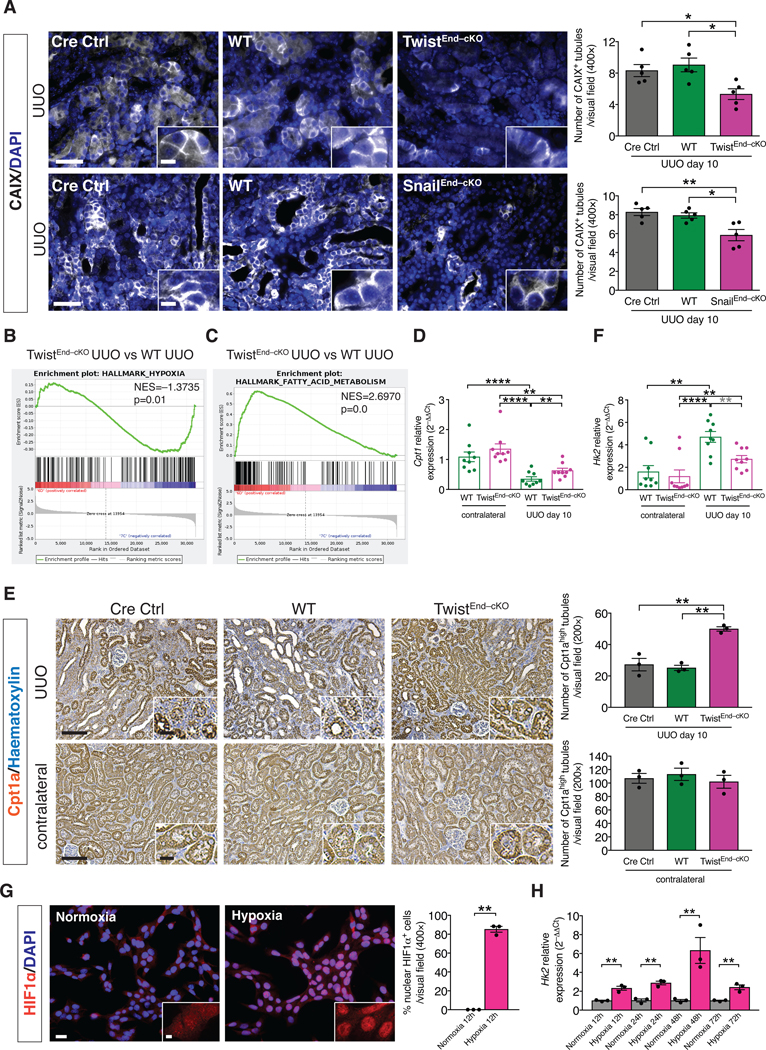
(A) Immunolabeling for the hypoxia marker CAIX of kidneys from the indicated experimental groups and respective quantification. All groups, n = 5 mice for each group. Scale bars: 50 μm. Insets: 12.5 μm. DAPI: nuclei. (B–C) GSEA enrichment plots of Hallmark gene dataset associated with hypoxia and fatty acid metabolism in the kidneys of TwistEnd–cKO UUO compared to WT UUO mice. All groups, n = 3 mice for each group. (D) Relative transcript levels of the metabolism-related gene Cpt1 (which encodes carnitine palmitoyltransferase 1A) in the kidneys of the indicated experimental groups. All groups, n = 9 mice for each group. (E) Immunohistochemistry analysis of the FAO marker Cpt1a in kidneys from the indicated experimental groups and respective quantification. All groups, n = 3 mice for each group. Scale bars: 100 μm. Insets: 25 μm. (F) Relative transcript levels of the metabolism-related gene Hk2 (which encodes hexokinase 2) in the kidneys of the indicated experimental groups. All groups, n = 9 mice for each group. (G) Immunolabeling for the hypoxia transcription factor HIF1α and relative quantification in MCT cells exposed to normoxia or hypoxia for 12 hrs. All groups, n = 3 biological replicates for each group. Scale bar: 20 μm. Insets: 5 μm. (H) Relative transcript level of Hk2 in MCT cells exposed to normoxia or hypoxia for 12 to 72 hrs. All groups, n = 3 biological replicates for each group. Data are presented as mean ± s.e.m. One-way analysis of variance (ANOVA) with Tukey post-hoc analysis with gray stars indicating the use of unpaired two-tailed t-test. Unpaired two-tailed Student’s t test (G–H). *P < 0.05, **P < 0.01, ****P < 0.0001.
Hypoxia is a known driver of metabolic perturbations [49]. RNAseq and GSEA analysis revealed that the top enriched pathways in TwistEnd–cKO and SnailEnd–cKO compared to the WT UUO kidneys were associated with metabolism, specifically fatty acid metabolism and oxidative phosphorylation (Fig. 5C and fig. S28A and B). Defective fatty acid oxidation (FAO) in renal tubular epithelial cell has been reported in both human CKD and in experimental models of kidney fibrosis [50], and our data indicate a positive correlation between decreased intratubular hypoxia, improved FAO metabolism and amelioration of fibrosis. We further evaluated the metabolic changes associated with EndMT-induced hypoxia in kidney fibrosis, specifically in tubular epithelial cells (TECs). Immunohistochemistry and qPCR analyses for rate-limiting enzymes of FAO (Cpt1a) suggested that FAO was impaired in TECs in fibrotic kidneys (Fig. 5D–E), as previously reported [50]. Conversely, expression of FAO-associated genes was higher in TwistEnd–cKO and SnailEnd–cKO UUO kidneys than in WT UUO kidneys (Fig. 5D–E and fig. S26D and H, S27B and F and S28C–E). In addition, the expression of the gene encoding HK2, a rate limiting enzyme during glycolysis, was increased in fibrotic kidneys compared to contralateral control kidneys, an effect that was less pronounced in TwistEnd–cKO and SnailEnd–cKO UUO kidneys (Fig. 5F, fig. S26E and I and S27C and G). These results suggest that suppression of EndMT preserves the expression of metabolic genes in fibrotic kidneys and preserves FAO in TECs.
We next exposed MCT mouse proximal tubular epithelial cells [51] to hypoxic conditions (1% O2). The hypoxic response of MCT cells was evident after 12 hours of 1% O2 exposure, as demonstrated by the nuclear stabilization of the hypoxia transcription factor HIF1α (Fig. 5G) and increased expression of the HIF1α-target gene Bnip3 (fig. S28F). Expression of glycolysis genes such as Hk2, Pfkl Pkm, Pdk1, Ldha and Slc2a1 was significantly induced during hypoxia (Fig. 5H and fig. S29A). Conversely, FAO genes such as Cpt1, Cpt2 and Acox1 were downregulated (fig. S29B). Associated with the hypoxia-induced increase in glycolytic gene expression, production and export of lactate was increased, as demonstrated by the increased expression of Slc16a3 (which encodes monocarboxylate transporter 4) and the increased concentration of lactate in the conditional media of MCT cells exposed to hypoxia (fig. S29A and C). These results support that hypoxia in TECs alters their metabolism by enhancing glycolysis while suppressing FAO. To investigate the functional consequences of increased glycolysis in kidney fibrosis, we inhibited glycolysis in vivo by treating mice with the glycolysis inhibitor, 3-bromopyruvate (3BP). Pharmacological targeting of glycolysis led to tubular health protection and inhibition of fibrosis, as demonstrated by the increase in the number of healthy tubules and decreased interstitial collagen in the UUO kidneys of 3BP-treated mice (fig. S29D).
EndMT-induced metabolic alterations of TECs relies in part on increased Myc activity
RNA-seq analysis has identified the presence of a glycolytic signature in human fibrotic kidneys, and the transcription factor MYC is a driver of these glycolytic transcriptional changes [52]. We evaluated whether Myc plays a role in the EndMT-mediated reprogramming of TEC metabolism. Expression analysis by qPCR in MCT cells revealed a 2 to 3–fold increase of c-Myc transcript levels in cells exposed to hypoxia compared to normoxic cells (Fig. 6A). GSEA analysis indicated a transcriptional enrichment of Myc target genes in WT compared to the TwistEnd–cKO and SnailEnd–cKO UUO kidneys, therefore supporting a positive correlation between intratubular hypoxia and Myc expression (Fig. 6B). In fact, the nuclear expression of Myc was increased in TECs in UUO kidneys at both 3 and 10 days after surgery, compared to those in contralateral control kidneys (Fig. 6C, fig. S26F and J and fig. S27D and H). This epithelial upregulation of Myc was significantly decreased in the UUO kidneys of TwistEnd–cKO and SnailEnd–cKO mice compared to UUO WT control mice (Fig. 6C).
Figure 6. Hypoxia-induced upregulation of Myc supports kidney fibrosis.
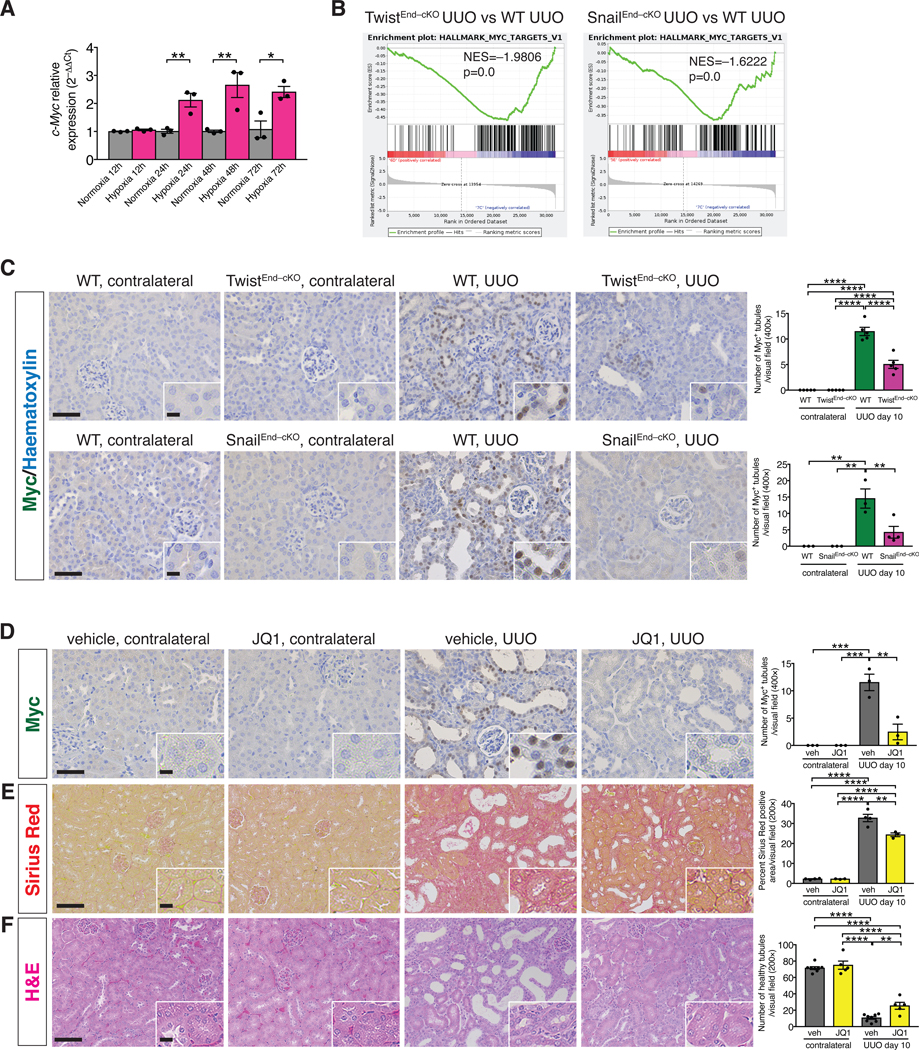
(A) Relative transcript level of c-Myc in MCT cells exposed to normoxia or hypoxia for 12 to 72 hrs. All groups, n = 3 biological replicates for each group. (B) GSEA enrichment plots of Hallmark gene dataset associated with Myc transcriptional targets in the kidneys of TwistEnd–cKO and SnailEnd–cKO UUO mice compared to WT UUO mice. All groups, n = 3 mice for each group. (C) Immunohistochemistry analysis of Myc in kidneys from the indicated experimental groups and respective quantification. WT and TwistEnd–cKO n = 5 mice; WT n = 3 mice, SnailEnd-cKO contr. n = 3 mice and SnailEnd-cKO UUO n = 4 mice. (D–F) Representative images of Myc immunohistochemistry (D), Sirius Red (E), and H&E (F) staining and respective quantification of UUO and contralateral kidneys from the indicated experimental groups. Vehicle n = 3–8 mice for each group, JQ1 n = 3–5 mice for each group. Scale bars: 50 μm (C–D) and 100 μm (E–F). Insets: 12.5 μm (C–D) and 25 μm (E–F). Data are presented as mean ± s.e.m. One-way analysis of variance (ANOVA) with Tukey post-hoc analysis (C–F). Unpaired two-tailed Student’s t test (A). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To test whether Myc plays a functional role in mediating EndMT-induced epithelial reprogramming of metabolism in kidney fibrosis, we pharmacologically targeted Myc in mice by employing the bromodomain inhibitor JQ1. JQ1 treatment was effective in supressing Myc expression (Fig. 6D) and kidney fibrosis (Fig. 6E), and was associated with decreased collagen deposition and improved tubular epithelial health (Fig. 6E–F and fig. S29E). We also genetically deleted c-Myc in TECs by breeding mice with a c-Myc conditional allele (MycL/L) with those expressing Cre under the proximal TEC–specific promoter, gamma glutamine transferase (γGTCre), to generate WT (γGTCre–;MycL/L) and Myc conditionally deleted mice (γGTCre+;MycL/L – hereafter referred to as MycγGT–cKO mice) (fig. S30A). Successful genetic deletion of Myc was confirmed by the detection of a ~500bp PCR product corresponding to the Cre-mediated recombined Myc allele in the kidneys of MyccKO kidneys compared to WT control[53] (fig. S30B). Immunohistochemical analysis showed that Myc levels were reduced in the TECs of MycγGT–cKO UUO kidneys (fig. S30C). Conditional deletion of Myc in proximal TECs decreased interstitial fibrosis and improved tubular health (Fig. 7A–B and fig. S30D–F). RNAseq performed on the whole UUO kidneys from MycγGT–cKO and WT mice confirmed the downregulation of Myc transcriptional targets in the MycγGT–cKO kidneys (Fig. 7C) and highlighted a strong enrichment for genes related to fatty acid metabolism and oxidative phosphorylation (Fig. 7D). This result was validated by the increase in the number of Cpt1a+ TECs detected by immunohistochemistry in the MycγGT–cKO UUO kidneys compared to WT kidneys (Fig. 7E and fig. S32C). Conversely, a downregulation of the glycolytic transcriptional signature (as determined by GSEA analysis) and a decrease in the number of HK2+ TECs (as detected by kidney immunostaining) were observed in MycγGT–cKO UUO kidneys compared to WT kidneys (Fig. 7D and F, and fig. S32D), supporting that suppression of Myc in TECs suppressed glycolysis and preserved FAO metabolism.
Figure 7. Genetic deletion of Myc in proximal tubular epithelial cells ameliorates kidney fibrosis and preserves metabolic homeostasis.
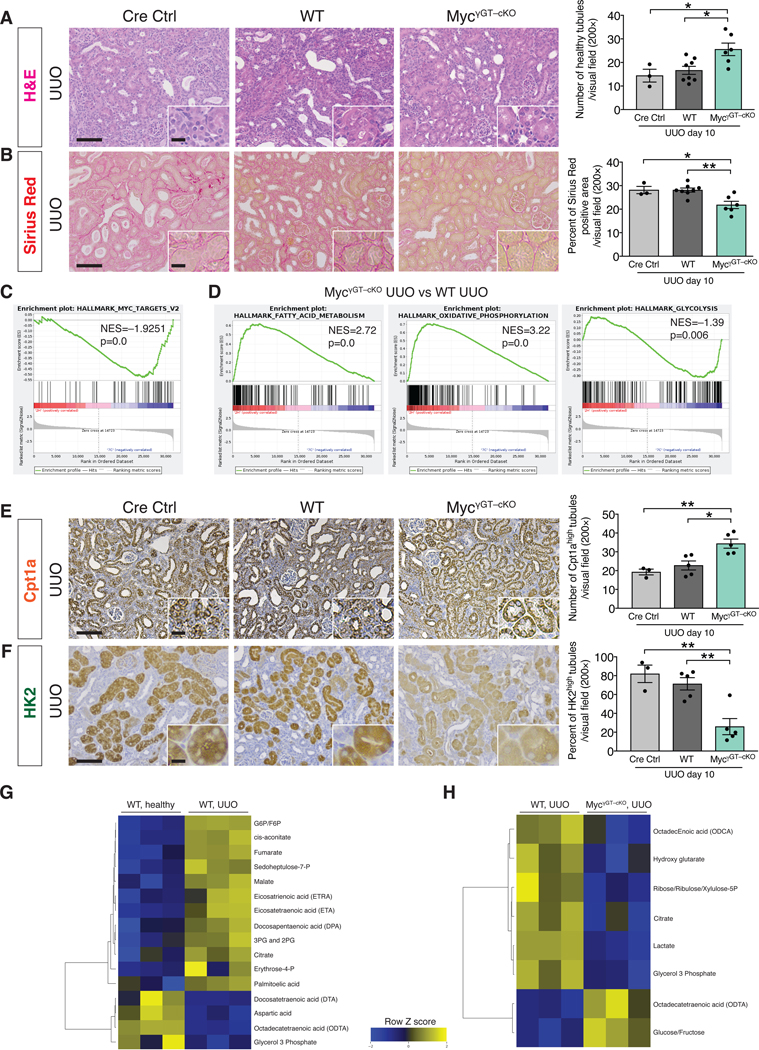
(A) Representative H&E images and quantification of the number of healthy tubules in UUO kidneys from the indicated experimental groups. Cre Ctrl n = 3 mice, WT n = 8 mice, MycγGT–cKO n = 6 mice. (B) Representative images of Sirius Red staining and quantification of Sirius Red positive area in UUO kidneys from the indicated experimental groups. Cre Ctrl n = 3 mice, WT n = 8 mice, MycγGT–cKO n = 6 mice. (C) GSEA enrichment plots of Hallmark gene dataset associated with Myc transcriptional targets in the kidneys of MycγGT–cKO UUO compared to WT UUO mice. All groups, n = 3 mice for each group. (D) GSEA enrichment plots of Hallmark gene dataset associated with fatty acid metabolism and oxidative phosphorylation in the kidneys of MycγGT–cKO UUO compared to WT UUO mice. All groups, n = 3 mice for each group. (E) Cpt1a immunohistochemistry in UUO kidneys from the indicated experimental groups and respective quantification. Cre Ctrl n = 3 mice, WT n = 5 mice, MycγGT–cKO n = 5 mice. (F) HK2 immunohistochemistry in UUO kidneys from the indicated experimental groups and respective quantification. Cre Ctrl n = 3 mice, WT n = 5 mice, MycγGT–cKO n = 5 mice. Scale bars: 100 μm. Insets: 25 μm. (G–H) Heat maps showing the altered metabolites (FDR < 0.25) in WT UUO kidneys compared to healthy kidneys (G) and in MycγGT–cKO UUO compared to WT UUO kidneys (H). Columns represent individual mouse kidney samples and rows refer to individual metabolites. Shades of yellow represent increased abundance of a metabolite and shades of blue represent decreased abundance of a metabolites. Data are presented as mean ± s.e.m. One-way analysis of variance (ANOVA) with Tukey post-hoc analysis. *P < 0.05, **P < 0.01.
We next validated these results by performing targeted metabolomics to assess fatty acids, glycolysis, and TCA cycle associated metabolites in healthy, WT UUO, and MycγGT–cKO UUO kidneys (Fig.7G and Fig. 7H). A set of metabolites displayed a distinct and signicant association with fibrotic kidneys compared to healthy kidneys (FDR < 0.25, Fig. 7G). Specifically, an accumulation of fatty acids (eicosatrienoic acid, eicosatetraenoic acid, docosapentaenoic acid and palmitoleic acid) was detected in fibrotic kidneys compared to healthy kidnesy, possibly reflecting their reduced utilization by the FAO pathway. Glycolysis and TCA intermediate metabolites (G6P, F6P, 3PG and 2PG, fumarate, amalate and citrate) were more abundant in the fibrotic kidneys compared to healthy kidneys, supporting an upregulation of the glycolytic pathway (Fig. 7G). In contrast, fibrotic kidneys with tubular epithelial deletion of Myc displayed decreased abundance of glycolysis intermediates (lactate) and metabolites derived from glycolysis intermediates (glycerol 3 phosphate, ribose/ribulose/xylulose 5P), and a concomitant accumulation of glucose/fructose, when compared to WT fibrotic kidneys (Fig. 7H).
Activated fibroblasts (myofibroblasts) participate in kidney fibrosis, and Myc expression in these stromal cells promotes renal fibrosis [52, 54]. To gain insight into the role of Myc in activated myofibroblasts in fibrotic kidneys, we genetically deleted c-Myc in myofibroblasts by breeding MycL/L mice with mice expressing Cre under the αSMA promoter (αSMACre) (fig. S31A–B). WT (αSMACre–; MycL/L) and myofibroblast-specific Myc knockout mice (αSMACre+;MycL/L – hereafter referred to as MycSMA–cKO mice) were phenotypically unremarkable compared to the WT littermate controls, and the mice were challenged with UUO (fig. S31C–E). Myc ablation in myofibroblasts of MycSMA–cKO mice did not impact the emergence of fibrosis or improve tubular epithelial health (fig. S31C–E and fig. S32A). Cpt1a immunostaining was also not altered in the MycSMA–cKO mice when compared to control mice (fig. S32B). Collectively, these results suggest that Myc upregulation in myofibroblasts does not play a rate-limiting role in the emergence of kidney fibrosis. Moreover, these data support that EndMT and the associated vascular damage drive fibrosis by affecting TEC metabolism through altered Myc expression, whereas altered Myc expression in myofibroblasts largely does not contribute to the development of fibrosis.
Discussion
EndMT was identified as a mediator of cardiac fibrosis in 2007, which was the first description of EndMT in adult organisms and in relation to pathological tissue remodeling [3]. TGFβ1 can induce EndMT in distinct pathologies [8, 23]. Notch, Wnt, NF-kB, TNFα, ET-1 and Cav-1 signaling pathways as well as hypoxia, oxidative stress, metabolic alterations and shear stress forces represent EndMT-inducing stimuli (reviewed in [8]). In vitro studies suggest that the transcription factors Snail and Twist can play a role in mediating TGFβ1 induced EndMT [17, 18, 45]. EndMT is recognized as a driver for atherosclerosis progression, and the extent of EndMT correlates with plaque instability [6, 7]. In other types of fibrotic diseases, such as pulmonary [55], hepatic [56] and intestinal [57] fibrosis, EndMT is a pathogenic determinant. In addition, EndMT contributes to the pathogenesis of fibrodysplasia ossificans progressiva [58], and to the development of the vascular malformations distinctive of cerebral cavernous malformation disease [59]. In renal fibrosis, multiple studies have identified features of EndMT in both experimental models of kidney fibrosis [4, 9, 11, 26] and in biopsies from CKD patients [12–14, 16]. In these studies, EndMT is associated with the development of fibrosis and is predominantly described as a transdifferentiation program that can generate fibroblasts, but the functional consequences of EndMT activation had not been investigated.
To define the functional, rate limiting contribution of EndMT to kidney fibrosis, we engineered mice to genetically target the acquisition of the EndMT program. Specifically, we probed whether the transcriptional factors that specify mesenchymal lineage, Twist and Snail, played a direct role in endothelial cells in the emergence of EndMT in kidney fibrosis, using multiple mouse models of kidney fibrosis. Our results provide evidence for a functional role of Twist and Snail in inducing EndMT in kidney fibrosis, and the conditional loss of Twist and Snail in endothelial cells was sufficient to limit EndMT and kidney fibrosis. These loss-of-function experiments highlight that the EndMT program is an injury response program of endothelial cells, which results in a loss of endothelial cell identity and acquisition of mesenchymal features. Such hybrid cells contribute to the loss of endothelial cell-cell junctions and vascular integrity, resulting in leaky capillaries and blood vessels. Compromised blood vessels lead to extravasation of blood proteins, including albumin, encouraging inflammation into this area of injury. Compromised vascular integrity also leads to tissue hypoxia and induces tubular epithelial cells stress. Hypoxia-damaged tubular epithelial cells launch many molecular pathways including elevation of glycolysis and suppression of FAO [31, 60]. Although the use of 3BP to suppress glycolysis could impact many distinct cell types in kidney fibrosis, suppression of glycolysis improved tubular health. We demonstrated that hypoxia mediated glycolysis in tubular epithelial cells was facilitated at least partially by Myc upregulation. Myc in turn upregulates many genes that encode enzymes in the glycolytic pathway, including HK2 and LDHA [61]. Suppression of EndMT led to a decrease in Myc expression, and genetic deletion of Myc in in TECs or pharmacological inhibition of Myc limited kidney fibrosis. Genetic deletion of Myc in fibroblasts did not impact fibrosis, supporting that kidney fibrosis is a disease of epithelial parenchyma damage, whose protection leads to suppression of fibrosis. An interesting corollary finding from these experiments is that Myc-associated metabolic reprograming of activated fibroblasts was not required for their accumulation and participation in renal fibrosis.
A study exploring the mechanistic link between tubular epithelial metabolic defects and induction of renal fibrosis highlighted that the metabolic defects induced by compromised mitochondrial integrity in injured TECs can directly regulate inflammation by activating the STING pathway [62]. Specifically, the authors reported cytokines differentially expressed in the metabolic defects of TECs. We noted similar upregulation in fibrotic kidneys and an attenuation of these transcripts in fibrotic kidneys from the EndMT–cKO mice, supporting that the EndMT-induced, hypoxia-mediated metabolic reprogramming of TECs share similar mechanisms [62].
Collectively, our study demonstrated that EndMT impacts kidney fibrosis by disrupting vascular integrity and inducing a detrimental cascade of hypoxia-induced metabolic reprograming in TECs. Our study identifies communication between endothelial and epithelial cells that facilitates kidney fibrosis and could potentially lead to new therapies.
Materials and Methods
Animal studies.
Cdh5Cre, Tie1Cre and Rosa26-LSL-EYFP mice were purchased from Jackson Laboratories. Cdh5CreERT2 mice were kindly provided by Dr. Chenghua Gu (Harvard Medical School, Boston). c-MycLoxP/LoxP mice were kindly provided by Dr. John Sedivy (Brown University, Providence). γGTCre, αSMACre, Twist1LoxP/LoxP and Snai1LoxP/LoxP mice were previously described [24, 26]. Twist1LoxP/LoxP and Snai1LoxP/LoxP mice were bred with the Cdh5CreERT2 mice to generate mice with the tamoxifen inducible, endothelial–specific deletion of Twist or Snai1. Eight week-old male and female mice were intraperitoneally (i.p.) injected with 2 mg of tamoxifen (Sigma, T5648) dissolved in corn oil (Sigma, C8267) for five consecutive days. Seven days after the last injection, mice were subjected to surgery or treatment. Twist1LoxP/LoxP and Snai1LoxP/LoxP mice were also bred with Cdh5Cre and with Tie1Cre mice to generate mice with the constitutive deletion of Twist or Snail in the endothelial cells. 9–10 week old male and female mice were used for the experiments. UUO was performed as previously described [63] and UUO mice were euthanized 3 and 10 days after surgery. Age-matched sham animals underwent the same procedure without ligation of the ureter. Folic acid–induced nephropathy was performed by daily i.p. injection of 150 mg/Kg body weight of folic acid (Sigma, F8758) dissolved in 150 mM sodium bicarbonate and mice were euthanized 3 days after last injection. c-MycLoxP/LoxP mice were bred with γGTCre and αSMACre mice to generate mice with the constitutive deletion of c-Myc in proximal tubular epithelial cells and in aSMA+ myofibroblasts, respectively. 9–12 week-old male and female mice were subjected to UUO and mice were euthanized 10 days after surgery. For 3BP treatment, mice were injected i.p. daily with 0.2 mg/ml 3BP in PBS, or with vehicle (PBS), on day 1 after surgery until euthanasia 7 days after UUO. For JQ1 treatment, mice were injected i.p. daily with 300 μl of 5 mg/ml JQ1 in PBS or with vehicle (PBS), on day 1 after surgery until euthanasia 10 days after UUO.
BUN measurement was performed on mouse serum samples collected at the time of the sacrifice using the QuantiChrom Urea assay kit (Bioassay system, DIUR-100) according to the manufacturer’s instructions. To assess permeability, mice were retro–orbitally injected with 1 mg of FITC–conjugated Dextran (70 kDa, Sigma) 1 minute before euthanasia. Mice whose kidneys were used for flow cytometry analysis were intracardially perfused with PBS right after euthanasia. All mice were on mixed C57/BL6, 129sv and BALB/c genetic background and littermates with similar genetic background were analyzed. All mice were screened for Twist1, Snai1 or Myc recombination PCRs to exclude the presence of a full body recombined allele. All animal procedures were reviewed and approved by the M.D. Anderson Cancer Center Institutional Animal Care and Use Committee. Investigators were not blinded for group allocation, but were blinded for the assessment of the phenotypic outcome assessed by histological analyses. No randomization method was used and no animals were excluded from the analysis.
Human biopsies.
Sections obtained from discarded nephrectomy speciments, de-identified (without any protected health information) were collected at the Beth Israel Deaconess Medical Center (Boston) through an IRB exempt protocol.
Histology and histopathology.
Kidneys were fixed in 10% neutral buffered formalin, embedded in paraffin and 5-μm thick sections were cut. Sections were stained with haematoxylin and eosin (H&E, Leica). Picrosirius red staining for collagen was performed using 0.1% picrosirius red (Direct Red 80, Sigma and Picric acid, Electron Microscopy Science) and counterstained with Weigert’s hematoxylin. MTS was performed using Gomori’s Trichrome Stain Kit (38016SS2, Leica Biosystems). The extent of renal injury was estimated using morphometric assessment of tubular damage and interstitial fibrosis. To evaluate the protection from tubular damage, five 200× visual fields were randomly selected for each UUO kidney and three for each contralateral kidney, and the number of healthy tubules was manually counted using the count tool of Adobe Photoshop. Tubules were defined as healthy when the dimension, structure, relative nucleus–cytoplasm disposition, integrity of the brush border and the basal membrane resembled those of normal tubules from healthy kidneys. For the analysis of the interstitial fibrosis, five 200× visual fields for each UUO kidney and three for each contralateral kidney were also randomly selected for MTS and picrosirius red stained kidney sections and interstitial fibrosis was manually evaluated by a grid intersection analysis using Adobe Photoshop. Representative images were acquired with a Leica DM 1000 LED microscope and an MC120 HD Microscope Camera with Las V4.4 Software (Leica).
Immunohistochemistry and immunofluorescence.
Formalin–fixed, paraffin–embedded kidney sections were deparaffinized and re–hydrated. Antigen retrieval was performed at 98 °C for 15 min in 10 mM citrate buffer pH 6 (microwave). For CAIX, c-Myc and Cpt1a staining, antigen retrieval was performed at 98 °C for 30 min in Tris–EDTA buffer (10 mM Tris base, 1 mM EDTA, 0.05% Tween–20) pH 9. The tissue sections were incubated with 4% cold water fish skin (CWFS) gelatin (Aurion) in TBS for 1 h before the overnight incubation with the primary antibody. The following day tissue sections were first incubated with the appropriate HRP-conjugated secondary antibodies according to the manufacturer’s instructions and then incubated with the selected fluorophore. The following antibodies were used: albumin–HRP (Bethyl, A90–134P, 1:200), αSMA–Cy3 (Sigma, C6198, 1:200), collagen I (AbDSerotec, 131001, 1:500; GBI Po–link Goat Polymer; Perkin–Elmer TSA–Cy3 1:50), CD31 (Bethyl, 00055, 1:200; anti-rabbit AlexaFluor 647), CD31 (Dianova, DIA310, 1:100; GBI Po–link Rat Polymer; Perkin–Elmer TSA–FITC 1:50), CAIX (Novus Biologicals, NB100–417, 1:1000; Life Technologies SuperPicture HRP–Polymer or Biocare rabbit-on-rodent HRP polymer; Perkin–Elmer TSA–Cy3 1:100), Cpt1a (Abcam, ab234111, 1:1000), c-Myc (Sigma, 395R-14, 1:100), HK2 (Cell Signaling, 2867, 1:400), ZO-1 (Abcam, ab221547, 1:500; Biocare rabbit-on-rodent HRP polymer; Perkin–Elmer TSA–Opal570 1:100). c-Myc, Cpt1a, and HK2 immunohistochemistry was processed using rabbit-on-rodent HRP polymer (Biocare) for 30 min and visualized by analyzing DAB positivity and hematoxylin counterstaining. DAPI was used for nuclear staining in IF staining.
Five representative images for each UUO kidney and three for each contralateral kidney were acquired. Immunofluorescence images were acquired with a Demo Axio Observer Z1 motorized inverted microscope with an Axiocam 506 monochrome camera and ZEN software (Zeiss), or with a LSM800 confocal laser scanning microscope and analyzed with ZEN software (Zeiss). Brigh-field images were acquired with Leica DM 1000 LED microscope and an MC120 HD Microscope Camera with Las V4.4 Software (Leica). The percentage of collagen 1 and αSMA positive area per visual field (200×) was quantified with ImageJ software. CAIX, c-Myc and Cpt1a staining were quantified by manually counting the number of positive tubules per visual field (200× or 400×) using the count tool of Adobe Photoshop. Interstitial albumin was manually evaluated by a grid intersection analysis using Adobe Photoshop. Peritubular capillary density was analyzed in the albumin immunohistochemistry images by using the quantification method described in [64].
For the dextran permeability assay, FITC-conjugated dextran was directly visualized on formalin-fixed, paraffin-embedded sections. De-paraffinization, re-hydration and antigen retrieval was performed as described above. Slides were blocked with 5% BSA-PBS for 1 hour, incubated overnight with anti-CD31 (Dianova, DIA310, 1:50) and subsequently with anti-rat AlexaFluor 647 (Invitrogen, 1:250) for 1 hour. DAPI was used for nuclear staining. Five representative images for each UUO kidney and three for each contralateral kidney were acquired with a LSM800 confocal laser scanning microscope and analyzed with ZEN software (Zeiss). A kidney from a mouse not injected with FITC-Dextran was used as a negative control to set the threshold of the autofluorescence background in the FITC channel. Quantification of the FITC-Dextran positive area per visual field (400×) was performed with ImageJ software. In order for the printed images to accurately represent the quality of the high-resolution screens, the color brightness of all immunofluorescence images was modified using Adobe Photoshop and was equally applied across the entire image and equally increased in all the acquired images. All images were acquired at 300 d.p.i. (or higher) resolution.
For the multiplexed EndMT staining, sections were serially stained with CD31 (Dianova, DIA310, 1:100; GBI Po–link Rat Polymer; Perkin–Elmer TSA–Cy5.5 1:50), YFP (Aves Lab, GFP-1020, 1:5000; GBI chicken HRP–Polymer; Perkin–Elmer TSA–Cy5 1:200) and αSMA (Dako, M0851, 1:400; GBI Klear Mouse Polymer; Perkin–Elmer TSA–Cy3 1:1000). Slides were scanned at 200× magnification, using the PerkinElmer Vectra multispectral slide scanning system. 5–8 fields were randomly selected for each mouse. Quantification was performed using PerkinElmer InForm Analysis Software. Pseudocoloring and non-linear adjustment (γ–changes) were applied in order for the printed images to accurately represent the quality of the high-resolution screens.
YFP visualization and immunofluorescence.
Mouse kidneys were fixed in 4% paraformaldehyde overnight at 4 °C and equilibrated in 30% sucrose overnight at 4 °C. Kidneys were then embedded in OCT compound, and 5-μm-thick frozen sections were blocked for 1 h with 5% normal goat serum and immunostained overnight with anti–αSMA–Cy3 (Sigma-Aldrich, C6198, 1:200). Slides were then mounted with Vectashield Mounting Medium with DAPI (Vectashield) and a glass coverslip and visualized using a YFP and RFP fluorescent filter. The numbers of double YFP+ and αSMA+ cells per visual field (400×) were counted in 5 fields of view. For hypoxia analysis, MCT cells adhered to coverslips were fixed with 4% PFA for 15 min at room temperature, permeabilized with 0.1% Triton X-100 for 10 min at room temperature and blocked in 5% goat serum-PBS for 1 h, and incubated with anti-HIF1α (Cayman, 10006421, 1:100) for 1 h. Cells were then incubated with anti-rabbit AlexaFluor 594 (Invitrogen, 1:300) for 1 h and with DAPI for nuclear staining. For CD8 staining, 10 μm-thick frozen sections from freshly OCT-embedded frozen kidneys were fixed for 10 min with ice-cold acetone, blocked for 1 h with 4% CWFG in PBS and immunostained overnight with anti-CD8 (BD Pharmingen, 550281, 1:50). The following day sections were incubated for 1 h with anti-rat AlexaFluor 594 (Invitrogen, 1:250) and then mounted with mounting media with DAPI (Sigma). CD8 staining was quantified by hand counting the number of CD8+ lymphocytes per visual field (400×). Representative pictures were taken at 400× magnification with Demo Axio Observer.Z1 motorized inverted microscope with an Axiocam 506 monochrome camera and ZEN software (Zeiss).
Cell culture and hypoxia treatment.
MCT mouse proximal tubular epithelial cells [51] were cultured in DMEM medium supplemented with 10% heat-inactivated FBS and penicillin/streptomycin (100 μg/ml) at 37°C and 5% CO2. The cells were STR profiled by IDEXX BioAnalytics (Columbia, MO) and were routinely tested and found to be free of mycoplasma. For hypoxia experiments, cells were cultured in 0.1% FBS-DMEM and incubated at 37°C, 5% CO2 and 1% O2 for 12, 24, 48 and 72 hours.
Lactate assay.
Lactate concentrations were determined using a L-Lactate Assay Kit (Eton Bioscience) according to the manufacturer’s protocol. Briefly, 50 μl of medium was mixed with 50 μl of Lactate Assay solution and incubated for 30 min at 37°C. The reaction was then stopped by adding 50 μl of 0.5M acetic acid. Colorimetric measurements were performed using a spectrophotometric multiwell plate reader.
DNA extraction and recombination PCR.
Genomic DNA was extracted from frozen kidneys using the QIAamp DNA Micro Kit (Qiagen) according to the manufacturer’s instructions. 100 ng of the purified DNA was used as template for the Twist, Snail and c-Myc recombination PCRs, using the following primers: Twist 114 5’-CCGGATCTATTTGCATTTTACCATGGGTCATC-3’, Twist 115 5’-CCTCTACCTGACCGTTAGATGGACTCGG-3’; Snail P1 5’-CTGCCAGGTGGGAAGGACT-3’, Snail P7 5’-CTAAGCCCCACAAAGCACCT-3’; c-Myc Del-S 5’- TCGCGCCCCTGAATTGCTAGGAA-3’, c-Myc Del-AS 5’- TGCCCAGATAGGGAGCTGTGATACTT-3’. PCR products were then resolved in a 2% agarose gel containing 0.005% ethidium bromide and imaged using InGenius3 gel imager and Genesys software (Syngene), or alternatively analyzed using QIAxcel capillary electrophoresis (Qiagen).
RNA extraction and quantitative real-time PCR analysis.
MCT cells and one eighth of each contralateral and UUO kidneys was homogenized in TRIzol reagent (Invitrogen) and total RNA was extracted according to the manufacturer’s directions. 2 μg of the extracted RNA were incubated with RQ1 DNase (Promega) at 37°C for 30 min and at 65°C for 10 min for enzyme deactivation. cDNA synthesis was performed using the Applied Biosystems cDNA synthesis kit according to the manufacturer’s directions. Quantitative PCR was performed to analyze the gene expression profiles of the listed genes using SYBR Green PCR master Mix in a QuantStudio 7 Flex Real-time PCR system (Applied Biosystems), and measurements were standardized to the expression of the housekeeping gene Gapdh, or to the expression of 18s or Arp36b4 genes (for metabolism-related genes). The expression data is presented as fold change (2–ΔΔCt). The ΔCt was used to measure statistical significance in the observed changes. Genes and primers sequences are listed in table S1.
RNA-Sequencing of mouse kidney samples.
Total RNA was extracted with TRIzol from 3 mice per each experimental group. RNA concentration was measured by Nanodrop and RNA quality was assessed using the Bioanalyzer (Agilent). Samples with a RIN score between 7.7 and 9.8 were submitted for sequencing. RNAseq libraries were generated using the Illumina TrueSeq stranded Whole Transcriptome RNAseq Library kit and high-output sequencing was carried out on Illumina NextSeq500, in the 2×75bp paired-end sequence reads configuration. Base calling was performed with bcl2fastq-1.8.4, RNA quality control was assessed with RNA-SeQC_v1.1.7.jar and the reference genome mm10 was used for alignment. Quantification of gene reads was performed with HTSEQ count, normalization and differential expression analysis were performed with DESEQ2 (p < 0.01, FDR < 0.05). All analyses were performed under R/3.6.0 environment. Gene Set Enrichment Analysis was performed with GSEA 2–2.2.1. RNAseq data have been deposited in GEO (https://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE144701.
Metabolomics.
Targeted metabolomics was performed on frozen mouse kidney samples to quantitatively measure metabolites associated with glycolysis, TCA and fatty acids. Extraction protocols and methods have been previously described [65–67]. Briefly, to extract these metabolites, 25 mg of kidney tissue was homogenized in 1:4 ice-cold water:methanol mixture containing an equimolar mixture of internal standard compounds. Subsequently, metabolic extraction was performed by using sequential application of ice-cold organic and aqueous solvents (water:methanol:chloroform:water; ratio 1:4:3:1), deproteinization and drying of the extract. The latter was resuspended in injection solvent and analyzed by liquid chromatography–coupled to mass spectrometry (LC-MS, Agilent 6495 triple quadruple mass spectrometer coupled with UPLC).
Flow cytometry.
To characterize immune infiltration, half of each contralateral and UUO kidneys was minced digested in a 5 ml mixture of 0.1 mg/ml Liberase (Roche, 05401020001) and 0.2 mg/ml DNase I (Roche, 10104159001) in RPMI medium. Kidneys were first dissociated using the GentleMACS octo dissociator (Miltenyil Biotech) and then digested at 37 °C for 25 min, shaking at 150 rpm. The tissue lysate was filtered through a 100-nm mesh and 5 ml of 10 mM EDTA pH 8, 10% FBS RPMI was added. Spleen samples were directly mashed through a 100-nm mesh with 5 ml of 10% FBS RPMI. Kidney and spleen samples were washed first with 10% RPMI and then with 2% FBS PBS. The resulting single-cell suspension was divided in half and subjected to the immunostaining for two immune cells panels. Panel 1 included the fixable viability dye eFluor 780 (eBioscience, 65–0865-14, 1:1000 in PBS), anti-CD45.2 Pacific Blue (Bio Legend 103126, 1:100), anti-CD3 phycoerythrin (PE)-Cy7 (eBioscience 25–0031-82, 1:200), anti-CD4 BV605 (Bio Legend 100548, 1:200), anti-CD8 BV650 (Bio Legend 100742, 1:200), anti-CD11b BV711 (BD 563168, 1:400), anti-CD49b PE (eBioscience 12–5971-81, 1:100), anti-FoxP3 A700 (eBio 56–5773-82, 1:50), anti-PD-L1 APC (Bio Legend 124312, 1:100), anti-PD1 PerCP-Cy5.5 (Bio Legend 135208, 1:100), anti-Ki67 A488 (BD 558616, 1:100). Panel 2 included fixable viability dye eFluor 780 (eBioscience, 65–0865-14, 1:1000), anti-CD45.2 Pacific Blue (Bio Legend 103126, 1:100), anti-CD3 A700 (eBioscience 56–0032-82, 1:50), anti-CD11b BV711 (BD 563168, 1:400), anti-Ly6C APC (BD 560595, 1:200), anti-Ly6G PE-Cy7 (BD 560601, 1:200), anti-CD19 BV650 (Bio Legend 115541, 1:100), anti-CD11c eFluor615 (eBioscience 42–0114-82, 1:50), anti-PD-L1 PE (eBioscience 12–5982-83, 1:100), anti-PD1 PerCP-Cy5.5 (Bio Legend 135208, 1:100), anti-Ki67 A488 (BD 558616, 1:100). Surface marker antibodies were diluted in 2% FBS-PBS, 1/5 final volume BV buffer (BD 563794), 50 μg/ml CD16/32 block (TONBO biosciences, 40–0161-M001) and were incubated with samples on ice for 30 min. To stain intracellular markers (Ki67 and FoxP3), cells were fixed and permeabilized with the FoxP3 staining buffer set (eBioscience, 00–5523-00), according to manufacturer’s instructions and subsequently incubated for 30 min on ice with the antibodies diluted in FoxP3 permeabilization buffer. Finally, cells were fixed with 4.2% formaldehyde (BD Cytofix, 554655), washed and resuspended in FACS staining buffer (1% FBS, 0.5 mM EDTA in PBS).
To characterize endothelial markers, half of each contralateral and UUO kidneys was digested as described above. The resulting single cell suspension was subjected to the following panel of antibodies for endothelial markers: anti-CD31 FITC (BD Bioscience, 558738, 1:100), anti-Tie2 PE (eBioscience 12–598782, 1:100) and anti-VEGFR2 (Chemicon, MAB-1669, 1:100; anti-rat AlexaFluor 647, Invitrogen, 1:100). Surface antibodies were diluted in 1% BSA-PBS and intracellular marker staining was performed as described above. Cells were fixed with 4.2% formaldehyde (BD Cytofix, 554655), washed and resuspended in FACS staining buffer (1% FBS, 0.5 mM EDTA in PBS). Samples were analyzed using the LSR Fortessa X-20 cell analyzer and data were analyzed using FlowJo software. Doublets were gated out using forward scatter width/height and side scatter width/height event characteristics.
Statistical analysis.
Statistical analyses were performed by using one-way ANOVA or unpaired two-tailed Student’s t-test with GraphPad Prism (GraphPad Software version 7 and version 8). Where variance was not equal (as determined by an F-test) Welch’s correction for unequal variances was applied. Error bars represent s.e.m. or s.d., as defined in the figure legends. For immunostaining quantification, multiple visual fields were averaged to produce a single value for each animal, which was then averaged again to represent the mean bar for the group in each graph. Statistical significance was defined as P < 0.05.
Supplementary Material
Supplementary Figure 1. Generation and validation of a tamoxifen-inducible mouse model to genetically delete Twist1 or Snai1 in endothelial cells.
Supplementary Figure 2. Evaluation of kidneys of healthy mice with tamoxifen-inducible conditional deletion of Twist1 or Snai1 in endothelial cells.
Supplementary Figure 3. Conditional deletion of Twist1 in endothelial cells reduces UUO-induced interstitial renal fibrosis.
Supplementary Figure 4. Conditional deletion of Snai1 in endothelial cells improves renal fibrosis and tubular health after UUO.
Supplementary Figure 5. Conditional deletion of Snai1 in endothelial cells limits renal interstitial fibrosis after UUO.
Supplementary Figure 6. Sham-operated mice do not display renal disease.
Supplementary Figure 7. Conditional deletion of Twist1 in endothelial cells ameliorates kidney fibrosis 3 days after UUO.
Supplementary Figure 8. Conditional deletion of Snai1 in endothelial cells ameliorates kidney fibrosis 3 days after UUO.
Supplementary Figure 9. Conditional deletion of Snai1 in endothelial cells protects against FA-induced kidney fibrosis.
Supplementary Figure 10. Fibrotic kidneys from mice with endothelial deletion of Twist1 or Snai1 display reduced inflammation.
Supplementary Figure 11. Effect of EndMT inhibition by Twist1 deletion on immune cell composition of fibrotic kidneys 10 days after UUO.
Supplementary Figure 12. Effect of EndMT inhibition by Snai1 deletion on immune cell composition in fibrotic kidneys 10 days after UUO.
Supplementary Figure 13. Inhibition of EndMT by Twist1 deletion in endothelial cells reduces the infiltration of CD8+ T cells in fibrotic kidneys 3 days after UUO.
Supplementary Figure 14. Inhibition of EndMT by Snai1 deletion reduces the infiltration of CD8+ T cells in fibrotic kidneys 3 days after UUO.
Supplementary Figure 15. Generation of Cdh5Cre and Tie1Cre–driven mouse models to genetically delete Twist1 or Snai1 in endothelial cells.
Supplementary Figure 16. Mice with conditional deletion of Twist1 or Snai1 in Cdh5-expressing endothelial cells are healthy.
Supplementary Figure 17. Mice with conditional deletion of Twist1 or Snai1 in Tie1-expressing endothelial cells are healthy.
Supplementary Figure 18. Constitutive Cdh5Cre–driven conditional deletion of Twist1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 19. Constitutive Cdh5Cre–driven conditional deletion of Snai1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 20. Tie1Cre–driven conditional deletion of Twist1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 21. Tie1Cre–driven conditional deletion of Snai1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 22. Validation of Twist1 and Snai1 deletion in Cdh5Cre and Cdh5CreERT2 models.
Supplementary Figure 23. Generation of endothelial-specific lineage traced alleles for analysis of EndMT in kidney fibrosis.
Supplementary Figure 24. Lineage tracing analysis of EndMT in kidney fibrosis.
Supplementary Figure 25. Vascular leakage and hypoxia are not detected in TwistEnd–cKO or SnailEnd–cKO contralateral kidneys.
Supplementary Figure 26. Characterization of hypoxia, Myc, and metabolic markers in WT and TwistEnd–cKO fibrotic kidneys 3 days after UUO.
Supplementary Figure 27. Characterization of hypoxia, Myc, and metabolic markers in WT and SnailEnd–cKO fibrotic kidneys 3 days after UUO.
Supplementary Figure 28. Hypoxia induces a glycolytic metabolic switch in tubular epithelial cells.
Supplementary Figure 29. Pharmacological targeting of glycolysis and Myc improves kidney fibrosis.
Supplementary Figure 30. Generation and validation of a mouse model to genetically delete Myc in proximal tubular epithelial cells.
Supplementary Figure 31. Conditional deletion of Myc in myofibroblasts does not affect kidney fibrosis.
Supplementary Figure 32. Conditional deletion of Myc in myofibroblasts does not improve renal metabolism during fibrosis.
Supplementary Table 1. List of real-time qPCR primers.
Data File S1. Source data.
Acknowledgments
Cdh5CreERT2 mice were kindly provided by Dr. Chenghua Gu (Harvard Medical School, Boston), c-MycLoxP/LoxP mice were kindly provided by Dr. John Sedivy (Brown University, Providence), Snai1LoxP/LoxP mice were kindly provided by Dr. Stephen J. Weiss (University of Michigan) and Twist1LoxP/LoxP mice were kindly provided by Dr. Richard R. Behringer (UT MDACC) via the Mutant Mouse Regional Resource Center (MMRRC) repository. JQ1 was a kind gift from Dr. James Bradner (Novartis). RNA sequencing was performed by the Sequencing and ncRNA Core Facility at MD Anderson Cancer Center. We thank C.G. Liu for performing RNAseq. Targeted metabolomics was performed by the Metabolomics Core Facility at Baylor College of Medicine (Houston, TX). We wish to thank J.L. Carstens for help with TSA staining, E. Ramirez for help with flow cytometry and immunotyping, S. Yang for designing Snail recombination PCR, O. Volpert, and J. Kim for advising on ZO-1 immunofluorescence, F. Grande Kugeratski for advising on HIF1α immunofluorescence, L. Gibson and R. Deliz-Aguirre for help with breeding and genotyping mice, and K. Vadnagara for technical help.
Funding
This work was primarily supported with funds from the University of Texas MD Anderson Cancer Center (UT MDACC) and the Beth Israel Deaconess Medical Center to R.K. The Metabolomics Core at Baylor College of Medicine is supported with funds from NIH (P30 CA125123), CPRIT Proteomics and Metabolomics Core Facility (RP170005), Dan L. Duncan Cancer Center and partially supported by the NIH/NCI R01CA220297 and NIH/NCI R01CA216426 to N.P.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Data and materials availability
RNAseq data have been deposited in GEO (https://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE144701. All other data needed to evaluate the conclusions in the paper are present in the paper or in the Supplementary Materials. Source data pertaining to all main and supplementary figures are available in the Data File S1.
References and Notes
- 1.Dejana E, Hirschi KK, and Simons M, The molecular basis of endothelial cell plasticity. Nat Commun, 2017. 8: p. 14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg LM and Markwald RR, Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res, 1995. 77(1): p. 1–6. [DOI] [PubMed] [Google Scholar]
- 3.Zeisberg EM, et al. , Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med, 2007. 13(8): p. 952–61. [DOI] [PubMed] [Google Scholar]
- 4.Zeisberg EM, et al. , Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol, 2008. 19(12): p. 2282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeisberg EM, et al. , Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res, 2007. 67(21): p. 10123–8. [DOI] [PubMed] [Google Scholar]
- 6.Chen PY, et al. , Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest, 2015. 125(12): p. 4514–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evrard SM, et al. , Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun, 2016. 7: p. 11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piera-Velazquez S and Jimenez SA, Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol Rev, 2019. 99(2): p. 1281–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Qu X, and Bertram JF, Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol, 2009. 175(4): p. 1380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basile DP, et al. , Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol, 2011. 300(3): p. F721–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curci C, et al. , Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol Dial Transplant, 2014. 29(4): p. 799–808. [DOI] [PubMed] [Google Scholar]
- 12.Li L, et al. , C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/beta-catenin signaling pathway in diabetic kidney disease. Metabolism, 2015. 64(5): p. 597–610. [DOI] [PubMed] [Google Scholar]
- 13.Peng H, et al. , ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci Rep, 2016. 6: p. 20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, et al. , Role of endothelial-to-mesenchymal transition induced by TGF-beta1 in transplant kidney interstitial fibrosis. J Cell Mol Med, 2017. 21(10): p. 2359–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng Y, et al. , Blocking protein phosphatase 2A signaling prevents endothelial-to-mesenchymal transition and renal fibrosis: a peptide-based drug therapy. Sci Rep, 2016. 6: p. 19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu-Dubois YC, et al. , Markers of Endothelial-to-Mesenchymal Transition: Evidence for Antibody-Endothelium Interaction during Antibody-Mediated Rejection in Kidney Recipients. J Am Soc Nephrol, 2016. 27(1): p. 324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokudo T, et al. , Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci, 2008. 121(Pt 20): p. 3317–24. [DOI] [PubMed] [Google Scholar]
- 18.Medici D, Potenta S, and Kalluri R, Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J, 2011. 437(3): p. 515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahmoud MM, et al. , Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep, 2017. 7(1): p. 3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun JX, et al. , SNAI1, an endothelial-mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis, 2018. 21(3): p. 635–652. [DOI] [PubMed] [Google Scholar]
- 21.Zhang B, et al. , Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int J Mol Med, 2018. 42(1): p. 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, et al. , Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes, 2010. 59(10): p. 2612–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xavier S, et al. , Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol, 2015. 26(4): p. 817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovisa S, et al. , Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med, 2015. 21(9): p. 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grande MT, et al. , Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med, 2015. 21(9): p. 989–97. [DOI] [PubMed] [Google Scholar]
- 26.LeBleu VS, et al. , Origin and function of myofibroblasts in kidney fibrosis. Nat Med, 2013. 19(8): p. 1047–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins DF, et al. , Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest, 2007. 117(12): p. 3810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimura K, et al. , Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol, 2008. 295(4): p. F1023–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballermann BJ and Obeidat M, Tipping the balance from angiogenesis to fibrosis in CKD. Kidney Int Suppl (2011), 2014. 4(1): p. 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi YJ, et al. , Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: Altered expression of vascular endothelial growth factor. Human Pathology, 2000. 31(12): p. 1491–1497. [DOI] [PubMed] [Google Scholar]
- 31.Lan R, et al. , Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol, 2016. 27(11): p. 3356–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabelink TJ and Carmeliet P, Renal metabolism in 2017: Glycolytic adaptation and progression of kidney disease. Nat Rev Nephrol, 2018. 14(2): p. 75–76. [DOI] [PubMed] [Google Scholar]
- 33.Li M, et al. , Elevated aerobic glycolysis in renal tubular epithelial cells influences the proliferation and differentiation of podocytes and promotes renal interstitial fibrosis. Eur Rev Med Pharmacol Sci, 2018. 22(16): p. 5082–5090. [DOI] [PubMed] [Google Scholar]
- 34.Rowe I, et al. , Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med, 2013. 19(4): p. 488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen YT, et al. , Generation of a Twist1 conditional null allele in the mouse. Genesis, 2007. 45(9): p. 588–92. [DOI] [PubMed] [Google Scholar]
- 36.Wynn TA, Cellular and molecular mechanisms of fibrosis. J Pathol, 2008. 214(2): p. 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang PM, Nikolic-Paterson DJ, and Lan HY, Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol, 2019. 15(3): p. 144–158. [DOI] [PubMed] [Google Scholar]
- 38.Turner JE, et al. , Tissue-Resident Lymphocytes in the Kidney. J Am Soc Nephrol, 2018. 29(2): p. 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu ZQ, et al. , A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat Commun, 2014. 5: p. 3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mammoto T, et al. , Twist1 controls lung vascular permeability and endotoxin-induced pulmonary edema by altering Tie2 expression. PLoS One, 2013. 8(9): p. e73407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, et al. , Endothelial TWIST1 promotes pathological ocular angiogenesis. Invest Ophthalmol Vis Sci, 2014. 55(12): p. 8267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mammoto T, et al. , Role of Twist1 Phosphorylation in Angiogenesis and Pulmonary Fibrosis. Am J Respir Cell Mol Biol, 2016. 55(5): p. 633–644. [DOI] [PubMed] [Google Scholar]
- 43.Mahmoud MM, et al. , TWIST1 Integrates Endothelial Responses to Flow in Vascular Dysfunction and Atherosclerosis. Circ Res, 2016. 119(3): p. 450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mammoto T, et al. , Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-beta-Smad Signaling. Am J Respir Cell Mol Biol, 2018. 58(2): p. 194–207. [DOI] [PubMed] [Google Scholar]
- 45.Cooley BC, et al. , TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med, 2014. 6(227): p. 227ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel J, et al. , Accelerated Endothelial to Mesenchymal Transition Increased Fibrosis via Deleting Notch Signaling in Wound Vasculature. J Invest Dermatol, 2018. 138(5): p. 1166–1175. [DOI] [PubMed] [Google Scholar]
- 47.Guerrot D, et al. , Progression of renal fibrosis: the underestimated role of endothelial alterations. Fibrogenesis Tissue Repair, 2012. 5(Suppl 1): p. S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haase VH, Mechanisms of hypoxia responses in renal tissue. J Am Soc Nephrol, 2013. 24(4): p. 537–41. [DOI] [PubMed] [Google Scholar]
- 49.Wheaton WW and Chandel NS, Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol, 2011. 300(3): p. C385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang HM, et al. , Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med, 2015. 21(1): p. 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haverty TP, et al. , Characterization of a Renal Tubular Epithelial-Cell Line Which Secretes the Autologous Target Antigen of Autoimmune Experimental Interstitial Nephritis. Journal of Cell Biology, 1988. 107(4): p. 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lemos DR, et al. , Interleukin-1beta Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J Am Soc Nephrol, 2018. 29(6): p. 1690–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Alboran IM, et al. , Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity, 2001. 14(1): p. 45–55. [DOI] [PubMed] [Google Scholar]
- 54.Shen Y, et al. , c-Myc promotes renal fibrosis by inducing integrin alphav-mediated transforming growth factor-beta signaling. Kidney Int, 2017. 92(4): p. 888–899. [DOI] [PubMed] [Google Scholar]
- 55.Hashimoto N, et al. , Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol, 2010. 43(2): p. 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dufton NP, et al. , Dynamic regulation of canonical TGFbeta signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat Commun, 2017. 8(1): p. 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rieder F, et al. , Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol, 2011. 179(5): p. 2660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Medici D, et al. , Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med, 2010. 16(12): p. 1400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maddaluno L, et al. , EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature, 2013. 498(7455): p. 492–6. [DOI] [PubMed] [Google Scholar]
- 60.Leonard MO, et al. , The role of HIF-1 alpha in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J Biol Chem, 2003. 278(41): p. 40296–304. [DOI] [PubMed] [Google Scholar]
- 61.Goetzman ES and Prochownik EV, The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front Endocrinol (Lausanne), 2018. 9: p. 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung KW, et al. , Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab, 2019. 30(4): p. 784–799 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugimoto H, et al. , Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med, 2012. 18(3): p. 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Babickova J, et al. , Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int, 2017. 91(1): p. 70–85. [DOI] [PubMed] [Google Scholar]
- 65.Zhang L, et al. , MNX1 Is Oncogenically Upregulated in African-American Prostate Cancer. Cancer Res, 2016. 76(21): p. 6290–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wangler MF, et al. , Peroxisomal biogenesis is genetically and biochemically linked to carbohydrate metabolism in Drosophila and mouse. PLoS Genet, 2017. 13(6): p. e1006825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vantaku V, et al. , Multi-omics Integration Analysis Robustly Predicts High-Grade Patient Survival and Identifies CPT1B Effect on Fatty Acid Metabolism in Bladder Cancer. Clin Cancer Res, 2019. 25(12): p. 3689–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Generation and validation of a tamoxifen-inducible mouse model to genetically delete Twist1 or Snai1 in endothelial cells.
Supplementary Figure 2. Evaluation of kidneys of healthy mice with tamoxifen-inducible conditional deletion of Twist1 or Snai1 in endothelial cells.
Supplementary Figure 3. Conditional deletion of Twist1 in endothelial cells reduces UUO-induced interstitial renal fibrosis.
Supplementary Figure 4. Conditional deletion of Snai1 in endothelial cells improves renal fibrosis and tubular health after UUO.
Supplementary Figure 5. Conditional deletion of Snai1 in endothelial cells limits renal interstitial fibrosis after UUO.
Supplementary Figure 6. Sham-operated mice do not display renal disease.
Supplementary Figure 7. Conditional deletion of Twist1 in endothelial cells ameliorates kidney fibrosis 3 days after UUO.
Supplementary Figure 8. Conditional deletion of Snai1 in endothelial cells ameliorates kidney fibrosis 3 days after UUO.
Supplementary Figure 9. Conditional deletion of Snai1 in endothelial cells protects against FA-induced kidney fibrosis.
Supplementary Figure 10. Fibrotic kidneys from mice with endothelial deletion of Twist1 or Snai1 display reduced inflammation.
Supplementary Figure 11. Effect of EndMT inhibition by Twist1 deletion on immune cell composition of fibrotic kidneys 10 days after UUO.
Supplementary Figure 12. Effect of EndMT inhibition by Snai1 deletion on immune cell composition in fibrotic kidneys 10 days after UUO.
Supplementary Figure 13. Inhibition of EndMT by Twist1 deletion in endothelial cells reduces the infiltration of CD8+ T cells in fibrotic kidneys 3 days after UUO.
Supplementary Figure 14. Inhibition of EndMT by Snai1 deletion reduces the infiltration of CD8+ T cells in fibrotic kidneys 3 days after UUO.
Supplementary Figure 15. Generation of Cdh5Cre and Tie1Cre–driven mouse models to genetically delete Twist1 or Snai1 in endothelial cells.
Supplementary Figure 16. Mice with conditional deletion of Twist1 or Snai1 in Cdh5-expressing endothelial cells are healthy.
Supplementary Figure 17. Mice with conditional deletion of Twist1 or Snai1 in Tie1-expressing endothelial cells are healthy.
Supplementary Figure 18. Constitutive Cdh5Cre–driven conditional deletion of Twist1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 19. Constitutive Cdh5Cre–driven conditional deletion of Snai1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 20. Tie1Cre–driven conditional deletion of Twist1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 21. Tie1Cre–driven conditional deletion of Snai1 reduces interstitial fibrosis and improves tubular health.
Supplementary Figure 22. Validation of Twist1 and Snai1 deletion in Cdh5Cre and Cdh5CreERT2 models.
Supplementary Figure 23. Generation of endothelial-specific lineage traced alleles for analysis of EndMT in kidney fibrosis.
Supplementary Figure 24. Lineage tracing analysis of EndMT in kidney fibrosis.
Supplementary Figure 25. Vascular leakage and hypoxia are not detected in TwistEnd–cKO or SnailEnd–cKO contralateral kidneys.
Supplementary Figure 26. Characterization of hypoxia, Myc, and metabolic markers in WT and TwistEnd–cKO fibrotic kidneys 3 days after UUO.
Supplementary Figure 27. Characterization of hypoxia, Myc, and metabolic markers in WT and SnailEnd–cKO fibrotic kidneys 3 days after UUO.
Supplementary Figure 28. Hypoxia induces a glycolytic metabolic switch in tubular epithelial cells.
Supplementary Figure 29. Pharmacological targeting of glycolysis and Myc improves kidney fibrosis.
Supplementary Figure 30. Generation and validation of a mouse model to genetically delete Myc in proximal tubular epithelial cells.
Supplementary Figure 31. Conditional deletion of Myc in myofibroblasts does not affect kidney fibrosis.
Supplementary Figure 32. Conditional deletion of Myc in myofibroblasts does not improve renal metabolism during fibrosis.
Supplementary Table 1. List of real-time qPCR primers.
Data File S1. Source data.